

- Department of Neuroscience, University of Minnesota, Minneapolis, MN, USA
Flickering light dilates retinal arterioles and increases retinal blood flow, a response termed functional hyperemia. This response is diminished in diabetic patients even before the appearance of overt clinical retinopathy. The loss of functional hyperemia could deprive retinal neurons of oxygen and nutrients, possibly exacerbating the development of diabetic retinopathy. We have tested whether inhibiting inducible nitric oxide synthase (iNOS) reverses the loss of functional hyperemia in diabetic rat retinas in vivo. Changes in retinal arteriole diameter were measured following diffuse flickering light stimulation in control rats, streptozotocin-induced type 1 diabetic rats and diabetic rats treated with aminoguanidine (AG; an iNOS inhibitor), either acutely via IV injection or chronically in drinking water. Flickering light-evoked large arteriole dilations (10.8 ± 1.1%) in control rats. This response was diminished by 61% in diabetic animals (4.2 ± 0.3%). Both acute and chronic treatment with AG restored flicker-induced arteriole dilations in diabetic rats (8.8 ± 0.9 and 9.5 ± 1.3%, respectively). The amplitude of the corneal electroretinogram b-wave was similar in control and diabetic animals. These findings demonstrate that inhibiting iNOS with AG is effective in preventing the loss of, and restoring, normal functional hyperemia in the diabetic rat retina. Previous work has demonstrated the efficacy of iNOS inhibitors in slowing the progression of diabetic retinopathy. This effect could be due, in part, to a restoration of functional hyperemia.
Introduction
Diabetic retinopathy, a leading cause of blindness in the developed world, has a complex pathology that affects both neuronal and vascular elements of the retina (Mizutani et al., 1996; Barber et al., 1998; Antonetti et al., 2006; Fletcher et al., 2007). In the healthy retina, flickering light induces dilation of retinal vessels and an increase in blood flow, a response termed functional hyperemia. This response brings additional oxygen and nutrients to meet the increased metabolic demands of active neurons (Polak et al., 2002; Riva et al., 2005). A reduction in functional hyperemia is one of the earliest retinal changes observed in diabetic patients (Garhofer et al., 2004; Mandecka et al., 2007; Bek et al., 2008; Nguyen et al., 2009; Pemp et al., 2009). The cause and the possible consequences of this reduction in flicker-induced vasodilation are not known, although it could possibly contribute to the development of diabetic retinopathy.
We have previously shown that nitric oxide (NO) modulates neurovascular signaling pathways in the retina. Both light and glial stimulation evoke retinal vasodilation when NO levels are low, but vasodilation is reduced and vasoconstriction enhanced when NO levels are raised (Metea and Newman, 2006). Interestingly, several laboratories have shown an upregulation of the inducible form of nitric oxide synthase (iNOS) in the diabetic retina (Du et al., 2002; Mishra and Newman, 2010), leading to high NO levels (Kowluru et al., 2000). This increase in tissue NO concentration could alter neuron-to-vessel signaling, leading to reduced functional hyperemia in diabetic patients.
A specific inhibitor of iNOS, aminoguanidine (AG), has indeed been shown to slow the progression of diabetic retinopathy in humans and in animal models of diabetes (Kern and Engerman, 2001; Bolton et al., 2004). These beneficial effects of AG have largely been attributed to the inhibition of advanced glycation end-product (AGE) formation (Brownlee et al., 1986). However, findings from multiple studies suggest that AG could also be acting by inhibiting iNOS (Kern et al., 2000; Kowluru et al., 2000; Kern and Engerman, 2001; Mishra and Newman, 2010), especially in early stages of the disease.
Using a streptozotocin-induced rat model of type 1 diabetes, we have now examined whether AG can reverse the disruption of functional hyperemia in the retinas of diabetic rats in vivo. We find that flicker-induced arteriole dilation is substantially reduced in diabetic rat retinas, as it is in patients with diabetic retinopathy. This reduction can be reversed by administering AG to the diabetic rats, either acutely via IV injection or chronically in drinking water.
Materials and Methods
This study was approved by the University of Minnesota Institutional Animal Care and Use Committee according to the National Institutes of Health Guidelines for experimental animals.
