1
Department of Biochemistry and Molecular Biology, Institute of Neurosciences of Castilla- Leon, University of Salamanca, Salamanca, Spain
2
Molecular and Genetics Unit, Institute of Child Health, University College London, London, UK
3
Department of Chemical Pathology, Great Ormond Street Hospital, London, UK
Approximately 15 years ago we reported that cytochrome c oxidase (CcO) was persistently inhibited as a consequence of endogenous induction and activation of nitric oxide (•NO) synthase-2 (NOS2) in astrocytes. Furthermore, the reactive nitrogen species implicated was peroxynitrite. In contrast to the reversible inhibition by •NO, which occurs rapidly, in competition with O2, and has signaling regulatory implications, the irreversible CcO damage by peroxynitrite is progressive in nature and follows and/or is accompanied by damage to other key mitochondrial bioenergetic targets. In purified CcO it has been reported that the irreversible inhibition occurs through a mechanism involving damage of the heme a3-CuB binuclear center leading to an increase in the Km for oxygen. Astrocyte survival, as a consequence of peroxynitrite exposure, is preserved due to their robust bioenergetic and antioxidant defense mechanisms. However, by releasing peroxynitrite to the neighboring neurons, whose antioxidant defense can, under certain conditions, be fragile, activated astrocytes trigger bioenergetic stress leading to neuronal cell death. Thus, such irreversible inhibition of CcO by peroxynitrite may be a plausible mechanism for the neuronal death associated with neurodegenerative diseases, in which the activation of astrocytes plays a crucial role.
After the discovery of the potential biological roles of nitric oxide (•NO) (Ignarro et al., 1987
; Palmer et al., 1987
), its implication as a key player during glutamatergic neurotransmission was shortly reported (Garthwaite et al., 1988
). By activating soluble guanylate cyclase (sGC), •NO thus participates in the signaling cascade leading to activation of cyclic GMP-dependent protein kinases (Garthwaite, 2008
). Due to its multiple targets, it is now clear that •NO has a critical role for tissue survival, including the brain (Almeida et al., 1999
; Pacher et al., 2007
; Calabrese et al., 2009
). In 1994, four independent laboratories reported that •NO, either exogenously added to several preparations containing mitochondria, or endogenously produced in intact cells, via the inducible form of •NO synthase (NOS2), inhibited cytochrome c oxidase (CcO), the terminal complex of the mitochondrial respiratory chain (Bolaños et al., 1994
; Brown and Cooper, 1994
; Cleeter et al., 1994
; Schweizer and Richter, 1994
). However, in the three laboratories that used exogenous •NO, the effect on CcO was found to be reversible (Brown and Cooper, 1994
; Cleeter et al., 1994
; Schweizer and Richter, 1994
) and in competition with O2 (Brown and Cooper, 1994
; Schweizer and Richter, 1994
), whereas in the other study (Bolaños et al., 1994
), which relied upon endogenous/sustained •NO formation, inhibition of CcO activity was found to be progressive and persistent.
