

 Priyenka Thapa1
Priyenka Thapa1
- 1 Center for Translational Neuroscience, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR, USA
- 2 Department of Physical Therapy, University of Central Arkansas, Conway, AR, USA
The P13 potential is the rodent equivalent of the P50 potential, which is an evoked response recorded at the vertex (Vx) 50 ms following an auditory stimulus in humans. Both the P13 and P50 potentials are only present during waking and rapid eye movement (REM) sleep, and are considered to be measures of level of arousal. The source of the P13 and P50 potentials appears to be the pedunculopontine nucleus (PPN), a brainstem nucleus with indirect ascending projections to the cortex through the intralaminar thalamus, mediating arousal, and descending inhibitory projections to the caudal pontine reticular formation (CPRF), which mediates the auditory startle response (SR). We tested the hypothesis that intracranial microinjection (ICM) of glutamate (GLU) or GLU receptor agonists will increase the activity of PPN neurons, resulting in an increased P13 potential response, and decreased SR due to inhibitory projections from the PPN to the CPRF, in freely moving animals. Cannulae were inserted into the PPN to inject neuroactive agents, screws were inserted into the Vx in order to record the P13 potential, and electrodes inserted into the dorsal nuchal muscle to record electromyograms and SR amplitude. Our results showed that ICM of GLU into the PPN dose-dependently increased the amplitude of the P13 potential and decreased the amplitude of the SR. Similarly, ICM of N-methyl-d-aspartic acid or kainate into the PPN increased the amplitude of the P13 potential. These findings indicate that glutamatergic input to the PPN plays a role in arousal control in vivo, and changes in glutamatergic input, or excitability of PPN neurons, could be implicated in a number of neuropsychiatric disorders with the common symptoms of hyperarousal and REM sleep dysregulation.
Introduction
The P50 potential is a midlatency auditory evoked response recorded at the vertex (Vx) and occurs approximately 40–70 ms following an auditory click stimulus (Picton et al., 1974). The P50 potential is present during waking and rapid eye movement (REM) sleep, but not during slow-wave sleep (SWS; Erwin and Buchwald, 1986), and is blocked by the cholinergic receptor antagonist scopolamine, suggesting that may be mediated, at least in part, by cholinergic neurons (Buchwald et al., 1991). The P13 potential in the freely moving rat occurs 11–15 ms following a click stimulus, is only present during waking and REM sleep, is blocked by scopolamine (Miyazato et al., 1995), and appears to be the rodent equivalent to the human P50 potential (Miyazato et al., 1995, 1996, 1999a,c; Teneud et al., 2000). Furthermore, both the P13 and P50 potentials are considered to be measures of level of arousal (Garcia-Rill et al., 2002).
The source of the P13 potential has been localized to the pedunculopontine nucleus (PPN), which is the cholinergic arm of the reticular activating system (RAS). PPN neurons are most active during waking and REM sleep (Datta and Siwek, 2002), that is, when the P13 potential is manifested. Intracranial microinjection (ICM) of inhibitory neuroactive agents into the PPN decreased the amplitude of the P13 potential in a dose-dependent manner (Miyazato et al., 1999b; Teneud et al., 2000; Mamiya et al., 2005). Furthermore, decreasing arousal levels through the use of anesthetics, ethanol, or head injury decreased the amplitude of the P13 potential (Miyazato et al., 1999a).
The PPN receives glutamatergic input locally and from other mesopontine nuclei. ICM of glutamate (GLU) into the PPN of the freely moving rat increased waking and REM sleep (Datta et al., 2001b). However, ICM into this nucleus of the glutamate receptor agonist N-methyl-D-aspartic acid (NMDA) specifically increased waking (Datta et al., 2001a), while the glutamate receptor agonist kainate (KA) specifically increased REM sleep (Datta, 2002). On the contrary, ICM of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and some metabotropic receptor agonists into the PPN had no effect on sleep-wake control (Datta, 2002). The PPN contains separate populations of cholinergic, glutamatergic, and GABAergic neurons (Wang and Morales, 2009), and a majority of PPN neurons project to the intralaminar thalamus (ILT), including the parafascicular nucleus (Pf; Sofroniew et al., 1985; Hallanger et al., 1987). Descending projections of the PPN include efferents to postural and locomotion control systems, including the caudal pontine reticular formation (CPRF), which mediates the auditory startle response (SR; Davis, 1984; Semba and Fibiger, 1992; Koch and Schnitzler, 1997). Inhibitory cholinergic projections from the PPN to the CPRF serve to modulate the response of CPRF neurons following an auditory stimulus (Hoffman and Ison, 1980; Koch and Schnitzler, 1997).
We tested the hypothesis that ICM of GLU or its agonists will increase the activity of PPN neurons, resulting in increased arousal, as manifested by an increase in the amplitude of the P13 potential following an auditory stimulus. Furthermore, increased activity in the PPN will result in increased inhibition of the CPRF, and a decrease in the SR following an auditory stimulus.
Materials and Methods
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences, and were in agreement with the National Institutes of Health guidelines for the care and use of laboratory animals.
Surgical Preparation
Adult male Harlan Sprague Dawley rats (220–300 g, n = 32) were anesthetized with ketamine (60 mg/kg) and xylazine (10 mg/kg) IM. Males were used to avoid potential effects of the estrous cycle on the responses observed. Anesthesia was maintained such that the withdrawal reflex to paw pinch was absent, and body temperature was maintained at 37°C by a water circulating heating pad and rectal thermometer with automatic heat lamp. Animals were placed in a stereotaxic holder using hollow ear bars that protected the middle ear from damage. After reflection of the scalp and right temporalis muscle, coordinates were marked for electrode placement (Paxinos and Watson, 1998).
Holes were drilled in the skull with a manual micro pin vise, and two stainless steel screws (1/8″ long, size #0–80) for recording potentials were inserted epidurally at the Vx (midway between virtual lambda and bregma, 1.0 mm lateral from the midline, bilaterally). The Vx potential was recorded with reference to a screw inserted into the right frontal sinus. An additional screw was inserted in the left occipital bone and cemented to the skull for reinforcement of the structure. Two pairs of insulated wires with 1 mm exposed ends for recording electromyograms (EMGs) were inserted into the dorsal nuchal muscle. A ground wire was placed subcutaneously in the dorsal neck. Electrodes were connected by Teflon insulated wires to a nine-pin receptacle cemented to the skull with dental resin and acrylic mixture (Stoelting Co., IL, USA). EMG wires and the ground were connected to the same receptacle. Dental cement was then used to fasten the receptacle to the skull and to insulate and protect the connections.
In addition, a pair of 25 gage stainless steel hypodermic tubing guide cannulae (18 mm length) was implanted bilaterally. Holes just large enough to allow passage of the hypodermic tubing were drilled into the skull at coordinates 1.0 mm anterior of the interaural line and 1.9 mm lateral of the midline (Paxinos and Watson, 1998). Cannulae were inserted into the holes and a stereotaxic micropositioner was used to position the lower end of each cannula at 6.0 mm above the horizontal plane passing through the interaural line (1.0 mm dorsal to the PPN). This placement was used in order to minimize cell damage in the PPN. The tops of the cannulae extended about 1 cm above the top of the skull and were cemented in place so that approximately 0.5 cm of the cannulae extended above the cement. The end of each cannula was fitted with a plastic cap and stylette to close off the open end of the cannula and prevent internal blockage of the cannula.
The skin was closed with suture around the receptacle. After surgery, animals were given 0.2 mL Penicillin G (IM), and 5 mL saline (subcutaneous), to prevent infection and dehydration. Additionally, 0.2 mL Bupronex was administered for pain management. Animals were placed in an intensive care neonatal incubator maintained at 37°C during recovery. Rats were housed individually to prevent damage of the receptacle in a vivarium with a 12/12 light dark schedule, with lights on from 6 AM to 6 PM. Food and water were available ad libitum, and animals recovered for 7 days.
Recording Procedures
We used a sound-attenuating chamber (40 cm × 40 cm × 40 cm internal dimensions) with two speakers centered on opposite walls 10.6 cm above the floor to create a diffuse sound field within the chamber. Auditory stimuli, in the form of 100 μs rectangular pulse clicks, were presented at various decibel levels by a Grass Instruments Click-Tone module (Grass Instruments, Model S10ASCM, West Warwick, RI, USA) to the speakers. Measurements were made in the chamber using a sound level meter set for a 32- to 10,000-Hz range and in averaging mode. The click stimulus was produced at 99.9 Hz (fastest setting on the S10CTCM) in the recording chamber with the door open. Measurements were: (1) 103 dB sound pressure level (SPL) at the speaker on one side and 102 dB SPL on the other, (2) 84 dB SPL at the center of the chamber, and 82 dB SPL at the back corners, all at 4.25 in above the floor of the recording chamber. All stimulus levels were well above the threshold of the P13 potential that was measured as ∼70 dB SPL at the center of the chamber (Miyazato et al., 1995, 1996, 1999a,c; Teneud et al., 2000). The chamber was equipped with a fan that provided both continuous airflow and white noise background (60 dB SPL), and was lit by a 12-V, 20-W bulb. Wires from a commutator swivel (Dragonfly, Model SL-10, Ridgeley, WV, USA) at the top of the chamber were connected to a plug that was inserted into the nine-pin receptacle affixed to the animal’s skull.
Recordings began after a 7-day recovery period. Rats were accommodated to the chamber before and after surgery. All responses were recorded from unrestrained, alert rats before and after treatment with saline (control, CTL), GLU, NMDA, or KA. During some experiments, animals were pretreated with the NMDA receptor antagonist 2R-amino-5-phosphonovaleric acid (AP5) or the KA receptor antagonist γ-D-glutamylaminomethylsulfonic acid (GAMS) 15 min prior to ICM of receptor agonists into the PPN. Recordings were made from the Vx and neck muscles (EMG). Individual waveform acquisition began 50 ms before the first set of clicks to ensure adequate baseline acquisition and continued 655 ms after the first stimulus. Click stimuli were presented every 5 s until 32 potentials were acquired for averaging using SuperScope software (GW Instruments, Somerville, MA, USA).
Following a 15-min acclimation period to the chamber, three control recordings of evoked potentials and SRs began for each animal in each session. Experimental recordings began following injection at 3 min intervals for 15 min (to detect rapid changes at the beginning of each treatment), and then at 10 min intervals for 30 min (45 min total).
