
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 28 February 2012
Sec. Neuro-Otology
Volume 3 - 2012 | https://doi.org/10.3389/fneur.2012.00020
This article is part of the Research Topic New Understanding of Plasticity in the Vestibular System: From Bench to Bed Side View all 12 articles
 Yiwen Zheng1,2
Yiwen Zheng1,2 Sangeeta Balabhadrapatruni1,2 Jean Ha Baek1,2 Phoebe Chung1 Catherine Gliddon1 Ming Zhang3
Sangeeta Balabhadrapatruni1,2 Jean Ha Baek1,2 Phoebe Chung1 Catherine Gliddon1 Ming Zhang3 Cynthia L. Darlington1,2 Ruth Napper2,3 Michael Strupp4 Thomas Brandt4
Cynthia L. Darlington1,2 Ruth Napper2,3 Michael Strupp4 Thomas Brandt4 Paul F. Smith1,2*
Paul F. Smith1,2*Previous studies in humans have shown that bilateral loss of vestibular function is associated with a significant bilateral atrophy of the hippocampus, which correlated with the patients’ spatial memory deficits. More recently, patients who had recovered from unilateral vestibular neuritis have been reported to exhibit a significant atrophy of the left posterior hippocampus. Therefore, we investigated whether bilateral vestibular deafferentation (BVD) would result in a decrease in neuronal number or volume in the rat hippocampus, using stereological methods. At 16 months post-BVD, we found no significant differences in hippocampal neuronal number or volume compared to sham controls, despite the fact that these animals exhibited severe spatial memory deficits. By contrast, using bromodeoxyuridine (BrdU) as a marker of cell proliferation, we found that the number of BrdU-labeled cells significantly increased in the dentate gyrus of the hippocampus between 48 h and 1 week following BVD. Although a substantial proportion of these cells survived for up to 1 month, the survival rate was significantly lower in BVD animals when compared with that in sham animals. These results suggest a dissociation between the effects of BVD on spatial memory and hippocampal structure in rats and humans, which cannot be explained by an injury-induced increase in cell proliferation.
Many studies in animals and humans have shown that loss of vestibular function, especially complete bilateral vestibular loss, can impair spatial memory (e.g., Stackman and Herbert, 2002; Wallace et al., 2002; Russell et al., 2003a; Brandt et al., 2005; Zheng et al., 2006, 2007, 2009a,b; Smith et al., 2009, 2010; Baek et al., 2010; Besnard et al., 2012). This spatial memory impairment is likely to be related to hippocampal dysfunction, since both place cell responses (Stackman et al., 2002; Russell et al., 2003b) and theta rhythm have been reported to be abnormal (Russell et al., 2006; Neo et al., 2012; Tai et al., 2012; but see Stackman et al., 2002 for conflicting evidence regarding theta rhythm). On the other hand, CA1 and dentate gyrus (DG) field potentials and long-term potentiation (LTP) in vivo were not significantly affected by bilateral vestibular loss (Zheng et al., 2010a).
Studies in humans have shown that hippocampal volume can be modulated by spatial memory experience. For example, London taxi drivers were reported to have increased hippocampal volume compared to controls (Maguire et al., 2000). Consistent with this finding, Hüfner et al. (2010) reported structural changes in the hippocampi of professional dancers and slackliners (who traverse a tightrope which is not held completely taut), who have unusual spatial memory experience, including specific vestibular stimulation. While dancers are subjected to extensive angular acceleration, slackliners experience considerable linear acceleration. Hüfner et al. found that trained subjects exhibited a smaller anterior volume, and a larger posterior volume, in the hippocampal formation, although they showed no difference in spatial memory compared to controls, according to the virtual Morris water maze test. These studies suggest that spatial memory experience may regulate the volume of different regions of the human hippocampus. By contrast, Brandt et al. (2005) reported that patients with bilateral vestibular loss, 5–10 years following the surgery, exhibited a bilateral atrophy of the hippocampus, of approx. 17%, which was correlated with spatial memory deficits in a virtual Morris water maze task. Patients with unilateral vestibular neurectomy did not exhibit such hippocampal atrophy (Hüfner et al., 2007). However, zu Eulenburg et al. (2010) reported that patients who had recovered from unilateral vestibular neuritis, exhibited a significant decrease in the volume of the left posterior hippocampus, irrespective of the laterality of the vestibular neuritis. By contrast, in rats, Besnard et al. (2012) found no significant difference in hippocampal volume following bilateral intratympanic injections of sodium arsanilate.
Despite the increasing body of evidence that links bilateral vestibular damage, learning and memory impairment, and hippocampal dysfunction, the underlying mechanisms remain to be elucidated. Using a rat model of complete bilateral vestibular deafferentation (BVD), the present study investigated the volume and total number of neurons in subregions of the hippocampus at 16 months following BVD. In addition, time course changes in hippocampal cell proliferation following BVD were also quantified.
Six male Wistar rats were used in this part of the study and were randomly allocated to the BVD (n = 3) and sham surgery (n = 3) conditions and sacrificed at 16 months post-surgery. We considered that these smaller sample sizes were justified in this particular case, based on the precision of stereological analysis, which involves minimal measurement error and therefore reduces variation (Gundersen and Jensen, 1987; West and Gundersen, 1990; Zhang et al., 2005). Although averages across animals were used in the Section “Statistical Analyses” (see below), error in the estimates for individual animals contributes to the overall variation around the group means, and objective and precise counting procedures can minimize this error. In addition, the two surgery conditions combined with five hippocampal subregions resulted in a 2 × 5 factorial design, which further increased statistical power (Cohen, 1992; Zheng et al., 2011; see Statistical Analyses).
At 14 months post-surgery, these animals were tested on a foraging task, in which they had been shown to exhibit severe spatial memory deficits (Baek et al., 2010; Figure 1).
