Department of Neurobiology, Center for Glial Biology in Medicine, Atomic Force Microscopy and Nanotechnology Laboratories, Civitan International Research Center, Evelyn F. McKnight Brain Institute, University of Alabama, Birmingham, AL, USA
Astrocytes play an important role in cell–cell signaling in the mammalian central nervous system. The ability of astrocytes to communicate with surrounding cells through gap-junctional coupling or signaling via the release of transmitters makes characterization of these cells difficult in vitro and even more so in vivo. To simplify the complexity of common in vitro systems, introduced by intercellular communication between astrocytes, we developed a novel cell culturing method, in which purified rat visual cortical astrocytes were grown in spatially defined cell-adhesion wells which we termed micropits. We showed that astrocytes cultured in micropit regions were viable and exhibited similar characteristics of Ca2+ dynamics and astrocytic marker expression to those of cells cultured in non-micropit regions. Examination of intracellular Ca2+ oscillations in solitary astrocytes cultured in micropits revealed less variable oscillations than those of non-micropit grouped astrocytes, which were in contact with their neighbors. Solitary cells in micropit regions can undergo ATP-mediated astrocyte-microglia signaling, demonstrating that this culturing method can also be used to investigate glial–glial interactions in a spatially well-defined microenvironment.
Astrocytes, a subtype of glial cells, play an important role in cell–cell signaling in the mammalian brain. Unlike neurons, astrocytes lack electrical excitability and display a form of excitability based on changes in intracellular Ca2+ levels that can be coupled to Ca2+-dependent exocytotic release of neurotransmitter such as glutamate and ATP. Evidence from the past decade has revealed that this exocytotic astrocytic communication pathway may play a role in synaptic neurotransmission in both physiological (reviewed in Haydon and Carmignoto, 2006
) and pathophysiological (reviewed in Jabs et al., 2008
; Rossi et al., 2007
) conditions.
Much of our knowledge about the functions of astrocytes comes from studies in cell culture systems and in acute brain slices, while some of the functional roles of astrocytes in vivo have only been recently examined (reviewed in Tian et al., 2006
). Although it is important to examine the functions of astrocytes in vivo, one needs to be cautious about the interpretation of data from in vivo studies, where the system is inherently complex. Some of the complexity observed in such studies can be attributed to astrocytic interactions with other cell types such as neurons, microglia and vasculature, and also to the intercellular communication among astrocytes via gap-junctional coupling and/or extracellularly released neurotransmitters. Without a clear understanding and characterization of solitary astrocytes, we might not be able to fully appreciate their functional contribution when working with other cells in vivo. Thus, we designed a new culturing approach to reduce intercellular communication to characterize astrocytes in a simplistic manner.
Striped micropatterned substrates generated with the use of microfabricated-polydimethylsiloxane (PDMS) molds have been used previously to control the growth and localization of cells in cell culture systems (Takano et al., 2002
). In this study, we showed that our micropattern cell culturing method using circular micropits can control the growth and localization of cells. Astrocytes cultured by this approach were viable. They exhibited similar characteristics in terms of astrocytic marker expression and Ca2+ dynamics to those of astrocytes grown in groups in a confluent monolayer, although solitary micropit astrocytes displayed less variability of Ca2+ oscillations. The cells cultured in micropits showed a low incidence and degree of intercellular communication with cells in the adjacent micropits. This culturing approach allows for reduced complexity in order to characterize solitary or small networks of astrocytes, and can also be used to study glial–glial interaction to better understand the role that astrocytes may play in the mammalian central nervous system.
Mold Fabrication
The master mold was designed using Autodesk Inventor (Autodesk Inc, San Rafael, CA). The mold was fabricated from a polytetrafluoroethylene (Teflon) block using an Ikegai TV-4 computer controlled four-axis milling machine (Ikegai Corporation, Japan) driven by a Faunc 18M controller (Faunc America, Hoffman Estates, Illinois) using Mastercam software (CNC Software, Tolland, CT). The holes corresponding to the micropits (see section below on Micropatterned Coverslip Preparation) were routed with a 50 μm cobalt micro-drill bit (cat# 53570DIN, Titex Precision Cutting Tools, Frankfurt, Germany).
The Teflon mold served as the master template for casting polydimethylsiloxane (PDMS) molds (Figure 1
). To make the PDMS mold, 10 parts of silicone elastomer base were mixed with 1 part of silicone elastomer curing agent (Sylgard 182; Dow Corning, Midland, MI). The silicone elastomer mixture was then poured onto the Teflon master template and was subjected to a degassing procedure by sonication for 60 min to remove gas bubbles trapped in the mixture. The silicone elastomer mixture was allowed to cure at 70°C for 6 h and the finished PDMS molds were used to generate micropatterned coverslips.
Micropatterned Coverslip Preparation
The micropatterned coverslip preparation scheme is shown in Figure 2
. A round borosilicate glass coverslip (diameter = 12 mm; thickness = 130–160 μm; Erie Scientific Co. D-262 custom glass purchased through Fisher Scientific cat# 12-545-82) pre-coated with polyethyleneimine (PEI, 1 mg/ml) was placed onto the PDMS mold with the PEI-coated side facing the mold (Figures 2
A,B). A 40-g weight was placed on top of the PEI-coated coverslip located on the PDMS mold for 5 min; this step is critical since it ensures that the pins of the mold will make good contact with the glass coverslip and prevent the agarose from covering the PEI-coated cell adhesion surface. A droplet of boiling agarose (2% w/v in water; EMD, Gibbstown, NJ) was added to one end of a channel, designed into the mold to aid in this step, while suction was simultaneously applied at the other end with a pipette tip attached to a vacuum line (Figure 2
C). This step allowed the agarose to fill the void between the PDMS mold and the glass coverslip (Figure 2
C). The agarose was allowed to cool down for at least 20 min before coverslips were removed from the mold. These micropatterned coverslips were then used for subsequent cell culturing procedures. In a subset of experiments examining the spatial relationship between the agarose of the micropatterned coverslip and the astrocytes in micropits, we dissolved rhodamine-6G (1.5 μM; Acros Organics, New Jersey) in agarose.
Atomic Force Microscopy (AFM) Imaging
A Nanoscope E with a J scanner (132 μm × 132 μm maximum scanning area; 5 μm height) (Digital Instruments, Santa Barbara, CA) and Si3N4 gold-coated cantilevers with integral tips (Veeco Nano Probes™ tips, Santa Barbara, CA) were used in contact mode to obtain images of micropits at room temperature (22–26°C). Prior to imaging, micropatterned agarose on coverslips was dehydrated by the addition of increasing concentrations of ethanol (50%, 70% and 95% each for 5 min) and then air dried as previously described (Parpura et al., 1993
). The depth of the micropits was determined by monitoring the voltage used to control the piezo (height mode), while surface features were revealed using deflection mode, where the change in voltage of the photodetector due to cantilever movement is monitored. The AFM imaging force was adjusted to <5 nN to avoid damaging of the dehydrated agarose.
Cell Cultures
Cells were cultured as described previously (Montana et al., 2004
). Briefly, visual cortices were dissected from 0–2-day-old Sprague Dawley rats and treated with papain (20 I.U./ml; Sigma) in Hank’s Balanced Salt Solution (HBSS) (Invitrogen) for 1 h at 37°C. The tissue was washed with HBSS and incubated with trypsin inhibitor (type II-O, 10 mg/ml, Sigma) in HBSS for 5 min. After an additional wash with HBSS, the tissue was triturated in culture medium containing α-minimum essential medium (α-MEM, Invitrogen) supplemented with: 10% (v/v) fetal bovine serum (Hyclone), 20 mM glucose, 2 mM L-glutamine, 1 mM sodium pyruvate, 14 mM sodium bicarbonate, penicillin (100 I.U./ml) and streptomycin (100 μg/ml), pH 7.35. The resulting cell suspension was applied to culture flasks and maintained in culture medium at 37°C in a 5% CO2/95% air incubator for 4–7 days in which cells in the flask reached ~50–60% confluence. At that time cultured cells were submitted to an astrocyte purification procedure. Flasks were shaken twice on a horizontal orbital shaker at 260 rpm, first for 1.5 h, and then after two exchanges with culture medium, shaken again for 18 h. Cells that remained adhered at the bottom of the flask were detached using trypsin (10,000 Nα-benzoyl-L-arginine ethyl ester hydrochloride units/ml; Sigma-Aldrich, St. Louis, MO) dissolved in HBSS and pelleted by centrifugation at 100 × g for 10 min. The resulting cell pellet was resuspended in culture medium and cells were then plated onto micropatterned coverslips. This culturing procedure yields astrocytic culture with purity >99% as confirmed by glial fibrillary acidic protein (GFAP) immunolabeling and accumulation of the dipeptide β-Ala-Lys conjugated to 7-amino-4-methylcoumarin-3-acetic acid (AMCA)(20 μM at 37°C for 2 h) (Montana et al., 2004
). The culture was microglia-free as revealed by the lack of labeling with FITC-conjugated isolectin B4 (see below) and also neuron-free as confirmed by the lack of synaptotagmin I immunoreactivty (Parpura et al., 1994
). To examine the viability of astrocytes, we used the vital dye calcein. The micropatterned coverslips containing astrocytes were incubated in culture medium with 5 μg/ml calcein acetoxymethyl (AM) ester at 37°C for 30 min. The dye was allowed to de-esterify for 30 min at room temperature in normal external solution containing the cell permeant nuclear stain Hoechst 33342 (5 μg/ml; Invitrogen). These coverslips were rinsed in normal external solution and visualized using a standard FITC (calcein) and DAPI (Hoechst) filter sets (Chroma, Rockingham, VT). The normal external solution contained (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.4).
Microglia were isolated from primary cultures in flasks (>2 weeks) by gently tapping the flask to dislodge microglia. The resulting cell suspension was pelleted by centrifugation at 100 × g for 10 min. The pellet was then resuspended and used either for purity assessment or fluo-3 loading (see Ca2+ Imaging section below). The purity of the microglial culture was tested by live-cell labeling with FITC-conjugated isolectin B4 (Sigma-Aldrich). After microglia isolation, the resulting cell pellet was resuspended in FITC-conjugated isolectin B4 (10 μg/ml; 30 min at room temperature). Following incubation with FITC-conjugated isolectin B4, cells were plated on PEI-coated coverslips, rinsed with external solution and then visualized using a FITC filter set. This isolation procedure yielded microglia cells with purity >99%.