Potentials and SRs were amplified using Grass preamplifiers (Grass Instruments, Model P511K, West Warwick, RI, USA) with high impedance probes (Grass HIP5). Vx signals were amplified 20 K times and filtered at 1 Hz–0.1 kHz before being digitized using an InstruNet model 100 B (GW Instruments, Somerville, MA, USA) analog to digital converter. EMGs were amplified 5K times, filtered at 300 Hz–10 kHz and rectified before averaging to obtain the SR. Recordings were digitized at a rate of 10 kHz and stored. Animals were tested between 8 AM and 1 PM to control for possible time of day effects.
ICM Techniques
After pre-injection recordings were made, animals received bilateral injections using a 32-gage stainless steel tube (19 mm length) to extend 1.0 mm beyond the lower end of the guide cannulae (dorsal boundary of the PPN). Vehicle for all injected substances was 0.9% physiological saline. The injectors were connected to a 5-μL Hamilton (Hamilton Comp., Reno, NV, USA) syringe by a 10-cm flexible polyethylene (PE) tube (0.38 mm ID). Before injection, the PE tubing was checked to ensure a firm seal around the syringe to prevent leakage from the tip of the PE tube. A small air bubble was introduced into the PE tubing to monitor the movement of fluid during injection. Concentrations of injected solutions were adjusted so that each injection was 0.1 μL.
Injections were delivered over 10–20 s while the animals were gently held. The junction of the tubing and the injector was also sealed and checked for leakage during the injection. The pressure from 0.1 μL injection was negligible and unlikely to disturb the seal of the PE tubing. The animal was returned to the recording chamber within 2 min of the beginning of the injections, and recordings began immediately.
All concentrations used were based on previous reports of tolerable and non-excitotoxic ICM concentrations that did not cause excessive agitation or locomotion (Datta and Siwek, 1997; Datta et al., 2001a,b; Datta, 2002; Kobayashi et al., 2004). GLU was injected in concentrations of 0.06, 0.16, 0.30, and 0.48 nmol. AP5 and GAMS were tested at 0.16, 0.30, 0.48 nmol. NMDA was tested at 0.025, 0.05, 0.10, 0.30, and 1.00 mmol. KA was tested at 10, 20, and 30 μmol. Injections of saline, GLU, AP5, GAMS, NMDA, or KA were carried out in random order, and no animal was treated more than once/week (to obtain complete washout of treatment) or given more than five neuroactive injections overall (to ensure prevention of desensitization or additive neuronal excitotoxicity).
Threshold Determination
The threshold for the P13 potential has been described in rats (Miyazato et al., 1995, 1999b,c), and determined by delivering click stimuli at intensities of 50–103 dB. This procedure was used to confirm that the animals’ hearing was not affected by preparatory procedures, and to verify that all animals used exhibited similar sensitivity to the auditory stimuli. This determination was made once for each animal used in these experiments. Previous studies in our lab have shown that the average threshold for eliciting P13 potentials for SD rats is 70 ± 2 dB SPL, and animals that varied more than ±5 dB SPL were excluded from testing (Miyazato et al., 1995, 1996, 1999a,c; Teneud et al., 2000). All auditory stimuli for testing were delivered at 103 dB to ensure suprathreshold activation of the P13 potential.
Measurement Techniques
Measurement of the P13 potential amplitude was made from the largest positive deflection between 11 and 13 ms (latency) to its beginning (foot) or the peak of the preceding negative deflection (NB). Trials in which the foot or NB of the P13 potential was indiscernible or absent, measurement was made from baseline at 0 ms. The time to peak of the P13 potential is 4–5 ms (Miyazato et al., 1995). The maximum amplitude of the rectified and averaged EMG above prestimulus baseline was measured as the SR at its peak latency of 6–9 ms.
In experiments involving acute treatments, the two most similar of the three pretreatment control values were averaged and designated as 100%. Subsequent post-injection amplitudes were calculated as a percentage of the averaged control recording. Thus, each test response was normalized to its own controls.
To control for variability among control values between animals, we also employed a “difference” measurement technique for assessing the effects of treatment on the P13 potential response during and after treatment. For this measurement, the amplitude of the control P13 potential was subtracted from the amplitude of the post-treatment P13 potential for each time point. This allowed for the effect of the treatment to be expressed in terms of “direction” (positive or negative) and degree (actual difference in microvolts).
Data Analysis and Statistical Methods
In these studies, each animal was injected with either saline (CTL) or one of four drug concentrations. Individual animals were then recorded at specific and consecutive time points following injection. In addition, each animal was tested multiple times (once/week) and received a different injection treatment in each testing session (no more than five neuroactive compounds and six testing sessions total). Therefore, there were multiple sampling measures for each animal. Because of the experimental design, repeated measures analysis of variance (ANOVA) was an appropriate statistical model because it is a type of factorial experiment that is capable of examining and comparing response trends over time and adjusting for multiple sampling error.
The amplitude and habituation of the P13 potential and SR for each animal was analyzed. Data were tested for normality using the Kolmogorov–Smirnov test. Repeated measures ANOVA was used to determine main effects and interactions of drug condition and time, where drug condition and time post-injection were designated independent variables. The study evaluated influences of drug condition and time post-injection on the P13 potential and SR amplitude. For this purpose, the averages of the response to saline were compared to the averages of the response to drug at each time point with amplitude designated the repeated measure. Significant main effects were tested with post hoc within subject comparisons. The Newman–Keuls test was used for its strength of analysis of unequal number of samples between groups as well as repeated measures.
One-way ANOVA was applied to the experimental paradigm to compare the difference in means between CTL samples and experimental samples at each time point, as well as differences between time points, for each individual drug concentration group. For example, one-way ANOVA was used to compare response means of GLU 0.06 nmol CTL vs. GLU 0.06 nmol 5 min, GLU 0.06 nmol 10 min, etc. Two-way ANOVA was applied to the experimental paradigm to compare the difference in means between samples of a specific time point of one experimental group and the same time point of a separate experimental ground. For example, two-way ANOVA was used to compare response means of GLU 0.06 nmol 10 min post-injection vs. saline 10 min post-injection, etc.
Data sets that failed the normality test were analyzed using the Kruskal–Wallis test followed by Dunn’s Multiple Comparisons post-tests. Differences were considered significant at p-values ≤ 0.05. Depending on the complexity of the analysis required, GB-State®, GraphPad InStat3®, or SAS® software was used for data analysis.
Power Analysis
We based our calculations on eight animals per ground using previously published data. Four similar P13 potential studies (Miyazato et al., 1999a,c, 2000a,b) showed maximum differences of three to fourfold (average = 69%) between the control and experimental treatments with an average SE of 9.2%. Thus, for p = 0.05 we had a power of 0.95 to detect these robust differences (0.89 for p = 0.01) between control and experimental treatments, and only a 5% chance of committing a type II error in the proposed studies.
Histological Procedures
After the completion of experiments, the site of intracranial injections was verified with electrolytic lesion. For lesioned subjects, the animal was anesthetized, and 19 mm insulated electrode (except 1 mm at the tip) was inserted into each guide cannulae. A lesion at the injection site (1 mm beyond the tip of the cannula) was created by applying a 0.5-mA DC current to the electrode for 2 s using a Grass stimulator (Model S88 with constant current unit). After lesioning, the animal was perfused transcardially using heparinized saline and 4% paraformaldehyde in 1.0 M phosphate buffered solution. After fixation, the brain was carefully removed from the skull and immersed in 20% sucrose overnight. The brainstem was dissected free and frozen in medium for sectioning using a Vibratome Ultra Pro 5000 Cyrostat.
Nicotinamide Adenine Dinucleotide Phosphate Diaphorase
Twenty micromolar sections were cut in the sagittal plane and collected in phosphate buffer containing 1 mM MgCl and processed histochemically for reduced nicotinamide adenine dinucleotide phosphate (NADPH)-diaphorase (which selectively labels PPN neurons; Vincent et al., 1983). Histological reconstruction of the locations of the NADPH-diaphorase-positive neurons in relation to the lesion allowed the injection site to be located, and confirmed that injections had been made into the PPN as defined by the region of NADPH-diaphorase-positive neurons. Data from animals in which the injection site overlapped the NADPH-diaphorase-positive neurons were correlated with the electrophysiological data, and only these animals were included in analysis.
Results
Injection Sites, P13 Potential, and Startle Response
Figure 1A shows a representative Vx recording of the P13 potential and SR from a single animal following ICM of saline. Note the presence of the P13 potential (filled circle) and SR (filled star). Following ICM of 0.16 nmol GLU into the PPN (Figure 1B), the P13 amplitude increased (open circle), and the SR decreased (open star) compared to saline. Injection sites were similar to those previously reported in our lab (Miyazato et al., 1999b, 2000a; Teneud et al., 2000). Figure 1C shows a photomicrograph of a sagittal section through the PPN, which is labeled using NADPH-diaphorase histochemistry. The injection cannula protruded beyond the guide cannula, with its tip at the dorsal edge of the PPN (X).
