 Linda J. Larson-Prior
Linda J. Larson-Prior Yo-El Ju2
Yo-El Ju2 James E. Galvin
James E. Galvin- 1Department of Radiology, Washington University School of Medicine, St. Louis, MO, USA
- 2Department of Neurology, Washington University School of Medicine, St. Louis, MO, USA
- 3Departments of Neurology, New York University Langone School of Medicine, New York, NY, USA
- 4Department of Psychiatry, New York University Langone School of Medicine, New York, NY, USA
- 5Department of Population Health, New York University Langone School of Medicine, New York, NY, USA
Subcortical circuits mediating sleep–wake functions have been well characterized in animal models, and corroborated by more recent human studies. Disruptions in these circuits have been identified in hypersomnia disorders (HDs) such as narcolepsy and Kleine–Levin Syndrome, as well as in neurodegenerative disorders expressing excessive daytime sleepiness. However, the behavioral expression of sleep–wake functions is not a simple on-or-off state determined by subcortical circuits, but encompasses a complex range of behaviors determined by the interaction between cortical networks and subcortical circuits. While conceived as disorders of sleep, HDs are equally disorders of wake, representing a fundamental instability in neural state characterized by lapses of alertness during wake. These episodic lapses in alertness and wakefulness are also frequently seen in neurodegenerative disorders where electroencephalogram demonstrates abnormal function in cortical regions associated with cognitive fluctuations (CFs). Moreover, functional connectivity MRI shows instability of cortical networks in individuals with CFs. We propose that the inability to stabilize neural state due to disruptions in the sleep–wake control networks is common to the sleep and cognitive dysfunctions seen in hypersomnia and neurodegenerative disorders.
Introduction
The brain is a complex dynamic system in which interactions on multiple temporal and spatial scales enable adaptive behaviors appropriate to environmental stimuli. These interactions are accomplished not only by specific network activities that produce organismal responses to stimuli but also by the general state of the system, which is most clearly represented in the shift of state from wake to sleep. Thus, system-wide dysfunctions can occur both in the networks responsible for specific functional responses to the external world and in the less well understood networks responsible for the maintenance of and switching between neural states.
The normal neural state transition from wake to sleep is defined by changes in scalp-recorded electroencephalogram (EEG) that exhibit a stereotypic progression through a full nocturnal sleep bout (Figure 1) (1). The transitional state from wake to sleep is characterized by a shift in EEG spectral content in which alpha (8–12 Hz) band power is reduced as theta (4–7 Hz) power increases. Behaviorally, subjects are drowsy and physically relaxed, although when questioned they do not report being asleep. Following this transitional period, as subjects descend to true sleep (N2) the scalp EEG exhibits an increase in low frequency power and characteristic spindles of sleep (7–14 Hz). Subjects then descend into slow wave sleep (SWS, N3) that is characterized by the presence of large amplitude slow (0.5–4 Hz) delta frequency waves on scalp EEG. Stages N1–N3 comprise non-rapid eye-movement (NREM) sleep and will cyclically alternate with rapid-eye-movement (2) sleep through the sleep bout. REM sleep exhibits an “active” pattern similar to that of wake, and is characterized by the distinct eye-movements from which its name was derived, peripheral atonia, and behavioral quiescence.
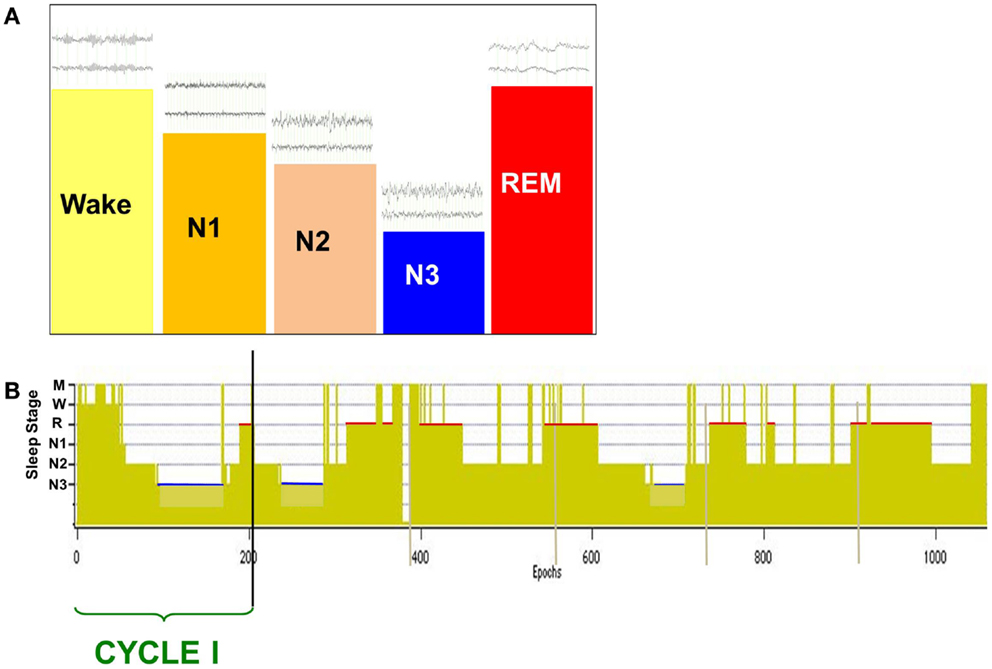
Figure 1. Scalp-recorded EEG defines the neural and behavioral states of wake and sleep (A), which are charted over a full nocturnal sleep bout in a hypnogram (B). The descent to sleep is characterized by changes in EEG frequency content and amplitude [above state bars in (A)], with deep sleep (slow wave sleep, N3) characterized by large amplitude slow waves (0.5–4 Hz). Over the nocturnal sleep bout, SWS (N3) duration is reduced [blue bars in (B)] while REM sleep duration increases [red bars in (B)] and is generally longest just prior to waking. A cycle is defined by the shift from REM sleep to another sleep stage, and normal sleepers generate two to seven cycles per night.
In 1949, Moruzzi and Magoun reported that stimulation of the brainstem reticular core produced changes in the EEG akin to those seen in arousal (3). Following the description of REM sleep by Aserinsky and Kleitman (4), studies in animal models showed the importance of the brainstem in the generation of this sleep stage (1, 5). Subsequent studies explored the neurotransmitter systems involved in the cyclic alternation of REM and NREM sleep, as well as brainstem regions active during wake, pointing to an important role for the brainstem reticular core in the control of sleep and waking. These studies clarified many mechanisms of the induction and maintenance of normal sleep, and the control of both circadian (24-h) and ultradian (90–120 min cycle of NREM/REM) rhythm generation in both animal and human subjects (1, 6–14).
Disruptions of sleep–wake and circadian cycling commonly accompany neuropsychiatric and neurodegenerative disease (15, 16). Such disruptions range from changes in the duration of nighttime sleep or specific sleep periods to disorders of circadian patterning such as seen in “sundowning” in Alzheimer’s Disease (AD) patients (15–19). In addition to overt disruption of nighttime sleep, and often considered solely a concomitant of the loss of sleep, daytime cognitive function may also be adversely impacted in these disorders. While the mechanisms by which neurocognitive and neurobehavioral dysfunction interact with sleep and circadian rhythm disruptions are currently unknown, there is clear overlap between sleep regulating regions and neurotransmitter and neural network systems affected in these disorders (15, 17, 20) that points to the potential for complex interactions between sleep and cognition.
In 1917, Constantin von Economo proposed a neurophysiological substrate for the control of the neural state transition from wake to sleep based upon the clinical and neuropathological features of a disorder in which patients exhibited abnormal sleep/wake rhythms (21, 22). This disorder, which von Eonomo termed encephalitis lethargica (EL), exhibited two subtypes with opposite effects on sleep/wake rhythms; one in which sleep duration was prolonged and intruded on waking periods and another in which patients had reduced sleep durations and difficulty in initiating or maintaining sleep (21, 23). Regardless of subtype, patients reported excessive daytime sleepiness (EDS). His observations led him to postulate the presence of an active sleep regulatory system centered in the hypothalamus (21). Von Economo’s hypothesis that the ventral hypothalamic region housed a sleep center while posterior hypothalamic regions generated the wake-state has informed studies of the neurobiology of sleep and arousal since its initial description, with later studies confirming a major role for hypothalamus in the regulation of sleep and wake (24–29).
The expression of these biological rhythms in the cortex was the focus of seminal studies by Mercia Steriade and his colleagues (30–32), who provided evidence of the role of thalamo-cortical circuits in the generation of the EEG signatures of sleep. This work was extended to show the importance of thalamus in generating EEG rhythms (33–35) while pointing out that the full expression of these rhythms required the interaction of both thalamus and cortex (30–32, 36–41). While the cortex has been suggested to play a role in the decentralized control of the homeostatic sleep drive (42–44), the current consensus puts the sleep/wake control center in subcortical circuits. However, regardless of its role in the primary control of sleep, thalamo-cortical circuitry has a clear and critical role in the regulation of cognitive and behavioral aspects of sleep and waking. Thus, the reintegration of this circuitry in theories of neural state regulation is necessary if we are to gain a true understanding of the role of sleep disregulation in pathological neural and cognitive states.
Brief Overview of Hypersomnia Disorders
Hypersomnia disorder is an umbrella term for a group of disorders in which the primary characteristic is EDS in the face of normal or longer than normal nocturnal sleep (45). Hypersomnia disorders (HDs) are recognized as primary disorders of sleep, and it is the lack of refreshing sleep – sleep that results in a wake period in which the patient feels alert and motivated – that often drives sufferers to seek medical assistance. Yet it must be recognized that disorders of hypersomnia are equally disorders of wake, as it is the waking state in which patients report the greatest distress due to cognitive, social, or workplace dysfunction.
While most research examining neurobiological and neurophysiological substrates of these disorders have focused on the neural circuitry that produces and maintains sleep and wake, it is the interactions of these sleep-related circuits with those functioning in wake-state arousal, and how these interactions influence cognition and behavior, that must ultimately be explained if effective therapies are to be developed.
Narcolepsy
Narcolepsy is a disorder in which sleep intrudes on daily activity while nocturnal sleep is frequently fragmented, and is classified by the ICDS-2 as a hypersomnia of central origin (46). Narcolepsy is clinically defined by a short sleep latency and two or more sleep onset REM periods (SOREMPs) during a multiple sleep latency test (MSLT) in which individuals are given four to five standardized daytime nap opportunities (46, 47). Two forms of narcolepsy are recognized; narcolepsy with cataplexy, currently named narcolepsy/hypocretin (HCRT) deficiency disorder (47), and narcolepsy without cataplexy. Narcolepsy with cataplexy is due to low levels of hypocretin-1, which can be confirmed by measurement in cerebrospinal fluid (47), and it has a strong linkage to human leukocyte antigens (48) with HLA-DBQ1*0602 mutations found in 90% of tested patients (49–51).
In 1998, two laboratories announced the discovery of a new hypothalamic peptide, one reporting its importance in feeding [orexin (ORX); (52)] and the other focused on its role in wake and sleep [HCRT; (53)]. The discovery that narcolepsy with cataplexy resulted from loss of ORX/HCRT-containing neurons in the posterior lateral hypothalamic area (pLHA) provided a fuller understanding of the symptomology of this disorder (54–56), and supported an early hypothesis put forward by von Ecomono (21). In this disorder sleep-to-wake transitions are unstable, as patients are generally unable to maintain consolidated sleep during the main nocturnal sleep period, and unable to maintain wake during the normal wake period. Individuals with narcolepsy have early onset of REM sleep; including at sleep onset, during the main sleep period and during naps, indicating a defect in the normal progression of sleep stages. Additional REM phenomena frequently associated with narcolepsy are also due to an instability between wake and REM: hallucinations in sleep–wake transitions (dreaming imagery of REM with awareness of wake), sleep paralysis (paralysis of REM sleep with awareness of wake), and REM sleep behavior disorder (dream imagery of REM sleep with muscle tonus of wake).
