 Bat-Chen R. Avraham-Lubin1,2 Olga Dratviman-Storobinsky1 Shimrit Dadon-Bar El1
Bat-Chen R. Avraham-Lubin1,2 Olga Dratviman-Storobinsky1 Shimrit Dadon-Bar El1 Murat Hasanreisoglu3
Murat Hasanreisoglu3 Nitza Goldenberg-Cohen1,2,4*
Nitza Goldenberg-Cohen1,2,4*- 1 The Krieger Eye Research Laboratory, Felsenstein Medical Research Center, Petah Tiqwa, Israel
- 2 Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- 3 Department of Ophthalmology, Rabin Medical Center, Petah Tiqwa, Israel
- 4 Pediatric Unit, Schneider Children’s Medical Center of Israel, Petah Tiqwa, Israel
The study investigated the therapeutic effect of hyperbaric oxygen (HBO) on anterior ischemic optic neuropathy in a rodent model (rAION). rAION was laser-induced in one eye of 63 mice. The fellow (uninjured) eye served as an internal control. Thirty-three mice underwent two 90-min sessions of 100% oxygen (2 atm) treatment immediately following injury and one session daily thereafter for up to 14 days. The remaining mice were untreated. Retinas were harvested at different time points, and mRNA levels of various genes were analyzed by real-time polymerase chain reaction and histologic study. Untreated mice: day 1 post-rAION – SOD-1 (oxidative-stress-related) decreased to 82% of control (uninjured eye) levels (P < 0.05), Caspase-3 (proapoptotic) decreased to 89%, Bcl-xL mildly increased (117%; all NS); day 3 – HO-1 and endothelial nitric oxide synthase (eNOS; ischaemia-related) decreased to 74%, and Bcl-2-associated X protein, Caspase-3, and B-cell lymphoma 2 (Bcl-2; apoptotic) increased by 170, 120, and 111%, respectively (all NS); day 21 – HO-1 increased to 222% (NS) and eNOS decreased to 48% (P < 0.05). Treated mice: day 1 – SOD-1 and Caspase-3 remained unchanged, Bcl-2 and Bcl-xL mildly increased (112 and 126% respectively); day 3 – HO-1 and eNOS increased, apoptosis-related gene decreased; day 21 – SOD-1 decreased whereas eNOS increased (P < 0.05), and HO-1 increased to a lesser degree than without treatment. None of the oxygen-treated animals had retinal ganglion cell loss or a decrease in Thy-1 expression. In conclusion, HBO treatment after rAION induction influences the expression of apoptosis-related genes as well as oxidative-stress-induced and ischaemia-related genes and may exert a neuroprotective effect.
Introduction
Anterior ischemic optic neuropathy (AION) and pressure-induced glaucoma primarily affect the retinal ganglion cells (RGCs; Kaushik et al., 2003; Levin, 2003). Several mechanisms are known to mediate programmed death (apoptosis) of the RGCs, namely, deprivation of neurotrophic factors, alterations in gene expression, and production of reactive oxygen species. However, RGC death does not occur immediately after axonal injury, and a significant number of cells survive even several days, suggesting the involvement of pro-survival factors as well. This assumption is supported by studies showing that axotomy leads to molecular events that are potentially destructive to RGCs but, at the same time, induce changes that are potentially protective against cellular injury (Levin, 1999). Identifying the processes that underlie the regulation of cell survival may lead to innovative therapeutic methods of neuroprotection (Levin, 1999).
The mechanisms of RGC death have been examined in different models of optic nerve transection and crush (Bonfanti et al., 1996; Chaudhary et al., 1999; Kermer et al., 1999), acute retinal ischemia and reperfusion (Buchi, 1992; Adachi et al., 1996; Selles-Navarro et al., 1996), and chronic elevation of intraocular pressure (IOP; Morrison et al., 1997; Mittag et al., 2000). Recently, we characterized a rodent model of AION (rAION) wherein axonal stroke was induced by selective illumination of the optic disk in the presence of a photoactive substance (Bernstein et al., 2003; Kaushik et al., 2003). The retinal blood supply remained intact. Because the final common pathway of axonal damage is RGC death, this model made it possible to investigate the molecular changes that occur in the retina following damage to the optic nerve, with and without treatment.
The kinetics of the signaling pathways involved in apoptosis after retinal ischemia have recently been investigated (Produit-Zengaffinen et al., 2009). Extensive molecular studies revealed that members of the B-cell lymphoma 2 (Bcl-2) gene family play a critical role in the activation and control of ganglion cell death (Nickells et al., 2008) and that Bcl-2-related family members are differently regulated in the early and late phases after an ischemic retinal insult (Produit-Zengaffinen et al., 2009). Similar to primary RGC degeneration, secondary degeneration following optic nerve damage was characterized by activation of Caspase-3, downregulation of the pro-survival genes Bcl-2 and Bcl-xL, and upregulation of the proapoptotic genes Bcl-2-associated X protein (Bax) and Bad (Levkovitch-Verbin et al., 2010).
Oxygen is a critical substrate in the alleviation of hypoxia, anoxia, and ischemia. However, it also exerts deleterious effects during reperfusion of previously ischemic tissues. Adding to this paradox is a spate of pioneering works identifying hyperoxygenation (hyperoxia) and its metabolites as solely and purposefully yielding cellular and clinical benefits, particularly in ischemic reperfusion injuries (Yogaratnam et al., 2006). These effects were further improved by a novel technology, called hyperbaric oxygen (HBO), which involves the administration of oxygen at supra-atmospheric pressures in high concentrations (Yogaratnam et al., 2006). Although the concept of HBO was introduced in the mid-twentieth century, it is only in the past decade or so that its therapeutic potential has been put to advantage for purposes of cellular tolerance and neuroprotection.