Induction of Diabetes
Two month old male Long–Evans rats (Harlan) were made diabetic by injecting streptozotocin (STZ; 70 mg/kg in citrate buffer, IP; Sigma; Mishra and Newman, 2010). Successful induction of diabetes (blood glucose > 250 mg/dl; OneTouch Ultra, LifeScan) was tested 3 days later. Animals were given supplemental insulin (1.5 U Lantus insulin glargine subcutaneously, thrice a week; sanofi-aventis U.S.) as previously described (Du et al., 2002). Blood glucose during the course of diabetes, monitored biweekly, averaged 535 ± 16 mg/dl (29.7 ± 0.9 mmol/l) for diabetic rats and 543 ± 12 mg/dl (30.2 ± 0.7 mmol/l) for diabetic rats treated with aminoguanidine (Sigma) in drinking water.
In vivo rat preparation
The in vivo rat preparation has been described previously (Srienc et al., 2010). Surgery was performed under 2% isoflurane anesthesia. Core body temperature was continuously monitored and maintained at 37°C (TC-1000 Temperature Controller, CWE). The left femoral vein and artery were cannulated for drug administration and monitoring of blood pressure, respectively, and a tracheotomy performed for artificial ventilation. The animal was placed in a modified stereotaxic holder and the right pupil dilated with 1% atropine sulfate (Alcon Laboratories). Corneal refraction was neutralized with gonioscopic prism solution and a contact lens (5.4 mm fundus lens, Ocular Instruments). The holder containing the rat was placed below an upright microscope.
Anesthesia was maintained during the experiment by continuously infusing α-chloralose-HBC-complex (55 mg/kg/h; Sigma). Animals were artificially ventilated (30–50 breaths/min; CWE SAR-830-P) and paralyzed with gallamine triethiodide (20 mg/kg bolus; 20 mg/kg/h; Sigma) to prevent eye movements. Mean arterial blood pressure (MABP), blood oxygen saturation (sO2), and pH were maintained within physiological limits (100–125 mmHg, 92–97%, and 7.35–7.45, respectively). MABP and sO2 were not different between experimental groups: control, 111.3 ± 2.8 mmHg and 95.7 ± 0.8%; diabetic, 122.2 ± 4.4 mmHg and 92.7 ± 1.5%; diabetic treated with AG IV, 115.4 ± 1.1 mmHg and 95.2 ± 0.8%; and diabetic treated with AG in water, 116.6 ± 2.5 mmHg and 94.8 ± 0.6% (P > 0.05). Following experiments, animals were euthanized by IV injection of 40 mM KCl (1 ml/kg).
Light Stimulation
Retinal photoreceptors were stimulated with a 12 Hz flickering diffuse white light (Riva et al., 2005). Stimulus trials were separated by 2 min periods in the dark. Light from a fiber optic illuminator was gated with an electromechanical shutter and focused onto the globe at a 45° angle through a fiber bundle. The illuminance of the light was 12 klux at the surface of the globe.
Measurement of Flicker-Induced Dilation
The retina was imaged with a 4× objective and an Olympus FluoView 1000 laser scanning confocal microscope. Blood vessels were labeled by IV injection of dextran fluorescein isothiocyanate (2,000,000 MW, 3% solution, 1–3 ml; Sigma). The luminal diameters of first order arterioles were measured with confocal line scans drawn perpendicular to the vessel (Srienc et al., 2010) and luminal diameter vs. time graphs calculated with a custom MatLab routine. Flicker-induced dilations were composed of transient and sustained phases. Response amplitude was measured using the first transient peak and the three largest responses averaged to calculate peak response. Diabetic animals developed partial cataracts, a common occurrence in STZ-treated animals (Perry et al., 1987). These cataracts did not interfere with line scan measurements of vessel diameter but prevented us from using laser speckle flowmetry (Srienc et al., 2010) to monitor blood velocity.
Electroretinogram Measurement
The corneal electroretinogram (ERG) electrode was attached to the center of a custom-made plastic light diffuser placed over the cornea. The reference electrode was placed under the skin on the right cheek, 1 cm from the eye. The retina was dark-adapted for 15 min prior to recording ERGs. Ten millisecond flashes of diffuse white light were focused onto the diffuser. A series of flashes (intensities −3 to 0 log units) was employed to determine the ERG b-wave intensity-response relation, with 0 log unit corresponding to an illuminance of 120 klux at the surface of the diffuser. ERGs were averaged to improve signal-to-noise ratio. Animals displaying noisy ERGs were excluded from analysis. ERGs were filtered (1–100 Hz bandpass) and b-wave amplitude was measured from the negative a-wave trough to the positive b-wave peak.
