 Jorge C. Kattah
Jorge C. Kattah
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 01 March 2018
Sec. Neuro-Otology
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00046
This article is part of the Research Topic Bilateral Vestibulopathy - Current Knowledge and Future Directions to Improve its Diagnosis and Treatment View all 20 articles
Background: Previous series of bilateral vestibular loss (BVL) identified numerous etiologies, but surprisingly, a cause in a significant number of cases remains unknown. In an effort to understand possible etiology and management strategies, a global effort is currently in progress. Here, I contribute my 10-year experience with both acute and chronic BVL during the 2007–2017 decade.
Methods: This is a retrospective review of the charts and EMR of patients diagnosed with BVL in the last 10 years. Following Institutional IRB approval, we identified 57 patients with a diagnosis of BVL and utilized the current diagnostic criteria listed by the Barany society (1). The inclusion criteria included patients with BVL of any cause, within an age span older than 18 and a neuro-otologic examination supporting the clinical impression of BVL.
Results: During the current decade 2007–2017, I identified two broad categories of BVL (acute and chronic) in 57 patients; only 41 of them had records available. The etiology includes: idiopathic: n = 9, Wernicke’s encephalopathy n = 11, superficial siderosis n = 3, paraneoplastic syndrome: n = 3, bilateral vestibular neuritis (recurrent AVS lasting days without cochlear symptoms) n = 3, simultaneous ototoxicity of aminoglycoside and chemotherapy toxicity n = 2, MELAS n = 2, Meniere’s disease treated with intra-tympanic streptomycin in one ear n = 1, acute phenytoin intoxication: n = 1, combined chronic unilateral tumor-related vestibulopathy and new contralateral vestibular neuritis (this patient presented with Betcherew’s phenomenon) n = 1, bilateral AICA stroke n = 1, mixed spinocerebellar ataxia type 3, n = 2 and CANVAS n = 2.
Conclusion: This cohort included a 28% overall incidence of acute and subacute BVL; among them, 65% improved with intervention. In the thiamine deficiency group, specifically, the vestibular function improved in 80% of the patients. Even though acute, subacute, or chronic showed slightly asymmetric horizontal-VOR gain loss, it never did cause spontaneous, primary straight gaze horizontal nystagmus. n = 39/41 patients had abnormal manual HIT, n = 26/41 BVL patients tested with video head impulse immediately after manual testing showed decreased VOR gain, including two with covert saccades. Two thiamine patients with positive bedside pretreatment manual HIT, tested after treatment with high-dose thiamine showed improved VOR. In acute thiamine deficiency, the horizontal VOR was abnormal and the vertical was either normal or mildly decreased. This series favored a neurologic cause of BVL. Finally, 20% of the chronic cases were idiopathic.
Bilateral vestibular loss is uncommon with an estimated incidence of 28/100,000 (1); however, despite advances in early diagnosis, and significant progress in vestibular testing, and imaging, parallel to molecular screening for genetic and immune BVL phenotypes, a significant number of patients remain idiopathic (2–4). One important and potentially preventable cause of BVL is exposure to ototoxic drugs with typical subacute onset of symptoms. Awareness of potential ototoxicity from antibiotics and close monitoring of patients with infections without other viable treatment led to preventive clinical measures and the use of serum levels to identify an early threshold of ototoxicity (5). In addition, I identified in these cohort patients with acute BVL due to thiamine deficiency and other less common causes. This group displayed overlapping symptoms and signs with the BVL but had acute onset, affected a specific group individuals at risk of nutritional deficiency and had additional clinical findings; timely recognition of this syndrome may lead to recovery. Although awareness of these higher risk patients is greater than 40 years (6–8). In this retrospective study, we analyze their clinical characteristics as a group (9–11). Here, I report my experience with 41 acute and subacute/chronic BVL patients studied during the past decade.
The University of Illinois College of Medicine IRB approved this study. We report the result of a retrospective chart review of patients with BVL diagnosed between years 2007 and 2017, 57 patients had a final diagnosis of BVL at our Center. Here, I classified this cohort according to the duration of the symptoms, the first group comprised either acute (less than 24-h), or subacute (less than 1 month) duration; and a second group with long-standing symptoms (greater than 1 month), I utilized the recently published diagnostic criteria from the Barany Society (12); only 41 of them had records available. Inclusion criteria included comparatively better static than dynamic visual acuity (VA) (more than 3-line deterioration), truncal imbalance, positive manual head impulse test in horizontal canal planes, and in some cases, positive vertical canal compromise, in these cases, we confirmed decreased VOR gain (below: 0.7). Bilaterally reduced caloric response with maximum slow phase velocity of <5°/s and torsional chair with decreased gain (<0.6), phase lead greater than 68° and bilaterally low time constants of the step rotational induced nystagmus (<5 s). All patients underwent a neurologic and vestibular clinical evaluation that included a mental status assessment to assess orientation and determine level of awareness and ability to follow the examination protocol.
