 Laurent Carteron
Laurent Carteron Pierre Bouzat
Pierre Bouzat Mauro Oddo
Mauro Oddo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 13 November 2017
Sec. Neurotrauma
Volume 8 - 2017 | https://doi.org/10.3389/fneur.2017.00601
This article is part of the Research Topic Monitoring Pathophysiology in the Injured Brain View all 19 articles
Cerebral microdialysis (CMD) allows bedside semicontinuous monitoring of patient brain extracellular fluid. Clinical indications of CMD monitoring are focused on the management of secondary cerebral and systemic insults in acute brain injury (ABI) patients [mainly, traumatic brain injury (TBI), subarachnoid hemorrhage, and intracerebral hemorrhage (ICH)], specifically to tailor several routine interventions—such as optimization of cerebral perfusion pressure, blood transfusion, glycemic control and oxygen therapy—in the individual patient. Using CMD as clinical research tool has greatly contributed to identify and better understand important post-injury mechanisms—such as energy dysfunction, posttraumatic glycolysis, post-aneurysmal early brain injury, cortical spreading depressions, and subclinical seizures. Main CMD metabolites (namely, lactate/pyruvate ratio, and glucose) can be used to monitor the brain response to specific interventions, to assess the extent of injury, and to inform about prognosis. Recent consensus statements have provided guidelines and recommendations for CMD monitoring in neurocritical care. Here, we summarize recent clinical investigation conducted in ABI patients, specifically focusing on the role of CMD to guide individualized intensive care therapy and to improve our understanding of the complex disease mechanisms occurring in the immediate phase following ABI. Promising brain biomarkers will also be described.
Cerebral microdialysis (CMD) has progressively evolved from a tool for clinical research into an additional brain monitoring modality to guide neurointensive care (1, 2). Evidence has accrued over the last years that CMD monitoring—in combination with other modalities such as intracranial pressure (ICP) and brain tissue PO2 (PbtO2), so called multimodal monitoring—may help guiding individualized intensive care therapy of comatose brain-injured patients, mainly after traumatic brain injury (TBI) and aneurysmal subarachnoid hemorrhage (SAH) (3, 4). Clinical utility of CMD has been particularly shown for the management of “secondary” cerebral insults, i.e., the number of pathological events that occur in the early phase following acute brain injury (ABI). The use of CMD has contributed to better define therapeutic thresholds for several routine interventions, such as cerebral perfusion pressure (CPP) optimization, oxygen therapy, red blood cell transfusion (RBCT), and metabolic control (blood glucose and nutrition). Exploration of the injured brain with CMD has also greatly contributed to better understand important post-injury mechanisms—such as energy dysfunction, hyperglycolysis, cortical spreading depressions, subclinical seizures, or brain edema—and to identify potential novel biomarkers of injury and prognosis. Recent reviews focused on specific technical aspects related to CMD monitoring, both in terms of the catheters and microdialyzate analyser technology (1). The scope of this review was to summarize recent clinical investigation conducted in neurocritical care patients, aiming to discuss the role of CMD to guide individualized intensive care therapy and to improve our understanding of the complex disease mechanisms occurring in the immediate phase following severe brain injury. We also describe emerging data on the potential utility of CMD to assess novel biomarkers of injury, as well as its role in interventional and pharmacological studies. We mainly focused our review on clinical studies published during the last 5 years (January 2012 to September 2017) and performed in patients with ABI, including TBI, SAH, and ICH.
In clinical practice, CMD biomarkers (generally sampled every hour and immediately analyzed at the bedside) should always be interpreted in the context of monitor location, type of injury, and patient clinical condition. Based on accrued clinical data over the last decade linking glucose and lactate/pyruvate (L/P) ratio with principal outcomes after ABI, compared to glutamate and glycerol, the 2015 CMD Consensus proposed to interpret CMD biomarkers in a tiered fashion and to use primarily CMD L/P ratio and glucose as step 1 to guide clinical interventions (2). Abnormalities of CMD L/P ratio and glucose reflect the complex pathophysiology underneath ABI; therefore, correct interpretation require integration of other monitored variables such as ICP and PbtO2.
Elevated CMD lactate and L/P ratio may be a marker of inadequate cerebral blood flow (CBF) and/or oxygen delivery. In this context, dramatic increases may be observed, which are associated with a concomitant decrease in CMD pyruvate and glucose. Given that cerebral circulation and/or oxygenation are impaired, ICP/CPP and/or PbtO2 values will be abnormal.
However, CMD lactate and L/P ratio may be elevated because of other mechanisms than ischemia or hypoxia (5). Cerebral energy dysfunction/failure has been described despite CBF and brain tissue oxygenation being normal (6, 7), whereby elevations of CMD lactate and L/P ratio may be predominantly attributable to increased glycolysis or mitochondrial dysfunction (impairment of oxygen utilization or cytopathic hypoxia) (8, 9). In this context, pyruvate may be normal or elevated, and elevations of CMD lactate and L/P ratio are of a lesser extent than during frank ischemia/hypoxia.
Low CMD glucose, therefore, may be related to cerebral energy dysfunction (10). On the other hand, apart from cerebral causes (ischemia/hypoxia or energy dysfunction), inadequate systemic glucose, because of intensive insulin therapy to maintain strict glycemic control, may cause further reductions of CMD glucose (11, 12).
To direct individualized intensive care therapy, it is therefore important to consider CMD L/P ratio rather than lactate alone, to look for dynamic changes and trends of both CMD L/P ratio and glucose, and finally to take into account additional monitor modalities (ICP/PbtO2), according to the modern paradigm of multimodality monitoring (13, 14).
Interpretation of absolute values is also dependent on probe location in an area of normal-appearing vs. around a lesion (e.g., hematoma or contusion) (2, 15). Also, a recent study in SAH patients suggests that delayed cerebral ischemia may be detected only when the probe is located within a brain area later affected by secondary infarction, which may justify the use of implantation guidelines (16).
In Figure 1, we propose an algorithm for interpretation of CMD abnormalities, centered on low CMD glucose as starting point of the clinical reasoning.
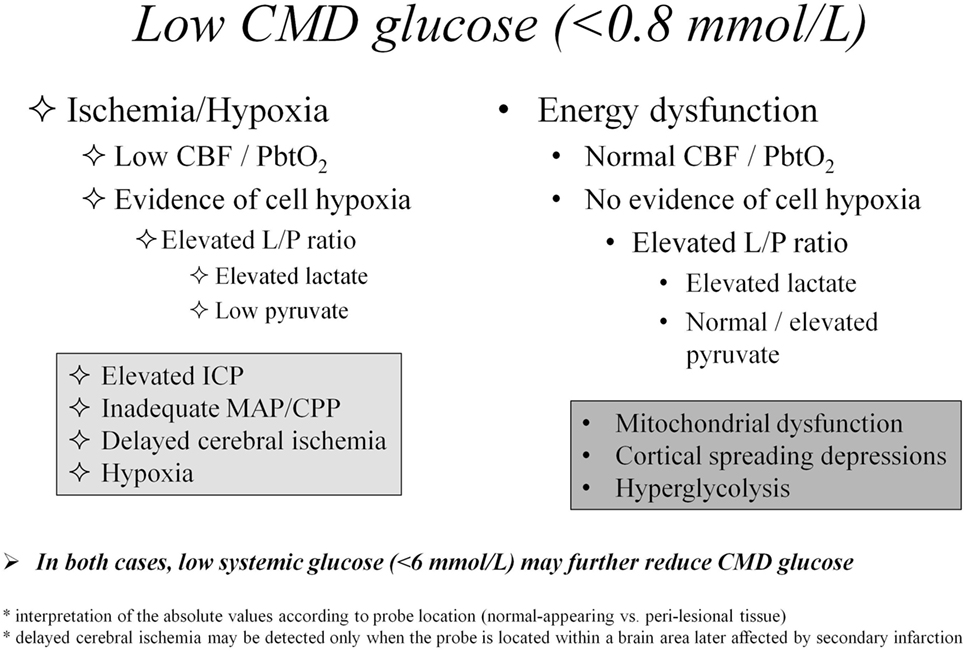
Figure 1. Differential diagnosis of cerebral metabolic abnormalities based on cerebral microdialysis. Abbreviations: CBF, cerebral blood flow; CMD, cerebral microdialysis; CPP, cerebral perfusion pressure; ICP, intracranial pressure; L/P, lactate/pyruvate; MAP, mean arterial pressure; PbtO2, brain tissue oxygen pressure.