Statistics
One-way ANOVA followed by one-tailed Dunnett’s post hoc test was used to calculate statistical significance for percent dilation data and two-tailed Dunnett’s post hoc test for resting diameter data. Homoscedastic two-tailed Student’s t-test was used to compare ERG data. Data presented as mean ± SEM. α = 0.05.
Results
We evaluated flicker-induced retinal arteriole dilation in vivo in diabetic and age-matched control rats. Experiments were conducted 7 months after induction of diabetes. The retina was stimulated with a diffuse 12 Hz flickering light and the luminal diameter of arterioles measured with confocal line scans (Figure 1). In age-matched control animals, light stimulation evoked pronounced arteriole dilations composed of an initial transient dilation followed by a sustained response (Figure 2). Flicker-evoked dilations in control retinas averaged 10.8 ± 1.1% (n = 6; summarized in Figure 3A). The dilatory response was greatly diminished in diabetic animals (Figure 2), where responses averaged 4.2 ± 0.3% (n = 6), only 39% of control responses (P < 0.001; Figure 3A).
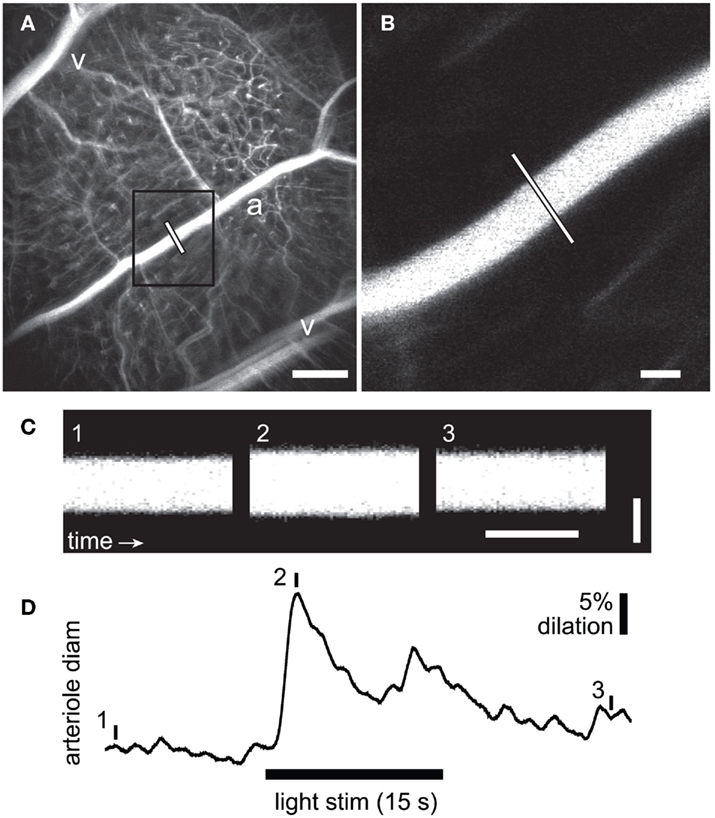
Figure 1. Measurement of flicker-induced changes in retinal arteriole diameter using confocal line scans. (A) Low power confocal image of retinal vessels. Arterioles (a) can be distinguished from venules (v) by their branching pattern and relative size. The diameter of the arteriole was measured with confocal line scans (white line). Scale bar, 250 μm. (B) High magnification image of the boxed area in A showing the region of the arteriole where line scans were taken. Scale bar, 25 μm. (C) Segments of a line scan image from the arteriole in (B). The vertical bars in (D) indicate the times at which the three segments were taken. Distance is represented vertically (scale bar, 25 μm) and time horizontally (scale bar, 100 ms). A diffuse flickering light-evoked vessel dilation, indicated by the widening of the vessel in segment 2. (D) Time course of the flicker-induced arteriole dilation. The onset of the flickering light (black bar at bottom) evoked an initial transient dilation followed by a smaller secondary dilation.