Patients with a history of alcohol dependence or nutritional deficiency underwent a specific protocol. The acute cases had a vestibular and ocular motor examination at bedside including manual and video head impulse (vHIT) followed by a neurologic examination. I repeated these tests after the intravenous administration of 500 mg of thiamine intravenously (9, 11). I ordered MRI of the brain in all cases. In patients with a subacute history, we measured serum thiamine levels and began oral thiamine replacement.
When the history suggested BVL, I implemented the following protocol: static followed by dynamic VA, direct ophthalmoscopy, visual field testing, oculomotor and cranial nerve testing, nystagmus analysis if present (both with fixation and with fixation block), manual head impulse in all six-canal planes. Truncal posture including Romberg test with eyes open and closed and balance while standing on a foam cushion. I tested gait and tandem gait whenever possible. Acute patients unable to stand had their ability to sit at the side of the bed with the arms crossed and eyes closed. At this point, I tested fine motor limb coordination, muscle tone, strength, and pathologic reflexes. We concluded the examination with nystagmus provocative maneuvers (routinely hyperventilation, mastoid vibration, Valsalva maneuver, horizontal head shaking, and positional testing). Simultaneous visual vestibulo-ocular reflex (VVOR) testing was performed only in our most recent cases.
If the clinical assessment suggested BVL, the work-up ordered included pure tone audiometry and vestibular testing including: Nystagmus recordings, bithermal caloric stimulation, torsion swing chair testing, and the vHIT; I did not order additional otolith function tests. At least one test of vestibular function in each case. Ideally, bithermal caloric testing; the vHIT measure different vestibular receptor frequencies and thus preferably in all patients, particularly if Meniere’s disease is suspected (4, 13, 14). VOR testing with the torsion/swing chair was performed in a few patients as well; however, several patients in this cohort underwent only one test of vestibular function, due to cost or availability issues. After 2012, all patients with BVL had manual and video HIT. With two exceptions, I tested acute cases at the bedside. MRI of the cerebellopontine angle included pre- and post-contrast images. Temporal bone CT scan with if necessary and routine blood tests to screen for metabolic abnormalities, vasculitis, autoimmune disorders, syphilis and Lyme’s serology, thiamine, folate and vitamin B12 serum levels and cerebrospinal fluid studies in selective patients with BVL and additional neurologic abnormalities. In addition, patients with BVL and additional neurologic abnormalities underwent testing in search of an autoimmune and paraneoplastic ataxia.
Follow-up longer than 1 year or greater was accomplished in n = 31/41 patients, some have been followed for ~10 years; this follow-up allowed us to assess the clinical course and the effect of medical treatment if any and assessment of the contribution of a defined Physical Therapy rehabilitation protocol.
I made a diagnosis of BVL in 57 patients at our Center; only 41 of them had records available (Tables 1 and 2). The average age was 58, with a range of 19–86; the gender distribution was 21 females and 20 males. The clinical course was characterized by a slowly progressive bilateral isolated vestibular or cochleovestibular loss in 29 patients and an acute vestibular syndrome in eight and acute sequential in four patients. BVL was associated with simultaneous CNS compromise in 28 and was isolated vestibular or cochleovestibular loss in 13 patients. Bilateral mixed cochleovestibular loss was present in three patients with superficial siderosis and two with MELAS, unilateral postsurgical deafness (unilateral jugular paraganglioma resection), and unilateral Meniere’s (post-unilateral transtympanic streptomycin injection).