As for reference values, L/P ratio >25 is considered abnormal (impaired cerebral oxidative metabolism), while L/P ratio >40 is the critical level above which brain energy crisis is defined. The reference level for CMD glucose is still debated, but probably lies at 1 (±0.15) mmol/L (17).
The CMD technique allows semicontinuous monitoring of cerebral glucose metabolism and of the interactions between blood and brain glucose in humans under conditions of varying glycemia (18). Glucose is the main substrate for the brain. However, in the aftermath of injury, the brain’s ability to use glucose may be reduced (19). Cerebral extracellular glucose may be limited (10, 20), therefore, enabling adequate glucose supply in ABI patients appears crucial to attenuate further brain damage (21). Following the two large single-center studies by van Den Berghe and colleagues in the early 2000 (22, 23), suggesting that tight glycemic control may benefit general critically ill patients, Vespa and colleagues were the first to show that actually this so-called intensive insulin strategy was associated with an increased prevalence of low CMD glucose and elevated LPR (24). This CMD study was concomitant to another outcome study by the Leuven’s group showing that, at the contrary, strict glycemic control may also benefit the outcome of neurointensive care patients (25). Additional CMD studies from several groups subsequently confirmed the seminal clinical investigation by Vespa and colleagues, showing that indeed strict glycemic control might reduce cerebral glucose availability and aggravate cerebral energy dysfunction (11, 26–31). Given the results of the multicentre NICE-SUGAR study, which did not confirm substantial outcome benefit for intensive vs. moderate blood glucose control both in the general ICU population (32, 33), and in the post hoc analysis of neurotrauma patients (34), a strategy of liberal glycemic control (7–10 mmol/L) was generally felt as safer in critically neurological patients by international recommendations (35). Indeed, using a cross-over design that alternated tight to moderate glycemic control, Vespa confirmed previous findings that intensive insulin therapy was associated with increased metabolic distress, as judged by lower CMD glucose and higher CMD L/P ratio during tight glycemia (12).
The glycemic control controversy illustrates how CMD monitoring has contributed to the actual progresses of intensive care therapies, and how physiologically oriented studies may influence our practice, especially in the field of neurointensive care where “true” evidence-based medicine derived from RCT is often lacking. A recent example of such approach was provided by the Innsbruck group led by Helbok: the authors found that rapid effective institution of enteral nutrition was associated with an increase in CMD glucose that was directly dependent on the magnitude of increase of blood glucose (36), reinforcing the recommendations for the early institution of enteral feeding in neurointensive care patients.
The Consensus on CMD suggests the use of CMD monitoring for the detection and treatment of low cerebral glucose, and to guide systemic glucose management and insulin use (2).
CMD markers—such as glucose and L/P ratio—may be good surrogate markers of CBF, and indeed this has recently been confirmed by several clinical studies combining microdiaylsis with brain imaging, both in patients with SAH (37–39) and TBI (40). A recently published small observational cohort study illustrated the potential value of CMD monitoring to help detecting cerebral hypoperfusion in comatose aSAH patients, in whom, the clinical examination was unreliable (37). This study stressed the importance of following dynamic trends over time of both CMD L/P ratio and glucose for the timely detection of secondary cerebral ischemic insults. It also confirmed the potential value of CMD biomarkers to avoid low CPP by adjusting CPP thresholds individually in comatose ABI patients (16, 41–43). Indeed, Bouzat and colleagues found that the addition of CMD (in combination with PbtO2) to ICP monitoring significantly improved the accuracy of detecting secondary hypoperfusion in patients with severe TBI (40).
The use of CMD monitoring to optimize CCP in order to prevent/avoid ischemia is recognized as potentially clinically useful for TBI and SAH patients by the Consensus on CMD (2).
Whether restrictive or more liberal thresholds for hemoglobin and RBCT should be used in neurointensive care is still debated, given the lack of randomized clinical trials in this setting. It is possible that the therapeutic approach may vary individually, according to the extent of injury; therefore, patients with more severe brain insults may benefit from higher hemoglobin (Hgb) levels (44, 45). Indeed, low Hgb <9 g/dL was shown to be associated with increased CMD markers of cerebral ischemia (elevated L/P ratio and low CMD glucose) (46, 47). The question is whether enhancing cerebral oxygen transport with RBCT may reduce cerebral damage: RBCT might improve PbtO2 in the majority (although not all) of patients (48, 49); however, improved PbtO2 did not translate into a clinically relevant benefit on cellular metabolism, as quantified by the non-significant amelioration of CMD L/P ratio (50, 51).
In various subsets of critically ill patients, including those with ABI, increasing inspired fraction of oxygen (FiO2) to achieve arterial hyperoxia (arterial partial pressure of oxygen, PaO2, >150 mmHg) was associated with worse outcome (52). Whether or not hyperoxia is beneficial after ABI remains controversial. Physiological studies testing the effect of hyperoxia on CMD biomarkers were conducted predominantly on TBI patients. Improving PbtO2 by way of normobaric hyperoxia may reduce L/P ratio (53, 54), although this effect seems of limited clinical relevance (55). When using CMD glutamate as a marker of increased excitotoxicity, Quintard and colleagues found an association between normobaric hyperoxia and increased cerebral glutamate (56). Recently, two prospective single-center trials brought additional important insights. Ghosh and colleagues, testing 120-min normobaric hyperoxia challenge in the acute phase (24–72 h) of TBI (16 patients; using an advanced multimodal monitoring, including PbtO2, CMD, near-infrared spectroscopy, and transcranial Doppler) found that hyperoxia was associated with an improvement of L/P ratio, as well as all other oxygenation and perfusion parameters, consistent with increased aerobic cerebral metabolism and better cellular redox state (57). Vidal-Jorge and colleagues in an elegant study using CMD to sample biomarkers of oxidative stress (8-iso-Prostaglandin F2α) found that increasing FiO2 to 1.0 for 4 h resulted in marked reduction in both CMD lactate and CMD L/P ratio only in patients with more severe injury, as defined by a CMD lactate >3.5 mmol/L, but did not change energy metabolism in the whole group of patients (58). Furthermore, hyperoxia caused a significant increase in 8-iso-PGF2α in patients in whom oxidative stress was detected at baseline, but not in those without (58).
Rockswold and colleagues, using a Phase II observational design, found that hyperbaric oxygen therapy [1 h at 1.5 atmospheres absolute (ATA)], followed by 3-h normobaric hyperoxia (100% FiO2 at 1.0 ATA) was effective in improving CMD L/P ratio and glycerol after TBI, both in relatively uninjured brain as well as in peri-contusional tissue; tissue benefit translated into better outcome in this study (59).