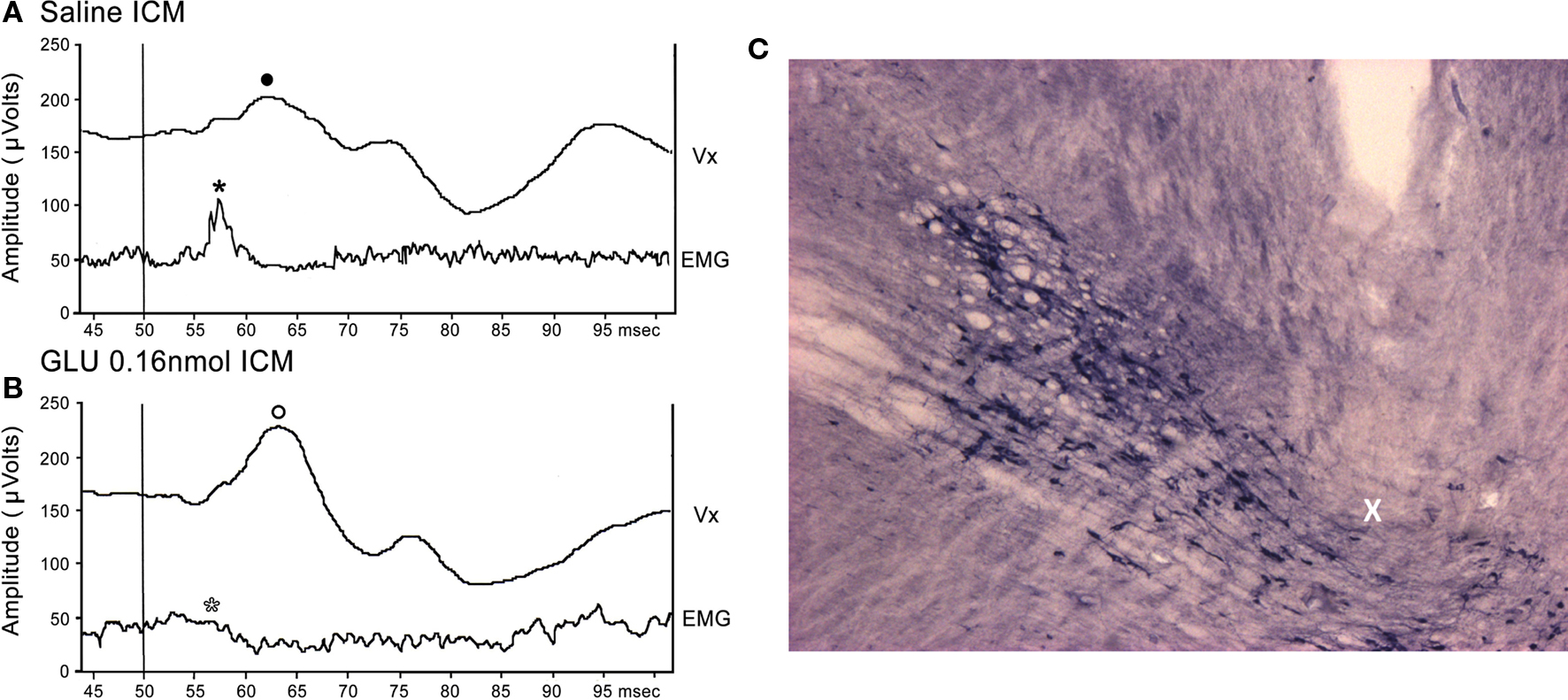
Figure 1. Representative P13 potential and SR recording: (A) Representative recordings of the P13 potential and SR from a single animal following ICM of saline. Note the presence of the P13 potential (filled circle) and SR (filled star). (B) Following ICM of 0.16 nmol GLU into the PPN (Figure 1B), the P13 potential amplitude increased (open circle) and SR amplitude decreased (open star) compared to saline. (C) Photomicrograph of a sagittal section through the PPN. The injection cannula protruded beyond the guide cannula, with its tip at the dorsal edge of the PPN (X).
Effects of GLU on the P13 Potential and SR
To test the effects of GLU ICM into the PPN on the amplitude of the P13 potential, saline and four concentrations of GLU were injected into the PPN and P13 potential amplitude was measured following auditory stimulation. Figure 2A shows the mean (calculated as the difference in amplitude at each time point from its own pre-injection CTL) and SE of the amplitude of the P13 potential following ICM of saline (n = 17), 0.06 nmol GLU (n = 10), 0.16 nmol GLU (n = 9), 0.30 nmol GLU (n = 8), and 0.48 nmol GLU (n = 8). Two-way repeated measures ANOVA with factors of time and drug concentration and post hoc testing comparing saline vs. each concentration of GLU revealed a statistically significant increase in the P13 potential amplitude compared to saline following 0.48 nmol GLU at 10 min (p < 0.05) and 15 min (p < 0.01, F = 3.13, df = 7) post-injection. Further statistical testing was performed to compare the CTL P13 potential amplitude (before ICM of GLU) to the amplitude following ICM at each time point (Figure 2B). One-way repeated measures ANOVA and post hoc testing comparing CTL vs. each concentration of GLU revealed a statistically significant increase in P13 potential amplitude between CTL and 0.16 nmol at 15 min (p < 0.05, F = 2.91, df = 8) post-injection; between CTL and 0.30 nmol at 5 and 45 min (p < 0.05) and 10–35 min (p < 0.01, F = 5.42, df = 7) post-injection; and between CTL and 0.48 nmol at 10, 15, and 25 min (p < 0.01, F = 9.64, df = 7) post-injection. ICM of saline did not result in any significant change in P13 potential amplitude at any time point (ns, F = 0.83, df = 16).
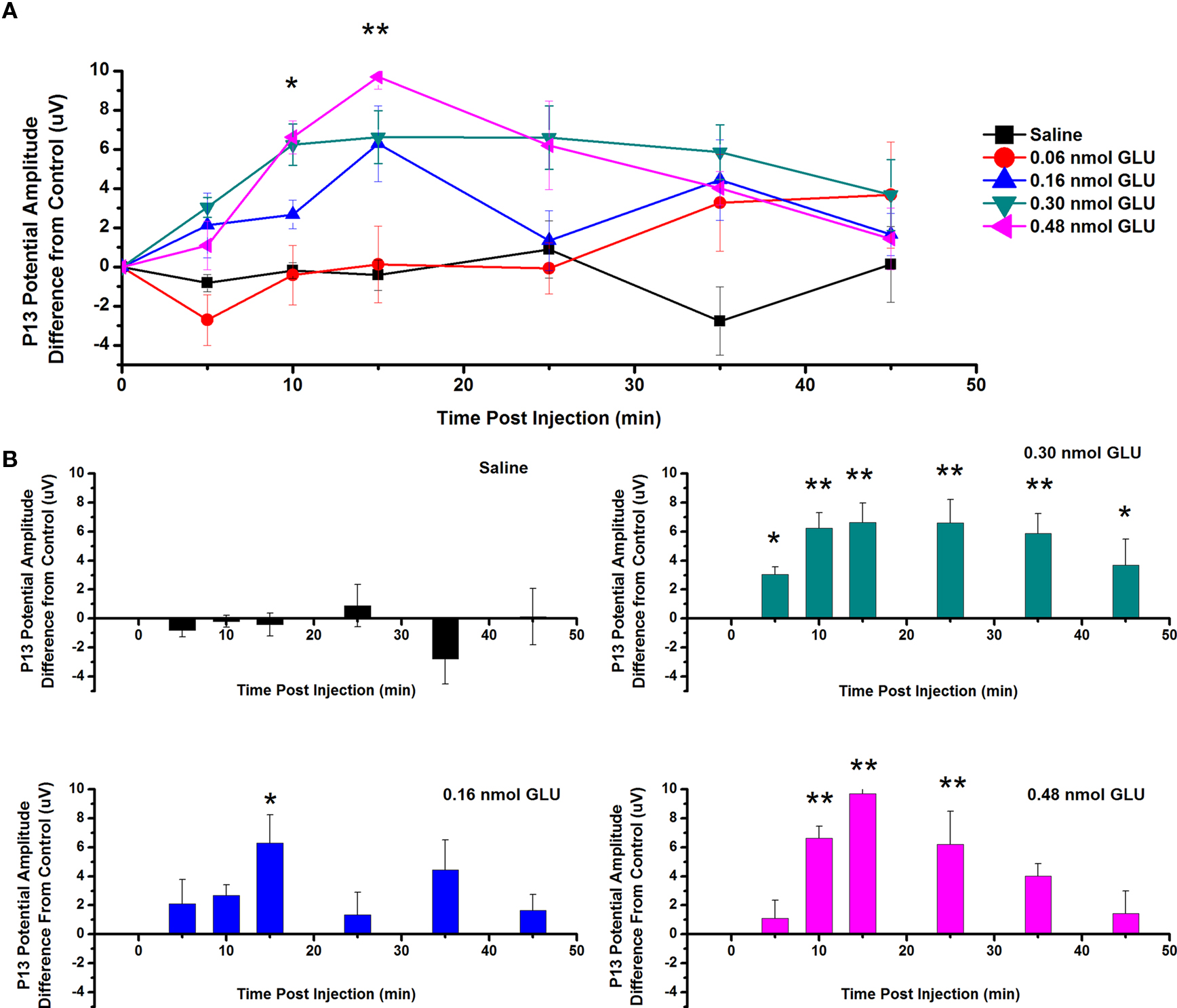
Figure 2. Intracranial microinjection of GLU into the PPN increased the amplitude of the P13 potential: (A) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL) and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 17), 0.06 nmol GLU (red, n = 10), 0.16 nmol GLU (blue, n = 9), 0.30 nmol GLU (green, n = 8), and 0.48 nmol GLU (pink, n = 8). Significance is denoted for two-way ANOVA between groups comparison. Note the significant increases in P13 potential amplitude compared to saline at 10 and 15 min after 0.48 nmol GLU (*p < 0.05, **p < 0.01). (B) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL), and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 17), 0.16 nmol GLU (blue, n = 9), 0.30 nmol GLU (green, n = 8), and 0.48 nmol GLU (pink, n = 8). Significance is denoted for one-way ANOVA for within subject comparison. Note the significant increases in P13 potential amplitude compared to CTL at 15 min after 0.16 nmol GLU; 5–45 min after 0.30 nmol GLU; and 10, 15, and 25 min after 0.48 nmol GLU (*p < 0.05, **p < 0.01).
The effect of GLU microinjection into the PPN on SR amplitude was also measured (Figure 3). Figure 3A shows the mean and SE of the amplitude of the SR following ICM of saline (n = 9), 0.06 nmol GLU (n = 7), 0.16 nmol GLU (n = 7), 0.30 nmol GLU (n = 7), and 0.48 nmol (n = 7). Two-way repeated measures ANOVA with factors of time and drug concentration and post hoc testing comparing saline vs. each concentration of GLU revealed a statistically significant decrease in SR amplitude following 0.16 nmol GLU at 5 min (p < 0.05, F = 2.25, df = 7) post-injection, and 0.30 nmol GLU at 5 min (p < 0.05, F = 2.23, df = 7) post-injection. Further statistical testing was performed to compare the CTL SR amplitude (before ICM of GLU) to the amplitude following ICM at each time point (Figure 3B). One-way repeated measures ANOVA and post hoc testing comparing CTL vs. each concentration of GLU revealed a statistically significant decrease in SR amplitude between CTL and 0.16 nmol GLU at 5 and 25 min (p < 0.05, F = 2.59, df = 7) post-injection and between CTL and 0.03 nmol GLU at 5, 15, and 25 min (p < 0.05, F = 2.34, df = 7) post-injection. ICM of saline did not result in any statistically significant change in SR amplitude.