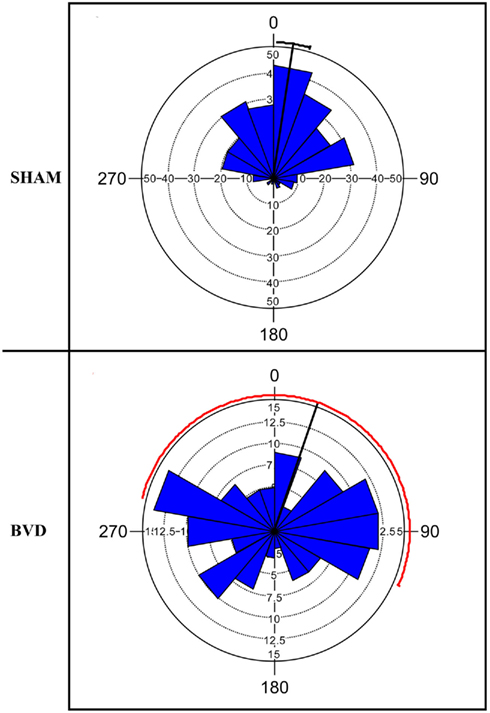
Figure 1. Rose diagram showing the initial heading angles of the sham and BVD animals for the dark training at 14 months post-op. The plots represent the animals’ directions on the homeward route in a foraging task in which they had to remember the location of a home base in darkness. In the plots, 0° represents the correct direction or heading angle. The mean vector is indicated by the black line and 95% confidence interval (CI) for the mean is indicated by the line extending either side. The 95% CI values for the BVD animals were unreliable due to low concentration of vectors, hence the red line. The inner circles (dotted line) indicate the number of observations for the given vectors (blue triangles). Notice that the BVD animals’ heading angles were distributed equally around 360°. Reproduced from Baek et al. (2010) with permission.
The surgery was conducted under a general anesthetic of ketamine hydrochloride (760 μg/kg, s.c.), medetomidine hydrochloride (300 μg/kg, s.c.), and atropine sulfate (80 μg/kg, s.c.). The wound margins were anesthetized locally with xylocaine. Under an operating microscope, the tympanic membrane was exposed using a retro-auricular approach and the tympanic membrane, malleus, and incus were removed. The stapedial artery was cauterized and the horizontal and anterior semicircular canal ampullae drilled open. The contents of the canal ampullae and the utricle and saccule were aspirated and the surgical opening of the temporal bone was sealed with dental cement. Carprofen (5 mg/kg, s.c.) was used for post-operative analgesia and atipamezole hydrochloride (5 mg/kg, s.c.) was used to reverse the effect of medetomidine hydrochloride. Our previous studies have shown that the BVD surgical procedure produces a complete and permanent lesion of the vestibular labyrinth with no damage beyond the temporal bone (Zheng et al., 2006).
Sham surgery consisted of exposing the temporal bone and removing the tympanic membrane without producing a vestibular lesion. This procedure provided a partial auditory control that involved damage to the tympanic membrane only with no other surgical trauma. All other procedures such as anesthesia and recovery remained the same as for the lesioned animals. All procedures were approved by the University of Otago Committee on Ethics in the Care and Use of Laboratory Animals (AEC 07/06).
At 16 months following the surgery, the rats were anesthetized with sodium pentobarbitone (100 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde. The brain was removed from the cranial cavity and post-fixed with 4% paraformaldehyde containing 30% sucrose for 4 h. The brain was then cryoprotected in 30% sucrose overnight at 4°C and frozen by completely covering it with O.C.T. compound and then immersing it in n-hexane solution in liquid nitrogen. The frozen tissue was stored at −20°C until required.
Forty micrometer sagittal sections throughout the hippocampus were collected using a random, systematic design. Briefly, the first section was selected by a random number generator and then every eighth section throughout the hippocampus was systematically collected. The sections were placed in a 24 well plate containing 30% sucrose with 30% ethylene glycol in a 0.1-M phosphate buffer solution (PBS) and stored at −20°C until use. Sections were collected from the left and right hippocampi in a counterbalanced fashion so that any left/right differences were controlled for in the design.
The sections were washed with PBS and incubated in cresyl violet (0.01%) for 30 min. The sections were then washed with distilled water, transferred onto glass microscope slides, dehydrated, and coverslipped with mounting media.
The optical fractionator method was used to estimate the neuronal number in different subregions of the hippocampus (West et al., 1991). Cell counting was carried out in five different subregions: (1) the granule cells of the DG; (2) the hilar cells; (3) the pyramidal cell layer of CA2/CA3; (4) the pyramidal cell layer of CA1; and (5) the subiculum. All of the stereological analysis was conducted blind to the experimental treatment groups. The sections were first viewed with a 4× objective lens, and the boundaries of each cell layer were outlined and recorded. Then the cells were counted under a 100× oil immersion objective using a fractionator scheme (Gundersen, 1986, 1988; West et al., 1991; Korbo et al., 1996), which involved counting cells in a known fraction of the thickness and the area of the region of interest in a known fraction of the sections throughout the hippocampus. A BH2 Olympus Optical Dissector microscope with programmable stepping motors attached was used to move the sections in x, y steps through a raster pattern covering each subregion of interest. The area of a step was 100 μm × 100 μm in the DG, 80 μm × 80 μm in the hilus and CA3, 70 μm × 70 μm in CA1, and 120 μm × 120 μm in the subiculum. A counting frame generated by a CAST-GRID software system (Olympus) was superimposed on the image at each step. The areas of the counting frames used were 129 μm2 in the DG, 387 μm2 in the hilus, 344 μm2 in CA3, 129 μm2 in CA1, and 473 μm2 in the subiculum. The height of the count frame was 10 μm. The top focal plane of the counting frame was positioned at 3 μm below the upper surface of the section and neuronal nuclei that came into focus within the counting frame through a depth of 10 μm were counted with the unbiased counting rules. Cells were counted as neurons if they had a clearly defined neuronal cytoplasm and a membrane-bound nucleus. Using this sampling scheme, an average of 175 neuronal nuclei was counted in each subregion with a coefficient of error (CE) of less than 0.10.