Overall, CMD has evolved over time as a tool that may help guiding individualized targeted therapy at the bedside in ABI patients and to test the physiologic response to a specific intervention (Table 1).
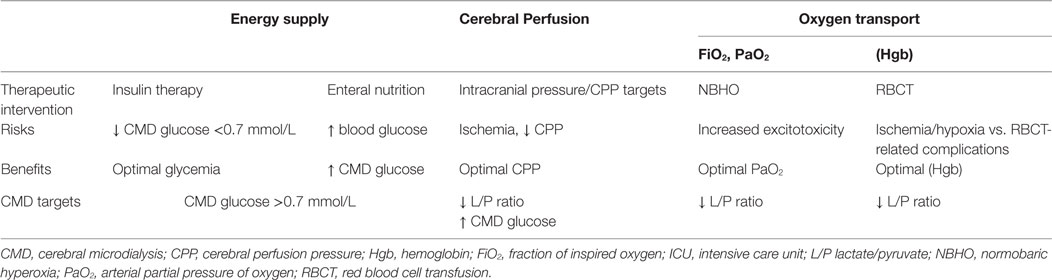
Table 1. Examples of ICU interventions guided by CMD.
Although it was not validated so far in large multicentre studies, CMD biomarkers such as CMD L/P ratio and glucose are associated with patient prognosis, at least in TBI patients (60). Therefore, it is conceivable to use CMD metabolites as surrogate outcome endpoints to test therapeutic efficacy in Phase II clinical trials.
Examples of therapies tested in studies using CMD biomarkers as surrogate outcome endpoints include:
– nitric oxide synthase inhibition (61)
– recombinant human interleukin-1 receptor antagonist (62)
– antiepileptic drugs (63, 64)
– focally perfused succinate (65)
– intravenous hypertonic lactate (66, 67)
– sedation (68).
Measuring the concentrations of drug molecules in the brain extracellular fluid appears superior to cerebrospinal fluid or plasma to test the ability to effectively deliver pharmacological agents across the blood–brain barrier into the brain and is an important step in the development of central nervous system therapies. CMD sampling can give valuable pharmacokinetic information of variations with time in drug concentrations of brain interstitial tissue versus plasma and may help in designing future therapies (69, 70), or to test drug penetration of several pharmacologic agents, such antimicrobials (71, 72) or antiepileptic drugs (63, 64).
Alterations of cerebral perfusion/oxygenation (73–75) and brain energy metabolism (9, 19, 20, 76–82) are important determinants of ABI. However, additional mechanisms are implicated in post-injury pathophysiology and CMD has contributed to elucidate some of these mechanisms (Figure 2). In this context, CMD catheters with larger membrane cut-off (100 kDa) than the standard ones (20 kDa) may have great utility for the identification and bedside follow-up of biomarkers of injury (e.g., cytokines, metallo-proteases) and recovery (e.g., markers of neurodegeneration) in specific pathologies (70, 83).
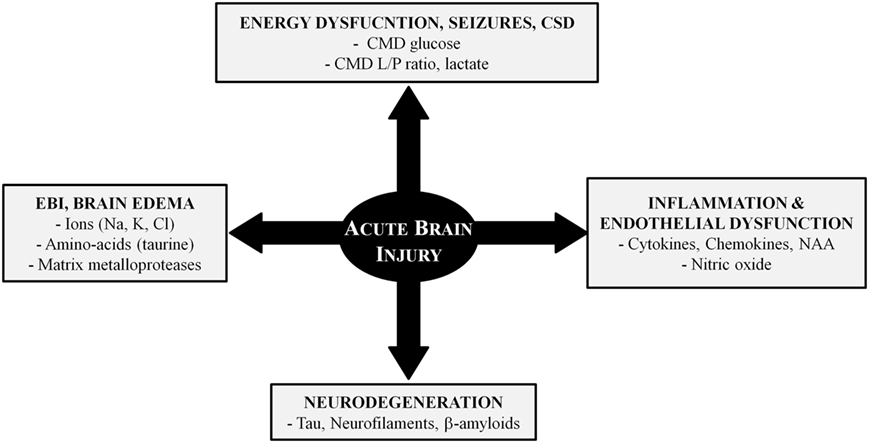
Figure 2. Pathophysiology of acute brain injury: the role of cerebral microdialysis. Abbreviations: CMD, cerebral microdialysis; CSD, cortical spreading depressions; EBI, early brain injury; L/P, lactate/pyruvate; NAA, n-acetyl aspartate.
Non-convulsive seizures and pseudo-periodic discharges might amplify secondary cerebral damage in the setting of ABI: using an elegant approach combining CMD with surface and intra-cortical electro-encephalography, Vespa and colleagues recently established a mechanistic link between seizures and metabolic crisis (84). This study is another example of how CMD can be used to monitor complex and concealed mechanisms but also to test the efficacy of future interventions aimed at specifically targeting seizure suppression.
Along the same line, pathological spreading depressions, which are frequently seen in TBI and SAH patients (85), cause significant local cerebral metabolic disturbances (reduced CMD glucose, elevated CMD LPR, and glutamate) (86–88); therefore, it is conceivable to use CMD as target for future interventional trials aimed at specifically treating spreading depressions.
Microdialysis studies have contributed to better characterize the exact nature of cerebral edema in different pathologies and to differentiate between cellular (or cytotoxic) and vasogenic edema. Alterations in the ionic profile of the extracellular space [main electrolytes (Na+, K+, Cl−) and amino-acids like taurine] correlate with cellular edema in patients with diffuse injury after TBI (89–92). Matrix metalloproteases (MMP) are important pathogenic determinants of blood–brain barrier breakdown and vasogenic edema: using 100 kDa catheters, which allows sampling of larger molecules, elevated CMD MMP have been observed in patients with focal parenchymal hemorrhages following TBI and SAH (93–97). These physiology studies contribute to better refine future treatments of brain edema, according to the specific pathology.
Using CMD has allowed the exploration of cytokine and chemokine profile after ABI (98–101), as well as to follow the dynamic changes in brain extracellular fluid of other biomarkers of inflammation (102), oxidative stress (NAA, isoprostane) (103, 104), and endothelial dysfunction (nitric oxide) (105), which may also be potential surrogate endpoints for interventional studies (58). Two recent scoping systematic reviews have addressed the potential value of microdialysis cytokines in severe TBI and poor-grade SAH (106, 107): although preliminary studies support feasibility of measurements and associations of CMD cytokines with tissue and neurophysiologic outcomes, evidence is very limited and further larger studies need to be conducted.
Markers of axonal degeneration—such as tau, β-amyloid, neurofilament light-chain (NfL), and neurofilament heavy chain (NfH)—have been the focus of recent clinical investigation, often in combination with magnetic resonance imaging, to better characterize posttraumatic axonal injury acutely in the intensive care unit (108–113). Preliminary data also established a potential link between tau protein and early brain injury following SAH (114, 115). Providing the reproducibility of these biomarkers is confirmed in larger scale studies, such approach holds great promise for early prognostication (to complement clinical and radiological information) and for a pathology-based patient selection to optimize future pharmacological interventional studies.
Table 2 summarizes main results of clinical CMD studies and their potential implications and clinical utility.