Immunocytochemistry
Unless otherwise stated, all solutions used in immunocytochemistry experiments were made with phosphate buffered saline (PBS) containing (in mM): 137 NaCl, 2.7 KCl, 10 Na2HPO4·7H2O, and 1.8 KH2PO4 (pH 7.2). Cell cultures on micropatterned coverslips were fixed with 4% (w/v) paraformaldehyde at room temperature for 30 min and permeabilized using 0.25% Triton X-100 for 10 min. Cells were then incubated with 10% (v/v) goat serum in PBS, to prevent non-specific binding, for 30 min followed by incubation overnight (>12 h) at 4°C with monoclonal primary antibodies to label plasma membrane glutamate aspartate transporter (GLAST) (1:1000 dilution; Millipore, Billerica, MA), GFAP (1:500; MP Biomedicals, Solon, OH), or glutamine synthetase (GS) (1:250; Millipore). After three washes with PBS, cells were incubated with a TRITC-conjugated secondary antibody (1:200; Millipore) at room temperature for 1 h. Cell cultures on micropatterned coverslips were mounted onto a glass microscope slide in glycerol containing 1% (w/v) n-propyl gallate to prevent photobleaching, and visualized using a standard TRITC filter set (Chroma). Parallel controls were performed in which the primary antibodies were omitted to test for non-specific binding of the secondary antibody.
Stimulation of Astrocytes
We used two methods to evoke increases in intracellular Ca2+ concentration ([Ca2+]i) in astrocytes: mechanical stimulation via a glass microelectrode (Charles et al., 1993
; Hua et al., 2004
) or application of agonist via a puffer pipette (Parpura et al., 1994
). Mechanical stimulation of astrocytes was achieved by transient contact with the cells, monitored by pipette resistance using −20 mV, 10 ms square pulses at 50 Hz delivered by a patch-clamp amplifier (PC-ONE; Dagan) equipped with a whole-cell head stage (PC-ONE-30; 1 GΩ). Pipette resistances measured 1.5–2.6 MΩ when pipette tips were filled with and immersed in external solution, which increased to 1.8–5.0 MΩ during transient contact with astrocytes lasting for approximately 1 s. Agonist stimulation of astrocytes was achieved by delivering pharmacological agents including glutamate (100 μM; Sigma-Aldrich), adenosine 5’-triphosphate (ATP) (100 μM; Sigma-Aldrich), bradykinin (BK) (1 μM; Sigma-Aldrich), and norepinephrine (NE) (100 μM; Sigma-Aldrich) via a puffer pipette connected to the PicoSpritzer II pressure ejection system (~70 kPa; Parker Instrumentation, Cleveland, OH).
Ca2+ Imaging
The intracellular Ca2+ levels in astrocytes were monitored using the Ca2+ indicator, fluo-3, by modification of a previously described procedure (Parpura et al., 1994
). Cells were loaded at room temperature in normal external solution, containing fluo-3 AM (10 μg/ml; Invitrogen) and pluronic acid (0.025% w/v; Invitrogen), for 30 min. After washing in normal external solution, the Ca2+ indicator was permitted to de-esterify for 30 min at room temperature in external solution containing Hoechst 33342, which allowed visualization of nuclei to select solitary astrocytes (Figures 6
–7
,9 and 11
) or small networks of these cells (Figure 11
) in the micropit culture. Coverslips containing dye-loaded cells were mounted onto a recording chamber and imaged. A FITC filter set was used to monitor fluo-3 fluorescence and a DAPI filter set was used for Hoechst. All experiments were performed at room temperature (22–26°C). Data were background subtracted using a fluorescence image acquired in a region of coverslip without cells. Fluo-3 intensity changes of entire astrocytes were expressed as dF/Fo (%), where dF represents the change of fluorescence after cell stimulation and Fo represents the fluorescence of the cell before stimulation. Astrocytes were considered responsive to a stimulus if dF/Fo values in two consecutive images were higher than the Fo + 3SDs.
In a subset of experiments examining the astrocyte-microglia interaction in micropits, we loaded astrocytes plated on micropatterned coverslips with Fura Red AM (2 μM; Invitrogen) and pluronic acid (0.025% w/v) for 20 min at 37°C. Dye was allowed to de-esterify for 20 min while cells were kept at room temperature in normal external solution. Coverslips containing Fura Red loaded astrocytes were mounted to the imaging chamber and fluo-3 preloaded microglial cells were added to the imaging chamber. We then waited ~10 min for microglia to settle onto the bottom before we proceeded with the experiment. The pre-loading of microglia in suspension was done at 37°C for 20 min in culture media containing fluo-3 AM (10 μg/ml) and pluronic acid (0.025% w/v), and then cells were pelleted by centrifugation at 100 × g for 10 min. The resulting cell pellet containing fluo-3 loaded microglia was resuspended in normal external solution and then the dye was permitted to de-esterify for 30 min at room temperature. The fluo-3 pre-loaded microglia were then used for experiments. The fluorescence emissions of the Ca2+ indicators from microglia and astrocytes were simultaneously monitored using a FITC Long pass filter set (exciter: 465–495 nm; dichroic: 505 DCLP; emitter: 515 nm long pass) where the fluo-3 fluorescence emission shows green (emission max ~526 nm), while Fura Red fluorescence emission shows red (emission max ~660 nm).
Extracellular Glutamate Imaging
We used an enzymatic assay that has been previously used to optically monitor the extracellular glutamate dynamics (Hua et al., 2004
; Innocenti et al., 2000
). This assay is based on monitoring NADH fluorescence that is produced when glutamate is converted to α-ketoglutarate by the enzyme L-glutamate dehydrogenase (GDH), while nicotinamide adenine dinucleotide (NAD+) is concomitantly converted to NADH. Given that GDH and NAD+ are added to the solution in which the astrocytes are bathed, glutamate released in the extracellular milieu can be detected as an increase in NADH fluorescence.
Glutamate imaging was accomplished using a standard DAPI filter set. Astrocytes were bathed in an enzymatic assay solution containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 5 glucose, 1 NAD+ (Sigma), and GDH (59 I.U./ml; Sigma), pH 7.35. Every experiment was preceded by a sham run (same exposure time but no mechanical stimulus applied) on cells bathed in solution lacking GDH and NAD+. This was used to correct for photobleaching and background subtraction in all imaging data. The reduction of fluorescence resulting from photobleaching in the sham run exhibited the same time course as in the matching experimental run. Data were expressed as dF/Fo (%), where Fo represents the fluorescence level before stimulation, whereas dF represents the change in fluorescence surrounding the astrocytes.
Light Microscopy
For experiments with cultured astrocytes, visualization was done using an inverted microscope (TE 300; Nikon) equipped with differential interference contrast (DIC) and wide-field epifluorescence. Immunocytochemistry and Ca2+ imaging experiments were performed using a 60X Plan Apo oil-immersion objective (1.4 NA; Nikon). For extracellular glutamate imaging experiments, we used a 40X SFluor oil-immersion objective (1.3 NA; Nikon). We also used a 20X plan-Fluor (0.5 NA; Nikon) objective: (1) when monitoring the intracellular Ca2+ levels of cells in two micropits simultaneously, (2) for examining calcein dye loading, (3) for visualization of Hoechst 33342 labeling in astrocytes, and (4) for imaging of cross-sections of the PDMS mold. Images were captured using a CoolSNAP-HQ cooled CCD camera (Roper Scientific) driven by V++ imaging software (Digital Optics, Auckland, New Zealand) which was also used for image processing and data analysis.
Spinning disc confocal imaging was performed on an Olympus IX81 microscope equipped with a Disc Scanning Unit (DSU) and a 60X Plan Apo objective (1.4 NA; Olympus). Top view images of the PDMS mold were captured using a color CCD camera (Fisher catalog# 12-563-1; Westover Scientific, Woodinville, WA) mounted on a Leica GZ6 stereomicroscope (Bannockburn, IL) and driven by LabView/IMAQ (National Instruments, Austin, TX).
Ca2+ Oscillations Analysis
Ca2+ transients from background subtracted fluo-3 emission traces were declared Ca2+ peaks (Table 1
) if over two consecutive images their dF values exceeded the fluorescence emission value of a 9-point moving average +2 standard deviations. The frequency components in intracellular Ca2+ concentration ([Ca2+]i) oscillations in astrocytes were determined by spectral analysis written for use in MATLAB (The Mathworks, Natick, MA) as previously described (Aizman et al., 2001
; Uhlen, 2004
; Uhlen et al., 2000
). Briefly, the ATP-evoked portion of the [Ca2+]i signal from a single cell was filtered, centered, and trend-corrected by computing Gauss least-squares approximation to subtract the trend component from the original data. The dominant frequency peak was determined by comparing the relative power of the peaks in the power spectrum. The relative power was calculated by taking the integral of a peak divided by integral of the entire power spectrum. We further considered only peaks if their power exceeded 5% of total power and if their frequency was above the cut-off frequency of 11 mHz, imposed by the length of agonist application (180 s) and the Nyquist-Shannon sampling theorem.
Statistical Analysis
The levels of [Ca2+]i upon various astrocytic stimulation procedures and expression levels of astrocytic makers in immunocytochemistry experiments were compared between astrocytes cultured in micropit and non-micropit area using Student’s t-tests. Glutamate release due to mechanical stimulation was assessed using paired t-tests. The effect of suramin on Ca2+ dynamics was assessed by Student’s t-tests. Statistical significance is defined as p < 0.05. Data were expressed as mean ± standard error of mean (SEM).
PDMS Mold Design and Micropit Characterization
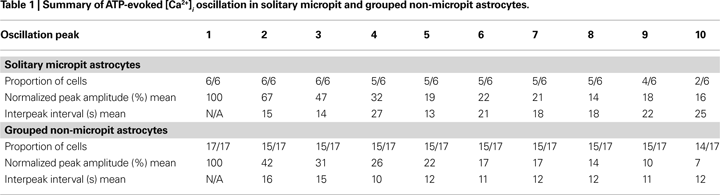
We designed a cell culturing approach to limit intercellular communication pathways to study and characterize solitary astrocytes or small networks of these cells in culture. The PDMS mold served as the master template to micropattern a non cell-adhesive substrate (agarose) onto glass coverslips pre-coated with a cell-adhesive substrate (PEI). The design and specifications of the PDMS mold are shown in Figures 1
A–C. The PDMS mold consists of two raised bilateral circular segments with an arrangement of pins between them consisting of a 39 × 15 array flanked by an 11-pin column on each side (Figure 1
D). The segments and pins are 50 μm in height, while the pins are 75 μm in diameter and positioned 300 μm apart from each other from center to center (Figure 1
B). The height of the outer rim, from the segment to the top of the rim, is 200 μm (Figure 1
E) and this distance is slightly higher than the thickness (130–160 μm) of the glass coverslip to ensure proper positioning of the glass coverslip on the PDMS mold. The specifications of the PDMS mold were verified, as shown in the bright field images in Figures 1
E–G, and the measurements of the mold were consistent with the designed specifications.