As with other HD, narcolepsy patients complain of memory problems and difficulties with concentration and attention (57–59). Attentional deficits, particularly in vigilance tasks, have also been reported (58, 60, 61). Attentional deficits seem to be an effect of the fluctuations of arousal that accompany this disorder, more than a deficit in attentional control in general (58, 59). These lapses of attention result in impaired vigilance over long periods that can be compensated by deploying attention in repeated shorter bouts (62).
The etiology of narcolepsy is currently unknown (63), although recent studies have provided strong experimental support for autoimmune etiology (64–67). Narcolepsy has been reported as secondary to tumors (68), head trauma (68–70), and immune-related disorders (67, 71, 72).
Kleine–Levin Syndrome
The International Classification of Sleep Disorders (ICDS-2) recognizes idiopathic and recurrent hypersomnia as distinct entities (46). The most common recurrent hypersomnia is Kleine–Levin Syndrome (KLS), a rare disorder that predominantly affects adolescent boys and is characterized by bouts of hypersomnolence during which the patient also exhibits one of the following: cognitive or mood disturbances, compulsive eating, hypersexuality, or disinhibition behaviors (47, 73). Behavioral, sleep, and mood symptoms remit in interictal periods (74).
Cognitive disturbances, unlike behavioral and mood disturbances, have been reported to outlast ictal periods (73–76). Depression and anxiety are common in this population (77, 78), and recent studies suggest that there are long-term deficits in memory and visuospatial function (73, 75, 76, 79). As these mood and cognitive symptoms are similar to those reported for idiopathic HD (iHSD), the pathophysiological mechanisms by which they are generated is expected to be similar.
Neuroimaging studies have provided some clues as to the genesis of cognitive and mood disruptions in this population. Although structural neuroimaging is generally read as normal in KLS, widespread abnormalities have been reported during ictal periods based on functional neuroimaging; with reduced blood flow to thalamus, hypothalamus, basal ganglia, and cortex (73, 80) together with hypometabolism in hypothalamus and cortex (81). Studies have shown hyperactivation in the thalamus of KLS patients during performance of a working memory task using fMRI (75, 76) that significantly differed from the activation seen in healthy control subjects and correlated with performance deficits in KLS patients.
The pathophysiology of the disorder is unknown and its diagnosis remains based upon symptomology (73, 76, 81). Interestingly, and in common with EL, a viral infection potentially associated with a subsequent autoimmune response has been proposed as a causative agent and two autopsy cases have reported inflammatory infiltrates in hypothalamus and thalamus (73, 77, 82).
Hypersomnolence Disorder
Hypersomnolence disorder (47) may include only non-refreshing sleep despite nocturnal sleep durations (idiopathic) but is more frequently coexistent with other neurological, psychological, mental, and sleep disorders (83, 84). EDS in spite of normal or long duration nocturnal sleep is characteristic of the disorder though insufficient to define it (45–47). A characteristic deterioration in waking function and general alertness is generally remarked and symptom duration must exceed 3 months with a sleep onset latency of <10 min for a clinical diagnosis to be made.
Idiopathic hypersomnia (iHSD) occurs in two forms: (1) long sleep duration, in which patients sleep in excess of 10 h per day while reporting non-refreshing sleep and EDS, and (2) normal sleep duration where sleep bouts are within normal range but EDS and non-refreshing sleep remain primary complaints. Patients report constant fatigue or a constant lack of alertness during wake periods more than sleepiness (85–87) together with great difficulty in waking after sleep. Sleep efficiency is good in these patients, and REM sleep onset latencies and durations are generally within normal range. However, changes in nocturnal sleep architecture during polysomnography (PSG) have been reported, including a reduction in the amount of SWS in the main sleep bout with normal homeostatic reduction (87–89).
In addition to non-refreshing sleep, patients report memory and attention deficits and commonly present with digestive system disorders, depression, and anxiety (84, 86). In a recent study based on self-reported cognitive changes, 79% of patients with hypersomnolence disorder (HSD) reported memory problems that included frequent forgetfulness (86). A recent study assessing sustained attention in a range of HSD patients found impaired vigilance that did not differ significantly between patient groups (90), suggesting that this is a common feature of HSD. Mood disorders are also commonly reported by individuals with iHSD, with prevalence between 15 and 25% in this population (78, 91) during asymptomatic periods. The prevalence of depressive symptoms during symptomatic episodes is greater (82), with up to 40% of patients exhibiting symptoms. The pathophysiological mechanisms for mood and cognitive symptoms in iHSD have yet to be specifically explored, although they are similar to those reported for KLS.
Neurobiological Substrates of Sleep and Waking
Subcortical Network Interactions in Sleep and Waking
Control of the sleep–wake cycle depends upon a widely distributed and complex neural system, many components of which have been shown to be affected by HD. The hypothalamus represents a primary control center in the regulation of this system; acting as the interface between circadian, energetic, sleep, and autonomic circuits that are all modulated in sleep (92–94). The circadian system provides information critical to the function of the sleep–wake control system, contributing one arm of the two-process model of sleep regulation (95) and interacting closely with subcortical regions to link this to the ultradian sleep–wake rhythm (96, 97). In the following, we provide a brief overview of the subcortical brain circuits involved in the regulation and control of sleep and wake together with the interactions between these systems and the cortex.
The preoptic region of the hypothalamus is currently recognized as the major sleep-promoting brain region (27–29, 98–100), with the ventrolateral preoptic area (VLPO) and the median preoptic nucleus (MnPO) providing inhibitory drive to brain regions engaged in the induction and maintenance of the waking state (Figure 2). Recent studies have pointed to an important role for melanin concentrating hormone (MCH) neurons of the lateral hypothalamic area (LHA) in sleep-promotion (101–105). MCH neurons co-release gamma-amino butyric acid (GABA) in wake-promoting regions, thus promoting sleep (106, 107). MCH neurons are active in both NREM and REM sleep and there is evidence suggestive of a specific role in the control of REM sleep, perhaps together with GABAergic neurons found interspersed with them in the lateral hypothalamus (101, 106, 108).
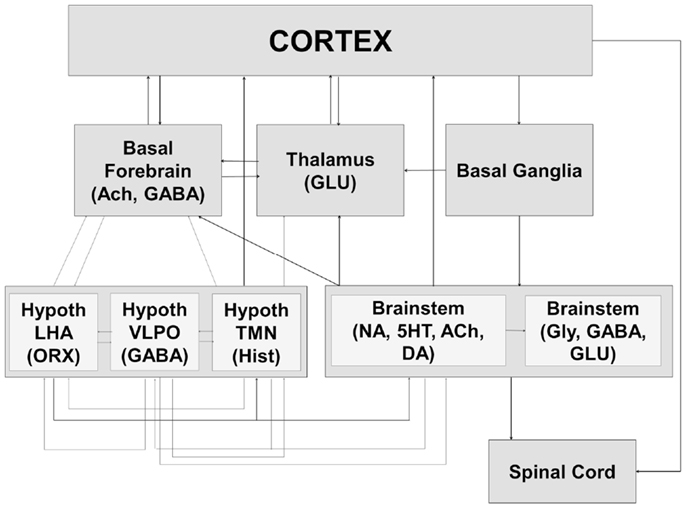
Figure 2. The sleep–wake cycle is controlled by a widely distributed and complex neural system that includes interacting components of the brainstem, hypothalamus, basal ganglia, basal forebrain, and thalamus. The interactions between these systems result in both global changes in neural state and their behavioral expression at the level of the spinal cord and brainstem (motor atonia or phasic bursts, autonomic responses) as well as the cortex (conscious awareness of external environment). Ach, acetylcholine; GABA, gamma-amino butyric acid; GLU, glutamate; Hypoth, hypothalamus; LHA, lateral hypothalamic area; ORX, orexin; VLPO, ventrolateral preoptic area; TMN, tuberomammillary nucleus; Hist, histamine; NA, noradrenaline; 5HT, serotonin; DA, dopamine; Gly, glycine.
Orexin producing cells in the LHA have been shown to play a major role in the induction and maintenance of the waking state (109–112) while linking autonomic and metabolic centers (113), thus acting as a major integrative system (Figure 3). Sleep-promoting MCH and GABA neurons are interspersed in the LHA with wake-promoting ORX neurons, providing for rapid mutual inhibition in state transitions. The waking state is generated by the inhibition of hypothalamic sleep-promoting centers together with excitation of wake-promoting centers in the hypothalamus, brainstem, and basal forebrain (Figures 2 and 3). Wake centers of the hypothalamus include both ORX cells of LHA and histaminergic (HIST) neurons of the tuberomammillary nucleus (TMN). The brainstem reticular activating system (RAS) represents the primary control system for wake (6, 26, 114–117) and consists of acetylcholine (Ach)-containing neurons in the pedunculopontine (PPT) and laterodorsal tegmental (LDT) nuclei, noradrenaline (NE)-containing cells in the locus coeruleus (LC), serotonergic (5HT) neurons of the dorsal and median raphe nuclei (RN), glutamatergic cells of the subcoeruleus complex (SCC), and dopaminergic cells of the ventral periaqueductal gray (vPAG) that are reciprocally connected to wake-promoting hypothalamic centers. Considered by some a rostral extension of the RAS, the basal forebrain includes a small population of Ach cells that are active in both wake and REM sleep, playing an important role in the generation of desynchronized electrical activity of the cortex in both states (118, 119). Basal forebrain neurons receive input from both hypothalamic and brainstem wake centers (Figure 2) and may represent a key mediating center of cortical arousal (120).
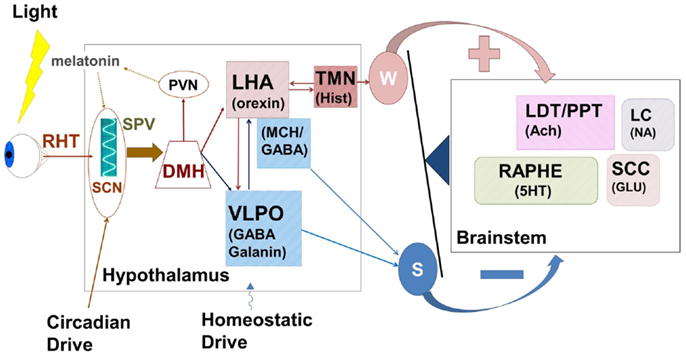
Figure 3. The hypothalamus represents a central control center for the interaction between circadian rhythms paced by the suprachiasmatic nucleus (SCN) and the sleep homeostat. Complex feedback loops within the hypothalamus, and between hypothalamus and brainstem wake-promoting centers control the putative “switch” between wake and sleep. Wake-promoting (red) signals serve to enhance activation of brainstem centers comprising the ascending reticular activating system while sleep-promoting (blue) centers inhibit this activation. In normal sleep, the state switch is rapid (<5 min) and the new state is quickly stabilized. RHT, retino-hypothalamic tract; SPV, subparaventricular zone; SCN, supra charismatic nucleus; DMH, dorsomedial hypothalamic nucleus; PVN, paraventricular hypothalamic nucleus; LHA, lateral hypothalamic area; VLPO, ventrolateral peropic area; TMN, tuberomammillary nucleus; LDT/PPT, laterodorsal tegmental/peduncular pontine tegmentum; LC, locus coeruleus; SCC, sub-coerulear complex; GABA, gamma-amino butyric acid; MCH, melanin concentrating hormone; Hist, histamine; Ach, acetylcholine; 5HT, serotonin; NA, noradrenaline; Glu, glutamate; W, wake; S, sleep.