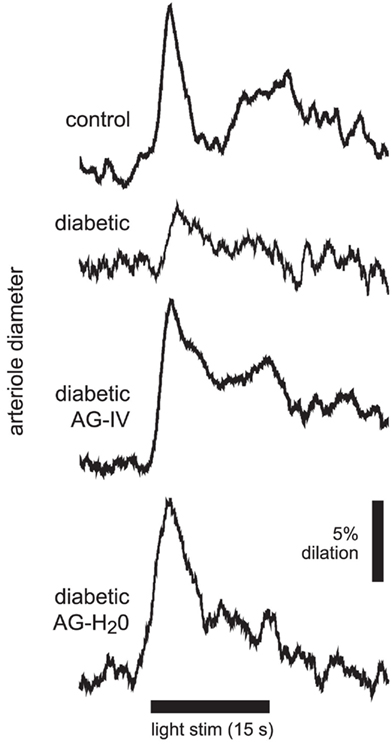
Figure 2. Flicker-induced dilation of retinal arterioles in control and diabetic animals. A diffuse flickering light (black bar at bottom) evoked a prominent arteriole dilation in a control animal, but a much smaller dilation in a diabetic animal. Treatment with the iNOS inhibitor aminoguanidine (AG) restored flicker-induced arteriole dilations in diabetic animals to control levels when given intravenously (diabetic AG-IV), or in drinking water (diabetic AG-H2O).
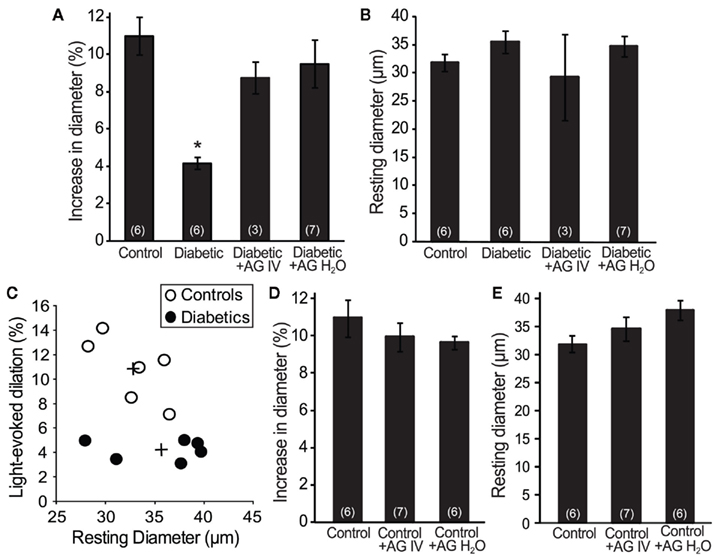
Figure 3. Aminoguanidine treatment reversed the loss of flicker-induced arteriole dilation in diabetic animals. (A) The reduction in flicker-induced vasodilation in diabetic animals was restored by treatment with AG (either IV or in H2O). (B) The resting arteriole diameter was similar in all experimental groups. (C) The amplitude of flicker-evoked dilation in diabetic animals was independent of resting arteriole diameter. +Indicates mean values for the control and diabetic groups. (D) AG did not alter flicker-induced vasodilations in control animals. (E) AG did not alter the resting arteriole diameter in control animals. Numbers in parenthesis indicate number of animals. *Indicates P < 0.001.
Increased tissue concentration of NO, resulting from iNOS upregulation, may be responsible for the reduction in flicker-induced dilations. We thus evaluated the effect of the iNOS inhibitor AG on flicker-induced arteriole dilations in vivo. AG was administered in two ways. We injected AG intravenously (100 mg/kg) to test the effect of acute AG treatment. Flicker-induced changes in arteriole diameter were measured within 4 h of AG injection. We also tested the effect of chronic AG treatment by adding AG to the drinking water of diabetic rats (500 mg/l; Hammes et al., 1991) starting 1 week after successful induction of diabetes and continuing until the experiments were conducted. Both treatment paradigms reversed the loss of flicker-induced arteriole dilation in diabetic animals (Figure 2). Flicker-induced dilations in the AG treatment groups averaged 8.8 ± 0.9% (n = 3) for IV administration and 9.5 ± 1.3% (n = 7) for drinking water administration, both significantly larger than in untreated diabetic rats (P < 0.02; Figure 3A). Neither treatment group differed from controls (P > 0.2).
The loss of neurovascular coupling observed in diabetic animals could be due to a change in the resting diameter of the arterioles. Vessels that are more dilated at rest would not be able to dilate as much in response to light. However, there was no significant difference in the average resting arteriole diameter between groups in our study. The resting diameter of retinal arterioles was 32.7 ± 1.4 μm (n = 6) in control animals, 35.6 ± 2.0 μm (n = 6) in diabetic animals (P > 0.7), and 29.3 ± 7.6 μm (n = 3; P > 0.7) and 34.8 ± 2.1 μm (n = 7; P > 0.8) in diabetic animals treated with AG IV and in drinking water, respectively (Figure 3B). The smaller flicker-evoked vasodilation observed in diabetic retinas was independent of the resting arteriole diameter, as demonstrated by a scatter plot of percent dilation vs. resting arteriole diameter (Figure 3C).
