 Julián Benito-León
Julián Benito-León Martin Laurence
Martin Laurence
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Neurol., 16 October 2017
Sec. Multiple Sclerosis and Neuroimmunology
Volume 8 - 2017 | https://doi.org/10.3389/fneur.2017.00535
Multiple sclerosis (MS) is a chronic inflammatory disorder of the central nervous system. Infectious triggers of MS are being actively investigated. Substantial evidence supports the involvement of the Epstein-Barr virus (EBV), though other viruses, bacteria, protists, and fungi are also being considered. Many links between fungi and diseases involving chronic inflammation have been found recently. Evidence linking MS and fungi is reviewed here. The HLA-DRB1*15 allele group is the most important genetic risk factor of MS, and is a risk factor in several other conditions linked to fungal infections. Many biomarkers of MS are consistent with fungal infections, such as IL-17, chitotriosidase, and antibodies against fungi. Dimethyl fumarate (DMF), first used as an industrial fungicide, was recently repurposed to reduce MS symptoms. Its mechanisms of action in MS have not been firmly established. The low risk of MS during childhood and its moderate association with herpes simplex virus type 2 suggest genital exposure to microbes (including fungi) should be investigated as a possible trigger. Molecular and epidemiological evidence support a role for infections such as EBV in MS. Though fungal infections have not been widely studied in MS, many lines of evidence are consistent with a fungal etiology. Future microbiome and serological studies should consider fungi as a possible risk factor for MS, and future clinical studies should consider the effect of fungicides other than DMF on MS symptoms.
Multiple sclerosis (MS) is the most common cause of non-traumatic neurological disability in young adults (1, 2). It is a complex heterogeneous inflammatory disorder characterized by nerve axons in the central nervous system (CNS) losing their myelin sheath (3). Understanding this process appears to be the largest obstacle in developing effective therapies.
The best established risk factors of MS are Epstein-Barr virus (EBV) seropositivity (4–6), major histocompatibility complex class II HLA-DRB1*15 alleles (7), smoking (8), vitamin D deficiency (9), young adulthood (10), and female sex (11). MS is considered an autoimmune disease in which the role of infections is actively debated (4, 12), though proof of autoimmunity is currently lacking.
Numerous infectious agents are suspected of triggering MS, and emerging evidence suggests links between established MS and gut microbiota (12). Though most studies focus on bacteria, fungi may also play an important role (13). Many links between fungi and diseases involving idiopathic inflammation have been found recently. Candida in the gut is associated with psoriasis (14, 15) and Crohn’s disease (16, 17). Antibodies against fungi are a risk factor of psoriasis (18, 19), and fungicides improve symptoms (20). Antibodies against fungal mannoproteins are a risk factor of ankylosing spondylitis (21), systemic lupus erythematosus (22, 23), sarcoidosis (24), and Crohn’s disease (25).
Few groups have considered a role for fungi in MS. In 1981, Truss reported the resolution of symptoms in five MS cases following antifungal therapy (26). In 2008, Ramos and colleagues reported finding serum antibodies against Candida in seven out of eight MS patients, while finding none in 10 healthy controls (27). In 2010, this association was replicated in a larger case-control study (28), and again in 2013 in a small case-control study that linked anti-Candida antibodies in the cerebrospinal fluid (CSF) to MS (29). In this review, we analyze the evidence linking MS to a possible fungal etiology.
The HLA-DRB1*15 allele group is the strongest genetic risk factor of MS (7). Of the three common HLA-DRB1*15 alleles, HLA-DRB1*1501 and HLA-DRB1*1503 are associated with MS, while HLA-DRB1*1502 is not (7). These three alleles are very similar: as compared to HLA-DRB1*1501, HLA-DRB1*1502 substitutes valine for glycine at position 86, and HLA-DRB1*1503 substitutes tyrosine for histidine at position 30 (7). HLA-DRB1*1501 and HLA-DRB1*1503 are two of many alleles forming half of the HLA-DR protein complex, which holds peptides collected by antigen-presenting cells (monocytes, macrophages, dendritic cells, and B cells) for presentation to CD4+ T cells. They thus play a key role in determining which antigens induce a T cell mediated immune response, as well as which antigens warrant naive B cell maturation into memory B cells.
The HLA-DRB1*15 allele group is an important risk factor in many other inflammatory conditions, some of which are suspected of having a fungal etiology (Table 1). Allergic bronchopulmonary aspergillosis (ABPA) is caused by an abnormal immune response against usually benign fungi which are often present in the airways (30). The HLA-DRB1*15 alleles associated with ABPA match those associated with MS: HLA-DRB1*1501 and HLA-DRB1*1503, but not HLA-DRB1*1502 (30, 31). Pulmonary sarcoidosis is suspected of being caused by an immune response to fungal antigens in the airways (32). Granulomatous prostatitis can be caused by fungal infections, though most cases are idiopathic (33); the recent discovery of a fungicidal protein in the prostate (34) suggests a fastidious fungal infection is present in this site (35, 36). Uveitis can be caused by different infectious agents, but in most cases none can be found (37). Uveitis is associated with various idiopathic inflammatory diseases including ankylosing spondylitis, Behçet’s disease, sarcoidosis, and Crohn’s disease (37); it is also associated with MS, and often coincides with MS onset (38). Several studies have linked idiopathic uveitis with fungal infections (Table 1).
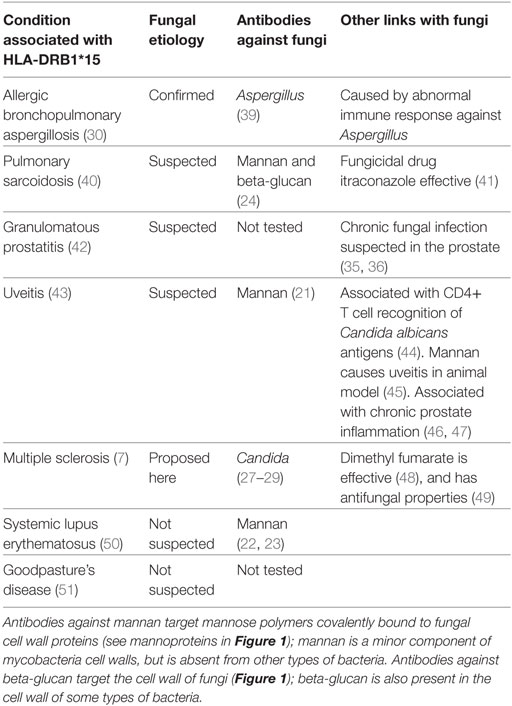
Table 1. Inflammatory conditions associated with HLA-DRB1*15.
As first proposed for ABPA (30), the HLA-DRB1*15 allele group appears to increase immune sensitivity to fungi: this likely protects the host from certain infections, while sometimes causing chronic inflammation due to exposure to usually benign fungi such as Aspergillus fumigatus.
HLA-DRB1*15 increases the risk of various conditions either caused or suspected of being caused by exposure to fungi (Table 1). This suggests MS risk might also be increased by exposure to fungi. All conditions in Table 1 that were studied for antibodies against fungi showed a positive association. This includes three studies which found that antibodies against various Candida species are associated with MS (27–29). With the exception of ABPA, it remains unclear how such antibodies can increase the risk of any of these diseases. Are memory B cells recognizing fungal epitopes directly involved, or are they a biomarker of another mechanism? The elimination of B cells in MS rapidly improves symptoms (52), suggesting they are directly involved, though the association with fungi remains speculative.
Candida species are the most common fungus on human mucosal surfaces, typically colonizing the host asymptomatically (53). Nearly half of adults have Candida in the mouth (53) or gut (14), and a majority of women have had genital exposure (54). Though the hypothesis of Candida infections reaching the CNS and directly causing MS has been proposed (27, 29), it appears unlikely that this would have gone unnoticed.
Antibodies against Candida indicate a past fungal infection produced memory B cells recognizing fungal epitopes. Memory B cells which recognize both a fungal epitope and an epitope in the CNS could explain the link between Candida antibodies and MS risk. As EBV infects naive B cells in mucosal surfaces exposed to fungi, “forbidden” memory B cells targeting fungal epitopes could be produced (55). Though speculative, this mechanism would explain three important observations. First, HLA-DRB1*15’s association with MS could be due to an increased probability of producing an immune response against fungi (Tables 1 and 2). Second, EBV’s association with MS could be due to naive B cells differentiating into “forbidden” memory B cells because they were exposed to both fungal antigens and EBV during affinity maturation (Table 3), resulting in the inadvertent recognition of an epitope in the CNS (55). Third, women’s elevated risk of MS (11) could be explained by higher antibody titers against Candida albicans measured in healthy women as compared to men, suggesting a higher probability of fungal infections in women perhaps due to genital exposure (56).
