 J. Elliott Robinson
J. Elliott Robinson Stephanie M. Wolfe
Stephanie M. Wolfe Kathleen Kaiser-Rogers
Kathleen Kaiser-Rogers Robert S. Greenwood
Robert S. Greenwood- 1School of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC, USA
- 2Division of Child Neurology, Department of Neurology, University of North Carolina School of Medicine, Chapel Hill, NC, USA
- 3Department of Pathology and Laboratory Medicine, University of North Carolina School of Medicine, Chapel Hill, NC, USA
Hemiconvulsion–hemiplegia–epilepsy syndrome (HHE) is a rare outcome of prolonged hemiconvulsion that is followed by diffuse unilateral hemispheric edema, hemiplegia, and ultimately hemiatrophy of the affected hemisphere and epilepsy. Here, we describe the case of a 3-year-old male with a 1;3 translocation leading to a terminal 1q43q44 deletion and a terminal 3p26.1p26.3 duplication that developed HHE after a prolonged febrile seizure and discuss the pathogenesis of HHE in the context of the patient’s complex genetic background.
Case Report
The patient is a 3-year-old male who was found in bed experiencing a tonic–clonic seizure with left eye deviation. The duration of the seizure was unknown given that his mother had last seen him at least 3–4 h prior to the event. The patient was noted by his mother to be febrile to 102.5°F prior to witnessed seizure activity; upon arrival of EMS, his temperature was 100.5°F. He was transported to a local hospital, seizures were acutely controlled with lorazepam and levetiracetam, and he was transitioned to his home zonisamide regimen. Initially, the patient was assumed to have Todd’s paralysis, but when he failed to return to baseline, an MRI was performed on day 2 that showed thinned and truncated corpus collosum, diffusely increased T2/FLAIR signal intensity throughout the cortices of the left hemispheric gray matter with associated restriction diffusion, left hemispheric edema, 6.8 mm midline shift, effacement of the left ventricle, and dilation of the right ventricle. He was transferred to our tertiary hospital for neurosurgical management. At the time of admission, the patient was minimally responsive and displayed right-sided hemiparesis and gaze deviation, increased tone bilaterally, left papilledema, left orbital swelling, and asymmetric pupils who were reactive to light. vEEG was significant for diffuse slowing of the L hemisphere without epileptiform discharges.
The patient’s past medical history was significant for hypotonia, global developmental delays, febrile seizures, breath holding spells, small stature, and patent foramen ovale. The child was born at 41 weeks to non-consanguinous parents following an uncomplicated delivery. He was found to have IUGR, a two vessel cord, ear indentations, branchial cleft cyst, small phallus, and thrombocytopenia. The patient developed apnea and staring spells during the first week of life, and MRI performed on postnatal day 8 did not reveal structural abnormalities or infarcts. Subsequent magnetic resonance angiography (MRA) demonstrated an asymmetrically small right transverse sinus that was patent. Karyotype and chromosome microarray analysis were performed, and the patient was found to carry an unbalanced 1;3 translocation that results in a 6.9-Mb deletion of the distal long arm of chromosome 1 containing 22 genes and an 8.6-Mb duplication of the distal short arm of chromosome 3 that contains 15 genes [46,XY,t(1;3)(q43;p26.1). arr[hg18] 1q43q44(240,287,612-247,190,999)x1,3p26.3p26.1(35,332-8,625,632)x3] (Table 1). Suspecting an association with the patient’s known chromosomal abnormality, the literature was reviewed, and another patient with similar symptoms and a smaller overlapping deletion involving 1q43q44 was identified (1) (Figure 1).