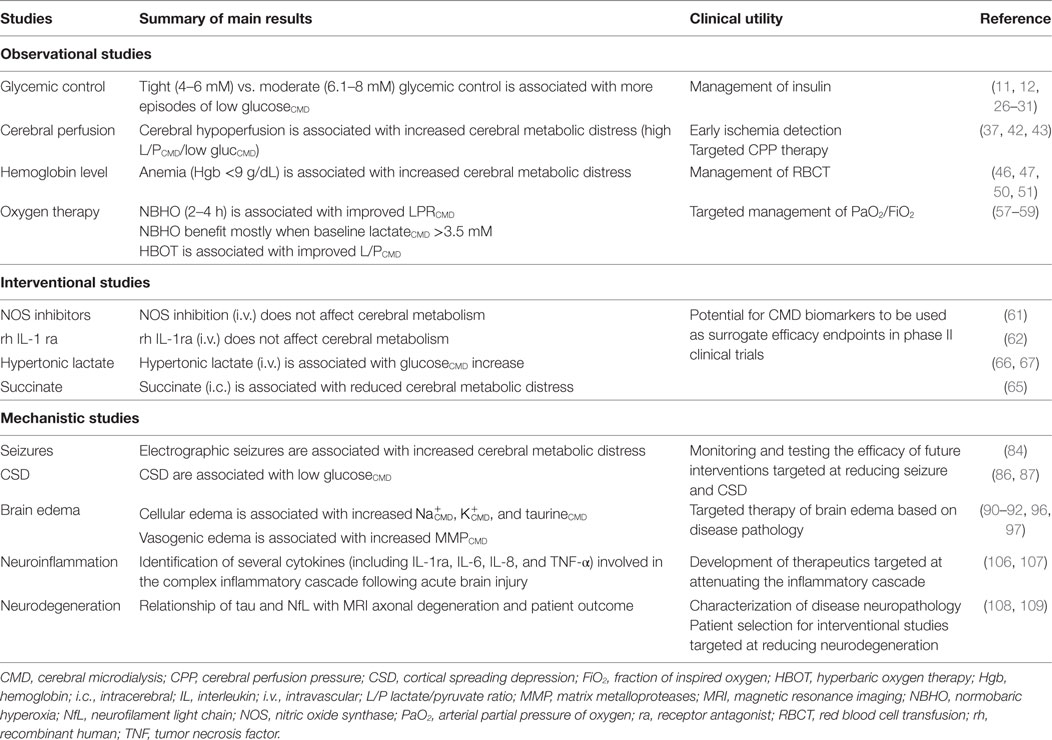
Table 2. Summary of clinical CMD studies.
Barriers to the widespread implementation of CMD are numerous, including costs, human resources, and the complexity of the technique (especially with respect to 100 kDa catheters) (1). These barriers may explain why CMD monitoring is still not in use in the majority of centers, as judged by a recent National survey on multimodal monitoring conducted in the UK (116). Recent consensus guidelines for the use of CMD in acute brain pathologies (2, 15) and the increased application of CMD in other acute contexts, e.g., anoxic-ischemic (117) or hepatic encephalopathy (118), may contribute to a broader implementation of this technique. The future of CMD is constantly evolving: technical refinements and the potential for automated near real-time continuous measurements may increase the performance and the accuracy of the technique (119–121), thereby facilitating the utilization in the intensive care unit.
Cerebral microdialysis is an important neuromonitoring tool that is increasing used at the bedside in combination with ICP and PbtO2 to guide therapy individually in brain-injured patients. Recent consensus on microdialysis monitoring may help optimizing protocols for microdialysis implementation in neurocritical care. Over the last decade, clinical investigation using microdialysis have contributed to better understand pathogenic mechanisms involved in secondary brain damage, such as cerebral edema, energy dysfunction, cortical spreading depression, neuroinflammation, and help refining novel therapeutic approaches, and drug effects on downstream targets. Future improvements of CMD technology may further enhance applicability.
LC drafted the manuscript and Table 1. PB drafted the manuscript and the figures. MO drafted and revised the manuscript, and drafted the Figures and Table 2.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by grants from the Swiss National Science Foundation (grant nr 32003B_155957, to MO), the Société Française d’Anesthésie et de Réanimation (SFAR, to LC), and the “Fondation des Gueules Cassées” (grant nr 41_2015, to LC).
1. Carpenter KL, Young AM, Hutchinson PJ. Advanced monitoring in traumatic brain injury: microdialysis. Curr Opin Crit Care (2017) 23(2):103–9. doi:10.1097/MCC.0000000000000400
2. Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med (2015) 41(9):1517–28. doi:10.1007/s00134-015-3930-y
3. Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on multimodality monitoring in neurocritical care: a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Neurocrit Care (2014) 21(Suppl 2):S1–26. doi:10.1007/s12028-014-0041-5
4. Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on multimodality monitoring in neurocritical care: a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Intensive Care Med (2014) 40(9):1189–209. doi:10.1007/s00134-014-3369-6
5. Diringer MN, Zazulia AR, Powers WJ. Does ischemia contribute to energy failure in severe TBI? Transl Stroke Res (2011) 2(4):517–23. doi:10.1007/s12975-011-0119-8
6. Chen HI, Stiefel MF, Oddo M, Milby AH, Maloney-Wilensky E, Frangos S, et al. Detection of cerebral compromise with multimodality monitoring in patients with subarachnoid hemorrhage. Neurosurgery (2011) 69(1):53–63. doi:10.1227/NEU.0b013e3182191451
7. Sahuquillo J, Merino MA, Sánchez-Guerrero A, Arikan F, Vidal-Jorge M, Martínez-Valverde T, et al. Lactate and the lactate-to-pyruvate molar ratio cannot be used as independent biomarkers for monitoring brain energetic metabolism: a microdialysis study in patients with traumatic brain injuries. PLoS One (2014) 9(7):e102540. doi:10.1371/journal.pone.0102540
8. Sala N, Suys T, Zerlauth JB, Bouzat P, Messerer M, Bloch J, et al. Cerebral extracellular lactate increase is predominantly nonischemic in patients with severe traumatic brain injury. J Cereb Blood Flow Metab (2013) 33(11):1815–22. doi:10.1038/jcbfm.2013.142
9. Vespa P, Bergsneider M, Hattori N, Wu HM, Huang SC, Martin NA, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab (2005) 25(6):763–74. doi:10.1038/sj.jcbfm.9600073
10. Patet C, Quintard H, Suys T, Bloch J, Daniel RT, Pellerin L, et al. Neuroenergetic response to prolonged cerebral glucose depletion after severe brain injury and the role of lactate. J Neurotrauma (2015) 32(20):1560–6. doi:10.1089/neu.2014.3781
11. Oddo M, Schmidt JM, Carrera E, Badjatia N, Connolly ES, Presciutti M, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med (2008) 36(12):3233–8. doi:10.1097/CCM.0b013e31818f4026
12. Vespa P, McArthur DL, Stein N, Huang SC, Shao W, Filippou M, et al. Tight glycemic control increases metabolic distress in traumatic brain injury: a randomized controlled within-subjects trial. Crit Care Med (2012) 40(6):1923–9. doi:10.1097/CCM.0b013e31824e0fcc
13. Citerio G, Oddo M, Taccone FS. Recommendations for the use of multimodal monitoring in the neurointensive care unit. Curr Opin Crit Care (2015) 21(2):113–9. doi:10.1097/MCC.0000000000000179
14. Oddo M, Villa F, Citerio G. Brain multimodality monitoring: an update. Curr Opin Crit Care (2012) 18(2):111–8. doi:10.1097/MCC.0b013e32835132a5
15. Hutchinson P, O’Phelan K; Participants in the International Multidisciplinary Consensus Conference on Multimodality Monitoring. International multidisciplinary consensus conference on multimodality monitoring: cerebral metabolism. Neurocrit Care (2014) 21(Suppl 2):S148–58. doi:10.1007/s12028-014-0035-3
16. Tholance Y, Barcelos GK, Perret-Liaudet A, Omar E, Carrillon R, Grousson S, et al. Placing intracerebral probes to optimise detection of delayed cerebral ischemia and allow for the prediction of patient outcome in aneurysmal subarachnoid haemorrhage. J Cereb Blood Flow Metab (2017) 37(8):2820–32. doi:10.1177/0271678X16675880
17. Sánchez-Guerrero A, Mur-Bonet G, Vidal-Jorge M, Gándara-Sabatini D, Chocrón I, Cordero E, et al. Reappraisal of the reference levels for energy metabolites in the extracellular fluid of the human brain. J Cereb Blood Flow Metab (2017) 37(8):2742–55. doi:10.1177/0271678X16674222
18. Abi-Saab WM, Maggs DG, Jones T, Jacob R, Srihari V, Thompson J, et al. Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: effects of hyperglycemia and hypoglycemia. J Cereb Blood Flow Metab (2002) 22(3):271–9. doi:10.1097/00004647-200203000-00004
