 Jijun Wan1* Hafsa Mamsa1 Janine L. Johnston2,3,4 Elizabeth L. Spriggs5,6,7 Harvey S. Singer8 David S. Zee8 Alhamza R. Al-Bayati1 Robert W. Baloh1,9 Joanna C. Jen1 and CINCH Investigators
Jijun Wan1* Hafsa Mamsa1 Janine L. Johnston2,3,4 Elizabeth L. Spriggs5,6,7 Harvey S. Singer8 David S. Zee8 Alhamza R. Al-Bayati1 Robert W. Baloh1,9 Joanna C. Jen1 and CINCH Investigators
- 1 Department of Neurology, University of California, Los Angeles, Los Angeles, CA, USA
- 2 Section of Neurology, Department of Medicine, University of Manitoba, Winnipeg, MB, Canada
- 3 Department of Ophthalmology, University of Manitoba, Winnipeg, MB, Canada
- 4 Department of Otolaryngology, University of Manitoba, Winnipeg, MB, Canada
- 5 Clinical Biochemistry and Genetics, Diagnostic Services of Manitoba, Winnipeg, MB, Canada
- 6 Department of Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, MB, Canada
- 7 Department of Pediatrics and Child Health, University of Manitoba, Winnipeg, MB, Canada
- 8 Department of Neurology, Johns Hopkins University, Baltimore, MD, USA
- 9 Department of Surgery, University of California, Los Angeles, Los Angeles, CA, USA
Episodic ataxia (EA) syndromes are heritable diseases characterized by dramatic episodes of imbalance and incoordination. EA type 2 (EA2), the most common and the best characterized subtype, is caused by mostly nonsense, splice site, small indel, and sometimes missense mutations in CACNA1A. Direct sequencing of CACNA1A fails to identify mutations in some patients with EA2-like features, possibly due to incomplete interrogation of CACNA1A or defects in other EA genes not yet defined. Previous reports described genomic deletions between 4 and 40 kb in EA2. In 47 subjects with EA (26 with EA2-like features) who tested negative for mutations in the known EA genes, we used multiplex ligation-dependent probe amplification to analyze CACNA1A for exonic copy number variations. Breakpoints were further defined by long-range PCR. We identified distinct multi-exonic deletions in three probands with classic EA2-like features: episodes of prolonged vertigo and ataxia triggered by stress and fatigue, interictal nystagmus, with onset during infancy or early childhood. The breakpoints in all three probands are located in Alu sequences, indicating errors in homologous recombination of Alu sequences as the underlying mechanism. The smallest deletion spanned exons 39 and 40, while the largest deletion spanned 200 kb, missing all but the first three exons. One deletion involving exons 39 through 47 arose spontaneously. The search for mutations in CACNA1A appears most fruitful in EA patients with interictal nystagmus and onset early in life. The finding of large heterozygous deletions suggests haploinsufficiency as a possible pathomechanism of EA2.
Introduction
Episodic ataxia (EA) syndromes are heritable diseases characterized by dramatic episodes of imbalance and incoordination (Jen et al., 2007). EA is clinically and genetically heterogeneous, with at least seven subtypes described to date. EA type 2 (EA2), the most common and the best characterized subtype, is defined by mutations in CACNA1A, which encodes Cav2.1, the main subunit of the P/Q type voltage-gated calcium channel (Ophoff et al., 1996). Clinically, EA2 is characterized by recurrent vertigo and ataxia lasting hours commonly triggered by physical exertion or emotional stress. EA2 typically presents early in life and is variably associated with interictal nystagmus and progressive ataxia. Direct sequencing has identified point or small indel mutations that span the entire gene, most of which are nonsense, splice site, or frameshift mutations predicted to lead to nonsense-mediated mRNA decay or truncated proteins to cause loss of channel function (Jen et al., 2007).
There are still many patients who exhibit EA2-like clinical features in whom no mutation has been identified; this lack of success in finding mutations has often been attributed to incomplete interrogation of the CACNA1A gene or genetic heterogeneity (Denier et al., 1999; Jen et al., 2004; Graves and Hanna, 2008; Mantuano et al., 2010). The use of alternative techniques led to recent reports of multi-exonic deletions of 4–40 kb in EA2 (Riant et al., 2008, 2010; Labrum et al., 2009). The exact breakpoints were ascertained in five individuals (Riant et al., 2008, 2010); all involved Alu elements, which are short interspersed elements around 300 bases in length that comprise the most abundant repetitive sequences, an estimated 10%, in the human genome and are known to mediate recombination (Cordaux and Batzer, 2009). All index patients harboring large deletions presented before age 30 (Riant et al., 2008, 2010; Labrum et al., 2009). Similarly, the yield for mutation screening by sequencing in those with late-onset EA is low, with genetic confirmation of EA2 in only two patients with onset after age 50 (Imbrici et al., 2005; Cuenca-Leon et al., 2009).