The implications of the reversible interaction of CcO with •NO for cell signaling and physiology are of paramount importance (Levonen et al., 2001
; Brookes et al., 2002
; Cooper, 2002
; Brown, 2007
; Taylor and Moncada, 2009
), although there still remain some issues that may require further validation (Garthwaite, 2008
). For instance, the concomitant bioenergetic stress following CcO inhibition by NO increases the AMP:ATP ratio that, by stimulating the AMP-activated protein kinase (AMPK), switches on glycolysis (Almeida et al., 2004
). In addition, a reduction in cytochrome c is followed by the inhibition of CcO by •NO, and this enhances mitochondrial superoxide formation  that activates signaling pathways involved in cytoprotection (Moncada and Erusalimsky, 2002
; Moncada and Bolanos, 2006
) and hypoxic effects (Palacios-Callender et al., 2007
). Moreover, during •NO-mediated interaction with CcO, O2− formation is observed even before an inhibition of respiration is observed (Palacios-Callender et al., 2004
; Jacobson et al., 2005
). Interestingly, reduced cytochrome c can also react with •NO to produce the nitroxyl anion (NO−) species (Sharpe and Cooper, 1998b
; Parihar et al., 2008
). Furthermore, either
that activates signaling pathways involved in cytoprotection (Moncada and Erusalimsky, 2002
; Moncada and Bolanos, 2006
) and hypoxic effects (Palacios-Callender et al., 2007
). Moreover, during •NO-mediated interaction with CcO, O2− formation is observed even before an inhibition of respiration is observed (Palacios-Callender et al., 2004
; Jacobson et al., 2005
). Interestingly, reduced cytochrome c can also react with •NO to produce the nitroxyl anion (NO−) species (Sharpe and Cooper, 1998b
; Parihar et al., 2008
). Furthermore, either  and •NO (Blough and Zafiriou, 1985
), or O2 and NO− (Sharpe and Cooper, 1998b
; Parihar et al., 2008
), can spontaneously react to form the very unstable pro-oxidant peroxynitrite anion (ONOO−). Thus, through the interaction of •NO with CcO, mitochondria become both the target of •NO and the site of peroxynitrite formation. However, as unraveled by other laboratories, extra-mitochondrial formation of peroxyntirite, for instance through the reaction of •NO with plasma membrane NADPH-derived O2− (Bal-Price et al., 2002
; Mander and Brown, 2005
), can also occur.
and •NO (Blough and Zafiriou, 1985
), or O2 and NO− (Sharpe and Cooper, 1998b
; Parihar et al., 2008
), can spontaneously react to form the very unstable pro-oxidant peroxynitrite anion (ONOO−). Thus, through the interaction of •NO with CcO, mitochondria become both the target of •NO and the site of peroxynitrite formation. However, as unraveled by other laboratories, extra-mitochondrial formation of peroxyntirite, for instance through the reaction of •NO with plasma membrane NADPH-derived O2− (Bal-Price et al., 2002
; Mander and Brown, 2005
), can also occur.
that activates signaling pathways involved in cytoprotection (Moncada and Erusalimsky, 2002
; Moncada and Bolanos, 2006
) and hypoxic effects (Palacios-Callender et al., 2007
). Moreover, during •NO-mediated interaction with CcO, O2− formation is observed even before an inhibition of respiration is observed (Palacios-Callender et al., 2004
; Jacobson et al., 2005
). Interestingly, reduced cytochrome c can also react with •NO to produce the nitroxyl anion (NO−) species (Sharpe and Cooper, 1998b
; Parihar et al., 2008
). Furthermore, either and •NO (Blough and Zafiriou, 1985
), or O2 and NO− (Sharpe and Cooper, 1998b
; Parihar et al., 2008
), can spontaneously react to form the very unstable pro-oxidant peroxynitrite anion (ONOO−). Thus, through the interaction of •NO with CcO, mitochondria become both the target of •NO and the site of peroxynitrite formation. However, as unraveled by other laboratories, extra-mitochondrial formation of peroxyntirite, for instance through the reaction of •NO with plasma membrane NADPH-derived O2− (Bal-Price et al., 2002
; Mander and Brown, 2005
), can also occur.Regardless of the specific source of formation, peroxyntirite can easily diffuse through membranes due to its high lipid solubility (Lim et al., 2008
), and hence it can reach targets distant from its site of production. In fact, peroxynitrite is implied in damages to several cellular components, including essential mitochondrial proteins such as aconitase (Castro et al., 1994
; Gardner et al., 1997
; Tortora et al., 2007
), NADH dehydrogenase (Radi et al., 1994
; Borutaite et al., 2000
), succinate dehydrogenase (Radi et al., 1994
), α-ketoglutarate dehydrogenase (Park et al., 1999
), amongst other mitochondrial targets (Radi et al., 2002
), DNA (Inoue and Kawanishi, 1995
; Uppu et al., 1996
; Cantoni and Guidarelli, 2008
) and lipids (Patel and Darley-Usmar, 1996
). It is now also very well accepted that peroxynitrite can be formed in vivo (Beal, 2002
), and is likely to modify or damage a still undetermined number of proteins (Liaudet et al., 2009
). The exact mechanisms whereby peroxynitrite interacts with proteins are not known. The most common reactions involve nitration of tyrosine on its position 3 (Radi, 2004
) and oxidation of sulfhydryls (Radi et al., 1991
), both of which can modify critical structures of proteins leading to change in activity (most likely inactivation). In fact, immunodetection of 3-nitrotyrosines in proteins of biological samples is now considered a good reflection of peroxynitrite formation (Greenacre and Ischiropoulos, 2001
; Bolanos et al., 2004
). Furthermore, critical proteins have been shown to be both nitrosylated (i.e., nitrated on tyrosine residues) and oxidized (i.e., sulfhydryl oxidation), such as mitochondrial NADH dehydrogenase (Landino, 2008
), giving rise to a complex way of modulation of enzyme activity that still requires further elucidation.
