
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 15 May 2018
Sec. Multiple Sclerosis and Neuroimmunology
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00340
 Sara Gil-Perotin1,2*
Sara Gil-Perotin1,2* Jéssica Castillo-Villalba1
Jéssica Castillo-Villalba1 Joan Carreres-Polo3Arantxa Navarré-Gimeno4Javier Mallada-Frechín5
Joan Carreres-Polo3Arantxa Navarré-Gimeno4Javier Mallada-Frechín5 Francisco Pérez-Miralles1,2Francisco Gascón4Carmen Alcalá-Vicente1,2
Francisco Pérez-Miralles1,2Francisco Gascón4Carmen Alcalá-Vicente1,2 Laura Cubas-Nuñez1
Laura Cubas-Nuñez1 Bonaventura Casanova-Estruch1,2
Bonaventura Casanova-Estruch1,2
The clinical diagnosis of patients with autoantibodies directed to conformational myelin oligodendrocyte glycoprotein MOG-IgG, can be challenging because of atypical clinical presentation. MOG-IgG seropositivity has been reported in several demyelinating diseases, including relapsing opticospinal syndromes [in the neuromyelitis optica spectrum disorders (NMOSD) and less frequently, in multiple sclerosis (MS)], but it has rarely been associated with the progressive course of disease. To contribute to the characterization of MOG-related demyelination, we describe the case of a patient with progressive demyelinating opticospinal disease, IgG-oligoclonal bands (OCB), and serum MOG-IgG.
The clinical diagnosis of patients with autoantibodies directed to conformational myelin oligodendrocyte glycoprotein MOG-IgG, can be challenging because of atypical clinical presentation (1–3). MOG-IgG seropositivity has been reported in several demyelinating diseases, including relapsing opticospinal syndromes [in the neuromyelitis optica spectrum disorders (NMOSD) and less frequently, in multiple sclerosis (MS)], but it has rarely been associated with the progressive course of disease (4). To contribute to the characterization of MOG-related demyelination, we describe the case of a patient with progressive demyelinating opticospinal disease, IgG-oligoclonal bands (OCB), and serum MOG-IgG.
A white man of western European ancestry presented in 1997 with loss of visual acuity at age 26 although he did not seek medical assistance. He recovered ad integrum. Three years later, he experienced an episode of bilateral optic neuritis (ON). Symptoms went into complete remission after intravenous methylprednisolone. Since then, there was constant deterioration of gait without relapses despite intensive physical therapy and a diagnosis of a progressive phase of MS was made in 2003. We first attended the patient in 2010. He had received interferon and he was taking glatiramer acetate at that time. The gait problem persisted (he required two walking aids to walk more than 50 m) and was associated with mild dysphagia. Neurological examination showed dysarthria, central nystagmus, moderate dysmetria, symmetrical paraparesis with spasticity, and left facial and body hypoesthesia. He had preserved urine and bowel control. An expanded disability status scale (EDSS) score of 6.0 was calculated. Specific tests to exclude other causes were performed in our hospital despite the previous diagnosis of MS. An analysis of OCB in the cerebrospinal fluid (CSF) found IgG-OCB, but not IgM-OCB. Serum AQP4-IgG were not detected in serial samples. Brain and spinal cord MRI (FLAIR, TSE T2 sequence, PD, and T1 post gadolinium) was performed (Table 1; Figure 1). Brain MRI showed cortical, juxtacortical, and multiple periventricular lesions at the level of the lateral ventricles (Dawson’s fingers) with predominant infratentorial lesions (thalami, pons, and cerebellum). In the spinal cord, two nodular lesions (<2 vertebral segments) were observed at the level of C2–C3. There were no gadolinium-enhanced lesions (GEL). The patient fulfilled Barkhoff’s criteria, presenting with IgG-OCB, but not AQP4-IgG, and MS was considered the most likely diagnosis. Treatment with mitoxantrone [5 doses of 12 mg/m2 (20 mg)] had little effect on progression. In 2013, compassionate use of rituximab was initiated, although EDSS was 6.5 because of impairment of ataxia. Posterior MRI (in 2016) showed new lesions in the brain, cerebellum, brainstem, and spinal cord (in C4–C5, C5–C6, and C7) with cortical brain atrophy. T2-hyperintensity could be observed in the left corticospinal tract at the pons level as a sign of Wallerian degeneration. Despite signs of progressive demyelination, GELs were not detected.

Table 1. Number of lesions in T2-weighted MRI.