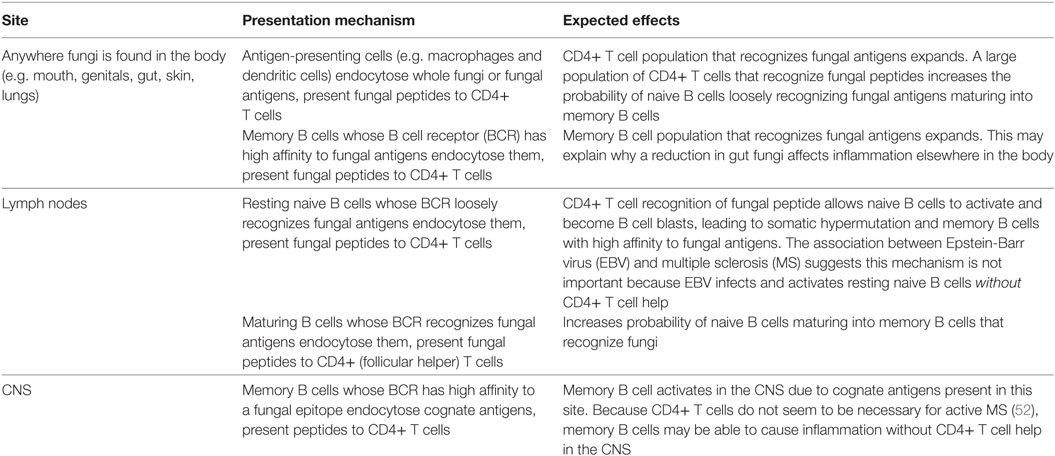
Table 2. Sites where HLA-DRB1*15 could be presenting fungal antigen peptides to CD4+ T cells, increasing the probability of memory B cells recognizing a “forbidden” epitope in the central nervous system (CNS).
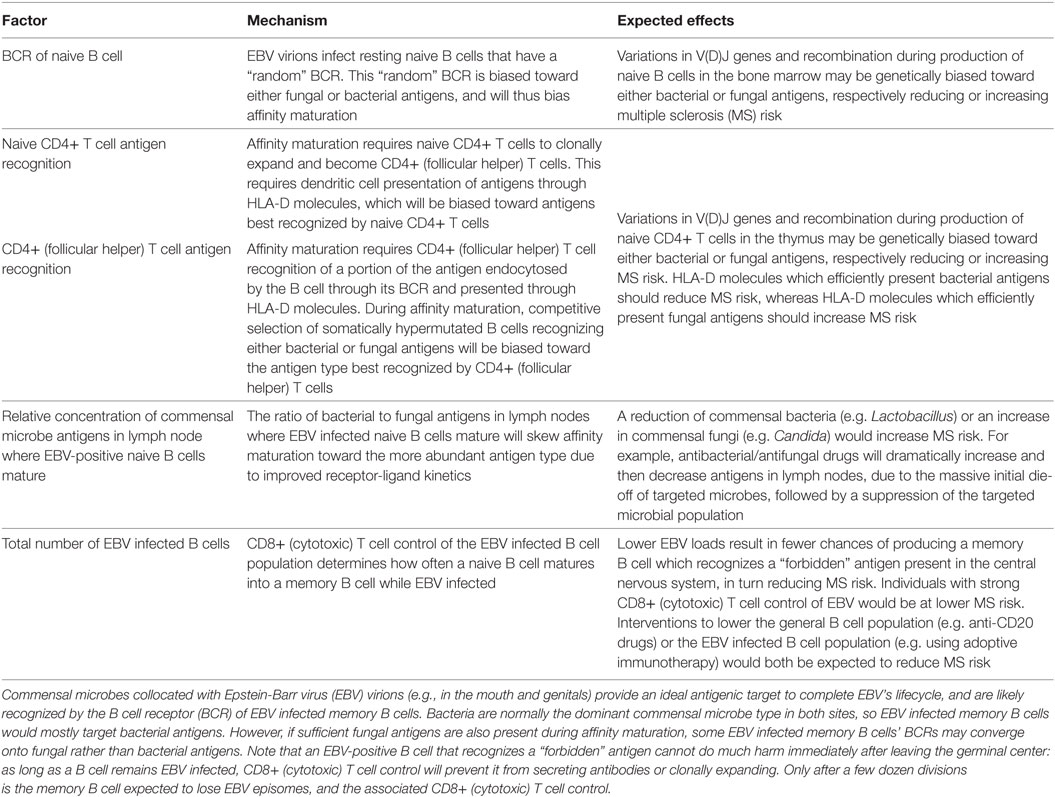
Table 3. Factors affecting the number of EBV infected memory B cells recognizing fungal antigens.
Mannoproteins are a ubiquitous component of fungal cell walls composed of protein-bound branched mannose polymers (Figure 1). Mammalian glycoproteins rarely have terminal mannose (57), making mannoproteins good antigens (58, 59). Innate immune recognition of mannoproteins by leukocytes occurs through macrophage mannose receptor (MMR) uptake, followed by presentation of mannoprotein peptides to CD4+ T cells using HLA-D molecules (58–60). As compared to cell walls of Gram-negative bacteria or Saccharomyces cerevisiae (a benign fungus), C. albicans mannoproteins induce strong production of IL-17 through this process (60). The proposed functions of the MMR include the clearance of fungi, bacteria, viruses, and homeostasis of human glycoproteins (61). The MMR is expressed by macrophages in active MS lesions, but not in controls or inactive disease (62). This suggests infectious agents inside the CNS may be contributing to inflammation in MS, or alternatively that human cell debris elicit MMR expression.
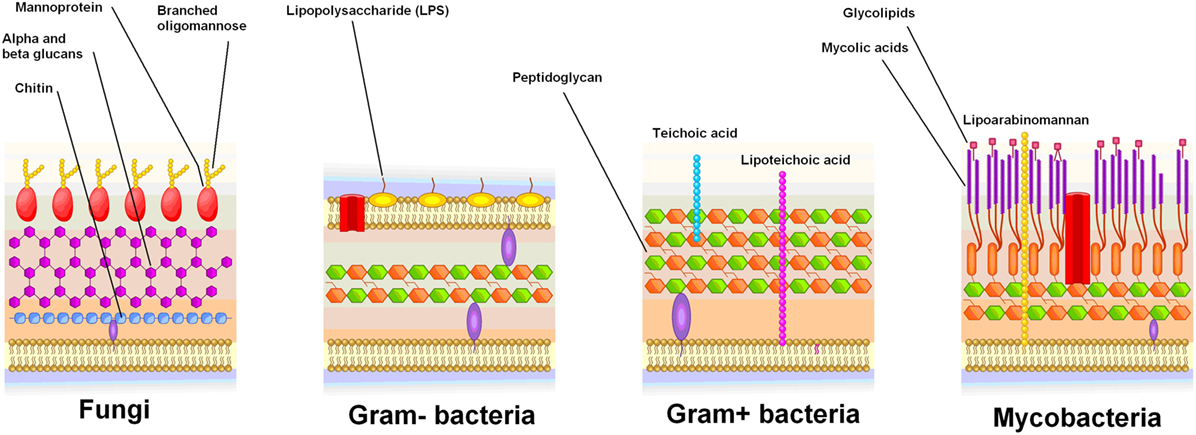
Figure 1. Cell wall structure of fungi and bacteria. Mannoproteins are specific to fungi, and are highly antigenic.
Mannose-binding lectin (MBL) is an innate immune defense protein synthesized in the liver, which binds to terminal mannose, N-acetylglucosamine, and fucose on microbial cell walls (63), and initiates the lectin complement pathway by activating mannose-binding protein-associated serine protease 2 (MASP-2) (64). MBL also binds to defective human glycoproteins lacking protective terminal residues such as galactose (57). Functional MBL/MASP-2 complexes are elevated in the plasma of MS cases as compared to controls, but not in the CSF (65). This suggests the immune response against infectious agents, such as bacteria or fungi, outside the CNS may be contributing to MS risk.
IL-17 is a cytokine produced by T cells (especially Th17 cells) and innate immune system cells (66) in response to intracellular and extracellular (67) bacterial and fungal infections (68, 69). For example, chronic Helicobacter pylori infections of the stomach induce IL-17 production (70). Genetic defects in the IL-17 pathway inhibit the clearance of fungal infections such as C. albicans (71).
IL-17 is also elevated in many idiopathic inflammatory diseases which do not have known infectious etiologies, such as psoriasis, ankylosing spondylitis, systemic lupus erythematosus, Crohn’s disease, and MS (72, 73). Thus IL-17 seems to play a role in chronic inflammation where infectious stimuli are lacking. Note that some of these conditions may have an as-yet-undiscovered infectious etiology, as became apparent for gastritis once H. pylori was discovered (74).