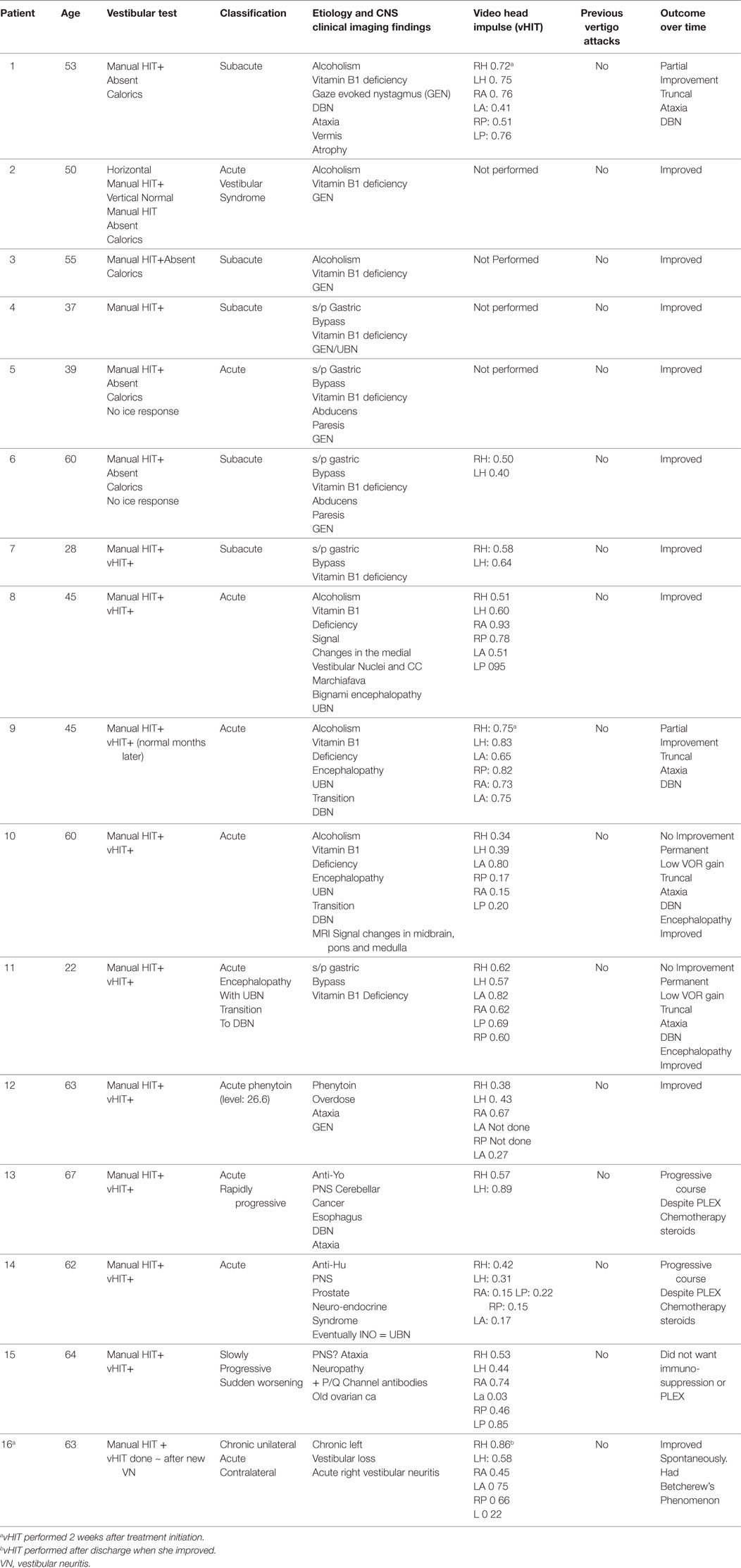
Table 1. Acute and subacute bilateral vestibulopathy.
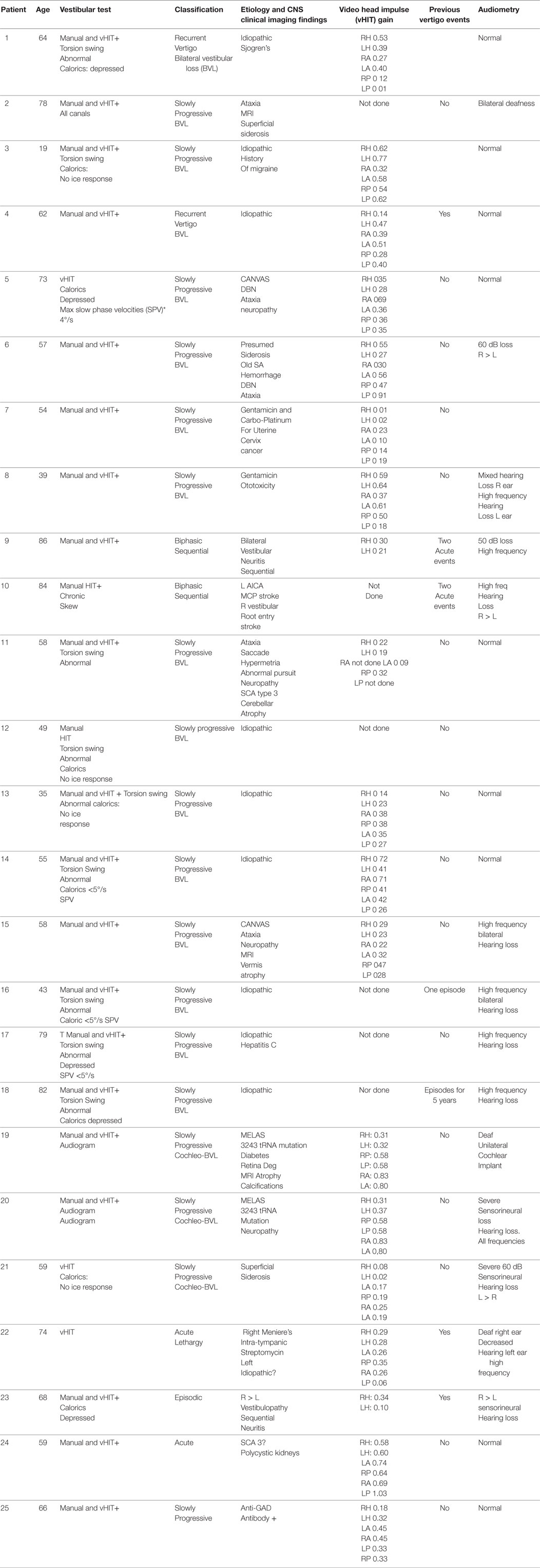
Table 2. Chronic bilateral vestibulopathy.