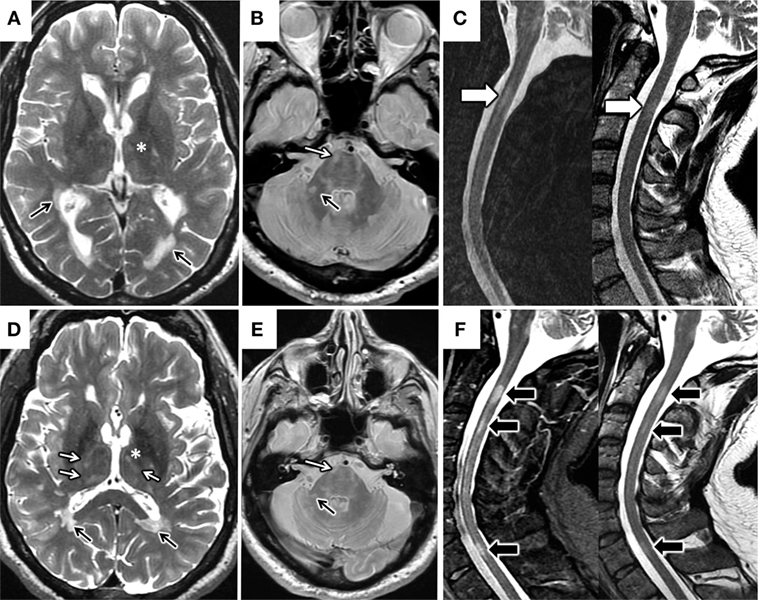
Figure 1. Changes in brain and spinal cord MRI. MRI 2010 (A–C). Axial T2 FSE MRI image of the brain (A) demonstrates multiple periventricular lesions in the lateral ventricles (black arrows) and scarce thalamic lesions (*). Axial proton-density-weighted MRI image (B) shows predominant infratentorial lesions (white arrow in pons and black arrow in right middle cerebellar peduncle). Sagittal STIR and T2 FSE MRI images of the spinal cord (C) highlight the presence of a short segment spinal cord lesion (white arrow in C2). MRI 2016 (D–F). Axial T2 FSE MRI image (D) shows increased in the number of thalamic lesions (* and white arrows) and stability of the periventricular lesions (black arrows). Axial proton-density-weighted MRI image (E) shows stability of the infratentorial lesions respect to 2010 (arrows). Sagittal STIR and T2 FSE MRI images of cervical spinal cord (F) demonstrate new short segment lesions affecting C3 and C7 levels respect (black arrows).
At last visit, in 2017, his EDSS was 7.5, he was not capable of walking, had lost urine control, and had severe impairment of swallowing. Retrospectively, the analysis of MOG-IgG1 in the serum obtained in 2010 with a validated cell-based assay in our laboratory gave a positive result (titration 1:320) that was confirmed by an external center. We did not test CSF because the threshold of sensitivity of detection at titers under 1:640 (5). A diagram of the clinical course and administered treatments is displayed as Figure 2.
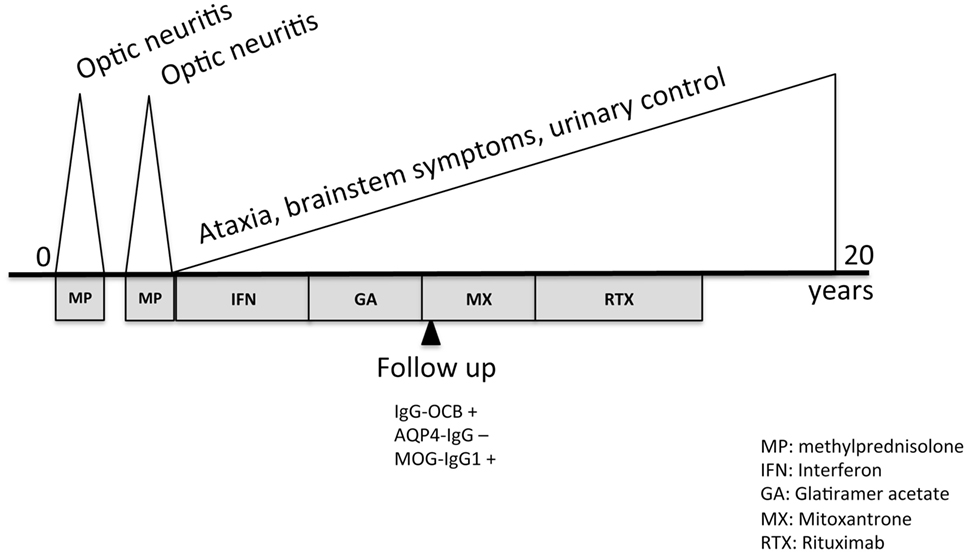
Figure 2. Timeline of clinical course and treatment.