In this study, we searched for genomic rearrangement in CACNA1A in patients with onset of EA before age 30 in whom no mutations in EA genes have been identified.
Subjects and Methods
Patients presenting with episodes of ataxia that began before age 30 underwent interview, neurological examination, laboratory investigation, neuroimaging studies, and genetic screening. This study was approved by the institutional review boards, and all subjects or their guardians provided written consent. A subset of the patients was also enrolled in the EA arm of the Consortium for the Clinical Investigation of Neurological Channelopathies (CINCH) study, a multicentered study sponsored by the Rare Diseases Clinical Research Network supported by the National Institutes of Health.
There were 47 unrelated subjects with onset of EA before the age of 30 years in whom we could not identify mutations in the known EA1, EA2, and EA6 genes. Among these subjects, four exhibited EA1-like features characterized by brief ataxia episodes with interictal myokymia. Twenty-six subjects presented with EA2-like features. One subject had EA6-like features, with episodic ataxia, seizures, alternating hemiplegia beginning during infancy. There were 16 subjects who experienced EA without interictal nystagmus or myokymia but had other eye movement abnormalities, movement disorder, or sleep disturbance.
We searched for genomic rearrangement in CACNA1A in 47 EA patients without known mutations by multiplex ligation-dependent probe amplification (MLPA; Schouten et al., 2002) using Kit P279 (MRC-Holland) on an ABI 3730 DNA Analyzer (Applied Biosystems) and processed the data by GeneMarker (Softgenetics).
Mindful of Alu-mediated chromosomal recombination (Riant et al., 2008, 2010; Cordaux and Batzer, 2009) and the high density (30%) of Alu sequences spanning CACNA1A, we identified Alu elements in CACNA1A by bioinformatic mining (genome.ucsc.edu) and prioritized candidate Alu elements with high similarity by pairwise alignment bl2seq (NCBI http://www.ncbi.nlm.nih.gov/). The breakpoints were ascertained by sequencing.