The etiology distribution (Tables 1 and 2) includes: idiopathic: 9, Wernicke’s encephalopathy: 11, superficial siderosis: 3, paraneoplastic syndrome: 3, bilateral vestibular neuritis (recurrent AVS lasting days without cochlear symptoms): 3, simultaneous ototoxicity of aminoglycoside and chemotherapy toxicity: 2, MELAS: 2, Meniere’s disease treated with intratympanic streptomycin in one ear: 1, acute phenytoin intoxication: 1, combined chronic unilateral tumor-related vestibulopathy and new contralateral vestibular neuritis (this patient presented with Betcherew’ s phenomenon): 1, bilateral AICA stroke: 1 (Figure 1), spinocerebellar ataxia type 3: second CANVAS: 2.
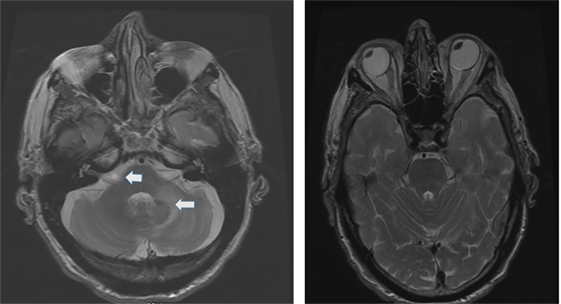
Figure 1. Axial T2 MRI: left panel shows a small area of increased signal intensity due to a small stroke involving the lateral pons, near the root entry of the right vestibular nerve (smaller arrow) and a larger stroke involving the left lateral pons and middle cerebellar peduncle (left panel larger arrow). Notice the conjugate ocular deviation of the eye to the right, coinciding with the slow phase of the nystagmus related to the second lacunar stroke in the right pons.
All patients underwent clinical testing as described in the protocol with the following result. Dynamic VA was abnormal in all patients. In five patients with primary gaze up (UBN) or downbeat (DBN), the nystagmus was a cofactor associated with impaired VA. We performed the manual HIT in all patients and recorded the vHIT in 30 patients examined when the vHIT device was first available to us. 39 patients had an abnormal manual HIT and two had covert saccades and low VOR gain (Figure 2). Prior to 2012, the patients underwent conventional bithermal caloric testing and 15 of them had absent caloric responses with conventional temperature stimulation, even when we utilized ice water stimulation. In few cases, standard caloric stimulation generated a weak horizontal nystagmus with slow phase velocities (SPV) of <5°/s. In some instances, the patients had caloric testing prior to our evaluation and we did not repeat it. Eight patients underwent torsion/swing chair testing decreased VOR gain and demonstrated phase lead when measured; the per-rotational and post-rotational nystagmus time constant was decreased (<5 s). Even though we did not routinely compare test of vestibular function in this series, we found them to be generally concordant in this small series (Figure 2).