Hyperbaric oxygen has been found to be a useful adjunct in several models of renal, skeletal, and myocardial infarction (Yogaratnam et al., 2006), and brain ischemic reperfusion injury (Hou et al., 2007; Singhal, 2007; Solmazgul et al., 2007; Vidigal et al., 2007; Eschenfelder et al., 2008), by improving oxygen perfusion to the ischemic tissues. It currently serves as a primary or adjuvant mode of treatment for various acute or chronic disorders. Eye diseases are still among the off-label applications of HBO despite cumulative evidence of its safety and efficacy in patients with retinal artery occlusion, cystoid macular edema secondary to retinal vein occlusion, scleral thinning and necrosis after pterygium surgery, orbital rhino-cerebral mucomycosis, non-healing corneal edema, and anterior segment ischemia (Oguz and Sobaci, 2008). Its possible benefit in some blinding diseases was noted as well. In a review of its uses in the eye, Butler et al. (2008) suggested various ocular disorders that may potentially benefit from HBO treatment, such as central retinal artery occlusion, radiation optic neuropathy, and selected cases of ischemic optic neuropathy. However, except in a few cases (Boschetti et al., 2006; Levy and Miller, 2006), findings for radiation-induced neuropathy have been disappointing (Danesh-Meyer, 2008). Beiran et al. (1995) successfully treated two patients with AION using HBO. The same group (Beiran et al., 2001) and others (Arnold and Levin, 2002) reported that when applied early, HBO improved visual outcome in patients with retinal arterial occlusion.
No methodologic investigations of HBO have been conducted to date in ischemic retinas, although earlier findings from our center highlighted the involvement of apoptosis in the cell loss consequent to central retinal artery occlusion (Goldenberg-Cohen et al., 2008). Prompted by the encouraging results in the literature, we sought to investigate the neuroprotective effect of HBO in a rAION.
Materials and Methods
Experimental Animals
Male wild-type C57/Bl6 mice aged 10–12 weeks were obtained for the study from Harlan Laboratories (Jerusalem). All mice were maintained and handled in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the NIH guidelines. All animal protocols were approved by the Animal Research Committee of Rabin Medical Center.
rAION Induction
Rodent model of AION was induced as previously described (Goldenberg-Cohen et al., 2005). In brief, 63 mice were anesthetized with ketamine 80 mg/kg and xylazine 4 mg/kg. A plastic custom-designed fundus corneal contact lens was used for direct in vivo visualization of the retina and optic nerve head. After intravenous administration of 0.03 mL of 2.5 mM rose bengal in phosphate-buffered saline, the optic nerve head was illuminated with argon green laser (532 nm) at the following specifications: 200 μm spot size, 50 mW power, constant duration (0.1 s). rAION was induced in the right eye of each animal; the fellow (uninjured) eye served as an internal control.
HBO Treatment
Immediately after rAION induction, 33 mice were placed in a HBO chamber specially designed for this experiment (Figure 1) and exposed to two sessions of 100% oxygen (2 atm) for 90 min. Thereafter treatment was repeated once daily for up to 14 days. The remaining 30 mice were untreated and served as the comparison group.

Figure 1. Hyperbaric oxygen chamber designed for mice. The mice can be seen through the windows during treatment.
Histological Examination
Sixteen mice were used for histological analysis. After euthanization at 1, 3, 7, or 21 days, both eyes were enucleated and embedded in paraffin or fixed in 4% formaldehyde, placed overnight in 30% sucrose dissolved in phosphate-buffered saline at 4°C, and embedded in OCT compound (Tissue-Tek, sakura finerek). Three sections (6 μm each) from each eye were mounted on slides and stained with hematoxylin and eosin for light microscopy analysis.
Apoptosis Assay
Longitudinal cross-sections from the paraffin-embedded eyes were cut 6 μm thick for in situ TdT-mediated dUTP nick end-labeling assay (TUNEL; Roche Diagnostics, Mannheim, Germany); results were analyzed with a fluorescence microscope (Fluoview X; Olympus, Tokyo, Japan) at 580 nm wavelength. The mean number of TUNEL-positive cells per slide was determined (three consecutive sections per slide, one slide for every 10 sections of 6 μm, total five slides per eye), with attention to the internal retinal layers. Findings were compared over time between the injured (right) and uninjured (left) eyes of the individual mice and between the injured eyes of the mice that underwent HBO treatment or were untreated.
Gene Expression
Forty-seven mice were used for molecular analysis. Immediately after euthanization at 1, 3, or 21 days following rAION induction, the retinas were frozen in liquid nitrogen. Total RNA was extracted with TRIzol™ reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA), followed by reverse-transcription into cDNA using random hexamers, according to the manufacturer’s protocol (Amersham Biosciences, UK), and MMLV-reverse transcriptase (Promega, Madison, WI, USA). cDNA was analyzed with real-time polymerase chain reaction (PCR) using the Sequence Detection System (ABI Prism 7900; Applied Biosystems, Inc. Foster City, CA, USA). The expression of the following genes was measured: superoxide dismutase-1 (SOD-1), heme-oxygenase-1 (HO-1), endothelial nitric oxide synthase (eNOS), vascular endothelial growth factor (VEGF), Bcl-2, Bax, Bcl-2-like protein 1 (Bcl-xL), cysteine–aspartic acid protease-3 (Caspase-3), and thymocyte cell surface antigen-1 (Thy-1); cDNA input levels were normalized against mouse beta actin (ACTB). Primer pairs of the oxidative-stress-, ischaemia-, and angiogenesis-related genes were kindly provided by Joseph A. Garcia, MD, PhD, Department of Internal Medicine, University of Texas Southern Medical Center, Dallas, TX, USA (Scortegagna et al., 2003; Ding et al., 2005). Reactions were performed in a 20-μL volume containing 4 μL cDNA, 1 μL each of forward and reverse primers, and buffer included in the Master Mix (SYBRR Green I; Applied Biosystems, Inc.). The primers are listed in Table 1. PCR cycling conditions were as follows: initial denaturation at 50°C for 2 min; followed by denaturation at 95°C for 2 min; followed by 40 cycles of denaturation at 95°C for 15 s; and annealing and extension at 60°C for 30 s. Duplicate transcriptase-based quantitative PCR (RT-QPCR) reactions were performed for each gene to minimize individual tube variability, and an average was taken for each time point. Standard curves were obtained using untreated mouse cDNA for each gene PCR assay. The results were quantified by a comparative Ct method, also known as the 2−ΔΔCt method (Giulietti et al., 2001), where:
ΔΔCt = ΔCt(sample) - ΔCt(reference).