To locate the breakpoints for the deletion in patient #946, we performed long-range PCR based on the manufacturer’s protocol (Takara Bio), with 10 forward primers between exon 1 and exon 4 and four reverse primers after exon 47. Primers were designed ∼10 kb apart as guided by the distribution of Alu elements, numbering more than 80 in the 134 kb candidate region for the 5′ breakpoint. Primer sequences are available on request. Long-range PCR narrowed the candidate region, for which all Alu elements were identified and aligned with those downstream of CACNA1A, as analyzed by bl2seq (Table 1).
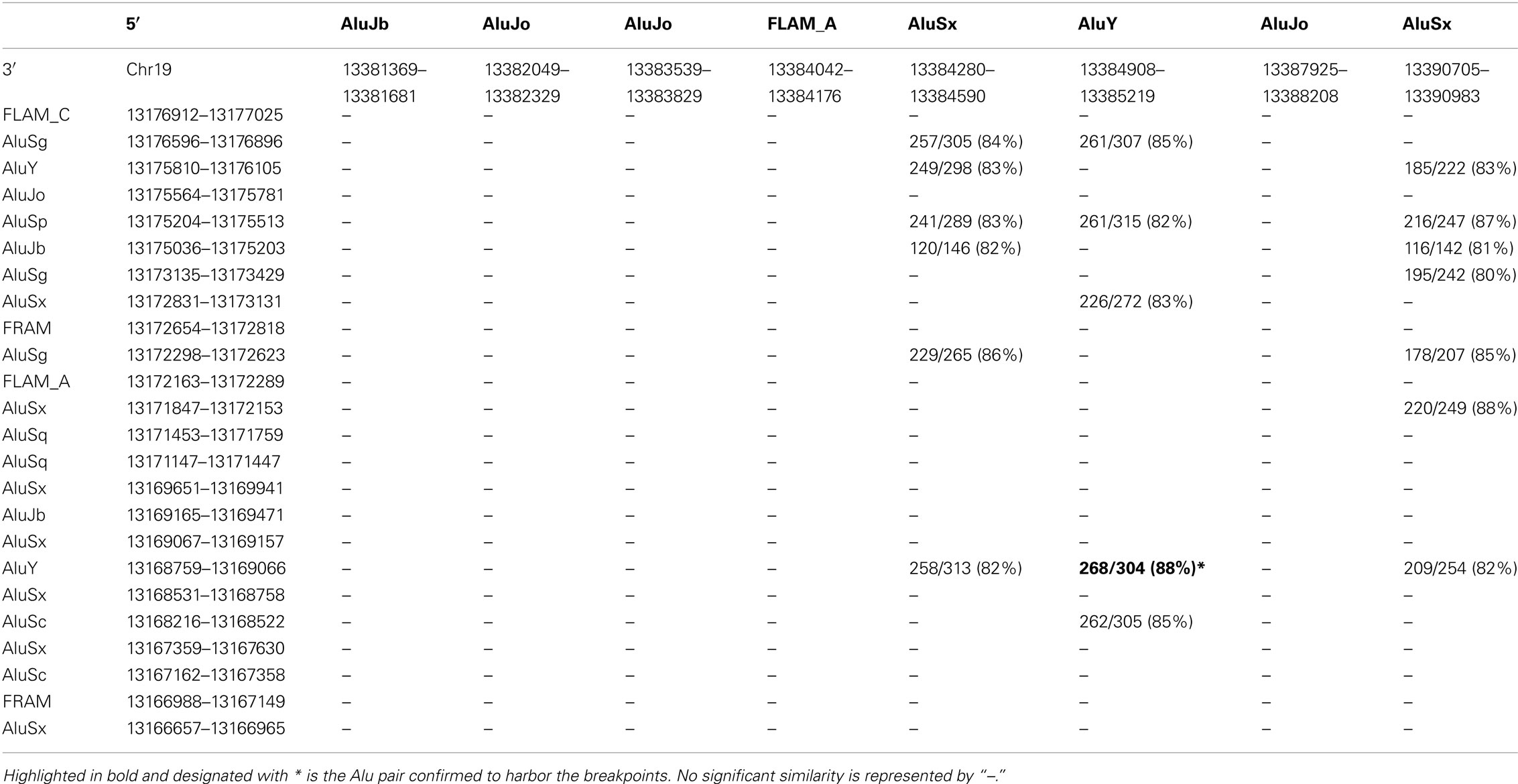
Table 1. Identifying likely breakpoints in Pt#946 by pairwise alignment of Alu sequences in the 5′ candidate region within intron 3 and the 3′ candidate region downstream of exon 47 of the CACNA1A gene, represented by the number of identical sequences (percentage).
Genotyping. In Pt #444 and her father, we typed a series of microsatellite markers on 19p flanking CACNA1A in addition to other microsatellite markers on multiple chromosomes.
Results
We observed a 50% reduction in the signal for multiple MLPA probes (Figure 1) in three unrelated probands with classic EA2-like features: episodes of prolonged vertigo and ataxia triggered by stress and fatigue, interictal nystagmus, with onset during infancy or early childhood (Table 2).
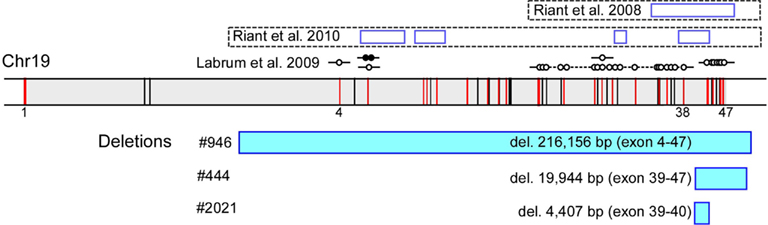
Figure 1. Newly identified and previously reported EA2-causing genomic rearrangements in CACNA1A that vary in size and location. In a diagram of the genomic structure of CACNA1A on chr 19 spanning approximately 300, 000 base pairs (shaded in gray), the vertical bars represent exons, and those in red harbor target sequences for the MLPA probes. The locations of exons 1, 4, 38, and 47 are specified. The rectangles highlighted in blue below the genomic structure of CACNA1A represent missing MLPA probes in the patients, labeled with the sizes and deleted exons. The open rectangles above the genomic structure of CACNA1A represent previously described deletions, with established boundaries, in five unrelated kindreds with EA2 (Riant et al., 2008, 2010), along with additional instances of genomic rearrangements without breakpoint ascertainment by D:Labrum et al., 2009; open circle – missing MLPA probe; closed circle – duplicated MLPA probe.
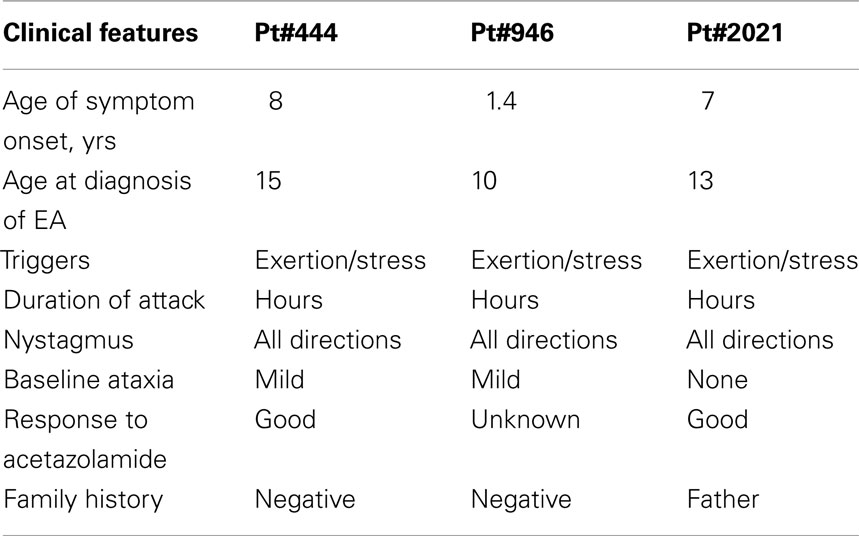
Table 2. Clinical features.
