 Lena H. Nguyen
Lena H. Nguyen Angélique Bordey
Angélique Bordey- 1Department of Neurosurgery, Yale School of Medicine, Yale University, New Haven, CT, United States
- 2Department of Cellular & Molecular Physiology, Yale School of Medicine, Yale University, New Haven, CT, United States
Hyperactivation of the mechanistic target of rapamycin complex 1 (mTORC1) due to mutations in genes along the PI3K-mTOR pathway and the GATOR1 complex causes a spectrum of neurodevelopmental disorders (termed mTORopathies) associated with malformation of cortical development and intractable epilepsy. Despite these gene variants’ converging impact on mTORC1 activity, emerging findings suggest that these variants contribute to epilepsy through both mTORC1-dependent and -independent mechanisms. Here, we review the literature on in utero electroporation-based animal models of mTORopathies, which recapitulate the brain mosaic pattern of mTORC1 hyperactivity, and compare the effects of distinct PI3K-mTOR pathway and GATOR1 complex gene variants on cortical development and epilepsy. We report the outcomes on cortical pyramidal neuronal placement, morphology, and electrophysiological phenotypes, and discuss some of the converging and diverging mechanisms responsible for these alterations and their contribution to epileptogenesis. We also discuss potential therapeutic strategies for epilepsy, beyond mTORC1 inhibition with rapamycin or everolimus, that could offer personalized medicine based on the gene variant.
Introduction
mTORopathies comprise several neurodevelopmental disorders, including tuberous sclerosis complex (TSC), focal cortical dysplasia type II (FCDII), hemimegalencephaly (HME), and polyhydramnios, megalencephaly, and symptomatic epilepsy (PMSE) syndrome, among others. These disorders are characterized by hyperactivation of the mechanistic target of rapamycin (mTOR) signaling, malformation of cortical development (MCD), and intractable epilepsy (Crino, 2015). Recent advances in genomics have identified genetic mutations in regulators of the mTOR pathway as a common molecular etiology for TSC, FCDII, HME, and PMSE, thus emphasizing a central role for mTOR in the pathogenesis of these disorders (Marsan and Baulac, 2018; Muhlebner et al., 2019). The mTOR signaling pathway regulates cell growth and metabolism via two protein complexes, mTORC1 and mTORC2. Here, we focus on mTORC1 because all the identified pathogenic gene variants thus far impinge on mTORC1 activity, and information on mTORC2 activity is limited for these neurodevelopmental disorders.
The canonical mTORC1 signaling pathway consists of phosphoinositide 3-kinase (PI3K), the activity of which is negatively regulated by phosphatase and tensin homolog (PTEN), AKT, tuberous sclerosis complex 1/2 (TSC1/2) protein complex, Ras homolog enriched in brain (RHEB), and mTORC1 (Dibble and Cantley, 2015) (Figure 1). mTORC1 signaling in response to cellular nutrients is additionally regulated by the GAP activity toward Rags 1 (GATOR1) protein complex (Bar-Peled and Sabatini, 2014). To date, numerous pathogenic gene variants in the PI3K-mTOR pathway and GATOR1 complex have been identified as genetic causes leading to mTORC1 hyperactivity, MCD, and epilepsy, and more will undoubtfully emerge with increasing accessibility to advanced genomic tools (Marsan and Baulac, 2018; Muhlebner et al., 2019). The role of mTOR signaling pathway in the brain has been the subject of many reviews (e.g., Lasarge and Danzer, 2014; Lipton and Sahin, 2014; Takei and Nawa, 2014; Switon et al., 2017) and is therefore not extensively discussed here. Rather, the aim of this review is to examine neuronal molecular and functional alterations resulting from distinct PI3K-mTOR and GATOR1 gene variants that converge on mTORC1 activation and discuss the potential mechanisms leading to epileptogenesis due to these pathological variants.