The total number of neurons (N) in each subregion was estimated using the equation:
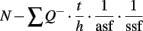
where Q− is the total number of cells actually counted, asf is the fraction of the counting frame area [a (frame)] to the area of the step [a(x, y) step] and ssf is the fraction of the number of sections sampled to the total number of sections throughout the hippocampus. The height (h) of the counting frame is also a fraction of the thickness of the section (t).
The volume of each subregion of the hippocampus or the whole hippocampus was estimated by a point (step) count method using the same sampling scheme: Volume(V) = Σp·a(p)·t·1/ssf, where Σp = Total number of points (steps) in each subregion or the whole hippocampus, a(p) = the area associated with each point (step), t = the thickness of the section.
Twenty-eight Wistar rats were used in this part of the study. Bromodeoxyuridine (BrdU), a marker for DNA replication or repair, was injected (300 mg/kg, i.p.) at 24 h (n = 2), 48 h (n = 1), or 72 h (n = 3) after the sham surgery and 24 h (n = 3), 48 h (n = 3), 72 h (n = 3), or 1 week (n = 3) after the BVD surgery. The animals were sacrificed at 24 h after BrdU injection for assessing cell proliferation. The results from the three sham groups were pooled since there were no significant differences between them. In addition, the survival of BrdU+ cells was investigated by injecting BrdU at 72 h after sham or BVD surgery and sacrificing the animals at 4 weeks after BrdU injection, using an additional five sham and five BVD animals. Small sample sizes were considered appropriate due to the use of stereological cell counting and also because the sham and four BVD groups generated a five factor design with a total of 28 degrees of freedom, thereby increasing statistical power (Zheng et al., 2011; see Statistical Analyses).
Rats were overdosed with sodium pentobarbital and perfused transcardially with 4% paraformaldehyde. The brains were dissected out, post-fixed, and frozen. Forty micrometer serial sections throughout the hippocampus were collected according to a random, systematic sampling design for free floating immunolabeling.
Sections were treated with 4 M HCl and Proteinase K (1:4000) at 37°C and neutralized with Borax at room temperature. A mouse monoclonal anti-BrdU antibody (Dako, 1:40) was incubated with the sections at 4°C overnight followed by a goat anti-mouse HRP conjugated secondary antibody (1:200) incubation for 2 h at room temperature. The immunolabeling was visualized using a DAB kit.
For each animal, every BrdU+ nucleus was counted under a 63× oil objective lens throughout the thickness of the section in one set of the serial sections collected. Since the DG, but not other areas (CA1, CA2, CA3, hilus, and subiculum) of the hippocampus, is one of the major neurogenic regions of the CNS (see Abdipranoto et al., 2008 for a review), we counted the number of BrdU+ nuclei in the DG separately and grouped BrdU counting in the other areas of the hippocampus together. The side of the hippocampus examined was counterbalanced in both the lesioned and sham groups. The total number of BrdU+ cells was then estimated using a modified fractionator method (Malberg et al., 2000; Nixon and Crews, 2004; Yang et al., 2008; Zheng et al., 2011).
Statistical analyses were performed in SPSS 19.0. The data were first tested for normality and homogeneity of variance, and, if necessary, natural log transformed. Two-way ANOVAs followed by Tukey’s post hoc tests were performed in order to compare the BVD and the sham animals for different hippocampal subregions and the interaction between surgery and subregion (Rice, 2007). Partial η2 values were used in addition, to estimate the strength of the effect of the factors on the dependent variables (Gamst et al., 2008). In order to estimate the survival rate of proliferating cells, the number of BrdU+ cells at 4 weeks was expressed as a ratio of the average number of proliferating cells in the BVD or sham groups at 72 h post-op. P < 0.05 was considered significant.
Because of the lack of previous studies using the same methods, we could calculate statistical power only using the data from the present study. We therefore estimated observed (i.e., post hoc) power using SPSS19 for an α rate of 0.05 with the ANOVA designs we employed (Gamst et al., 2008). Although observed power for the effect of surgery on neuronal number and volume, where no significant differences were actually found, was only 14.2 and 26.6%, respectively, estimated power for subregion differences was 100% in both cases, suggesting that the ANOVAs were powerful enough to detect differences where they existed. Furthermore, in the case of the cell proliferation data, the observed power was 90.6% for surgery, 96.2% for subregion, and 100% for the interaction between surgery and subregion. These power analyses indicate that, despite the small sample sizes, the use of factorial designs provided sufficient power to detect differences where they existed.
When examined under the light microscope, no changes in hippocampal gross morphology were identifiable in BVD rats (Figures 2A,B) The total number of neurons estimated in the subregions of the hippocampus of the six rats were analyzed and the corresponding mean CE are listed in Table 1. There was no significant difference in the total number of neurons between the BVD and sham animals (Figure 2C), no significant interaction between surgery and subregion, but, as expected, a large and significant difference across the five hippocampal subregions [F(4,20) = 141.71, P = 0.000; Figure 2C]. The partial η2 for subregion was 0.97, indicating that it had a major effect on neuronal number.