Though IL-17 has been implicated in the clearance of both bacterial and fungal infections (67, 75), its production is readily induced by C. albicans mannoproteins as compared to bacterial antigens (60). In a mouse model of arthritis stimulated by the injection of bacterial antigens into knee-joints, the addition of minute amounts of fungal antigens skewed the T cell immune response toward Th17 cells, tripling IL-17 concentration in the joints (76).
Though IL-17 is unambiguously associated with both fungal infections and MS, this association only provides weak circumstantial evidence of a causative link between MS and fungi, as IL-17 is also associated with bacterial infections and many idiopathic inflammatory diseases. Since IL-17 production is very sensitive to fungal antigens (60), even low fungal loads inside the CNS could provoke a Th17 immune response, especially in the presence of other inflammatory stimuli (76).
Chitotriosidase is a protein produced by activated macrophages, which hydrolyzes chitin (77). Chitin is a polysaccharide present in fungal cell walls, yet absent from bacterial and mammalian cells (Figure 1) (77). The exact function of chitotriosidase in the clearance of fungal infections is unclear (77). Macrophage chitotriosidase synthesis is stimulated by chitin, but not by other immunostimulatory compounds such as lipopolysaccharide or zymosan (78), suggesting that it is part of the innate immune response against chitin-bearing microbes (77).
Elevated chitotriosidase levels occur in a variety of diseases, both infectious and non-infectious (79). Chitotriosidase is thought to be a biomarker of macrophage activation, and may have functions unrelated to chitin (79). Elevated chitotriosidase in the CSF is an important biomarker of MS (80–82). This suggests a fungal infection inside the CNS may be contributing to inflammation in MS, or alternatively that macrophages produce chitotriosidase in response to non-chitin stimuli.
Calprotectin is a broad-spectrum antimicrobial protein complex produced mainly by neutrophils, but also by monocytes, macrophages, endothelial, and epithelial cells, which is released during infections or inflammation (83). Fungi are particularly susceptible to calprotectin, with minimum inhibitory concentrations of ~16 μg/ml as compared to 64–256 μg/ml for bacteria (84). Elevated calprotectin has been reported in many autoimmune diseases such as Crohn’s disease and systemic lupus erythematosus (85). Calprotectin levels in the CSF are markedly elevated during MS flare-ups (86), suggesting an infection may be present inside the CNS.
Dimethyl fumarate (DMF) is an inexpensive fungicide (49) long used to protect consumer goods from fungi (87). In 2013, DMF was repurposed to treat MS in the United States of America (48). The New England Journal of Medicine nicknamed this the “Poison Chair” treatment, referring to its continued use as a fungicide on furniture, which sometimes causes cutaneous irritation (88). DMF had previously been approved in Germany to treat psoriasis (88). No consensus exists as to what its mechanisms of action are in MS or psoriasis. Anti-inflammatory interaction with the nuclear factor (erythroid-derived 2)-like 2 pathway has been proposed to explain the efficacy of DMF in reducing MS symptoms (89), though other immunomodulatory mechanisms continue to be investigated (90).
Several antifungal drugs have been shown to improve psoriasis symptoms: nystatin (20, 91–93), ketoconazole (94, 95), and itraconazole (96, 97). Elevated antibodies against Malassezia furfur (18, 19) and C. albicans (19) are risk factors of psoriasis, as is Candida colonization of the gut (14, 15). Cutaneous exposure to lysed M. furfur cells triggers psoriasis in susceptible individuals (98). These links between psoriasis and fungi suggest DMF’s antifungal properties may be important. The paucity of studies linking antifungal compounds other than DMF with a reduction in MS symptoms prevents a similar conclusion from being reached for MS: if future studies demonstrate the efficacy of many antifungal drugs in MS, this would provide strong evidence of a fungal etiology.
The effect of oral nystatin on psoriasis is surprising, as it is not absorbed (99), thus cannot reach fungi beyond the gut. Yet oral nystatin has been repeatedly shown to reduce psoriasis symptoms (20, 91–93), despite never reaching sites of psoriatic inflammation. Perhaps Candida colonization of the gut causes the proliferation of lymphocytes recognizing antigens on the skin (100, 101)—in other words an id reaction (102) stimulated by fungi in the gut. Unlike in psoriasis, the long-term use of oral nystatin has not been widely studied in MS. It would be interesting to know if it reduces MS symptoms as reported by Truss (26): this would suggest fungi outside the CNS are contributing to MS.
The following example illustrates the four main hypothesized steps required to produce memory B cells recognizing an MS-causing antigen present in the CNS. They are based on our recent review of EBV and MS, which concluded that EBV coerces memory B cells to recognize commensal microbe antigens (55), resulting in “forbidden” memory B cells recognizing an antigen in the CNS, as originally proposed by Pender (103, 104). This process requires a memory B cell to encounter similar epitopes three times: first in the mouth (where the memory B cell originates), then anywhere in the body (where the memory B cell clonally expands), and finally in the CNS (where the memory B cell recognizes a “forbidden” antigen and induces MS-causing inflammation). The antigens, fungal species, and organs chosen in this example are for illustrative purposes only—the production of “forbidden” MS-causing memory B cells may occur in a number of different ways.
Benign commensal fungi frequently colonize the oral cavity and respiratory system, including Aspergillus species (105). Aspergillus cells are recognized by CD4+ T cells following HLA-D antigen presentation by phagocytes. This is particularly efficient in HLA-DRB1*1501 and HLA-DRB1*1503 carriers, and leads to a population of activated CD4+ T cells recognizing Aspergillus antigens in the lymph nodes of the mouth (Waldeyer’s ring).
Meanwhile, a resting naive B cell near Waldeyer’s ring is infected by an EBV virion: this occurs often in healthy EBV carriers (106). Once infected, this naive B cell migrates to a germinal center within Waldeyer’s ring, performs affinity maturation and emerges as an isotype-switched memory B cell containing a few dormant EBV episomes (106). EBV hides from the immune system within these long-lived memory B cells, maintaining a persistent infection for the rest of the host’s life (106). Eventually these memory B cells reactivate and differentiate into plasma cells in the mouth by finding a cognate antigen which is also recognized by CD4+ T cells (55, 106). EBV virions are then produced in these plasma cells, from which they can infect other hosts and maintain a stable EBV-positive memory B cell population in the same host (106).
Under EBV-free circumstances, a resting naive B cell only activates and goes through affinity maturation if its B cell receptor (BCR) has a decent affinity to an abundant antigen, and if it receives CD4+ T cell help for a peptide present in this antigen (106). When EBV infected, a naive B cell will activate and go through affinity maturation despite filling neither of these two conditions (106): its BCR will be honed to recognize any abundant antigen present in the lymph node (55). Which antigen is selected matters little for EBV, as long as the memory B cell eventually differentiates into a plasma cell in the mouth (55). This means commensal microbe antigens present in Waldeyer’s ring and recognized by CD4+ T cells in this site will be selected (55).
When present, CD4+ T cells recognizing Aspergillus antigens will skew epitope recognition of EBV-infected memory B cells toward fungal antigens by providing CD4+ T cell help during affinity selection in the light zone of the germinal center. Antibodies against the mannose polymer portion of fungal mannoproteins have been associated with many autoimmune diseases (Table 1), so these are good antigen candidates (Figure 1). Mannoproteins provide a high avidity target for memory B cells because of their branched mannose polymer structure: mannose termini attached to the same protein can simultaneously bind to each arm of a BCR (Figure 2). Their protein portion can be recognized by CD4+ T cells, giving memory B cells the CD4+ T cell help necessary to differentiate into a plasma cell in the mouth and shed EBV virions.
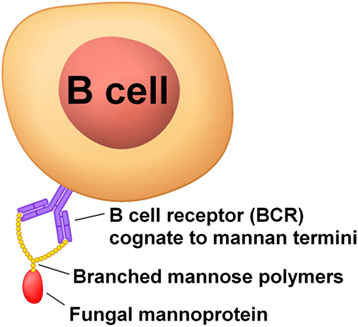
Figure 2. Mannoproteins have a high avidity to B cell BCRs because of the branched nature of their mannose polymers.
After completing affinity maturation, the EBV-infected memory B cell recirculates in peripheral blood and in the lymphatic system (106). During this time, no protein-coding genes from any of the EBV episomes in the memory B cell’s nucleus are expressed: this is essential for EBV’s survival because EBV-infected memory B cells are sought by a large number of T cells: ~3% of CD8+ T cells recognize EBV proteins (107).
In order to maintain long-term immunity, the memory B cell periodically divides—about 2.7% of memory B cells are dividing at any given time (106). During memory B cell division, EBV episomes must be replicated and partitioned between daughter cells. This is accomplished by expressing a single EBV gene (EBNA-1); the EBNA-1 protein is only present during cellular division to reduce the probability of the memory B cell being killed by CD8+ T cells while it is not dividing (106).