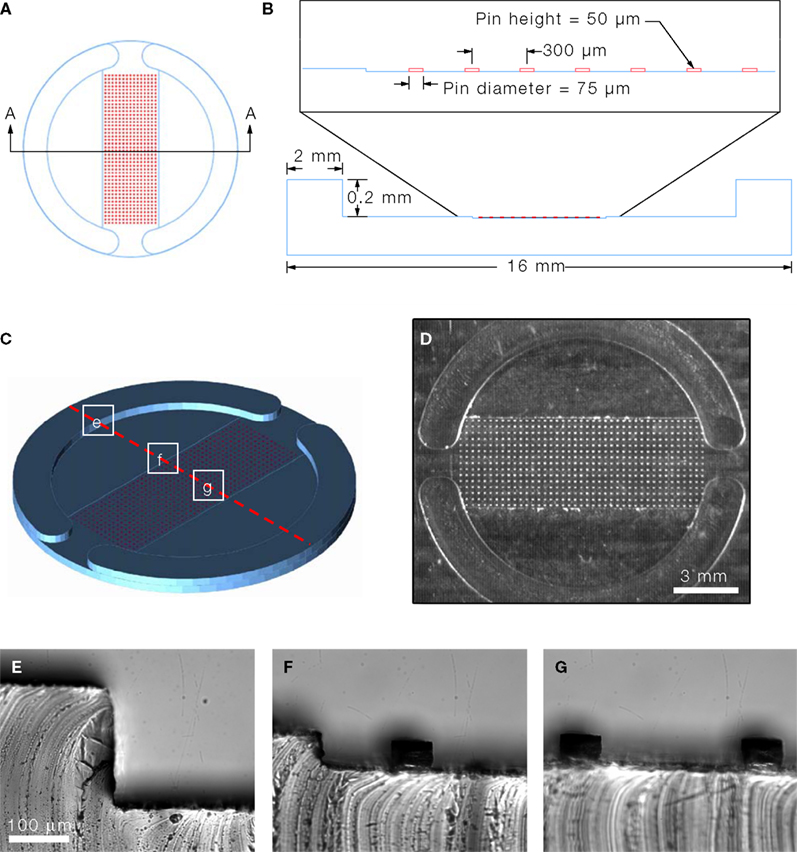
Figure 1. PDMS mold design. (A) A Computer-Aided Design (CAD) image of the PDMS mold. (B) The cross-section [A–A line in (A)] of the designed PDMS mold with its specifications. (C) 3-D CAD image of the PDMS mold design. The mold was cut and cross-sectional images were taken at the positions indicated by the boxed regions along the dashed red line and the corresponding bright field images are shown in (E–G). (D) A top view image of the PDMS mold. (E) The height of the outer rim is 200 μm. (F) The pins are 75 μm in diameter and with a height of 50 μm consistent with designed specification. (G) The inter-pin distance from center to center is 300 μm. Scale bar in (E) applies to panels (E–G).
The micropatterned coverslips were generated by micropatterning agarose on a PEI-coated coverslip (Figure 2
). This approach has previously been used to control the localization of cells in defined microdomains (Takano et al., 2002
). Here we utilized agarose as a cell adhesion inhibitor to generate well-defined circular cell adhesive surfaces, which we termed micropits, to control the localization and growth of cells in culture as illustrated in the schematic diagram in Figure 2
E. Cells plated on these micropatterned coverslips can either grow in the micropit or non-micropit regions (Figure 2
D). While other available methods have been used to control the localization and growth of cells in micropatterned cell adhesive domains (Bekkers and Stevens, 1991
; Chen et al., 1997
; Recknor et al., 2004
; Takano et al., 2002
), their micropatterning approaches were not designed to limit intercellular communication pathways which is the goal behind our PDMS mold design. We spaced the micropits 300 μm apart and this arrangement was tested for the extent of intercellular communication between the cells in different micropits (see Results below). The surface area (SA) of each micropit is 4417 μm2. Since there are 607 micropits per micropatterned coverslip, the total micropit SA corresponds to ~2.4% of the total SA of a PEI-coated glass coverslip (~113 mm2). The SA in the non-micropit area (two segmental regions of the coverslip) corresponds to ~53% of the SA of the coverslip. Astrocytes grown on permissive substrates can divide and grow to reach a confluent layer of cells since these cells display cell-to-cell contact inhibition, whereas cells in micropits are spatially confined by the area of permissive substrate which limits their growth. The rationale behind co-culturing cells on the non-micropit regions is to utilize these cells as support cells for cells cultured in micropits since they can provide glial-trophic factors and also to comparatively study micropit vs. non-micropit cells grown on the same coverslip.
Figure 2. Micropatterned coverslip preparation scheme.
(A) A polyethyleneimine (PEI)-coated coverslip was placed on top the PDMS mold with the PEI-coated side facing the PDMS mold as shown in (B).
(C) Boiling agarose (2% w/v in water) was added to one of the open ends while gentle suction was applied at the other end to draw agarose into the channel until it was completely filled. The agarose was allowed to cool down and then the micropatterned coverslip was removed from the PDMS mold. (D) The micropatterned coverslip consists of a micropit array in the middle portion with non-pit segmental regions on each side. Plating and culturing astrocytes on these micropatterned coverslips yields cells grown in the micropit regions as shown in the schematic diagram in (E) as well as grouped cells in the two non-micropit regions on either side.
We characterized the micropit domains of the micropatterned coverslips using Atomic Force Microscopy (AFM) (Figure 3
). The AFM images of a micropit in deflection mode (Figure 3
A), height mode (Figure 3
B), and topographic representation (Figure 3
C) showed that the base of the micropit is flat and mostly devoid of agarose. While the AFM allowed us to examine the surface topography of the micropit, it can not disclose the entire pit due to tip convolution (Keller and Franke, 1993
). For example, the rim of the micropit as shown in Figure 3
C does not appear to be vertical due to the opening angle of the pyramidal tip integrated on a cantilever (see Figure 3
D). Here, the opening angle of the AFM tip is ~59°, which is in good agreement with the manufacturer’s specification. It should be noted that the measurable angle of the pit wall along the direction of the scan differs from one side to another owing due to the cantilever tilt in scanning direction. The smoothness of the cell adhesion surface of the micropit was examined and we found that the root mean square of height was 45.1 ± 9.6 nm (n = 6). We next examined the dimensions of the micropit (Figure 3
D). We found the micropit diameter was 79 μm (the horizontal distance between cursors), while the depth was ~1 μm (vertical distance between cursors). The measured depth of the micropit was ~1 μm instead of 50 μm like the pins because the micropatterned coverslips were dehydrated for the AFM which caused shrinkage of the agarose. Thus, the depth of the micropit corresponds to ~2% of the design height of 50 μm under hydrated conditions and altogether the measurements of the micropits were consistent with the PDMS mold design.
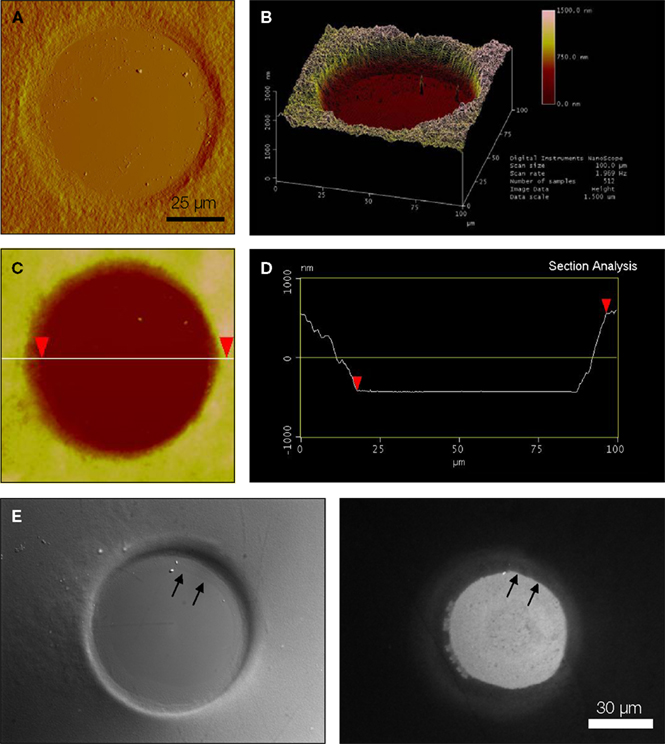
Figure 3. Characterization of micropits. Panels (A) and (B) are atomic
force microscopy (AFM) images of a micropit in deflection mode and height mode, respectively. (B) The line shown in the height mode AFM image corresponds to the scanning direction of the AFM tip across the sample and the red cursors correspond to those in (D). (C) A topographic representation of the micropit. (D) The section analysis of the micropit shows the micropit
with a diameter of 79 μm and a vertical distance of 998 nm. The vertical distance of the micropit measured with AFM is smaller than 50 μm since the 2% (w/v) agarose sample is dehydrated for use in the AFM. (E) The PEI-coated cell-adhesion surface of the micropit labeled with FITC-conjugated heparin as shown in DIC image (left panel) and epifluorescence image visualized with a FITC filter set (right panel). The strong labeling of PEI-coated surface with
FITC-conjugated heparin shows that heparin can bind to the cell-adhesion surface of the micropit but not in regions of the coverslip covered by agarose. There is a radial gradient of heparin binding evident near the edge of the micropit indicated by two arrows where there is a decay in fluorescence intensity until it reaches the background level near the border of
the micropit.
To determine whether any agarose could leak under the pin into the base of the micropit area forming a thin-layer which could prevent cell adhesion, we applied FITC-conjugated heparin, a poly anionic molecule that binds to the positively charged PEI coated glass surface (Figure 3
E). We found that the surface of the micropit was mainly devoid of agarose and thus available for cell adhesion, as indicated by labeling with FITC-conjugated heparin (right panel of Figure 3
E). Furthermore, our results demonstrated that FITC-conjugated heparin can effectively be used to label the cell adhesion surface of the micropatterned coverslip and this assay can be routinely used to test the quality of micropatterned coverslips for cell culturing.