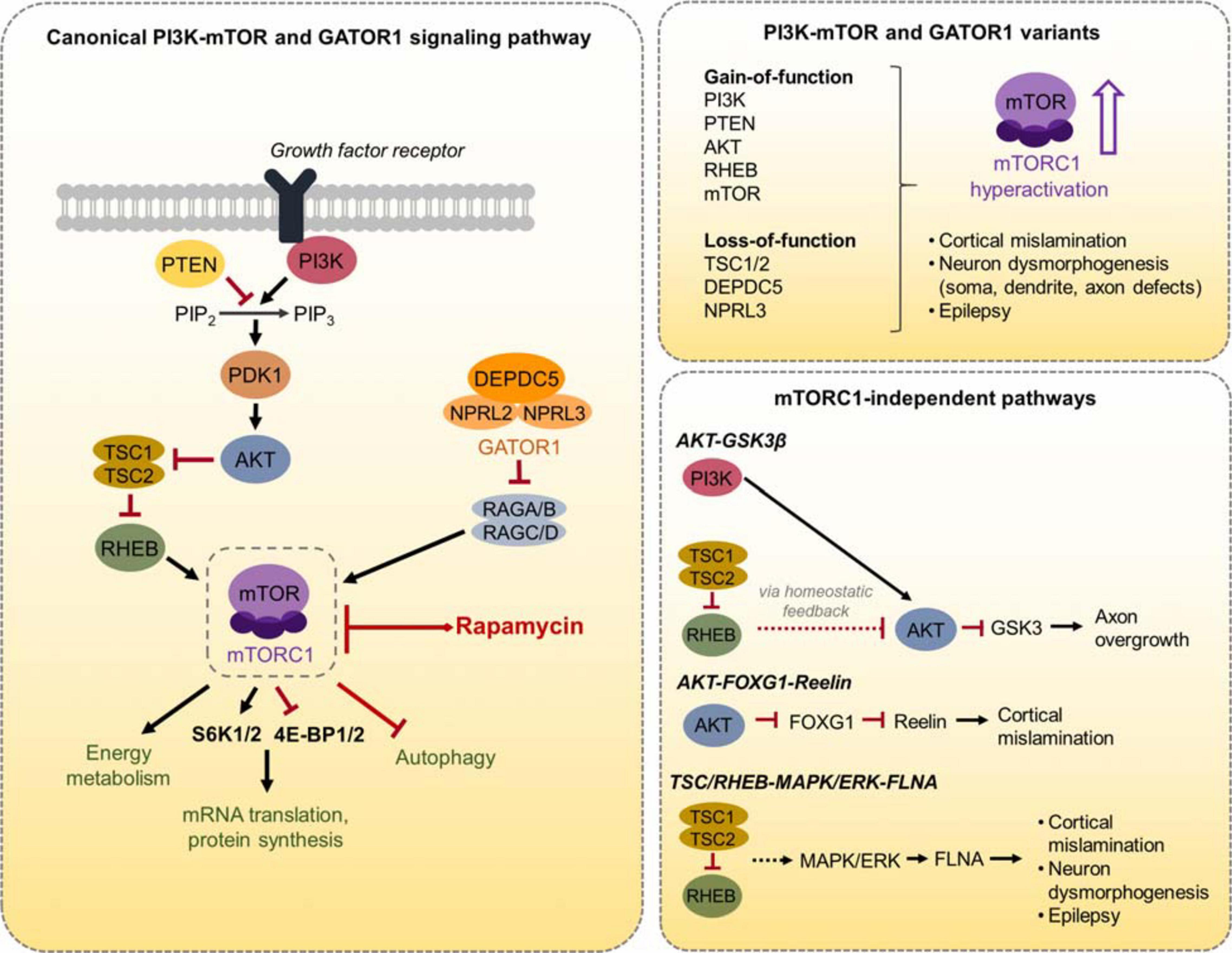
Figure 1. Schematic diagram of the canonical PI3K-mTOR and GATOR1 signaling pathway and mTORC1-independent pathways. (Left) The canonical PI3K-mTOR signaling pathway, the GATOR1 complex, and several mTORC1 downstream effectors are shown. mTORC1 regulates mRNA translation and protein synthesis via direct phosphorylation of S6K1/2 and 4E-BP1/2. mTORC1 also regulates energy metabolism and autophagy, as well as other processes not shown here. (Right, top) A summary of the PI3K-mTOR and GATOR1 gene variants discussed in this review is depicted. These variants converge on mTORC1 hyperactivation, leading to cortical mislamination, neuron dysmorphogenesis, and epilepsy. (Right, bottom) The three mTORC1-independent pathways that are discussed in this review and the associated phenotypes are depicted.