Unlike human chromosomes, EBV episome replication and partitioning is not very reliable, and after a few dozen divisions, some daughter cells become EBV-free (108). Once EBV-free, daughter memory B cells are no longer sought by T cells, so their longevity, clonal expansion, and differentiation into plasma cells cease being constrained by immune surveillance of EBV proteins.
When EBV first infects the host, up to half of memory B cells contain EBV episomes (106). This high viral load is due to a lack of T cells recognizing EBV proteins (106). As adaptive immunity is acquired over the following year, the fraction of memory B cells containing EBV episomes drops ten thousand-fold and stabilizes at about half a million memory B cells (or 0.001%) (106). For the rest of the host’s life, a small number of EBV-positive memory B cells are produced in the mouth to replace those eliminated by immune surveillance and other forms of attrition (106). A small subset of the EBV-positive memory B cell pool is expected to target a “forbidden” antigen and survive long enough to become EBV-free (55). However, this population of memory B cells is likely too small to cross the blood–brain barrier and find a cognate antigen in the CNS without first clonally expanding.
To clonally expand, the now EBV-free “forbidden” memory B cell that we have been following from the mouth must find a cognate antigen and an activated CD4+ T cell that recognizes a peptide from this antigen. Lymph nodes exposed to fungal antigens from the gastrointestinal tract and genitals provide the best environment for this chance event to occur—especially when these sites are colonized with Candida. CD4+ T cells which readily recognize fungal antigens should increase the probability of this chance event happening: such cells are associated with idiopathic uveitis (44), though no similar study could be found for MS. Antibodies against Candida species can be used as a proxy for CD4+ T cell sensitivity to fungal antigens: they are strongly associated with MS risk (27–29).
Fungal colonization is thus expected to be an MS risk factor because it allows “forbidden” memory B cells to clonally expand after shedding their EBV episomes. To the best of our knowledge, no study has directly measured fungal colonization in relation to MS. The efficacy of the oral fungicide DMF suggests fungal colonization of the gut may be an important contributor to the clonal expansion of “forbidden” MS-causing memory B cells.
As the now large EBV-free “forbidden” memory B cell population recirculates through the blood and lymphatic system, a small fraction of these cells will reach the CNS. Once in the CNS, these cells will find an antigen cognate to their BCR, enabling the chain of events leading to demyelination. Eliminating these memory B cells should improve MS symptoms, as suggested by the efficacy of anti-CD20 drugs (52). The exact nature of the antigen found by “forbidden” memory B cells in the CNS is not known, though mannose polymers are plausible candidates.
Because fungi are eukaryotes, they have many genes which are similar to human genes: molecular mimicry due to an epitope conserved within opisthokonta could explain why memory B cells recognizing a fungal epitope also recognize a human epitope. Alternatively, there could be an elusive fungal infection in the CNS against which immune tolerance is lost in the presence of these memory B cells. Several histological reports are consistent with a fungus in the human brain, though definite proof is currently lacking (109–117).
The events in the example above account for the two best established MS risk factors: HLA-DRB1*15 and EBV seropositivity, as previously described (55). They also account for the association with antibodies against Candida (and thus female sex), and for the efficacy of DMF and anti-CD20 drugs in MS. However, they do not account for three other risk factors: age at onset (young adulthood), smoking, and herpes simplex virus type 2 (HSV-2) seropositivity (118–121). These three additional risk factors suggest a sexually acquired infection is involved.
Multiple sclerosis is rare in children, followed by a sudden increase in risk in adolescence (10). Nearly all types of infections (including EBV and fungi) are very common in children, except sexually transmitted infections (STIs). No known MS risk factors account for the low incidence of MS in children. If the set of infectious agents etiologically involved in MS does not include an STI, why is the rate of MS so low in childhood?
While Candida is very common in children (53), vaginal Candida infections are rare in girls (54). The distribution of the age at first vulvovaginal candidiasis episode and MS onset match well (10, 54). Perhaps genital exposure to Candida species can trigger MS. Adult women have higher antibody titers against Candida than men, for which vulvovaginal candidiasis is the simplest explanation (56). This difference may contribute to women’s increased risk of MS as compared to men’s. Because antibodies against Candida are common in children (122), humoral immune responses to genital Candida infections cannot directly explain the very low incidence of MS in children. Only if the humoral immune responses in these sites were different, producing “forbidden” memory B cells in the genitals, but not in the mouth or gut, could vulvovaginal candidiasis explain the age at onset of MS. Perhaps naive B cells in genitalia are simultaneously exposed to EBV virions and Candida antigens, whereas combined exposure is rare in the mouth and gut. This hypothesis would be plausible for Candida in the gut (where EBV virions are generally absent), but not for Candida in the mouth (which is replete with EBV virions). While genital Candida exposure may well be an MS risk factor, it cannot account for the low incidence of MS in children.
Two diseases have low incidence in children, peak incidence in young adults, and moderate associations with smoking and HSV-2: cervical cancer, respectively OR = ~1.8 (123) and OR = ~1.6 (124) and MS, respectively OR = ~1.5 (8) and OR = ~1.6 (121). We now know that cervical cancer is not caused by HSV-2 and smoking—these two risk factors act as surrogates for oncogenic human papilloma virus (HPV) exposure, and their association with cervical cancer disappears after controlling for oncogenic HPV (124–128).
Multiple sclerosis’ association with young adulthood, smoking, and HSV-2 seropositivity remains unexplained. In 2002, Hawkes hypothesized that an as-yet-unrecognized STI was causing MS based on three observations (129). First, the distribution of the age at onset of common STIs approximately matches MS (10, 130). Second, MS “clusters” have been reported on small islands where soldiers were posted during World War II (129). Third, many known STIs disseminate throughout the body and cause chronic neurological symptoms through demyelination, such as syphilis (131), human T-lymphotropic virus (132), and human immunodeficiency virus (133). A fourth observation was added by Hawkes in 2006: HSV-2 seropositivity is associated with MS (121). We add a fifth observation: smoking increases MS risk, possibly by acting as a surrogate for an STI.
Though the existence of an as-yet-unrecognized STI may seem unlikely, this hypothesis has been proposed to explain the sexual risk factors of prostate cancer (134–136) (currently no strong candidate) (137) and reactive arthritis (138) (strongest candidate is Chlamydia trachomatis, found in ~13% of cases and ~2.4% of controls) (139). Prostate cancer has recently been linked to an intracellular fungal infection (35, 36). Antibodies against fungi have been linked to reactive arthritis symptoms, especially uveitis (21). Older studies have found antibodies against a component of the prostate and prostatic inflammation in reactive arthritis (140–143) and uveitis (46, 47, 144) patients. Finally, HLA-DRB1*15 and antibodies against a component of the prostate have been linked to granulomatous prostatitis (42, 145).
Together, these studies suggest genital exposure to fungi may play an important etiological role in prostate disease, reactive arthritis, and MS. Could the unexplained histological evidence of fungal cells in the CNS (110, 111, 113, 117) and in the prostate (146, 147) be caused by a fungal infection spreading from the genitals after sexual debut? If so, loss of immune tolerance to this putative infection could be a plausible cause of prostate disease, reactive arthritis, uveitis, and MS.
Varied molecular and epidemiological evidence supports a role for infections in MS (Table 4). EBV is the most strongly associated infection, though the underlying mechanisms are not firmly established (55). Unlike EBV (148), MS is rare in children (10), suggesting EBV is insufficient to cause MS on its own. Genital exposure to an infection involved in MS could solve the puzzling lag between childhood EBV infections and MS onset (129).