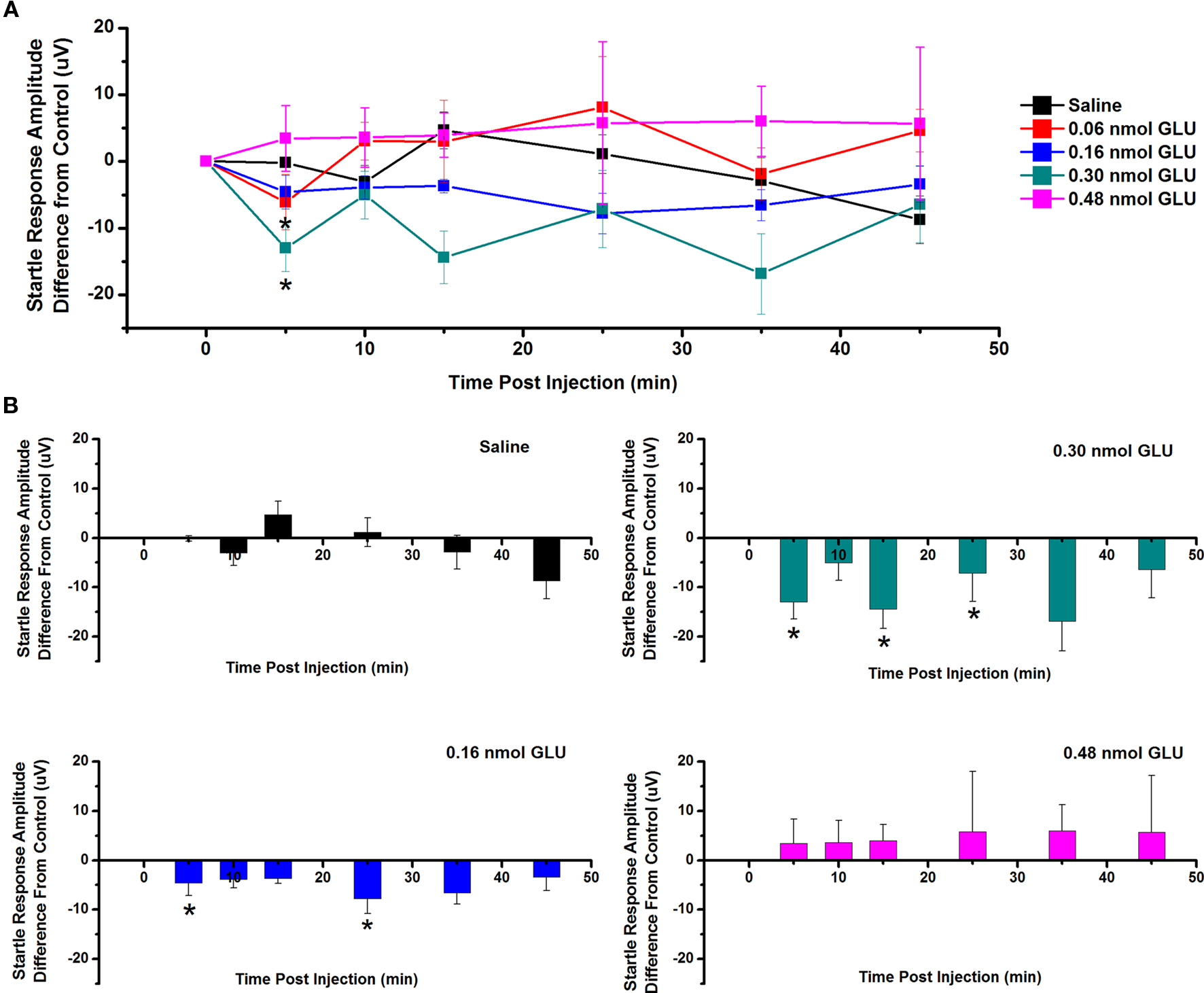
Figure 3. Intracranial microinjection of GLU into the PPN decreased the amplitude of the SR: (A) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL) and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 9), 0.06 nmol GLU (red, n = 7), 0.16 nmol GLU (blue, n = 7), 0.30 nmol GLU (green, n = 7), and 0.48 nmol GLU (pink, n = 7). Significance is for two-way ANOVA between groups comparison. Note the significant decreases in SR amplitude compared to saline at 5 min following 0.16 and 0.30 nmol GLU (*p < 0.05, **p < 0.01). (B) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL), and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 9), 0.16 nmol GLU (blue, n = 7), 0.30 nmol GLU (green, n = 7), and 0.48 nmol GLU (pink, n = 7). Significance is denoted for one-way ANOVA for within subject comparison. Note the significant decrease in SR amplitude compared to CTL at 5 and 25 min following 0.16 nmol GLU; and 5, 15, and 25 min following 0.30 nmol GLU (*p < 0.05, **p < 0.01).
Effects of NMDA on the P13 Potential and SR
To test the effects of ICM of NMDA into the PPN on P13 potential amplitude, saline and five concentrations of NMDA were injected into the PPN, and P13 response amplitude was measured following auditory stimuli. Figure 4A shows the mean and SE of the amplitude of the P13 potential following ICM of saline (n = 17), 0.025 mmol NMDA (n = 8), 0.05 mmol NMDA (n = 12), 0.10 mmol NMDA (n = 8), 0.30 mmol NMDA (n = 8), and 1.00 mmol NMDA (n = 8). Two-way repeated measures ANOVA with factors of time and drug concentration and post hoc testing comparing saline vs. each concentration of NMDA revealed a statistically significant increase in the P13 potential amplitude following 1.00 mmol NMDA at 10, 15, and 35 min (p < 0.01, F = 2.66, df = 7) post-injection. Further statistical testing was performed to compare the CTL P13 potential amplitude (before ICM of NMDA) to amplitude following ICM at each time point (Figure 4B). One-way repeated measures ANOVA and post hoc testing comparing CTL vs. each concentration of NMDA revealed a statistically significant increase in P13 potential amplitude between CTL and 0.10 mmol NMDA at 10 and 15 min (p < 0.05, F = 2.31, df = 7) post-injection; between CTL and 0.30 mmol NMDA at 10 and 15 min (p < 0.05, F = 3.31, df = 7) post-injection; and between CTL and 1.0 mmol NMDA at 10 and 15 min (p < 0.01, F = 4.82, df = 7) post-injection. ICM of saline did not result in any significant change in P13 potential amplitude at any time point (ns, F = 0.83, df = 16).
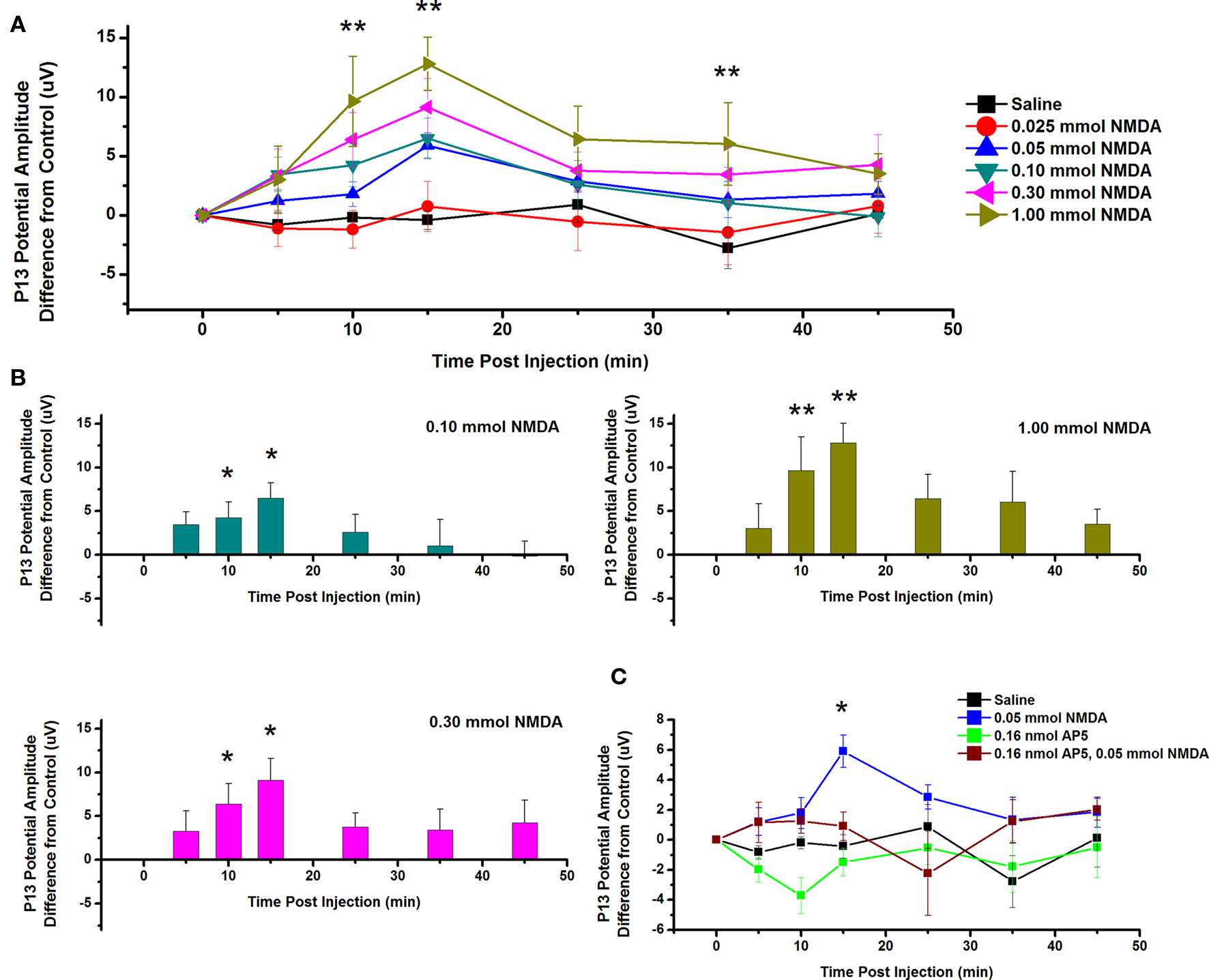
Figure 4. Intracranial microinjection of NMDA into the PPN increased the amplitude of the P13 potential: (A) Mean amplitude difference (μV) from CTL amplitude before injection (CTL) and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 17), 0.025 mmol NMDA (red, n = 8), 0.05 mmol NMDA (blue, n = 12), 0.10 mmol NMDA (green, n = 8), 0.30 mmol NMDA (pink, n = 8), and 1.00 mmol NMDA (yellow, n = 8). Significance is denoted for two-way ANOVA between groups comparison. Note the significant increases in P13 potential amplitude compared to saline at 10, 15, and 35 min following 1.00 mmol NMDA (*p < 0.05, **p < 0.01). (B) Mean amplitude difference (μV) from CTL amplitude before injection (CTL) and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 17), 0.10 mmol NMDA (green, n = 8), 0.30 mmol NMDA (pink, n = 8), and 1.00 mmol NMDA (yellow, n = 8). Significance is denoted for one-way ANOVA within subject comparison. Note the significant increase in P13 potential amplitude compared to CTL at 10 and 15 min following 0.10 mmol NMDA; 10 and 15 min following 0.30 mmol NMDA; and 10 and 15 min following 1.00 mmol NMDA (*p < 0.05, **p < 0.01). (C) Mean amplitude difference (μV) from CTL amplitude before injection (CTL) and following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 17), 0.05 mmol NMDA (blue, n = 12), 0.16 nmol AP5 (green, n = 6), and 0.05 NMDA after pretreatment with 0.16 nmol AP5 (red, n = 5). Significance is denoted for two-way ANOVA between groups comparison. Note the significant increase in amplitude of the P13 potential following 0.05 mmol NMDA compared to saline, and NMDA pretreated with AP5 (*p < 0.05, **p < 0.01).
