
- Biomedicine Business Area, National Security Technology Department, Johns Hopkins University Applied Physics Laboratory, Laurel, MD, USA
Due to complex injurious environment where multiple blast effects interact with the body parallel, blast-induced neurotrauma is a unique clinical entity induced by systemic, local, and cerebral responses. Activation of autonomous nervous system; sudden pressure increase in vital organs such as lungs and liver; and activation of neuroendocrine–immune system are among the most important mechanisms that contribute significantly to molecular changes and cascading injury mechanisms in the brain. It has been hypothesized that vagally mediated cerebral effects play a vital role in the early response to blast: this assumption has been supported by experiments where bilateral vagotomy mitigated bradycardia, hypotension, and apnea, and also prevented excessive metabolic alterations in the brain of animals exposed to blast. Clinical experience suggests specific blast–body–nervous system interactions such as (1) direct interaction with the head either through direct passage of the blast wave through the skull or by causing acceleration and/or rotation of the head; and (2) via hydraulic interaction, when the blast overpressure compresses the abdomen and chest, and transfers its kinetic energy to the body’s fluid phase, initiating oscillating waves that traverse the body and reach the brain. Accumulating evidence suggests that inflammation plays important role in the pathogenesis of long-term neurological deficits due to blast. These include memory decline, motor function and balance impairments, and behavioral alterations, among others. Experiments using rigid body- or head protection in animals subjected to blast showed that head protection failed to prevent inflammation in the brain or reduce neurological deficits, whereas body protection was successful in alleviating the blast-induced functional and morphological impairments in the brain.
Introduction
The history of blast injuries coincides with the history of explosives and modern warfare. Blast effects from high explosives have been described in the World Wars I and II as well as in the military actions of the twentieth and current centuries. For several reasons, the wars of the twenty-first century brought a new injury pattern: (1) the overwhelming majority of injuries are caused by explosions or blasts by rocket-propelled grenades, improvised explosive devices (IEDs), and land mines (Ritenour et al., 2010); (2) the mortality caused by explosion is relatively low due to improved interceptive properties of body armors, effectiveness of treatments, and promptness of medical evacuation, among others; and (3) due to increased survivability, the rate of severe, long-term deficits shows increasing trend. Based on the U.S. Defense and Veterans Brain Injury Center (DVBIC) analysis, approximately 180,000 U.S. military service members had a diagnosis of traumatic brain injury (TBI) in the period of 2001–2010. The overwhelming distribution of mild TBI (mTBI) was caused by blast. Because many servicemen with potential TBI remain undiagnosed or have delayed diagnosis, this number could be potentially higher (Terrio et al., 2009).
The complexity of the injurious environment generated by an explosion has been long recognized (Zuckerman, 1940). The injuries caused by multiple effects of the blast wave have been identified as: (a) primary blast injury cased by the blast wave itself; (b) secondary injury caused by the fragments of debris propelled by the explosion; (c) tertiary injury due to the acceleration of the body or part of the body by the blast wind and the sudden deceleration when it hits a ground or surrounding object; and (d) flash burns or toxic gas inhalation due to the intense heat of the explosion (Owen-Smith, 1981). It has been long posited that primary blast, e.g., blast overpressure generated during an explosion typically affects gas-containing organs through main mechanisms such as spalling, implosion, inertia, and pressure differences. On the other hand, the possibility of blast-induced neurotrauma (BINT) remained underestimated due to an outdated dogma that neurologic impairments caused by primary blasts are rare since the skull provides excellent protection for the brain (Rossle, 1950). Interestingly, despite several detailed reports published in 1940s portraying neurological symptoms in soldiers exposed to blast and describing underlying clinical findings and morphological changes in the brain not seen before (Stewart and Russel, 1941; Garai, 1944), the medical community ignored the pre-existing knowledge and attributed neurologic impairments due to blast to rare cases of air emboli in cerebral blood vessels (Clemedson, 1956).
While many servicemen with TBI experience remission of their symptoms, over one-third suffers at least one mTBI-related symptom at the post-deployment health assessment (Terrio et al., 2009). Immediately after exposure, individuals exposed to blast reported memory loss for events before and after explosion, confusion, impaired sense of reality, and reduced decision-making ability (Cernak et al., 1999a,b; Martin et al., 2008; Warden et al., 2009). Later, sometimes months and years after blast exposure, patients with BINT may experience irritability; memory and speech problems (reduced verbal fluency, working memory, and executive functioning; Nelson et al., 2009); headache; dizziness and balance problems (Hoffer et al., 2010); as well as psychological impairments such as depression and post-traumatic stress disorder (Terrio et al., 2009; Peskind et al., 2010). While many question the possibility that primary blast could have damaging effects to the brain, there is a higher-level agreement about the aspects of BINT that sets aside this pathological condition from civilian TBI, including high rates of sensory impairment, increased sensitivity toward pain issues, multiple organ involvement, and strong emotional context in which the injury occurred (French, 2010).
Thus, to be able to develop reliable and accurate diagnostic tools, efficient and timely treatments, and successful preventive measures, we have to identify what makes BINT potentially different from civilian TBI, and specify the main elements involved in the complexity of BINT.
Basic Mechanisms of Bint
Traumatic brain injury of all types and etiologies is caused by mechanical factors interacting with the brain either directly or indirectly. It is a complex process that consists of four overlapping phases: primary injury; evolution of the primary injury; secondary or additional injury; and regeneration (Reilly, 2001; Cernak, 2005). In civilian, non-blast TBI, the primary injury develops as a consequence of a direct interaction between the mechanical force and the head. The predominate injury causes are (1) direct contusion of the brain from the skull as a consequence of a direct hit; (2) brain contusion caused by a movement against rough interior surfaces of the skull, and/or indirect contusion of the brain opposite to the sate of the impact, i.e., coup–contrecoup; (3) shearing and stretching of the brain tissue due to a motion of the brain structures relative to each other or the skull; and (4) vascular consequences of the impact such as subdural hematoma originating from ruptured bridging blood vessels between brain and dura mater; reduced cerebral blood flow caused by intracranial pressure or infarction (Greve and Zink, 2009); and brain edema developing as a consequence of increased permeability of cerebral vasculature.
In BINT, these primary injury categories can be induced by: (a) fragments of the environment generated by explosion (so called “secondary blast effect”) and impacting the head causing either blunt or penetrating head trauma; or (b) when explosion propels (i.e., accelerates) the body through the air and the brain is contused either due to the acceleration/deceleration (coup–contrecoup), or a hit from the surrounding objects or ground (so called “tertiary blast effect”). Nevertheless, in addition to these primary injury categories seen in civilian environment, primary blast is a unique injurious factor that is exclusively generated by explosion. The shockwave or blast wave, comprised of a high-pressure front that compresses the surrounding air and is followed by a negative pressure phase, is the major determinant of primary blast-induced injury. It travels with a high velocity, often thousands km/h (Owen-Smith, 1981), and damages its surroundings in few milliseconds. Primary blast envelops the whole body, and interacts with all organs and organ systems, including the brain, simultaneously (Cernak and Noble-Haeusslein, 2010). The main mechanisms by which primary blast may cause brain injury can be direct or indirect. A direct passage of the blast wave through the skull might cause bruising, acceleration, or rotation of the brain. On the other hand, as the front of the blast wave connects with the elastic body wall, it compresses the abdomen and chest transferring its kinetic energy inside the body, to the body’s fluid phase (i.e., blood). The resulting “rippling” effect generates oscillating waves in the blood that traverse the body delivering the kinetic energy of the blast wave to organs, including the brain, remote from the initial point of coupling. Once delivered, that kinetic energy initiates functional and morphological alterations in distinct, mainly deep brain structures (Cernak et al., 1999a; Warden et al., 2009; Peskind et al., 2010). This second, indirect interactive pathway is unique for BINT, and underlies its immense complexity as compared to non-blast TBI.