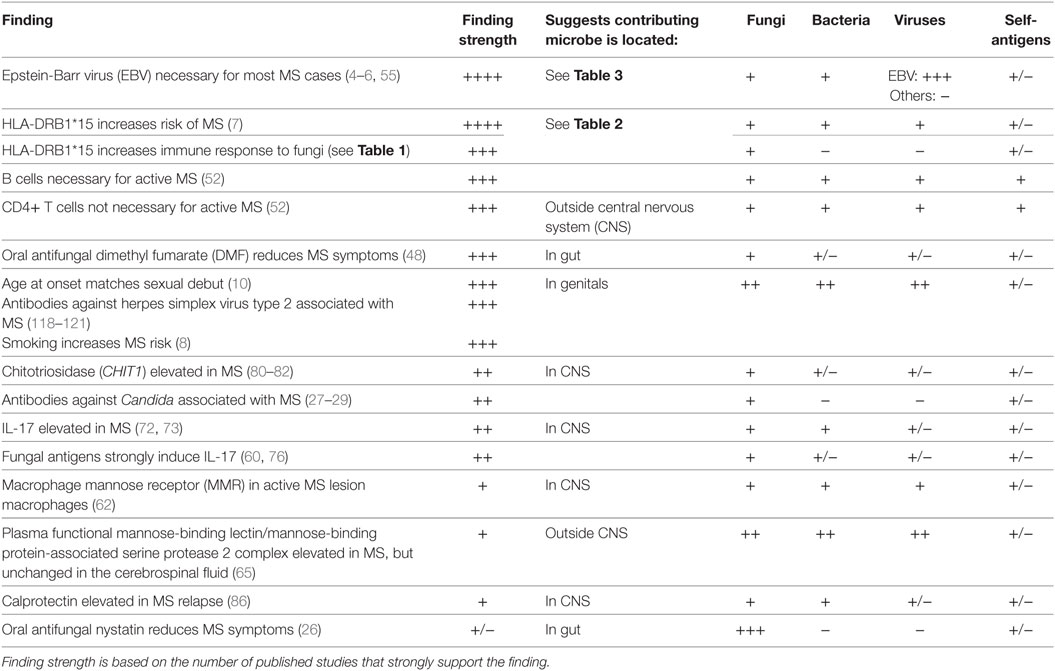
Table 4. Summary of findings linking multiple sclerosis (MS) to various infection types and recognition of self-antigens.
The links between various idiopathic inflammatory conditions and antibodies against fungi (Table 1) could be explained by memory B cells accidentally recognizing benign antigens due to the simultaneous presence of EBV virions and fungal antigens near the same lymph node (55). Since fungi are eukaryotes, cross-reactive antibodies between fungal and human proteins are plausible. Alternatively, an elusive fungal infection could be present in the CNS, though there is currently no strong evidence supporting this. Excluding the presence of non-abundant fungi in the CNS is not trivial: the Th17 immune response is extremely sensitive to fungal antigens, suggesting even minute quantities could result in a robust response (60, 76).
Evidence presented here is also consistent with pathways shared between the immune response to fungi and MS inflammation, which means links with fungi could be coincidental. To establish causality, either many antifungal drugs must be shown to improve MS symptoms or case-control studies must show a strong association with specific fungal species. Neither condition is met today.
Thorough microbiome studies of the CNS, mouth, gut, and genitalia in association with MS should be run. Particular attention should be given to fungal infections which generally do not affect children, such as vulvovaginal candidiasis. A very recent study strongly suggests many microbes present in humans have yet to be discovered (149). Older molecular surveys of oral (105) and genital (150) fungi in healthy individuals found many novel species: the most common fungus found in the vagina was not a Candida species as expected, but rather an unknown species detected in about 25% of healthy individuals (150).
The authors both contributed in drafting and reviewing the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
JB-L is supported by the National Institutes of Health, Bethesda, MD, USA (NINDS #R01 NS39422), the Commission of the European Union (grant ICT-2011-287739, NeuroTREMOR), the Ministry of Economy and Competitiveness (grant RTC-2015-3967-1, NetMD—platform for the tracking of movement disorder), and the Spanish Health Research Agency (grant FIS PI12/01602 and grant FIS PI16/00451).
1. Benito-León J, Martin E, Vela L, Villar M, Felgueroso B, Marrero C, et al. Multiple sclerosis in Mostoles, central Spain. Acta Neurol Scand (1998) 98:238–42. doi:10.1111/j.1600-0404.1998.tb07302.x
2. Browne P, Chandraratna D, Angood C, Tremlett H, Baker C, Taylor BV, et al. Atlas of multiple sclerosis 2013: a growing global problem with widespread inequity. Neurology (2014) 83:1022–4. doi:10.1212/WNL.0000000000000768
3. Stadelmann C, Wegner C, Brück W. Inflammation, demyelination, and degeneration-recent insights from MS pathology. Biochim Biophys Acta (2011) 1812:275–82. doi:10.1016/j.bbadis.2010.07.007
4. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol (2007) 61:288–99. doi:10.1002/ana.21117
5. Pakpoor J, Disanto G, Gerber JE, Dobson R, Meier UC, Giovannoni G, et al. The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: a meta-analysis. Mult Scler J (2012) 19:162–6. doi:10.1177/1352458512449682
6. Almohmeed YH, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and multiple sclerosis. PLoS One (2013) 8:e61110. doi:10.1371/journal.pone.0061110
7. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: a comprehensive review. J Autoimmun (2015) 64:13–25. doi:10.1016/j.jaut.2015.06.010
8. Handel AE, Williamson AJ, Disanto G, Dobson R, Giovannoni G, Ramagopalan SV. Smoking and multiple sclerosis: an updated meta-analysis. PLoS One (2011) 6:e16149. doi:10.1371/journal.pone.0016149
9. Ascherio A, Munger KL, Simon KC. Vitamin D and multiple sclerosis. Lancet Neurol (2010) 9:599–612. doi:10.1016/S1474-4422(10)70086-7
10. Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain (2006) 129:606–16. doi:10.1093/brain/awl007
11. Orton S-M, Herrera BM, Yee IM, Valdar W, Ramagopalan SV, Sadovnick AD, et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurol (2006) 5:932–6. doi:10.1016/S1474-4422(06)70581-6
12. Venkatesan A, Johnson RT. Infections and multiple sclerosis. Handb Clin Neurol (2014) 122:151–71. doi:10.1016/B978-0-444-52001-2.00007-8
13. Hube B. From commensal to pathogen: stage-and tissue-specific gene expression of Candida albicans. Curr Opin Microbiol (2004) 7:336–41. doi:10.1016/j.mib.2004.06.003
14. Waldman A, Gilhar A, Duek L, Berdicevsky I. Incidence of Candida in psoriasis – a study on the fungal flora of psoriatic patients. Mycoses (2001) 44:77–81. doi:10.1046/j.1439-0507.2001.00608.x
15. Sepahi S, Riahi-Zanjani B, Ghorani A. The role of Candida albicans in the pathogenesis of psoriasis vulgaris: systematic literature review. Rev Clin Med (2016) 3(3):122–7. doi:10.22038/RCM.2016.6485
16. Gerard R, Sendid B, Colombel J-F, Poulain D, Jouault T. An immunological link between Candida albicans colonization and Crohn’s disease. Crit Rev Microbiol (2015) 41:135–9. doi:10.3109/1040841X.2013.810587
17. Hoarau G, Mukherjee P, Gower-Rousseau C, Hager C, Chandra J, Retuerto M, et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. MBio (2016) 7:e1250–1216. doi:10.1128/mBio.01250-16
18. Squiquera L, Galimberti R, Morelli L, Plotkin L, Milicich R, Kowalckzuk A, et al. Antibodies to proteins from Pityrosporum ovale in the sera from patients with psoriasis. Clin Exp Dermatol (1994) 19:289–93. doi:10.1111/j.1365-2230.1994.tb01197.x
19. Liang Y, Wen H, Xiao R. Serum levels of antibodies for IgG, IgA, and IgM against the fungi antigen in psoriasis vulgaris. Hunan Yi Ke Da Xue Xue Bao (2003) 28:638–40.
20. Ascioglu O, Soyuer U, Aktas E. Improvement of psoriasis with oral nystatin. In: Tümbay E, Seeliger HPR, Anğ Ö, editors. Candida and Candidamycosis. Federation of European Microbiological Societies Symposium Series. Boston, MA: Springer (1991). Vol. 50. p. 279–81.