We first provide an overview of the distinct PI3K-mTOR or GATOR1 gene variants and introduce different animal models aimed to recapitulate the disease phenotypes. In particular, we discuss the advantages of in utero electroporation (IUE)-based models to replicate the genetic mosaicism as well as the focal nature of MCD observed in TSC and FCDII. We then review the current literature on IUE-based animal models of TSC/FCDII targeting cortical pyramidal neurons, which at this time comprises 33 studies summarized in Table 1. We compare the outcomes of distinct PI3K-mTOR and GATOR1 gene variants on the structural alterations that are pathological hallmarks of TSC/FCDII, namely cell misplacement and neuronal hypertrophy. We discuss these findings in the context of major molecular processes downstream mTORC1 that have known contributions to these specific alterations, such as cap-dependent translation. We next compare the electrophysiological properties and synaptic activities of neurons expressing the distinct gene variants. We focus on cell autonomous mechanisms of neuronal hyperexcitability, although non-cell autonomous changes in the surrounding environment such as decreased interneuron density (Zhong et al., 2021), altered connectivity of non-targeted, contralateral neurons (Onori et al., 2020), and increased vascular density (Zhang et al., 2019) have been reported in these models. One major finding we discuss is the abnormal expression of an mTORC1-dependent hyperpolarization-activated cyclic nucleotide-gated (HCN) channel isoform 4, HCN4, which provides a cyclic AMP (cAMP)-dependent mode of hyperexcitability in mutant pyramidal neurons expressing constitutively active Rheb (Hsieh et al., 2016). However, it remains uncertain whether these channels are present in neurons expressing the other gene variants. Both structural and functional changes will be discussed in a broader context for how they contribute to epileptogenesis. We then diverge from an mTOR-centric view by highlighting specific parallel molecular players and pathways that are additionally altered as a consequence of some, but not all, gene variants. We particularly examine the activities of AKT-Glycogen synthase kinase 3 beta (GSK3β) on axon growth, AKT-Forkhead box G1 (FOXG1)-reelin on neuronal migration, and TSC/RHEB-mitogen-activated protein kinase (MAPK)/ERK-filamin A (FLNA) on neuronal dysmorphogenesis and seizures. Finally, we discuss how these novel findings can lead to new treatment options in addition to using classical mTOR inhibitors such as rapamycin and everolimus.
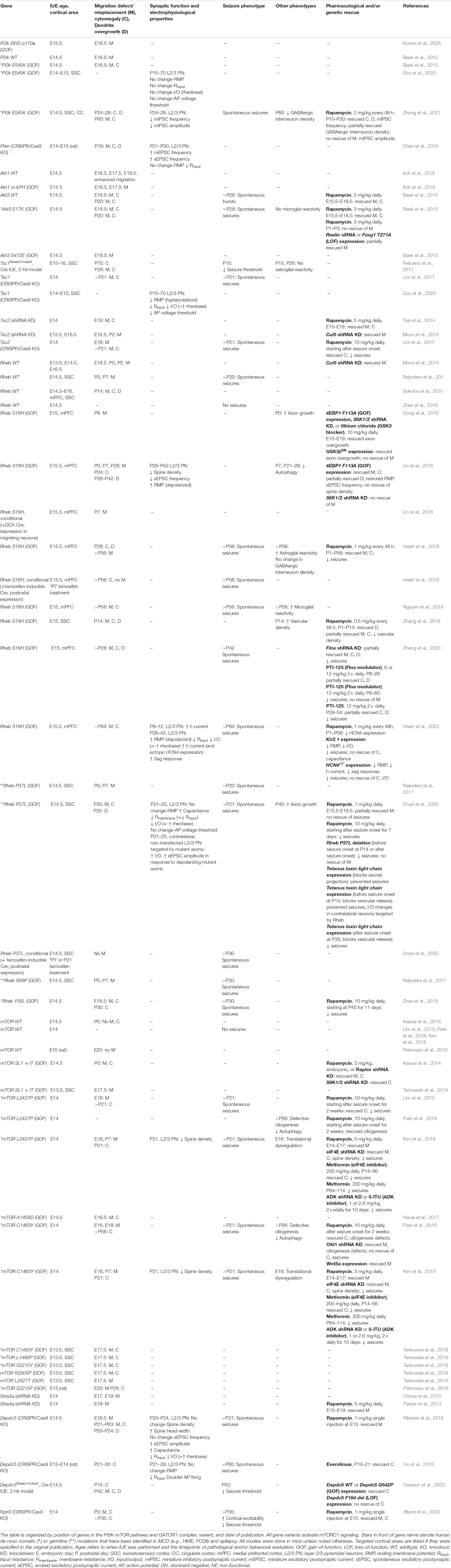
Table 1. Summary of IUE-based rodent models of mTORopathies.