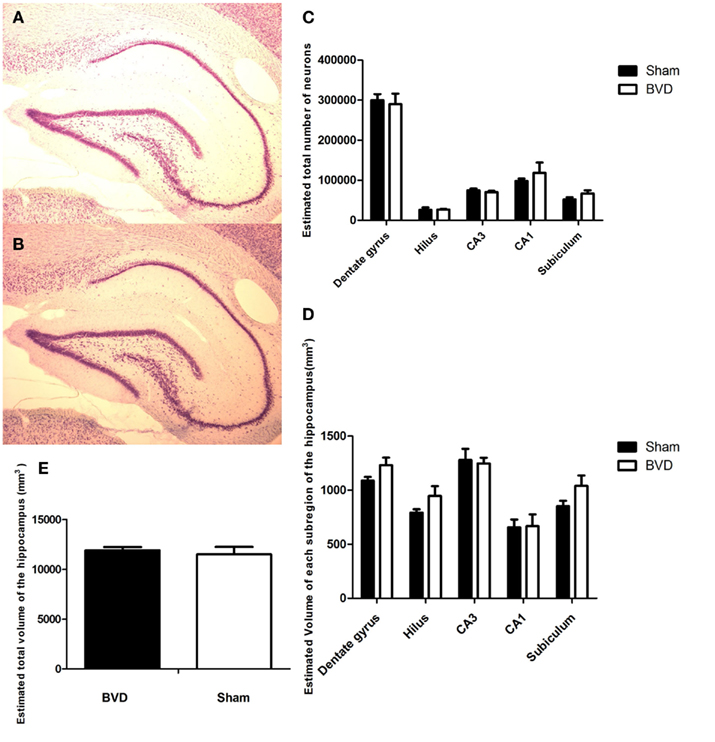
Figure 2. Cresyl violet stained sections showing no difference in the morphology of the cells in the CA3 region in the hippocampus between the sham (A) and (B) BVD animals. (C) Estimated total number of neurons in the subregions of the hippocampus in animals at 16 months post-BVD or sham surgery. (D) Estimated volume (in cubic millimeter) of the subregions of the hippocampus in animals at 16 months post-BVD or sham surgery. (E) Estimated total volume of the entire hippocampus (in cubic millimeter) in animals at 16 months post-BVD or sham surgery. Bars show means + 1 SEM.
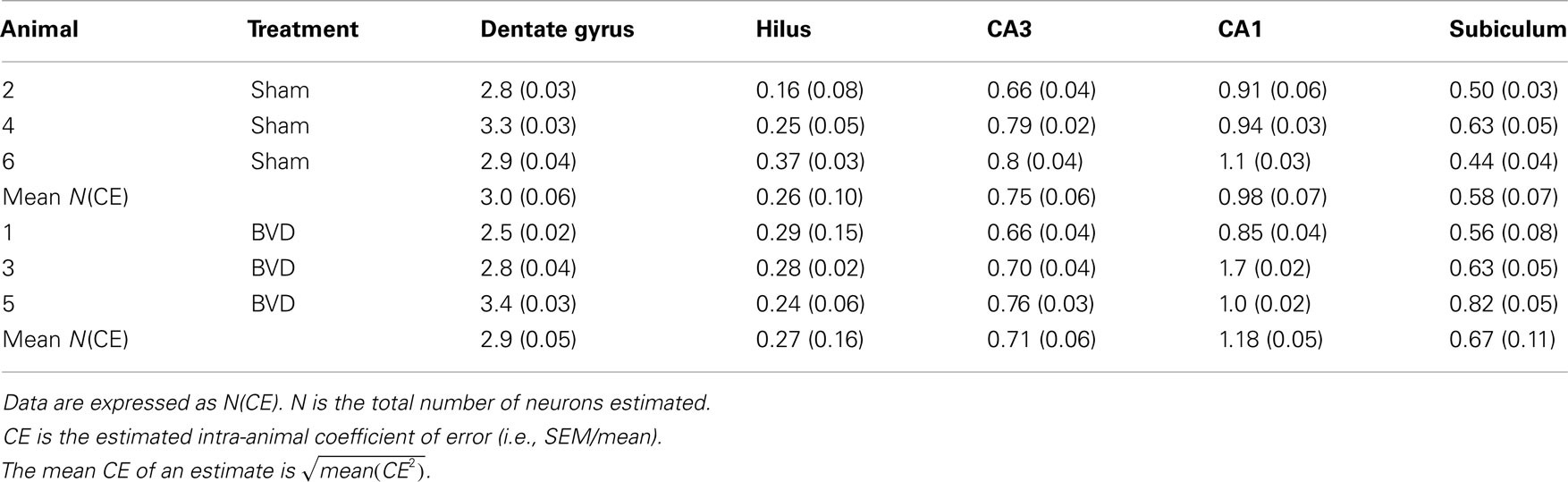
Table 1. Total number of neurons in different subregions of hippocampus (×105).
Analysis of the volume data showed that there was no significant difference between the BVD and sham animals, no significant interaction between surgery and subregion, but a large and significant difference in volume across the five hippocampal subregions [F(4,20) = 20.83, P = 0.000; Figure 2D]. The partial η2 for subregion was 0.81, indicating that it had a large effect on volume.
Figure 2D shows the volume of each subregion of the hippocampus and Figure 2E the volume of the whole hippocampus of the six rats analyzed.
Following BVD, there was a time-dependent increase in the number of cells containing the proliferation marker, BrdU, in the DG, compared to sham animals (Figure 3). The BrdU+ve nuclei were observed not only in the subgranular layer of the DG, but also in other areas of the hippocampus in both sham and BVD rats (Figures 3A–E). There were significant surgery [F(4,28) = 4.65, P = 0.005] and subregion effects [F(1,28) = 15.0, P = 0.001], with a significant interaction between surgery and subregion [F(4,28) = 14.57, P = 0.000]. However, the partial η2 values for surgery and subregion were only 0.40 and 0.35, respectively; nonetheless, η2 for the interaction between surgery and subregion was 0.68, indicating that it had a large effect on cell proliferation.
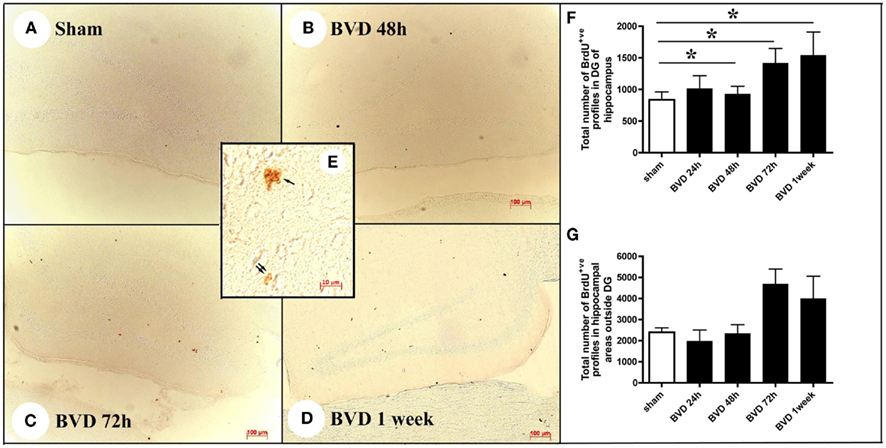
Figure 3. Examples of BrdU immunostaining in sham animals (A) and animals at 48 h (B), 72 h (C), and 1 week (D) following BVD. Inserted picture (E) showing a cluster of BrdU+ nuclei in the subgranular layer of the DG (single arrow) and two BrdU+ nuclei in the hilus of the hippocampus (double arrow) under high magnification. (F,G) Showing time course changes in the total number of BrdU+ nuclei estimated in the DG and other areas of the hippocampus, respectively. Data are expressed as mean ± SEM.