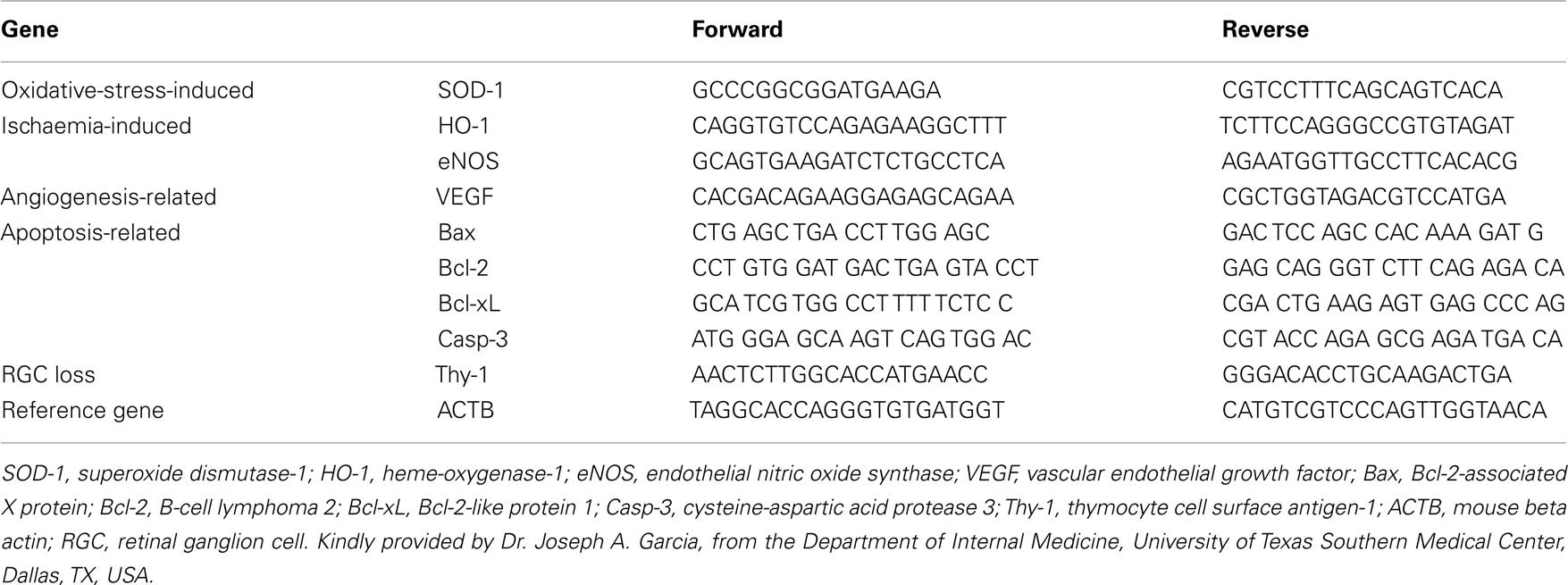
Table 1. Primer sequences used for RT-PCR analysis.
Statistical Analysis
The results were statistically analyzed with the SPSS (SPSS Inc., Chicago, IL, USA) for Windows, version 15.0.1. Mean gene expression was compared between groups by two-way ANOVA. A P-value of less than 0.05 was considered statistically significant.
Results
Histological Changes in the Retina
In the untreated rAION-injured eyes, light microscopy study revealed cell loss in the RGC layer already at 3 days following rAION induction (14% loss). Maximal loss of 27% was noted at 21 days (Figure 2).
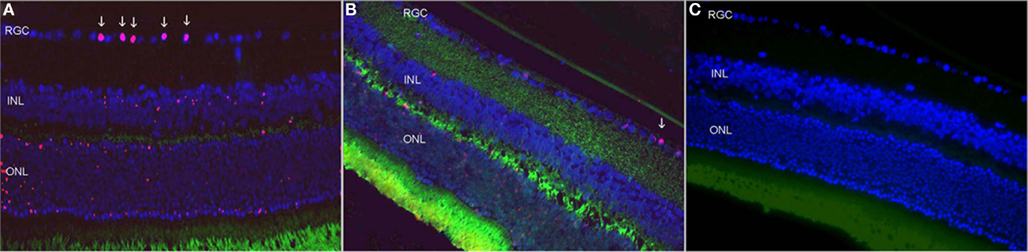
Figure 2. Retinal ganglion cells apoptosis 3 days after rAION induction. (A) Untreated eye after rAION induction. Note the TUNEL-positive cells (red) mainly in the RGC layer. (B) HBO-treated rAION. Note the minimal apoptosis compared to [(A) ×40]. (C) Uninjured eye; no apoptosis.
The HBO-treated rAION eyes showed only 8% cell loss at 21 days. The inner retinal layers were slightly edematous after treatment.
Apoptosis Assay
On TUNEL assay of the untreated rAION-injured eyes, apoptosis was noted in the ganglion cell layer. During the first 3 days after rAION induction, only a limited number of TUNEL-positive RGCs were found. Maximal apoptosis was detected at 7 days after rAION induction. When findings were calculated for the whole eye, we counted 180 positive RGCs at 3 days after rAION induction and an additional 600 positive RGCs at 7 days (Figure 3).
Figure 3. Retinal ganglion cells loss following rAION induction. (A) Untreated eye 21 days after rAION induction. Note the RGC loss (arrows). (B) HBO-treated eye. Note the absence of RGC loss (×40). (C) Uninjured eye (H&E).
In the HBO-treated eyes with rAION, apoptotic RGCs were less abundant. Maximal apoptosis was detected 3 days after rAION induction. When findings were calculated for the whole eye, we counted 97 TUNEL-positive cells in the RGC layer 3 days after rAION induction, and an additional 164 cells at 7 days.
Gene Expression
Thy-1
Untreated rAION-injured eyes. Thy-1-gene expression gradually decreased, and by 21 days after rAION induction, levels measured 73% of levels in the non-injured (left) eyes.
HBO-treated rAION-injured eyes. Thy-1 gene expression remained unchanged relative to levels in the uninjured eyes (NS) (Tables 2 and 3).