Cells of the LHA provide glutamatergic and orexinergic inputs to brainstem wake-promoting regions of the RAS and receive inhibitory drive from the VLPO. Sleep onset is initiated by activation of the sleep-promoting VLPO, which acts to inhibit both RAS and LHA. The balance between sleep-promotion and wake-promotion is accomplished by a feedback mechanism that enables the relatively rapid switch in state with a mechanism resembling an electronic switch; an analogy that led Saper and his colleagues to develop the flip–flop switch model of this state transition (26, 121). This mechanism provides for the rapid stabilization of a newly entered state, but disruption of this balance can lead to state instability where inappropriate state switches can occur.
Under normal conditions, sleep initiation moves the system into NREM sleep, following which cyclic alternations between NREM and REM sleep develop (Figure 1B) that constitute the ultradian sleep rhythm. A normal sleeper will exhibit two to seven such cycles over a nocturnal sleep bout. It is generally agreed that mesopontine brainstem nuclei contain the regulatory circuitry for the stereotypic alternations of NREM and REM sleep. Glutamatergic neurons of the SCC are proposed to be the primary inducer of REM sleep (122, 123), with current studies suggesting that hypothalamic MCH cells act to stabilize this state (106). SCC innervation of cholinergic neurons of the LDT/PPN and basal forebrain result in the desynchronized EEG characteristic of REM while inhibitory drive to wake-promoting areas such as DRN and LC aid in state stabilization. Skeletal muscle atonia is a unique characteristic of normal REM sleep and is due to SCC excitation of medullary reticular centers that act to inhibit spinal motor neurons (122–124).
In a normal nocturnal sleep bout, there is a gradual reduction in the duration of NREM sleep periods with the longest period occurring in early cycles and the late cycles frequently containing only N2 sleep. In contrast, REM sleep durations are longest in the final cycles of the nocturnal sleep bout. The reduction in N3 content over the night represents the reduction in sleep drive that is currently conceived as a homeostatic regulatory mechanism.
Sleep homeostasis is the process by which sleep propensity increases over the wake period and is dissipated during the sleep period. Early observations pointed to a strong tie between prior wake time and deep NREM sleep (125–128). The proposal of a two-process control system for the regulation of sleep and waking was put forward by Borbely and Acherman (127, 129) in a model where circadian and homeostatic processes interact to maintain and regulate sleep/wake cycling. Substantial support for this model has accrued, leading to general acceptance of its central tenets, which include the importance of slow wave activity (SWA) as a marker of both sleep need and its dissipation (126, 128, 130–134). While no central regulatory center for sleep homeostasis has been defined, a number of studies have implicated circulating neuroactive molecules as potential mediators of homeostatic control (43, 135–138).
Thalamo-Cortical Network Interactions in Arousal and Sleep
Based on a series of studies pointing to the importance of the rostral brainstem in arousal (3, 139, 140) and REM sleep (141, 142), the majority consensus among physiologists in the 1990s was that the brainstem reticular system controlled the oscillatory network responsible for wake and sleep. A large body of evidence followed these studies, reporting the importance of brainstem cholinergic systems in the behavioral and electrophysiological expression of wake and arousal. The discovery that the thalamus did not present a passive, quiescent response to the shift from wake to sleep, but instead exhibited dual processing led to the conception of sleep as a process in which the thalamus acted to “gate out” external information and thus prevent arousal (143).
A different role for the thalamus, and thalamo-cortical interaction, was provided by seminal studies investigating electrophysiological rhythms in the cortex during sleep and wake. In a series of studies investigating the role of the thalamo-cortical circuitry in sleep, Steriade and his colleagues (30–32) introduced a new, ultraslow (<1 Hz) cortical rhythm distinct from SWS that was generated in cortical neurons and projected to thalamus (32) where it served to organize the slower sleep rhythms of spindles and slow waves (30). Emphasizing its cortical origin, studies showed that thalamic lesions did not abolish the rhythm (143, 144) and that the cortex itself maintains SWA even in an isolated slice preparation (38). Further, the ultraslow rhythm is also seen in thalamic nuclei, most strongly in the reticulothalamic cells that have been shown responsible for the generation of the spindles of sleep (32). The importance of these seminal papers was in the understanding that thalamo-cortical interactions are ultimately the generators of the major sleep rhythms recorded at the scalp by which sleep states are defined. Thus, the thalamus acts as a major integrating center, generating the rhythms of both wake and sleep in concert with a widespread network encompassing brainstem, hypothalamus, and cortex. In the current understanding of sleep–wake circuitry, sleep is a state actively generated by a large and complex neural network.
In keeping with current theories of brain network function in sleep, the thalamus has been shown to continue to transmit external information to the cortex in both NREM and REM states (145–153). However, while information continues to be transmitted from the thalamus to the cortex, there are distinct differences between wake-state responses and those seen in either NREM or REM sleep. During NREM sleep, neuroimaging studies have shown that higher order cortical regions show significantly reduced or absent responses to stimuli (146, 147, 150) while responses in primary cortices appear to remain near to those noted in wake (145, 152). Further, the timing of inputs relative to thalamo-cortical waveforms characterizing NREM sleep significantly impacts the degree to which further processing occurs (150). In REM sleep, where cortical activity resembles that of wake, recent reports suggest that more complex processing may occur than seen in deep NREM sleep (149, 153) while yet remaining suppressed relative to that of wake.
While further research is required to fully describe the complex network interactions resulting in normal sleep/wake transitioning, it is increasingly clear that the hypothalamus and thalamus represent critical integration and control centers by which these states are fully expressed. As noted by von Economo and illustrated by the clear instability of state in narcolepsy and other HDs, the hypothalamus plays a critical role in the transitions between and maintenance of the states of wake and sleep. Yet, Llinas and Steriade (39) point to the thalamus as the fundamental determinant of system state, and this view is upheld by studies showing that thalamic ablation leads to a pathologically prolonged state of wake in both animals and humans (154–156). To complicate matters further, as research focuses on the role of diffusible somnogens as potential mediators of the homeostatic sleep drive (43, 135–138), some are suggesting a central role for the neocortex in the control of sleep and waking. This suggestion has received some support from recent studies showing that sleep may not be globally exhibited, but occur locally in specific cortical areas even as the organism displays behavioral wake (157–161).
Increasing evidence thus points to a widespread and highly connected network that acts in concert with the circadian rhythms generated in the suprachiasmatic nucleus of the hypothalamus to not only control the cycling of neural state between wake and sleep, but to integrate that state with metabolic and physiological systems sharing the same circadian timing (92, 96, 97). At present, recognition of the complex and redundant anatomical linkages by which the thalamo-cortical, hypothalamic, subcortical, and brainstem sleep/wake control centers interact (Figures 4 and 5) leads to a better understanding of the huge array of behavioral and physiological responses that could result from dysfunction at any level of this network.
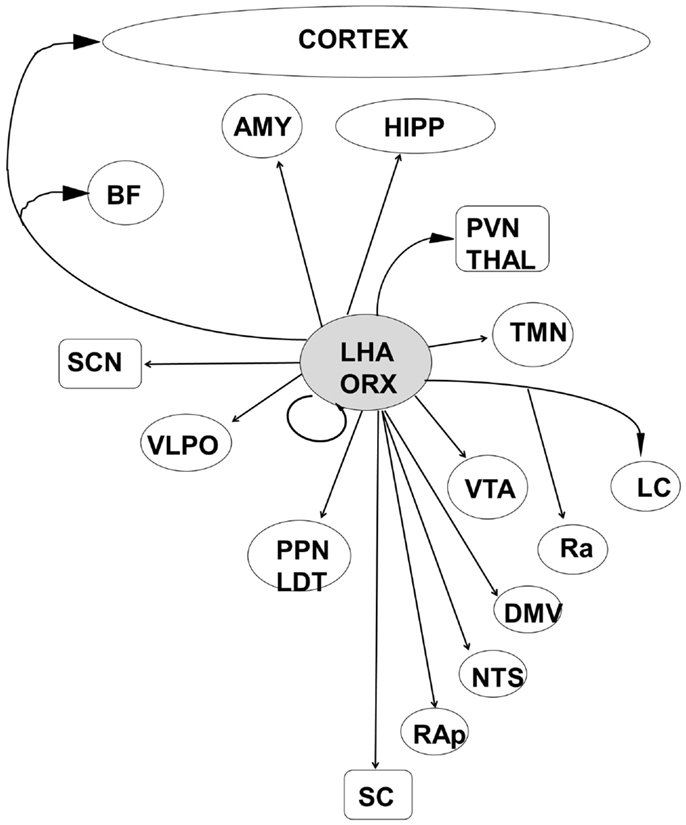
Figure 4. The orexinergic cells of the posterior lateral hypothalamic area are well placed to integrate information from metabolic and autonomic centers with both sleep–wake and circadian control systems. LHA, lateral hypothalamic area; ORX, orexin/hypocretin; HIPP, hippocampal formation; AMY, amygdala; BF, basal forebrain; PVN Thal, paraventricular nucleus of the thalamus; SCN, suprachiasmatic nucleus; VTA, ventral tegmental area; VLPO, ventrolateral preoptic area; TMN, tuberomammillary nucleus; LC, locus coeruleus; Ra, Raphe nuclei; PPN/LDT, laterodorsal tegmental/peduncular pontine tegmentum; NTS, nucleus tractus solitaries; DMV, dorsal motor nucleus of the vagus; RAp, raphe pallidus; SC, spinal cord.
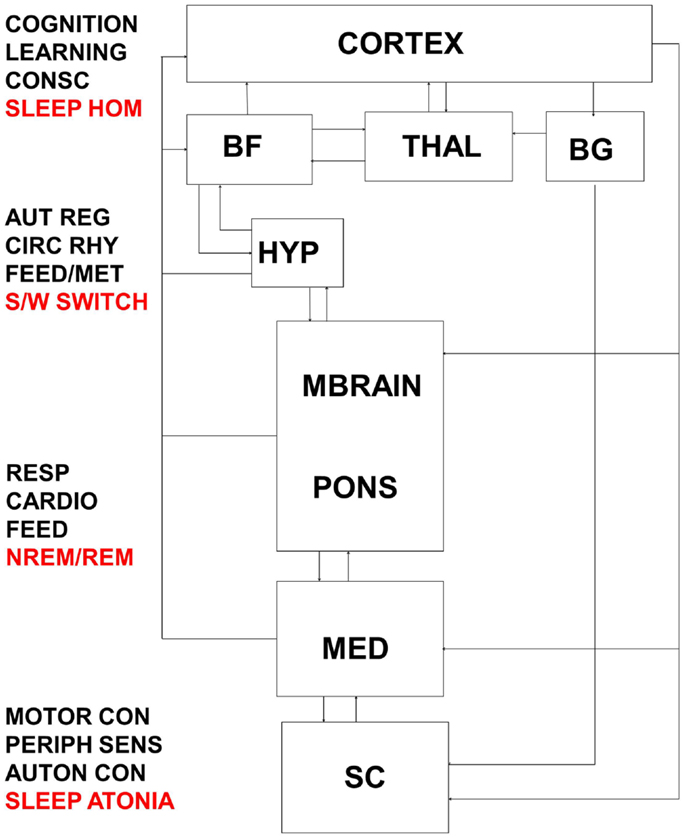
Figure 5. Interactions between thalamo-cortical, hypothalamic, subcortical, brainstem, and spinal control centers are highly complex and result in the integrated response of metabolic, autonomic, and cognitive systems to daily sleep–wake rhythms. Dysfunction at multiple levels in this system would reverberate to encompass broadly distributed symptomology. BF, basal forebrain; THAL, thalamus; BG, basal ganglia; HYP, hypothalamus; MBRAIN, midbrain; MED, medulla; SC, spinal cord; CONSC, consciousness; SLEEP HOM, sleep homeostasis; AUT REG, autonomic regulation; CIRC RHY, circadian rhythm; FEED/MET, feeding/metabolism; S/W switch, sleep/wake switch; RESP, respiratory control; CARDIO, cardiovascular control; FEED, feeding; NREM/REM, NREM/REM circuit controlling switching between sleep states; MOTOR CON, motor control; PERIPH SENS, peripheral sensation; AUTON CON, autonomic control.