Systemic Effects of Bint
Blast-induced neurotrauma is caused by multiple, interwoven mechanisms of systemic, local, and cerebral responses to blast exposure, often interacting with the body simultaneously (Cernak et al., 1991, 1996b; Cernak and Noble-Haeusslein, 2010; Figure 1). While the importance of multi-system, multi-organ response to blast exposure is more obvious in moderate-to-severe brain injuries, it is often neglected in the case of mild BINT. Nevertheless, accumulating clinical and experimental evidence show that systemic and local alterations initiated by blast significantly influence the brain’s response, thus contribute to the pathobiology of acute and/or chronic deficits due to blast.
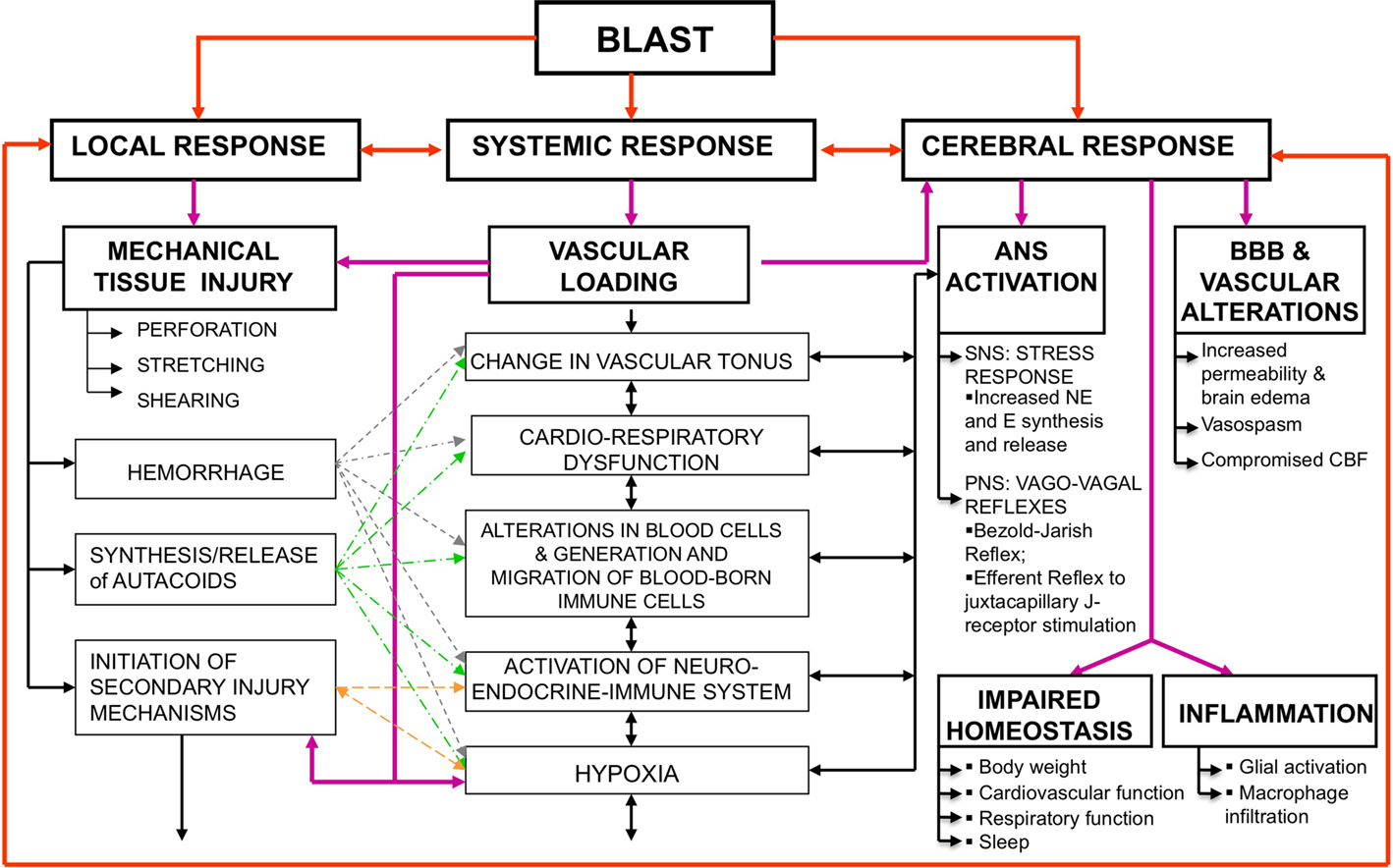
Figure 1. Simultaneous activation of systemic, local, and cerebral responses to blast exposure, and interactive mechanisms causing or contributing to the pathobiology of BINT. Abbreviations: ANS, autonomous nervous system; BBB, blood brain barrier; BINT, blast-induced neurotrauma; CBF, cerebral blood flow; E, epinephrine; NE, norepinephrine; SNS, sympathetic nervous system; PNS, parasympathetic nervous system.
Immediate Activation of the Autonomous Nervous System
In the experiments using rabbits, the animals were exposed to a blast overpressure generated by a compressed air-driven shock tube and causing mild-to-moderate blast lung injury. Aiming to analyze the effects of instantaneous activation of parasympathetic nervous system (PSNS) on the early response to blast, one subset of the animals was subjected to pulmonary deafferentation performed by bilateral dissection of the vagus 2 h before a blast exposure. Measured 30 min after blast exposure, brain edema and significant metabolic disturbances were found in the brain including decreased glucose and adenosine-triphosphate (ATP) concentrations (Cernak et al., 1996b); increased calcium and reduced magnesium concentrations as well as reduced magnesium/calcium ratio (Cernak et al., 1995). Bilateral vagotomy successfully mitigated bradycardia, hypotension, and apnea caused by blast; prevented extreme metabolic alterations and brain edema; but failed to eliminate oxidative stress in the brain due to blast (Cernak et al., 1996b).
It was hypothesized that hyperinflation of the lungs caused by blast overpressure stimulates the juxtacapillary J-receptors located in the alveolar interstitium and innervated by vagal fibers, triggering a vago-vagal reflex that leads to apnea followed by tachypnea, bradycardia, and hypotension (Zuckerman, 1940; Cernak et al., 1996b). In addition, hypoxia/ischemia caused by the pulmonary vagal reflex stimulates chemoreceptors in the left ventricle and subsequently activates a cardiovascular decompressor Bezold–Jarish reflex leading to significant parasympathetic efferent discharge to the heart (Zucker, 1986). This, in turn, further deepens heart rate reduction (bradycardia) and peripheral blood vessel dilatation resulting in lowering blood pressure and eventually contributing to cerebral hypoxemia (Cernak et al., 1996a).
Na+, K+-ATPase During Early Post-Traumatic Phase
Based on previous studies showing that blast exposure induces distinct pathological alterations in the brain including impaired energy metabolism (Cernak et al., 1995, 1996b), the activity of sodium–potassium ATPase (Na+, K+-ATPase; E.C.3.6.1.3.) was measured in the brainstem and erythrocyte membranes of rabbits exposed to blast. The aim was to establish the comparability of the enzyme changes as an indirect measure of systemic-cerebral response interaction during the early post-traumatic period after blast (Cernak et al., 1997). The Na+, K+-ATPase, i.e., sodium pump, is a ubiquitous plasma membrane enzyme that catalyzes the movement of K+ into cells in exchange for Na+, sustaining a gradient for Na+ in to and for K+ out of the cell (Skou and Esmann, 1992). The resulting gradient is used as an energy source for the de- and re-polarization of the membrane potential, regulation of cytoplasmic ionic composition, and for transepithelial transport. The sodium pump is regulated by numerous factors including several circulating hormones (Lopina, 2000; Lima et al., 2008). Since Na+, K+-ATPase is very sensitive to free radical reactions (Petrushanko et al., 2007) and impairments of energy metabolism (Silver and Erecinska, 1997), its activity can indicate membrane response to an insult indirectly giving insight into the efficiency of compensatory mechanisms and the level of membrane dysfunction.