19. Glenn TC, Martin NA, Horning MA, McArthur DL, Hovda DA, Vespa P, et al. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J Neurotrauma (2015) 32(11):820–32. doi:10.1089/neu.2014.3483
20. Vespa PM, McArthur D, O’Phelan K, Glenn T, Etchepare M, Kelly D, et al. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: a microdialysis study. J Cereb Blood Flow Metab (2003) 23(7):865–77. doi:10.1097/01.WCB.0000076701.45782.EF
21. Xu Y, McArthur DL, Alger JR, Etchepare M, Hovda DA, Glenn TC, et al. Early nonischemic oxidative metabolic dysfunction leads to chronic brain atrophy in traumatic brain injury. J Cereb Blood Flow Metab (2009) 30(4):883–94. doi:10.1038/jcbfm.2009.263
22. Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, et al. Intensive insulin therapy in the medical ICU. N Engl J Med (2006) 354(5):449–61. doi:10.1056/NEJMoa052521
23. van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, et al. Intensive insulin therapy in critically ill patients. N Engl J Med (2001) 345(19):1359–67. doi:10.1056/NEJMoa011300
24. Vespa P, Boonyaputthikul R, McArthur DL, Miller C, Etchepare M, Bergsneider M, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med (2006) 34(3):850–6. doi:10.1097/01.CCM.0000201875.12245.6F
25. Van den Berghe G, Schoonheydt K, Becx P, Bruyninckx F, Wouters PJ. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology (2005) 64(8):1348–53. doi:10.1212/01.WNL.0000158442.08857.FC
26. Helbok R, Schmidt JM, Kurtz P, Hanafy KA, Fernandez L, Stuart RM, et al. Systemic glucose and brain energy metabolism after subarachnoid hemorrhage. Neurocrit Care (2010) 12(3):317–23. doi:10.1007/s12028-009-9327-4
27. Kurtz P, Claassen J, Helbok R, Schmidt J, Fernandez L, Presciutti M, et al. Systemic glucose variability predicts cerebral metabolic distress and mortality after subarachnoid hemorrhage: a retrospective observational study. Crit Care (2014) 18(3):R89. doi:10.1186/cc13857
28. Kurtz P, Claassen J, Schmidt JM, Helbok R, Hanafy KA, Presciutti M, et al. Reduced brain/serum glucose ratios predict cerebral metabolic distress and mortality after severe brain injury. Neurocrit Care (2013) 19(3):311–9. doi:10.1007/s12028-013-9919-x
29. Magnoni S, Tedesco C, Carbonara M, Pluderi M, Colombo A, Stocchetti N. Relationship between systemic glucose and cerebral glucose is preserved in patients with severe traumatic brain injury, but glucose delivery to the brain may become limited when oxidative metabolism is impaired: implications for glycemic control. Crit Care Med (2012) 40(6):1785–91. doi:10.1097/CCM.0b013e318246bd45
30. Schlenk F, Graetz D, Nagel A, Schmidt M, Sarrafzadeh AS. Insulin-related decrease in cerebral glucose despite normoglycemia in aneurysmal subarachnoid hemorrhage. Crit Care (2008) 12(1):R9. doi:10.1186/cc6776
31. Schlenk F, Nagel A, Graetz D, Sarrafzadeh AS. Hyperglycemia and cerebral glucose in aneurysmal subarachnoid hemorrhage. Intensive Care Med (2008) 34(7):1200–7. doi:10.1007/s00134-008-1044-5
32. NICE-SUGAR Study Investigators, Finfer S, Chittock DR, Su SY, Blair D, Foster D, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med (2009) 360(13):1283–97. doi:10.1056/NEJMoa0810625
33. NICE-SUGAR Study Investigators, Finfer S, Liu B, Chittock DR, Norton R, Myburgh JA, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med (2012) 367(12):1108–18. doi:10.1056/NEJMoa1204942
34. NICE-SUGAR Study Investigators for the Australian and New Zealand Intensive Care Society Clinical Trials Group and the Canadian Critical Care Trials Group, Finfer S, Chittock D, Li Y, Foster D, Dhingra V, et al. Intensive versus conventional glucose control in critically ill patients with traumatic brain injury: long-term follow-up of a subgroup of patients from the NICE-SUGAR study. Intensive Care Med (2015) 41(6):1037–47. doi:10.1007/s00134-015-3757-6
35. Badjatia N, Vespa P; Participants of the International Multi-disciplinary Consensus Conference on Multimodality Monitoring. Monitoring nutrition and glucose in acute brain injury. Neurocrit Care (2014) 21(Suppl 2):S159–67. doi:10.1007/s12028-014-0036-2
36. Kofler M, Schiefecker AJ, Beer R, Gaasch M, Rhomberg P, Stover J, et al. Enteral nutrition increases interstitial brain glucose levels in poor-grade subarachnoid hemorrhage patients. J Cereb Blood Flow Metab (2017). doi:10.1177/0271678X17700434
37. Patet C, Quintard H, Zerlauth JB, Maibach T, Carteron L, Suys T, et al. Bedside cerebral microdialysis monitoring of delayed cerebral hypoperfusion in comatose patients with poor grade aneurysmal subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry (2017) 88(4):332–8. doi:10.1136/jnnp-2016-313766
38. Rostami E, Engquist H, Howells T, Ronne-Engström E, Nilsson P, Hillered LT, et al. The correlation between cerebral blood flow measured by bedside xenon-CT and brain chemistry monitored by microdialysis in the acute phase following subarachnoid hemorrhage. Front Neurol (2017) 8:369. doi:10.3389/fneur.2017.00369
39. Rostami E, Engquist H, Johnson U, Howells T, Ronne-Engström E, Nilsson P, et al. Monitoring of cerebral blood flow and metabolism bedside in patients with subarachnoid hemorrhage – a xenon-CT and microdialysis study. Front Neurol (2014) 5:89. doi:10.3389/fneur.2014.00089
40. Bouzat P, Marques-Vidal P, Zerlauth JB, Sala N, Suys T, Schoettker P, et al. Accuracy of brain multimodal monitoring to detect cerebral hypoperfusion after traumatic brain injury. Crit Care Med (2015) 43(2):445–52. doi:10.1097/CCM.0000000000000720
41. Dizdarevic K, Hamdan A, Omerhodzic I, Kominlija-Smajic E. Modified Lund concept versus cerebral perfusion pressure-targeted therapy: a randomised controlled study in patients with secondary brain ischaemia. Clin Neurol Neurosurg (2012) 114(2):142–8. doi:10.1016/j.clineuro.2011.10.005
42. Ko SB, Choi HA, Parikh G, Helbok R, Schmidt JM, Lee K, et al. Multimodality monitoring for cerebral perfusion pressure optimization in comatose patients with intracerebral hemorrhage. Stroke (2011) 42(11):3087–92. doi:10.1161/STROKEAHA.111.623165
43. Schmidt JM, Ko SB, Helbok R, Kurtz P, Stuart RM, Presciutti M, et al. Cerebral perfusion pressure thresholds for brain tissue hypoxia and metabolic crisis after poor-grade subarachnoid hemorrhage. Stroke (2011) 42(5):1351–6. doi:10.1161/STROKEAHA.110.596874