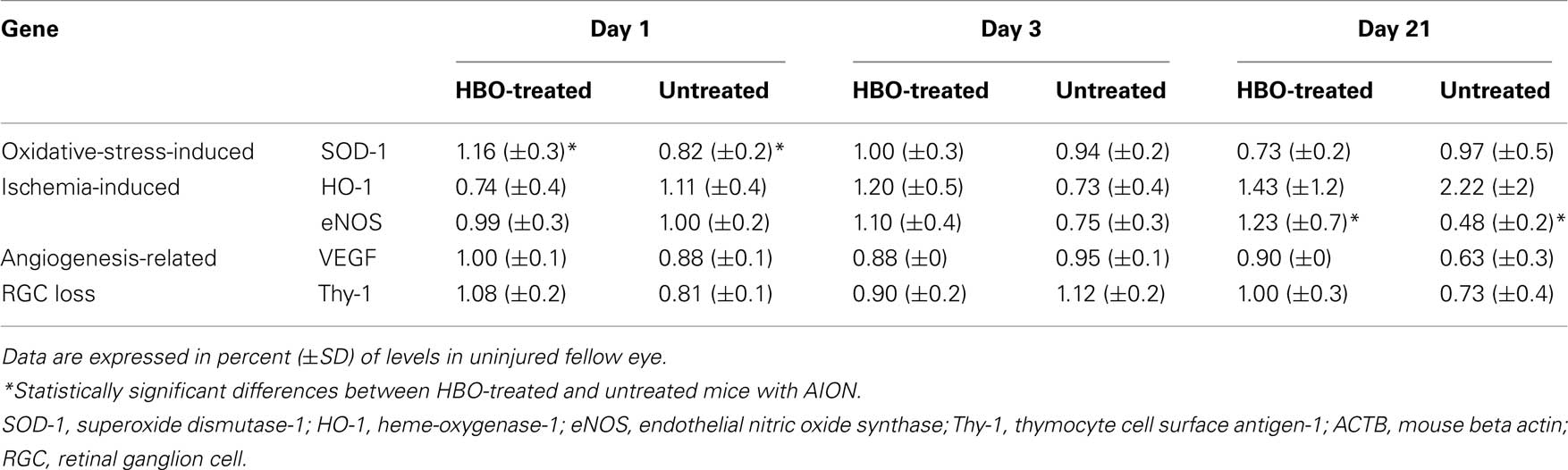
Table 2. Expression of oxidative-stress-induced, ischaemia-induced, and angiogenesis-related genes following rAION induction at various time points, with and without HBO treatment.
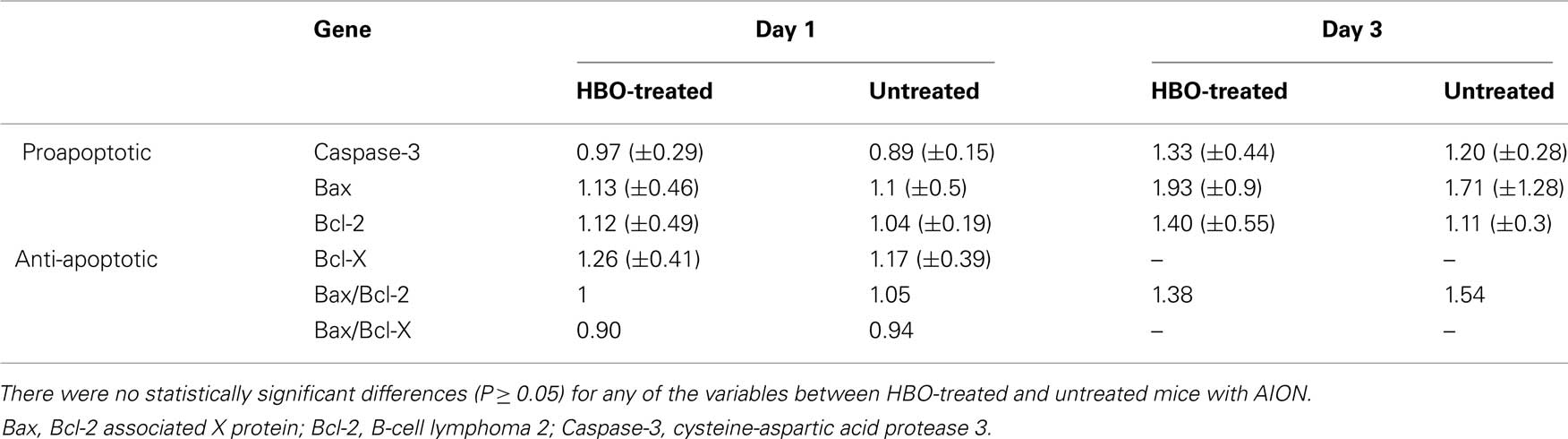
Table 3. Expression of apoptosis-related genes following rAION induction at various time points, with and without HBO treatment.
SOD-1
Untreated rAION-injured eyes. Expression of the oxidative-stress-induced gene, SOD-1, decreased to 82% of the level in the uninjured (left) eye on day 1 after injury (P < 0.05) and returned to the uninjured level by day 3.
HBO-treated rAION-injured eyes. SOD-1 gene expression increased to 116% of levels in the uninjured eyes on day 1 (P < 0.05), returned to the level in the uninjured eyes on day 3 (like in the untreated rAION eyes), and decreased to 73% of uninjured eye levels on day 21 (NS).
HO-1 and eNOS
Untreated rAION-injured eyes. Expression of the ischemia-related genes, HO-1 and eNOS, showed no change on day 1 and decreased to 73 and 75% of levels in the uninjured (left) eyes, respectively on day 3. On 21 days, HO-1 expression increased to 222% of levels in the uninjured eyes and eNOS levels decreased to 48% of uninjured eye levels (P = 0.01).
HBO-treated rAION-injured eyes. On day 1, HO-1 decreased to 74% of levels in the uninjured eye, whereas eNOS expression showed no change. On day 3, the expression of both genes increased, to 120 and 110% of levels in the uninjured eye, respectively (NS). On day 21, levels of HO-1 and eNOS increased to 143 and 123% (P = 0.04), respectively.
VEGF
Untreated rAION-injured eyes. VEGF decreased to 88% of levels in the uninjured eyes on day 1. On day 3, there was no change from the levels in the uninjured eyes. On day 21, levels dropped to 63% of uninjured eye levels (NS).
HBO-treated rAION-injured eyes. There was no significant change in VEGF levels at 1, 3, and 21 days after injury induction (100, 88, and 90%, respectively).