Pi3K-mTor and Gator1 Gene Variants Resulting in Malformation of Cortical Development and Epilepsy
Early evidence for aberrant mTORC1 activation in TSC, FCDII, and HME, was identified by histological analysis of brain specimens from patients who underwent surgery for epilepsy (Baybis et al., 2004; Miyata et al., 2004; Ljungberg et al., 2006; Aronica et al., 2007). mTORC1 hyperactivation was also later demonstrated in patients with the rare neurodevelopmental disorder, PMSE (Puffenberger et al., 2007). These disorders collectively termed mTORopathies, share key pathological hallmarks, including MCD with cortical enlargement, mislamination, gliosis, and the presence of dysmorphic, cytomegalic neurons (Crino, 2015). TSC, FCDII, and HME are primarily distinguished based on the extent of the affected cortical regions, with HME having broader MCDs involving as much as whole cerebral hemispheres while TSC and FCDII are characterized by focal MCDs. These mTORopathies are associated with frequent seizures and are the most common causes for childhood intractable epilepsy (Harvey et al., 2008; Crino, 2015).
The mTOR pathway is a ubiquitously expressed signaling pathway that is involved in many important cellular processes, including cell growth, protein synthesis, autophagy, metabolism, and lysosome biogenesis (Saxton and Sabatini, 2017). In the brain, mTOR signaling controls cortical development and neural-specific functions, such as synaptic plasticity and learning and memory (Lipton and Sahin, 2014; Takei and Nawa, 2014). As a result, dysregulation of mTOR signaling has been implicated in several neurodevelopmental and neuropsychiatric disorders (Costa-Mattioli and Monteggia, 2013). The mTOR protein is an evolutionary conserved serine/threonine kinase. Association of mTOR with the Raptor adaptor protein characterizes mTORC1, a rapamycin-sensitive complex that regulates protein synthesis via direct phosphorylation of the translational regulators p70 S6 kinase 1 and 2 (S6K1/2) and eIF4E-binding protein 1 and 2 (4E-BP1/2) (Ma and Blenis, 2009; Saxton and Sabatini, 2017). Levels of phosphorylated S6K1/2 [or its downstream effector, ribosomal protein S6 (RPS6)] and 4E-BP1/2 are reliably used as readouts for mTORC1 activity. mTORC1 regulation of autophagy and metabolism occurs via additional downstream effectors (Saxton and Sabatini, 2017).
The best-characterized upstream regulation of mTORC1 activity is through the PI3K-AKT signaling pathway (Dibble and Cantley, 2015). PI3K is a lipid kinase that is activated by cell-surface receptors in response to extracellular cues such as growth factors. PI3K activates AKT, which directly phosphorylates and inactivates the TSC1/TSC2 complex. Inactivation of TSC1/2, which functions as GTPase-activating proteins (GAPs), results in activation of the GTP-binding protein RHEB that in turn activates mTORC1. The lipid phosphatase PTEN works as a negative regulator of mTORC1 signaling by counteracting PI3K function and thereby opposing downstream activation of mTORC1. In addition to PI3K-AKT signaling, recent studies have identified the GATOR1 complex, which consists of DEP domain containing 5 (DEPDC5) and the Nitrogen permease regulator 2-like (NRPL2) and 3-like (NPRL3) proteins, as a negative regulator of mTORC1 activity in response to cellular amino acid levels (Bar-Peled and Sabatini, 2014). GATOR1 functions as GAPs that inhibit mTORC1 activity by inactivating the Rag GTPases. When amino acid levels are low, GATOR1 signals to inhibit mTORC1. Conversely, when amino acid levels are adequate, GATOR1 releases mTORC1 inhibition to promote cell growth. Thus, there are two major molecular branches, PI3K/PTEN-AKT-TSC-RHEB and GATOR1, that control mTORC1 activity (Figure 1).