Astrocytic Culture in Micropits
We next examined the occupancy of the micropatterned coverslips by astrocytes and the morphology of these cells (Figures 4
A,B). Solitary astrocytes can be cultured in micropits where they expand to cover most of the available cell-adhesion surface of the micropit as shown in the DIC image of a live solitary astrocyte in a micropit (Figure 4
A). The astrocyte in the micropit assumed a polygonal morphology similar to that of cells cultured in non-micropit regions. The spatial relationship of the cell to the micropit was examined using scanning confocal microscopy (Figure 4
B). In this experiment, the astrocyte was visualized by incubating it with β Ala-Lys-Nε-AMCA peptide (Figure 4
B; DAPI channel, blue), which is selectively taken up by astrocytes via the PepT2 transporter (Dieck et al., 1999
). This allowed for examination of the cytoplasmic volume of the cell. The astrocyte in the micropit was hemi-spherical and it covered the majority of the cell adhesive surface in the micropit. To label the surrounding micropit, the agarose used was doped with unconjugated rhodamine 6G (Figure 4
B; TRITC channel-red). Some of this dye was able to diffuse out of the agarose and, consequently, can be seen within the astrocyte as punctate stain most likely reflecting its endocytosis by the astrocyte.
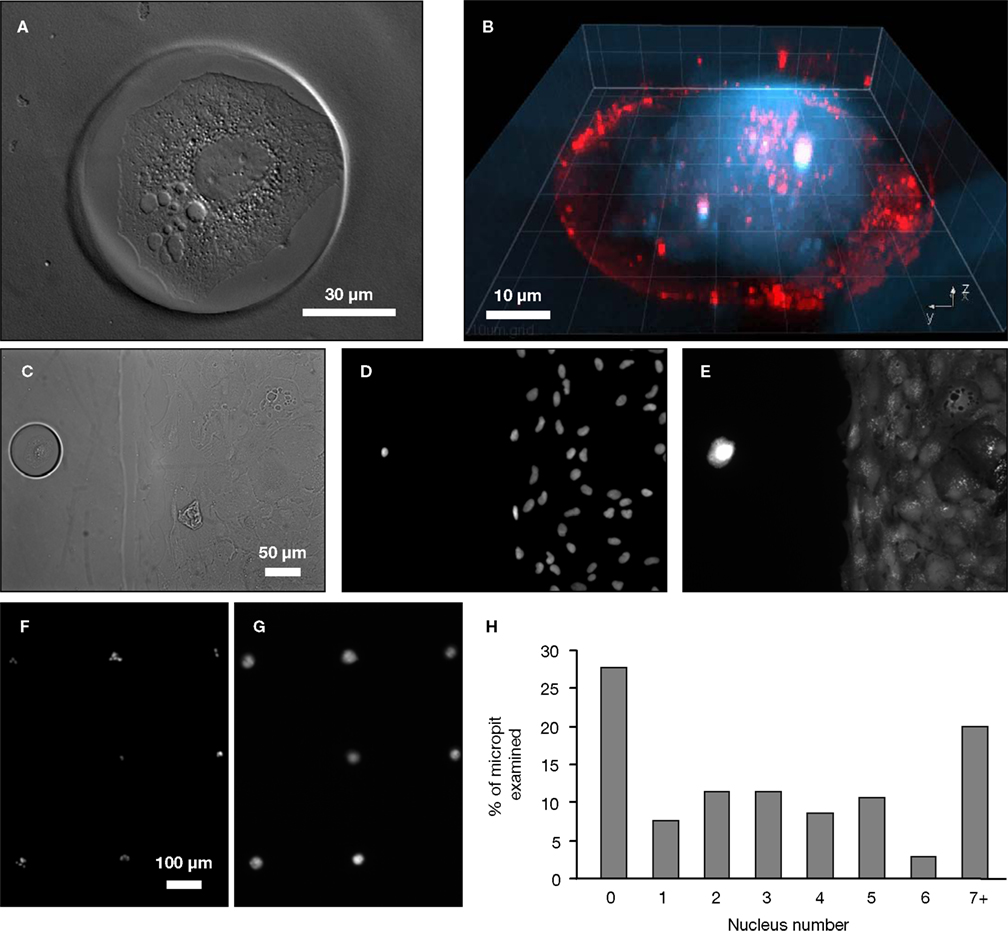
Figure 4. Cell cultures on micropatterned coverslips. (A) A DIC image of a live solitary astrocyte in a micropit. This cell has a polygonal morphology and it covers the majority of the cell adhesive surface. (B) A confocal image of a live solitary astrocyte that had transported the fluorescently labeled dipeptide β-Ala-Lys-AMCA into the cell resulting in a diffuse blue labeling of the cytoplasm. Rhodamine-containing agarose was used to visualize the boundaries of the micropit. The non-conjugated rhodamine which had diffused out of the agarose was endocytosed by the astrocyte causing red punctate labeling within the cytoplasm of the cell. (C–G) Viability of cultured astrocytes. Panels (E) and (G) show the viability of cells cultured on micropatterned coverslips examined using the vital dye calcein. (E) An image of calcein dye loading and retention in a solitary micropit cell and in grouped non-micropit cells. The corresponding bright field image (C) and epifluorescence image of nuclei labeling with Hoechst 33342 (D) are shown. Small networks of cells were found in micropits as shown in (F) where more than one nucleus present in the micropits was shown by Hoechst 33342 labeling; the corresponding image of calcein-loaded cells is shown in (G). (H) A frequency histogram of the number of astrocytes occupying each micropit (n = 210) as revealed by the nucleus/cell number. Scale bar in (C) applies to panels (D–E), while scale bar in (F) applies to (G).
To test the viability of astrocytes cultured in micropits, we loaded cells with calcein AM and examined the dye retention in the astrocytes (Figures 4
C,E,G). We found that culturing cells in micropits did not alter their viability since accumulation of this vital dye in micropit cells was comparatively similar to that of non-micropit cells (Figures 4
E,G). Since we are interested in studying small networks of astrocytes as well as solitary cells using these micropits, we counted the number of astrocytes that resided within each micropit by staining their nuclei with cell permeant Hoechst 33342 (Figures 4
C,D,F). Indeed, solitary astrocytes and small networks of astrocytes can be cultured within micropits (Figures 4
F,H). At a plating cell density of 1 × 104 cells/ml, we found that ~72% of micropits examined were occupied by cells, while ~28% were not (Figure 4
H). In micropits occupied by cells, ~44% of micropits contained small networks of cell with 2–6 astrocytes, ~8% contained solitary astrocytes, and ~20% contained clusters of seven or more cells. Since the area of the micropit dictates the number of cells that can adhere to it, it appears that the area designed could accommodate for a maximum of six cells which were well attached directly to the micropit surface. In micropits that contained clusters of seven or more cells, only small portions of the cells in the cluster were able to adhere to the cell-adhesion surface of the micropit. Such cells could not be selected for further experimentation because we could not differentiate individual cell responses within the cell clusters.
After confirming that astrocytes cultured in micropits were viable, we then examined whether culturing astrocytes in micropits would alter their typical gene expression profile. To test this, we performed immunocytochemistry experiments to label for the astrocytic markers: GFAP, GLAST, and glutamine synthetase. We found that astrocytes cultured in micropits expressed all three astrocytic markers examined (Figure 5
). Astrocytes cultured in non-micropit regions of the micropatterned coverslip showed heterogeneity in expression of the astrocytic markers within a subpopulation of cells (Figures 5
A–C, bottom right panels) and such heterogeneity was also present in small networks of cell within micropits (Figures 5
A–C, middle right panels). Quantitative analysis of fluorescence intensity showed that only GFAP fluorescence displayed a significant difference between micropit and non-micropit cells (Figure 5
C, graph). The increase of fluorescence intensity for GFAP labeling in micropit astrocytes might be due to the difference in thickness of the cells cultured in micropits versus those in non-micropit regions. If a micropit cell is thicker then it would appear to have more labeling for GFAP per unit area. Alternatively, it could be that the increase fluorescence represents the higher expression of GFAP regularly seen in reactive astrocytes (see Discussion).
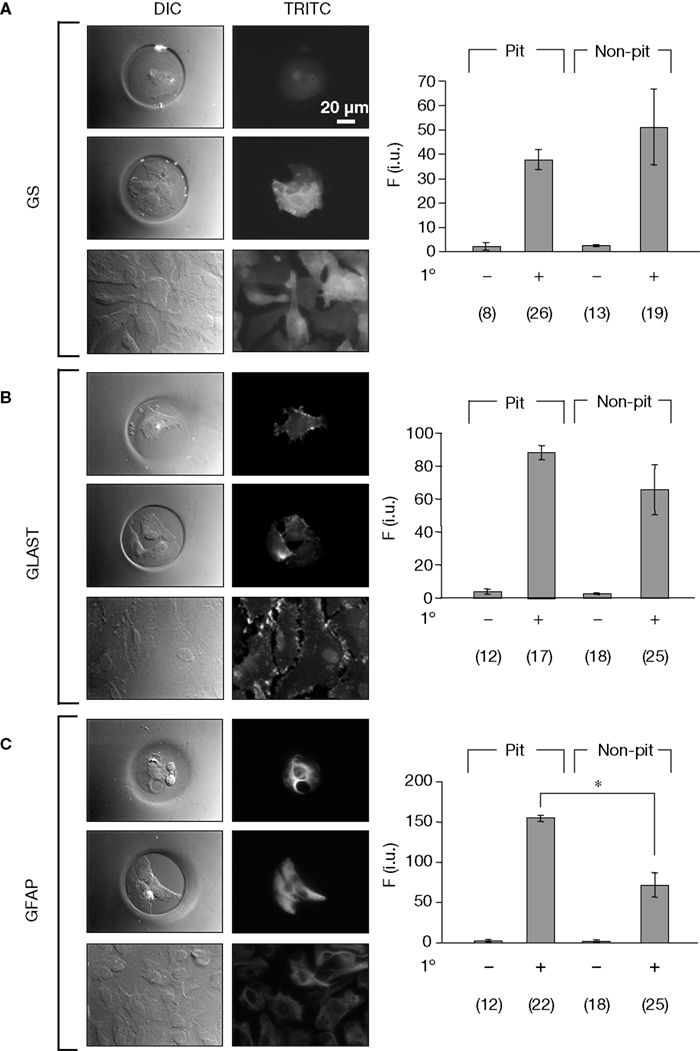
Figure 5. Astrocytes in micropits express astrocytic markers. Astrocytes were labeled using primary antibodies against glutamine synthetase (GS), plasma membrane glutamate aspartate transporter (GLAST), or glial fibrillay acidic protein (GFAP) followed by TRITC-conjugated secondary antibodies (A–C). The images in each panel show fluorescence of solitary cells in micropits (top), small networks of cells in micropits (middle), and grouped non-micropit cells (bottom) with the corresponding differential interference contrast (DIC) images shown on the left. The bar graphs on the right of each panel show the quantification of fluorescence intensity (F) in intensity units (i.u.) within the camera’s dynamic range (0–4095) shown as mean ± SEM; 1° indicates the presence (+) or absence (−) of primary antibody against the astrocytic marker. The numbers of cells tested are shown in parentheses. The GFAP labeling was significantly higher in micropit astrocytes compared to non-micropit astrocytes (Student’s t-test; *p < 0.05).