Caspase-3
Untreated rAION-injured eyes. Expression of the proapoptotic gene, Caspase-3, decreased on day 1–89% of levels in the uninjured eyes and increased on day 3–120%.
HBO-treated rAION-injured eyes. Caspase-3 levels remained at the level of the uninjured eyes on day 1 (97%) and increased to 133% of uninjured eye levels on day 3.
Bax
Untreated rAION-injured eyes. Expression of the proapoptotic Bax gene showed only a mild change (110%) on day 1 and a more significant change, to 171% of uninjured eye levels, on day 3.
HBO-treated rAION-injured eyes. Bax expression increased from 113% of uninjured eye levels on day 1 to 193% on day 3 (NS).
Bcl-2 and Bcl-X
Untreated rAION-injured eyes. Expression of the anti-apoptotic Bcl-2 and Bcl-X genes did not change on day 1. On day 3, Bcl-2 mildly increased, to 111% of uninjured eye levels (NS). The Bax/Bcl-2 ratio showed no change on day 1 and increased to 1.54 on day 3. The Bax/Bcl-x ratio showed no change on day 1 only.
HBO-treated rAION-injured eyes. Bcl-2 increased to 140% of levels in the uninjured eyes on day 3. The Bax/Bcl-2 ratio was 1.38.
Discussion
The present study shows that HBO treatment is effective in preserving the RGCs after the induction of rAION. We found that injured mice treated with HBO had less cell apoptosis than untreated mice, with non-significant differences in the expression of apoptosis-related genes. We characterized the changes in genes related to oxidative-stress, ischemia, and angiogenesis in the injured retinas of the untreated and HBO-treated mice.
The rAION is a rodent model of white matter stroke, resembling central nervous system stroke, with similar characteristics to AION in humans (Bernstein et al., 2003; Goldenberg-Cohen et al., 2005; Chen et al., 2008). Studies in rats have shown that oxygen, when applied early and in high doses, serves as a highly neuroprotective molecule in transient focal cerebral ischemia (Hou et al., 2007; Eschenfelder et al., 2008). These findings suggest that hyperoxia, acting via multiple direct and indirect mechanisms, could serve as a therapeutic strategy to salvage acutely ischemic central nervous system tissue. Of the available oxygen delivery methods, HBO appears to be the most potent (Singhal, 2007). Three previous clinical failures of HBO in patients with stroke (Singhal, 2007) were apparently attributable to methodologic technicalities, such as delayed time to therapy, inadequate sample size, and use of excessive chamber pressures (Singhal, 2007). Therefore, in the present study, using a model of rAION induction, we treated the mice three times within the first 24 h after AION induction and once per day thereafter for another 14 days. The chamber pressure was 2 atm, as used in the clinic. Each group consisted of at least seven mice. Furthermore, the previous clinical trials did not assess the long-term benefit of HBO in patients with tissue reperfusion. The development of the mouse model of rAION (Bernstein et al., 2003; Goldenberg-Cohen et al., 2005, 2009), and a more recent primate model (Chen et al., 2008), has made it possible to compare the histological effect of HBO and the accompanying changes in expression of the relevant genes over time. This has important clinical implications for the combination of hyperoxygenation with neuroimaging and thrombolysis in the treatment of victims of ischemic stroke (Singhal, 2007).
The neuroprotective effect of HBO in the brain has been explained molecularly by the prevention of cell apoptosis, which in turn preserves brain tissue and promotes neurologic functional recovery (Yin et al., 2003). Our TUNEL findings of a rescue effect of HBO on the RGC in a model of optic nerve stroke are supported by an earlier study of a rodent model of focal cerebral ischaemia wherein HBO treatment abolished DNA fragmentation and reduced the number of TUNEL-positive cells in the brain (Yin et al., 2003). This diminished the infarct area and led to an improvement in neurologic scores 7 days after reperfusion. Others noted that the neuroprotective effect of HBO was dose-dependent and that HBO yielded better results than normobaric oxygen in terms of both clinical outcome and infarct volume reduction (Eschenfelder et al., 2008). Similar neuroprotective effects of HBO in ischemia-reperfusion injury were reported in skeletal muscle (Vidigal et al., 2007) and intestine (Bertoletto et al., 2007). In the ischemic reperfusion model of skeletal muscle, HBO treatment, applied during different periods, prevented interstitial hemorrhage, development of a neutrophil infiltrate, and cellular necrosis (Vidigal et al., 2007). However, unlike our study and studies in the brain (Yin et al., 2003), there was no reduction in the apoptosis index in the muscle. In the study of rat intestinal mucosa, apoptosis was reduced when HBO was administered early, during intestinal ischemia, but not when it was administered later, during ischaemia-reperfusion (Bertoletto et al., 2007). In our study, mice underwent HBO treatment immediately after rAION induction, and then again daily for up to 14 days.
We examined several upstream apoptosis factors, Bcl-2, Bcl-x, and Bax, in the mitochondrial pathways, but none of the changes following HBO treatment were statistically significant. Following the induction of retinal ischemia, as measured by an increase in IOP, some earlier studies found no change in Bcl-2 expression and protein levels (Kaneda et al., 1999), whereas others reported an increase in Bcl-2 protein levels which was maintained for at least 24 h (Produit-Zengaffinen et al., 2009). In the present study, following rAION induction, Bcl-2, and Bcl-x levels increased mildly in the untreated mice and more in the HBO-treated mice, and further increased on day 3. Bax increased on day 3 as well, more in the HBO-treated mice. This finding for the proapoptotic Bax gene is in line with previous studies in a rat model of ischemic brain (Isenmann et al., 1997). However, histologically (TUNEL assay), apoptosis decreased on days 3 and 7 in the HBO-treated eyes (P < 0.05), with preservation of the RGC on day 21 (P < 0.05). By contrast, a study of a rat model of middle cerebral artery occlusion reported no change in Bax expression and an increase in Bcl-2 (Li et al., 2009). We did not expect changes in expression of Caspase-3 gene levels, as it is activated in the protein form, although it mildly increased in both groups on day 3 (NS).