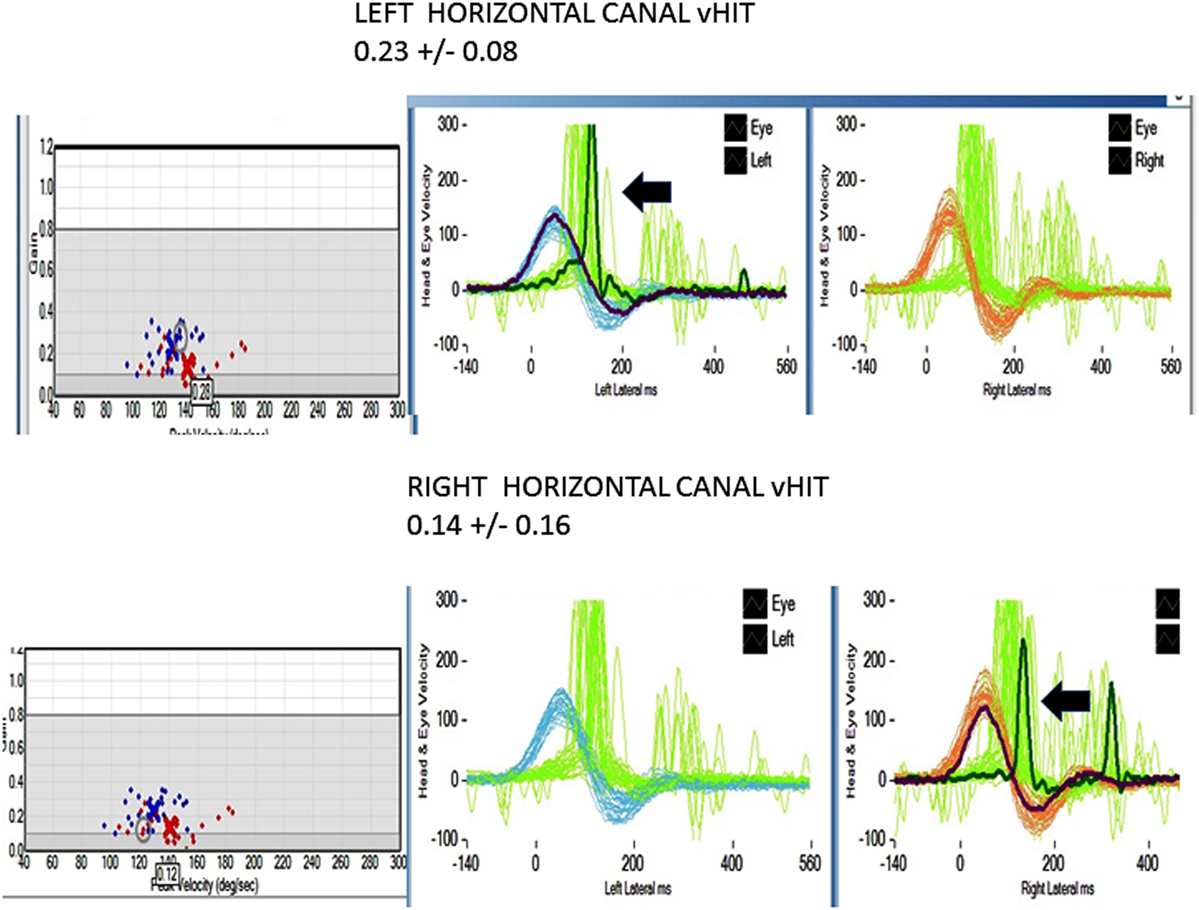
Figure 2. Horizontal video head impulse (h vHIT). Upper panel left h a-VOR gain is low 0.23 ± 0.08 (normal >0.7–1.0). The arrow points to a covert, corrective saccade, which was not apparent during the manual HIT. Lower panel right h a-VOR gain is low (0.14 ± 0.16). The arrow points to a covert, corrective saccade, which was not apparent during the manual HIT. The vertical canal gain was also low (not shown).
In the patients with BVL due to Wernicke’s encephalopathy, the pretreatment seem thiamine levels were low in n = 8/11 cases, the remaining three had an infusion of high-dose intravenous thiamine without preceding serum level. Four of them had abnormal MRI findings typical for Wernicke’s encephalopathy.
In this cohort, BVL in n = 9/41 (~20%) was idiopathic as noted in previous reports (2–4). In addition, this series is biased toward BVL associated with a neurologic disease and thus differs from previous BVL series; therefore, the etiology frequency in our population must be interpreted with this fact in mind; however, we identified several BVL patients in the context of acute and subacute neurologic syndromes in n = 16/41 (>25%).
Recently developed diagnostic criteria provided precise clinical and laboratory signs of BVL (12). I utilized the proposed protocol in this retrospective series. Search for the BVL diagnostic gold standard test is a still matter of debate; because caloric testing, rotational testing, and manual or vHIT test lack 100% specificity/sensitivity, clinicians may use one or more of these modalities to confirm the clinically suspected diagnosis (3, 4, 15). However, the combination of head impulse test, dynamic VA, and Romberg testing while standing on a rubber foam cushion is a practical initial step in subacute and chronic to effectively select patients requiring additional investigation (16). I implemented further quantitative testing thereafter (4). In acute patients, the testing protocol varied and included dynamic VA and both manual and vHIT in all six semicircular canal (SCC) planes. I paid special attention to comparing the horizontal versus vertical canal VOR gain; in central, BVL lesions preferentially affecting the MVN and suggests thiamine deficiency as a potential cause (8, 10, 17), which may be confirmed with pretreatment serum thiamine levels and a favorable response to vitamin supplementation and normal diet (8–10, 17). Similarly, selective peripheral vestibulopathies may spare the anterior canal VOR (18). The value of testing the VOR gain in plane of all six-SCC cannot be overemphasized. I did not identify in this series, a single case of acute simultaneous, bilateral peripheral vestibulopathy.