Patient #444 began suffering episodic imbalance and vertigo at age 8 years. Vertigo was preceded by non-throbbing neck pain and was accompanied and followed by nausea and repeated vomiting. She also noted blurred vision not corrected by refraction, but denied oscillopsia or diplopia. Episodes occurred daily or every other day and lasted two or more hours. She also experienced severe motion sickness and frequent throbbing headaches not temporally related to episodic dizziness. The patient was noted to have nystagmus at the age of 6 months. Developmental milestones were appropriate. Family history is positive for migraine in her father and paternal grandmother. Physical examination showed impaired pursuit, downbeat nystagmus in primary gaze, and multidirectional gaze-evoked nystagmus, with stiff and wide-based gait. Two MRIs were normal. The patient has responded well to acetazolamide 250 mg twice or three times daily, with complete resolution of dizziness but not her headaches (Figure 2).
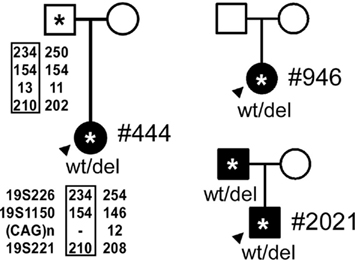
Figure 2. The pedigrees for the probands, indicated by arrowheads. (*, DNA sample obtained; closed symbol, affected; circle – female; square – male). Genotyping of Patient #444 and her father with 19p markers (from 5′ to 3′, 19S226, 19S1150, polymorphic CAG repeats, and 19S221) are shown with the pedigree. Haplotypes shared between the patient and the father are highlighted by rectangles.
Patient #946 presented at age 17 months with recurrent attacks of vertigo and ataxia associated with severe nausea and vomiting. During the attacks, her balance is severely impaired with slurring of speech such that she needs to lie down to sleep. The attacks typically last for several hours, and they are triggered by exertion or stress, or they can occur abruptly without any apparent cause. Her development is delayed: walking at around 17 months, and speaking her first words around 3 years. She is in special education. Family history is negative for similar symptoms of recurrent vertigo and ataxia. Mother has migraine. Her examination showed impaired pursuit, horizontal and downward gaze-evoked nystagmus, dysdiadochokinesis, and mild difficulty in tandem walking. Routine blood work, EEG, auditory brainstem evoked responses, and brain MRI were all normal. She had tried various different medications including carbamazepine, periactin, meclizine, and promethazine without improvement. She has not yet tried acetazolamide (Figure 2).
Patient #2021 had the onset of episodic dizziness associated with imbalance, nausea, occasional emesis, and tinnitus at the age of 7 years. Episodes are triggered by exercise, stress, and anxiety, persist for 20–120 min, and are relieved by stopping activities, lying down, or sleeping. The patient is active in sports, playing soccer, and wrestling. Feeling “overheated” while participating in these activities often triggers an event. During the episodes of dizziness, he feels warm, has a sense of moving, his speech slurs, and his limbs feel like “rubber” or “jello.” His past medical history is notable for frequent ear infections, associated with a transient bilateral hearing impairment and speech delay of approximately 6 months. Childhood development was otherwise normal, academically he has done well through high school, and he has been treated for depression. Family history is significant with his father having a similar, but milder, condition that began in childhood and was triggered by exercise. MRI and MRA studies in the father were normal. On physical examination, the patient had impaired smooth pursuits, saccadic dysmetria, gaze-evoked nystagmus, but otherwise steady gait and coordination. Treatment with acetazolamide produced some benefit (Figure 2).
We confirmed multi-exonic deletions by ascertaining the breakpoints (Figure 3). In Patient #444, the heterozygous deletion spanned exons 39 through 47 (Figures 1 and 3) to predict a truncated protein, if translated, missing the C-terminus (Figure 4). Her father was not found to have this same deletion; and genotyping of microsatellite markers on multiple chromosomes of Patient #444 and her father confirmed paternity. The genotyping results of 19p markers (from 5′ to 3′, 19S226, 19S1150, polymorphic CAG repeats in exon 47, and 19S221) are shown with the pedigree. Specifically, sequencing of the polymorphic CAG repeats in Patient #444 confirmed the deletion of one of the two alleles for exon 47; the absence of either paternal alleles of 11 and 13 repeats in the patient suggests that the deletion occurred de novo on a paternal allele of CACNA1A (Figure 2).