The identification of autoantibodies directed to conformational MOG (6, 7) in several demyelinating diseases (8) has encouraged clinicians to link their presence to patients with a characteristic disease pathogenesis or clinical course (9–12). MOG is expressed in the myelin sheath and the oligodendrocyte membrane and antibodies against MOG have been detected in the sera and CSF of patients with acute disseminated encephalomyelitis, NMOSD, relapsing isolated ON, and, more rarely, in MS. Technical limitations to detect the autoantibodies and borderline manifestations in seropositive patients with opticospinal disease has so far prevented the detailed characterization of MOG pathologies (3). However, recent multicentre studies with large numbers of MOG-IgG positive patients, have added more information regarding prognosis and disease course (13–15). The present case report describes a MOG-IgG-positive patient with progressive neurological deterioration after two episodes of ON. He was diagnosed as having progressive MS with brainstem predominance and myelitis, which evolved poorly despite aggressive immunosuppressive therapies. MOG-IgG-related entities are considered part of the NMOSD (16–19) and progressive deterioration has been described rarely (4, 13). Because of its atypical presentation, in this case it is interesting to analyze whether the assessed clinical progression was the result of ongoing deterioration secondary to inflammation, to a decrease in the reserve capacity of the brain upon formation of large pyramidal lesions (20), or both, or a deconditioning phenomenon. On the one hand, cumulative cortical atrophy was evident and new lesions in the spinal cord during MRI follow-up, without relapses, implied persistent disease activity. Moreover, the positivity for MOG-IgG in 2010, 13 years after the disease onset (in the absence of clinical attacks) supported the presence of active disease (1, 5, 21). On the other hand, GELs were not described in the MRI as hyperintensities in T2-weighted images in any of the studies, suggesting that chronic inflammation was not related to blood–brain barrier disruption. Left corticospinal tract degeneration in the pons could also have played a role in the progression of his disability, expressed as increasing spasticity, and loss of dexterity. However, there was no brainstem or myelitis clinical attack identified in the clinical course that could have caused an acute pyramidal lesion to blame. Instead, gait worsened progressively. Deconditioning, as an expression of deterioration because of lack of physical activity has been described in MS as related to chronic fatigue (22). In the present case, deconditioning was not a likely to be an influencing factor because of the intensive in-hospital physical therapy program and years of private physical therapy.
MRI findings with brainstem involvement shared some features with NMOSD and other cases of MOG-IgG (23). Our patient, in addition, presented with multiple brain lesions (periventricular, cortical, and juxtacortical) and short segment spinal cord lesions, more characteristic of MS. A high percentage of MOG-IgG-positive patients fulfilled criteria for both MS and NMOSD (13), but considering the latest McDonald criteria (24), that include IgG-OCB as a new criterion for dissemination in time, our patient fits better into the definition of MS. Supporting this, biopsy specimens from patients with MOG-IgG displayed features of pattern II MS with myelin injury (25–27) instead of the astrocytopathy that characterizes NMO. Nevertheless, it has yet to be elucidated whether there is enough data to define a separate entity distinct from MS or NMOSD. MOG-IgG positivity was initially shown to imply a benign prognosis (28). However, recent reports are consistent with more relapsing behavior and absence of response to certain therapies (13), mostly when MOG-IgG persist (1). In the case described in this report, corticoids served to recover visual acuity after ON, but afterward first-line therapies did not halt the clinical course and had to be switched to more aggressive treatments, including anti-CD20 therapies, with no effect on the progression of disability. We do not know whether anti-CD20 therapies had been used earlier, the disease course could have been slowed. Altogether, the present case emphasizes the need for MOG-IgG detection in atypical cases of MS (progressive or not) to design the therapeutic strategy and further investigation necessary to clarify the pathogenesis of MOG pathologies.
Data available from the authors upon request.
Written informed consent was obtained from the participant for participate in our study and for the publication of this case report in accordance with the Declaration of Helsinki and all research was conducted following legal and ethical requirements at the Research Institute of the Hospital La Fe and was approved by its Institutional Review Board.
BC-E and SG-P wrote the manuscript. SG-P, JC-V, and LC-N set up and performed cell-based assay. AN-G, JM-F, CA-V, FP-M, FG, and JC-P assisted the patient in the study. All authors made a critical revision of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank A. Saiz and E. Meinl for kindly helping in validating our MOG detection technique.
This work was funded by Health Institute Carlos III PI13/00663 (BC-E) and Rio Hortega CM12/00014 (SG-P), and predoctoral fellowship from ONCE fundation (LC-N).