21. Maillet J, Ottaviani S, Tubach F, Roy C, Nicaise-Rolland P, Palazzo E, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA) in spondyloarthritis: prevalence and associated phenotype. Joint Bone Spine (2016) 83(6):665–8. doi:10.1016/j.jbspin.2015.10.011
22. Dai H, Li Z, Zhang Y, Lv P, Gao X. Elevated levels of serum antibodies against Saccharomyces cerevisiae mannan in patients with systemic lupus erythematosus. Lupus (2009) 18:1087–90. doi:10.1177/0961203309105131
23. Mankaï A, Sakly W, Thabet Y, Achour A, Manoubi W, Ghedira I. Anti-Saccharomyces cerevisiae antibodies in patients with systemic lupus erythematosus. Rheumatol Int (2013) 33:665–9. doi:10.1007/s00296-012-2431-3
24. Suchankova M, Paulovicova E, Paulovicova L, Majer I, Tedlova E, Novosadova H, et al. Increased antifungal antibodies in bronchoalveolar lavage fluid and serum in pulmonary sarcoidosis. Scand J Immunol (2015) 81:259–64. doi:10.1111/sji.12273
25. Dotan I, Fishman S, Dgani Y, Schwartz M, Karban A, Lerner A, et al. Antibodies against laminaribioside and chitobioside are novel serologic markers in Crohn’s disease. Gastroenterology (2006) 131:366–78. doi:10.1053/j.gastro.2006.04.030
27. Ramos M, Pisa D, Molina S, Rábano A, Juarranz Á, Carrasco L. Fungal infection in patients with multiple sclerosis. Open Mycol J (2008) 2:22–8. doi:10.2174/1874437000802010022
28. Benito-Leon J, Pisa D, Alonso R, Calleja P, Diaz-Sanchez M, Carrasco L. Association between multiple sclerosis and Candida species: evidence from a case-control study. Eur J Clin Microbiol Infect Dis (2010) 29:1139–45. doi:10.1007/s10096-010-0979-y
29. Pisa D, Alonso R, Jimenez-Jimenez FJ, Carrasco L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur J Clin Microbiol Infect Dis (2013) 32(6):795–801. doi:10.1007/s10096-012-1810-8
30. Chauhan B, Santiago L, Hutcheson PS, Schwartz HJ, Spitznagel E, Castro M, et al. Evidence for the involvement of two different MHC class II regions in susceptibility or protection in allergic bronchopulmonary aspergillosis. J Allergy ClinImmunol (2000) 106:723–9. doi:10.1067/mai.2000.109913
31. Chauhan B, Knutsen AP, Hutcheson PS, Slavin RG, Bellone CJ. T cell subsets, epitope mapping, and HLA-restriction in patients with allergic bronchopulmonary aspergillosis. J Clin Invest (1996) 97:2324. doi:10.1172/JCI118675
32. Tercelj M, Salobir B, Rylander R. β-glucan in the lymph nodes in sarcoidosis and in Kveim-Siltzbach test reagent. Sarcoidosis Vasc Diffuse Lung Dis (2017) 34:130–5.
33. Yurkanin JP, Ahmann F, Dalkin BL. Coccidioidomycosis of the prostate: a determination of incidence, report of 4 cases, and treatment recommendations. J Infect (2006) 52:e19–25. doi:10.1016/j.jinf.2005.04.017
34. Edstrom Hagerwall AM, Rydengard V, Fernlund P, Morgelin M, Baumgarten M, Cole AM, et al. beta-Microseminoprotein endows post coital seminal plasma with potent candidacidal activity by a calcium- and pH-dependent mechanism. PLoS Pathog (2012) 8:e1002625. doi:10.1371/journal.ppat.1002625
35. Stott-Miller M, Wright JL, Stanford JL. MSMB gene variant alters the association between prostate cancer and number of sexual partners. Prostate (2013) 73(16):1803–9. doi:10.1002/pros.22719
36. Sutcliffe S, De Marzo AM, Sfanos KS, Laurence M. MSMB variation and prostate cancer risk: clues towards a possible fungal etiology. Prostate (2014) 74:569–78. doi:10.1002/pros.22778
37. Barisani-Asenbauer T, Maca SM, Mejdoubi L, Emminger W, Machold K, Auer H. Uveitis-a rare disease often associated with systemic diseases and infections-a systematic review of 2619 patients. Orphanet J Rare Dis (2012) 7:57. doi:10.1186/1750-1172-7-57
38. Messenger W, Hildebrandt L, Mackensen F, Suhler E, Becker M, Rosenbaum JT. Characterisation of uveitis in association with multiple sclerosis. Br J Ophthalmol (2015) 99:205–9. doi:10.1136/bjophthalmol-2014-305518
39. Wang JL, Patterson R, Rosenberg M, Roberts M, Cooper BJ. Serum IgE and IgG antibody activity against Aspergillus fumigatus as a diagnostic aid in allergic bronchopulmonary aspergillosis. Am Rev Respir Dis (1978) 117:917–27. doi:10.1164/arrd.1978.117.5.917
40. Voorter CE, Drent M, Van Den Berg-Loonen EM. Severe pulmonary sarcoidosis is strongly associated with the haplotype HLA-DQB1*0602-DRB1*150101. Hum Immunol (2005) 66:826–35. doi:10.1016/j.humimm.2005.04.003
41. Terčelj M, Salobir B, Zupancic M, Rylander R. Antifungal medication is efficient in the treatment of sarcoidosis. Ther Adv Respir Dis (2011) 5:157–62. doi:10.1177/1753465811401648
42. Alexander RB, Mann DL, Borkowski AA, Fernandez-Vina M, Klyushnenkova EN, Kodak J, et al. Granulomatous prostatitis linked to HLA-DRB1*1501. J Urol (2004) 171:2326–9. doi:10.1097/01.ju.0000127759.10293.fa
43. Tang WM, Pulido JS, Eckels DD, Han DP, Mieler WF, Pierce K. The association of HLA-DR15 and intermediate uveitis. Am J Ophthalmol (1997) 123:70–5. doi:10.1016/S0002-9394(14)70994-8
44. Cozon GJN, Mbitikon-Kobo FM, Fatoohi F, Spire M, Grange J-D, Kodjikian L. Abnormal cellular reactivity to microbial antigens in patients with uveitis. Invest Ophthalmol Vis Sci (2008) 49:2526–30. doi:10.1167/iovs.07-1454
45. Brown BR, Lee EJ, Snow PE, Vance EE, Iwakura Y, Ohno N, et al. Fungal-derived cues promote ocular autoimmunity through a Dectin-2/Card9-mediated mechanism. Clin Exp Immunol (2017). doi:10.1111/cei.13021
46. Catterall R, Perkins ES. Uveitis and urogenital disease in the male. Br J Ophthalmol (1961) 45:109. doi:10.1136/bjo.45.2.109
47. Dark A, Morton R. Acute anterior uveitis in men. Association with chronic prostatitis. Br J Ophthalmol (1968) 52:907. doi:10.1136/bjo.52.12.907
48. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med (2012) 367:1098–107. doi:10.1056/NEJMoa1114287
49. Sahajwalla CG. Antimicrobial Vaginal Dosage Forms Employing Dimethyl Fumarate or Propionic Acid [PhD dissertation]. Portland, OR:Oregon State University (1983).
50. Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: a comprehensive review. J Autoimmun (2015) 64:125–36. doi:10.1016/j.jaut.2015.08.004
51. Phelps RG, Rees AJ. The HLA complex in Goodpasture’s disease: a model for analyzing susceptibility to autoimmunity. Kidney Int (1999) 56:1638–53. doi:10.1046/j.1523-1755.1999.00720.x
52. Disanto G, Morahan J, Barnett M, Giovannoni G, Ramagopalan S. The evidence for a role of B cells in multiple sclerosis. Neurology (2012) 78:823–32. doi:10.1212/WNL.0b013e318249f6f0
54. Geiger AM, Foxman B, Gillespie BW. The epidemiology of vulvovaginal candidiasis among university students. Am J Public Health (1995) 85:1146–8. doi:10.2105/AJPH.85.8_Pt_1.1146
55. Laurence M, Benito-León J. Epstein-Barr virus and multiple sclerosis: updating Pender’s hypothesis. Mult Scler Relat Disord (2017) 16:8–14. doi:10.1016/j.msard.2017.05.009
56. Severance EG, Gressitt KL, Stallings CR, Katsafanas E, Schweinfurth LA, Savage CL, et al. Candida albicans exposures, sex specificity and cognitive deficits in schizophrenia and bipolar disorder. NPJ Schizophr (2016) 2:16018. doi:10.1038/npjschz.2016.18
57. Saevarsdóttir S, Vikingsdottir T, Valdimarsson H. The potential role of mannan-binding lectin in the clearance of self-components including immune complexes. Scand J Immunol (2004) 60:23–9. doi:10.1111/j.0300-9475.2004.01437.x
58. Levitz SM, Specht CA. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res (2006) 6:513–24. doi:10.1111/j.1567-1364.2006.00071.x
59. Levitz SM. Th17 cells bounce off the fungal wall. Cell Host Microbe (2009) 5:311–3. doi:10.1016/j.chom.2009.04.004
60. van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, Koenen HJ, Cheng S-C, Joosten I, et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe (2009) 5:329–40. doi:10.1016/j.chom.2009.02.006
61. Taylor PR, Gordon S, Martinez-Pomares L. The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol (2005) 26:104–10. doi:10.1016/j.it.2004.12.001
62. Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, Van Der Valk P, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation (2013) 10:1. doi:10.1186/1742-2094-10-35
63. Ezekowitz RA. Role of the mannose-binding lectin in innate immunity. J Infect Dis (2003) 187:S335–9. doi:10.1086/374746
64. Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol (2003) 40:423–9. doi:10.1016/S0161-5890(03)00155-X
65. Kwok JY, Vaida F, Augst RM, Denise YY, Singh KK. Mannose binding lectin mediated complement pathway in multiple sclerosis. J Neuroimmunol (2011) 239:98–100. doi:10.1016/j.jneuroim.2011.08.018
66. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10:479–89. doi:10.1038/nri2800
67. Curtis MM, Way SS. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology (2009) 126:177–85. doi:10.1111/j.1365-2567.2008.03017.x
68. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol (2007) 8:639–46. doi:10.1038/ni1467