Adult male rabbits were randomly assigned to three groups: control, mild-to-moderate, and moderate-to-severe blast injury groups. The blast injury groups were anesthetized and subjected to a blast overpressure generated by a compressed air-driven shock tube. During the early post-traumatic period (i.e., 30 min post-exposure), arterial pressure, electrocardiogram (ECG) and electroencephalogram (EEG) activities were measured. At the end of the 30-min observation period, arterial blood samples were collected, the animals sacrificed, and brain structures harvested. Light-microscopy examinations of brain samples stained with Hematoxylin–Eosin, Trichrom Masson, and van Gieson Elastica were also performed. Erythrocyte plasma membranes (Dodge et al., 1963; Mrsulja et al., 1993) and brainstem homogenates (Enseleit et al., 1984; Cernak et al., 1997) for the Na+, K+-ATPase assays were prepared as previously described. Briefly, the reaction mixture for the Na+, K+-ATPase assay contained 5.0 mM/L MgCl2, 130.0 mM/L NaCl, 20.0 mM/L KCl, and 45.0 mM/L Tris–HCl buffer, pH 7.4. The reaction was started by the addition of ATP (disodium salt, vanadium free) to a final concentration of 0.1, 0.5, 1.0, 1.5, 3.0, and 5.0 mM, and stopped after a 60 min incubation period with the addition of the ice cold HClO4. The activity obtained in the presence of 2 mM ouabain (without the NaCl and KCl) was attributed to Mg2+-ATPase. Na+, K+-ATPase activity was calculated as a difference between the total ATPase activity (Na+, K+, Mg2+-dependent) and Mg2+-ATPase activity. Released inorganic phosphate (Pi) was measured by the method of Fiske and Subbarow (1925). Enzyme specific activity was expressed as nmol Pi released per mg of protein per min. All assays were performed in duplicate and the mean was used for statistical analysis. In this experiment, protein was measured by the method of (Lowry et al., 1951), with bovine serum albumin used as standard. The Michaelis–Menten constant (Km) and the maximum reaction rate (Vmax) of Na+, K+-ATPase were calculated using the Lineweaver–Burk double reciprocal plot.
Blast exposure caused the characteristic fall in mean arterial pressure, bradycardia, fast and shallow respiration, and significant increases in both frequency and amplitude of the EEG activity as compared to control animals (25 versus 15–18 Hz, and 22 versus 10 μV, respectively). The neuropathology analysis showed no hemorrhages or areas of contused brain tissue. The development of brain edema as well as the sodium pump changes demonstrated a graded response to the shockwave intensity, i.e., more prominent in the moderate-to-severe blast group as compared to the animals subjected to mild-to-moderate intensity blast. In all tested groups Na+, K+-ATPase activity in plasma membrane was substrate (ATP) dependent, such that the lowest activity was obtained at 0.1 mM, whereas the highest activity was at 1–1.5 mM concentrations of the substrate, ATP. At these substrate concentrations, the activity of the Na+, K+-ATPase was significantly increased in the erythrocyte membranes and the brainstem as compared to control values.
Changes in Na+, K+-ATPase have been shown being time-dependent (Sztriha et al., 1987), and in the present study the activity Na+, K+-ATPase was measured at the time when significant neuro-endocrine changes occur in the brain with TBI. Indeed, complex alterations such as the over-activation or inhibition of neurotransmitter synthesis, release, or transport have been reported during the early period after TBI (McIntosh et al., 1994). Among the most important neuro-endocrine changes, an increase in norepinephrine (NE) synthesis and metabolism have been reported in animal experiments inducing transient cerebral ischemia (Globus et al., 1989; Bhardwaj et al., 1990), focal cortical lesions (Pappius, 1991), and fluid-percussion TBI (McIntosh et al., 1994) during the first hours after the insult. Several mechanisms have been suggested to explain the NE activation of the enzyme: one involves specific receptors or a non-specific chelating action related to the catechol group that would relieve the inhibition by divalent cations such as Ca2+ (Godfraind et al., 1974; Adam-Vizi et al., 1979; Phillis and Wu, 1981), while the other describes a possibility that NE removes an endogenous inhibitory factor present in the cytoplasm (Hernandez, 1992). A dose-dependent response of the Na+, K+-ATPase to dopamine and NE concentrations in mouse brain synaptosomes have been confirmed by Desaiah and Ho (1977) reporting that these catecholamines modified the behavior of the enzyme kinetics against ATP, producing an increase in Vmax. Our previous studies have shown a direct relationship between injury severity and blood concentrations of epinephrine and NE in soldiers with military gunshot or missile (thus explosive) injuries (Savic et al., 1995) as well as in the brain of rats exposed to whole-body blast during the early post-traumatic phase.
Taken together, we hypothesize that the increased activity and Vmax of the Na+, K+-ATPase in our study may be related to the catecholamine surge in the brain. Additionally, the edema formation developed in the brainstem parallel with the increase in the Na+, K+-ATPase activity could be explained as a result of Na+-influx, bearing in mind that abrupt increase of cytoplasmic Na+ in the brain tissue has been shown after trauma both in vitro (Young et al., 1986; Sheldon et al., 2004; Wang et al., 2009) and in vivo (Young et al., 1986). Moreover, the role of the Na+-influx in the pathobiology of acute vasogenic edema was demonstrated by in vivo sodium MRI imaging in both clinical and experimental studies of brain edema development compared with signal in health brain tissue (Turski et al., 1986).
Importance of the Indirect Blast–Brain Interacting Pathway in Long-Term Neurological Deficits
In an attempt to link systemic and cerebral inflammation as one of the potential mechanisms leading to long-term neurological deficits caused by blast, we performed real-time, in vivo imaging of myeloperoxidase (MPO) activity of activated phagocytes in mice exposed to mild intensity blast, during a 1-month post-injury period. Namely, migration and accumulation of polymorphonuclear leukocytes (PMNs) are among the major hallmarks of the host response to injuries (Maslinska and Gajewski, 1998; Toft et al., 2003; Menezes et al., 2008). Locally, activated PMNs release antimicrobial and inflammatory mediators, such as MPO, among others. MPO is a key inflammatory enzyme secreted by activated PMNs and macrophages/microglia, capable of generating highly reactive oxygen species to cause additional damage in various pathological conditions such as cerebral ischemia (Weston et al., 2007; Breckwoldt et al., 2008); ischemic heart conditions (Loria et al., 2008); inflammatory bowel disease (Naito et al., 2007); and kidney (Malle et al., 2003) and lung (Kinnula, 2005) pathologies, among others. Based on its importance in inflammatory processes and as a reliable indicator of PMN presence in tissues (Arnhold and Flemmig, 2010), MPO has been widely used as an inflammatory marker of both acute and chronic conditions (Werner and Szelenyi, 1992; Breckwoldt et al., 2008; Faith et al., 2008).