Post hoc tests showed that in the DG there were significant differences in the number of BrdU+ve nuclei between sham and BVD groups at 48 h (T = −3.86, P = 0.02), 72 h (T = −6.99, P = 0.000), and 1 week post-op. (T = −5.64, P = 0.0002), with no significant difference at 24 h (Figures 3F,G). However, for the other areas of the hippocampus, the sham condition was not significantly different to the BVD groups.
Furthermore, a substantial proportion of these BrdU+ve cells survived at least for 4 weeks. However, there was a significantly lower survival rate in BVD animals compared to sham controls [F(2,16) = −7.69, P = 0.01]. Survival rates were also significantly lower in the DG compared to other areas of the hippocampus [F(1,16) = 8.12, P = 0.01], with no significant interaction between treatment and region (Figure 4).
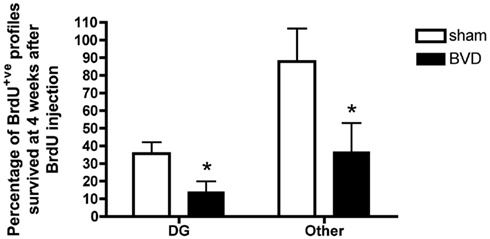
Figure 4. Survival rate of BrdU+ cells in different subregions of the hippocampus. Data are expressed as mean ± SEM.
The results of this study show that, unlike humans with BVD, who exhibit a significant bilateral atrophy of the hippocampus (Brandt et al., 2005) and with unilateral vestibular neuritis who exhibit a significant atrophy of the left posterior hippocampus (zu Eulenburg et al., 2010), at 16-months post-BVD rats did not exhibit a significant decrease in either hippocampal volume or neuronal number in the different hippocampal subregions. These rats did exhibit spatial memory deficits which were similar to the human patients with BVD (Baek et al., 2010; see Figure 1). Recently, Besnard et al. (2012) analyzed hippocampal volume using MRI and found no significant difference in rats treated with intratympanic sodium arsanilate compared to sham-treated controls. Our results extend their observation to show that there was also no hippocampal neuronal loss following BVD.
It could be argued that the neuronal numbers might be overestimated since cresyl violet also stains the basket and glial cells in the granule and pyramidal cell layers. However, glial cells are very easy to exclude from counting based on their smaller size, large nuclei, and sparse cytoplasm (Ling et al., 1973). Although basket cells are morphologically similar to the surrounding neurons, they comprise less than 1% of the neurons in these layers (Seress, 1978). Therefore, including the basket cells should not have significantly affected the estimation of the total number of neurons.
Given the profound hyperactivity reported in rats with BVD (Goddard et al., 2008; Baek et al., 2010; Stiles et al., 2012), one possible explanation for the lack of hippocampal atrophy and neuronal loss might be that BVD increased cell proliferation, since exercise has been shown to increase hippocampal cell proliferation and neurogenesis (Olson et al., 2006). Interestingly, in this study we found a significant increase in the number of BrdU+ve cells in the DG of the hippocampus following BVD, in a time-dependent manner. Although BrdU has been widely used as a marker of cell proliferation, BrdU can also be incorporated into damaged DNA during the process of DNA repair (Landgren and Curtis, 2011). Therefore, we cannot exclude the possibility that the increase in BrdU labeling following BVD reflects DNA repair processes rather than cell proliferation. However, the fact that a substantial proportion of these BrdU+ve cells survived, at least up to 1 month post-BVD, and no neuronal loss was found, suggests possible cell proliferation rather than DNA repair. Indeed, peripheral nerve damage-induced cell proliferation has been reported in the brainstem vestibular nucleus and cochlear nucleus following vestibular and cochlear lesions, respectively (Tighilet et al., 2007; Dutheil et al., 2009; Zheng et al., 2010b, 2011). While both the vestibular nucleus and cochlear nucleus are only one synapse away from the peripheral damage, the present results suggest that changes in cell proliferation can be induced in the hippocampus, a site that is several synapses away from the damage, following BVD. It is also noteworthy that a large proportion of BrdU+ve cells were located in areas other than the DG, the neurogenic region of the hippocampus. This raises the question of to what extent this cell proliferation reflects neurogenesis as opposed to gliogenesis? Although BVD induced a significant increase in the number of BrdU+ve cells at acute time points, the survival rate of these cells was much lower than that in sham rats. This suggests that significantly more BrdU+ve cells had been eliminated in BVD rats than in sham rats. In theory, this would result in neuronal loss in BVD rats. However, this was not the case. Further investigations are necessary to elucidate the underlying mechanisms. However, the decrease in the number of surviving BrdU+ve cells may explain the spatial memory deficits in these rats. It has been suggested that the generation of newborn neurons in the DG may be involved in spatial memory, since these cells have a lower threshold for LTP and are therefore suitable to form functional synaptic connections that could provide the framework for spatial memory to occur (refer to Aimone et al., 2010; Lucassen et al., 2010 for review).