69. van de Veerdonk FL, Gresnigt MS, Kullberg BJ, Van Der Meer JW, Netea M. Th17 responses and host defense against microorganisms: an overview. BMB Rep (2009) 42(12):776–87. doi:10.5483/BMBRep.2009.42.12.776
70. Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C, Rizzo A, et al. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol (2008) 38:470–8. doi:10.1002/eji.200737635
71. Huppler AR, Bishu S, Gaffen SL. Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy. Arthritis Res Ther (2012) 14:1. doi:10.1186/ar3893
72. Yamada H. Current perspectives on the role of IL-17 in autoimmune disease. J Inflamm Res (2010) 3:33–44. doi:10.2147/JIR.S6375
73. Tabarkiewicz J, Pogoda K, Karczmarczyk A, Pozarowski P, Giannopoulos K. The role of IL-17 and Th17 lymphocytes in autoimmune diseases. Arch Immunol Ther Exp (2015) 63:435–49. doi:10.1007/s00005-015-0344-z
74. Dooley CP, Cohen H, Fitzgibbons PL, Bauer M, Appleman MD, Perez-Perez GI, et al. Prevalence of Helicobacter pylori infection and histologic gastritis in asymptomatic persons. N Engl J Med (1989) 321:1562–6. doi:10.1056/NEJM198912073212302
75. Hernández-Santos N, Gaffen SL. Th17 cells in immunity to Candida albicans. Cell Host Microbe (2012) 11:425–35. doi:10.1016/j.chom.2012.04.008
76. Marijnissen RJ, Koenders MI, Van De Veerdonk FL, Dulos J, Netea MG, Boots AM, et al. Exposure to Candida albicans polarizes a T-cell driven arthritis model towards Th17 responses, resulting in a more destructive arthritis. PLoS One (2012) 7:e38889. doi:10.1371/journal.pone.0038889
77. Vega K, Kalkum M. Chitin, chitinase responses, and invasive fungal infections. Int J Microbiol (2011) 2012:10. doi:10.1155/2012/920459
78. Gorzelanny C, Pöppelmann B, Pappelbaum K, Moerschbacher BM, Schneider SW. Human macrophage activation triggered by chitotriosidase-mediated chitin and chitosan degradation. Biomaterials (2010) 31:8556–63. doi:10.1016/j.biomaterials.2010.07.100
79. Elmonem MA, Van Den Heuvel LP, Levtchenko EN. Immunomodulatory effects of chitotriosidase enzyme. Enzyme Res (2016) 2016:2682680. doi:10.1155/2016/2682680
80. Czartoryska B, Fiszer U, Lugowska A. Chitotriosidase activity in cerebrospinal fluid as a marker of inflammatory processes in neurological diseases. J Lab Med (2001) 25:77–81. doi:10.1515/labm.2001.25.3-4.77
81. Sotgiu S, Barone R, Arru G, Fois M, Pugliatti M, Sanna A, et al. Intrathecal chitotriosidase and the outcome of multiple sclerosis. Mult Scler (2006) 12:551–7. doi:10.1177/1352458506070614
82. Verbeek M, Notting E, Faas B, Claessens-Linskens R, Jongen P. Increased cerebrospinal fluid chitotriosidase index in patients with multiple sclerosis. Acta Neurol Scand (2010) 121:309–14. doi:10.1111/j.1600-0404.2009.01242.x
83. Stríz I, Trebichavský I. Calprotectin – a pleiotropic molecule in acute and chronic inflammation. Physiol Res (2004) 53:245–53.
84. Steinbakk M, Naess-Andresen C, Fagerhol M, Lingaas E, Dale I, Brandtzaeg P. Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet (1990) 336:763–5. doi:10.1016/0140-6736(90)93237-J
85. Foell D, Roth J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum (2004) 50:3762–71. doi:10.1002/art.20631
86. Berg-Hansen P, Vandvik B, Fagerhol M, Holmøy T. Calprotectin levels in the cerebrospinal fluid reflect disease activity in multiple sclerosis. J Neuroimmunol (2009) 216:98–102. doi:10.1016/j.jneuroim.2009.09.006
87. Searle NE, Tisdale WH. Pest Control. U.S. Patent and Trademark Office, Washington, DC (1940). U.S. Patent No 2,218,181.
88. Ropper AH. The “poison chair” treatment for multiple sclerosis. N Engl J Med (2012) 367:1149–50. doi:10.1056/NEJMe1209169
89. Linker RA, Lee D-H, Ryan S, Van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain (2011) 134:678–92. doi:10.1093/brain/awq386
90. Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci U S A (2016) 113:4777–82. doi:10.1073/pnas.1603907113
91. Crutcher N, Rosenberg EW, Belew PW, Skinner RB Jr, Eaglstein NF, Baker SM. Oral nystatin in the treatment of psoriasis. Arch Dermatol (1984) 120:435–6. doi:10.1001/archderm.120.4.435a
92. Ganor S. Treatment of early psoriasis lesions with a oral amphotericin B or nystatin. Int J Dermatol (1988) 27:420–420. doi:10.1111/j.1365-4362.1988.tb02398.x
93. Buslau M, Hänel H, Holzmann H. The significance of yeasts in seborrheic eczema. Hautarzt (1989) 40:611–3.
94. Rosenberg EW, Belew PW. Improvement of psoriasis of the scalp with ketoconazole. Arch Dermatol (1982) 118:370–1. doi:10.1001/archderm.118.6.370
95. Farr P, Marks J, Krause L, Shuster S. Response of scalp psoriasis to oral ketoconazole. Lancet (1985) 326:921–2. doi:10.1016/S0140-6736(85)90853-0
96. Faergemann J. Treatment of Sebopsoriasis with Itraconazole: behandlung von Sebopsoriasis mit Itraconazole. Mycoses (1985) 28:612–8. doi:10.1111/j.1439-0507.1985.tb02094.x
97. Kose O, Erbil H, Gur A. Oral itraconazole for the treatment of seborrhoeic dermatitis: an open, noncomparative trial. J Eur Acad Dermatol Venereol (2005) 19:172–5. doi:10.1111/j.1468-3083.2005.01090.x
98. Lober CW, Belew PW, Rosenberg EW, Bale G. Patch tests with killed sonicated microflora in patients with psoriasis. Arch Dermatol (1982) 118:322–5. doi:10.1001/archderm.1982.01650170036019
99. Larson JL, Wallace TL, Tyl RW, Marr MC, Myers CB, Cossum PA. The reproductive and developmental toxicity of the antifungal drug Nyotran® (liposomal nystatin) in rats and rabbits. Toxicol Sci (2000) 53:421–9. doi:10.1093/toxsci/53.2.421
101. Pauls K, Schön M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P, et al. Role of integrin α E (CD103) β 7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J Invest Dermatol (2001) 117:569–75. doi:10.1046/j.0022-202x.2001.01481.x
102. Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics (2011) 128:e453–7. doi:10.1542/peds.2010-2757
103. Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol (2003) 24:584–8. doi:10.1016/j.it.2003.09.005
104. Tracy SI, Kakalacheva K, Lünemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. Persistence of Epstein-Barr virus in self-reactive memory B cells. J Virol (2012) 86:12330–40. doi:10.1128/JVI.01699-12
105. Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog (2010) 6:e1000713. doi:10.1371/journal.ppat.1000713
106. Thorley-Lawson DA. EBV persistence-introducing the virus. Curr Top Microbiol Immunol. (2015) 390:151–209. doi:10.1007/978-3-319-22822-8_8
107. Hislop AD, Taylor GS. T-cell responses to EBV. Curr Top Microbiol Immunol. (2015) 391:325–53. doi:10.1007/978-3-319-22834-1_11
108. Nanbo A, Sugden A, Sugden B. The coupling of synthesis and partitioning of EBV’s plasmid replicon is revealed in live cells. EMBO J (2007) 26:4252–62. doi:10.1038/sj.emboj.7601853
109. Howard J, Pilkington GJ. Fibronectin staining detects micro-organisms in aged and Alzheimer’s disease brain. Neuroreport (1992) 3:615–8. doi:10.1097/00001756-199207000-00018
110. Castellani RJ, Siedlak SL, Fortino AE, Perry G, Ghetti B, Smith MA. Chitin-like polysaccharides in Alzheimer’s disease brains. Curr Alzheimer Res (2005) 2:419–23. doi:10.2174/156720505774330555
111. Sotgiu S, Musumeci S, Marconi S, Gini B, Bonetti B. Different content of chitin-like polysaccharides in multiple sclerosis and Alzheimer’s disease brains. J Neuroimmunol (2008) 197:70–3. doi:10.1016/j.jneuroim.2008.03.021
112. Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimer Dis (2014) 41:301–11. doi:10.3233/JAD-132681
113. Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Rodal I, et al. Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int J Biol Sci (2015) 11:546. doi:10.7150/ijbs.11084
114. Pisa D, Alonso R, Juarranz A, Rábano A, Carrasco L. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimer Dis (2015) 43:613–24. doi:10.3233/JAD-141386
115. Pisa D, Alonso R, Rábano A, Rodal I, Carrasco L. Different brain regions are infected with fungi in Alzheimer’s disease. Sci Rep (2015) 5:15015. doi:10.1038/srep15015
116. Pisa D, Alonso R, Rábano A, Carrasco L. Corpora amylacea of brain tissue from neurodegenerative diseases are stained with specific antifungal antibodies. Front Neurosci (2016) 10:86. doi:10.3389/fnins.2016.00086
117. Pisa D, Alonso R, Rábano A, Horst MN, Carrasco L. Fungal enolase, β-tubulin, and chitin are detected in brain tissue from Alzheimer’s disease patients. Front Microbiol (2016) 7:1772. doi:10.3389/fmicb.2016.01772
118. Catalano LW. Herpesvirus hominis antibody in multiple sclerosis and amyotrophic lateral sclerosis. Neurology (1972) 22:473–473. doi:10.1212/WNL.22.5.473
119. Ferrante P, Castellani P, Barbi M, Bergamini F. The Italian cooperative multiple sclerosis case-control study: preliminary results on viral antibodies. Ital J Neurol Sci (1987) Suppl 6:45–50.