Briefly, mice were anesthetized, mounted in supine position to the animal holder secured inside the driven section of the helium-driven shock tube, and exposed to mild intensity shock wave (measured rupture pressure: 183 ± 14 kPa, i.e., 26.5 ± 2.1 psig; measured total pressure: 103 kPa, i.e., 14.9 psig) causing 5% mortality (Cernak et al., 2010). Subsets of animals (n = 5) were exposed to: (1) whole-body blast; (2) blast with torso (chest and abdomen) protection using a custom-made rigid Plexiglas “body armor,” while the head exposed; or (3) blast with head protection using a custom-made Plexiglas “helmet” covering skull, face, and the neck of the animal while the torso exposed (Figure 2). Ten minutes before imaging, mice were injected with XenoLight Rediject Inflammation probe (Caliper Life Sciences; Hopkinton, MA, USA) at 200 mg/kg (150 μL/mouse) intraperitoneally. The XenoLight Rediject Inflammation probe is a chemiluminescent reagent in a ready-to-use format that allows for longitudinal tracking of MPO level and inflammation status, in vivo, in a variety of disease models. Bioluminescence imaging (BLI) was performed using the IVIS® Imaging System 3-D Series (Caliper Life Sciences, Hopkinton, MA, USA). During the imaging, the animals were anesthetized with a gas mixture (isoflurane:nitrous oxide:oxygen at 1:66:33% proportions, respectively) using the integrated system for gas anesthesia. The duration of imaging was 5 min. To validate substrate injection quality, fluorescence images were taken right before BLI, using the 745-nm excitation and 800-nm emission filter set, and exposure time between 1 and 5 s. Photons were quantified using Living Image software (Caliper Life Sciences, Hopkinton, MA, USA). Sham control animals did show only low-intensity localized bioluminescence at the injection site (results not shown).

Figure 2. Experimental setting for establishing the importance of blast-head and blast–body interactive pathways in the pathobiology of BINT. (A) whole-body blast exposure without protection; (B) head blast exposure with torso (chest and abdomen) protection; and (C) torso blast exposure with head protection.
Figure 3 shows the distribution of increased bioluminescence in mice subjected to whole-body blast exposure imaged during the 1-month observation period. One day after blast exposure, the highest MPO activity is found in the gastrointestinal tract, with clearly outlined transverse and descending colon. Additionally, enhanced signal is observed bilaterally in diaphragmal mediastinal parts of the lungs as well as in the brain. Interestingly, the PMNs activation showed a declining trend 3 days post-exposure followed by intensified MPO activity in lungs, gastrointestinal tract and the brain measured at 7, 14, and 30 days after blast. It is noteworthy that the most intense bioluminescence signal measured in the brain was found 1 month after injury.
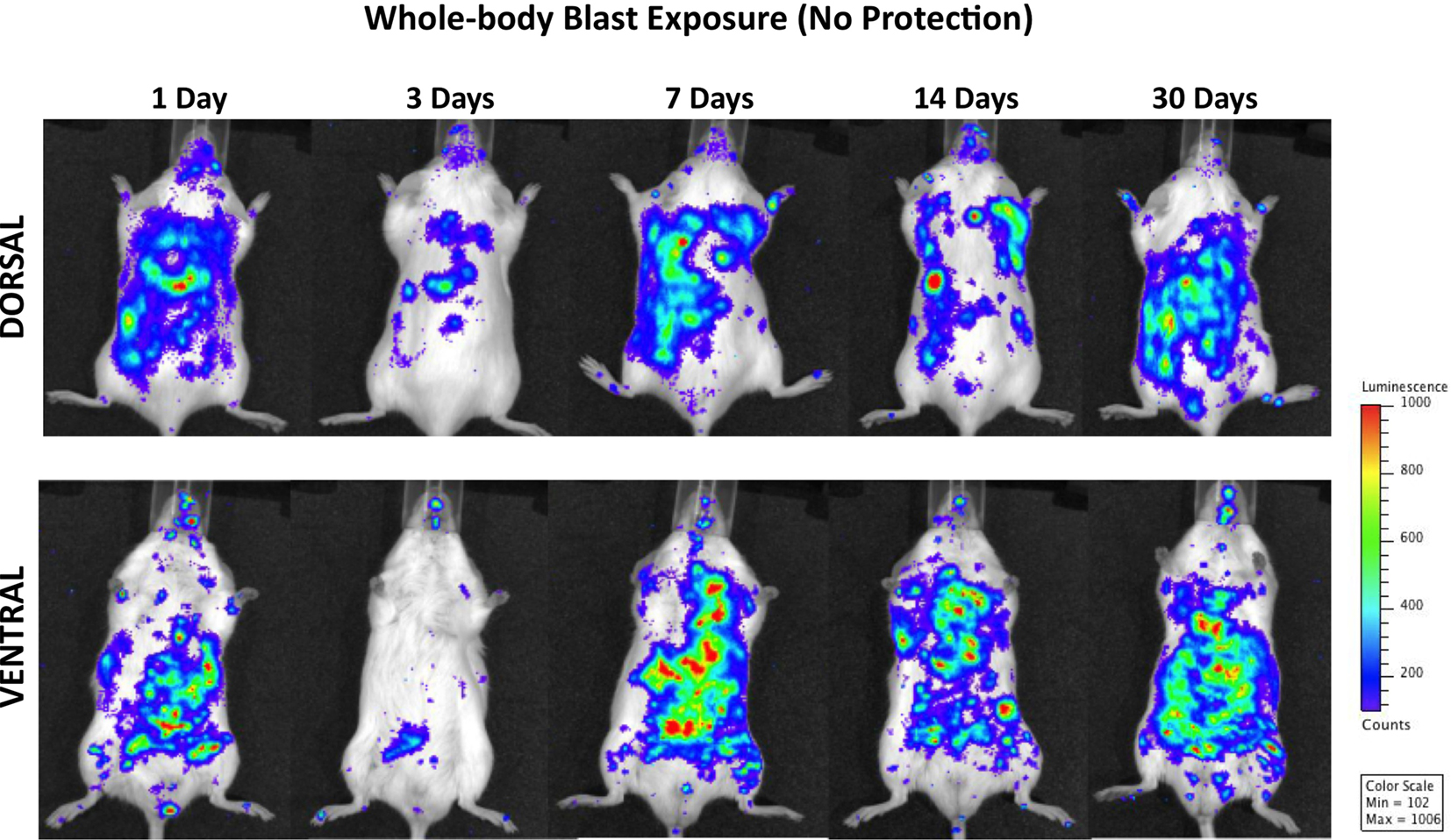
Figure 3. Distribution of increased bioluminescence showing MPO activity in mice subjected to whole-body blast exposure, imaged during the 1-month observation period. The representative photographs show the same animal imaged in both dorsal and ventral positions at the given time point after blast exposure. The intensity of bioluminescence was scaled based on the photon counts. The nose cone that can be discerned on the photos is part of the anesthesia device that is the integral part of the IVIS® imaging system 3-D series, and does not represent a head cover.
Figure 4 demonstrates MPO activity in animals subjected to blast with head protection. Because the torso was exposed to blast, the finding of the activation of PMNs in lungs and gastrointestinal tract, similar to the changes observed in animals with whole-body blast exposure, is not surprising. The temporal and spatial profiles of MPO activity were also comparable, i.e., a declining trend at 3 days post-trauma followed by progressively increasing signal peaking at 1 month after blast. However, regardless of the head protection, MPO was significantly increased in the brain, with the highest activity at 14 and 30 days post-injury suggesting chronic inflammation. Importantly, the increase in MPO activity observed in mice with head protection was equal to corresponding changes found in the brain of animals exposed to whole-body blast without head protection, suggesting that head protection did not prevent inflammation in the brain and implying the importance of inflammatory cells of systemic origin in the pathobiology of blast-induced inflammatory processes in the brain.