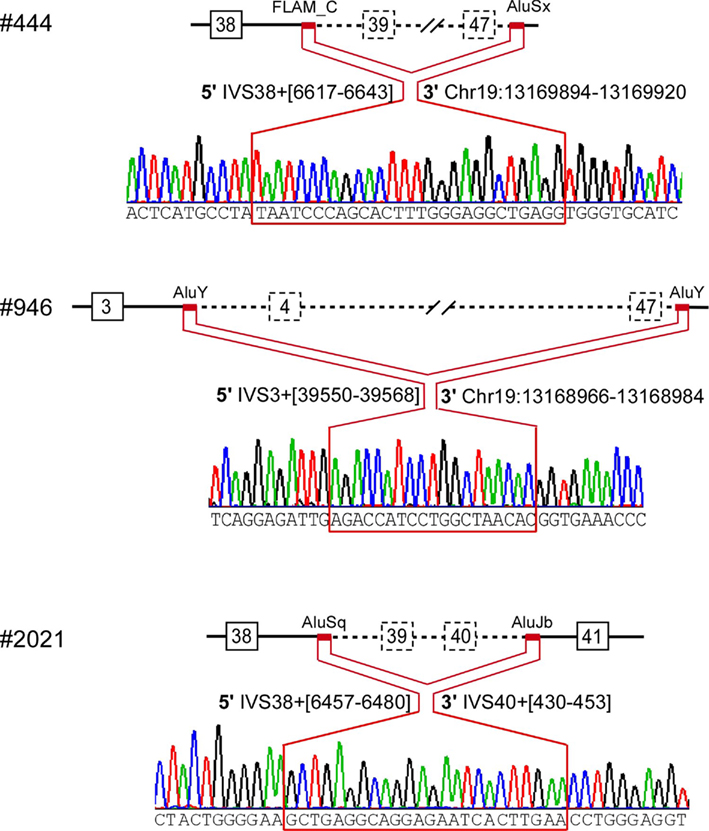
Figure 3. Breakpoint ascertainment. The red lines highlight the stretch of identical sequence shared by the 5′ and 3′ Alu elements that mediated homologous recombination, with specific nucleotide positions for the 5′ and for the 3′ Alu elements. The 3′ breakpoints downstream of the CACNA1A gene are labeled based on the March 2006 human reference sequence (hg18, NCBI Build 36.1).
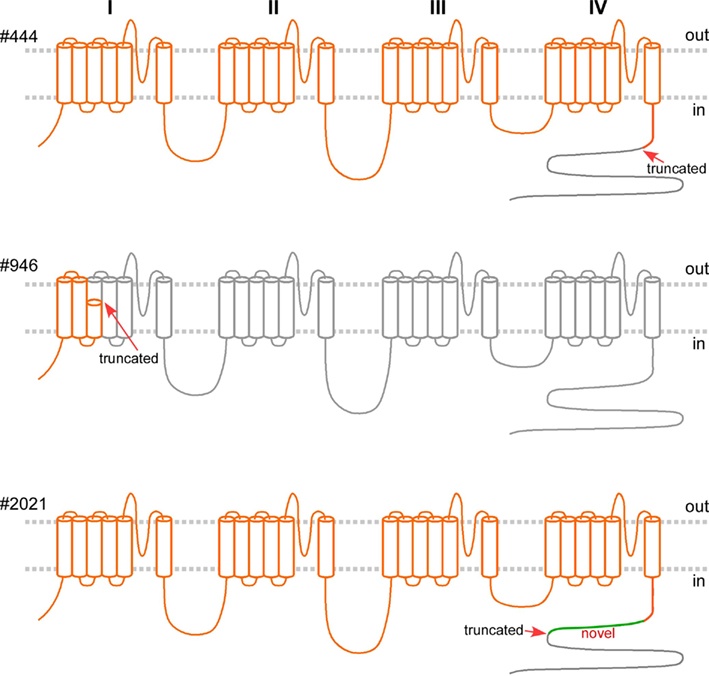
Figure 4. Predicted gene products of the mutated alleles, if translated, in orange, superimposed on the wild type Cav2.1 (in gray) spanning 2510 amino acid residues, with four homologous domains, each with six transmembrane segments. The intracellular C-terminus is largely missing in #444 (with truncation at residue 1917, arrow). The predicted gene product of the mutated allele in Pt #946 encodes only the first three transmembrane segments. The gene product in Pt#2021 of the mutated allele with the deletion of exon 39–40, if translated, is predicted to comprise 79 novel residues (in green) following residue 1917, missing the tail (gray).
The largest deletion, missing all but the first three exons, was found in Patient #946 (Figures 1 and 3). DNA samples from her parents were not available for testing. The candidate region of the 5′ breakpoint included sequences downstream of exon 1 and upstream of exon 4, which spanned 134 kb. Identifying the most highly homologous Alu sequences in the candidate regions facilitated the identification of the breakpoints (Table 1).
We confirmed the 200 kb deletion, for which the 5′ breakpoint resided in an Alu sequence in intron 3 that harbored a long stretch of identical sequences and shared high similarity (88%) with the 3′ breakpoint in an Alu sequence 10 kb downstream of CACNA1A (Table 1; Figure 3). If made, the predicted mutant gene product constitutes only three transmembrane segments (Figure 4).