AG treatment could affect flicker-induced dilations in controls animals, but this was not the case in our experiments. AG administration had no effect on flicker-induced dilations in controls. Vasodilations averaged 10.0 ± 0.8% (n = 7; P > 0.3) for IV administration and 9.6 ± 0.4% (n = 6; P > 0.2) for drinking water administration (Figure 3D). AG was administered in drinking water for 2 months prior to experiments. The resting arteriolar diameter of control animals treated with AG acutely (via IV injection) was 34.5 ± 2.1 μm (n = 7; P > 0.6) and chronically (in drinking water) was 37.9 ± 1.8 μm (n = 6; P = 0.1), neither statistically different from untreated controls (Figure 3E).
The loss of neurovascular coupling could also be due to a decrease in light-evoked neuronal activity in diabetic animals, caused either by a loss of neuronal responsiveness or by neuronal death. We evaluated retinal responsiveness by monitoring the b-wave of the ERG, which primarily reflects ON bipolar cell activity. A series of ERGs were recorded to light flashes of increasing intensities (Figure 4A). There was no difference in the maximal b-wave amplitude between control and diabetic animals: 0.22 ± 0.01 mV (n = 7) vs. 0.24 ± 0.03 mV (n = 3), respectively (P > 0.4; Figures 4B,C). The sensitivity of the retina, evaluated by the light intensity that evoked a half-maximal b-wave response, was also similar: 0.012 ± 0.003 in controls vs. 0.014 ± 0.007 in diabetic rats (P > 0.7, where an intensity value of 1 corresponds to 0 log unit intensity; Figures 4B,D). These results indicate that overall neuronal activity was not reduced in the retinas of our diabetic rats.
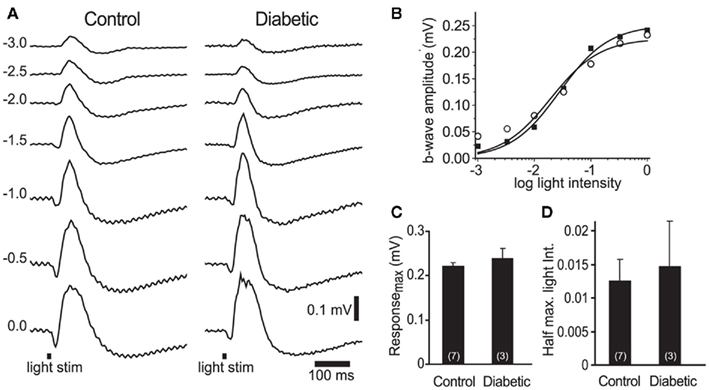
Figure 4. Electroretinogram recordings from control and diabetic animals were similar. (A) Intensity series of ERG traces from a control and a diabetic animal to 10 ms light flashes (black bars at bottom) of increasing intensities (numbers indicate intensity in log units). (B) Intensity-response relations for the control (open circles) and diabetic (black squares) series shown in (A). (C,D) The maximum b-wave response (C) and the light intensity evoking a half-maximal b-wave response (D) were similar in control and diabetic animals (P > 0.4). The data were fit with the Rushton–Naka relation (Naka and Rushton, 1966).
Discussion
Our results demonstrate that the iNOS inhibitor AG, delivered either IV or in drinking water, reverses the loss of functional hyperemia in an animal model of type 1 diabetes. Although AG also inhibits the formation of AGEs (Brownlee et al., 1986), it is likely that AG acts to restore functional hyperemia by blocking iNOS. Our findings demonstrate that the loss of light-evoked vasodilation is reversed within 4 h following IV administration of AG. AG could not act this rapidly if it functioned by reducing formation of AGEs. In addition, we showed previously that the loss of light-evoked vasodilations in an ex vivo isolated retina preparation were also restored rapidly by AG, within 30 min (Mishra and Newman, 2010). A second selective iNOS inhibitor, 1400W, was also effective in reversing the loss of functional hyperemia in this ex vivo preparation. Previous work (Metea and Newman, 2006) has demonstrated that NO, the product of iNOS, inhibits light-evoked vasodilations in healthy retinas. Together, these results strongly suggest that AG functions to reverse the loss of functional hyperemia by inhibiting iNOS and lowering NO.