Cognitive Aspects of Arousal and Sleep
The complex functional integration of changes in behavioral state indexed by the alternation of wake and sleep is generally conceived as binary – the system is either in one state or the other. While changes in behavioral state may indeed show pathological binary state changes, as in the intrusion of REM sleep on wake seen in narcolepsy with cataplexy; such binary shifts in state are far less common in other HDs, where they frequently present as lapses of attention that could be considered neural states intermediate to wake and sleep.
Such intermediate states are well recognized in the attentional lapses seen with day-dreaming, boredom, or drifting off due to mild sleepiness (162, 163). Cognitive lapses have been the source of studies investigating transportation safety, where attentional lapses are a common cause of accidents (164–166). While lapses in attention are common during wake-state periods, they are exacerbated by sleep loss (167–169) where brief sleep intrusions (microsleeps) are identifiable on EEG (170).
Neuroimaging studies have pointed to neural substrates for such attentional lapses, identifying the importance of the thalamus in both the maintenance of wake and in the allocation of attentional focus under conditions of high cognitive load (168, 170). When sleep deprived, subjects showed increased thalamic activity in response to a visual attention task (168) relative to activation during rested task performance. A study looking at the neural correlates of microsleeps during a visual tracking task reported decreased thalamic activity during microsleep periods (170).
A number of studies have identified two counteracting brain networks (171–174), one most active during alert arousal [“task-positive,” (171, 173, 175, 176)] and the other dominant during periods of quiet waking or internally directed mentation [“default mode network (DMN),” (171, 177–180)]. When subjects are actively engaged in task performance, DMN activity is reduced (177, 181) while that of the “task-positive” network is enhanced (182, 183). During natural sleep (48) and under conditions of sleep deprivation (184), the anticorrelated activity noted between these two networks is reduced. While the level of anticorrelation between task-positive and DMN networks shows substantial inter-individual variability (172, 185), its maintenance reduces variability in task performance (172). In keeping with these results, brief lapses in attention during task performance have been linked to increased DMN activity (162).
Keeping in mind both the dynamic nature and the connectional complexity of the brain networks (186) controlling wake, sleep, and their intermediate states, these data suggest that dysfunction within these networks can be expressed across the full state space of the organism, resulting in diverse biobehavioral abnormalities (Figure 5). Disorders of sleep, such as the HDs, result in disordered cognitive and physiological function together with changes in the normal alternation of wake and sleep states. It is equally the case that disorders of cognition, as seen in neurodegenerative disorders such as AD and Dementia with Lewy Bodies (DLB), result in disordered sleep that includes abnormal alternations between wake and sleep states.
Fluctuations of Cognition
Cognitive fluctuations are spontaneous alterations in cognition, attention, and arousal (187) in which EDS is a prominent component and may include inappropriate sleep periods or decreased responsiveness during normal waking hours (188–190). Individuals with cognitive fluctuations (CFs) not only exhibit a higher propensity to fall asleep (hypersomnia), they also transition from a less alert to a more alert state spontaneously. Thus, the attentional lapses characteristic of CF are a manifestation of a general propensity toward inappropriate alterations in brain state.
Cognitive fluctuations are a core diagnostic criterion of DLB (188, 191), and are also seen in AD, Parkinson’s Disease (PD), and 1–3% of non-demented individuals (192–194). In DLB, CFs are more likely to be associated with daytime sleepiness, lethargy, and sleeping than in AD or vascular dementias (188, 189). Functionally, CFs result in worse clinical dementia ratings and are associated with poorer neuropsychological performance (195), greater functional impairment (193), poorer quality of life, and increased caregiver burden (196).
Diagnosis of CF generally relies upon clinical assessment (192), although caregiver reports (187, 188, 192) may also be useful. The hypothesis that fluctuations of performance on attentional tasks would reflect clinically defined and more long-term CFs has been tested in a number of studies (193, 197) with mixed results. A recent study investigating the relationship between daytime sleepiness and cognitive performance in DLB and PD patients (197), using maintenance of wake to define alertness levels, reported that CFs and level of alertness may be independent of one another, a suggestion endorsed by the study of Escandon and colleagues (195).
Electroencephalogram and neuroimaging data from several groups support the hypothesis that fluctuations reflect abnormal functional brain network interactions. Cortical slowing is a common feature of dementia, with a decrease in alpha-band amplitude in DLB accompanied by a loss of functional alpha coupling between frontal and temporal regions (198–201). While spectral abnormalities are common to a number of neurological disorders, including AD, recent studies have pointed to higher amplitude delta and theta rhythms in DLB relative to AD (199) that, together with other differences in the inter-relationships between magneto-electrical cortical rhythms may provide biomarkers of neurodegenerative disorders to aid in early diagnosis and development of therapeutics (200, 202).
Neuroimaging studies of DLB report metabolic and blood flow reductions in parietal, frontal, and occipital cortices together with gray matter atrophy that is predominant in parieto-temporal regions (203–209). Occipital lobe dysfunction identified using both emission tomography and functional magnetic resonance imaging techniques, has been associated with poor visuospatial performance and visual hallucinations in some studies (203, 207, 210). While fewer studies have focused on the brain regions or networks underlying fluctuating levels of alertness, a single photon emission tomography (SPECT) study reported an association of CFs in DLB with increased perfusion in thalamus (211), a finding partially supported by findings of hyperperfusion in both thalamus and striatum in DLB patients in whom fluctuation status was not reported (212). Additionally, regional deficits in cerebral blood flow in the precuneus and occipital lobes have been reported to differentiate DLB from AD (210, 213, 214). Using diffusion tensor imaging (DTI), loss of white matter integrity in the posterior cingulate and visual association areas has been reported in DLB (215, 216). More recent studies have focused on the role of neural network interactions, with the understanding that disparate brain regions interact to produce different brain states and activities. Increased functional connectivity between precuneus, putamen, and parietal cortex has been reported (217) with a second study reporting increases in connectivity between posterior cingulate regions and thalamus, globus pallidus, and anterior cingulate (218). Using multivariate analytic techniques in SPECT, a recent study has reported that decreased activity in bilateral parietal and parieto-temporal regions distinguished DLB from AD. While more studies are needed, particularly in regard to fluctuations of alertness, these studies point to a pattern of deficits in regional connectivity, metabolism, and blood flow that include areas important in the allocation and maintenance of attention, including the frontal cortex, parietal cortex, posterior cingulate, and precuneus. While data are mixed concerning changes in thalamic activity levels during rest, and studies addressing changes in thalamic connectivity in DLB or CFs are lacking, changes in thalamic function in patients with CF have been reported (211, 212).
Conclusion
Over the past two decades, the central importance of sleep to both physiological and mental health has become increasingly clear. The understanding that sleep is both a local and a global phenomenon (157, 159, 160, 219) not fully constrained to the nocturnal sleep bout but locally apparent even in wake (158, 220) has provided a strong basis for the proposal that its effects on wake-state cognitive function are strong. The growing evidence of the importance of sleep to cognitive function (221–225) suggests that prolonged sleep disregulation may be a major factor in long-term cognitive decline, particularly when coupled with normal changes related to increasing age.
While the neural basis for disruptions in the normal alternation between wake and sleep state differs between HDs and the CFs seen in DLB, those changes impact the distributed and complex network controlling those states, while studies focused on CFs have yet to examine subcortical interactions that may provide evidence of such associations. The brain must maintain a balance between dynamic stability and instability; stability so as to recapitulate states and behaviors with proven efficacy, and instability so as to rapidly transition between states in response to unexpected or novel inputs. While neuroimaging studies provide important information on those brain networks involved in behavior, it is worth noting that these networks are dynamically regulated such that regional network membership shifts on millisecond timescales (186, 226–228). Further, as reported by Hellyer and colleagues (186), one property of these dynamic interactions may be the stabilization of network interactions during wake-state behaviors in which attention must be focused on behavioral tasks to provide optimal performance. Thus, we propose here that reductions in the ability to stabilize network interactions may underlie both disturbances in cognitive function that accompany sleep abnormalities and the disruptions in sleep that accompany neuropathological cognitive function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported in part by the Knight Alzheimer’s Disease Research Center of Washington University School of Medicine (LLP), the McDonnell Center for System Neuroscience (LLP), AG008051 and AG040211 awarded by the National Institutes of Health (JEG).
References
1. McCarley RW, Greene RW, Rainnie D, Portas CM, editors. Brainstem neuromodulation and REM sleep. Semin Neurosci (1995) 7:341–54. doi: 10.1006/smns.1995.0037
2. Klauer SG, Dingus TA, Neale VL, Sudweeks JD, Ramsey DJ. The Impact of Driver Inattention on Near-Crash/Crash Risk: An Analysis Using the 100-car Naturalistic Driving Study Data. Transportation Research Board Report HS-810 594 (2006). 1–190.
3. Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol (1949) 1(1):455–73. doi:10.1016/0013-4694(49)90219-9
4. Aserinsky E, Kleitman N. Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science (1953) 118(3062):273–4. doi:10.1126/science.118.3062.273
5. Datta S, MacLean RR. Neurobiological mechanisms for the regulation of mammalian sleep-wake behavior: reinterpretation of historical evidence and inclusion of contemporary cellular and molecular evidence. Neurosci Biobehav Rev (2007) 31(5):775–824. doi:10.1016/j.neubiorev.2007.02.004
6. Munk MH, Roelfsema PR, König P, Engel AK, Singer W. Role of reticular activation in the modulation of intracortical synchronization. Science (1996) 272(5259):271–4. doi:10.1126/science.272.5259.271
7. Capece ML, Baghdoyan HA, Lydic R. Carbachol stimulates [35S] Guanylyl 5’-(γ-Thio)-triphosphate binding in rapid eye movement sleep-related brainstem nuclei of rat. J Neurosci (1998) 18(10):3779–85.
8. Baghdoyan H, Lydic R. M2 muscarinic receptor subtype in the feline medial pontine reticular formation modulates the amount of rapid eye movement sleep. Sleep (1999) 22:835–48.
9. Portas CM, Bjorvatn B, Ursin R. Serotonin and the sleep/wake cycle: special emphasis on microdialysis studies. Prog Neurobiol (2000) 60(1):13–35. doi:10.1016/S0301-0082(98)00097-5
10. Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci (2002) 3(8):591–605.