1. Hyun J-W, Woodhall MR, Kim S-H, Jeong IH, Kong B, Kim G, et al. Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J Neurol Neurosurg Psychiatry (2017) 88:811–7. doi:10.1136/jnnp-2017-315998
2. Juryńczyk M, Weinshenker B, Akman-Demir G, Asgari N, Barnes D, Boggild M, et al. Status of diagnostic approaches to AQP4-IgG seronegative NMO and NMO/MS overlap syndromes. J Neurol (2016) 263:140–9. doi:10.1007/s00415-015-7952-8
3. Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm (2015) 2:e62. doi:10.1212/NXI.0000000000000062
4. Jarius S, Ruprecht K, Stellmann JP, Huss A, Ayzenberg I, Willing A, et al. MOG-IgG in primary and secondary chronic progressive multiple sclerosis: a multicenter study of 200 patients and review of the literature. J Neuroinflammation (2018) 15:88. doi:10.1186/s12974-018-1108-6
5. Di Pauli F, Mader S, Rostásy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol (2011) 138:247–54. doi:10.1016/j.clim.2010.11.013
6. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med (1999) 5:170–5. doi:10.1038/5532
7. O’Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med (2007) 13:211–7. doi:10.1038/nm1488
8. Reindl M, Di Pauli F, Rostásy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol (2013) 9:455–61. doi:10.1038/nrneurol.2013.118
9. Kim S-M, Woodhall MR, Kim J-S, Kim S-J, Park KS, Vincent A, et al. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm (2015) 2:e163. doi:10.1212/NXI.0000000000000163
10. Pröbstel A-K, Rudolf G, Dornmair K, Collongues N, Chanson J-B, Sanderson NSR, et al. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflammation (2015) 12:46. doi:10.1186/s12974-015-0256-1
11. Ramanathan S, Reddel SW, Henderson A, Parratt JDE, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm (2014) 1:e40. doi:10.1212/NXI.0000000000000040
12. Ramanathan S, Dale RC, Brilot F. Anti-MOG antibody: the history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev (2016) 15:307–24. doi:10.1016/j.autrev.2015.12.004
13. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation (2016) 13:280. doi:10.1186/s12974-016-0719-z
14. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry (2018) 89:127–37. doi:10.1136/jnnp-2017-316880
15. Reindl M. Clinical course of MOG antibody-associated recurrent demyelinating diseases. J Neurol Neurosurg Psychiatry (2018) 89:118. doi:10.1136/jnnp-2017-317249
16. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology (2012) 79:1273–7. doi:10.1212/WNL.0b013e31826aac4e
17. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol (2014) 71:276–83. doi:10.1001/jamaneurol.2013.5857
18. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology (2014) 82:474–81. doi:10.1212/WNL.0000000000000101
19. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology (2015) 85(2):177–89. doi:10.1212/WNL.0000000000001729
20. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain (2009) 132:1175–89. doi:10.1093/brain/awp070
21. Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, et al. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: a comparative study. Brain (1999) 122(Pt 11):2047–56. doi:10.1093/brain/122.11.2047
22. MacAllister WS, Krupp LB. Multiple sclerosis-related fatigue. Phys Med Rehabil Clin N Am (2005) 16:483–502. doi:10.1016/j.pmr.2005.01.014
23. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson APD, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler (2016) 22:470–82. doi:10.1177/1352458515593406
24. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol (2018) 17:162–73. doi:10.1016/S1474-4422(17)30470-2
25. Jarius S, König FB, Metz I, Ruprecht K, Paul F, Brück W, et al. Pattern II and pattern III MS are entities distinct from pattern I MS: evidence from cerebrospinal fluid analysis. J Neuroinflammation (2017) 14:171. doi:10.1186/s12974-017-0929-z
26. Kaneko K, Sato DK, Nakashima I, Nishiyama S, Tanaka S, Marignier R, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry (2016) 87:1257–9. doi:10.1136/jnnp-2015-312676
27. Spadaro M, Gerdes LA, Mayer MC, Ertl-Wagner B, Laurent S, Krumbholz M, et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol (2015) 2:295–301. doi:10.1002/acn3.164
Keywords: recurrent inflammatory optic neuropathy, NMO, multiple sclerosis, progression, spinal cord, cell-based assay, myelin oligodendrocyte glycoprotein
Citation: Gil-Perotin S, Castillo-Villalba J, Carreres-Polo J, Navarré-Gimeno A, Mallada-Frechín J, Pérez-Miralles F, Gascón F, Alcalá-Vicente C, Cubas-Nuñez L and Casanova-Estruch B (2018) Progressive Demyelination in the Presence of Serum Myelin Oligodendrocyte Glycoprotein-IgG: A Case Report. Front. Neurol. 9:340. doi: 10.3389/fneur.2018.00340
Received: 14 March 2018; Accepted: 30 April 2018;
Published: 15 May 2018
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Anne-Katrin Pröbstel, University of California, San Francisco, United StatesCopyright: © 2018 Gil-Perotin, Castillo-Villalba, Carreres-Polo, Navarré-Gimeno, Mallada-Frechín, Pérez-Miralles, Gascón, Alcalá-Vicente, Cubas-Nuñez and Casanova-Estruch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Gil-Perotin, c2FyYS5nYXJjaWFAdXYuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.