The loss of functional hyperemia could be due to a decrease in light-evoked neuronal activity. However, our ERG experiments suggest that light-evoked neuronal activity was not diminished in our diabetic animals. Several previous animal studies have demonstrated a decrease in ERG amplitude in early stages of diabetic retinopathy (Barber et al., 1998; Phipps et al., 2004; Antonetti et al., 2006; Fletcher et al., 2007). There are a number of reasons why our ERG results might differ from these earlier studies. Many ERG studies have been conducted using albino strains whose retinas are susceptible to light damage, compounding the effects of diabetic retinopathy. Our experiments are conducted in pigmented Long–Evans rats, a strain that displays a much lower retinal inflammatory response up to 4 months after induction of diabetes by streptozotocin, compared to changes observed in albino Sprague-Dawley retinas (Kirwin et al., 2009). We also treated our rats with supplemental insulin, which could slow the loss of the ERG in diabetic animals.
The reduced flicker-induced vasodilation we observed could also be due to a loss of vascular responsiveness. This does not appear to be the case. A recent study demonstrated that vascular reactivity to exogenous NO stimulation is unchanged in diabetic patients (Pemp et al., 2009). In addition, we demonstrated in an earlier study using the ex vivo retina preparation that prostaglandin E2-induced dilation of retinal arterioles remains unchanged in diabetic animals (Mishra and Newman, 2010). The decrease in flicker-induced vasodilation could also be due to an increase in the resting diameter of the vessels. However, there was no significant difference in resting diameter in control and diabetic groups, and the reduced dilation in diabetic animals was independent of resting vessel diameter (Figure 3C).
Instead, the loss of flicker-induced vasodilation is likely caused by altered neurovascular coupling in diabetic retinas. Recent evidence suggests that flicker-induced vasodilation in the retina as well as neuronal activity-dependent vasodilation in the brain, is largely mediated by glial cells. Release of transmitters from active neurons induces Ca2+ increases in glial cells that, in turn, release vasodilatory agents onto blood vessels (Metea and Newman, 2006; Attwell et al., 2010). In the retina, NO modulates this signaling pathway by an as-yet unidentified mechanism, whereby high tissue NO results in reduced vasodilation and enhanced vasoconstriction (Metea and Newman, 2006). The high NO levels found in the diabetic retina (Kowluru et al., 2000) due to increased iNOS expression (Du et al., 2002; Mishra and Newman, 2010) likely inhibit the glial release of dilatory agents, disrupting the coupling between neuronal activity and vasodilation. It is likely that AG acts by inhibiting iNOS and lowering retinal NO levels, thus allowing normal neurovascular signaling to occur. Our finding that AG does not alter functional hyperemia in control animals supports this view, as iNOS expression is minimal in the healthy retina. The oxygenation response to carbogen inhalation challenge, another vascular response that is diminished in diabetic retinas, is also restored by iNOS inhibitors (Berkowitz et al., 2004).
Light-evoked vasodilation in the retina, as well as neuronal activity-dependent vasodilation in the brain, is believed to be important for meeting the increased demand of active neurons for oxygen and glucose (Polak et al., 2002; Riva et al., 2005). A disruption of this coupling may have damaging effects due to the resulting hypoxia, hypoglycemia, or accumulation of harmful metabolites. Such adverse effects could potentially be offset by an increase in resting vessel diameter, and such increases have been reported in some studies of early stage diabetic retinopathy (Klein et al., 2004; Roy et al., 2011). However, a loss of functional hyperemia has been reported in diabetic patients who showed no increase in resting vessel diameter (Garhofer et al., 2004; Bek et al., 2008). Our results also show no increase in resting diameter in diabetic animals at the 7 month post-induction stage. Further research is needed to determine whether the loss of functional hyperemia in the diabetic retina results in adverse physiological consequences.