44. Badenes R, Oddo M, Suarez JI, Antonelli M, Lipman J, Citerio G, et al. Hemoglobin concentrations and RBC transfusion thresholds in patients with acute brain injury: an international survey. Crit Care (2017) 21(1):159. doi:10.1186/s13054-017-1748-4
45. Oddo M, Levine JM, Kumar M, Iglesias K, Frangos S, Maloney-Wilensky E, et al. Anemia and brain oxygen after severe traumatic brain injury. Intensive Care Med (2012) 38(9):1497–504. doi:10.1007/s00134-012-2593-1
46. Kurtz P, Schmidt JM, Claassen J, Carrera E, Fernandez L, Helbok R, et al. Anemia is associated with metabolic distress and brain tissue hypoxia after subarachnoid hemorrhage. Neurocrit Care (2010) 13(1):10–6. doi:10.1007/s12028-010-9357-y
47. Oddo M, Milby A, Chen I, Frangos S, MacMurtrie E, Maloney-Wilensky E, et al. Hemoglobin concentration and cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke (2009) 40(4):1275–81. doi:10.1161/STROKEAHA.108.527911
48. Leal-Noval SR, Rincón-Ferrari MD, Marin-Niebla A, Cayuela A, Arellano-Orden V, Marín-Caballos A, et al. Transfusion of erythrocyte concentrates produces a variable increment on cerebral oxygenation in patients with severe traumatic brain injury: a preliminary study. Intensive Care Med (2006) 32(11):1733–40. doi:10.1007/s00134-006-0376-2
49. Smith MJ, Stiefel MF, Magge S, Frangos S, Bloom S, Gracias V, et al. Packed red blood cell transfusion increases local cerebral oxygenation. Crit Care Med (2005) 33(5):1104–8. doi:10.1097/01.CCM.0000162685.60609.49
50. Kurtz P, Helbok R, Claassen J, Schmidt JM, Fernandez L, Stuart RM, et al. The effect of packed red blood cell transfusion on cerebral oxygenation and metabolism after subarachnoid hemorrhage. Neurocrit Care (2016) 24(1):118–21. doi:10.1007/s12028-015-0180-3
51. Zygun DA, Nortje J, Hutchinson PJ, Timofeev I, Menon DK, Gupta AK. The effect of red blood cell transfusion on cerebral oxygenation and metabolism after severe traumatic brain injury. Crit Care Med (2009) 37(3):1074–8. doi:10.1097/CCM.0b013e318194ad22
52. Helmerhorst HJ, Roos-Blom MJ, van Westerloo DJ, de Jonge E. Association between arterial hyperoxia and outcome in subsets of critical illness: a systematic review, meta-analysis, and meta-regression of cohort studies. Crit Care Med (2015) 43(7):1508–19. doi:10.1097/CCM.0000000000000998
53. Tisdall MM, Tachtsidis I, Leung TS, Elwell CE, Smith M. Increase in cerebral aerobic metabolism by normobaric hyperoxia after traumatic brain injury. J Neurosurg (2008) 109(3):424–32. doi:10.3171/JNS/2008/109/9/0424
54. Tolias CM, Reinert M, Seiler R, Gilman C, Scharf A, Bullock MR. Normobaric hyperoxia-induced improvement in cerebral metabolism and reduction in intracranial pressure in patients with severe head injury: a prospective historical cohort-matched study. J Neurosurg (2004) 101(3):435–44. doi:10.3171/jns.2004.101.3.0435
55. Nortje J, Coles JP, Timofeev I, Fryer TD, Aigbirhio FI, Smielewski P, et al. Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: preliminary findings. Crit Care Med (2008) 36(1):273–81. doi:10.1097/01.CCM.0000292014.60835.15
56. Quintard H, Patet C, Suys T, Marques-Vidal P, Oddo M. Normobaric hyperoxia is associated with increased cerebral excitotoxicity after severe traumatic brain injury. Neurocrit Care (2015) 22(2):243–50. doi:10.1007/s12028-014-0062-0
57. Ghosh A, Highton D, Kolyva C, Tachtsidis I, Elwell CE, Smith M. Hyperoxia results in increased aerobic metabolism following acute brain injury. J Cereb Blood Flow Metab (2017) 37(8):2910–20. doi:10.1177/0271678X16679171
58. Vidal-Jorge M, Sanchez-Guerrero A, Mur-Bonet G, Castro L, Rădoi A, Riveiro M, et al. Does normobaric hyperoxia cause oxidative stress in the injured brain? A microdialysis study using 8-iso-prostaglandin F2alpha as a biomarker. J Neurotrauma (2017) 34(19):2731–42. doi:10.1089/neu.2017.4992
59. Rockswold SB, Rockswold GL, Zaun DA, Liu J. A prospective, randomized Phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J Neurosurg (2013) 118(6):1317–28. doi:10.3171/2013.2.JNS121468
60. Timofeev I, Carpenter KL, Nortje J, Al-Rawi PG, O’Connell MT, Czosnyka M, et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain (2011) 134(Pt 2):484–94. doi:10.1093/brain/awq353
61. Stover JF, Belli A, Boret H, Bulters D, Sahuquillo J, Schmutzhard E, et al. Nitric oxide synthase inhibition with the antipterin VAS203 improves outcome in moderate and severe traumatic brain injury: a placebo-controlled randomized Phase IIa trial (NOSTRA). J Neurotrauma (2014) 31(19):1599–606. doi:10.1089/neu.2014.3344
62. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab (2014) 34(5):845–51. doi:10.1038/jcbfm.2014.23
63. Carpenter KL, Timofeev I, Nortje J, Czosnyka M, Pickard JD, Hutchinson PJ. A microdialysis study of oral vigabatrin administration in head injury patients: preliminary evaluation of multimodality monitoring. Acta Neurochir Suppl (2012) 114:271–6. doi:10.1007/978-3-7091-0956-4_53
64. Shannon RJ, Timofeev I, Nortje J, Hutchinson PJ, Carpenter KL. Monitoring vigabatrin in head injury patients by cerebral microdialysis: obtaining pharmacokinetic measurements in a neurocritical care setting. Br J Clin Pharmacol (2014) 78(5):981–95. doi:10.1111/bcp.12414
65. Jalloh I, Helmy A, Howe DJ, Shannon RJ, Grice P, Mason A, et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab (2017) 37(7):2626–38. doi:10.1177/0271678X16672665
66. Bouzat P, Sala N, Suys T, Zerlauth JB, Marques-Vidal P, Feihl F, et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med (2014) 40(3):412–21. doi:10.1007/s00134-013-3203-6
67. Quintard H, Patet C, Zerlauth JB, Suys T, Bouzat P, Pellerin L, et al. Improvement of neuroenergetics by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J Neurotrauma (2016) 33(7):681–7. doi:10.1089/neu.2015.4057
68. Tanguy M, Seguin P, Laviolle B, Bleichner JP, Morandi X, Malledant Y. Cerebral microdialysis effects of propofol versus midazolam in severe traumatic brain injury. J Neurotrauma (2012) 29(6):1105–10. doi:10.1089/neu.2011.1817
69. Shannon RJ, Carpenter KL, Guilfoyle MR, Helmy A, Hutchinson PJ. Cerebral microdialysis in clinical studies of drugs: pharmacokinetic applications. J Pharmacokinet Pharmacodyn (2013) 40(3):343–58. doi:10.1007/s10928-013-9306-4
70. Thelin EP, Carpenter KL, Hutchinson PJ, Helmy A. Microdialysis monitoring in clinical traumatic brain injury and its role in neuroprotective drug development. AAPS J (2017) 19(2):367–76. doi:10.1208/s12248-016-0027-7