Next, the NMDA receptor antagonist AP5 was injected before NMDA. Figure 4C shows the mean and SE of the amplitude of the P13 potential following ICM of saline (n = 17), 0.05 nmol NMDA (n = 12), 0.16 nmol AP5 (n = 6), and 0.05 mmol NMDA pretreated with 0.16 nmol AP5 (n = 5). Two-way ANOVA with factors of time and drug and post hoc testing revealed a statistically significant increase in P13 potential amplitude following 0.05 mmol NMDA at 15 min (p < 0.05, F = 2.57, df = 7) post-injection. Based on previous analysis, one-way repeated measures ANOVA and post hoc testing comparing CTL vs. 0.05 mmol NMDA revealed a statistically significant increase in P13 potential amplitude at 15 min (p < 0.05, F = 3.05, df = 11) post-injection. One-way ANOVA comparing CTL vs. AP5/NMDA revealed no statistically significant difference in P13 potential amplitude at any time point (ns, F = 1.77, df = 4). Therefore, ICM of NMDA increased the amplitude of the P13 potential, and this effect was blocked by pretreatment with the antagonist AP5.
The effect of ICM of NMDA into the PPN on SR amplitude was also measured. Figure 5A shows the mean and SE of the amplitude of the SR following saline (n = 9), 0.025 mmol NMDA (n = 5), 0.05 mmol NMDA (n = 7), 0.10 mmol NMDA (n = 6), 0.30 mmol NMDA (n = 6), and 1.00 mmol NMDA (n = 4). Two-way repeated measures ANOVA with factors of time and drug concentration comparing saline vs. each concentration of NMDA revealed no statistically significant decrease in SR amplitude between saline and 0.025 mmol NMDA (ns, F = 0.77, df = 7), 0.05 mmol NMDA (ns, F = 0.64, df = 7), 0.10 mmol NMDA (ns, F = 0.76, df = 7), 0.30 mmol NMDA (ns, F = 0.72, df = 7), or 1.0 mmol NMDA (ns, F = 1.63, df = 7) at any time point. Further statistical testing was performed to compare the CTL SR amplitude (before ICM of NMDA) to amplitude following ICM at each time point (Figure 5B). One-way repeated measures ANOVA comparing CTL vs. each concentration of NMDA revealed no statistically significant decrease in SR amplitude between CTL and 0.10 mmol NMDA (ns, F = 1.29, df = 6); between CTL and 0.30 mmol NMDA (ns, F = 0.91, df = 5); or between CTL and 1.00 mmol NMDA (ns, F = 0.79, df = 3). ICM of saline did not result in any significant change in SR amplitude (ns, F = 0.94, df = 16). Therefore, ICM of NMDA into the PPN does not result in any statistically significant change in the SR following an auditory stimulus. However, there does seem to be a decreasing trend in SR amplitude 10 min post-injection. This may not have reached significance due to the small amplitude of the change and the variability between subjects.
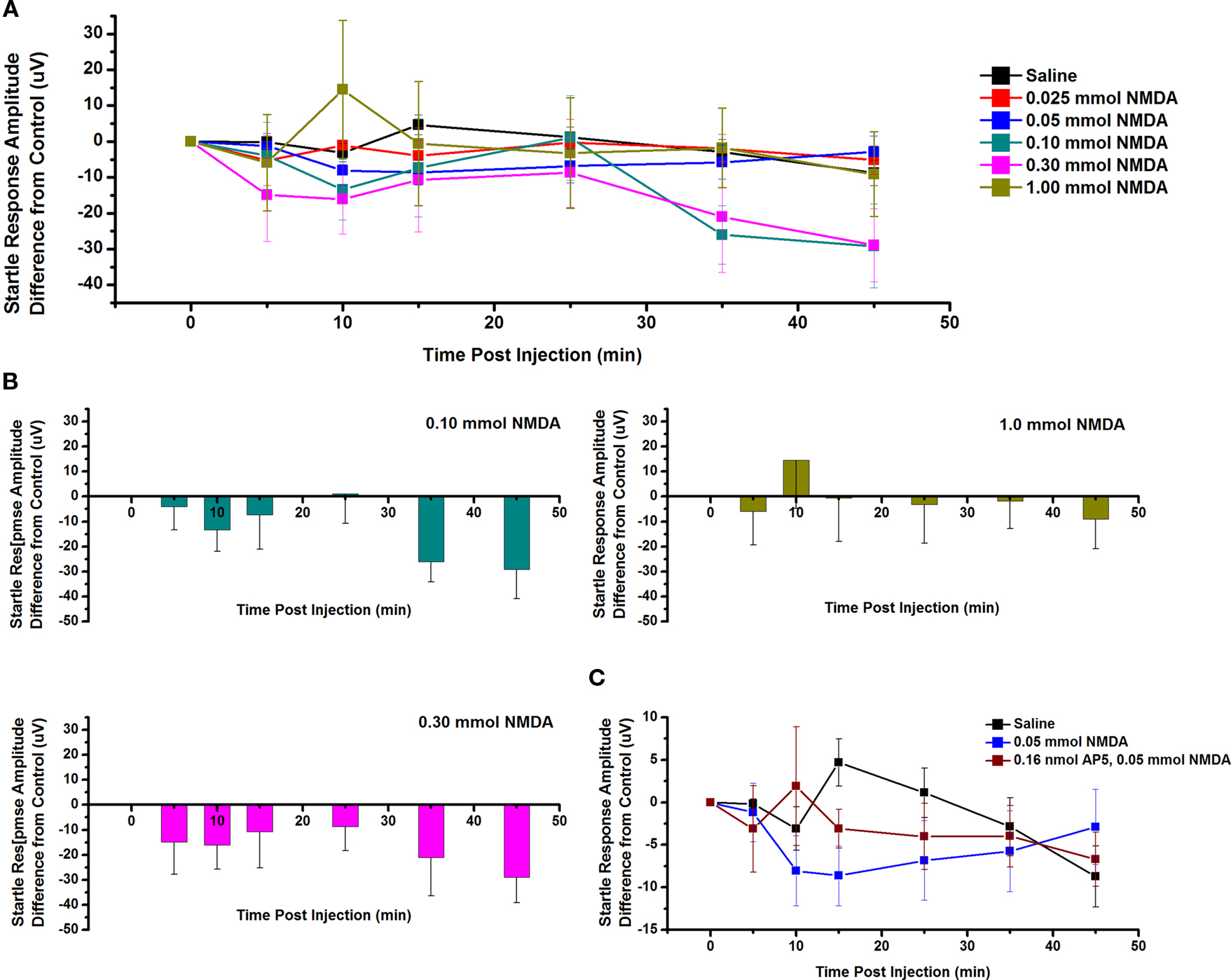
Figure 5. Intracranial microinjection of NMDA into the PPN increased the amplitude of the SR: (A) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL) and following injections at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 9), 0.025 mmol NMDA (red, n = 5), 0.05 mmol NMDA (blue, n = 12), 0.10 mmol NMDA (green, n = 6), 0.30 mmol NMDA (pink, n = 6), and 1.00 mmol NMDA (yellow, n = 9). Note the numerical decrease in SR amplitude following 0.05, 0.10, and 0.30 mmol NMDA at 10 min post-injection. (B) Note the decrease in SR amplitude compared to CTL following 0.10 mmol NMDA at 10 min and following 0.30 mmol NMDA at 5, 10, and 15 min post-injection. (C) Mean amplitude (μV) from CTL amplitude recorded before injection (CTL) and immediately following injection at 5, 10, 15, 25, 35, and 45 min. Averages after saline (black, n = 9), 0.05 mmol NMDA (blue, n = 12), and 0.05 mmol NMDA pretreated with 0.16 nmol AP5 (red, n = 4). Note the decrease in the SR amplitude following 0.05 mmol NMDA alone compared to saline and this decrease was partially blocked by pretreatment with 0.16 nmol AP5. However, this was not statistically significant.
Next, the NMDA receptor antagonist AP5 was added before NMDA. Figure 5C shows the SR amplitude following injection of saline (n = 9), 0.05 mmol NMDA (n = 7), and 0.05 mmol NMDA pretreated with 0.16 nmol AP5 (n = 4). Two-way ANOVA with factors of time and drug revealed no statistical difference at any time point between AP5/NMDA and saline or NMDA alone. Based on previous analysis, one-way repeated measures ANOVA comparing CTL vs. 0.05 mmol NMDA revealed no statistically significant decrease in SR amplitude. One-way ANOVA comparing CTL vs. AP5/NMDA also revealed no statistical difference at any time point (ns, F = 0.64, df = 3). However, it did appear as if NMDA alone decreased the amplitude of the SR and this was blocked by pretreatment with AP5 at 10, 15, and 35 min.
Effects of KA on the P13 Potential and SR
To test the effects of ICM of KA into the PPN on P13 potential amplitude, saline and four concentrations of KA were injected into the PPN and P13 response amplitude was measured following auditory stimuli. Figure 6A shows the mean and SE of the amplitude of the P13 potential following ICM of saline (n = 17), 10 μmol KA (n = 8), 15 μmol KA (n = 8), 20 μmol KA (n = 9), and 30 μmol KA (n = 4). Two-way repeated measures ANOVA with factors of time and drug concentration and post hoc testing comparing saline vs. each concentration of KA revealed a statistically significant increase in P13 potential amplitude following 15 μmol KA at 5 min (p < 0.05, F = 2.23, df = 7) post-injection; 20 μmol KA at 5 and 45 min (p < 0.05) and 25 and 35 min (p < 0.01, F = 2.73, df = 7) post-injection; 30 μmol at 5, 45, and 55 min (p < 0.05) and 10, 25, 35 min (p < 0.01, F = 3.04, df = 7) post-injection. Further statistical testing was performed to compare the CTL P13 potential amplitude (before ICM of KA) to the amplitude following ICM at each time point (Figure 6B). One-way repeated measures ANOVA and post hoc testing comparing CTL vs. each concentration of KA revealed statistically significant increases in P13 potential amplitude between CTL and 15 μmol KA at 5 min (p < 0.05, F = 2.68, df = 8) post-injection; between CTL and 20 μmol KA at 5, 35, and 45 min (p < 0.05) and 25 min (p < 0.01, F = 5.44, df = 8) post-injection; and between CTL and 30 μmol at 5 min (p < 0.05, F = 2.06, df = 3) post-injection. ICM of saline did not result in any significant change in P13 potential amplitude at any time point (ns, F = 0.83, df = 16).