The smallest deletion, spanning exons 39 and 40, was found in Patient #2021 and subsequently his symptomatic father (Figures 1 and 3). This deletion is predicted to cause a shift in the open reading frame to introduce 79 novel residues downstream of residue 1917 and a premature stop codon (Figure 4).
The breakpoints in all three probands involved Alu sequences: AluY-AluY in #946; FLAM_C-AluSx in #444; and AluSq-AluJb in #2021 (Figure 3). The Alu element pairs shared high similarity, with a length of identical stretch of nucleotides ranging from 24 to 48 and an overall similarity of 76–88%.
Discussion
We focused our genetic investigation on individuals with onset of EA early in life. We identified novel large genomic deletions in three unrelated subjects. All three deletions resulted from homologous recombination of Alu elements, as previously observed by Riant et al. (2008, 2010). The deletion in Pt #444 arose de novo. Because the parents were asymptomatic, we suspected that the deletion in Pt #946 likely also occurred spontaneously; however, DNA samples from the parents were not available for testing.
Riant et al. (2010) used the quantitative multiplex polymerase chain reaction of short fluorescent fragments method to probe every exon to identify large genomic deletions in 4 of 27 probands with typical EA2-like features. Labrum et al. (2009) using MLPA with partial but incomplete probes for CACNA1A identified genomic duplication or deletion in 10 of 53 subjects with EA2 or its allelic disorder familial hemiplegic migraine. Of note, in five of the subjects, the deletions involved the same exons 39–47; none had a positive family history. We also used MLPA to identify new deletions in 3 of 26 probands with EA2-like clinical features in a cohort of 47 subjects with EA onset before age 30. In Pt #444, we demonstrated that the genomic rearrangement involving the deletion of exons 39–47 arose spontaneously from the paternal allele. Whether genomic rearrangement of CACNA1A may contribute to late-onset EA await further analysis.
The genomic rearrangements in CACNA1A described thus far are highly variable in size and location (Figure 1; Riant et al., 2008, 2010; Labrum et al., 2009). Given the high density of Alu elements estimated to occupy approximately 30% of the genomic sequence of CACNA1A, we expect many more instances of large genomic rearrangements elsewhere in this gene. Our findings further emphasize that genomic rearrangement is an important cause of EA2 and should be included in the genetic analysis of CACNA1A.
Our observations highlight challenges in the clinical diagnosis and genetic definition of episodic ataxia. Perhaps because of its rarity, EA is under-recognized and under-diagnosed. In each of the three subjects described in this report, it took several years and extensive clinical testing to reach the clinical diagnosis of episodic ataxia, which was followed by another several years before the genetic confirmation of EA2. There remain many patients with EA2-like clinical features without mutations in the EA2 gene, suggesting genetic heterogeneity although the possibility of undetected intronic or promoter mutations in the CACNA1A gene still exists.
While MLPA is relatively straightforward to perform and analyze, there was extensive random variation of single probe intensity, especially for exon 47. The absence of Alu elements in intron 46 suggests that the deletion of exon 47 alone may be unlikely, as confirmed by sequencing of the polymorphic CAG repeats in exon 47 in samples with low signal for exon 47 but normal signal for exon 46. The distribution of Alu sequences may be considered in developing additional MLPA probes for CACNA1A.
Although there is general consensus that most of the mutations that cause EA2 result in loss of calcium channel function, it is not clear whether they cause disease by haploinsufficiency or a dominant negative effect. Understanding the disease mechanism may help guide treatment. Results from in vitro expression studies suggested a dominant negative effect (Jouvenceau et al., 2001; Page et al., 2004, 2010; Jeng et al., 2006; Mezghrani et al., 2008). Yet, these studies all used artificial cDNA constructs in heterologous cell expression systems that bypass nonsense-mediated mRNA decay, which is a stringent cellular mRNA surveillance mechanism that detects and degrades mRNAs with nonsense mutations to prevent the expression of truncated proteins. The recently described frameshift mutation c.165dupA predicted to lead to only a fragment of the N-terminal tail even if translated; this short peptide does not contain any motif for interaction with the auxiliary subunits or the wild type Cav2.1, leading the investigators to favor haploinsufficiency over a dominant negative effect (Mantuano et al., 2010). Similarly, the deletion of over 200 kb in Pt #946 is unlikely to lead to a stable gene product to exert a dominant negative effect on the gene product of the normal allele. The finding of genomic deletions of variable sizes in CACNA1A all causing similar clinical features suggests shared mechanisms for these deletions as null alleles through haploinsufficiency.