71. Frasca D, Dahyot-Fizelier C, Adier C, Mimoz O, Debaene B, Couet W, et al. Metronidazole and hydroxymetronidazole central nervous system distribution: 1. Microdialysis assessment of brain extracellular fluid concentrations in patients with acute brain injury. Antimicrob Agents Chemother (2014) 58(2):1019–23. doi:10.1128/AAC.01760-13
72. Poeppl W, Zeitlinger M, Donath O, Wurm G, Müller M, Botha F, et al. Penetration of doripenem in human brain: an observational microdialysis study in patients with acute brain injury. Int J Antimicrob Agents (2012) 39(4):343–5. doi:10.1016/j.ijantimicag.2011.11.019
73. Coles JP, Cunningham AS, Salvador R, Chatfield DA, Carpenter A, Pickard JD, et al. Early metabolic characteristics of lesion and nonlesion tissue after head injury. J Cereb Blood Flow Metab (2009) 29(5):965–75. doi:10.1038/jcbfm.2009.22
74. Coles JP, Fryer TD, Smielewski P, Chatfield DA, Steiner LA, Johnston AJ, et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab (2004) 24(2):202–11. doi:10.1097/01.WCB.0000103022.98348.24
75. Veenith TV, Carter EL, Geeraerts T, Grossac J, Newcombe VF, Outtrim J, et al. Pathophysiologic mechanisms of cerebral ischemia and diffusion hypoxia in traumatic brain injury. JAMA Neurol (2016) 73(5):542–50. doi:10.1001/jamaneurol.2016.0091
76. Carpenter KL, Jalloh I, Gallagher CN, Grice P, Howe DJ, Mason A, et al. (13)C-labelled microdialysis studies of cerebral metabolism in TBI patients. Eur J Pharm Sci (2014) 57:87–97. doi:10.1016/j.ejps.2013.12.012
77. Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci (2015) 9:112. doi:10.3389/fnins.2015.00112
78. Gallagher CN, Carpenter KL, Grice P, Howe DJ, Mason A, Timofeev I, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain (2009) 132(Pt 10):2839–49. doi:10.1093/brain/awp202
79. Glenn TC, Kelly DF, Boscardin WJ, McArthur DL, Vespa P, Oertel M, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab (2003) 23(10):1239–50. doi:10.1097/01.WCB.0000089833.23606.7F
80. Jalloh I, Carpenter KL, Grice P, Howe DJ, Mason A, Gallagher CN, et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: microdialysis studies using 1,2-(13)C2 glucose. J Cereb Blood Flow Metab (2015) 35(1):111–20. doi:10.1038/jcbfm.2014.177
81. Jalloh I, Carpenter KL, Helmy A, Carpenter TA, Menon DK, Hutchinson PJ. Glucose metabolism following human traumatic brain injury: methods of assessment and pathophysiological findings. Metab Brain Dis (2015) 30(3):615–32. doi:10.1007/s11011-014-9628-y
82. Jalloh I, Helmy A, Shannon RJ, Gallagher CN, Menon DK, Carpenter KL, et al. Lactate uptake by the injured human brain: evidence from an arteriovenous gradient and cerebral microdialysis study. J Neurotrauma (2013) 30(24):2031–7. doi:10.1089/neu.2013.2947
83. Hillered L, Dahlin AP, Clausen F, Chu J, Bergquist J, Hjort K, et al. Cerebral microdialysis for protein biomarker monitoring in the neurointensive care setting – a technical approach. Front Neurol (2014) 5:245. doi:10.3389/fneur.2014.00245
84. Vespa P, Tubi M, Claassen J, Buitrago-Blanco M, McArthur D, Velazquez AG, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol (2016) 79(4):579–90. doi:10.1002/ana.24606
85. Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med (2011) 17(4):439–47. doi:10.1038/nm.2333
86. Hinzman JM, Wilson JA, Mazzeo AT, Bullock MR, Hartings JA. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J Neurotrauma (2016) 33(19):1775–83. doi:10.1089/neu.2015.4226
87. Rogers ML, Leong CL, Gowers SA, Samper IC, Jewell SL, Khan A, et al. Simultaneous monitoring of potassium, glucose and lactate during spreading depolarization in the injured human brain – proof of principle of a novel real-time neurochemical analysis system, continuous online microdialysis. J Cereb Blood Flow Metab (2017) 37(5):1883–95. doi:10.1177/0271678X16674486
88. Sakowitz OW, Santos E, Nagel A, Krajewski KL, Hertle DN, Vajkoczy P, et al. Clusters of spreading depolarizations are associated with disturbed cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke (2013) 44(1):220–3. doi:10.1161/STROKEAHA.112.672352
89. Antunes AP, Schiefecker AJ, Beer R, Pfausler B, Sohm F, Fischer M, et al. Higher brain extracellular potassium is associated with brain metabolic distress and poor outcome after aneurysmal subarachnoid hemorrhage. Crit Care (2014) 18(3):R119. doi:10.1186/cc13916
90. Martínez-Valverde T, Sánchez-Guerrero A, Vidal-Jorge M, Torné R, Castro L, Gandara D, et al. Characterization of the ionic profile of the extracellular space of the injured and ischemic brain: a microdialysis study. J Neurotrauma (2017) 34(1):74–85. doi:10.1089/neu.2015.4334
91. Martínez-Valverde T, Vidal-Jorge M, Montoya N, Sánchez-Guerrero A, Manrique S, Munar F, et al. Brain microdialysis as a tool to explore the ionic profile of the brain extracellular space in neurocritical patients: a methodological approach and feasibility study. J Neurotrauma (2015) 32(1):7–16. doi:10.1089/neu.2014.3473
92. Kofler M, Schiefecker A, Ferger B, Beer R, Sohm F, Broessner G, et al. Cerebral taurine levels are associated with brain edema and delayed cerebral infarction in patients with aneurysmal subarachnoid hemorrhage. Neurocrit Care (2015) 23(3):321–9. doi:10.1007/s12028-015-0140-y
93. Guilfoyle MR, Carpenter KL, Helmy A, Pickard JD, Menon DK, Hutchinson PJ. Matrix metalloproteinase expression in contusional traumatic brain injury: a paired microdialysis study. J Neurotrauma (2015) 32(20):1553–9. doi:10.1089/neu.2014.3764
94. Helbok R, Schiefecker AJ, Beer R, Dietmann A, Antunes AP, Sohm F, et al. Early brain injury after aneurysmal subarachnoid hemorrhage: a multimodal neuromonitoring study. Crit Care (2015) 19:75. doi:10.1186/s13054-015-0809-9
95. Roberts DJ, Jenne CN, Léger C, Kramer AH, Gallagher CN, Todd S, et al. Association between the cerebral inflammatory and matrix metalloproteinase responses after severe traumatic brain injury in humans. J Neurotrauma (2013) 30(20):1727–36. doi:10.1089/neu.2012.2842
96. Roberts DJ, Jenne CN, Léger C, Kramer AH, Gallagher CN, Todd S, et al. A prospective evaluation of the temporal matrix metalloproteinase response after severe traumatic brain injury in humans. J Neurotrauma (2013) 30(20):1717–26. doi:10.1089/neu.2012.2841