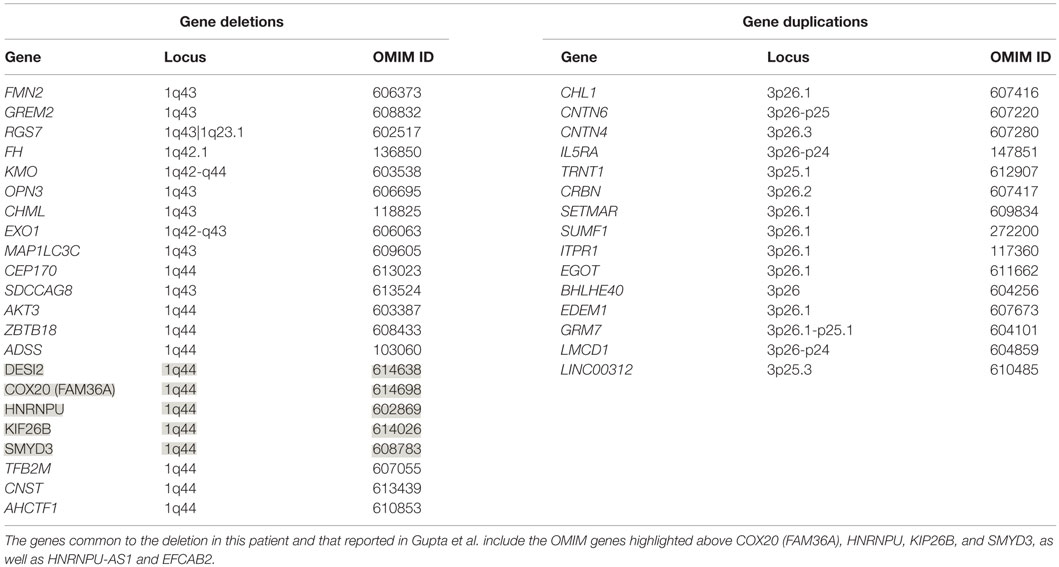
Table 1. Deleted and duplicated OMIM genes resulting from an unbalanced t(1;3)(q43;p26.1).
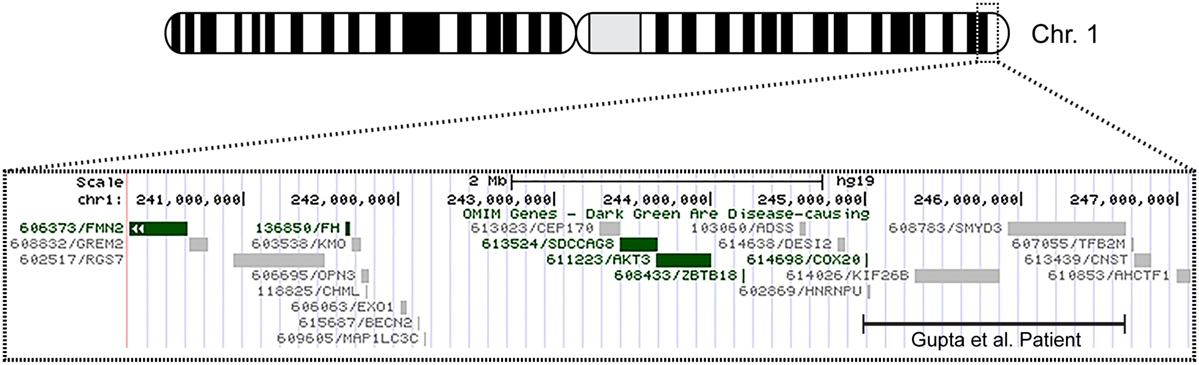
Figure 1. Overview of patient’s chromosome 1 deletion. Chromosome 1 ideogram displaying our patient’s 6.9 Mb deletion and the deletion observed in the patient described by Gupta et al. (1). The OMIM genes common to both deletions include COX20 (FAM36A), HNRNPU, HNRNPU-AS1, EFCAB2, KIP26B, and SMYD3.