The mechanism underlying HBO-induced reduction of ischemia/reperfusion injury may involve the suppression of neutrophil adhesion to the endothelial cell surface (Buras and Reenstra, 2007). Accordingly, prolonged inhibition of nitric oxide synthesis was found to lead to an oxidant-associated rise in neutrophic-endothelial adhesion (Niu et al., 1994). In the present study, HBO treatment reversed the decrease in eNOS expression seen after rAION induction in the untreated mice (Table 2). Similarly, in a previous study, nitrite and nitrate content increased by 2.2-fold in rats with cardiac infarct treated with normoxia plus hyperbaria or hyperoxia plus hyperbaria, and the NOS-3 protein level increased by 1.2-fold (Cabigas et al., 2006). The authors speculated that cardioprotection was directly dependent on NOS-mediated oxygen availability.
It is not clear how HBO affects the ischemia-induced pathophysiological responses, such as angiogenesis or skeletal muscle regeneration (Asano et al., 2007). In our study, the untreated rAION mice showed an immediate decrease in VEGF levels, which resolved by day 3 but reappeared on day 21, whereas the HBO-treated mice showed only a short, non-significant reduction in VEGF levels at all time points (Table 2). These data suggest that HBO exerts a neuroprotective effect by increasing VEGF levels. Early studies of optic nerve head damage in rodents reported transient leakage in the small vessels on fluorescein angiography (Goldenberg-Cohen et al., 2005). Following severe focal cerebral ischemia, HBO reduced blood–brain barrier (BBB) damage, and edema (Veltkamp et al., 2005). However, as there is no clear vasogenic edema in the optic nerve ischemic model, the HBO-treated mice did not show increased vascular permeability, and therefore, no significant change in VEGF levels. Other ischemic models of the hind limbs, but not the central nervous system, reported a slight increase in levels of VEGF mRNA (Asano et al., 2007), which was reversed with HBO treatment (Asano et al., 2007).
Heme-oxygenase-1 is known to exert a neuroprotective effect in ischemic conditions by regulating blood perfusion (Arai-Gaun et al., 2004). In our study, HO-1 expression increased already on day 3 after treatment. In the acute phase (day 1), HBO also prevented a decrease in SOD-1 (P < 0.05), which protects cells against oxidative-stress (Chan et al., 1994; Nishi et al., 2005). HBO-enhanced SOD-1 activity during particular periods of reperfusion has been reported in models of severe brain ischemia, indicating a potential benefit of treatment (Mrsic-Pelcic et al., 2004b). In a similar finding to ours, rats exposed to global cerebral ischemia for 20 min and treated with HBO within 6 h of reperfusion showed no change in SOD-1 expression (Mrsic-Pelcic et al., 2004a). However, unlike the present model, no treatment or delayed treatment was associated with the initial increase in SOD-1.
There are a few limitations to this study. HBO treatment was induced immediately after rAION induction, whereas in a clinical setting, treatment would likely be delayed at least by several hours if not a few days. Pilot clinical trials of neuroprotective agents have failed in part because of an inability to acquire subjects within 48 h of AION onset. Furthermore, even when given immediately after the acute event, we were not able to prove the relevance of HBO treatment in terms of molecular changes in AION. As opposed to the histological findings, most of the molecular differences in this study were not statistically significant. We have therefore described only trends, representing a possible effect of HBO treatment. However, it should be emphasized that only SOD-1 and eNOS levels were found to be significantly affected, at the early phase and the late phase, respectively, whereas none of the apoptosis-related genes showed a significant change.
Conclusion
Oxygenation has been widely applied in numerous experimental and clinical conditions (Barach, 2000). The main finding of this study is that in a rodent model of AION, RGC loss is reduced at 21 days after HBO treatment, suggesting that HBO may have a neuroprotective effect. Although our significant findings were limited to the histological but not the molecular effect, they may nevertheless have an important implications for the future development of treatments for acute ischemia-related visual loss in humans.
Acknowledgments
We thank Dr. Joseph A. Garcia, from the Department of Internal Medicine, University of Texas Southern Medical Center, Dallas, TX, USA, for kindly providing the primer sequences for this study; Dorit Karesh for the statistical analysis; and Gloria Ginzach for editing the manuscript. This study was partially supported by the Zanvyl and Isabelle Krieger Fund, Baltimore, MD, USA and the Eldor-Metzner Clinician-Scientist Award, Chief Scientist, Ministry of Health, Israel (NGC, grant number 3-3741). This work was presented in part at the Annual Meeting of the Association for Research in Vision and Ophthalmology, Fort Lauderdale, FL, USA, May 2008, and the Annual Meeting of the Israel Society for Eye and Vision Research, Neve-Ilan, Israel, March 2008. We thank Senior Technician Israel Shreger for designing and building the hyperbaric oxygen chamber.
References
Adachi, M., Takahashi, K., Nishikawa, M., Miki, H., and Uyama, M. (1996). High intraocular pressure-induced ischaemia and reperfusion injury in the optic nerve and retina in rats. Graefes Arch. Clin. Exp. Ophthalmol. 234, 445–451.
Arai-Gaun, S., Katai, N., Kikuchi, T., Kurokawa, T., Ohta, K., and Yoshimura, N. (2004). Heme oxygenase-1 induced in muller cells plays a protective role in retinal ischaemia-reperfusion injury in rats. Invest. Ophthalmol. Vis. Sci. 45, 4226–4232.
Arnold, A. C., and Levin, L. A. (2002). Treatment of ischemic optic neuropathy. Semin. Ophthalmol. 17, 39–46.
Asano, T., Kaneko, E., Shinozaki, S., Imai, Y., Shibayama, M., Chiba, T., Ai, M., Kawakami, A., Asaoka, H., Nakayama, T., Mano, Y., and Shimokado, K. (2007). Hyperbaric oxygen induces basic fibroblast growth factor and hepatocyte growth factor expression, and enhances blood perfusion and muscle regeneration in mouse ischemic hind limbs. Circ. J. 71, 405–411.
Barach, P. (2000). Management of the critically ill patient in the hyperbaric chamber. Int. Anesthesiol. Clin. 38, 153–166.
Beiran, I., Goldenberg, I., Adir, Y., Tamir, A., Shupak, A., and Miller, B. (2001). Early hyperbaric oxygen therapy for retinal artery occlusion. Eur. J. Ophthalmol. 11, 345–350.
Beiran, I., Rimon, I., Weiss, G., Pikkel, J., and Miller, B. (1995). Hyperbaric oxygenation therapy for ischemic optic neuropathy. Eur. J. Ophthalmol. 5, 285–286.