Intracellular Ca2+ Dynamics and Ca2+-Dependent Glutamate Release
Astrocytes exhibit a form of excitability based on changes in [Ca2+]i upon agonist stimulation (Charles et al., 1991
; Cornell-Bell et al., 1990
; Jensen and Chiu, 1990
). This Ca2+ excitability can be coupled to Ca2+-dependent release of glutamate (Parpura et al., 1994
) and ATP (Coco et al., 2003
). To study this, we quantitatively analyzed the stimulus-evoked Ca2+ responses in solitary micropit astrocytes and compared responses to those of single cells within grouped-astrocytes in non-micropit regions (Figure 6
). Two stimulation procedures were used to elicit [Ca2+]i elevations: (1) focal mechanical stimulation (Figure 6
A) and (2) local application of agonists via puffer pipette (Figures 6
B–E). Mechanical stimulation activates intracellular signaling transduction to elevate [Ca2+]i. This method provides stimulation with good spatiotemporal control (Charles et al., 1993
). In comparison, elevation of [Ca2+]i with agonist stimulation in astrocytes is dependent on the expression and activation of plasma membrane metabotropic or ionotropic receptors. While the source for the elevated [Ca2+]i in astrocytes comes primarily from the internal stores, extracellular Ca2+ may also contribute to [Ca2+]i elevation (Hua et al., 2004
; Malarkey et al., 2008
). We found that both mechanical and agonist stimulations caused an increase in [Ca2+]i in micropit and non-micropit astrocytes. The astrocytic Ca2+ responses of solitary astrocytes in micropits were generally similar to those of astrocytes in non-micropit regions in terms of peak and cumulative responses. However, the peak Ca2+ responses elicited by mechanical and norepinephrine stimuli were significantly lower in micropit astrocytes (Figures 6
A,D).
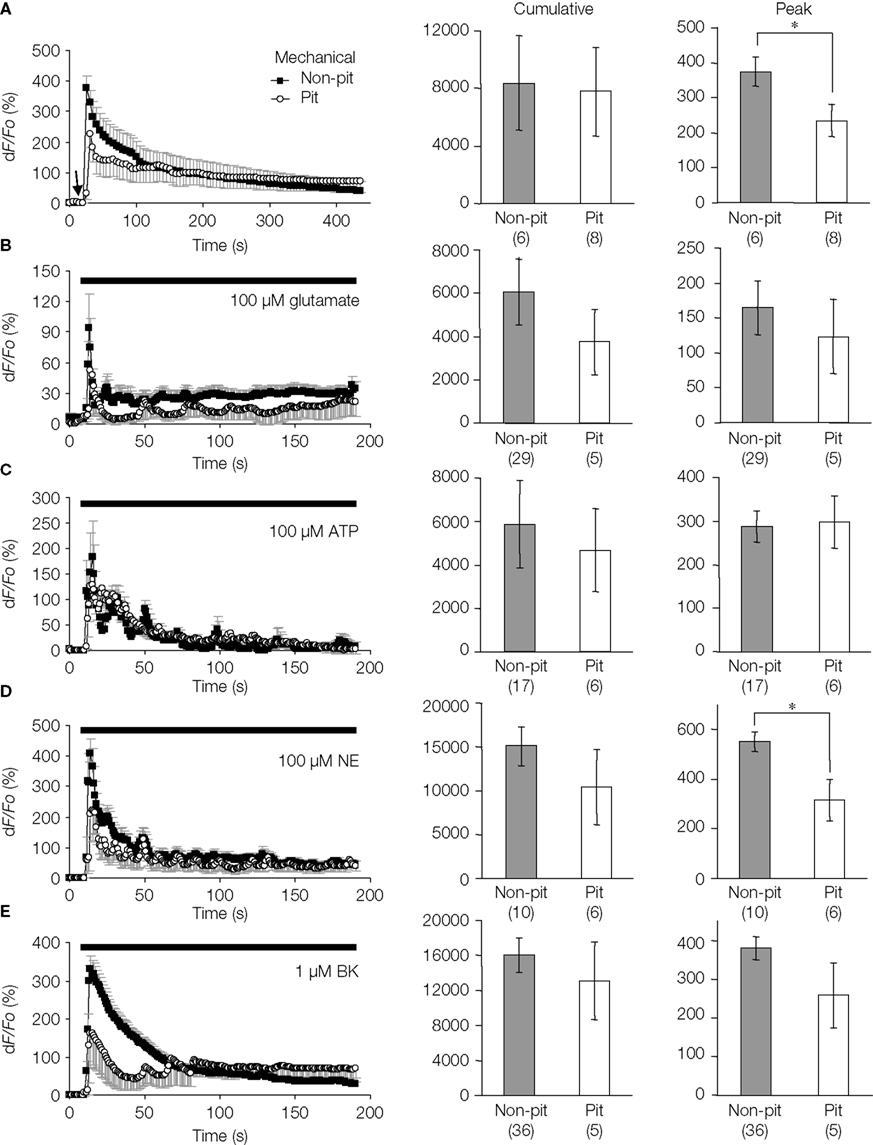
Figure 6. Intracellular Ca2+ dynamics of astrocytes on micropatterned coverslips. (A–E) Comparison between the intracellular Ca2+ dynamics of solitary astrocytes in micropits and single astrocytes grown within grouped astrocytes in non-micropit regions after mechanical or agonist stimulation. The graphs on the left side represent fluo-3 fluorescence changes expressed as dF/Fo (%), with the points and error bars representing the mean and SEM (shown only in one direction for clarity). The arrow in panel (A) denotes the time that mechanical stimulation was applied to the cell. Horizontal bars in graphs (B–E) indicate the application of agonists: ATP, adenosine 5’-triphosphate; NE, norepinephrine; BK, bradykinin. Two sets of bar graphs on the right show quantification of cumulative and peak intracellular Ca2+ responses in astrocytes grown in micropit and non-pit regions of coverslips. The numbers of cells tested are shown in parentheses. The peak Ca2+ responses in non-micropit cells were significantly higher (Student’s t-test; *p < 0.05) than in solitary micropit cells, upon mechanical (A) or NE stimulation (C).
We next tested if the agonist-evoked Ca2+ dynamics included oscillations. Indeed, stimulation with glutamate, ATP, and norepinephrine caused clear oscillatory increases in [Ca2+]i in solitary micropit astrocytes similar to those observed in grouped non-micropit astrocytes. These results are in good agreement with studies examining Ca2+ oscillations in grouped cells with intact intercellular communication pathways (Cornell-Bell et al., 1990
; Muyderman et al., 2001
; Pasti et al., 1995
, 2001
) and also solitary astrocytes in regular dissociated cell cultures (Hua et al., 2004
). To further examine [Ca2+]i oscillation patterns, we implemented spectral analysis of [Ca2+]i responses in a subset of astrocytes stimulated by ATP (Figure 7
). We find that the majority of solitary micropit astrocytes (4 out of 6; 67%) exhibited ATP-evoked [Ca2+]i oscillations having dampening of amplitude with either regular interpeak intervals (two astrocytes; e.g., Cell 1 in Figures 7
A,C) or some change in interpeak intervals (two astrocytes; e.g., Cell 2 in Figure 7
C). The power spectrum corresponding to the [Ca2+]i oscillation trace of Cell 1 showed one fundamental frequency at 82 mHz and two harmonic frequency at 164 and 246 mHz (Figure 7
E). The predictable exponential decay of the power spectral density at each of the harmonic frequencies indicates that the solitary cell only exhibited one dominant oscillation frequency. The power spectrum of Cell 2 showed the peaks of the power spectrum densities also decay exponentially similar to the cell that exhibited regular inter-peak interval (Figure 7
E). However, subsequent peaks after the dominant frequency did not fit as well as harmonics of the dominant frequency, in this case, most likely due to dampening of frequency. The remaining two solitary micropit astrocytes did not show dampening of peak amplitude/frequency; one of them showed two peaks of equal power, while the other one displayed a complex power spectrum with peaks at variable frequencies and variable powers, similar to power spectra seen in single cells within a group of astrocytes in non-micropit region. In contrast, the [Ca2+]i response and power spectrum of a single cell within a group of astrocytes in non-micropit region exhibited a greater degree of variability which might be due to cross-talk between astrocytes via the intercellular communication pathways that are present in grouped astrocytes as mentioned above. The major category of grouped astrocytes (8 out of 17; 47%) exhibited complex ATP-evoked [Ca2+]i oscillations (Figures 7
B,D) and also complex power spectrum with peaks at variable frequencies and variable powers (Figure 7
F). The remaining grouped astrocytes (53%) could be categorized between three equally represented types (three cells in each category): (1) astrocytes that displayed no significant power spectrum peaks above the cut-off frequency; (2) astrocytes showing only a single power spectrum peak (i.e., Cell 1 type, but lacking harmonics); and (3) astrocytes showing power spectrum peaks with amplitude decay and change in frequency (Cell 2 type). Further comparison of ATP-evoked [Ca2+]i elevations between solitary micropit and non-micropit astrocytes (Table 1
) indicates that the normalized peak Ca2+ responses in subsequent oscillation peaks were generally higher with longer interpeak intervals in solitary micropit astrocytes when compared to non-micropit astrocytes. Altogether, the astrocytes with intact intercellular communication pathways exhibited a more variable Ca2+ response when compared to solitary micropit astrocytes. Thus, the design of our micropattern cell culture approach achieved the goal of reducing the complexity due to intercellular communication among cells in groups.