11. Mistlberger RE. Circadian regulation of sleep in mammals: role of the suprachiasmatic nucleus. Brain Res Rev (2005) 49(3):429–54. doi:10.1016/j.brainresrev.2005.01.005
12. Saper CB, Cano G, Scammell TE. Homeostatic, circadian, and emotional regulation of sleep. J Comp Neurol (2005) 493(1):92–8. doi:10.1002/cne.20770
13. Fuller PM, Saper CB, Lu J. The pontine REM switch: past and present. J Physiol (2007) 584(3):735–41. doi:10.1113/jphysiol.2007.140160
14. McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med (2007) 8(4):302–30. doi:10.1016/j.sleep.2007.03.005
15. Staner L. Comorbidity of insomnia and depression. Sleep Med Rev (2010) 14(1):35–46. doi:10.1016/j.smrv.2009.09.003
16. Raggi A, Ferri R. Sleep disorders in neurodegenerative diseases. Eur J Neurol (2010) 17(11):1326–38.
17. Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci (2010) 11(8):589–99. doi:10.1038/nrn2868
18. Coogan AN, Schutová B, Husung S, Furczyk K, Baune BT, Kropp P, et al. The circadian system in Alzheimer’s disease: disturbances, mechanisms, and opportunities. Biol Psychiatry (2013) 74(5):333–9. doi:10.1016/j.biopsych.2012.11.021
19. Leistedt SJ, Coumans N, Dumont M, Lanquart JP, Stam CJ, Linkowski P. Altered sleep brain functional connectivity in acutely depressed patients. Hum Brain Mapp (2009) 30(7):2207–19. doi:10.1002/hbm.20662
20. Riemann D, Spiegelhalder K, Nissen C, Hirscher V, Baglioni C, Feige B. REM sleep instability-a new pathway for insomnia? Pharmacopsychiatry (2012) 45(5):167. doi:10.1055/s-0031-1299721
21. Triarhou LC. The percipient observations of Constantin von Economo on encephalitis lethargica and sleep disruption and their lasting impact on contemporary sleep research. Brain Res Bull (2006) 69(3):244–58. doi:10.1016/j.brainresbull.2006.02.002
22. Foley PB. Encephalitis lethargica and the influenza virus. II. The influenza pandemic of 1918/19 and encephalitis lethargica: epidemiology and symptoms. J Neural Transm (2009) 116(10):1295–308. doi:10.1007/s00702-009-0295-9
23. Reid AH, McCall S, Henry JM, Taubenberger JK. Experimenting on the past: the enigma of von Economo’s encephalitis lethargica. J Neuropathol Exp Neurol (2001) 60(7):663–70.
24. Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat Neurosci (2001) 4(7):732–8. doi:10.1038/89522
25. Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci (2003) 23(33):10691–702.
26. Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature (2005) 437(7063):1257–63. doi:10.1038/nature04284
27. Gvilia I, Xu F, McGinty D, Szymusiak R. Homeostatic regulation of sleep: a role for preoptic area neurons. J Neurosci (2006) 26(37):9426–33. doi:10.1523/JNEUROSCI.4827-05.2006
28. Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci (2008) 1129(1):275–86. doi:10.1196/annals.1417.027
29. Luppi PH, Fort P. What are the mechanisms activating the sleep-active neurons located in the preoptic area? Sleep Biol Rhythms (2011) 9(s1):59–64. doi:10.1111/j.1479-8425.2010.00464.x
30. Steriade M, Nuñez A, Amzica F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci (1993) 13(8):3266–83.
31. Steriade M, Nunez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci (1993) 13(8):3252–65.
32. Steriade M, Contreras D, Dossi RC, Nunez A. The slow (<1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci (1993) 13(8):3284–99.
33. Deleuze C, Huguenard JR. Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci (2006) 26(33):8633–45. doi:10.1523/JNEUROSCI.2333-06.2006
34. Crunelli V, Cope DW, Hughes SW. Thalamic T-type Ca2+ channels and NREM sleep. Cell Calcium (2006) 40(2):175–90. doi:10.1016/j.ceca.2006.04.022
35. Hughes SW, Crunelli V. Just a phase they’re going through: the complex interaction of intrinsic high-threshold bursting and gap junctions in the generation of thalamic α and θ rhythms. Int J Psychophysiol (2007) 64(1):3–17. doi:10.1016/j.ijpsycho.2006.08.004
36. Timofeev I, Steriade M. Low-frequency rhythms in the thakunus of intact-cortex and decorticated cats. J Neurophysiol (1996) 76(6).
37. Steriade M, Amzica F. Coalescence of sleep rhythms and their chronology in corticothalamic networks. Sleep Res Online (1998) 1(1):1.
38. Timofeev I, Grenier F, Bazhenov M, Sejnowski T, Steriade M. Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex (2000) 10(12):1185–99. doi:10.1093/cercor/10.12.1185
39. Llinás RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol (2006) 95(6):3297–308. doi:10.1152/jn.00166.2006
40. Destexhe A, Contreras D, Steriade M. Spatiotemporal analysis of local field potentials and unit discharges in cat cerebral cortex during natural wake and sleep states. J Neurosci (1999) 19(11):4595–608.
41. Achermann P, Borbely A. Low-frequency (<1 Hz) oscillations in the human sleep electroencephalogram. Neuroscience (1997) 81(1):213–22. doi:10.1016/S0306-4522(97)00186-3
42. Kilduff TS, Cauli B, Gerashchenko D. Activation of cortical interneurons during sleep: an anatomical link to homeostatic sleep regulation? Trends Neurosci (2011) 34(1):10–9. doi:10.1016/j.tins.2010.09.005
43. Porkka-Heiskanen T, Kalinchuk AV. Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev (2011) 15(2):123–35. doi:10.1016/j.smrv.2010.06.005
44. Coulon P, Budde T, Pape H-C. The sleep relay – the role of the thalamus in central and decentral sleep regulation. Pflügers Arch (2012) 463(1):53–71. doi:10.1007/s00424-011-1014-6
45. Ohayon MM. From wakefulness to excessive sleepiness: what we know and still need to know. Sleep Med Rev (2008) 12(2):129–41. doi:10.1016/j.smrv.2008.01.001
46. Medicine AAoS. The International Classification of Sleep Disorders – Revised. Chicago, IL: American Association of Sleep Medicine (2005). 297 p.
47. Association AP. The Diagnostic and Statistical Manual of Mental Disorders: DSM 5: bookpointUS; (2013).
48. Larson-Prior LJ, Power JD, Vincent JL, Nolan TS, Coalson RS, Zempel J, et al. Modulation of the brain’s functional network architecture in the transition from wake to sleep. Prog Brain Res (2011) 193:278–94. doi:10.1016/B978-0-444-53839-0.00018-1
49. Burgess CR, Scammell TE. Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci (2012) 32(36):12305–11. doi:10.1523/JNEUROSCI.2630-12.2012
50. Krahn LE, Pankratz VS, Oliver L, Boeve BF, Silber M. Hypocretin (orexin) levels in cerebrospinal fluid of patients with narcolepsy: relationship to cataplexy and HLA DQB1* 0602 status. Sleep (2002) 25(7):733–6.
51. Lin L, Hungs M, Mignot E. Narcolepsy and the HLA region. J Neuroimmunol (2001) 117(1):9–20. doi:10.1016/S0165-5728(01)00333-2
52. Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell (1998) 92(4):573–85. doi:10.1016/S0092-8674(02)09256-5
53. De Lecea L, Kilduff T, Peyron C, Gao X-B, Foye P, Danielson P, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A (1998) 95(1):322–7.
54. Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell (1999) 98(3):365–76. doi:10.1016/S0092-8674(00)81965-0
55. Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron (2000) 27(3):469–74. doi:10.1016/S0896-6273(00)00058-1
56. Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet (2000) 355(9197):39–40. doi:10.1016/S0140-6736(99)05582-8
57. Rieger M, Mayer G, Gauggel S. Attention deficits in patients with narcolepsy. Sleep (2003) 26(1):36–43.
58. Naumann A, Bellebaum C, Daum I. Cognitive deficits in narcolepsy. J Sleep Res (2006) 15(3):329–38. doi:10.1111/j.1365-2869.2006.00533.x
59. Bayard S, Langenier MC, De Cock VC, Scholz S, Dauvilliers Y. Executive control of attention in narcolepsy. PLoS One (2012) 7(4):e33525. doi:10.1371/journal.pone.0033525
60. Fulda S, Schulz H. Cognitive dysfunction in sleep disorders. Sleep Med Rev (2001) 5(6):423–45. doi:10.1053/smrv.2001.0157
61. Fronczek R, Middelkoop HA, Van Dijk J, Lammers GJ. Focusing on vigilance instead of sleepiness in the assessment of narcolepsy: high sensitivity of the Sustained Attention to Response Task (SART). Sleep (2006) 29(2):187.
62. Naumann A, Daum I. Narcolepsy: pathophysiology and neuropsychological changes. Behav Neurol (2003) 14(3):89–98. doi:10.1155/2003/323060
64. Hallmayer J, Faraco J, Lin L, Hesselson S, Winkelmann J, Kawashima M, et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet (2009) 41(6):708–11. doi:10.1038/ng.372
65. Cvetkovic-Lopes V, Bayer L, Dorsaz S, Maret S, Pradervand S, Dauvilliers Y, et al. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J Clin Invest (2010) 120(3):713. doi:10.1172/JCI41366
66. Sunmaila G. Narcolepsy: a review. Neuropsychiatr Dis Treat (2011) 7:507–18. doi:10.2147/NDT.S23624
67. Kornum BR, Faraco J, Mignot E. Narcolepsy with hypocretin/orexin deficiency, infections and autoimmunity of the brain. Curr Opin Neurobiol (2011) 21(6):897–903. doi:10.1016/j.conb.2011.09.003
68. Kanbayashi T, Sagawa Y, Takemura F, Ito S-U, Tsutsui K, Hishikawa Y, et al. The pathophysiologic basis of secondary narcolepsy and hypersomnia. Curr Neurol Neurosci Rep (2011) 11(2):235–41. doi:10.1007/s11910-011-0178-y
69. Castriotta RJ, Lai JM. Sleep disorders associated with traumatic brain injury. Arch Phys Med Rehabil (2001) 82(10):1403–6. doi:10.1053/apmr.2001.26081
70. Verma A, Anand V, Verma NP. Sleep disorders in chronic traumatic brain injury. J Clin Sleep Med (2007) 3(4):357.
71. Nishino S, Kanbayashi T, Fujiki N, Uchino M, Ripley B, Watanabe M, et al. CSF hypocretin levels in Guillain-Barre syndrome and other inflammatory neuropathies. Neurology (2003) 61(6):823–5. doi:10.1212/01.WNL.0000081049.14098.50
72. Brass SD, Duquette P, Proulx-Therrien J, Auerbach S. Sleep disorders in patients with multiple sclerosis. Sleep Med Rev (2010) 14(2):121–9. doi:10.1016/j.smrv.2009.07.005
73. Miglis MG, Guilleminault C. Kleine-Levin syndrome: a review. Nat Sci Sleep (2014) 6:19. doi:10.2147/NSS.S44750
74. Arnulf I, Lin L, Gadoth N, File J, Lecendreux M, Franco P, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol (2008) 63(4):482–93. doi:10.1002/ana.21333
75. Engström M, Landtblom A-M, Karlsson T. Brain and effort: brain activation and effort-related working memory in healthy participants and patients with working memory deficits. Front Hum Neurosci (2013) 7:140. doi:10.3389/fnhum.2013.00140