The molecular mechanism responsible for the irreversible inhibition of CcO by peroxyntirite was first investigated by Sharpe and Cooper (Sharpe and Cooper, 1998a
). They revealed a complex interaction between peroxyntirite and purified CcO causing an increase in the Km for O2 and a decrease in the Vmax of the enzyme, as well as spectra changes compatible with oxidation of the a3 center, partial reduction of cytochrome a, and irreversible damage to the CuA site (Sharpe and Cooper, 1998a
; Pearce et al., 1999
). The decrease in Vmax observed by Sharpe and Cooper (1998a)
is compatible with the 25% inhibition of the specific activity of COX in astrocytes activated to endogenously produce peroxynitrite (Bolaños et al., 1994
). Regardless the specific mechanism leading to CcO irreversible damage by peroxynitrite, it is now clear that this interaction may have a still non-conceived pathophysiological implications (Cooper et al., 2003
).
Due to its high-energy requirements to support ionic balance and neurotranmission (Hertz et al., 2007
; Nehlig and Coles, 2007
; Pellerin et al., 2007
; Cai and Sheng, 2009
), the central nervous system is one of the most vulnerable targets to mitochondrial impairment. However, the brain is a complex tissue formed by the organization of four well-differentiated neural cell types, including neurons, astrocytes, microglia and oligodendrocytes (Hertz et al., 2007
). Such cell types do not act in isolation, as a degree of active collaboration exists between the differing cells, e.g. astrocytes are involved in glutamate homeostasis and the trafficking of antioxidants to neuronal cells (Fellin, 2009
; Perea et al., 2009
). Furthermore, astrocytes and neurons establish a mutual paracrine-like communication system through the release of soluble and cell permeable factors, involving cytokines (Peuchen et al., 1997
), reactive oxygen and nitrogen species (RONS) (Whitney et al., 2009
) or substrate precursors (Pellerin et al., 2007) (Figure 1
).