Repeat MRI was performed on day 4 and showed worsening left-to-right subfalcine herniation anteriorly, patent arterial and venous supply, and left-sided diffusion restriction (Figure 2). Magnetic resonance spectroscopy (MRS) showed associated significant increase in lactate within the left cerebral hemisphere with decreased N-acetylaspartic acid, choline, and creatinine. A smaller but elevated lactate signal was observed in the right hemisphere. Due to worsening left hemispheric edema, the patient was started on intravenous mannitol on the third day of admission and transitioned to enteral sodium chloride 24 h later. Head CT on the sixth day of admission indicated that hemispheric swelling had stabilized, and repeat MRI/MRS on day 8 was essentially unchanged. Laboratory testing on this day revealed decreased total and free carnitine levels (14 and 16 nmol/mL, respectively); venous lactate was within normal limits (0.8 mmol/L). Given the patient’s history, clinical presentation, and MRI findings, he was diagnosed with hemiconvulsion–hemiplegia–epilepsy syndrome (HHE). The patient was seen in clinic approximately 2 months later after the completion of inpatient rehabilitation and displayed dense right hemiparesis with the ability to sit alone without support. His mother reported that his vocalizations and cognitive function had returned to baseline, and he was attempting to crawl again. At this time, the patient had not experienced a seizure since the inciting event.
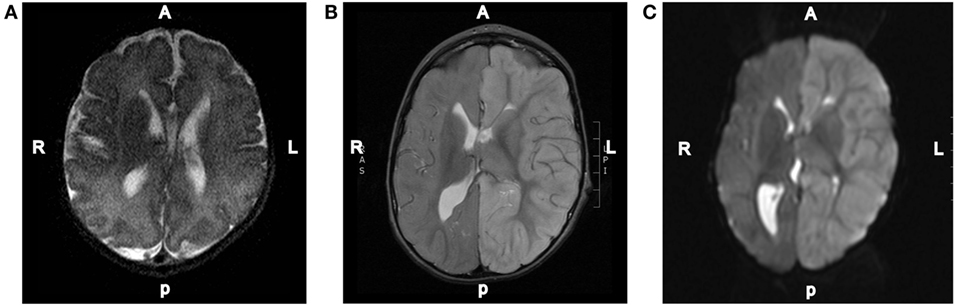
Figure 2. Representative MRI images. (A) T2-weighted axial brain MRI done of this patient at 3 days of age. (B) T2-weighted axial brain MRI done on day 2 of the hospital admission showing diffuse increased T2 signal intensity throughout the left hemispheric gray matter. (C) Diffusion-weighted axial brain MRI done on day 2 of the hospital admission showing diffuse left hemispheric cerebral edema.
Discussion
Hemiconvulsion–hemiplegia–epilepsy syndrome is a rare outcome of prolonged focal status epilepticus that typically occurs in children under 4 years of age, in the context of febrile illness. While the incidence is unknown, it has been reported to have declined during the last several decades (2). A 2012 review of all published cases by Auvin and colleagues (3) reveal that approximately 30% of cases have precipitating factors (e.g., cortical dysplasia, viral infection, and known genetic polymorphisms), while the rest are idiopathic in nature. Clinically, HHE is characterized by prolonged unilateral clonic seizure followed by diffuse unilateral hemispheric edema and the development of hemiplegia (4). Cytotoxic edema in HHE may be radiographically similar to acute ischemic stroke, except that it is not limited to discrete vascular territories and is associated with normal MRA. The resolution of hemispheric edema is usually followed by global cerebral hemiatrophy, recurrent epilepsy, and persistent hemiplegia. Early neuroradiographic findings include increased ipsilateral T2- and diffusion-weighted signals, decreased apparent diffusion coefficient (ADC), and, in some cases, midline or temporal lobe herniation (3). These findings are associated with increased lactate and reduced N-acetyl acetate (NAA) signals on MRS (3, 5). While the acute phase of HHE is characterized by ictal discharges on EEG, slow waves and spikes followed by higher amplitude delta wave slowing may be seen in the affected hemisphere following the seizure (3). Several postmortem pathological examinations of brain parenchyma from affected individual have been mixed; findings include diffuse cortical scarring, edema and necrosis of cortical layers III and V, axonal damage, and spongiosis within the temporal lobe (6–8).