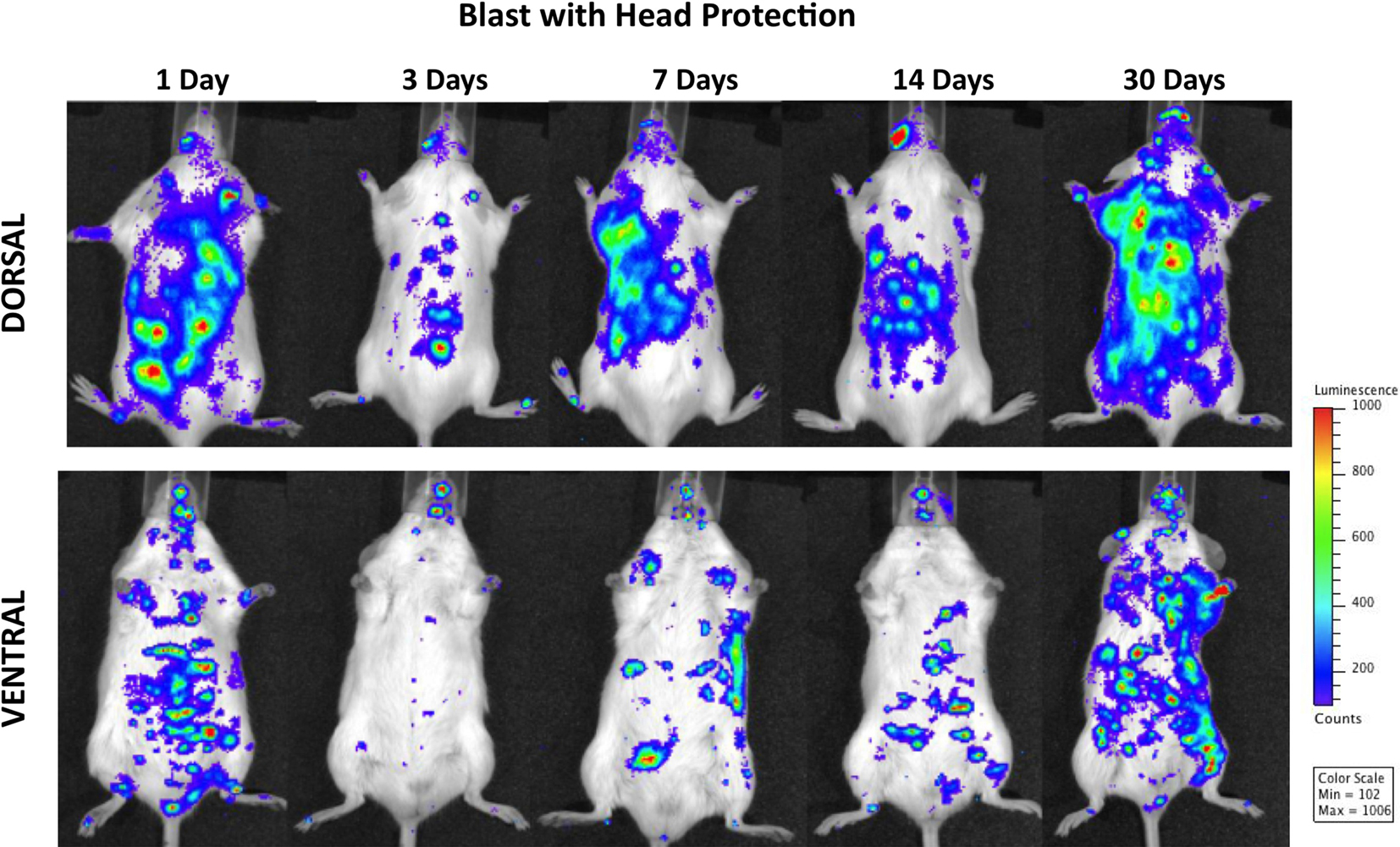
Figure 4. Distribution of increased bioluminescence showing MPO activity in mice subjected to blast with head protection, imaged during the 1-month observation period. The representative photographs show the same animal imaged in both dorsal and ventral positions at the given time point after blast exposure. The intensity of bioluminescence was scaled based on the photon counts. The nose cone that can be discerned on the photos is part of the anesthesia device that is the integral part of the IVIS® imaging system 3-D series, and does not represent a head cover.
It is also noteworthy that while head protection did not prevent inflammation in the brain, it did reduce MPO activity in the gastrointestinal tract. It is well known that vagal afferent neurons play an important role in gut–brain communication (Berthoud and Neuhuber, 2000; Danzer et al., 2004) responding to a variety of physiological stimuli relevant not only to food intake and regulation of digestive activity, but in the response to immunological and noxious stimuli also (Gaykema et al., 2007). Indeed, direct and indirect pathways that originate in the caudal brainstem and propagate immune-related information from the periphery have been shown to play important role in the integration of brain responses to infection and injury (Gaykema et al., 2007). Additionally, it has been suggested that part of the peripheral immune information can be conveyed via vagal pathway to the catecholaminergic neurons in the medullary visceral zone from where it is transferred to the central amygdaloid nucleus (Ge et al., 2001). Such an approach suggests a complex interaction of the autonomous nervous system components with immune system elements in the response to injury-induced inflammation. Thus, we hypothesize that protecting the head reduced the susceptibility of the brain toward incoming vagal information and subsequently decreased the intensity of the efferent feedback response to peripheral injury; this in turn led to diminished inflammation in the gastrointestinal tract and lungs after blast as compared to animals with whole-body blast exposure.
Figure 5 shows progressive development of inflammation in both peripheral organs/organ systems and the brain of animals subjected to blast with torso protection. Systemic inflammation and the intensity of MPO activation in the brain were significantly reduced throughout the 1 month post-injury period as compared to mice either with whole-body exposure to blast or those with head protection. Moreover, while the inflammation in the brain was evident at 24 h post-exposure and still present at the end of the observation period, its intensity was significantly less than in animals with whole-body exposure or with head protection.