Bernstein, S. L., Guo, Y., Kelman, S. E., Flower, R. W., and Johnson, M. A. (2003). Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Invest. Ophthalmol. Vis. Sci. 44, 4153–4162.
Bertoletto, P. R., Fagundes, D. J., De Jesus Simoes, M., Oshima, C. T., De Souza Montero, E. F., Simões, R. S., and Fagundes, A. T. (2007). Effects of hyperbaric oxygen therapy on the rat intestinal mucosa apoptosis caused by ischaemia-reperfusion injury. Microsurgery 27, 224–227.
Bonfanti, L., Strettoi, E., Chierzi, S., Cenni, M. C., Liu, X. H., Martinou, J.-C., Maffei, L., and Rabacchi, S. A. (1996). Protection of retinal ganglion cells from natural and axotomy-induced cell death in neonatal transgenic mice overexpressing bcl-2. J. Neurosci. 16, 4186–4194.
Boschetti, M., De Lucchi, M., Giusti, M., Spena, C., Corallo, G., Goglia, U., Ceresola, E., Resmini, E., Vera, L., Minuto, F., and Ferone, D. (2006). Partial visual recovery from radiation-induced optic neuropathy after hyperbaric oxygen therapy in a patient with Cushing disease. Eur. J. Endocrinol. 154, 813–818.
Buchi, E. R. (1992). Cell death in the rat retina after a pressure-induced ischaemia-reperfusion insult: an electron microscopic study. I. Ganglion cell layer and inner nuclear layer. Exp. Eye Res. 55, 605–613.
Buras, J. A., and Reenstra, W. R. (2007). Endothelial-neutrophil interactions during ischaemia and reperfusion injury: basic mechanisms of hyperbaric oxygen. Neurol. Res. 29, 127–131.
Butler, F. K. Jr., Hagan, C., and Murphy-Lavoie, H. (2008). Hyperbaric oxygen therapy and the eye. Undersea Hyperb. Med. 35, 333–387.
Cabigas, B. P., Su, J., Hutchins, W., Shi, Y., Schaefer, R. B., Recinos, R. F., Nilakantan, V., Kindwall, E., Niezgoda, J. A., and Baker, J. E. (2006). Hyperoxic and hyperbaric-induced cardioprotection: role of nitric oxide s ynthase 3. Cardiovasc. Res. 72, 143–151.
Chan, P. H., Epstein, C. J., Kinouchi, H., Kamii, H., Imaizumi, S., Yang, G., Chen, S. F., Gafni, J., and Carlson, E. (1994). SOD-1 transgenic mice as a model for studies of neuroprotection in stroke and brain trauma. Ann. N. Y. Acad. Sci. 738, 93–103.
Chaudhary, P., Ahmed, F., Quebada, P., and Sharma, S. C. (1999). Caspase inhibitors block the retinal ganglion cell death following optic nerve transection. Brain Res. Mol. Brain Res. 67, 36–45.
Chen, C. S., Johnson, M. A., Flower, R. A., Slater, B. J., Miller, N. R., and Bernstein, S. L. (2008). A primate model of nonarteritic anterior ischemic optic neuropathy. Invest. Ophthalmol. Vis. Sci. 49, 2985–2992.
Ding, K., Scortegagna, M., Seaman, R., Birch, D. G., and Garcia, J. A. (2005). Retinal disease in mice lacking hypoxia-inducible transcription factor-2alpha. Invest. Ophthalmol. Vis. Sci. 46, 1010–1016.
Eschenfelder, C. C., Krug, R., Yusofi, A. F., Meyne, J. K., Herdegen, T., Koch, A., Zhao, Y., Carl, U. M., and Deuschl, G. (2008). Neuroprotection by oxygen in acute transient focal cerebral ischaemia is dose dependent and shows superiority of hyperbaric oxygenation. Cerebrovasc. Dis. 25, 193–201.
Giulietti, A., Overbergh, L., Valckx, D., Decallonne, B., Bouillon, R., and Mathieu, C. (2001). An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods 25, 386–401.
Goldenberg-Cohen, N., Dadon, S., Avraham, B. R., Kramer, M., Hasanreisoglu, M., Eldar, I., Weinberger, D., and Bahar, I. (2008). Molecular and histological changes following central retinal artery occlusion in a mouse model. Exp. Eye Res. 87, 327–333.
Goldenberg-Cohen, N., Dadon-Bar-El, S., Hasanreisoglu, M., Avraham-Lubin, B. C., Dratviman-Storobinsky, O., Cohen, Y., and Weinberger, D. (2009). Possible neuroprotective effect of brimonidine in a mouse model of ischemic optic neuropathy. Clin. Experiment. Ophthalmol. 37, 718–729.
Goldenberg-Cohen, N., Guo, Y., Margolis, F., Cohen, Y., Miller, N. R., and Bernstein, S. L. (2005). Oligodendrocyte dysfunction after induction of experimental anterior optic nerve ischaemia. Invest. Ophthalmol. Vis. Sci. 46, 2716–2725.
Hou, H., Grinberg, O., Williams, B., Grinberg, S., Yu, H., Alvarenga, D. L., Wallach, H., Buckey, J., and Swartz, H. M. (2007). The effect of oxygen therapy on brain damage and cerebral pO(2) in transient focal cerebral ischaemia in the rat. Physiol. Meas. 28, 963–976.
Isenmann, S., Wahl, C., Krajewski, S., Reed, J. C., and Bahr, M. (1997). Up-regulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur. J. Neurosci. 9, 1763–1772.
Kaneda, K., Kashii, S., Kurosawa, T., Kaneko, S., Akaike, A., Honda, Y., Minami, M., and Satoh, M. (1999). Apoptotic DNA fragmentation and upregulation of Bax induced by transient ischaemia of the rat retina. Brain Res. 815, 11–20.
Kaushik, S., Pandav, S. S., and Ram, J. (2003). Neuroprotection in glaucoma. J. Postgrad. Med. 49, 90–95.
Kermer, P., Klocker, N., Labes, M., Thomsen, S., Srinivasan, A., and Bahr, M. (1999). Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett. 453, 361–364.
Levin, L. A. (1999). Intrinsic survival mechanisms for retinal ganglion cells. Eur. J. Ophthalmol. 9(Suppl. 1), S12–S16.