97. Sarrafzadeh A, Copin JC, Bengualid DJ, Turck N, Vajkoczy P, Bijlenga P, et al. Matrix metalloproteinase-9 concentration in the cerebral extracellular fluid of patients during the acute phase of aneurysmal subarachnoid hemorrhage. Neurol Res (2012) 34(5):455–61. doi:10.1179/1743132812Y.0000000018
98. Helmy A, Antoniades CA, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Principal component analysis of the cytokine and chemokine response to human traumatic brain injury. PLoS One (2012) 7(6):e39677. doi:10.1371/journal.pone.0039677
99. Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog Neurobiol (2011) 95(3):352–72. doi:10.1016/j.pneurobio.2011.09.003
100. Helmy A, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Sex and the cytokines: are there fundamental differences in response to brain injury? Neurosurgery (2011) 69(4):E1029–30. doi:10.1227/NEU.0b013e3182299839
101. Mellergard P, Sjogren F, Hillman J. The cerebral extracellular release of glycerol, glutamate, and FGF2 is increased in older patients following severe traumatic brain injury. J Neurotrauma (2012) 29(1):112–8. doi:10.1089/neu.2010.1732
102. Bache S, Rasmussen R, Rossing M, Hammer NR, Juhler M, Friis-Hansen L, et al. Detection and quantification of microRNA in cerebral microdialysate. J Transl Med (2015) 13:149. doi:10.1186/s12967-015-0505-1
103. Clausen F, Marklund N, Lewen A, Enblad P, Basu S, Hillered L. Interstitial F(2)-isoprostane 8-iso-PGF(2alpha) as a biomarker of oxidative stress after severe human traumatic brain injury. J Neurotrauma (2012) 29(5):766–75. doi:10.1089/neu.2011.1754
104. Shannon RJ, van der Heide S, Carter EL, Jalloh I, Menon DK, Hutchinson PJ, et al. Extracellular N-acetylaspartate in human traumatic brain injury. J Neurotrauma (2016) 33(4):319–29. doi:10.1089/neu.2015.3950
105. Tisdall MM, Rejdak K, Kitchen ND, Smith M, Petzold A. The prognostic value of brain extracellular fluid nitric oxide metabolites after traumatic brain injury. Neurocrit Care (2013) 19(1):65–8. doi:10.1007/s12028-011-9633-5
106. Zeiler FA, Thelin EP, Czosnyka M, Hutchinson PJ, Menon DK, Helmy A. Cerebrospinal fluid and microdialysis cytokines in aneurysmal subarachnoid hemorrhage: a scoping systematic review. Front Neurol (2017) 8:379. doi:10.3389/fneur.2017.00379
107. Zeiler FA, Thelin EP, Czosnyka M, Hutchinson PJ, Menon DK, Helmy A. Cerebrospinal fluid and microdialysis cytokines in severe traumatic brain injury: a scoping systematic review. Front Neurol (2017) 8:331. doi:10.3389/fneur.2017.00331
108. Magnoni S, Brody DL. New perspectives on amyloid-beta dynamics after acute brain injury: moving between experimental approaches and studies in the human brain. Arch Neurol (2010) 67(9):1068–73. doi:10.1001/archneurol.2010.214
109. Magnoni S, Esparza TJ, Conte V, Carbonara M, Carrabba G, Holtzman DM, et al. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain (2012) 135(Pt 4):1268–80. doi:10.1093/brain/awr286
110. Magnoni S, Mac Donald CL, Esparza TJ, Conte V, Sorrell J, Macrì M, et al. Quantitative assessments of traumatic axonal injury in human brain: concordance of microdialysis and advanced MRI. Brain (2015) 138(Pt 8):2263–77. doi:10.1093/brain/awv152
111. Marklund N, Farrokhnia N, Hanell A, Vanmechelen E, Enblad P, Zetterberg H, et al. Monitoring of beta-amyloid dynamics after human traumatic brain injury. J Neurotrauma (2014) 31(1):42–55. doi:10.1089/neu.2013.2964
112. Petzold A, Tisdall MM, Girbes AR, Martinian L, Thom M, Kitchen N, et al. In vivo monitoring of neuronal loss in traumatic brain injury: a microdialysis study. Brain (2011) 134(Pt 2):464–83. doi:10.1093/brain/awq360
113. Tsitsopoulos PP, Marklund N. Amyloid-beta peptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury: a review of experimental and clinical studies. Front Neurol (2013) 4:79. doi:10.3389/fneur.2013.00079
114. Helbok R, Schiefecker A, Delazer M, Beer R, Bodner T, Pfausler B, et al. Cerebral tau is elevated after aneurysmal subarachnoid haemorrhage and associated with brain metabolic distress and poor functional and cognitive long-term outcome. J Neurol Neurosurg Psychiatry (2015) 86(1):79–86. doi:10.1136/jnnp-2013-307326
115. Schiefecker AJ, Dietmann A, Beer R, Pfausler B, Lackner P, Kofler M, et al. Neuroinflammation is associated with brain extracellular TAU-protein release after spontaneous subarachnoid hemorrhage. Curr Drug Targets (2017) 18(12):1408–16. doi:10.2174/1389450117666160201111804
116. Wijayatilake DS, Talati C, Panchatsharam S. The monitoring and management of severe traumatic brain injury in the united kingdom: is there a consensus? A national survey. J Neurosurg Anesthesiol (2015) 27(3):241–5. doi:10.1097/ANA.0000000000000143
117. Hifumi T, Kawakita K, Yoda T, Okazaki T, Kuroda Y. Association of brain metabolites with blood lactate and glucose levels with respect to neurological outcomes after out-of-hospital cardiac arrest: a preliminary microdialysis study. Resuscitation (2017) 110:26–31. doi:10.1016/j.resuscitation.2016.10.013
118. Rivera-Espinosa L, Floriano-Sánchez E, Pedraza-Chaverrí J, Coballase-Urrutia E, Sampieri AI, Ortega-Cuellar D, et al. Contributions of microdialysis to new alternative therapeutics for hepatic encephalopathy. Int J Mol Sci (2013) 14(8):16184–206. doi:10.3390/ijms140816184
119. Dahlin AP, Purins K, Clausen F, Chu J, Sedigh A, Lorant T, et al. Refined microdialysis method for protein biomarker sampling in acute brain injury in the neurointensive care setting. Anal Chem (2014) 86(17):8671–9. doi:10.1021/ac501880u
120. Das C, Wang G, Sun Q, Ledden B. Multiplexed and fully automated detection of metabolic biomarkers using microdialysis probe. Sens Actuators B Chem (2017) 238:633–40. doi:10.1016/j.snb.2016.07.097
Keywords: microdialysis, traumatic brain injury, subarachnoid hemorrhage, cerebral metabolism, ischemia, hypoxia, biomarkers, neurointensive care
Citation: Carteron L, Bouzat P and Oddo M (2017) Cerebral Microdialysis Monitoring to Improve Individualized Neurointensive Care Therapy: An Update of Recent Clinical Data. Front. Neurol. 8:601. doi: 10.3389/fneur.2017.00601
Received: 19 May 2017; Accepted: 27 October 2017;
Published: 13 November 2017
Edited by:
Niklas Marklund, Lund University, SwedenReviewed by:
Raimund Helbok, Innsbruck Medical University, AustriaCopyright: © 2017 Carteron, Bouzat and Oddo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Oddo, bWF1cm8ub2Rkb0BjaHV2LmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.