Aminoguanidine has been previously assessed as a treatment for complications of diabetes in both animal models and in diabetic patients. A clinical study has demonstrated the beneficial effects of AG on retinopathy, where significantly fewer patients showed progression of retinopathy when treated with AG, compared to placebo-treated patients (Bolton et al., 2004). Several studies on animal models of diabetes have also demonstrated the efficacy of iNOS inhibitors in slowing the progression of diabetic retinopathy (Hammes et al., 1991; Kern et al., 2000; Kowluru et al., 2000; Kern and Engerman, 2001; Du et al., 2002). Early treatment with iNOS inhibitors has also been shown to be more efficacious in slowing the progression of the disease (Berkowitz et al., 2005). The beneficial effects of AG have generally been attributed to the inhibition of AGE formation. However, two studies have shown that AG prevents the development of diabetic retinopathy without significantly affecting AGE production (Kern et al., 2000; Kern and Engerman, 2001), suggesting that the principal action of AG in diabetic retinas is to inhibit iNOS. Supporting this view, diabetic retinopathy is also reduced in iNOS-deficient mice (Zheng et al., 2007). It remains to be determined whether the beneficial effect of iNOS inhibitors is due to a restoration of functional hyperemia. Targeting iNOS or its downstream signaling pathway with more selective inhibitors to reverse the loss of functional hyperemia may help to resolve this issue and may suggest new therapies for treating diabetic retinopathy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Elizabeth R. Seaquist, Tess Kornfield, and Steven Sullivan for comments on the manuscript, and Michael Burian for expert technical assistance. Supported by Fondation Leducq, the National Eye Institute (EY004077), and the Minnesota Medical Foundation.
References
Antonetti, D. A., Barber, A. J., Bronson, S. K., Freeman, W. M., Gardner, T. W., Jefferson, L. S., Kester, M., Kimball, S. R., Krady, J. K., LaNoue, K. F., Norbury, C. C., Quinn, P. G., Sandirasegarane, L., and Simpson, I. A. (2006). Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 55, 2401–2411.
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., MacVicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243.
Barber, A. J., Lieth, E., Khin, S. A., Antonetti, D. A., Buchanan, A. G., and Gardner, T. W. (1998). Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Invest. 102, 783–791.
Bek, T., Hajari, J., and Jeppesen, P. (2008). Interaction between flicker-induced vasodilatation and pressure autoregulation in early retinopathy of type 2 diabetes. Graefes Arch. Clin. Exp. Ophthalmol. 246, 763–769.
Berkowitz, B. A., Luan, H., Gupta, R. R., Pacheco, D., Seidner, A., Roberts, R., Liggett, J., Knoerzer, D. L., Connor, J. R., Du, Y., Kern, T. S., and Ito, Y. (2004). Regulation of the early subnormal retinal oxygenation response in experimental diabetes by inducible nitric oxide synthase. Diabetes 53, 173–178.
Berkowitz, B. A., Roberts, R., Luan, H., Peysakhov, J., Knoerzer, D. L., Connor, J. R., and Hohman, T. C. (2005). Drug intervention can correct subnormal retinal oxygenation response in experimental diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 46, 2954–2960.
Bolton, W. K., Cattran, D. C., Williams, M. E., Adler, S. G., Appel, G. B., Cartwright, K., Foiles, P. G., Freedman, B. I., Raskin, P., Ratner, R. E., Spinowitz, B. S., Whittier, F. C., and Wuerth, J. P. (2004). Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 24, 32–40.
Brownlee, M., Vlassara, H., Kooney, A., Ulrich, P., and Cerami, A. (1986). Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 232, 1629–1632.
Du, Y., Smith, M. A., Miller, C. M., and Kern, T. S. (2002). Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J. Neurochem. 80, 771–779.
Fletcher, E. L., Phipps, J. A., Ward, M. M., Puthussery, T., and Wilkinson-Berka, J. L. (2007). Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr. Pharm. Des. 13, 2699–2712.
Garhofer, G., Zawinka, C., Resch, H., Kothy, P., Schmetterer, L., and Dorner, G. T. (2004). Reduced response of retinal vessel diameters to flicker stimulation in patients with diabetes. Br. J. Ophthalmol. 88, 887–891.
Hammes, H. P., Martin, S., Federlin, K., Geisen, K., and Brownlee, M. (1991). Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc. Natl. Acad. Sci. U.S.A. 88, 11555–11558.
Kern, T. S., and Engerman, R. L. (2001). Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes 50, 1636–1642.
Kern, T. S., Tang, J., Mizutani, M., Kowluru, R. A., Nagaraj, R. H., Romeo, G., Podesta, F., and Lorenzi, M. (2000). Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest. Ophthalmol. Vis. Sci. 41, 3972–3978.
Kirwin, S. J., Kanaly, S. T., Linke, N. A., and Edelman, J. L. (2009). Strain-dependent increases in retinal inflammatory proteins and photoreceptor FGF-2 expression in streptozotocin-induced diabetic rats. Invest. Ophthalmol. Vis. Sci. 50, 5396–5404.