It must be recognized that differences in hearing between the BVD and sham rats may have contributed to the results observed in this study. While the BVD rats received a surgical labyrinthectomy which would have resulted in damage to the cochlea, sham animals had only the tympanic membrane removed, and therefore would have retained some auditory function. We used tympanic membrane removal in the sham animals as a partial control for hearing loss in the BVD animals, reasoning that if differences caused by the BVD were due simply to hearing loss, then sham animals should exhibit similar effects (Baek et al., 2010). It is conceivable that differences in hearing loss could have contributed to the observed results. However, acoustic trauma has recently been reported to inhibit cell proliferation and neurogenesis in the rat hippocampus (Kraus et al., 2010).
There are at least six possible explanations for the apparent discrepancy between the data on hippocampal volume in human and rat following BVD. First, it may be due to the difference in time post-BVD. In the Brandt et al. (2005) study, the patients were between 5 and 10 years post-BVD, whereas in the current rat study, the animals were at 16 months post-BVD. It is difficult to compare the life spans of rats and humans, but considering that 24 months of age is regarded as “aged” for a rat (Liu et al., 2010), 16 months of age is equivalent to a long-term time point. Given that the humans and rats both exhibited spatial memory deficits and were at long time intervals following BVD, the difference in time post-op. seems the least likely explanation for the discrepancy. Furthermore, Besnard et al. (2012) also observed no change in hippocampal volume at shorter time intervals following bilateral vestibular loss. Second, it may be due to the effect of sex. Brandt et al. (2005) analyzed brains from both males and females, whereas our study and that of Besnard et al. (2012) were restricted to males. However, Brandt et al. (2005) found no significant interaction between surgery status and sex; therefore, there was no evidence that the inclusion of females in the current study would have made any difference. Third, there may be a difference between the sides of the hippocampus. By analyzing sections from both the left and right hippocampus, we obscured differences that might have occurred on one side but not the other. Once again, however, Brandt et al. (2005) found a significant decrease in volume in both the left and right hippocampi. Furthermore, we counterbalanced our sampling of sections across the two sides; therefore this explanation does not seem likely. Besnard et al. (2012) analyzed the left and right hippocampi using MRI and also found no significant difference compared to sham controls. Fourth, it is interesting to consider potential differences between humans and rats in terms of their response to BVD. One obvious explanation for hippocampal atrophy in humans with BVD is that chronic stress from the disability leads to high circulating levels of cortisol. Short-term increases in cortisol have been reported in patients with vestibular disorders (Horii et al., 2007); however, long-term increases have been reported only in the case of Meniere’s disease and acoustic neuroma (Horner and Cazals, 2005; van Cruijsen et al., 2005). To date, every rat or guinea pig study that has examined corticosterone or cortisol levels (respectively) following unilateral or BVD has found no significant difference compared to sham controls up 5 months post-op. (Gliddon et al., 2003; Lindsay et al., 2005; Zhang et al., 2005; Russell et al., 2006; Zheng et al., 2008). It is conceivable that BVD causes a long-term increase in glucocorticoids only in humans, as a result of the psychosocial impact of living without a balance system. This might explain why humans but not rats exhibit hippocampal atrophy despite similar spatial memory deficits. If this is true, the important implication might be that hippocampal atrophy following BVD is not directly related to the dysfunction of place cells or theta activity, but is an indirect consequence of the difficulties that humans with bilateral vestibular loss experience in their life (Zingler et al., 2008). One important point is that the laboratory animals only concern is the lack of a functional vestibular system. Food, warmth and social contact are all provided within their home environment. It is possible that hippocampal atrophy may occur in vestibular-deficient animals in their natural environment where food, warmth and social interaction are not provided and the animal must obtain them. This hypothesis is consistent with the fact that hippocampal place cell activity is disrupted rapidly and reversibly after intratympanic injection of tetrodotoxin, long before major structural changes could take place in the hippocampus (Stackman et al., 2002). A fifth possibility is that the lack of hippocampal atrophy in rats is somehow related to their unusual locomotor hyperactivity following BVD, which is not seen in humans (Stiles et al., 2012). Locomotor activity has been reported to increase hippocampal cell proliferation and neurogenesis (Olson et al., 2006) and it is conceivable that it minimizes neuronal loss in rats following BVD.
A further possibility is that hippocampal atrophy in humans following BVD is partially due, not to a reduction in the number of neurons, but to other structural changes, such as an atrophy of dendritic trees. This hypothesis will require further investigation using Golgi labeling of dendrites.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Sangeeta Balabhadrapatruni and Jean Ha Baek were supported by University of Otago Ph.D. Scholarships. Yiwen Zheng was supported by a Sir Charles Hercus HRC Senior Research Fellowship. We would like to thank Prof. Robert Sloviter for his help in visually inspecting the cell layers of the hippocampus for possible changes in neuronal morphology. This research was supported by grants from the New Zealand Neurological Foundation.
Abdipranoto, A., Wu, S., Stayte, S., and Vissel, B. (2008). The role of neurogenesis in neurodegenerative diseases and its implications for therapeutic development. CNS Neurol. Disord. Drug Targets 7, 187–210.
Aimone, J. B., Deng, W., and Gage, F. H. (2010). Adult neurogenesis: integrating theories and separating functions. Trends Cogn. Sci. (Regul. Ed.) 14, 325–337.
Baek, J.-H., Zheng, Y., Darlington, C. L., and Smith, P. F. (2010). Evidence that spatial memory deficits in rats following bilateral vestibular loss may be permanent. Neurobiol. Learn. Mem. 94, 402–413.
Besnard, S., Machado, M. L., Vignaux, G., Boulouard, M., Coquerel, A., Bouet, V., Freret, T., Denise, P., and Lelong-Boulouard, V. (2012). Influence of vestibular input on spatial and nonspatial memory and on hippocampal NMDA receptors. Hippocampus. (in press).
Brandt, T., Schautzer, F., Hamilton, D., Brüning, R., Markowitsch, H. J., Kalla, R., Darlington, C. L., Smith, P. F., and Strupp, M. (2005). Vestibular loss causes hippocampal atrophy and impaired spatial memory in humans. Brain 128, 2732–2741.