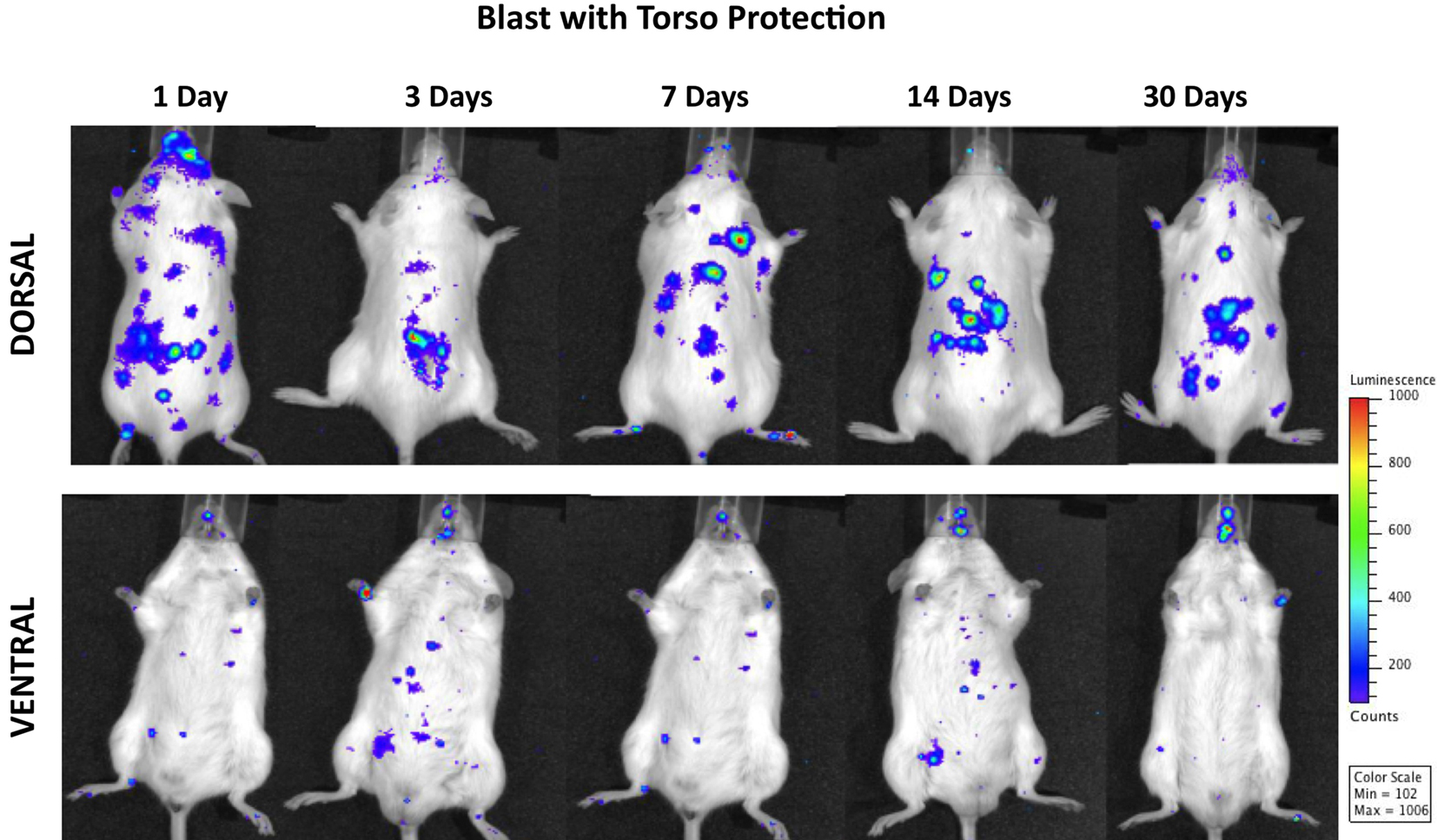
Figure 5. Distribution of increased bioluminescence showing MPO activity in mice subjected to blast with torso protection while the head exposed, imaged during the 1-month observation period. The representative photographs show the same animal imaged in both dorsal and ventral positions at the given time point after blast exposure. The intensity of bioluminescence was scaled based on the photon counts. The nose cone that can be discerned on the photos is part of the anesthesia device that is the integral part of the IVIS® imaging system 3-D series, and does not represent a head cover.
The fact that there was still MPO activation in the lungs regardless of the torso protection could be explained by pulmonary complications of the head injury such as neurogenic lung edema, seen both experimentally (Baumann et al., 2007; Irwin et al., 2008) and clinically (Dettbarn and Davidson, 1989; Fontes et al., 2003) after TBI. The roles of systemic sympathetic discharge, changes in intracranial pressure, and inflammation in central nervous system trigger zones have been suggested as essential mechanisms underlying the etiopathogenesis of neurogenic pulmonary edema (Sedy et al., 2008).
Taken together, the results of our study clearly demonstrate the importance of the indirect, i.e., blast–body interaction as well as the decisive role of autonomous nervous–neuroendocrine–immune systems interaction in the pathogenesis of BINT.
Discussion and Conclusions
Our findings demonstrate a multi-phase and multi-factorial nature of BINT where the mechanisms essential for the injury development and progress show significantly different phase-dependent trends. While acute alterations reflect compensatory attempts (such as increased activation of the sodium pump to potentially compensate for injury-induced impairments in electrolyte milieu or parasympathetic stimulation to counteract the sudden pressure increase in the lungs and heart), chronic changes such as inflammation can lead to irreversible degenerative pathologies. Thus, to establish reliable diagnostic tools capable of identifying BINT and its progress, we should fully understand and characterize the multiple phases of the brain response to blast exposure.
The Na+, K+-ATPase activity change is an excellent example demonstrating the duality of response mechanisms to injury where increased and reversible activity after various brain insults in animals during the acute (<3 h) post-injury period (MacMillan, 1982; Goldberg et al., 1984; Sztriha et al., 1987) is followed by reduced activity during the chronic post-injury period (Jovicic et al., 2008; Crema et al., 2010) potentially leading to irreversible neurodegenerative central nervous system disorders (Seddik et al., 1991; Kumar and Kurup, 2002; Lima et al., 2008). While the Na+, K+-ATPase activity is not disease specific, its phase-dependent activity change as well as its kinetic profile to ATP might be a useful indicator of the BINT development and progress. The fact that the sodium pump changes correspond well to the changes in the erythrocyte membranes makes measurements of the Na+, K+-ATPase even more feasible as diagnostic parameters for BINT. There is a long list of research tasks ahead of us that concerns the importance of the Na+, K+-ATPase in the pathobiology of the BINT including establishing temporal and spatial profile characterization of its activity and kinetic pattern in the brain structures parallel with the changes in the erythrocyte membrane, and testing the diagnostic values of identified alterations in predicting functional neurological (memory, cognition, motor, and behavior) deficits, among others.
The results showing that head protection does not prevent chronic inflammation and neurological deficits suggest the vital importance of the blast–body interactive pathway in the etiopathogenesis of BINT. This fact is extremely important in designing effective protective equipment for servicemen that would prevent blast-body coupling, thus prevent the systemically induced mechanisms of BINT, as well as protect the brain from the direct impact caused by blast.
We need well-organized studies that begin in the field and identify the blast environment in relation to immediate and delayed mTBI symptoms in servicemen. Further, those data should be shared with the basic and applied research community to understand the uniqueness of this injury and to develop adequate and sensitive diagnostic tools and successful treatment. Without knowing what and how the blast injury occurred, it will be difficult to develop timely, reliable and specific diagnosis, and treatments for our servicemen. This multifaceted task is extremely challenging, and its successful solution is possible only through highly synchronized multi-institutional and international research efforts.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Farid A. Ahmed significantly contributed to the Section “Importance of the Indirect Blast–Brain Interacting Pathway in Long-term Neurological Deficits” study. The study described in Section “Importance of the Indirect Blast–Brain Interacting Pathway in Long-term Neurological Deficits” was supported by the Johns Hopkins University Applied Physics Laboratory internal research and development (IR&D) funds. The sponsor did not influence the study design; data collection, analysis and interpretation of data; or the writing of the article.
References
Adam-Vizi, V., Vizi, E. S., and Horvath, I. (1979). Stimulation by noradrenaline of Na+K+ ATPase in different fractions of rat brain cortex. J. Neural Transm. 46, 59–69.
Arnhold, J., and Flemmig, J. (2010). Human myeloperoxidase in innate and acquired immunity. Arch. Biochem. Biophys. 500, 92–106.
Baumann, A., Audibert, G., McDonnell, J., and Mertes, P. M. (2007). Neurogenic pulmonary edema. Acta Anaesthesiol. Scand. 51, 447–455.
Berthoud, H. R., and Neuhuber, W. L. (2000). Functional and chemical anatomy of the afferent vagal system. Auton. Neurosci. 85, 1–17.
Bhardwaj, A., Brannan, T., Martinez-Tica, J., and Weinberger, J. (1990). Ischemia in the dorsal hippocampus is associated with acute extracellular release of dopamine and norepinephrine. J. Neural Transm. Gen. Sect. 80, 195–201.
Breckwoldt, M. O., Chen, J. W., Stangenberg, L., Aikawa, E., Rodriguez, E., Qiu, S., Moskowitz, M. A., and Weissleder, R. (2008). Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. U.S.A. 105, 18584–18589.
Cernak, I., Ignjatovic, D., Andelic, G., and Savic, J. (1991). Metabolic changes as part of the general response of the body to the effect of blast waves. Vojnosanit. Pregl. 48, 515–522.