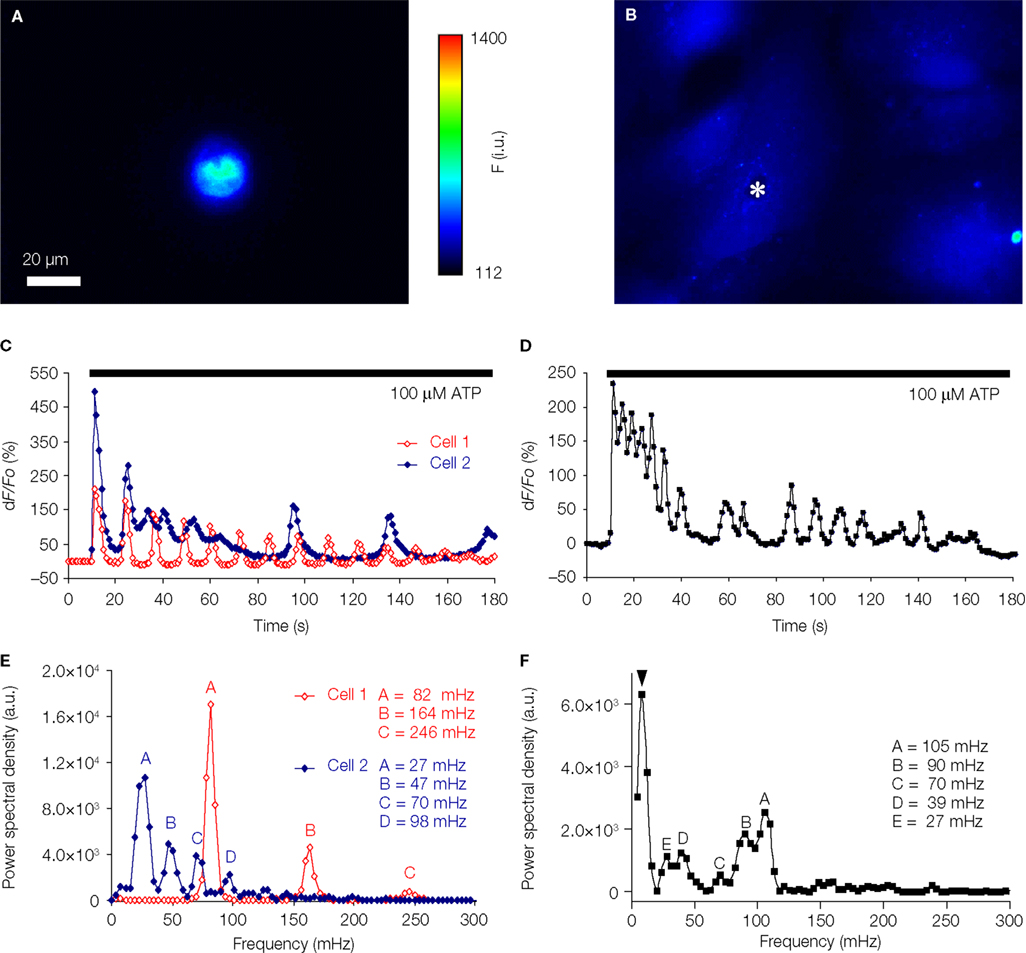
Figure 7. Solitary micropit astrocytes exhibit agonist-evoked [Ca2+]i oscillations. (A) A solitary micropit astrocyte and (B) grouped non-micropit astrocytes loaded with the Ca2+ indicator fluo-3. The pseudo color scale represents the fluorescence intensity (F) ranging 112–1400 intensity units (i.u.). The time-lapse sequence of the intracellular Ca2+ response of a solitary micropit astrocyte in (A) stimulated with ATP is shown in (C, Cell 1) and also in Supplementary Movie 1, while the time-lapse sequence or a single astrocyte [marked with asterisk in (B)] within group of astrocytes grown in non-micropit region is shown in (D) and also in Supplementary Movie 2. (C) Two examples of the intracellular Ca2+ response of a solitary micropit astrocyte stimulated with ATP. The [Ca2+]i oscillations from
Cell 1 (red trace) shows regular inter-peak intervals and a dampening in amplitude. The [Ca2+]i oscillations from Cell 2 (blue trace) showed dampening in peak amplitude as seen in Cell 1 and in addition it showed an increase in inter-peak intervals. (D) An example of ATP-evoked [Ca2+]i response of a single cell within the group of astrocytes in the non-micropit region. The intracellular Ca2+ response displays an oscillatory pattern with various inter-peak intervals. The power spectra in (E) and (F) show the frequency components of the oscillations corresponding to the Ca2+ signals in (C) and (D), respectively. (E) The power spectrum of Cell 1 shows only one dominant frequency of 82 mHz (peak A). Peaks B and C are 2nd and 3rd harmonics because their frequencies are multiple integers of the fundamental frequency. The power spectrum of Cell 2 shows a dominant frequency (peak A) at 27 mHz and subsequent peaks with decaying amplitude. (D) The power spectrum of the non-micropit astrocyte within the group of astrocytes was more complex with multiple peaks of various frequencies ranging from 27–105 mHz. The power spectrum density is given in arbitrary units (a.u.). Arrowhead in (F) indicates a peak that is below the cut-off frequency.
Astrocytes can undergo Ca2+-dependent release of gliotransmitters such as glutamate (Parpura et al., 1994
) and ATP (Coco et al., 2003
; Pangrsic et al., 2007
). Therefore, we tested the ability of solitary micropit astrocytes to undergo Ca2+-dependent release of glutamate induced by mechanical stimulation. The release of ATP from astrocytes was tested using astrocyte-microglia signaling (see below). To examine glutamate release from solitary micropit astrocytes, we used an optical approach that has been previously used to visualize mechanical stimulation-evoked glutamate release from astrocytes (Hua et al., 2004
; Innocenti et al., 2000
; Malarkey et al., 2008
). We imaged glutamate release at two different focal planes, and NADH fluorescence intensity was analyzed in the regions of interest (ROIs) adjacent to the cell (Figure 8
A) At focal plane A, which was at the level just above the rim of the micropit, the GDH enzyme in the presence of glutamate can convert NAD+ to NADH, which fluoresces when excited using a DAPI filter set. However, at focal plane B, which was at the level of the cell in the micropit just above the cell interface with the coverslip, the ROIs would fall within the agarose which is less accessible to GDH.
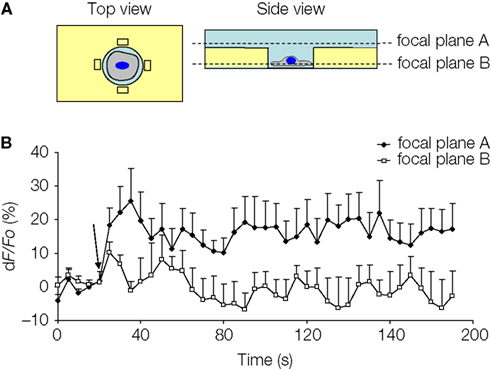
Figure 8. Solitary micropit astrocytes can release glutamate upon mechanical stimulation. (A) Schematic diagram of the experimental setup in top and side views. The regions of interest (ROIs; rectangles) we selected for this analysis were placed close to the edge of the micropit (top view). The side view showing the imaging planes (A and B) selected during GDH imaging experiments. (B) The time lapse of changes in NADH fluorescence expressed as dF/Fo (%). Each point on the graph represents the mean and SEM (shown only in one direction for clarity). The arrow denotes the time of mechanical stimulation. Upon stimulation, there was an increase in NADH fluorescence, reporting on extracellular glutamate, detected in the ROIs (extracellular space with external solution) when time lapse images were obtained in focal plane A (n = 5 cells) above the agarose. In parallel experiments, we detected glutamate release, albeit with reduced levels, in the ROIs when time lapse images were obtained in focal plane B (n = 5 cells) within the agarose. This reduced detection of glutamate release was most likely due to the restricted accessibility of agarose to GDH.
We verified that the ROIs at focal plane B reported on fluorescence changes within the agarose at the glass coverslip-agarose interface, by calculating the depth of field for the 40× objective using the equation:

where δ is the depth of field, n is the refractive index of the hydrated agarose (1.38) (Chen-Izu et al., 2006
; Hollingworth et al., 2000
), λ is the wavelength of emitted light (0.46 μm), and NA is the numerical aperture of the objective (1.3). The calculated depth of field was 0.75 μm. Owing to the thickness of agarose (50 μm), the ROIs at focal plane B thus reported on fluorescence changes within the agarose layer at the interface with the glass coverslip. Indeed, upon mechanical stimulation of the solitary micropit astrocytes, we found significant elevation of NADH fluorescence in the ROIs at both the focal plane A (n = 5; peak dF/Fo = 32 ± 7%; p < 0.05, paired t-test) and the focal plane B (n = 5; peak dF/Fo = 20 ± 5%; p < 0.05, paired t-test), indicating the ability of these cells to release glutamate (Figure 8
B). Consistent with the reduced accessibility of agarose gel to GDH, the fluorescence intensity in focal plane B was 62% of that recorded from focal plane A. Taken together, our data show that astrocytes in micropits exhibit intracellular Ca2+ responses to mechanical and agonist stimulations and that these cells can undergo consequential Ca2+-dependent glutamate release.
Astrocyte-Microglia Signaling
Communication from astrocytes to microglia has previously been shown to be ATP-mediated (Verderio and Matteoli, 2001
) and this form of signaling is important for the recruitment of microglia to sites of injury in the central nervous system (Davalos et al., 2005
; Kurpius et al., 2007
). Here, we tested whether this form of heterocellular signaling is intact in solitary micropit astrocytes co-inhabited by microglia (Figures 9
A,B). We performed focal mechanical stimulation to a solitary micropit astrocytes to cause Ca2+-dependent ATP release (Coco et al., 2003
; Pangrsic et al., 2007
) while observing changes in [Ca2+]i in adjacent microglial cells within the micropit. The [Ca2+]i in both cell types was monitored simultaneously by loading astrocytes with the Ca2+ indicator Fura Red, while fluo-3 was loaded in microglia. Mechanical stimulation of solitary micropit astrocytes caused a decrease in fluorescence in the stimulated astrocyte, and an increase in fluorescence in the adjacent microglia; both of these fluorescence intensity changes indicated an elevation of [Ca2+]i in these cells (Figure 9
B, left column and Figure 9
C, top graph). The peak Ca2+ response in Fura Red loaded astrocytes evoked by mechanical stimulation was −34 ± 5% (Figure 9
D). This value is comparable to an astrocytic response of ~200% in fluo-3 loaded astrocytes (Figure 6
A), when the difference in affinity for Ca2+ of two dyes is taken into account (Thomas et al., 2000
). In the microglial cell, after the initial increase of fluorescence there was a recovery followed by a decrease in fluorescence to levels below the initial intensity at rest (Figure 9
C, top graph). This could be caused by photobleaching of the fluo-3 or its loss via fusion of secretory vesicles containing fluo-3 (Bolsover and Silver, 1991
).