76. Engström M, Karlsson T, Landtblom A-M. Thalamic activation in the Kleine-Levin syndrome. Sleep (2013) 37(2):379–86.
77. Arnulf I, Rico TJ, Mignot E. Diagnosis, disease course, and management of patients with Kleine-Levin syndrome. Lancet Neurol (2012) 11(10):918–28. doi:10.1016/S1474-4422(12)70187-4
78. Dauvilliers Y, Lopez R, Ohayon M, Bayard S. Hypersomnia and depressive symptoms: methodological and clinical aspects. BMC Med (2013) 11(1):78. doi:10.1186/1741-7015-11-78
79. Landtblom AM, Dige N, Schwerdt K, Säfström P, Granerus G. Short-term memory dysfunction in Kleine-Levin syndrome. Acta Neurol Scand (2003) 108(5):363–7. doi:10.1034/j.1600-0404.2003.00171.x
80. Oliveira MM, Conti C, Prado GF. Pharmacological treatment for Kleine-Levin syndrome. Cochrane Datase Syst Rev (2013) 8:CD006685. doi:10.1002/14651858.CD00685.pub3
81. Haba-Rubio J, Prior JO, Guedj E, Tafti M, Heinzer R, Rossetti AO. Kleine-Levin syndrome: functional imaging correlates of hypersomnia and behavioral symptoms. Neurology (2012) 79(18):1927–9. doi:10.1212/WNL.0b013e318271f85a
82. Billiard M, Jaussent I, Dauvilliers Y, Besset A. Recurrent hypersomnia: a review of 339 cases. Sleep Med Rev (2011) 15(4):247–57. doi:10.1016/j.smrv.2010.08.001
84. Ohayon MM, Reynolds CF, Dauvilliers Y. Excessive sleep duration and quality of life. Ann Neurol (2013) 73(6):785–94. doi:10.1002/ana.23818
86. Vernet C, Leu-Semenescu S, Buzare MA, Arnulf I. Subjective symptoms in idiopathic hypersomnia: beyond excessive sleepiness. J Sleep Res (2010) 19(4):525–34. doi:10.1111/j.1365-2869.2010.00824.x
87. Pizza F, Ferri R, Poli F, Vandi S, Cosentino FI, Plazzi G. Polysomnographic study of nocturnal sleep in idiopathic hypersomnia without long sleep time. J Sleep Res (2013) 22(2):185–96. doi:10.1111/j.1365-2869.2012.01061.x
88. Sforza E, Gaudreau H, Petit D, Montplaisir J. Homeostatic sleep regulation in patients with idiopathic hypersomnia. Neurophysiol Clin (2000) 111(2):277–82. doi:10.1016/S1388-2457(99)00242-4
89. Weinhold SL, Seeck-Hirschner M, Nowak A, Goeder R, Baier PC. Wake-REM sleep transitions for measuring REM sleep disturbance: comparison between narcolepsy, idiopathic hypersomnia and healthy controls. Sleep Biol Rhythms (2011) 9(3):172–7. doi:10.1111/j.1479-8425.2011.00503.x
90. van Schie MK, Thijs RD, Fronczek R, Middelkoop HA, Lammers GJ, VAN DIJKJ. Sustained attention to response task (SART) shows impaired vigilance in a spectrum of disorders of excessive daytime sleepiness. J Sleep Res (2012) 21(4):390–5. doi:10.1111/j.1365-2869.2011.00979.x
91. Vernet C, Arnulf I. Idiopathic hypersomnia with and without long sleep time: a controlled series of 75 patients. Sleep (2009) 32(6):753.
92. Morin LP. Neuroanatomy of the extended circadian rhythm system. Exp Neurol (2013) 243:4–20. doi:10.1016/j.expneurol.2012.06.026
93. Burdakov D, Karnani MM, Gonzalez A. Lateral hypothalamus as a sensor-regulator in respiratory and metabolic control. Physiol Behav (2013) 121:117–24. doi:10.1016/j.physbeh.2013.03.023
94. Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest (2011) 121(6):2133. doi:10.1172/JCI46043
96. Miyamoto H, Nakamaru-Ogiso E, Hamada K, Hensch TK. Serotonergic integration of circadian clock and ultradian sleep-wake cycles. J Neurosci (2012) 32(42):14794–803. doi:10.1523/JNEUROSCI.0793-12.2012
97. Abbott SM, Arnold JM, Chang Q, Miao H, Ota N, Cecala C, et al. Signals from the brainstem sleep/wake centers regulate behavioral timing via the circadian clock. PLoS One (2013) 8(8):e70481. doi:10.1371/journal.pone.0070481
98. Fort P, Bassetti C, Luppi PH. Alternating vigilance states: new insights regarding neuronal networks and mechanisms. Eur J Neurosci (2009) 29(9):1741–53. doi:10.1111/j.1460-9568.2009.06722.x
99. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron (2010) 68(6):1023–42. doi:10.1016/j.neuron.2010.11.032
100. Benedetto L, Chase MH, Torterolo P. GABAergic processes within the median preoptic nucleus promote NREM sleep. Behav Brain Res (2012) 232(1):60–5. doi:10.1016/j.bbr.2012.03.033
101. Hassani OK, Henny P, Lee MG, Jones BE. GABAergic neurons intermingled with orexin and MCH neurons in the lateral hypothalamus discharge maximally during sleep. Eur J Neurosci (2010) 32(3):448–57. doi:10.1111/j.1460-9568.2010.07295.x
102. Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A (2009) 106(7):2418–22. doi:10.1073/pnas.0811400106
103. Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, et al. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci (2013) 33(25):10257–63. doi:10.1523/JNEUROSCI.1225-13.2013
104. Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, et al. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci (2013) 16(11):1637–43. doi:10.1038/nn.3522
105. Tsunematsu T, Ueno T, Tabuchi S, Inutsuka A, Tanaka KF, Hasuwa H, et al. Optogenetic manipulation of activity and temporally controlled cell-specific ablation reveal a role for MCH neurons in sleep/wake regulation. J Neurosci (2014) 34(20):6896–909. doi:10.1523/JNEUROSCI.5344-13.2014
106. Sapin E, Bérod A, Léger L, Herman PA, Luppi P-H, Peyron C. A very large number of GABAergic neurons are activated in the tuberal hypothalamus during paradoxical (REM) sleep hypersomnia. PLoS One (2010) 5(7):e11766. doi:10.1371/journal.pone.0011766
107. Fraigne JJ, Peever JH. Melanin-concentrating hormone neurons promote and stabilize sleep. Sleep (2013) 36(12):1767–68.
108. Lagos P, Monti JM, Jantos H, Torterolo P. Microinjection of the melanin-concentrating hormone into the lateral basal forebrain increases REM sleep and reduces wakefulness in the rat. Life Sci (2012) 90(23):895–9. doi:10.1016/j.lfs.2012.04.019
109. Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci (2007) 8(3):171–81. doi:10.1038/nrn2092
110. Hara J, Sakurai T. Interaction between sleep mechanisms and orexin neurons. Sleep Biol Rhythms (2011) 9(s1):38–43. doi:10.1111/j.1479-8425.2010.00466.x
111. Carter ME, Brill J, Bonnavion P, Huguenard JR, Huerta R, de Lecea L. Mechanism for Hypocretin-mediated sleep-to-wake transitions. Proc Natl Acad Sci U S A (2012) 109(39):E2635–44. doi:10.1073/pnas.1202526109
112. Alexandre C, Andermann ML, Scammell TE. Control of arousal by the orexin neurons. Curr Opin Neurobiol (2013) 23(5):752–9. doi:10.1016/j.conb.2013.04.008
113. de Lecea L. Hypocretins and the neurobiology of sleep-wake mechanisms. Prog Brain Res (2012) 198:15. doi:10.1016/B978-0-444-59489-1.00003-3
115. Lin J-S, Anaclet C, Sergeeva OA, Haas HL. The waking brain: an update. Cell Mol Life Sci (2011) 68(15):2499–512. doi:10.1007/s00018-011-0631-8
116. Qu W-M, Xu X-H, Yan M-M, Wang Y-Q, Urade Y, Huang Z-L. Essential role of dopamine D2 receptor in the maintenance of wakefulness, but not in homeostatic regulation of sleep, in mice. J Neurosci (2010) 30(12):4382–9. doi:10.1523/JNEUROSCI.4936-09.2010
117. Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci (2006) 26(1):193–202. doi:10.1523/JNEUROSCI.2244-05.2006
118. Sarter M, Bruno J. Cortical cholinergic inputs mediating arousal, attentional processing and dreaming: differential afferent regulation of the basal forebrain by telencephalic and brainstem afferents. Neuroscience (1999) 95(4):933–52. doi:10.1016/S0306-4522(99)00487-X
119. Deurveilher S, Semba K. Basal forebrain regulation of cortical activity and sleep-wake states: roles of cholinergic and non-cholinergic neurons. Sleep Biol Rhythms (2011) 9(s1):65–70. doi:10.1111/j.1479-8425.2010.00465.x
120. Arrigoni E, Mochizuki T, Scammell TE. Activation of the basal forebrain by the orexin/hypocretin neurones. Acta Physiol (2010) 198(3):223–35. doi:10.1111/j.1748-1716.2009.02036.x
121. Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature (2006) 441(7093):589–94. doi:10.1038/nature04767
122. Ramaligam V, Chen MC, Saper CB, Lu J. Perspectives on the rapid eye movement sleep switch in rapid eye movement sleep behavior disorder. Sleep Med (2013) 14(8):707–13. doi:10.1016/j.sleep.2013.03.017
123. Luppi P-H, Clément O, Fort P. Paradoxical (REM) sleep genesis by the brainstem is under hypothalamic control. Curr Opin Neurobiol (2013) 23(5):786–92. doi:10.1016/j.conb.2013.02.006
124. Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, et al. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One (2011) 6(10):e24998. doi:10.1371/journal.pone.0024998
125. Dijk D-J, Brunner DP, Borbély AA. EEG power density during recovery sleep in the morning. Electroencephalogr Clin Neurophysiol (1991) 78(3):203–14. doi:10.1016/0013-4694(91)90034-2
126. Brunner DP, Dijk D-J, Tobler I, Borbély AA. Effect of partial sleep deprivation on sleep stages and EEG power spectra: evidence for non-REM and REM sleep homeostasis. Electroencephalogr Clin Neurophysiol (1990) 75(6):492–9. doi:10.1016/0013-4694(90)90136-8
127. Achermann P, Dijk D-J, Brunner DP, Borbély AA. A model of human sleep homeostasis based on EEG slow-wave activity: quantitative comparison of data and simulations. Brain Res Bull (1993) 31(1):97–113. doi:10.1016/0361-9230(93)90016-5