Levin, L. A. (2003). Retinal ganglion cells and neuroprotection for glaucoma. Surv. Ophthalmol. 48(Suppl. 1), S21–S24.
Levkovitch-Verbin, H., Dardik, R., Vander, S., and Melamed, S. (2010). Mechanism of retinal ganglion cells death in secondary degeneration of the optic nerve. Exp. Eye Res. 91, 127–134.
Levy, R. L., and Miller, N. R. (2006). Hyperbaric oxygen therapy for radiation-induced optic neuropathy. Ann. Acad. Med. Singap. 35, 151–157.
Li, J. S., Zhang, W., Kang, Z. M., Ding, S. J., Liu, W. W., Zhang, J. H., Guan, Y. T., and Sun, X. J. (2009). Hyperbaric oxygen preconditioning reduces ischaemia-reperfusion injury by inhibition of apoptosis via mitochondrial pathway in rat brain. Neuroscience 159, 1309–1315.
Mittag, T. W., Danias, J., Pohorenec, G., Yuan, H. M., Burakgazi, E., Chalmers-Redman, R., Podos, S. M., and Tatton, W. G. (2000). Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Invest. Ophthalmol. Vis. Sci. 41, 3451–3459.
Morrison, J. C., Moore, C. G., Deppmeier, L. M., Gold, B. G., Meshul, C. K., and Johnson, E. C. (1997). A rat model of chronic pressure-induced optic nerve damage. Exp. Eye Res. 64, 85–96.
Mrsic-Pelcic, J., Pelcic, G., Peternel, S., Pilipovic, K., Simonic, A., and Zupan, G. (2004a). Effects of the hyperbaric oxygen treatment on the Na+, K+-ATPase and superoxide dismutase activities in the optic nerves of global cerebral ischaemia-exposed rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 667–676.
Mrsic-Pelcic, J., Pelcic, G., Vitezic, D., Antoncic, I., Filipovic, T., Simonic, A., and Zupan, G. (2004b). Hyperbaric oxygen treatment: the influence on the hippocampal superoxide dismutase and Na+, K+-ATPase activities in global cerebral ischaemia-exposed rats. Neurochem. Int. 44, 585–594.
Nickells, R. W., Semaan, S. J., and Schlamp, C. L. (2008). Involvement of the Bcl2 gene family in the signaling and control of retinal ganglion cell death. Prog. Brain Res. 173, 423–435.
Nishi, T., Maier, C. M., Hayashi, T., Saito, A., and Chan, P. H. (2005). Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischaemia. J. Cereb. Blood Flow Metab. 25, 1312–1324.
Niu, X. F., Smith, C. W., and Kubes, P. (1994). Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 74, 1133–1140.
Oguz, H., and Sobaci, G. (2008). The use of hyperbaric oxygen therapy in ophthalmology. Surv. Ophthalmol. 53, 112–110.
Produit-Zengaffinen, N., Pournaras, C. J., and Schorderet, D. F. (2009). Retinal ischaemia-induced apoptosis is associated with alteration in Bax and Bcl-x(L) expression rather than modifications in Bak and Bcl-2. Mol. Vis. 15, 2101–2110.
Scortegagna, M., Ding, K., Oktay, Y., Gaur, A., Thurmond, F., Yan, L. J., Marck, B. T., Matsumoto, A. M., Shelton, J. M., Richardson, J. A., Bennett, M. J., and Garcia, J. A. (2003). Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 35, 331–340.
Selles-Navarro, I., Villegas-Perez, M. P., Salvador-Silva, M., Ruiz-Gomez, J. M., and Vidal-Sanz, M. (1996). Retinal ganglion cell death after different transient periods of pressure-induced ischaemia and survival intervals. A quantitative in vivo study. Invest. Ophthalmol. Vis. Sci. 37, 2002–2014.
Solmazgul, E., Uzun, G., Cermik, H., Atasoyu, E. M., Aydinoz, S., and Yildiz, S. (2007). Hyperbaric oxygen therapy attenuates renal ischaemia/reperfusion injury in rats. Urol. Int. 78, 82–85.
Veltkamp, R., Siebing, D. A., Sun, L., Heiland, S., Bieber, K., Marti, H. H., Nagel, S., Schwab, S., and Schwaninger, M. (2005). Hyperbaric oxygen reduces blood–brain barrier damage and oedema after transient focal cerebral ischaemia. Stroke 36, 1679–1683.
Vidigal, J., Jose Fagundes, D., De Jesus Simoes, M., Oshima, C. T., Odashiro, A. N., Santos Simões, R., Negrini Fagundes, A. T., Taha, M. O., and De Souza Montero, E. F. (2007). Effect of different periods of hyperbaric oxygen on ischaemia-reperfusion injury of rat skeletal muscle. Microsurgery 27, 252–257.
Yin, D., Zhou, C., Kusaka, I., Calvert, J. W., Parent, A. D., Nanda, A., and Zhang, J. H. (2003). Inhibition of apoptosis by hyperbaric oxygen in a rat focal cerebral ischemic model. J. Cereb. Blood Flow Metab. 23, 855–864.
Keywords: hyperbaric oxygen therapy, neuroprotection, rAION, mouse model
Citation: Avraham-Lubin B-CR, Dratviman-Storobinsky O, El SD-B, Hasanreisoglu M and Goldenberg-Cohen N (2011) Neuroprotective effect of hyperbaric oxygen therapy on anterior ischemic optic neuropathy. Front. Neur. 2:23. doi: 10.3389/fneur.2011.00023
Received: 28 December 2010; Accepted: 30 March 2011;
Published online: 13 April 2011.
Edited by:
Valerie Purvin, Midwest Eye Institute, USAReviewed by:
Walter M. Jay, Loyola University Medical Center, USAPrem Subramanian, Johns Hopkins University School of Medicine, USA
Copyright: © 2011 Avraham-Lubin, Dratviman-Storobinsky, El, Hasanreisoglu and Goldenberg-Cohen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Nitza Goldenberg-Cohen, The Krieger Eye Research Laboratory, Felsenstein Medical Research Center, Beilinson Campus, Petah Tiqwa 49100, Israel. e-mail:bmNvaGVuMUBnbWFpbC5jb20=