Cernak, I., Malicevic, Z., Prokic, V., Zunic, G., Djurdjevic, D., Ilic, S., and Savic, J. (1997). Indirect neurotrauma caused by pulmonary blast injury: development and prognosis. Int. Rev. Armed Forces Med. Serv. 52, 114–120.
Cernak, I., Merkle, A. C., Koliatsos, V. E., Bilik, J. M., Luong, Q. T., Mahota, T. M., Xu, L., Slack, N., Windle, D., and Ahmed, F. A. (2010). The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. doi: 10.1016/j.nbd.2010.10.025. [Epub ahead of print].
Cernak, I., and Noble-Haeusslein, L. J. (2010). Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J. Cereb. Blood Flow Metab. 30, 255–266.
Cernak, I., Radosevic, P., Malicevic, Z., and Savic, J. (1995). Experimental magnesium depletion in adult rabbits caused by blast overpressure. Magnes. Res. 8, 249–259.
Cernak, I., Savic, J., Ignjatovic, D., and Jevtic, M. (1999a). Blast injury from explosive munitions. J. Trauma. 47, 96–103; discussion 103–104.
Cernak, I., Savic, J., Zunic, G., Pejnovic, N., Jovanikic, O., and Stepic, V. (1999b). Recognizing, scoring, and predicting blast injuries. World J. Surg. 23, 44–53.
Cernak, I., Savic, J., Malicevic, Z., Zunic, G., Radosevic, P., and Ivanovic, I. (1996a). Leukotrienes in the pathogenesis of pulmonary blast injury. J. Trauma 40, S148–S151.
Cernak, I., Savic, J., Malicevic, Z., Zunic, G., Radosevic, P., Ivanovic, I., and Davidovic, L. (1996b). Involvement of the central nervous system in the general response to pulmonary blast injury. J. Trauma 40, S100–S104.
Crema, L., Schlabitz, M., Tagliari, B., Cunha, A., Simao, F., Krolow, R., Pettenuzzo, L., Salbego, C., Vendite, D., Wyse, A. T., and Dalmaz, C. (2010). Na+, K+ ATPase activity is reduced in amygdala of rats with chronic stress-induced anxiety-like behavior. Neurochem. Res. 35, 1787–1795.
Danzer, M., Jocic, M., Samberger, C., Painsipp, E., Bock, E., Pabst, M. A., Crailsheim, K., Schicho, R., Lippe, I. T., and Holzer, P. (2004). Stomach–brain communication by vagal afferents in response to luminal acid backdiffusion, gastrin, and gastric acid secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G403–G411.
Desaiah, D., and Ho, I. K. (1977). Kinetics of catecholamine sensitive Na+-K+ ATPase activity in mouse brain synaptosomes. Biochem. Pharmacol. 26, 2029–2035.
Dettbarn, C. L., and Davidson, L. J. (1989). Pulmonary complications in the patient with acute head injury: neurogenic pulmonary edema. Heart Lung 18, 583–589.
Dodge, J. T., Mitchell, C., and Hanahan, D. J. (1963). The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 100, 119–130.
Enseleit, W. H., Domer, F. R., Jarrott, D. M., and Baricos, W. H. (1984). Cerebral phospholipid content and Na+, K+-ATPase activity during ischemia and postischemic reperfusion in the mongolian gerbil. J. Neurochem. 43, 320–327.
Faith, M., Sukumaran, A., Pulimood, A. B., and Jacob, M. (2008). How reliable an indicator of inflammation is myeloperoxidase activity? Clin. Chim. Acta 396, 23–25.
Fiske, C. H., and Subbarow, Y. (1925). The colorimetric determination of phosphorus. J. Biol. Chem. 66, 375–400.
Fontes, R. B., Aguiar, P. H., Zanetti, M. V., Andrade, F., Mandel, M., and Teixeira, M. J. (2003). Acute neurogenic pulmonary edema: case reports and literature review. J. Neurosurg. Anesthesiol. 15, 144–150.
French, L. M. (2010). Military traumatic brain injury: an examination of important differences. Ann. N. Y. Acad. Sci. 1208, 38–45.
Gaykema, R. P., Chen, C. C., and Goehler, L. E. (2007). Organization of immune-responsive medullary projections to the bed nucleus of the stria terminalis, central amygdala, and paraventricular nucleus of the hypothalamus: evidence for parallel viscerosensory pathways in the rat brain. Brain Res. 1130, 130–145.
Ge, X., Yang, Z., Duan, L., and Rao, Z. (2001). Evidence for involvement of the neural pathway containing the peripheral vagus nerve, medullary visceral zone and central amygdaloid nucleus in neuroimmunomodulation. Brain Res. 914, 149–158.
Globus, M. Y., Busto, R., Dietrich, W. D., Martinez, E., Valdes, I., and Ginsberg, M. D. (1989). Direct evidence for acute and massive norepinephrine release in the hippocampus during transient ischemia. J. Cereb. Blood Flow Metab. 9, 892–896.
Godfraind, T., Koch, M. C., and Verbeke, N. (1974). The action of EGTA on the catecholamines stimulation of rat brain Na-K-ATPase. Biochem. Pharmacol. 23, 3505–3511.
Goldberg, W. J., Watson, B. D., Busto, R., Kurchner, H., Santiso, M., and Ginsberg, M. D. (1984). Concurrent measurement of (Na+, K+)-ATPase activity and lipid peroxides in rat brain following reversible global ischemia. Neurochem. Res. 9, 1737–1747.
Greve, M. W., and Zink, B. J. (2009). Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. 76, 97–104.
Hoffer, M. E., Balaban, C., Gottshall, K., Balough, B. J., Maddox, M. R., and Penta, J. R. (2010). Blast exposure: vestibular consequences and associated characteristics. Otol. Neurotol. 31, 232–236.
Irwin, D. C., Subudhi, A. W., Klopp, L., Peterson, D., Roach, R., and Monnet, E. (2008). Pulmonary edema induced by cerebral hypoxic insult in a canine model. Aviat. Space Environ. Med. 79, 472–478.
Jovicic, M. E., Popovic, M., Nesic, K. J., Popovic, N., Pavlovic, S. J., and Rakic, L. (2008). Aging, aluminium and basal forebrain lesions modify substrate kinetics of erythrocyte membrane Na, K-ATPase in the rat. J. Alzheimers Dis. 14, 85–93.
Kinnula, V. L. (2005). Production and degradation of oxygen metabolites during inflammatory states in the human lung. Curr. Drug Targets Inflamm. Allergy 4, 465–470.
Kumar, A. R., and Kurup, P. A. (2002). Inhibition of membrane Na+-K+ ATPase activity: a common pathway in central nervous system disorders. J. Assoc. Physicians India 50, 400–406.
Lima, F. D., Souza, M. A., Furian, A. F., Rambo, L. M., Ribeiro, L. R., Martignoni, F. V., Hoffmann, M. S., Fighera, M. R., Royes, L. F., Oliveira, M. S., and de Mello, C. F. (2008). Na+, K+-ATPase activity impairment after experimental traumatic brain injury: relationship to spatial learning deficits and oxidative stress. Behav. Brain Res. 193, 306–310.
Lopina, O. D. (2000). Na+, K+-ATPase: structure, mechanism, and regulation. Membr. Cell Biol. 13, 721–744.
Loria, V., Dato, I., Graziani, F., and Biasucci, L. M. (2008). Myeloperoxidase: a new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm. 2008, 135625.
Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275.
MacMillan, V. (1982). Cerebral Na+, K+-ATPase activity during exposure to and recovery from acute ischemia. J. Cereb. Blood Flow Metab. 2, 457–465.