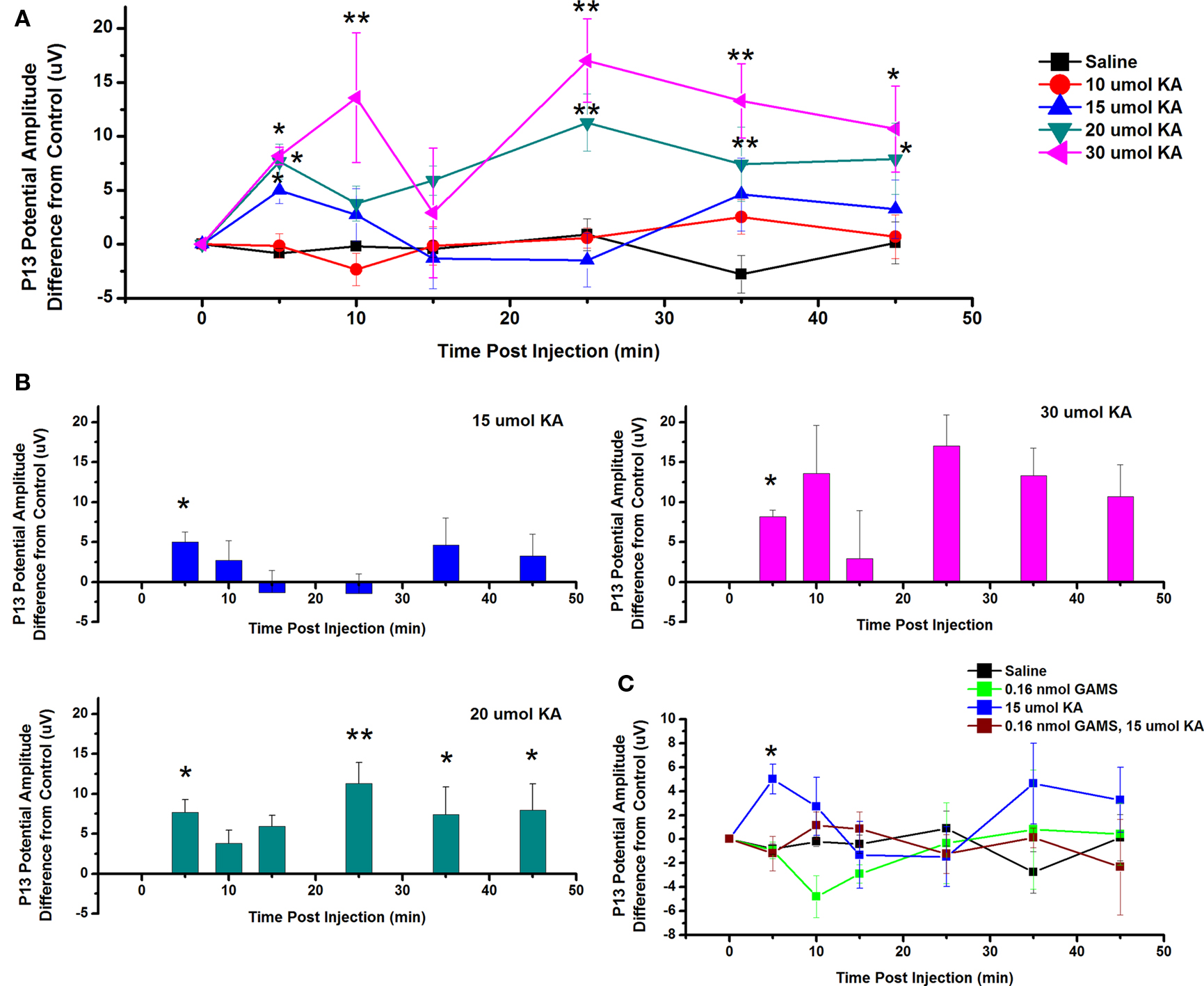
Figure 6. Intracranial microinjection of KA into the PPN increased the amplitude of the P13: (A) Mean amplitude difference (μV) from CTL recorded before injection (CTL) or 5, 10, 15, 25, 35, and 45 min following injection. Averages after saline (black, n = 17), 10 μmol KA (red, n = 8), 15 μmol KA (blue, n = 8), 20 μmol KA (green, n = 9), and 30 μmol KA (pink, n = 4). Significance is denoted for two-way ANOVA group comparison. Note the significant increases in P13 potential amplitude compared to saline at 5 min following 15, 20, and 30 μmol KA, 10 min following 30 μmol KA, 25 and 35 min following 20 and 30 μmol KA, and 35 min following 20 and 30 μmol KA (*p < 0.05, **p < 0.01). (B) Mean amplitude difference (μV) from CTL recorded before injection (CTL) or 5, 10, 15, 25, 35, and 45 min following injection. Averages after saline (black, n = 17), 15 μmol KA (blue, n = 8), 20 μmol KA (green, n = 9), and 30 μmol KA (pink, n = 4). Significance is denoted for one-way ANOVA within subject comparison. Note the significant increases in P13 potential amplitude compared to CTL at 5 min following 15 μmol KA; 5, 25, 35, and 45 min following 20 μmol KA; and at 5 min following 30 μmol KA (*p < 0.05, **p < 0.01). (C) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL) or 5, 10, 15, 25, 35, and 45 min following injection. Averages after saline (black, n = 17), 0.16 nmol GAMS (green, n = 4), 15 μmol KA (blue, n = 8), and 15 μmol KA pretreated with 0.016 nmol GAMS (red, n = 4). The P13 potential amplitude was significantly higher following 15 μmol KA alone than following ICM of saline or KA pretreated with GAMS at 5 min post-injection (*p < 0.05, **p < 0.01).
Next, the KA receptor antagonist GAMS was added before KA to determine if the increase in P13 potential amplitude could be blocked. Figure 6C shows the mean and SE of the amplitude of the P13 potential following ICM of saline (n = 17), 0.16 nmol GAMS (n = 4), 15 μmol KA (n = 8), and 15 μmol KA pretreated with 0.16 nmol GAMS (n = 4). Two-way ANOVA with factors of time and drug and post hoc testing revealed statistically significant increase in the P13 amplitude following KA compared to KA pretreated with GAMS at 5 min (p < 0.05, F = 2.12, df = 7) post-injection. Based on previous analysis, one-way repeated measures ANOVA and post hoc testing comparing CTL vs. 15 μmol KA revealed a statistically significant increase in P13 potential amplitude at 5 min (p < 0.05, F = 2.6828, df = 8). One-way ANOVA comparing CTL vs. GAMS/KA revealed no statistically significant difference in P13 potential amplitude at any time point. Therefore, ICM of KA increased the amplitude of the P13 potential and this effect is blocked by pretreatment with the antagonist GAMS.
The effect of ICM of KA into the PPN on SR amplitude was also measured. Figure 7A shows the mean and SE of the amplitude of the SR following saline (n = 9), 15 μmol KA (n = 2), and 20 μmol KA (n = 2). One-way repeated measures ANOVA comparing CTL vs. each concentration of KA revealed no statistically significant decrease in SR amplitude between CTL and any concentration of KA at any time point (Figure 7B). Figure 7C shows the SR amplitude following ICM of saline (n = 9), 15 μmol KA (n = 2), and 15 μmol KA pretreated with GAMS 0.16 nmol (n = 3). ICM of GAMS/KA did not result in any significant change in SR amplitude compared to CTL or KA alone at any time point, revealed by one and two-way ANOVA, respectively. However, there was clearly a trend toward decreased SR amplitude at some points, which appears to have been blocked by pretreatment with GAMS. Lack of statistical significance could be due to variability in the individual recordings.
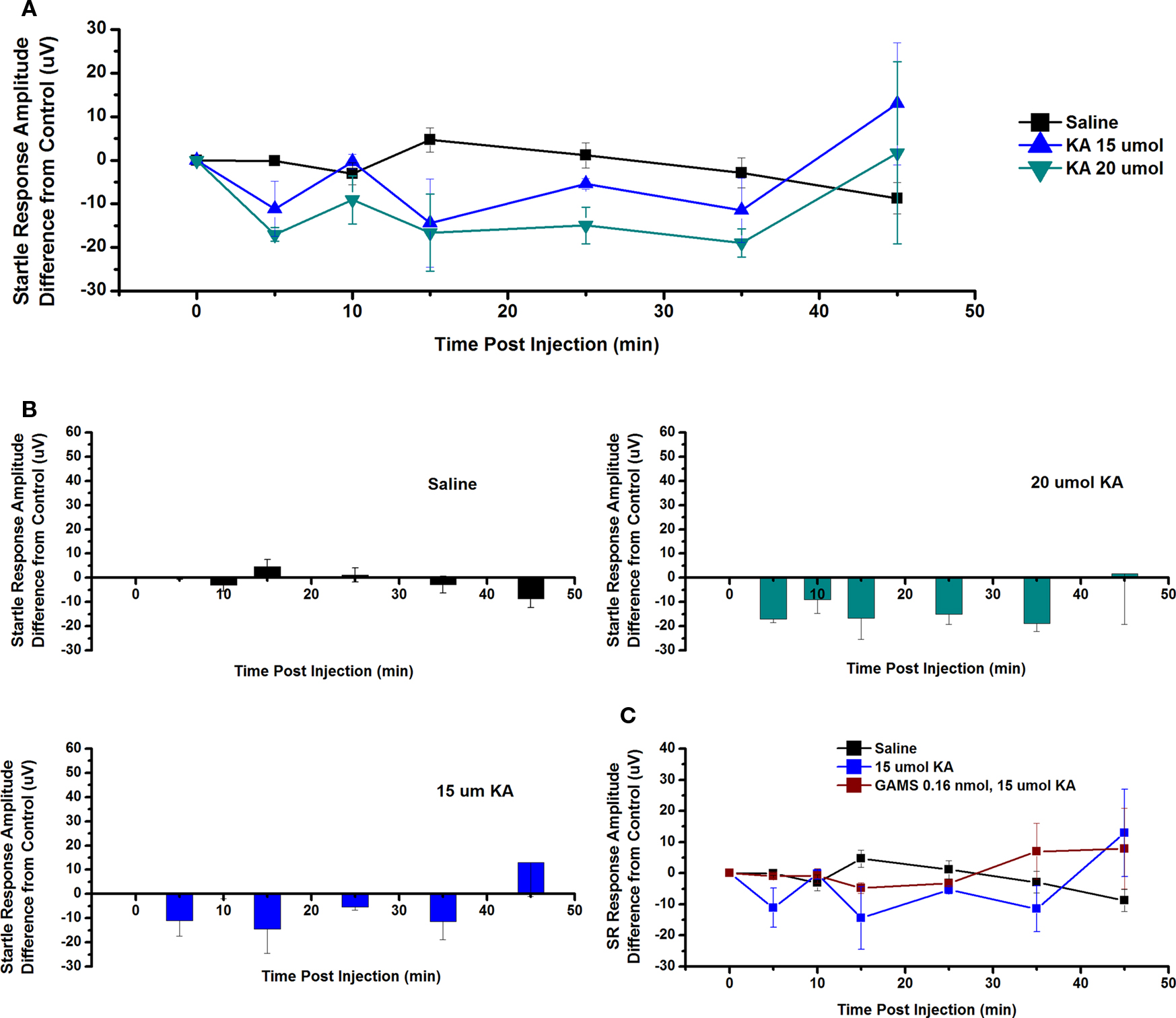
Figure 7. Intracranial microinjection of KA into the PPN decreased the SR amplitude, an Effect Blocked by Pretreatment with GAMS: (A) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL) and 5, 10, 15, 25, 35, and 45 min following injection. Averages after saline (black, n = 9), 15 μmol KA (blue, n = 2) and 20 μmol KA (green, n = 2). Note the numerical decreases in amplitude of the SR compared to saline after 5, 15, 25, and 35 min post-injection, but these responses were not significant. (B) Averages after saline (black, n = 9), 15 μmol KA (blue, n = 2) and 20 μmol KA (green, n = 2). Note the numerical decrease in SR amplitude at 5, 10, and 35 min following 15 μmol KA, and at 5, 10, 15, 25, and 35 min following 20 μmol KA. (C) Mean amplitude difference (μV) from CTL amplitude recorded before injection (CTL) and 5, 10, 15, 25, 35, and 45 min following injection. Averages after saline (black, n = 9), 15 μmol KA (blue, n = 2), and 15 μmol KA pretreated with 0.16 nmol GAMS (red, n = 3). Note the decrease in SR amplitude following 15 μmol KA and this was blocked by pretreatment with GAMS (results not significant).