The pathophysiology of HEE has not been elucidated but may involve neuronal injury secondary to excitotoxicity and inflammatory changes triggered by prolonged hemiconvulsion (3). In this framework, neuronal energy metabolism is perturbed during the ictal event (9) and leads to excitoxic cell injury that is worsened by the cytokine-mediated inflammatory milieu caused by concurrent febrile illness (4). Genetic factors, such as channelopathies secondary to mutation within SCN1A (10) or CACNA1A (11) or pro-thrombotic states due to the presence of factor V Leiden (12), protein S deficiency (12, 13), or the MTHFR C677T polymorphism (12), may predispose the patient to HHE by increasing the duration of ictal activity, lowering the febrile seizure threshold, and/or exacerbating oxidative stress during seizure activity. To date, it unknown why seizures are restricted to a single hemisphere, although it has been hypothesized that the slow kinetics of corpus callosum development may limit seizure generalization (14). Another important unanswered question is whether if specific infectious agents preferentially predispose to HHE. For example, infection with human herpes virus-6 or -7 (HHV-6 and HHV-7, respectively) has been linked to febrile seizure in the general population (15, 16); yet, there has only been one published case of HHV-7 detected in the CSF of an HHE patient (17).
For this patient, cytogenetic analysis revealed a 1q43q44 deletion and a 3p26.1p26.3 duplication. Analysis of the deleted portion reveals 22 missing genes on chromosome 1 (Table 1), several of which appear to be medically relevant. AKT3, which codes for a serine–threonine kinase that is active during brain development, has been associated with microcephaly, while abnormalities of the corpus collosum may involve ZBTB18 (ZNF238), whose product is a C2H2-type zinc finger protein that is expressed during embryogenesis and functions as a transcriptional repressor (18). Analysis of the duplication reveals three doses of 15 genes (Table 1). 3p26 duplications have not been widely described in the literature and are generally not associated with seizure. However, based on a single patient with developmental disability and epilepsy, as well as a 3p26.3 duplication that includes the CHL1 gene that codes for neural cell adhesion molecule L1-like protein, it has been proposed that this gene is a dosage sensitive one that may predispose to abnormal cognitive development (18).
Only five 1q44 deleted genes (Table 1) are the same in our patient and the patient with HHE reported by Gupta et al. (1), and only two genes, COX20 (FAM36A) and HNRNPU, have been linked to intellectual disability and epilepsy (18, 19). HNRNPU codes for heterogeneous nuclear ribonucleoprotein U, which is a ribosomal protein involved in pre-mRNA processing during development (18). The FAM36A protein, a human ortholog of COX20, facilitates assembly of cytochrome c oxidase (COX; Complex IV of the electron transport chain), and mutant FAM36A can result in decreased COX expression in patient fibroblasts and may limit mitochondrial capacity for oxidative phosphorylation (20, 21). Given the role of COX20 in proper cellular expression of COX and the observation that mitochondrial encephalopathy, lactic acidosis, and strokes (MELAS) is noted for producing rapid onset of MRI signal changes associated with cytotoxic edema (e.g., decreased ADC) without a vascular distribution like the changes seen in our patient, it is possible that mitochondrial dysfunction related to COX20 deletion may have exacerbated the patient’s symptoms. Metabolic workup during admission, however, was equivocal, and the patient’s presentation likely had a multifactorial etiology.