Malle, E., Buch, T., and Grone, H. J. (2003). Myeloperoxidase in kidney disease. Kidney Int. 64, 1956–1967.
Martin, E. M., Lu, W. C., Helmick, K., French, L., and Warden, D. L. (2008). Traumatic brain injuries sustained in the Afghanistan and Iraq wars. Am. J. Nurs. 108, 40–47.
Maslinska, D., and Gajewski, M. (1998). Some aspects of the inflammatory process. Folia Neuropathol. 36, 199–204.
McIntosh, T. K., Yu, T., and Gennarelli, T. A. (1994). Alterations in regional brain catecholamine concentrations after experimental brain injury in the rat. J. Neurochem. 63, 1426–1433.
Menezes, G. B., Rezende, R. M., Pereira-Silva, P. E., Klein, A., Cara, D. C., and Francischi, J. N. (2008). Differential involvement of cyclooxygenase isoforms in neutrophil migration in vivo and in vitro. Eur. J. Pharmacol. 598, 118–122.
Mrsulja, B. B., Stojadinovic, N., Maletic-Savatic, M., Stojanovic, T., Cvejic, V., Nenadovic, M., Paunovic, V. R., Marinkovic, D., and Kostic, V. S. (1993). Substrate erythrocyte membrane Na, K-ATPase: a potentially ubiquitous mechanism reflecting central nervous system pathology. Iugoslav. Physiol. Pharmacol. Acta 29, 115–119.
Naito, Y., Takagi, T., and Yoshikawa, T. (2007). Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J. Gastroenterol. 42, 787–798.
Nelson, L. A., Yoash-Gantz, R. E., Pickett, T. C., and Campbell, T. A. (2009). Relationship between processing speed and executive functioning performance among OEF/OIF veterans: implications for postdeployment rehabilitation. J. Head Trauma Rehabil. 24, 32–40.
Pappius, H. M. (1991). Brain injury: new insights into neurotransmitter and receptor mechanisms. Neurochem. Res. 16, 941–949.
Peskind, E. R., Petrie, E. C., Cross, D. J., Pagulayan, K., McCraw, K., Hoff, D., Hart, K., Yu, C. E., Raskind, M. A., Cook, D. G., and Minoshima, S. (2010). Cerebrocerebellar hypometabolism associated with repetitive blast exposure mild traumatic brain injury in 12 Iraq war Veterans with persistent post-concussive symptoms. Neuroimage. doi:10.1016/j.neuroimage.2010.04.008. [Epub ahead of print].
Petrushanko, I. Y., Bogdanov, N. B., Lapina, N., Boldyrev, A. A., Gassmann, M., and Bogdanova, A. Y. (2007). Oxygen-induced regulation of Na/K ATPase in cerebellar granule cells. J. Gen. Physiol. 130, 389–398.
Phillis, J. W., and Wu, P. H. (1981). Catecholamines and the sodium pump in excitable cells. Prog. Neurobiol. 17, 141–184.
Reilly, P. L. (2001). Brain injury: the pathophysiology of the first hours. “Talk and die revisited”. J. Clin. Neurosci. 8, 398–403.
Ritenour, A. E., Blackbourne, L. H., Kelly, J. F., McLaughlin, D. F., Pearse, L. A., Holcomb, J. B., and Wade, C. E. (2010). Incidence of primary blast injury in US military overseas contingency operations: a retrospective study. Ann. Surg. 251, 1140–1144.
Rossle, R. (1950). “Pathology of blast effects,” in German Aviation Medicine, World War II ed. USAF School of Aviation Medicine, (Washington, DC: Department of the Air Force), 1260–1273.
Savic, J., Lazarov, A., Malicevic, Z., Nikolic, J., and Vujinov, S. (1995). Relationship between early neuroendocrine response and severity of war injury according to the red cross classification. Vojnosanit. Pregl. 52, 107–117.
Seddik, Z., Habib, Y. A., and el Shamy, E. (1991). The prognostic value of the brain sodium–potassium ATPase enzyme concentration in head injury. Childs Nerv. Syst. 7, 135–138.
Sedy, J., Zicha, J., Kunes, J., Jendelova, P., and Sykova, E. (2008). Mechanisms of neurogenic pulmonary edema development. Physiol. Res. 57, 499–506.
Sheldon, C., Diarra, A., Cheng, Y. M., and Church, J. (2004). Sodium influx pathways during and after anoxia in rat hippocampal neurons. J. Neurosci. 24, 11057–11069.
Silver, I. A., and Erecinska, M. (1997). Energetic demands of the Na+/K+ ATPase in mammalian astrocytes. Glia 21, 35–45.
Stewart, O. W., and Russel, C. K. (1941). Injury to the central nervous system by blast. Lancet 1, 172–174.
Sztriha, L., Joo, F., Dux, L., and Boti, Z. (1987). Effects of systemic kainic acid administration on regional Na+, K+-ATPase activity in rat brain. J. Neurochem. 49, 83–87.
Terrio, H., Brenner, L. A., Ivins, B. J., Cho, J. M., Helmick, K., Schwab, K., Scally, K., Bretthauer, R., and Warden, D. (2009). Traumatic brain injury screening: preliminary findings in a US army brigade combat team. J. Head Trauma Rehabil. 24, 14–23.
Toft, P., Andersen, S. K., and Tonnesen, E. K. (2003). The systematic inflammatory response after major trauma. Ugeskr. Laeg. 165, 669–672.
Turski, P. A., Perman, W. H., Hald, J. K., Houston, L. W., Strother, C. M., and Sackett, J. F. (1986). Clinical and experimental vasogenic edema: in vivo sodium MR imaging. Work in progress. Radiology 160, 821–825.
Wang, J. A., Lin, W., Morris, T., Banderali, U., Juranka, P. F., and Morris, C. E. (2009). Membrane trauma and Na+ leak from Nav1.6 channels. Am. J. Physiol., Cell Physiol. 297, C823–C834.
Warden, D. L., French, L. M., Shupenko, L., Fargus, J., Riedy, G., Erickson, M. E., Jaffee, M. S., and Moore, D. F. (2009). Case report of a soldier with primary blast brain injury. Neuroimage 47(Suppl. 2), T152–T153.
Werner, U., and Szelenyi, I. (1992). Measurement of MPO activity as model for detection of granulocyte infiltration in different tissues. Agents Actions Spec No. C101–C103.
Weston, R. M., Jones, N. M., Jarrott, B., and Callaway, J. K. (2007). Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J. Cereb. Blood Flow Metab. 27, 100–114.
Young, W., DeCrescito, V., Flamm, E. S., Hadani, M., Rappaport, H., and Cornu, P. (1986). Tissue Na, K, and Ca changes in regional cerebral ischemia: their measurement and interpretation. Cent. Nerv. Syst. Trauma 3, 215–234.
Zucker, I. H. (1986). Left ventricular receptors: physiological controllers or pathological curiosities? Basic Res. Cardiol. 81, 539–557.
Keywords: blast, traumatic brain injury, autonomous nervous system, inflammation
Citation: Cernak I (2010) The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front. Neur. 1:151. doi: 10.3389/fneur.2010.00151
Received: 16 November 2010;
Accepted: 24 November 2010;
Published online: 10 December 2010.
Edited by:
Marten Risling, Karolinska Institutet, SwedenReviewed by:
Marten Risling, Karolinska Institutet, SwedenHans Lindå, Karolinska Institutet, Sweden
Copyright: © 2010 Cernak. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ibolja Cernak, Johns Hopkins University Applied Physics Laboratory, 11100 Johns Hopkins Road, Mail Stop MP2 N108, Laurel, MD 20723, USA. e-mail:aWJvbGphLmNlcm5ha0BqaHVhcGwuZWR1