Discussion
The aim of these studies was to determine how specific glutamate receptors in the PPN modulate arousal as manifested by the amplitude of the P13 potential in the freely moving animal. Major findings include (1) ICM of GLU into the PPN of the rat dose-dependently increased the amplitude of the P13 potential and decreased SR amplitude, and (2) ICM of the specific agonists NMDA and KA increased the amplitude of the P13 potential, which was blocked by pretreatment with the respective receptor antagonists. NMDA and KA also seemed to decrease the amplitude of the SR, but these results were not statistically significant.
The temporal relationship between the P13 potential and SR has been described previously (Miyazato et al., 1996). An early excitation of the SR occurs at 6–9 ms, followed by an inhibitory phase at 10–16 ms during the peak P13 response (Figure 1). In some animals, a late excitation of the SR at 18–22 ms was also observed. It has been proposed that the function of the SR is to activate descending reticulospinal systems in order to respond to novel input (Miyazato et al., 1996). The P13 potential inhibition of the SR was proposed to function to reset ongoing motor programs in order to mount a response (Miyazato et al., 1996).
GLU and Agonist Effects on the P13 Potential
The PPN sends cholinergic projections to the Pf and other nuclei in the ILT (Sofroniew et al., 1985; Hallanger et al., 1987). The P13 potential, recorded from the Vx, appears to reflect this output from the PPN (Miyazato et al., 1995, 1996, 1999a,c; Teneud et al., 2000). These results show that GLU dose-dependently increased the amplitude of the P13 potential. This increase was first induced by 0.16 nmol GLU and the peak effect was at 15 min post-injection. However, significant amplitude increases induced by higher concentrations of GLU were observed as early as 5 min post-injection. The effects of GLU on the amplitude of the P13 potential began to return to control levels by 25 min post-injection. Multiple GLU receptors may have contributed to the increase in P13 potential amplitude, and these receptors may be active at different time points. Thus the effects that were observed early (5 min) may be due to fast ionotropic receptors, whereas the effects seen later (15 min) may be due to slower acting ionotropic and perhaps metabotropic receptors.
Intracranial microinjection of the glutamate receptor agonists NMDA and KA into the PPN also dose-dependently increased the amplitude of the P13 potential. The increase induced by NMDA was first observed following 0.05 mmol NMDA and the peak effect was at 15 min post-injection. Furthermore, significant increases induced by higher concentrations of NMDA were observed as early as 10 min post-injection. Following KA, the increase in P13 potential amplitude was first induced following 15 μmol KA, and the peak effect was at 5 min post-injection and the effect waned by 15 min post-injection.
There are two aspects of the increase in P13 potential amplitude that are of particular interest. This first is that injection of KA at concentrations above 15 μmol produced significant increases in P13 potential amplitude as early as 5 min. This is supportive of the known functions of KA receptors to mediate fast excitatory postsynaptic currents via ionotropic channels (Hille, 2001).
The second observation of interest is that KA not only increased P13 potential amplitude rapidly, but also appeared to have a biphasic effect with significant increases at 25–45 min. That is, the initial increase occurred at 5 min, returned toward baseline by 10 and 15 min, and then increased again at 25 min for higher concentrations of KA at 20 and 30 μmol. Given that KA receptors desensitize quickly in response to KA application (Hille, 2001), the response is possibly due to secondary effects by metabotropic or NMDA receptors. The initial depolarization by KA receptor activation (increased amplitudes at 5 min) may have released the Mg block of voltage gated NMDA receptors, producing a secondary increase in P13 potential amplitude at 25 min. However, these later effects were not observed following NMDA alone, when the peak response was 15 min post-injection, and at 25 min the response was not significantly different from saline. Another possibility is metabotropic actions of KA receptors (Rodriguez-Moreno and Sihra, 2007), which are slower to activate and could account for the increase at 25 min.
GLU and Agonist Effects on SR
In addition to stimulating excitatory ascending projections from the PPN, GLU also stimulates inhibitory descending projections to the CPRF (Davis, 1984; Semba et al., 1990; Koch and Schnitzler, 1997), which modulate the SR. We previously reported that descending projections from the PPN tend to inhibit large neurons involved in mediating the inhibition of postural, usually extensor, muscles, but also exciting small neurons presumably involved in locomotion (Homma et al., 2002). This mechanism was proposed to participate in the atonia of paradoxical sleep, as well as in the modulation of the SR, in which inhibition of extensors (the SR is a flexor response) primes the system to “fight or flee.” ICM of GLU into the PPN resulted in a significant decrease in the amplitude of the SR as early as 5 min post-injection. It is interesting to note that ICM of GLU decreased the amplitude of the SR potential in a dose-dependent fashion (Figure 2) at concentrations of 0.06, 0.16, and 0.30 nmol. However, the highest dose of 0.48 nmol was not statistically different from saline. The PPN contains a number of GABAergic interneurons (Mena-Segovia et al., 2009; Wang and Morales, 2009). Perhaps an excess of GLU release into the PPN excites the numerous GABAergic interneurons present in the PPN, resulting in inhibition of output neurons, disinhibition of the neurons in the CPRF, and no change in the SR. Injection of the glutamate receptor agonists NMDA or KA into the PPN resulted in a general decrease in the amplitude of the SR as early as 5 min post-injection, although this decrease was not statistically significant. This suggests that the descending inhibitory effect by PPN may require activation of multiple types of glutamate receptors, including ionotropic and perhaps metabotropic receptors.
Disorders Involving Attentional and Postural Disturbances
Dysregulation of the RAS has been implicated in a number of psychiatric disorders characterized by an increase in vigilance and REM sleep drive (Garcia-Rill et al., 2003). The P50 potential (amplitude and/or habituation) and SR in humans has been described as potentiated in patients with Schizophrenia (decreased habituation), Post Traumatic Stress Disorder (decreased habituation), Parkinson’s (increased amplitude and decreased habituation), and Huntington’s (decreased amplitude and habituation) diseases ( Freedman et al., 1983; Buchwald et al., 1989; Gillette et al., 1997; Teo et al., 1997; Skinner et al., 1999; Arnfred et al., 2003; Uc et al., 2003), among other disorders. A population of intractable (but not outpatient) schizophrenic patients were found to have an increase in PPN cell number (Garcia-Rill et al., 1995). Therefore, it is reasonable to consider involvement of the RAS, specifically the PPN, in the disease process (Buchwald et al., 1989; Fein et al., 1994; Cancelli et al., 2006). Considering the above findings, perhaps agents that modulate glutamate release in the PPN, or control activity of PPN neurons, would have some therapeutic value for patients with disorders marked by changes in P50 potential amplitude and/or habituation.
Limitations and Technical Considerations
Glutamate activates metabotropic glutamate receptors (mGluR) and AMPA receptors as well as KA and NMDA receptors. The effects of metabotropic receptor activation are typically slower than those of ionotropic receptors due to their interactions with G-proteins and intracellular second messengers (Hille, 2001). It is possible that the longer lasting responses (>25 min) in P13 potential and SR amplitude produced by higher concentrations of GLU injections were due to mGluR activation in the PPN. However, data from other laboratories have reported non-significant contributions of mGluRs and AMPA receptors in the PPN to the generation and maintenance of waking and REM sleep (Datta and Siwek, 1997; Datta et al., 2001a,b; Datta, 2002). Future studies should, nevertheless, test the effects of mGluR or AMPA agonists and antagonists ICM into the PPN on P13 potential and SR amplitudes.
Summary and Conclusion
The overall goal of these studies was to determine how specific glutamate receptors within the PPN modulate arousal as manifested by P13 potential amplitude. Our findings indicate that activation of KA and NMDA receptors in the PPN resulted in an increase in the net activity of the cells and increased cholinergic output to both ascending thalamocortical projections and descending reticulospinal systems, as reflected by increased P13 potential amplitude and decreased SR amplitude. KA receptor activation in the PPN contributed to the fast, short duration increases in arousal and pre-attentional mechanisms and decreases in postural mechanisms. NMDA receptor activation in the PPN produced similar effects on arousal and postural modulation, however, their response was delayed and longer in duration compared to that of KA receptors. These findings provide insight to the excitatory glutamatergic regulation of the RAS and have implications for the changes in arousal that occur in a number of neuropsychiatric conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Supported by USPHS grants NS20246 and RR20146.