Klein, R., Klein, B. E., Moss, S. E., Wong, T. Y., Hubbard, L., Cruickshanks, K. J., and Palta, M. (2004). The relation of retinal vessel caliber to the incidence and progression of diabetic retinopathy: XIX: the Wisconsin Epidemiologic Study of Diabetic Retinopathy. Arch. Ophthalmol. 122, 76–83.
Kowluru, R. A., Engerman, R. L., and Kern, T. S. (2000). Abnormalities of retinal metabolism in diabetes or experimental galactosemia VIII. Prevention by aminoguanidine. Curr. Eye Res. 21, 814–819.
Mandecka, A., Dawczynski, J., Blum, M., Muller, N., Kloos, C., Wolf, G., Vilser, W., Hoyer, H., and Muller, U. A. (2007). Influence of flickering light on the retinal vessels in diabetic patients. Diabetes Care 30, 3048–3052.
Metea, M. R., and Newman, E. A. (2006). Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J. Neurosci. 26, 2862–2870.
Mishra, A., and Newman, E. A. (2010). Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia 58, 1996–2004.
Mizutani, M., Kern, T. S., and Lorenzi, M. (1996). Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Invest. 97, 2883–2890.
Naka, K. I., and Rushton, W. A. (1966). S-potentials from luminosity units in the retina of fish (Cyprinidae). J. Physiol. (Lond.) 185, 587–599.
Nguyen, T. T., Kawasaki, R., Wang, J. J., Kreis, A. J., Shaw, J., Vilser, W., and Wong, T. Y. (2009). Flicker light-induced retinal vasodilation in diabetes and diabetic retinopathy. Diabetes Care 32, 2075–2080.
Pemp, B., Garhofer, G., Weigert, G., Karl, K., Resch, H., Wolzt, M., and Schmetterer, L. (2009). Reduced retinal vessel response to flicker stimulation but not to exogenous nitric oxide in type 1 diabetes. Invest. Ophthalmol. Vis. Sci. 50, 4029–4032.
Perry, R. E., Swamy, M. S., and Abraham, E. C. (1987). Progressive changes in lens crystallin glycation and high-molecular-weight aggregate formation leading to cataract development in streptozotocin-diabetic rats. Exp. Eye Res. 44, 269–282.
Phipps, J. A., Fletcher, E. L., and Vingrys, A. J. (2004). Paired-flash identification of rod and cone dysfunction in the diabetic rat. Invest. Ophthalmol. Vis. Sci. 45, 4592–4600.
Polak, K., Schmetterer, L., and Riva, C. E. (2002). Influence of flicker frequency on flicker-induced changes of retinal vessel diameter. Invest. Ophthalmol. Vis. Sci. 43, 2721–2726.
Riva, C. E., Logean, E., and Falsini, B. (2005). Visually evoked hemodynamical response and assessment of neurovascular coupling in the optic nerve and retina. Prog. Retin. Eye Res. 24, 183–215.
Roy, M. S., Klein, R., and Janal, M. N. (2011). Retinal venular diameter as an early indicator of progression to proliferative diabetic retinopathy with and without high-risk characteristics in African Americans with type 1 diabetes mellitus. Arch. Ophthalmol. 129, 8–15.
Srienc, A. I., Kurth-Nelson, Z. L., and Newman, E. A. (2010). Imaging retinal blood flow with laser speckle flowmetry. Front Neuroenerg. 2:128.
Keywords: diabetic retinopathy, functional hyperemia, iNOS inhibitors
Citation: Mishra A and Newman EA (2012) Aminoguanidine reverses the loss of functional hyperemia in a rat model of diabetic retinopathy. Front. Neuroenerg. 3:10. doi: 10.3389/fnene.2011.00010
Received: 12 October 2011; Paper pending published: 13 November 2011;
Accepted: 28 December 2011; Published online: 10 January 2012.
Edited by:
Bruno Weber, University of Zurich, SwitzerlandReviewed by:
Angus M. Brown, University of Nottingham, UKTim Murphy, The University of British Columbia, Canada
Copyright: © 2012 Mishra and Newman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Eric A. Newman, Department of Neuroscience, University of Minnesota, Minneapolis, 6-145 Jackson Hall, 321 Church Street SE, Minneapolis, MN 55455, USA e-mail:ZWFuQHVtbi5lZHU=