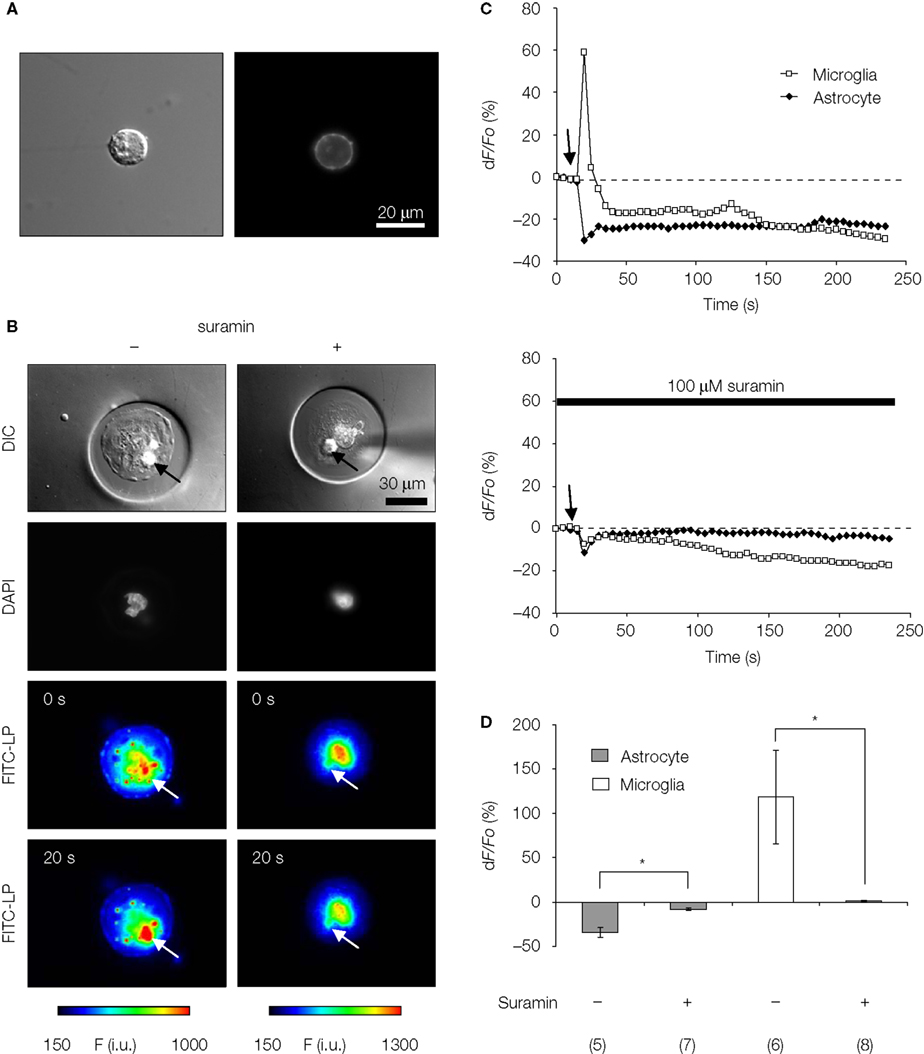
Figure 9. ATP-mediated astroglial-microglial signaling. (A) The purity of isolated microglia was confirmed with live-labeling of microglia with FITC-conjugated isolectin B4 as shown with the DIC image (left panel) and the epifluorescence image (right panel). (B) The nuclear staining with Hoechst 33342 (DAPI filter set) shows the presence of solitary astrocytes in each micropit, while the DIC images show the presence of one microglial cell (arrows) on top of the astrocyte in the micropit in each experiment. The pseudocolor images [linear representation of fluorescence intensity (F) ranging 150–1000 and 150–1300 intensity units (i.u.)] in the two lower panels were acquired with a FITC-long pass (FITC-LP) filter to examine the Ca2+ dynamics of astrocytes and microglia that were loaded with the Ca2+ indicator Fura Red and fluo-3, respectively. The arrows indicate the location of the microglial cells. In the experiment shown on the left image series that was performed in the absence of suramin, mechanical stimulation of the solitary micropit astrocyte caused a transient decrease in Fura Red fluorescence, while the microglial cell showed a transient increase in fluo-3 fluorescence; both intensity changes report on the increase in intracellular Ca2+ levels. These responses were reduced when cells were pre-incubated with suramin for 5 min, which was then kept throughout the entire experiment, to block purinergic receptors [right image series of panel (B)]. The right DIC image contains a shadow of the micropipette used for induction of mechanical stimulus. (C) Time lapse changes in fluorescence expressed as dF/Fo (%) corresponding to the experiments in (B). (D) Quantification of the peak Ca2+ responses of astrocytes and microglia in each experimental group with (+) or without (−) suramin. The numbers of cells tested are shown in parentheses. Bars represent means ± SEMs. Asterisks indicate statistical significance (Student’s t-test; p < 0.05).
To determine whether astrocyte-microglia intercellular communication is mediated by released ATP, we performed the same experimental procedure as above while blocking ATP receptors. Suramin (100 μM), a non-selective P2-receptor antagonist, was applied throughout the experiment and also during a pre-incubation period (5 min). The treatment with suramin completely abolished the astrocyte-microglial signaling (Figure 9
B, right column; Figure 9
C, bottom graph). When we examined the effect of suramin on the peak Ca2+ responses in stimulated solitary micropit astrocytes, suramin application significantly reduced the peak amplitude, indicating that this rise in astrocytic [Ca2+]i is mediated by the autocrine action of ATP (Figure 9
D). These results are in good agreement with the existence of purinergic astrocyte-microglia signaling (Verderio and Matteoli, 2001
) and autocrine effects of ATP released from astrocytes (Shiga et al., 2001
).
Diffusion of Gliotransmitters Between Micropits
In our design of the micropit mold, we considered intercellular communication between cells residing in neighboring micropits via extracellularly released gliotransmitters. Our goal was to create a mold design allowing for sufficient cell occupancy in the micropits (Figure 4
), while minimizing the intercellular communication between cells in neighboring micropits. We therefore modeled the diffusion of glutamate release from a solitary micropit astrocyte to determine the optimal inter-pit distance. In our diffusion model we treated the diffusion of glutamate from a single astrocyte in micropit as a point source of glutamate released into a semi-infinite volume (Crank, 1975
). Here, a general solution of 3D-diffusion equation is given by:

where M is the initial number of molecules at the point source, D is the diffusion coefficient, r is the distance from point source, C is the concentration of transmitter in a hemispherical shell at time t and distance r.
In previous studies that optically examined mechanical stimulation-evoked glutamate release from cultured astrocytes, the range of extracellular glutamate surrounding cells was found to be 1–100 μM (Innocenti et al., 2000
). Therefore, we chose 100 μM as the initial extracellular glutamate concentration within the hemispherical volume of the micropit, and this corresponds to an M of 6.8 × 1011 molecules at the point source in the simulation model. Although mechanical stimulation causes release of glutamate and ATP (Figures 8 and 9
), the diffusion of ATP is slower compared to glutamate, so we only modeled diffusion of glutamate (Figure 10
). The diffusion coefficient at 25°C in solution for ATP is 3.7 × 10−6 cm2/s (Hubley et al., 1996
) and 7.6 × 10−6 cm2/s for glutamate (Longsworth, 1953
).
In our diffusion model, at a distance of 300 μm away from the point source, which is comparable to the distance between the micropits (Figure 10
A) the peak glutamate concentration calculated is ~6 μM at 20 s after release (Figures 10
B,C). This implies that we should record some intercellular communication between astrocytes since they express functional glutamate and/or ATP receptors (Figures 6
B,C). Indeed, we confirmed that astrocytes in micropits respond to a puffer application of glutamate (8 out of 10 astrocytes tested; 100 μM; Figure 11
A, right panels) and ATP (5 out of 7 astrocytes tested; 100 μM). Thus, 76% of micropit astrocytes originating form the visual cortex respond to glutamate/ATP. In comparison, glutamate and ATP evoked Ca2+ responses occurred in ~80% and 90% of astrocytes in acute hippocampal slices at 10 and 100 μM of bath applied transmitter, respectively (see Figure 5 in Zur Nieden and Deitmer, 2006
). Therefore, the responsiveness of micropit astrocytes to ATP and glutamate are in good agreement with that of astrocytes in acute slices, especially when considered that the puffer application could lead to up to a 10-fold dilution of the transmitter when compared to bath application (Parpura et al., 1994
).
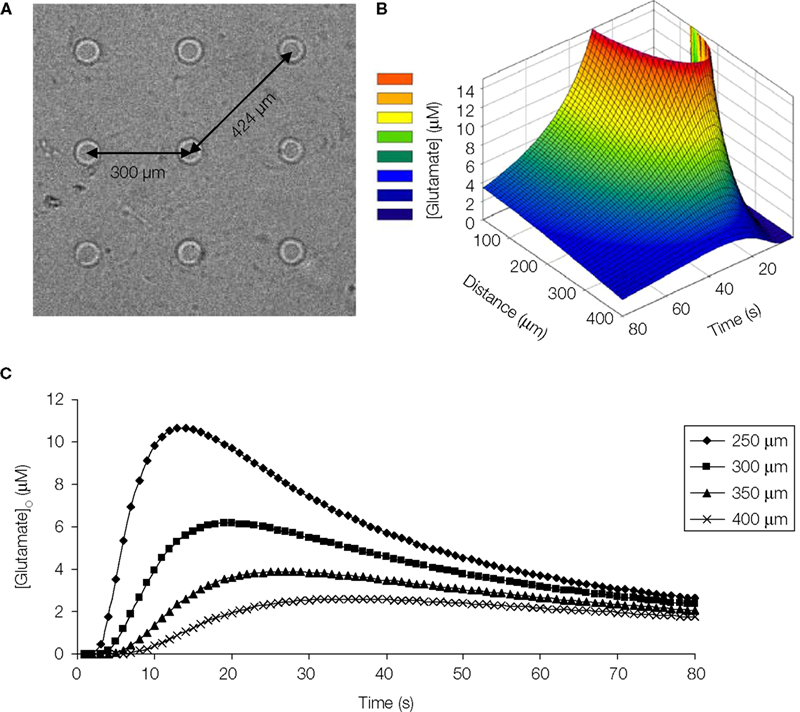
Figure 10. Diffusion model of point source glutamate release from a micropit. (A) Bright field image of micropit regions of the micropatterned coverslip. The shortest inter-pit distance (center to center) is 300 μm, while the diagonal distance to the nearest micropit is 424 μm. (B) Concentration profile of point source diffusion of glutamate (initial concentration of 100 μM glutamate) from a micropit. (C) The concentration profile of glutamate at various distances and times from the source.