References
Arnfred, S. M., Chen, A. C., Glenthoj, B. Y., and Hemmingsen, R. P. (2003). Normal p50 gating in unmedicated schizophrenia outpatients. Am. J. Psychiatry 160, 2236–2238.
Buchwald, J. S., Erwin, R. J., Read, S., Van Lancker, D., and Cummings, J. L. (1989). Midlatency auditory evoked responses: differential abnormality of P1 in Alzheimer’s disease. Electroencephalogr. Clin. Neurophysiol. 74, 378–384.
Buchwald, J. S., Rubinstein, E. H., Schwafel, J., and Strandburg, R. J. (1991). Midlatency auditory evoked responses: differential effects of a cholinergic agonist and antagonist. Electroencephalogr. Clin. Neurophysiol. 80, 303–309.
Cancelli, I., Cadore, I. P., Merlino, G., Valentinis, L., Moratti, U., Bergonzi, P., Gigli, G. L., and Valente, M. (2006). Sensory gating deficit assessed by P50/Pb middle latency event related potential in Alzheimer’s disease. J. Clin. Neurophysiol. 23, 421–425.
Datta, S. (2002). Evidence that REM sleep is controlled by the activation of brain stem pedunculopontine tegmental kainate receptor. J. Neurophysiol. 87, 1790–1798.
Datta, S., Patterson, E. H., and Spoley, E. E. (2001a). Excitation of the pedunculopontine tegmental NMDA receptors induces wakefulness and cortical activation in the rat. J. Neurosci. Res. 66, 109–116.
Datta, S., Spoley, E. E., and Patterson, E. H. (2001b). Microinjection of glutamate into the pedunculopontine tegmentum induces REM sleep and wakefulness in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R752–R759.
Datta, S., and Siwek, D. F. (1997). Excitation of the brain stem pedunculopontine tegmentum cholinergic cells induces wakefulness and REM sleep. J. Neurophysiol. 77, 2975–2988.
Datta, S., and Siwek, D. F. (2002). Single cell activity patterns of pedunculopontine tegmentum neurons across the sleep–wake cycle in the freely moving rats. J. Neurosci. Res. 70, 611–621.
Davis, M. (1984). “The mammalian startle response,” in Neural Mechanisms of Startle Behavior, ed. R. C. Eaton (New York: Plenum Press), 287–351.
Erwin, R., and Buchwald, J. S. (1986). Midlatency auditory evoked responses: differential effects of sleep in the human. Electroencephalogr. Clin. Neurophysiol. 65, 383–392.
Fein, G., Biggins, C., and van Dyke, C. (1994). The auditory P50 response is normal in Alzheimer’s disease when measured via a paired click paradigm. Electroencephalogr. Clin. Neurophysiol. 92, 536–545.
Freedman, R., Adler, L. E., Waldo, M. C., Pachtman, E., and Franks, R. D. (1983). Neurophysiological evidence for a defect in inhibitory pathways in schizophrenia: comparison of medicated and drug-free patients. Biol. Psychiatry 18, 537–551.
Garcia-Rill, E., Biedermann, J. A., Chambers, T., Skinner, R. D., Mrak, R. E., Husain, M., and Karson, C. N. (1995). Mesopontine neurons in schizophrenia. Neuroscience 66, 321–335.
Garcia-Rill, E., Kobayashi, T., and Good, C. (2003). The developmental decrease in REM sleep. Thalamus Relat. Syst. 2, 115–131.
Garcia-Rill, E., Skinner, R. D., Clothier, J., Dornhoffer, J., Uc, E., Fann, A., and Mamiya, N. (2002). The sleep state- dependent midlatency auditory evoked P50 potential in various disorders. Thalamus Relat. Syst. 2, 9–19.
Gillette, G. M., Skinner, R. D., Rasco, L. M., Fielstein, E. M., Davis, D. H., Pawelak, J. E., Freeman, T. W., Karson, C. N., Boop, F. A., and Garcia-Rill, E. (1997). Combat veterans with posttraumatic stress disorder exhibit decreased habituation of the P1 midlatency auditory evoked potential. Life Sci. 61, 1421–1434.
Hallanger, A. E., Levey, A. I., Lee, H. J., Rye, D. B., and Wainer, B. H. (1987). The origins of cholinergic and other subcortical afferents to the thalamus in the rat. J. Comp. Neurol. 262, 105–124.
Hoffman, H. S., and Ison, J. R. (1980). Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol. Rev. 87, 175–189.
Homma, Y., Skinner, R. D., and Garcia-Rill, E. (2002). Effects of pedunculopontine nucleus (PPN) stimulation on pontine reticular formation neurons in vitro. J. Neurophysiol. 87, 3033–3047.
Kobayashi, T., Skinner, R. D., and Garcia-Rill, E. (2004). Developmental decrease in REM sleep: the shift to kainate receptor regulation. Thalamus Relat. Syst. 2, 315–324.
Koch, M., and Schnitzler, H. U. (1997). The acoustic startle response in rats – circuits mediating evocation, inhibition and potentiation. Behav. Brain Res. 89, 35–49.
Mamiya, N., Buchanan, R., Wallace, T., Skinner, R. D., and Garcia-Rill, E. (2005). Nicotine suppresses the P13 auditory evoked potential by acting on the pedunculopontine nucleus in the rat. Exp. Brain Res. 164, 109–119.
Mena-Segovia, J., Micklem, B. R., Nair-Roberts, R. G., Ungless, M. A., and Bolam, J. P. (2009). GABAergic neuron distribution in the pedunculopontine nucleus defines functional subterritories. J. Comp. Neurol. 515, 397–408.
Miyazato, H., Skinner, R. D., Cobb, M., Andersen, B., and Garcia-Rill, E. (1999a). Midlatency auditory-evoked potentials in the rat: effects of interventions that modulate arousal. Brain Res. Bull. 48, 545–553.
Miyazato, H., Skinner, R. D., and Garcia-Rill, E. (1999b). Neurochemical modulation of the P13 midlatency auditory evoked potential in the rat. Neuroscience 92, 911–920.
Miyazato, H., Skinner, R. D., and Garcia-Rill, E. (1999c). Sensory gating of the P13 midlatency auditory evoked potential and the startle response in the rat. Brain Res. 822, 60–71.
Miyazato, H., Skinner, R. D., Crews, T., Williams, K., and Garcia-Rill, E. (2000a). Serotonergic modulation of the P13 midlatency auditory evoked potential in the rat. Brain Res. Bull. 51, 387–391.
Miyazato, H., Skinner, R. D., and Garcia-Rill, E. (2000b). Locus coeruleus involvement in the effects of immobilization stress on the p13 midlatency auditory evoked potential in the rat. Prog. Neuropsychopharmacol. Biol. Psychiatry 24, 1177–1201.
Miyazato, H., Skinner, R. D., Reese, N. B., Boop, F. A., and Garcia-Rill, E. (1995). A middle-latency auditory-evoked potential in the rat. Brain Res. Bull. 37, 247–255.
Miyazato, H., Skinner, R. D., Reese, N. B., Mukawa, J., and Garcia-Rill, E. (1996). Midlatency auditory evoked potentials and the startle response in the rat. Neuroscience 75, 289–300.
Picton, T. W., Hillyard, S. A., Krausz, H. I., and Galambos, R. (1974). Human auditory evoked potentials. I. Evaluation of components. Electroencephalogr. Clin. Neurophysiol. 36, 179–190.
Rodriguez-Moreno, A., and Sihra, T. S. (2007). Metabotropic actions of kainate receptors in the CNS. J. Neurochem. 103, 2121–2135.
Semba, K., and Fibiger, H. C. (1992). Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro- and antero-grade transport and immunohistochemical study. J. Comp. Neurol. 323, 387–410.
Semba, K., Reiner, P. B., and Fibiger, H. C. (1990). Single cholinergic mesopontine tegmental neurons project to both the pontine reticular formation and the thalamus in the rat. Neuroscience 38, 643–654.
Skinner, R. D., Rasco, L. M., Fitzgerald, J., Karson, C. N., Matthew, M., Williams, D. K., and Garcia-Rill, E. (1999). Reduced sensory gating of the P1 potential in rape victims and combat veterans with posttraumatic stress disorder. Depress. Anxiety 9, 122–130.
Sofroniew, M. V., Priestley, J. V., Consolazione, A., Eckenstein, F., and Cuello, A. C. (1985). Cholinergic projections from the midbrain and pons to the thalamus in the rat, identified by combined retrograde tracing and choline acetyltransferase immunohistochemistry. Brain Res. 329, 213–223.
Teneud, L., Miyazato, H., Skinner, R. D., and Garcia-Rill, E. (2000). Cholinergic modulation of the sleep state-dependent P13 midlatency auditory evoked potential in the rat. Brain Res. 884, 196–200.
Teo, C., Rasco, L., al-Mefty, K., Skinner, R. D., Boop, F. A., and Garcia-Rill, E. (1997). Decreased habituation of midlatency auditory evoked responses in Parkinson’s disease. Mov. Disord. 12, 655–664.
Uc, E. Y., Skinner, R. D., Rodnitzky, R. L., and Garcia-Rill, E. (2003). The midlatency auditory evoked potential P50 is abnormal in Huntington’s disease. J. Neurol. Sci. 212, 1–5.
Vincent, S. R., Satoh, K., Armstrong, D. M., and Fibiger, H. C. (1983). NADPH-diaphorase: a selective histochemical marker for the cholinergic neurons of the pontine reticular formation. Neurosci. Lett. 43, 31–36.
Keywords: arousal, glutamate, kainate, N-methyl-d-aspartic acid, pedunculopontine nucleus, P13 auditory evoked potential, startle response
Citation: Simon C, Wallace-Huitt T, Thapa P, Skinner RD and Garcia-Rill E (2011) Effects of glutamate receptor agonists on the P13 auditory evoked potential and startle response in the rat. Front. Neur. 2:3. doi: 10.3389/fneur.2011.00003
Received: 22 November 2010;
Paper pending published: 13 December 2010;
Accepted: 13 January 2011;
Published online: 27 January 2011.
Edited by:
Michael H. Chase, University of California Los Angeles School of Medicine, USAReviewed by:
Subimal Datta, Boston University School of Medicine, USAPablo Torterolo, Universidad de la República, Uruguay
Copyright: © 2011 Simon, Wallace-Huitt, Thapa, Skinner and Garcia-Rill. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Edgar Garcia-Rill, Center for Translational Neuroscience, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, 4301 West Markham Street, Slot 847, Little Rock, AR 72205, USA. e-mail:Z2FyY2lhcmlsbGVkZ2FyQHVhbXMuZWR1