120. Wandinger K, Jabs W, Siekhaus A, Bubel S, Trillenberg P, Wagner H, et al. Association between clinical disease activity and Epstein-Barr virus reactivation in MS. Neurology (2000) 55:178–84. doi:10.1212/WNL.55.2.178
121. Hawkes CH, Giovannoni G, Keir G, Cunnington M, Thompson EJ. Seroprevalence of herpes simplex virus type 2 in multiple sclerosis. Acta Neurol Scand (2006) 114:363–7. doi:10.1111/j.1600-0404.2006.00677.x
122. Savolainen J, Viander M, Koivikko A. IgE-, IgA-and IgG-antibody responses to carbohydrate and protein antigens of Candida albicans in asthmatic children. Allergy (1990) 45:54–63. doi:10.1111/j.1398-9995.1990.tb01084.x
123. Gandini S, Botteri E, Iodice S, Boniol M, Lowenfels AB, Maisonneuve P, et al. Tobacco smoking and cancer: a meta-analysis. Int J Cancer (2008) 122:155–64. doi:10.1002/ijc.23033
124. Dahlström LA, Andersson K, Luostarinen T, Thoresen S, Ögmundsdottír H, Tryggvadottír L, et al. Prospective seroepidemiologic study of human papillomavirus and other risk factors in cervical cancer. Cancer Epidemiol Biomarkers Prev (2011) 20:2541–50. doi:10.1158/1055-9965.EPI-11-0761
125. Mayberry RM. Cigarette smoking, herpes simplex virus type 2 infection, and cervical abnormalities. Am J Public Health (1985) 75:676–8. doi:10.2105/AJPH.75.6.676
126. Phillips AN, Smith GD. Cigarette smoking as a potential cause of cervical cancer: has confounding been controlled? Int J Epidemiol (1994) 23:42–9. doi:10.1093/ije/23.1.42
127. Franco E, Spence A. Commentary: smoking and human papillomavirus infection: the pursuit of credibility for an epidemiologic association. Int J Epidemiol (2008) 37:547–8. doi:10.1093/ije/dyn057
128. Vaccarella S, Herrero R, Snijders PJ, Dai M, Thomas JO, Hieu NT, et al. Smoking and human papillomavirus infection: pooled analysis of the international agency for research on cancer HPV prevalence surveys. Int J Epidemiol (2008) 37:536–46. doi:10.1093/ije/dyn033
129. Hawkes CH. Is multiple sclerosis a sexually transmitted infection? J Neurol Neurosurg Psychiatry (2002) 73:439–43. doi:10.1136/jnnp.73.4.439
131. Chahine LM, Khoriaty RN, Tomford WJ, Hussain MS. The changing face of neurosyphilis. Int J Stroke (2011) 6:136–43. doi:10.1111/j.1747-4949.2010.00568.x
132. Yamano Y, Sato T. Clinical pathophysiology of human T-lymphotropic virus-type 1-associated myelopathy/tropical spastic paraparesis. Front Microbiol (2012) 3:389. doi:10.3389/fmicb.2012.00389
133. Tan S, Guiloff R, Scaravilli F. AIDS-associated vacuolar myelopathy A morphometric study. Brain (1995) 118:1247–61. doi:10.1093/brain/118.5.1247
134. Strickler HD, Goedert JJ. Sexual behavior and evidence for an infectious cause of prostate cancer. Epidemiol Rev (2001) 23:144–51. doi:10.1093/oxfordjournals.epirev.a000781
135. Dennis LK, Dawson DV. Meta-analysis of measures of sexual activity and prostate cancer. Epidemiology (2002) 13:72–9. doi:10.1097/00001648-200201000-00012
136. Taylor ML, Mainous AG III, Wells BJ. Prostate cancer and sexually transmitted diseases: a meta-analysis. Fam Med (2005) 37:506–12.
137. Hrbacek J, Urban M, Hamsikova E, Tachezy R, Heracek J. Thirty years of research on infection and prostate cancer: no conclusive evidence for a link. A systematic review. Urol Oncol (2013) 31(7):951–65. doi:10.1016/j.urolonc.2012.01.013
138. Catterall R. Significance of non-specific genital infection in uveitis and arthritis. Lancet (1961) 278:739–42. doi:10.1016/S0140-6736(61)90688-2
139. Ostaszewska-Puchalska I, Zdrodowska-Stefanow B, Kuryliszyn-Moskal A, Bułhak-Kozioł V, Sokołowska M. Incidence of Chlamydia trachomatis infection in patients with reactive arthritis. Reumatologia (2015) 53:69. doi:10.5114/reum.2015.51505
140. Romanus R. Pelvo-spondylitis ossificans in the male and genitourinary infection. Acta Med Scand (1953) 145:178–241.
141. Mason RM, Murray RS, Oates JK, Young AC. Prostatitis and ankylosing spondylitis. Br Med J (1958) 1:748–51. doi:10.1136/bmj.1.5073.748
142. Olhagen B. Chronic uro-polyarthritis in the male. Acta Med Scand (1960) 168:339–45. doi:10.1111/j.0954-6820.1960.tb13455.x
143. Grimble A, Lessof MH. Anti-prostate antibodies in arthritis. Br Med J (1965) 2:263–4. doi:10.1136/bmj.2.5456.263
144. Grimble A. Auto-immunity to prostate antigen in rheumatic disease. J Clin Pathol (1964) 17:264–7. doi:10.1136/jcp.17.3.264
145. Cohen R. Method of Diagnosis of Prostate Cancer. U.S. Patent and Trademark Office, Washington, DC (2004). U.S. Patent No 6,677,157.
146. Guillan R, Zelman S. The incidence and probable origin of melanin in the prostate. J Urol (1970) 104:151–3. doi:10.1016/S0022-5347(17)61689-6
147. Seman G, Gallager HS, Johnson DE. Melanin-like pigment in the human prostate. Prostate (1982) 3:59–72. doi:10.1002/pros.2990030109
148. Balfour HH, Sifakis F, Sliman JA, Knight JA, Schmeling DO, Thomas W. Age-specific prevalence of Epstein-Barr virus infection among individuals aged 6–19 years in the United States and factors affecting its acquisition. J Infect Dis (2013) 208:1286–93. doi:10.1093/infdis/jit321
149. Kowarsky M, Camunas-Soler J, Kertesz M, De Vlaminck I, Koh W, Pan W, et al. Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc Natl Acad Sci U S A (2017) 114:9623–8. doi:10.1073/pnas.1707009114
Keywords: multiple sclerosis, fungal infections, dimethyl fumarate, memory B cells, Epstein-Barr virus, HLA-DRB1*15
Citation: Benito-León J and Laurence M (2017) The Role of Fungi in the Etiology of Multiple Sclerosis. Front. Neurol. 8:535. doi: 10.3389/fneur.2017.00535
Received: 29 July 2017; Accepted: 25 September 2017;
Published: 16 October 2017
Edited by:
Masaaki Murakami, Hokkaido University, JapanReviewed by:
Izumi Kawachi, Niigata University, JapanCopyright: © 2017 Benito-León and Laurence. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julián Benito-León, amJlbml0b2w2N0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.