Dutheil, S., Brezun, J. M., Leonard, J., Lacour, M., and Tighilet, B. (2009). Neurogenesis and astrogenesis contribution to recovery of vestibular functions in the adult cat following unilateral vestibular neurectomy: cellular and behavioral evidence. Neuroscience 164, 1444–1456.
Gamst, G., Meyers, L. S., and Guarino, A. J. (2008). Analysis of Variance Designs. Cambridge: Cambridge University Press.
Gliddon, C., Darlington, C. L., and Smith, P. F. (2003). Activation of the hypothalamic- pituitary-adrenal axis in the guinea pig following unilateral labyrinthectomy. Brain Res. 964, 306–310.
Goddard, M., Zheng, Y., Darlington, C. L., and Smith, P. F. (2008). Locomotor and exploratory behavior in the rat following bilateral vestibular deafferentation. Behav. Neurosci. 122, 448–459.
Gundersen, H. J. (1986). Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J. Microsc. 143, 3–45.
Gundersen, H. J., and Jensen, E. B. (1987). The efficiency of systematic sampling in stereology and its prediction. J. Microsc. 147, 229–263.
Horii, A., Uno, A., Kitahara, T., Mitani, K., Masumura, C., Kizawa, K., and Kubo, T. (2007). Effects of fluvoxamine on anxiety, depression and subjective handicaps of chronic dizziness patients with or without neuro-otologic diseases. J. Vestib. Res. 17, 1–8.
Horner, K. C., and Cazals, Y. (2005). Stress hormones in Meniere’s disease and acoustic neuroma. Brain Res. Bull. 66, 1–8.
Hüfner, K., Binetti, C., Hamilton, D. A., Stephan, T., Flanagin, V. L., Linn, J., Labudda, K., Markowitsch, H., Glasauer, S., Jahn, K., Strupp, M., and Brandt, T. (2010). Structural and functional plasticity of the hippocampal formation in professional dancers and slackliners. Hippocampus 21, 855–865.
Hüfner, K., Hamilton, D. A., Kalla, R., Stephan, T., Glasauer, S., Ma, J., Brüning, R., Markowitsch, H. J., Labudda, K., Schichor, C., Strupp, M., and Brandt, T. (2007). Spatial memory and hippocampal volume in humans with unilateral vestibular deafferentation. Hippocampus 17, 471–485.
Korbo, L., Ladefoged, O., Lam, H. R., Ostergaard, G., West, M. J., and Arlien-Soborg, P. (1996). Neuronal loss in hippocampus in rats exposed to toluene. Neurotoxicology 17, 359–366.
Kraus, K. S., Mitra, S., Jimenez, Z., Hinduja, S., Ding, D., Jiang, H., Gray, L., Lobarinas, E., Sun, W., and Salvi, R. J. (2010). Noise trauma impairs neurogenesis in the rat hippocampus. Neuroscience 167, 1216–1226.
Landgren, H., and Curtis, M. A. (2011). Locating and labeling neural stem cells in the brain. J. Cell. Physiol. 226, 1–7.
Lindsay, L., Liu, P., Gliddon, C., Zheng, Y., Smith, P. F., and Darlington, C. L. (2005). Cytosolic glucocorticoid receptor expression in the vestibular nucleus and hippocampus following unilateral vestibular deafferentation. Exp. Brain Res. 162, 309–314.
Ling, E. A., Paterson, J. A., Privat, A., Mori, S., and Leblond, C. P. (1973). Investigation of glial cells in semi-thin sections. I. Identification of glial cells in the brain of young rats. J. Comp. Neurol. 149, 43–72.
Liu, P., Zhang, H., Devaraj, R., Ganesalingam, G., and Smith, P. F. (2010). A multivariate analysis of the effects of aging on glutamate, GABA and arginine metabolites in the rat vestibular nucleus. Hear. Res. 269, 122–133.
Lucassen, P. J., Meerlo, P., Naylor, A. S., van Dam, A. M., Dayer, A. G., Fuchs, E., Oomen, C. A., and Czéh, B. (2010). Regulation of adult neurogenesis by stress, sleep disruption, exercise and inflammation: implications for depression and antidepressant action. Eur. Neuropsychopharmacol. 20, 1–17.
Maguire, E. A., Gadian, D. G., Johnsrude, I. S., Good, C. D., Ashburner, J., Frackowiak, R. S., and Frith, C. D. (2000). Navigation-related structural change in the hippocampi of taxi drivers. Proc. Natl. Acad. Sci. U.S.A. 97, 4398–4403.
Malberg, J., Eisch, A., Nestler, E., and Duman, R. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104–9110.
Neo, P., Carter, D., Zheng, Y., Smith, P. F., Darlington, C. L., and McNaughton, N. (2012). Septal elicitation of hippocampal theta rhythm did not repair the cognitive and emotional deficits resulting from vestibular lesions. Hippocampus. (in press).
Nixon, K., and Crews, F. (2004). Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J. Neurosci. 24, 9714–9722.
Olson, A. K., Eadie, B. D., Ernst, C., and Christie, B. R. (2006). Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus 16, 250–260.
Russell, N. A., Horii, A., Smith, P. F., Darlington, C. L., and Bilkey, D. K. (2003a). Bilateral peripheral vestibular lesions produce long-term changes in spatial learning in the rat. J. Vestib. Res. 13, 9–16.
Russell, N. A., Horii, A., Smith, P. F., Darlington, C. L., and Bilkey, D. K. (2003b). Long-term effects of permanent vestibular lesions on hippocampal spatial firing. J. Neurosci. 23, 6490–6498.
Russell, N. A., Horii, A., Smith, P. F., Darlington, C. L., and Bilkey, D. K. (2006). Lesions of the vestibular system disrupt hippocampal theta rhythm in the rat. J. Neurophysiol. 96, 4–14.
Seress, L. (1978). Pyramid-like basket cells in the granular layer of the dentate gyrus in the rat. J. Anat. 127, 163–168.
Smith, P. F., Brandt, T., Strupp, M., Darlington, C. L., and Zheng, Y. (2009). Balance before reason in rats and humans. Ann. N. Y. Acad. Sci. 1164, 127–133.