Genetic characterization of EA2 not only is important for diagnosis but also may dictate the choice of treatment. New therapeutic strategies are being developed to intervene at the level of splicing (Wilton et al., 2007), translation (Welch et al., 2007), and post-translational quality control (Williams et al., 2006) to restore the function of nonsense, splice site, or missense mutants. On the other hand, those patients with large genomic rearrangements in EA2 will likely benefit the most from medications with targets downstream from the mutant calcium channels to restore cerebellar neuronal function. Our patients have demonstrated modest responses to acetazolamide, which is a carbonic anhydrase inhibitor with no direct influence on calcium channels, and whose efficacy in EA and other neurological channelopathies has not been elucidated (Griggs et al., 1978). Recently, 4-aminopyridine, a voltage-gated potassium channel blocker, has emerged as a promising alternative to acetazolamide. 4-aminopyridine was found to be effective in stopping attacks in patients with EA (Strupp et al., 2004, 2011) as well as mutant mice (Weisz et al., 2005), likely by restoring the precision of cerebellar Purkinje cell pacemaking (Alvina and Khodakhah, 2010). An FDA-sponsored randomized trial is now underway on the efficacy and tolerability of 4-aminopyridine in EA2.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the National Institutes of Health R01NS064183 and U54NS059065.
Appendix
The Consortium for Clinical Investigation of Neurologic Channelopathies (CINCH), led by Robert C. Griggs, M. D., University of Rochester, is supported by NIH U54 NS059065. Episodic Ataxia Project Co-Principal Investigators: J. C. Jen, University of California, Los Angeles (UCLA), R. W. Baloh, UCLA, and R. C. Griggs, University of Rochester.
The members of CINCH involved in the recruitment of patients with episodic ataxia are, in alphabetical order: Anthony Amato (Brigham and Women’s Hospital), Richard Barohn (University of Kansas), Yoon-Hee Cha (UCLA), Tracey Graves (Institute of Neurology, UK), Angelika Hahn (London Health Science Centre, Canada), Michael G. Hanna (Institute of Neurology, UK), Kim Hart (University of Rochester), Laura Herbelin (University of Kansas), Barbara Herr (University of Rochester), Yunxia Wang (University of Kansas).
References
Alvina, K., and Khodakhah, K. (2010). The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. 30, 7258–7268.
Cordaux, R., and Batzer, M. A. (2009). The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703.
Cuenca-Leon, E., Banchs, I., Serra, S. A., Latorre, P., Fernandez-Castillo, N., Corominas, R., Valverde, M. A., Volpini, V., Fernandez-Fernandez, J. M., Macaya, A., and Cormand, B. (2009). Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J. Neurol. Sci. 280, 10–14.
Denier, C., Ducros, A., Vahedi, K., Joutel, A., Thierry, P., Ritz, A., Castelnovo, G., Deonna, T., Gerard, P., Devoize, J. L., Gayou, A., Perrouty, B., Soisson, T., Autret, A., Warter, J. M., Vighetto, A., Van Bogaert, P., Alamowitch, S., Roullet, E., and Tournier-Lasserve, E. (1999). High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 52, 1816–1821.
Graves, T. D., and Hanna, M. G. (2008). Episodic ataxia: SLC1A3 and CACNB4 do not explain the apparent genetic heterogeneity. J. Neurol. 255, 1097–1099.
Griggs, R. C., Moxley, R. T. III, Lafrance, R. A., and McQuillen, J. (1978). Hereditary paroxysmal ataxia: response to acetazolamide. Neurology 28, 1259–1264.
Imbrici, P., Eunson, L. H., Graves, T. D., Bhatia, K. P., Wadia, N. H., Kullmann, D. M., and Hanna, M. G. (2005). Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology 65, 944–946.
Jen, J., Kim, G. W., and Baloh, R. W. (2004). Clinical spectrum of episodic ataxia type 2. Neurology 62, 17–22.
Jen, J. C., Graves, T. D., Hess, E. J., Hanna, M. G., Griggs, R. C., Baloh, R. W., and CINCH Investigators. (2007). Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 130, 2484–2493.
Jeng, C. J., Chen, Y. T., Chen, Y. W., and Tang, C. Y. (2006). Dominant-negative effects of human P/Q-type Ca2+ channel mutations associated with episodic ataxia type 2. Am. J. Physiol. Cell Physiol. 290, C1209–C1220.
Jouvenceau, A., Eunson, L. H., Spauschus, A., Ramesh, V., Zuberi, S. M., Kullmann, D. M., and Hanna, M. G. (2001). Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet 358, 801–807.
Labrum, R. W., Rajakulendran, S., Graves, T. D., Eunson, L. H., Bevan, R., Sweeney, M. G., Hammans, S. R., Tubridy, N., Britton, T., Carr, L. J., Ostergaard, J. R., Kennedy, C. R., Al-Memar, A., Kullmann, D. M., Schorge, S., Temple, K., Davis, M. B., and Hanna, M. G. (2009). Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: implications for diagnostic testing. J. Med. Genet. 46, 786–791.