Data from recent sequencing and copy number variation studies suggest that heterozygous loss of any one of the five OMIM genes deleted in both our patient and the patient described by Gupta et al. is not sufficient to cause HHE. In each case, multiple sequence variants likely to result in loss of function (i.e., nonsense, frameshift, or splice site changes) were identified in the Exome Aggregation Consortium (ExAC) database. With the exception of KIF26B and SMYD3, no exon containing deletions involving these genes individually or en masse were identified in either the Children’s Hospital of Philadelphia (CHOP) database or the Database of Genomic Variants (DGV) for benign copy number variation. While these data suggest that heterozygous loss of any one of these genes is compatible with normal development, it is conceivable that haploinsufficiency may contribute to the development of HHE, possibly in conjunction with sequence and/or copy number variants located within the complementary genes on the non-deleted homolog or elsewhere within the genome. Further studies, including obtaining a muscle biopsy to assay COX function and sequencing non-deleted genomic regions on the normal chromosome 1 homolog for deleterious mutations, will be necessary to elucidate the etiology of this patient’s presentation.
Ethics Statement
The patient’s mother provided verbal and written consent for use of the patient’s personal health information in this case report in order to disseminate knowledge of the patient’s presentation to the broader medical community.
Author Contributions
JR and SW wrote the manuscript, JR prepared the figures, KK-R completed genomic analysis, and RG and KK-R edited the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by F30AA021312 to JR and U01HG006487 to RG.
References
1. Gupta R, Agarwal M, Boqqula VR, Phadke RV, Phadke SR. Hemiconvulsion-hemiplegia-epilepsy syndrome with 1q44 microdeletion: causal or chance association. Am J Med Genet A (2014) 164A(1):186–9. doi: 10.1002/ajmg.a.36198
2. Roger J, Dravet C, Bureau M. Unilateral seizures: hemiconvulsions-hemiplegia syndrome (HH) and hemiconvulsions-hemiplegia-epilepsy syndrome (HHE). Electroencephalogr Clin Neurophysiol Suppl (1982) 35:211–21.
3. Auvin S, Bellavoine V, Merdariu D, Delanoe C, Elmaleh-Berges M, Gressens P, et al. Hemiconvulsion-hemiplegia-epilepsy syndrome: current understandings. Eur J Paediatr Neurol (2012) 16(5):413–21. doi:10.1016/j.ejpn.2012.01.007
4. Tenney JR, Schapiro MB. Child neurology: hemiconvulsion-hemiplegia-epilepsy syndrome. Neurology (2012) 79(1):e1–4. doi:10.1212/WNL.0b013e31825dce5f
5. Freeman JL, Coleman LT, Smith LJ, Shield LK. Hemiconvulsion-hemiplegia-epilepsy syndrome: characteristic early magnetic resonance imaging findings. J Child Neurol (2002) 17(1):10–6. doi:10.1177/088307380201700103
6. Auvin S, Devisme L, Maurage CA, Soto-Ares G, Cuisset JM, Leclerc F, et al. Neuropathological and MRI findings in an acute presentation of hemiconvulsion-hemiplegia: a report with pathophysiological implications. Seizure (2007) 16(4):371–6. doi:10.1016/j.seizure.2007.01.009
7. Mori Y. Anatomopathology and pathogeny of the hemiconvulsion-hemiplegia-epilepsy syndrome. Part II. J Neurosurg Sci (1979) 23(1):1–22.