Analysis of our findings suggest that an acute etiology is more frequent than anticipated. Neurologic patients now undergo routinely manual HIT and vHIT testing in acute in-patient units. The referral population served at our Center represents a mixed small urban and rural population; however, the referral base is large, probably in the order of three million patients. To facilitate the discussion will divide our cohort in acute and subacute bilateral vestibular loss (BVL) (symptom evolution <1 month duration) (Table 1) and chronic vestibular loss (Table 2) evolving over a period greater than 1 month, but usually of longer duration. In the chronic BVL group, several patients had either isolated bilateral peripheral loss, cochleovestibular loss, or mixed peripheral/central vestibular loss in the context of a neurodegenerative process.
Among the 41 cases, 16 patients had an acute/subacute presentation (11 Wernicke’s encephalopathy (WE), three paraneoplastic syndromes, and two additional cases included one with acute phenytoin intoxication and one acute right vestibular neuritis and with chronic posttumor resection-related contralateral vestibulopathy, who displayed Betcherew’s phenomena) (Table 1) (19). In WE, the presumed location of the lesion in WE is the medial vestibular nucleus (8, 17, 20); however, at present, temporal bone studies in WE have not been performed to my knowledge; therefore, it is unknown if a co-existent peripheral vestibulopathy is also present. None of our acute BVL patients had spontaneous primary gaze horizontal nystagmus, except for the one sequential patient with Betcherew’s phenomenon. n = 15/16 patients with acute/subacute bilateral vestibulopathy had direction changing, symmetric horizontal gaze evoked nystagmus (GEN). UBN in straight-ahead fixation was noted in five patients with acute thiamine deficiency (four with encephalopathy and one without) and in one PNS patient. UBN switched to chronic DBN in three cases and resolved in two. UBN was present in one PNS and DBN in a second PNS patient.
In the acute cases, and in concert with the subacute and chronic BVL cases, primary gaze horizontal nystagmus was absent. Development of bilateral symmetric or minimally asymmetric horizontal vestibular loss, regardless of acuity does not cause fixation horizontal nystagmus (21). To explain the horizontal GEN, failure of the horizontal neural integrator is likely responsible; this is the most common type of nystagmus present in Wernicke’s Encephalopathy (20) and other brainstem lesions (22, 23). Several acute patients had vertical nystagmus in primary gaze; however, the precise location of the lesion responsible is unknown. Candidate locations could be the medulla (nucleus intercalatus and nucleus of Roller) (17, 24, 25), the pons (superior vestibular nucleus) (26), or the interstitial nucleus of Cajal (27).
The overall outcome of the acute BVL patient was favorable. Among 11 thiamine, six deficient patients responded to thiamine replacement and a normal diet with complete recovery and three of them had normal h-VOR gain but developed chronic DBN and gait ataxia.
In addition, the acute phenytoin intoxication patient slowly normalized the VOR gain once the medication was properly titrated; the patient with Betcherew’s’ phenomenon improved spontaneously over a few days. Two of three paraneoplastic syndrome patients treated with immunosuppression, cancer specific management had continuous neurologic and medical deterioration, and died, and one of them declined treatment.
The patients in this group comprised 25 patients (Table 2). Nine patients had isolated, idiopathic BVL, a number roughly comparable with previous BVL series (2–4, 28) Those patients with BVL due to ototoxic medications, together with those due to presumed sequential vestibular neuritis remain either clinically stationary, or are slowly improving through vestibular adaptation. One patient with bilateral sequential AICA strokes eventually had partial improvement after the second stroke (Figure 1); he was the only patient with chronic diplopia due to persistent, low-amplitude skew deviation.
In peripheral vestibulopathy, the manual HIT was abnormal in the plane of all canals tested, the vertical HIT in the plane of the posterior canal was, in our experience, easier to detect than the anterior canal, thus, often requiring vHIT for confirmation; however, the anterior canal VOR gain, as mentioned before, may be selectively spared in peripheral vestibulopathy (18). In previous series, exposure to ototoxic drugs is a frequent cause of BVL. Two patients in this series had BVL caused by exposure to gentamicin; curiously, both patients had gynecologic malignancies and had treatment with carboplatin and Taxol, they had discontinued all these medications when I first saw them. The lowest vHIT gain recorded in the series affected the ototoxic and idiopathic groups (near vestibular areflexia).
Patients with acute and those with chronic peripheral and central vestibular abnormalities had GEN, which was not present in isolated peripheral lesions. Eye movement abnormalities were frequent with central lesions (often diplopia due to extraocular muscle weakness, internuclear ophthalmoplegia, or skew deviation). We found additional eye movements abnormalities due to brainstem or cerebellar dysfunction. We did not test the VVOR routinely, thus, we cannot comment in its diagnostic value in this series; however, this would be another possible differentiating characteristic pointing to central localization and frequently present in CANVAS (29). In this series, the CANVAS patients had an abnormal VVOR (9). Sensorineural loss was more frequent with peripheral vestibulopathies.