Mantuano, E., Romano, S., Veneziano, L., Gellera, C., Castellotti, B., Caimi, S., Testa, D., Estienne, M., Zorzi, G., Bugiani, M., Rajabally, Y. A., Barcina, M. J., Servidei, S., Panico, A., Frontali, M., and Mariotti, C. (2010). Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J. Neurol. Sci. 291, 30–36.
Mezghrani, A., Monteil, A., Watschinger, K., Sinnegger-Brauns, M. J., Barrere, C., Bourinet, E., Nargeot, J., Striessnig, J., and Lory, P. (2008). A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J. Neurosci. 28, 4501–4511.
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, S. M., Lamerdin, J. E., Mohrenweiser, H. W., Bulman, D. E., Ferrari, M., Haan, J., Lindhout, D., van Ommen, G. J., Hofker, M. H., Ferrari, M. D., and Frants, R. R. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552.
Page, K. M., Heblich, F., Davies, A., Butcher, A. J., Leroy, J., Bertaso, F., Pratt, W. S., and Dolphin, A. C. (2004). Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J. Neurosci. 24, 5400–5409.
Page, K. M., Heblich, F., Margas, W., Pratt, W. S., Nieto-Rostro, M., Chaggar, K., Sandhu, K., Davies, A., and Dolphin, A. C. (2010). N terminus is key to the dominant negative suppression of Ca(V)2 calcium channels: implications for episodic ataxia type 2. J. Biol. Chem. 285, 835–844.
Riant, F., Lescoat, C., Vahedi, K., Kaphan, E., Toutain, A., Soisson, T., Wiener-Vacher, S. R., and Tournier-Lasserve, E. (2010). Identification of CACNA1A large deletions in four patients with episodic ataxia. Neurogenetics 11, 101–106.
Riant, F., Mourtada, R., Saugier-Veber, P., and Tournier-Lasserve, E. (2008). Large CACNA1A deletion in a family with episodic ataxia type 2. Arch. Neurol. 65, 817–820.
Schouten, J. P., McElgunn, C. J., Waaijer, R., Zwijnenburg, D., Diepvens, F., and Pals, G. (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 30, e57.
Strupp, M., Kalla, R., Claassen, J., Adrion, C., Mansmann, U., Klopstock, T., Freilinger, T., Neugebauer, H., Spiegel, R., Dichgans, M., Lehmann-Horn, F., Jurkat-Rott, K., Brandt, T., Jen, J. C., and Jahn, K. (2011). A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 77, 269–275.
Strupp, M., Kalla, R., Dichgans, M., Freilinger, T., Glasauer, S., and Brandt, T. (2004). Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology 62, 1623–1625.
Weisz, C. J., Raike, R. S., Soria-Jasso, L. E., and Hess, E. J. (2005). Potassium channel blockers inhibit the triggers of attacks in the calcium channel mouse mutant tottering. J. Neurosci. 25, 4141–4145.
Welch, E. M., Barton, E. R., Zhuo, J., Tomizawa, Y., Friesen, W. J., Trifillis, P., Paushkin, S., Patel, M., Trotta, C. R., Hwang, S., Wilde, R. G., Karp, G., Takasugi, J., Chen, G., Jones, S., Ren, H., Moon, Y. C., Corson, D., Turpoff, A. A., Campbell, J. A., Conn, M. M., Khan, A., Almstead, N. G., Hedrick, J., Mollin, A., Risher, N., Weetall, M., Yeh, S., Branstrom, A. A., Colacino, J. M., Babiak, J., Ju, W. D., Hirawat, S., Northcutt, V. J., Miller, L. L., Spatrick, P., He, F., Kawana, M., Feng, H., Jacobson, A., Peltz, S. W., and Sweeney, H. L. (2007). PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91.
Keywords: episodic ataxia, EA2, genomic rearrangement, CACNA1A, mutation
Citation: Wan J, Mamsa H, Johnston JL, Spriggs EL, Singer HS, Zee DS, Al-Bayati AR, Baloh RW, Jen JC and CINCH Investigators (2011) Large genomic deletions in CACNA1A cause episodic ataxia type 2. Front. Neur. 2:51. doi: 10.3389/fneur.2011.00051
Received: 29 April 2011; Paper pending published: 27 May 2011;
Accepted: 26 July 2011; Published online: 09 September 2011.
Edited by:
Ji Soo Kim, Seoul National University, South KoreaReviewed by:
Aasef G. Shaikh, Case Western Reserve University, USASeong-Hae Jeong, Chungnam National University Hospital, South Korea
Kwang-Dong Choi, Pusan National University Hospital, South Korea
Copyright: © 2011 Wan, Mamsa, Johnston, Spriggs, Singer, Zee, Al-Bayati, Baloh, Jen and Investigators. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jijun Wan, Department of Neurology, University of California, Los Angeles, 710 Westwood Plaza, Los Angeles, CA 90095-1769, USA. e-mail: jwan@mednet.ucla.edu