128. Dijk D-J, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci (1995) 15(5):3526–38.
129. Borbély AA, Achermann P. Concepts and models of sleep regulation: an overview. J Sleep Res (1992) 1(2):63–79. doi:10.1111/j.1365-2869.1992.tb00013.x
130. Finelli L, Baumann H, Borbély A, Achermann P. Dual electroencephalogram markers of human sleep homeostasis: correlation between theta activity in waking and slow-wave activity in sleep. Neuroscience (2000) 101(3):523–9. doi:10.1016/S0306-4522(00)00409-7
131. Knoblauch V, Kräuchi K, Renz C, Wirz-Justice A, Cajochen C. Homeostatic control of slow-wave and spindle frequency activity during human sleep: effect of differential sleep pressure and brain topography. Cereb Cortex (2002) 12(10):1092–100. doi:10.1093/cercor/12.10.1092
132. Van Dongen H, Rogers NL, Dinges DF. Sleep debt: theoretical and empirical issues*. Sleep Biol Rhythms (2003) 1(1):5–13. doi:10.1046/j.1446-9235.2003.00006.x
133. Vyazovskiy V, Achermann P, Tobler I. Sleep homeostasis in the rat in the light and dark period. Brain Res Bull (2007) 74(1):37–44. doi:10.1016/j.brainresbull.2007.05.001
134. Leemburg S, Vyazovskiy VV, Olcese U, Bassetti CL, Tononi G, Cirelli C. Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc Natl Acad Sci U S A (2010) 107(36):15939–44. doi:10.1073/pnas.1002570107
135. Krueger JM, Rector DM, Roy S, Van Dongen HP, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci (2008) 9(12):910–9. doi:10.1038/nrn2521
136. Landolt H-P. Sleep homeostasis: a role for adenosine in humans? Biochem Pharmacol (2008) 75(11):2070–9. doi:10.1016/j.bcp.2008.02.024
137. Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci (2009) 10(3):199–210. doi:10.1038/nrn2576
138. Yang YC, Hu CC, Huang CS, Chou PY. Thalamic synaptic transmission of sensory information modulated by synergistic interaction of adenosine and serotonin. J Neurochem (2013) 128:852–63. doi:10.1111/jnc.12499
140. Baghdoyan HA, Rodrigo-Angulo ML, McCarley RW, Hobson JA. Site-specific enhancement and suppression of desynchronized sleep signs following cholinergic stimulation of three brainstem regions. Brain Res (1984) 306(1):39–52. doi:10.1016/0006-8993(84)90354-8
141. Leonard C, Llinas R. Serotonergic and cholinergic inhibition of mesopontine cholinergic neurons controlling REM sleep: an in vitro electrophysiological study. Neuroscience (1994) 59(2):309–30. doi:10.1016/0306-4522(94)90599-1
142. Bier MJ, McCarley RW. REM-enhancing effects of the adrenergic antagonist idazoxan infused into the medial pontine reticular formation of the freely moving cat. Brain Res (1994) 634(2):333–8. doi:10.1016/0006-8993(94)91939-9
143. Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience (2000) 101(2):243–76. doi:10.1016/S0306-4522(00)00353-5
144. Fuller P, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol (2011) 519(5):933–56. doi:10.1002/cne.22559
145. Portas CM, Krakow K, Allen P, Josephs O, Armony JL, Frith CD. Auditory processing across the sleep-wake cycle: simultaneous EEG and fMRI monitoring in humans. Neuron (2000) 28(3):991–9. doi:10.1016/S0896-6273(00)00169-0
146. Massimini M, Rosanova M, Mariotti M. EEG slow (approximately 1 Hz) waves are associated with nonstationarity of thalamo-cortical sensory processing in the sleeping human. J Neurophysiol (2003) 89(3):1205–13. doi:10.1152/jn.00373.2002
147. Massimini M, Ferrarelli F, Huber R, Esser SK, Singh H, Tononi G. Breakdown of cortical effective connectivity during sleep. Science (2005) 309(5744):2228–32. doi:10.1126/science.1117256
148. Tononi G, Massimini M. Why does consciousness fade in early sleep? Ann N Y Acad Sci (2008) 1129(1):330–4. doi:10.1196/annals.1417.024
149. Massimini M, Ferrarelli F, Murphy M, Huber R, Riedner B, Casarotto S, et al. Cortical reactivity and effective connectivity during REM sleep in humans. Cogn Neurosci (2010) 1(3):176–83. doi:10.1080/17588921003731578
150. Schabus M, Dang-Vu TT, Heib DPJ, Boly M, Desseilles M, Vandewalle G, et al. The fate of incoming stimuli during NREM sleep is determined by spindles and the phase of the slow oscillation. Front Neurol (2012) 3:40. doi:10.3389/fneur.2012.00040
151. Hennevin E, Huetz C, Edeline J-M. Neural representations during sleep: from sensory processing to memory traces. Neurobiol Learn Mem (2007) 87(3):416–40. doi:10.1016/j.nlm.2006.10.006
152. Nir Y, Vyazovskiy VV, Cirelli C, Banks MI, Tononi G. Auditory responses and stimulus-specific adaptation in rat auditory cortex are preserved across NREM and REM sleep. Cereb Cortex (2013). doi:10.1093/cercor/bht328
153. Mazza S, Perchet C, Frot M, Michael GA, Magnin M, Garcia-Larrea L, et al. Asleep but aware? Brain Cogn (2014) 87:7–15. doi:10.1016/j.bandc.2014.02.007
154. Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol (2003) 2(3):167–76. doi:10.1016/S1474-4422(03)00323-5
155. Villablanca JR. Counterpointing the functional role of the forebrain and of the brainstem in the control of the sleep-waking system. J Sleep Res (2004) 13(3):179–208. doi:10.1111/j.1365-2869.2004.00412.x
156. Lugaresi E, Provini F, Montagna P. The neuroanatomy of sleep. Considerations on the role of the thalamus in sleep and a proposal for a caudorostral organization. Eur J Anat (2014) 8(2):85–93.
157. Rector DM, Schei JL, Van Dongen H, Belenky G, Krueger JM. Physiological markers of local sleep. Eur J Neurosci (2009) 29(9):1771–8. doi:10.1111/j.1460-9568.2009.06717.x
158. Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature (2011) 472(7344):443–7. doi:10.1038/nature10009
159. Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, et al. Regional slow waves and spindles in human sleep. Neuron (2011) 70(1):153–69. doi:10.1016/j.neuron.2011.02.043
160. Nobili L, Ferrara M, Moroni F, De Gennaro L, Russo GL, Campus C, et al. Dissociated wake-like and sleep-like electro-cortical activity during sleep. Neuroimage (2011) 58(2):612–9. doi:10.1016/j.neuroimage.2011.06.032
161. Sarasso S, Pigorini A, De Carli F, Ferrara M. Local aspects of sleep: observations from intracerebral recordings in humans. Neurobiol Circadian Timing (2012) 199:219. doi:10.1016/B978-0-444-59427-3.00013-7
162. Weissman D, Roberts K, Visscher K, Woldorff M. The neural bases of momentary lapses in attention. Nat Neurosci (2006) 9(7):971–8. doi:10.1038/nn1727
163. Cheyne JA, Carriere JS, Smilek D. Absent-mindedness: lapses of conscious awareness and everyday cognitive failures. Conscious Cogn (2006) 15(3):578–92. doi:10.1016/j.concog.2005.11.009
164. Edkins GD, Pollock CM. The influence of sustained attention on railway accidents. Accid Anal Prev (1997) 29(4):533–9. doi:10.1016/S0001-4575(97)00033-X
165. Moller HJ, Kayumov L, Shapiro CM. Microsleep episodes, attention lapses and circadian variation in psychomotor performance in a driving simulation paradigm. Proceedings of the Second International Driving Symposium on Human Factors in Driver Assessment, Training and Vehicle Design (2003). p. 130–7.
166. Roca J, Lupiáñez J, López-Ramón M-F, Castro C. Are drivers’ attentional lapses associated with the functioning of the neurocognitive attentional networks and with cognitive failure in everyday life? Transp Res Part F Traffic Psychol Behav (2013) 17:98–113. doi:10.1016/j.trf.2012.10.005
167. Lim J, Dinges DF. Sleep deprivation and vigilant attention. Ann N Y Acad Sci (2008) 1129(1):305–22. doi:10.1196/annals.1417.002
168. Tomasi D, Wang R, Telang F, Boronikolas V, Jayne M, Wang G-J, et al. Impairment of attentional networks after 1 night of sleep deprivation. Cereb Cortex (2009) 19(1):233–40. doi:10.1093/cercor/bhn073
169. Chee MW, Tan JC, Zheng H, Parimal S, Weissman DH, Zagorodnov V, et al. Lapsing during sleep deprivation is associated with distributed changes in brain activation. J Neurosci (2008) 28(21):5519–28. doi:10.1523/JNEUROSCI.0733-08.2008
170. Poudel GR, Innes CR, Bones PJ, Watts R, Jones RD. Losing the struggle to stay awake: divergent thalamic and cortical activity during microsleeps. Hum Brain Mapp (2014) 35(1):257–69. doi:10.1002/hbm.22178
171. Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci U S A (2005) 102(27):9673–8. doi:10.1073/pnas.0504136102
172. Kelly A, Uddin LQ, Biswal BB, Castellanos FX, Milham MP. Competition between functional brain networks mediates behavioral variability. Neuroimage (2008) 39(1):527–37. doi:10.1016/j.neuroimage.2007.08.008
173. Chai XJ, Castañón AN, Öngür D, Whitfield-Gabrieli S. Anticorrelations in resting state networks without global signal regression. Neuroimage (2012) 59(2):1420–8. doi:10.1016/j.neuroimage.2011.08.048
174. Fornito A, Harrison BJ, Zalesky A, Simons JS. Competitive and cooperative dynamics of large-scale brain functional networks supporting recollection. Proc Natl Acad Sci U S A (2012) 109(31):12788–93. doi:10.1073/pnas.1204185109
175. Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci (2007) 8(9):700–11. doi:10.1038/nrn2201
176. Fox MD, Corbetta M, Snyder AZ, Vincent JL, Raichle ME. Spontaneous neuronal activity distinguishes human dorsal and ventral attention systems. Proc Natl Acad Sci U S A (2006) 103(26):10046–51. doi:10.1073/pnas.0604187103
177. Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A (2001) 98(2):676–82. doi:10.1073/pnas.98.2.676
178. Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci U S A (2003) 100(1):253–8. doi:10.1073/pnas.0135058100
179. Mason MF, Norton MI, Van Horn JD, Wegner DM, Grafton ST, Macrae CN. Wandering minds: the default network and stimulus-independent thought. Science (2007) 315(5810):393–5. doi:10.1126/science.1131295
180. Andrews-Hanna JR, Reidler JS, Huang C, Buckner RL. Evidence for the default network’s role in spontaneous cognition. J Neurophysiol (2010) 104(1):322. doi:10.1152/jn.00830.2009
181. Mckiernan KA, Kaufman JN, Kucera-Thompson J, Binder JR. A parametric manipulation of factors affecting task-induced deactivation in functional neuroimaging. J Cogn Neurosci (2003) 15(3):394–408. doi:10.1162/089892903321593117
182. Vincent JL, Kahn I, Snyder AZ, Raichle ME, Buckner RL. Evidence for a frontoparietal control system revealed by intrinsic functional connectivity. J Neurophysiol (2008) 100(6):3328–42. doi:10.1152/jn.90355.2008
183. Spreng RN, Stevens WD, Chamberlain JP, Gilmore AW, Schacter DL. Default network activity, coupled with the frontoparietal control network, supports goal-directed cognition. Neuroimage (2010) 53(1):303–17. doi:10.1016/j.neuroimage.2010.06.016
184. De Havas JA, Parimal S, Soon CS, Chee MW. Sleep deprivation reduces default mode network connectivity and anti-correlation during rest and task performance. Neuroimage (2012) 59(2):1745–51. doi:10.1016/j.neuroimage.2011.08.026