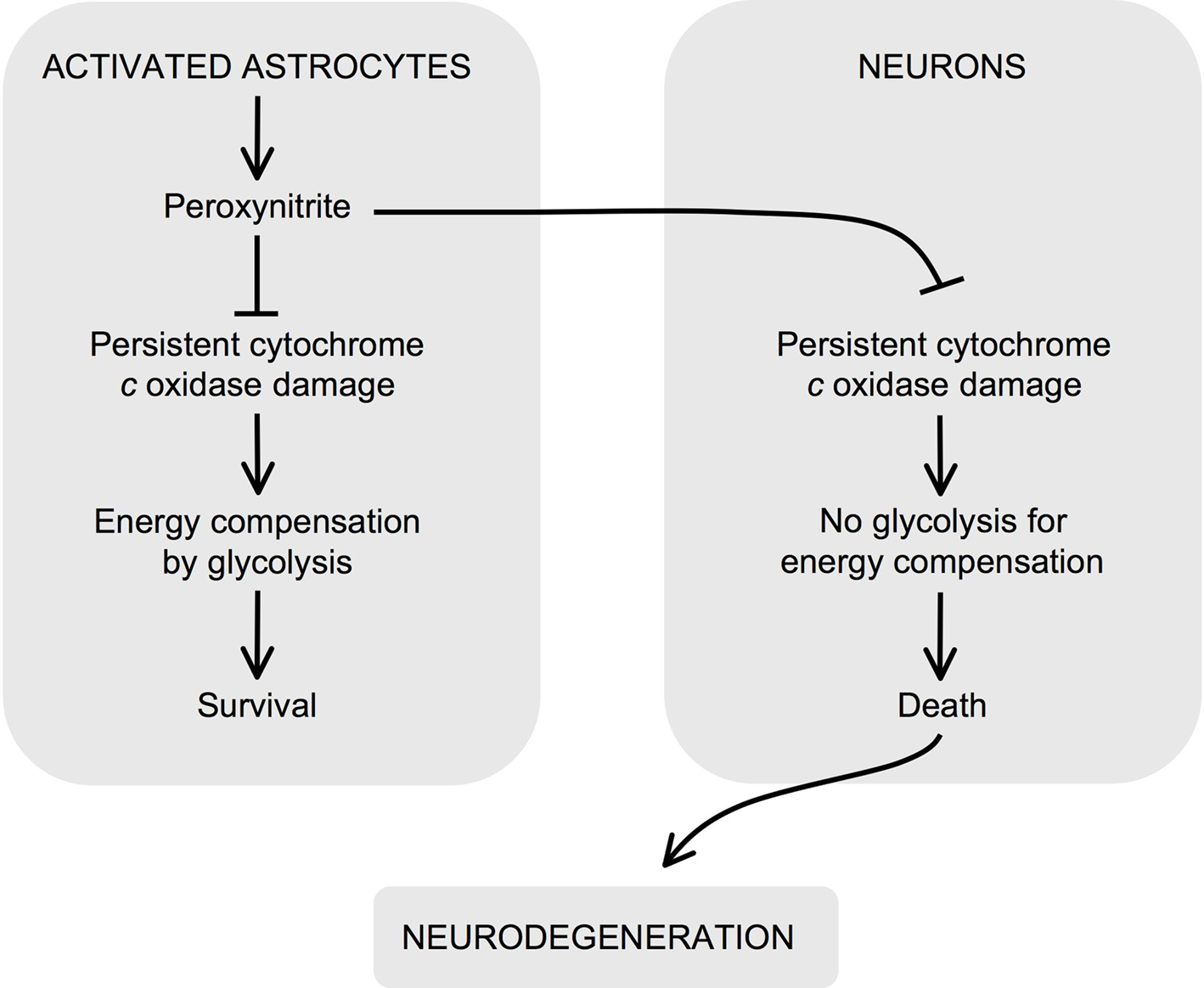
Figure 1. Role for irreversible inhibition of cytochrome c oxidase in neurodegeneration. Peroxynitrite can be formed in astrocytes upon activation. In astrocytes, peroxyntrite irreversibly damages cytochrome c oxidase, which causes mitochondrial dysfunction. However, these cells compensate the energy deficiency by activating glycolysis and survive. Peroxynitrite is a highly diffusible molecule, and hence it reaches neighboring neurons, where it irreversibly damages cytochrome c oxidase. In contrast to astrocytes, neurons cannot up-regulate the energy-compensating glycolysis and hence they die by bioenergetic crisis. Thus, the irreversible inhibition of cytochrome c oxidase by peroxynitrite has a critical negative effect on neuronal survival and may contribute to the propagation of neurodegeneration.