8. Berhouma M, Chekili R, Brini I, Kchir N, Jemel H, Bousnina S, et al. Decompressive hemicraniectomy in a space-occupying presentation of hemiconvulsion-hemiplegia-epilepsy syndrome. Clin Neurol Neurosurg (2007) 109(10):914–7. doi:10.1016/j.clineuro.2007.07.027
9. Toldo I, Calderone M, Boniver C, Dravet C, Guerrini R, Laverda AM. Hemiconvulsion-hemiplegia-epilepsy syndrome: early magnetic resonance imaging findings and neuroradiological follow-up. Brain Dev (2007) 29(2):109–11. doi:10.1016/j.braindev.2006.06.005
10. Sakakibara T, Nakagawa E, Saito Y, Sakuma H, Komaki H, Sugai K, et al. Hemiconvulsion-hemiplegia syndrome in a patient with severe myoclonic epilepsy in infancy. Epilepsia (2009) 50(9):2158–62. doi:10.1111/j.1528-1167.2009.02175.x
11. Yamazaki S, Ikeno K, Abe T, Tohyama J, Adachi Y. Hemiconvulsion-hemiplegia-epilepsy syndrome associated with CACNA1A S218L mutation. Pediatr Neurol (2011) 45(3):193–6. doi:10.1016/j.pediatrneurol.2011.04.010
12. Scantlebury MH, David M, Carmant L. Association between factor V Leiden mutation and the hemiconvulsion, hemiplegia, and epilepsy syndrome: report of two cases. J Child Neurol (2002) 17(9):713–7. doi:10.1177/088307380201700914
13. Mondal RK, Chakravorty D, Das S. Hemiconvulsion, hemiplegia, epilepsy syndrome and inherited protein S deficiency. Indian J Pediatr (2006) 73(2):157–9. doi:10.1007/BF02820211
14. Bahi-Buisson N, Kossorotoff M, Barnerias C, Boddaert N, Bourgeois M, Dulac O, et al. Atypical case of hemiconvulsions-hemiplegia-epilepsy syndrome revealing contralateral focal cortical dysplasia. Dev Med Child Neurol (2005) 47(12):830–4. doi:10.1111/j.1469-8749.2005.tb01089.x
15. Laina I, Syriopoulou VP, Daikos GL, Roma ES, Papageorgiou F, Kakourou T, et al. Febrile seizures and primary human herpesvirus 6 infection. Pediatr Neurol (2010) 42(1):28–31. doi:10.1016/j.pediatrneurol.2009.07.016
16. Teach SJ, Wallace HL, Evans MJ, Duffner PK, Hay J, Faden HS. Human herpesviruses types 6 and 7 and febrile seizures. Pediatr Neurol (1999) 21(4):699–703. doi:10.1016/S0887-8994(99)00068-5
17. Kawada J, Kimura H, Yoshikawa T, Ihira M, Okumura A, Morishima T, et al. Hemiconvulsion-hemiplegia syndrome and primary human herpesvirus 7 infection. Brain Dev (2004) 26(6):412–4. doi:10.1016/j.braindev.2003.12.003
18. Ballif BC, Rosenfeld JA, Traylor R, Theisen A, Bader PI, Ladda RL, et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet (2012) 131(1):145–56. doi:10.1007/s00439-011-1073-y
19. Thierry G, Beneteau C, Pichon O, Flori E, Isidor B, Popelard F, et al. Molecular characterization of 1q44 microdeletion in 11 patients reveals three candidate genes for intellectual disability and seizures. Am J Med Genet A (2012) 158A(7):1633–40. doi:10.1002/ajmg.a.35423
20. Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet (2013) 22(4):656–67. doi:10.1093/hmg/dds473
Keywords: hemiconvulsion–hemiparesis–epilepsy, 1q43q44 deletion, COX20, FAM36A, mitochondrial
Citation: Robinson JE, Wolfe SM, Kaiser-Rogers K and Greenwood RS (2016) Stroke-Like Presentation Following Febrile Seizure in a Patient with 1q43q44 Deletion Syndrome. Front. Neurol. 7:67. doi: 10.3389/fneur.2016.00067
Received: 03 February 2016; Accepted: 19 April 2016;
Published: 04 May 2016
Edited by:
Eugene R. Schnitzler, Loyola University Medical Center, USAReviewed by:
Tina Bregant, Children’s Hospital Ljubljana, SloveniaAnju Aggarwal, University College of Medical Sciences, India
Susan Cohen, Medical College of Wisconsin, USA
Copyright: © 2016 Robinson, Wolfe, Kaiser-Rogers and Greenwood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert S. Greenwood, Z3JlZW53b3ImI3gwMDA0MDtuZXVyb2xvZ3kudW5jLmVkdQ==