A summary of the prognosis in this 41-patiet cohort includes Recovery in n = 14/41 cases, partial recovery, and progressive deterioration in n = 12/41. Two of the paraneoplastic syndromes died within 6 months after the diagnosis, because of cancer management complications and six patients are wheelchair bound (BVL with neurodegenerative disorders and two with superficial siderosis), the remaining 15 cases remain stable as noted in previous series (28).
In conclusion, the incidence and distribution of BVL varies with the population that attends the different centers and the specialty of the clinicians evaluating BVL interests in acute BVL is increasing because of portable technology and the possibility of BVL improvement. Table 3 is a summary of the main vestibular findings identified in the two groups of BVL patients.
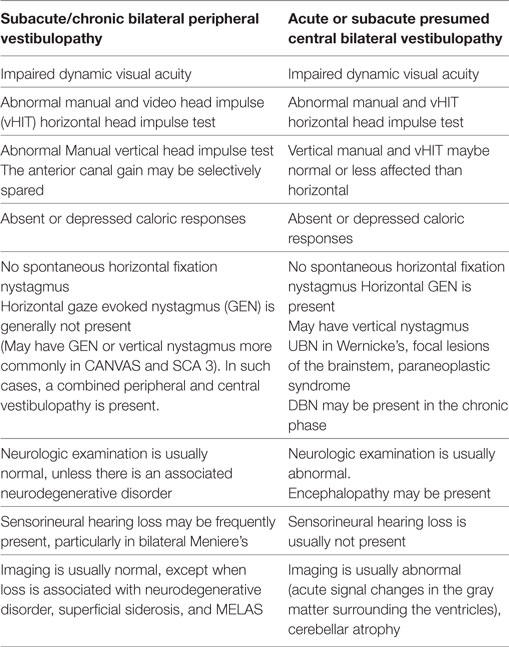
Table 3. Comparative findings in bilateral vestibular loss.
This study was approved by the University Of Illinois College Of Medicine IRB and follows the tenants of the declaration of Helsinski.
This is a single author paper. I examined each patient and conducted a retrospective review.
Otometrics corporation loaned research equipment (beta site in 2012). This unit is no longer being used.
The author acknowledges the members of the Balance Center at the Illinois Neurologic Institute: Cynthia Guede, APN, and Samantha Mueller, AuD who collaborated in the evaluation of these patients.
1. Ward BK, Agrawal Y, Hoffman HJ, Carey JP, Della Santina CC. Prevalence and impact of bilateral vestibular hypofunction: results from the 2008 US National Health Interview Survey. JAMA Otolaryngol Head Neck Surg (2013) 139:803–10. doi:10.1001/jamaoto.2013.3913
2. Zingler VC, Cnyrim C, Jahn K, Weintz E, Fernbacher J, Frenzel C, et al. Causative factors and epidemiology of bilateral vestibulopathy in 255 patients. Ann Neurol (2007) 61:524–32. doi:10.1002/ana.21105
3. Hain TC, Cherchi M, Yacovino DA. Bilateral vestibular loss. Semin Neurol (2013) 33:195–203. doi:10.1055/s-0033-1354597
4. Lucieer F, Vonk P, Guinand N, Stokroos R, Kingma H, van de Berg R. Bilateral vestibular hypofunction: insights in etiologies, clinical subtypes, and diagnostics. Front Neurol (2016) 7:26. doi:10.3389/fneur.2016.00026
5. Perletti G, Vral A, Patrosso MC, Marras E, Ceriani I, Willems P, et al. Prevention and modulation of aminoglycoside ototoxicity (review). Mol Med Rep (2008) 1:3–13. doi:10.3892/mmr.1.1.3
6. Ghez C. Vestibular paresis: a clinical feature of Wernicke’s disease. J Neurol Neurosurg Psychiatry (1969) 32:134–9. doi:10.1136/jnnp.32.2.134
7. Furman JM, Becker JT. Vestibular responses in Wernicke’s encephalopathy. Ann Neurol (1989) 26:669–74. doi:10.1002/ana.410260513
8. Choi KD, Oh SY, Kim HJ, Kim JS. The vestibulo-ocular reflexes during head impulse in Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry (2007) 78:1161–2. doi:10.1136/jnnp.2007.121061
9. Kattah JCDS, Pula JH, Mantokoudis G, Therani AS, Newman Toker DE. Vestibular signs in non-encephalopathic Wernicke’s disease. Neurol Clin Pract (2013) 3:460–7. doi:10.1212/01.CPJ.0000435749.32868.91
10. Akdal G, MacDougall HG, Chen L, Tanriverdizade T, Yigitaslan O, Halmagyi GM. Selective impairment of horizontal vestibulo-ocular reflexes in acute Wernicke’s encephalopathy. J Neurol Sci (2016) 365:167–8. doi:10.1016/j.jns.2016.04.013
11. Kattah JC. The spectrum of vestibular and ocular motor abnormalities in thiamine deficiency. Curr Neurol Neurosci Rep (2017) 17:40. doi:10.1007/s11910-017-0747-9