With regards to astrocytes, the presence of cytokines ( interferon-γ, interleukine-1ß or tumor necrosis factor-α), immunogenic neuronal proteins (S100ß) or other exogenous components such as viruses or bacterial endotoxins, such as lipopolysaccharide (LPS) trigger the transcription of a number of genes leading to a situation generally known as activation (Peuchen et al., 1997
). Activated astrocytes are characterized by certain small morphological changes (Hertz et al., 2007
) and by the release of RONS, including •NO and ONOO− (Peuchen et al., 1997
). There are now a number of human diseases associated with this type of neuroinflammatory picture. These include multiple sclerosis (Nair et al., 2008
), Alzheimer’s and Parkinson’s diseases (McGeer and McGeer, 2008
; Rodriguez et al., 2009
).
Our own previous data reported that activated atrocytes, which express NOS2 (Murphy et al., 1993
), continuously release a considerable amount of •NO (Brown et al., 1995
), which is responsible for their inhibition of CcO activity (Bolaños et al., 1994
). Moreover, the persistent inhibition of CcO (∼25% inhibition of complex activity) had functional consequences to mitochondrial respiration, since complete removal of •NO from the activated astrocytes significantly reduced (by ∼18%) their rate of oxygen consumption (Brown et al., 1995
). However, these cells were shown to be highly resistant to any survival problem triggered by such inhibition, and the identification of a NOS2-mediated increase in the rate of glucose conversion to lactate suggested that the ensuing mitochondrial bionergetic crisis was compensated by the up-regulation of glycolysis (Bolaños et al., 1994
). In fact, further investigation into this issue has revealed a specific mechanism linking mitochondrial inhibition to glycolysis up-regulation by NO involving the switching on of the 5′AMP-activated protein kinase–6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (isoform 3) pathway in astrocytes, but not in neurons (Almeida et al., 2004
). Thus, astrocytes are activated cells that can release large amounts of RONS without compromising their own survival. However, due to the lipid solubility nature of RONS, the neighboring neurons may become vulnerable targets.
In order to study the metabolic consequences of activated astrocytes to neurons, we used a neuronal-astrocyte co-culture system to more closely resemble the interaction between these two cells in vivo. Thus, astrocytes grown on the surface of a co-culture microwell membrane device were first challenged to induce NOS2 by incubation with interferon-γ and LPS. When activated astrocytes produced large amounts of •NO, they were posed on the top of a neuronal culture monolayer (Bolaños et al., 1996
) and further incubated for several time points, either in the presence or in the absence of the NOS inhibitor N-monomethyl-L-arginine (NMMA). We found that 24 h after the co-incubation period, neurons showed a NMMA-sensitive inhibition of CcO activity (Bolaños et al., 1996
) that, in contrast to the damage to other components of the mitochondrial respiratory chain, was time-dependent and permanent (Stewart et al., 2000
), causing a bioernergetic deficiency leading to cell death (Bolaños et al., 1996
). Thus, incubation of neurons to peroxynitrite-producing astrocytes for 24 h is followed by subsequent full recovery of COX activity; however, such recovery does not take place if the period of incubation was 48 h (Stewart et al., 2000
). These results suggest that COX would remain inhibited as long as peroxynitrite is present, hence respiration at COX recovers depending on the enzyme turnover rate. It should also be mentioned that other components of the mitochondrial respiratory chain are also damaged, though at much lower extent, by endogenous peroxynitrite in astrocytes, including succinate-cytochrome c oxidoreductase (Bolaños et al., 1994
; Hargreaves et al., 2007
), and NADH-ubiquinone oxidoreductase (Bolaños et al., 1994
). Moreover, these results clearly demonstrated that activated astrocytes can damage neighboring neurons through a mechanism in which •NO and/or •NO-derivatives are involved. However, it was intriguing that neurons were relatively resistant against activated astrocytes when compared with the high vulnerability of neurons if incubated, alone, with several pro-oxidant compounds (Bolaños et al., 1995
, 1996
). The role for antioxidant glutathione in neuroprotection against the neurotoxic effects of nitric oxide was thus first proposed (Bolaños et al., 1995
, 1996
).