185. Anderson JS, Ferguson MA, Lopez-Larson M, Yurgelun-Todd D. Connectivity gradients between the default mode and attention control networks. Brain Connect (2011) 1(2):147–57. doi:10.1089/brain.2011.0007
186. Hellyer PJ, Shanahan M, Scott G, Wise RJ, Sharp DJ, Leech R. The control of global brain dynamics: opposing actions of frontoparietal control and default mode networks on attention. J Neurosci (2014) 34(2):451–61. doi:10.1523/JNEUROSCI.1853-13.2014
187. Ferman T, Smith G, Boeve B, Ivnik R, Petersen R, Knopman D, et al. DLB fluctuations specific features that reliably differentiate DLB from AD and normal aging. Neurology (2004) 62(2):181–7. doi:10.1212/WNL.62.2.181
188. Metzler-Baddeley C. A review of cognitive impairments in dementia with Lewy bodies relative to Alzheimer’s disease and Parkinson’s disease with dementia. Cortex (2007) 43(5):583–600. doi:10.1016/S0010-9452(08)70489-1
189. Boddy F, Rowan EN, Lett D, O’Brien JT, McKeith IG, Burn DJ. Subjectively reported sleep quality and excessive daytime somnolence in Parkinson’s disease with and without dementia, dementia with Lewy bodies and Alzheimer’s disease. Int J Geriatr Psychiatry (2007) 22(6):529–35. doi:10.1002/gps.1709
190. Pao WC, Boeve BF, Ferman TJ, Lin S-C, Smith GE, Knopman DS, et al. Polysomnographic findings in dementia with Lewy bodies. Neurologist (2013) 19(1):1–6. doi:10.1097/NRL.0b013e31827c6bdd
191. McKeith I, Dickson D, Lowe J, Emre M, O’brien J, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies Third report of the DLB consortium. Neurology (2005) 65(12):1863–72. doi:10.1212/01.wnl.0000187889.17253.b1
192. Walker M, Ayre G, Perry E, Wesnes K, McKeith I, Tovee M, et al. Quantification and characterisation of fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease. Dement Geriatr Cogn Disord (2000) 11(6):327–35. doi:10.1159/000017262
193. Ballard C, Walker M, O’Brien J, Rowan E, McKeith I. The characterisation and impact of ‘fluctuating’cognition in dementia with Lewy bodies and Alzheimer’s disease. Int J Geriatr Psychiatry (2001) 16(5):494–8. doi:10.1002/gps.381
194. Perry RJ, Hodges JR. Attention and executive deficits in Alzheimer’s disease. A critical review. Brain (1999) 122(3):383–404. doi:10.1093/brain/122.3.383
195. Escandon A, Al-Hammadi N, Galvin JE. Effect of cognitive fluctuation on neuropsychological performance in aging and dementia. Neurology (2010) 74(3):210–7. doi:10.1212/WNL.0b013e3181ca017d
196. Lee DR, McKeith I, Mosimann U, Ghosh-Nodyal A, Thomas AJ. Examining carer stress in dementia: the role of subtype diagnosis and neuropsychiatric symptoms. Int J Geriatr Psychiatry (2013) 28(2):135–41. doi:10.1002/gps.3799
197. Bliwise DL, Scullin MK, Trotti LM. Fluctuations in cognition and alertness vary independently in dementia with Lewy bodies. Mov Disord (2014) 29(1):83–9. doi:10.1002/mds.25707
198. Franciotti R, Iacono D, Penna SD, Pizzella V, Torquati K, Onofrj M, et al. Cortical rhythms reactivity in AD, LBD and normal subjects: a quantitative MEG study. Neurobiol Aging (2006) 27(8):1100–9. doi:10.1016/j.neurobiolaging.2005.05.027
199. Bonanni L, Thomas A, Tiraboschi P, Perfetti B, Varanese S, Onofrj M. EEG comparisons in early Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease with dementia patients with a 2-year follow-up. Brain (2008) 131(3):690–705. doi:10.1093/brain/awm322
200. Babiloni C, De Pandis MF, Vecchio F, Buffo P, Sorpresi F, Frisoni GB, et al. Cortical sources of resting state electroencephalographic rhythms in Parkinson’s disease related dementia and Alzheimer’s disease. Neurophysiol Clin (2011) 122(12):2355–64. doi:10.1016/j.clinph.2011.03.029
201. Bosboom J, Stoffers D, Stam C, Van Dijk B, Verbunt J, Berendse H, et al. Resting state oscillatory brain dynamics in Parkinson’s disease: an MEG study. Neurophysiol Clin (2006) 117(11):2521–31. doi:10.1016/j.clinph.2006.06.720
202. Snaedal J, Johannesson GH, Gudmundsson TE, Blin NP, Emilsdottir AL, Einarsson B, et al. Diagnostic accuracy of statistical pattern recognition of electroencephalogram registration in evaluation of cognitive impairment and dementia. Dement Geriatr Cogn Disord (2012) 34(1):51–60. doi:10.1159/000339996
203. Minoshima S, Foster NL, Petrie EC, Albin RL, Frey KA, Kuhl DE. Neuroimaging in dementia with Lewy bodies: metabolism, neurochemistry, and morphology. J Geriatr Psychiatry Neurol (2002) 15(4):200–9. doi:10.1177/089198870201500405
204. Watson R, Blamire AM, O’Brien JT. Magnetic resonance imaging in Lewy body dementias. Dement Geriatr Cogn Disord (2009) 28(6):493–506. doi:10.1159/000264614
205. Nagahama Y, Okina T, Suzuki N, Matsuda M. Neural correlates of psychotic symptoms in dementia with Lewy bodies. Brain (2010) 133(2):557–67. doi:10.1093/brain/awp295
206. Perneczky R, Drzezga A, Boecker H, Ceballos-Baumann AO, Granert O, Förstl H, et al. Activities of daily living, cerebral glucose metabolism, and cognitive reserve in Lewy body and Parkinson’s disease. Dement Geriatr Cogn Disord (2008) 26(5):475–81. doi:10.1159/000167791
207. Taylor J-P, Firbank MJ, He J, Barnett N, Pearce S, Livingstone A, et al. Visual cortex in dementia with Lewy bodies: magnetic resonance imaging study. Br J Psychiatry (2012) 200(6):491–8. doi:10.1192/bjp.bp.111.099432
208. Kantarci K, Lowe VJ, Boeve BF, Weigand SD, Senjem ML, Przybelski SA, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging (2012) 33(9):2091–105. doi:10.1016/j.neurobiolaging.2011.09.024
209. Taylor J-P, Colloby SJ, McKeith IG, O’Brien JT. Covariant perfusion patterns provide clues to the origin of cognitive fluctuations and attentional dysfunction in Dementia with Lewy bodies. Int Psychogeriatr (2013) 25(12):1–12. doi:10.1017/S1041610213001488
210. Perneczky R, Drzezga A, Boecker H, Förstl H, Kurz A, Häussermann P. Cerebral metabolic dysfunction in patients with dementia with Lewy bodies and visual hallucinations. Dement Geriatr Cogn Disord (2008) 25(6):531–8. doi:10.1159/000132084
211. O’Brien JT, Firbank MJ, Mosimann UP, Burn DJ, McKeith IG. Change in perfusion, hallucinations and fluctuations in consciousness in dementia with Lewy bodies. Psychiatry Res (2005) 139(2):79–88. doi:10.1016/j.pscychresns.2005.04.002
212. Sato T, Hanyu H, Hirao K, Shimizu S, Kanetaka H, Iwamoto T. Deep gray matter hyperperfusion with occipital hypoperfusion in dementia with Lewy bodies. Eur J Neurol (2007) 14(11):1299–301. doi:10.1111/j.1468-1331.2007.01951.x
213. Colloby SJ, Fenwick JD, Williams DE, Paling SM, Lobotesis K, Ballard C, et al. A comparison of 99mTc-HMPAO SPET changes in dementia with Lewy bodies and Alzheimer’s disease using statistical parametric mapping. Eur J Nucl Med Mol Imaging (2002) 29(5):615–22. doi:10.1007/s00259-002-0778-5
214. Kemp PM, Hoffmann SA, Holmes C, Bolt L, Ward T, Holmes RB, et al. The contribution of statistical parametric mapping in the assessment of precuneal and medial temporal lobe perfusion by 99mTc-HMPAO SPECT in mild Alzheimer’s and Lewy body dementia. Nucl Med Commun (2005) 26(12):1099–106. doi:10.1097/00006231-200512000-00009
215. Lee JE, Park H-J, Park B, Song SK, Sohn YH, Lee JD, et al. A comparative analysis of cognitive profiles and white-matter alterations using voxel-based diffusion tensor imaging between patients with Parkinson’s disease dementia and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry (2010) 81(3):320–6. doi:10.1136/jnnp.2009.184747
216. Matsui H, Nishinaka K, Oda M, Niikawa H, Kubori T, Udaka F. Dementia in Parkinson’s disease: diffusion tensor imaging. Acta Neurol Scand (2007) 116(3):177–81. doi:10.1111/j.1600-0404.2006.00795.x
217. Galvin J, Price J, Yan Z, Morris J, Sheline Y. Resting bold fMRI differentiates dementia with Lewy bodies vs Alzheimer disease. Neurology (2011) 76(21):1797–803. doi:10.1212/WNL.0b013e31821ccc83
218. Kenny ER, Blamire AM, Firbank MJ, O’Brien JT. Functional connectivity in cortical regions in dementia with Lewy bodies and Alzheimer’s disease. Brain (2012) 135(2):569–81. doi:10.1093/brain/awr327
219. Murphy M, Huber R, Esser S, Riedner BA, Massimini M, Ferrarelli F, et al. The cortical topography of local sleep. Curr Top Med Chem (2011) 11(19):2438. doi:10.2174/156802611797470303
220. Vassalli A, Dijk DJ. Sleep function: current questions and new approaches. Eur J Neurosci (2009) 29(9):1830–41. doi:10.1111/j.1460-9568.2009.06767.x
221. Walker MP, Stickgold R. Overnight alchemy: sleep-dependent memory evolution. Nat Rev Neurosci (2010) 11(3):218. doi:10.1038/nrn2762-c1
222. Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev (2006) 10(1):49–62. doi:10.1016/j.smrv.2005.05.002
223. Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature (2004) 430(6995):78–81. doi:10.1038/nature02663
224. Yoo S-S, Hu PT, Gujar N, Jolesz FA, Walker MP. A deficit in the ability to form new human memories without sleep. Nat Neurosci (2007) 10(3):385–92. doi:10.1038/nn1851
225. Diekelmann S, Biggel S, Rasch B, Born J. Offline consolidation of memory varies with time in slow wave sleep and can be accelerated by cuing memory reactivations. Neurobiol Learn Mem (2012) 98(2):103–11. doi:10.1016/j.nlm.2012.07.002
226. Marzetti L, Della Penna S, Snyder AZ, Pizzella V, Nolte G, de Pasquale F, et al. Frequency specific interactions of MEG resting state activity within and across brain networks as revealed by the multivariate interactions measure. Neuroimage (2013) 79:172–83. doi:10.1016/j.neuroimage.2013.04.062
227. Deco G, Buehlmann A, Masquelier T, Hugues E. The role of rhythmic neural synchronization in rest and task conditions. Front Hum Neurosci (2011) 5:4. doi:10.3389/fnhum.2011.00004
Keywords: hypersomnia, cognitive fluctuations, sleep, review, brain networks
Citation: Larson-Prior LJ, Ju Y-E and Galvin JE (2014) Cortical–subcortical interactions in hypersomnia disorders: mechanisms underlying cognitive and behavioral aspects of the sleep–wake cycle. Front. Neurol. 5:165. doi: 10.3389/fneur.2014.00165
Received: 17 April 2014; Accepted: 18 August 2014;
Published online: 11 September 2014.
Edited by:
Maria Engström, Linköping University, SwedenReviewed by:
Karl Æ. Karlsson, Reykjavik University, IcelandPablo Torterolo, Universidad de la República, Uruguay
Copyright: © 2014 Larson-Prior, Ju and Galvin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda J. Larson-Prior, Departments of Radiology and Neurology, Washington University School of Medicine, 4525 Scott Avenue, Box 8225, St. Louis, MO 63110, USA e-mail: lindap@npg.wustl.edu