12. Strupp M, Kim JS, Murofushi T, Straumann D, Jen JC, Rosengren SM, et al. Bilateral vestibulopathy: diagnostic criteria consensus document of the classification committee of the Barany society. J Vestib Res (2017) 27:177–89. doi:10.3233/VES-170619
13. Perez N, Rama-Lopez J. Head-impulse and caloric tests in patients with dizziness. Otol Neurotol (2003) 24:913–7. doi:10.1097/00129492-200311000-00016
14. Burston A, Mossman S, Mossman B, Weatherall M. Comparison of the video head impulse test with the caloric test in patients with sub-acute and chronic vestibular disorders. J Clin Neurosci (2017) 47:294–8. doi:10.1016/j.jocn.2017.10.040
15. Furman JM, Kamerer DB. Rotational responses in patients with bilateral caloric reduction. Acta Otolaryngol (1989) 108:355–61. doi:10.3109/00016488909125539
16. Petersen JA, Straumann D, Weber KP. Clinical diagnosis of bilateral vestibular loss: three simple bedside tests. Ther Adv Neurol Disord (2013) 6:41–5. doi:10.1177/1756285612465920
17. Kattah JC, Guede C, Hassanzadeh B. The medial vestibular nuclei, a vulnerable target in thiamine deficiency. J Neurol (2017) 265(1):213–5. doi:10.1007/s00415-017-8670-1
18. Tarnutzer AA, Bockisch CJ, Buffone E, Weiler S, Bachmann LM, Weber KP. Disease-specific sparing of the anterior semicircular canals in bilateral vestibulopathy. Clin Neurophysiol (2016) 127:2791–801. doi:10.1016/j.clinph.2016.05.005
19. Zee DS, Preziosi TJ, Proctor LR. Bechterew’s phenomenon in a human patient. Ann Neurol (1982) 12:495–6. doi:10.1002/ana.410120519
21. Baloh RW, Halmagyi GM. Disorders of the Vestibular System. New York, Oxford: Oxford University Press (1996).
22. Leigh R, Zee DS. The Neurology of Eye Movements. 5th ed. New York, Oxford: Oxford University Press (2015).
23. Lee SH, Kim HJ, Kim JS. Ocular motor dysfunction due to brainstem disorders. J Neuroophthalmol (2017) 1–20. doi:10.1097/WNO.0000000000000583
24. Janssen JC, Larner AJ, Morris H, Bronstein AM, Farmer SF. Upbeat nystagmus: clinicoanatomical correlation. J Neurol Neurosurg Psychiatry (1998) 65:380–1. doi:10.1136/jnnp.65.3.380
25. Pierrot-Deseilligny C, Richeh W, Bolgert F. Upbeat nystagmus due to a caudal medullary lesion and influenced by gravity. J Neurol (2007) 254:120–1. doi:10.1007/s00415-006-0302-0
26. Pierrot-Deseilligny C, Milea D. Vertical nystagmus: clinical facts and hypotheses. Brain (2005) 128:1237–46. doi:10.1093/brain/awh532
27. Fisher A, Gresty M, Chambers B, Rudge P. Primary position upbeating nystagmus. A variety of central positional nystagmus. Brain (1983) 106(Pt 4):949–64. doi:10.1093/brain/106.4.949
28. Zingler VC, Weintz E, Jahn K, Mike A, Huppert D, Rettinger N, et al. Follow-up of vestibular function in bilateral vestibulopathy. J Neurol Neurosurg Psychiatry (2008) 79:284–8. doi:10.1136/jnnp.2007.122952
29. Szmulewicz DJ, Waterston JA, MacDougall HG, Mossman S, Chancellor AM, McLean CA, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci (2011) 1233:139–47. doi:10.1111/j.1749-6632.2011.06158.x
Keywords: bilateral vestibulopathy, acute bilateral vestibular loss, chronic bilateral, vestibular loss, neurologic disorders associated with bilateral vestibular loss
Citation: Kattah JC (2018) Clinical Characteristics and Etiology of Bilateral Vestibular Loss in a Cohort from Central Illinois. Front. Neurol. 9:46. doi: 10.3389/fneur.2018.00046
Received: 01 December 2017; Accepted: 18 January 2018;
Published: 01 March 2018
Edited by:
Alexander A. Tarnutzer, University of Zurich, SwitzerlandReviewed by:
Konrad P. Weber, University of Zurich, SwitzerlandCopyright: © 2018 Kattah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge C. Katttah, a2F0dGFoakB1aWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.