To test for intercellular communication between astrocytes in neighboring micropits, we performed mechanical stimulation of a solitary astrocyte in one micropit to evoke transmitter release while monitoring the Ca2+ responses of the cell(s) in adjacent micropits (Figures 11
A–D). Mechanical stimulation of a solitary micropit astrocyte elicited transient Ca2+ responses in the astrocytes in adjacent micropits in 5 out of 17 experiments (Figures 11
A,C). The peak Ca2+ responses of cells in the nearby micropits, responding to the transmitters released from the mechanically stimulated astrocyte, were lower than those elicited by subsequent direct agonist stimulation (Figure 11
C). The time interval between mechanically induced peak Ca2+ response of the solitary micropit astrocyte and the detectable initiation of the Ca2+ response in adjacent micropit cells ranged from 20 to 245 s; the peak to peak time of this interpit communication was 35–290 s. We did not find correlation between the mechanically-induced Ca2+ response (peak dF/Fo, cumulative dF/Fo or its duration) in the solitary micropit astrocyte and the Ca2+ response in adjacent micropit cells (unadjusted r2 less than 0.025). However, the reduction and delay in Ca2+ response in adjacent micropits is consistent with the diffusion of transmitters based on our diffusion model. Furthermore, inter-pit intercellular communication occurred in a lower proportion of astrocytes (29%; 5 out of 17 astrocytes tested) than the proportion of these cells (76%) capable of directly responding to glutamate/ATP. Taken together these results indicate that the inter-pit distance of 300 μm is effective in reducing the extent of paracrine signaling between cells in neighboring micropits.
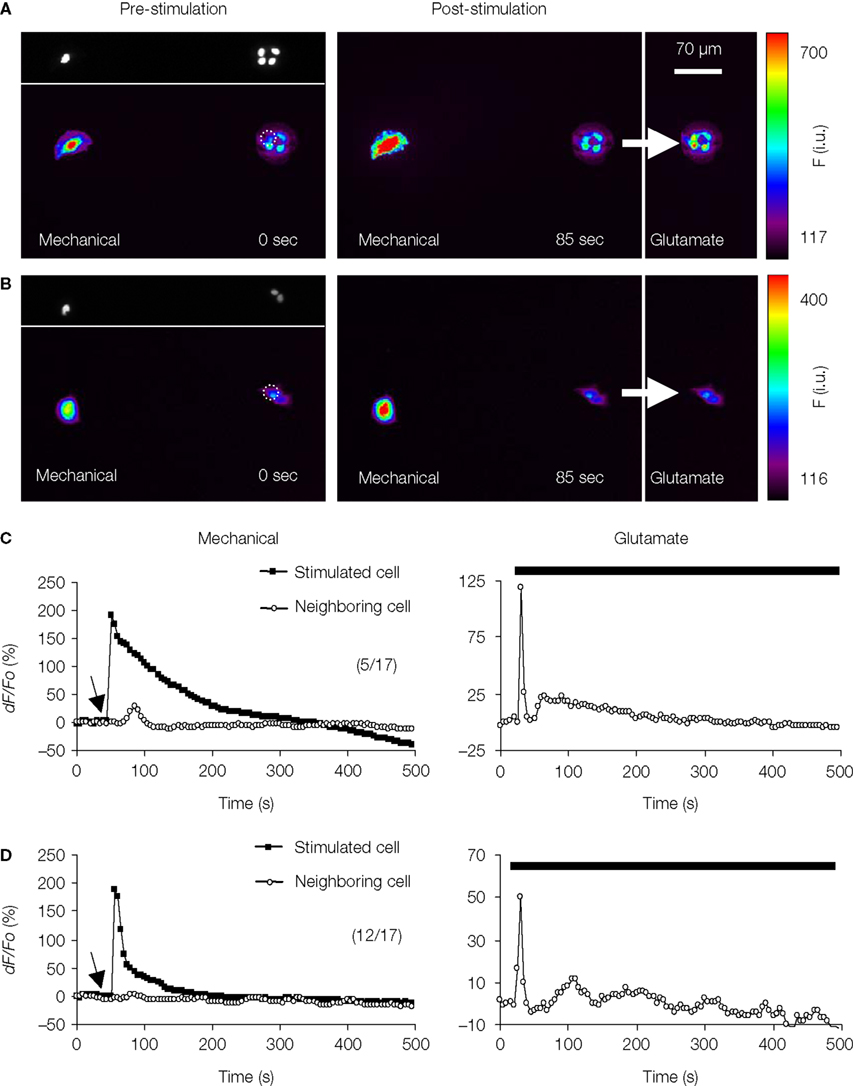
Figure 11. Examination of intercellular communication pathways between astrocytes in neighboring micropits. (A) Simultaneous monitoring of [Ca2+]i in astrocytes loaded with fluo-3 in two adjacent micropits. The top panel in (A) shows a solitary astrocyte in a micropit with four cells in a neighboring pit with nuclei stained with Hoechst 33342, while the lower panel shows the same cells loaded with fluo-3 in pseudocolor prior to stimulation (0 s). Mechanical stimulation of a solitary astrocyte in a micropit (leftmost cell) elicited an increase in [Ca2+]i in the cells in the nearest (right hand) micropit (85 s). After recovery, the cells in the right hand pit were stimulated (arrow) with glutamate (100 μM); the peak response is shown in far right image. (B) A representative experiment where mechanical stimulation (85 s) of the solitary astrocyte in the micropit (left) did not elicit any Ca2+ response in the cells in the adjacent micropit (right), even though these cells responded to subsequent stimulation with glutamate (arrow). Dotted circles in (A,B) indicate astrocytes within neighboring micropits that were used for time-lapse traces (C,D). The pseudo color scale represents the fluorescence intensity (F) ranging 117–700 and 116–400 intensity units (i.u.). All images represent raw data with pixel intensities within the camera’s dynamic range (0–4095). (C,D) Time lapse series of fluo-3 fluorescence of individual cells, corresponding to the experiments shown in (A,B) reporting on [Ca2+]i dynamics. The arrows (left panels) indicates when mechanical stimulation was applied, the bars (right panels) denote when control glutamate stimulation was applied. The numbers in parentheses indicate the number of cases in each condition.
In this study, we demonstrated that our method of micropatterning cell-adhesion substrate can be used to control the growth and localization of astrocytes. The astrocytes cultured in micropits were viable and exhibited similar properties as astrocytes in the non-micropit region of the micropatterned coverslip. It has been previously demonstrated that a decrease in the size of available cell-adhesive surface caused bovine epithelial cells to switch from growth to apoptosis (Chen et al., 1997
; Dike et al., 1999
). When the size of the cell-adhesive surface area was decreased below 3000 μm2, there was an increase in the fraction of cells that underwent apoptosis and a reduction in DNA synthesis. Accordingly, the micropit cell-adhesive surface area of 4417 μm2 we designed into the pattern is favorable for cellular growth and our data using calcein support this notion. Additionally, the micropit astrocytes exhibited similar expression of astrocytic markers as that in grouped astrocytes in non-micropit regions. The exception was GFAP, which appears to be upregulated in solitary astrocytes within micropits. Since upregulation of GFAP protein expression is a hallmark of reactive astrocytes, the increase in GFAP expression in micropit astrocytes may suggest that they are more reactive than those astrocytes grown in the non-micropit regions.
By designing the micropits to be 300 μm apart from each other, we reduced the extent of paracrine signaling between astrocytes in neighboring micropits. Additionally, by growing solitary astrocytes within micropits, it appears that the ATP-evoked [Ca2+]i oscillations in solitary micropit astrocytes become less variable and display more pronounced peaks than those of grouped astrocytes in non-micropit regions (Figure 7
). This finding may implicate a role for astrocytic connectivity in intracellular Ca2+ oscillations. The follow-up investigations determining the role of connectivity would require a systematic approach to compare Ca2+ dynamics in solitary vs. small networks (i.e., 2–6) of astrocytes grown in micropits. Consequently, the repertoire of molecules that may govern differences in Ca2+ oscillations in astrocytes could be investigated using micropit cultures. It is then tempting to speculate that possible correlations between Ca2+ oscillations/dynamics induced by various physiological stimuli and the morphology of the astrocytes, their protein expression, or their connectivity may be used to define functional classes of astrocytes (for review on basic principal of astrocytic classification, see Kimelberg, 2009
). Similarly, the isolation provided by the micropit system could be used to compare the properties of astrocytes isolated from various regions of the brain, especially that astrocytes have been shown to exhibit brain region-specific properties (Kipp et al., 2008
; Peters et al., 2005
; Zschocke et al., 2005
).
Micropit cultures can also be used to characterize glial–glial (astrocyte–microglia) interactions (Figure 9
). We showed that mechanical stimulation of astrocytes elicited ATP-mediated astrocytic-microglial signaling and these results were consistent with studies of cultured astrocytes with intact intercellular communication pathways (Verderio and Matteoli, 2001
). Similar pre-existing culturing systems using microislands have been used previously to study neuronal-glial bi-directional signaling, where a single neuron was cultured onto small groups of astrocytes (Mennerick and Zorumski, 1994
; Parpura and Haydon, 2000
). Although we have not systematically investigated the viability of neurons grown in micropits, we have observed live neurons in 8-day old mixed cultures originating from visual cortices (unpublished observation). Nonetheless, the advantage of micropit over microisland culture in studying glial-neuronal interactions is that with micropits it is possible to generate the most simplistic scenario to study astrocytic-neuronal signaling, where a single astrocyte interacts with a single neuron. Since single neurons can form synapses to themselves, autapses, and astrocytes can modulate synaptic transmission and plasticity (reviewed in Ni et al., 2007
), the use of this one neuron-one astrocyte culturing system could be utilized to better understand the bi-directional information flow between the components of the tripartite synapse (Araque et al., 1999
).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Daniel Adams from University of California at Riverside Microfabrication Facility for help with construction of master mold, Nathan Reiland PE for his help with CAD images, Dr. Umar Mohideen for lending us a J scanner, Dr. Wei Liu for his discussion on diffusion model and Dr. Yingchun Ni for help with the initial glutamate release measurements in micropit cultures. This work is supported by a grant from the National Institute of Mental Health (MH 069791 to VP) and the National Institute of Heath Neuroscience Training Program in Neurobiology of Cognition and Cognitive Disorders at University of Alabama, Birmingham (to RCR).
Supplementary Movies 1 and 2 can be found on the journal website at the following address: http://www.frontiersin.org/
neuroengineering/paper/10.3389/neuro.16/002.2008/.