Incubation of neurons with exogenous peroxynitrite triggered a dose-dependent persistent inhibition of CcO that is not observed in astrocytes at identical concentrations (Bolaños et al., 1995
). Such an effect was correlated with a deficiency in the intracellular concentrations of glutathione in neurons, being astrocytes highly resistant to such effect (Bolaños et al., 1995
). In contrast, when neurons were incubated in the presence of activated astrocytes, the limited effect on ATP concentrations and neuronal survival suggested the development of a protective mechanism not existing in the neurons when cultured alone (Bolaños et al., 1996
). By investigating this, we found that neurons increased their intracellular concentration of glutathione by the presence of either resting or activated astrocytes (Bolaños et al., 1996
), indicating the transfer of glutathione, or its direct precursor, from astrocytes to neurons. The mechanism and implications for the transfer of this astrocyte-neuronal glutathione precursors has been confirmed and studied in depth (Dringen et al., 1999
). In fact, co-incubation of neurons with glutathione-depleted astrocytes resulted in a much higher vulnerability of neurons against astrocyte-derived •NO and/or ONOO− (Gegg et al., 2005
), suggesting that the astrocyte antioxidant capacity must be preserved in order to efficiently protect neurons against damage. However, glutathione deficiency can also be damaging to the astrocytes themselves, since glutathione depletion dramatically increase the susceptibility of astrocytes to peroxynitrite at doses that are not damaging in normal astrocytes (Barker et al., 1996
). Thus, the role for intercellular glutathione exchange between neurons and astrocytes are currently considered to be a major target for neuroprotection (Vargas et al., 2008
; Miller et al., 2009
; Sandhu et al., 2009
).
In conclusion, whilst the reversible modulation of CcO by •NO is of paramount importance for the physiological regulation of cell functions, the persistent interaction of its endogenous derivative peroxynitrite with this mitochondrial complex may have profound and permanent consequences for cell survival. This is of particular importance in the central nervous system, since neurons are particularly vulnerable cells against mitochondrial impairment (Bolaños et al., 1995
), possibly due to their unability to up- regulate glycolysis and hence to compensate for any bioenergetic crisis (Bolaños et al., 1995
; Herrero-Mendez et al., 2009
). In addition, the glutathione-dependent fragile nature of neurons compromise their defense against the release of RONS from activated glial cells, which trigger permanent damage to CcO and also to other critical mitochondrial components. Thus, the irreversible inhibition of CcO by peroxynitrite may be a critical contributing factor for the neuronal death accompanying neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases (Ebadi and Sharma, 2003
; Atamna and Frey, 2007
; Fukui and Moraes, 2008
), in which CcO (Parker et al., 1990
) and glutathione deficiency (Perry et al., 1982
), are hallmarks.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Juan P. Bolaños is supported by the Ministerio de Ciencia e Innovación (SAF2007-61492 and Consolider-Ingenio CSD2007-00020, Spain) and by the Junta de Castilla y León (SA046A10-2; Grupo de Excelencia GR206 and Red de Terapia Celular y Medicina Regenerativa).
Cooper, C. E., Davies, N. A., Psychoulis, M., Canevari, L., Bates, T. E., Dobbie, M. S., Casley, C. S., and Sharpe, M. A. (2003). Nitric oxide and peroxynitrite cause irreversible increases in the K(m) for oxygen of mitochondrial cytochrome oxidase: in vitro and in vivo studies. Biochim. Biophys. Acta 1607, 27–34.
Uppu, R. M., Cueto, R., Squadrito, G. L., Salgo, M. G., and Pryor, W. A. (1996). Competitive reactions of peroxynitrite with 2’-deoxyguanosine and 7,8-dihydro-8-oxo-2’-deoxyguanosine (8-oxodG): relevance to the formation of 8-oxodG in DNA exposed to peroxynitrite. Free Radic. Biol. Med. 21, 407–411.