Smith, P. F., Darlington, C. L., and Zheng, Y. (2010). Move it or lose it: is stimulation of the vestibular system necessary for normal spatial memory? Hippocampus 20, 36–43.
Stackman, R. W., Clark, A. S., and Taube, J. S. (2002). Hippocampal spatial representations require vestibular input. Hippocampus 12, 291–303.
Stackman, R. W., and Herbert, A. M. (2002). Rats with lesions of the vestibular system require a visual landmark for spatial navigation. Behav. Brain Res. 128, 27–40.
Stiles, L., Zheng, Y., Darlington, C. L., and Smith, P. F. (2012). The D2 dopamine receptor and locomotor hyperactivity following bilateral vestibular deafferentation in the rat. Behav. Brain Res. 227, 150–158.
Tai, S. K., Ma, J., Ossenkopp, K.-P., and Leung, L. S. (2012). Activation of immobility-related hippocampal theta by cholinergic septohippocampal neurons during vestibular stimulation. Hippocampus. (in press).
Tighilet, B., Brezun, J., Sylvie, G., Gaubert, C., and Lacour, M. (2007). New neurons in the vestibular nuclei complex after unilateral vestibular neurectomy in the adult cat. Eur. J. Neurosci. 25, 47–58.
van Cruijsen, N., Dullaart, R. P. F., Wit, H. P., and Albers, F. W. J. (2005). Analysis of cortisol and other stress-related hormones in patients with Meniere’s disease. Otol. Neurotol. 26, 1214–1219.
Wallace, D. G., Hines, D. J., Pellis, S. M., and Whishaw, I. Q. (2002). Vestibular information is required for dead reckoning in the rat. J. Neurosci. 22, 10009–10017.
West, M. J., and Gundersen, H. J. (1990). Unbiased stereological estimation of the number of neurons in the human hippocampus. J. Comp. Neurol. 296, 1–22.
West, M. J., Slomianka, L., and Gundersen, H. J. (1991). Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 231, 482–497.
Yang, F., Wang, J.-C., Han, J.-L., Zhao, G., and Jiang, W. (2008). Different effects of mild and severe seizures on hippocampal neurogenesis in adult rats. Hippocampus 18, 460–468.
Zhang, R., Smith, P. F., and Darlington, C. L. (2005). An immunocytochemical and stereological study of cytosolic glucocorticoid receptors in the rat medial vestibular nucleus and effects of unilateral vestibular deafferentation. Acta Otolaryngol. 125, 1258–1264.
Zheng, Y., Balabhadrapatruni, S., Masumura, C., Munro, O., Darlington, C. L., and Smith, P. F. (2009a). Bilateral vestibular deafferentation causes deficits in a 5-choice serial reaction time task in rats. Behav. Brain Res. 203, 113–117.
Zheng, Y., Goddard, M., Darlington, C. L., and Smith, P. F. (2009b). Long-term deficits on a foraging task after bilateral vestibular deafferentation in rats. Hippocampus 19, 480–486.
Zheng, Y., Begum, S., Zhang, C., Fleming, K., Masumura, C., Zhang, M., Smith, P. F., and Darlington, C. L. (2011). Bromodeoxyuridine uptake reflecting DNA repair, neuronal de-differentiation or possible neurogenesis in the adult brainstem cochlear nucleus following bilateral cochlear lesions in the rat. Exp. Brain Res. 3/4, 477–487.
Zheng, Y., Darlington, C. L., and Smith, P. F. (2006). Impairment and recovery on a food foraging task following unilateral vestibular deafferentation in rats. Hippocampus 16, 368–378.
Zheng, Y., Goddard, M., Darlington, C. L., and Smith, P. F. (2007). Bilateral vestibular deafferentation impairs performance in a spatial forced alternation task in rats. Hippocampus 17, 253–256.
Zheng, Y., Goddard, M., Darlington, C. L., and Smith, P. F. (2008). The effects of bilateral vestibular deafferentation on anxiety-related behaviours in Wistar rats. Behav. Brain Res. 193, 55–62.
Zheng, Y., Mason-Parker, S. E., Logan, B., Darlington, C. L., Smith, P. F., and Abraham, W. C. (2010a). Hippocampal synaptic transmission and LTP in vivo are intact following bilateral vestibular deafferentation in the rat. Hippocampus 20, 461–468.
Zheng, Y., Masumura, C., Chung, P., Darlington, C. L., and Smith, P. F. (2010b). Cell proliferation and survival in the vestibular nucleus following bilateral vestibular deafferentation in the adult rat. Neurosci. Lett. 468, 85–88.
Zingler, V. C., Weintz, E., Jahn, K., Mike, A., Huppert, D., Rettinger, N., Brandt, T., and Strupp, M. (2008). Follow-up of vestibular function in bilateral vestibulopathy. J. Neurol. Neurosurg. Psychiatr. 79, 284–288.
Keywords: vestibular lesions, hippocampal volume, number estimation, cell proliferation, stereology, rat
Citation: Zheng Y, Balabhadrapatruni S, Baek JH, Chung P, Gliddon C, Zhang M, Darlington CL, Napper R, Strupp M, Brandt T and Smith PF (2012) The effects of bilateral vestibular loss on hippocampal volume, neuronal number, and cell proliferation in rats. Front. Neur. 3:20. doi: 10.3389/fneur.2012.00020
Received: 29 November 2011;
Paper pending published: 15 December 2011;
Accepted: 03 February 2012;
Published online: 28 February 2012.
Edited by:
Pierre-Paul Vidal, Université Rene Descartes, FranceReviewed by:
Pierre Denise, Université de Caen Basse Normandie, FranceCopyright: © 2012 Zheng, Balabhadrapatruni, Baek, Chung, Gliddon, Zhang, Darlington, Napper, Strupp, Brandt and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Paul F. Smith, Department of Pharmacology and Toxicology, School of Medical Sciences, University of Otago Medical School, Dunedin, New Zealand. e-mail:cGF1bC5zbWl0aEBzdG9uZWJvdy5vdGFnby5hYy5ueg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.