Genetic mutations leading to mTORC1 hyperactivity were first identified in the prototypical mTORopathy, TSC. Individuals with TSC have germline and somatic inactivating mutations in the TSC1 or TSC2 genes, leading to mTORC1 activation in multiple organs, including the brain (Kwiatkowski, 2003; Orlova and Crino, 2010). More recently, the emergence of advanced sequencing techniques led to the seminal discovery that FCDII and HME are caused by de novo brain somatic mutations in mTORC1 pathway genes that occur during neurodevelopment. Work by Lee et al. (2012) and Poduri et al. (2012) was first to demonstrate that brain somatic mutations in PI3K, AKT3, and mTOR are causal of HME. This was followed by crucial studies from Lim et al. (2015) and Nakashima et al. (2015)
showing that brain somatic mutations in mTOR cause FCDII. Brain somatic mutations in TSC1 or TSC2, in the absence of germline mutations, also lead to FCDII (Lim et al., 2017). Numerous studies have been published since and have been discussed in several review articles (Marsan and Baulac, 2018; Muhlebner et al., 2019). PMSE is caused by a homozygous deletion in the STE20-related kinase adaptorα (Strada) gene, a modulator of mTORC1 signaling via the AMP-activated protein kinase (AMPK)-TSC1/2 pathway (Puffenberger et al., 2007; Orlova et al., 2010). To date, pathogenic variants in 14 distinct genes along the PI3K-mTOR pathway and GATOR1 complex, including PI3K, PTEN, AKT3, TSC1, TSC2, RHEB, MTOR, STRADA, DEPDC5, NPRL2, NPRL3, KPTN, SZT2, and TBC1D7, have been linked to MCD and epilepsy (Kim and Lee, 2019; Crino, 2020). Mutations in some genes (i.e., MTOR) occur much more frequently than in others (i.e., KPTN, SZT2, and TBC1D7) (D’Gama et al., 2017; Baldassari et al., 2019; Sim et al., 2019). Given that all of these mutations converge on mTORC1 leading to its hyperactivation, pharmacological intervention with mTOR inhibitors (e.g., rapamycin and its analog everolimus) has been proposed as a therapeutic option for epilepsy associated with these MCDs. In 2018, the rapamycin analog everolimus was US Food and Drug Administration (FDA)-approved to treat seizures in individuals with TSC (French et al., 2016). The efficacy of everolimus in controlling seizures in individuals with FCDII is currently assessed in a clinical trial (ClinicalTrials.gov identifier: NCT03198949).
Designing Animal Models of mTORopathies
Hyperactive mTORC1 signaling leads to cellular and network alterations resulting in MCD and epilepsy (Lasarge and Danzer, 2014; Represa, 2019). However, the precise mechanisms remain elusive. Many animal models of mTORopathies have been established to aid in understanding the impact of mTORC1 hyperactivation on brain development and epilepsy. Brain-specific, conditional transgenic mice, such as Pten, Tsc1, Tsc2, and Depdc5 knockout and mTOR gain-of-function mutation, exhibit key human mTORopathy phenotypes, including enlarged cortex, ectopic neuronal placement, neuronal hypertrophy, gliosis, and epilepsy (e.g., Kassai et al., 2014; Ostendorf and Wong, 2015; Yuskaitis et al., 2018). By contrast, global homozygous transgenic mice with constitutive mTOR activation are embryonic lethal. Newer rodent models using IUE to focally express or suppress mTORC1 pathway regulating genes in the cortex (discussed more below), as well as Tsc2 and Depdc5 zebrafish models (Kim S. H. et al., 2011; de Calbiac et al., 2018; Swaminathan et al., 2018), also recapitulate many of the mTORopathy phenotypes. Recent models also include patient-derived induced pluripotent stem cells (IPSCs) and cortical organoids, which provide the advantage of studying mTOR function in human cell populations (Blair et al., 2018; Andrews et al., 2020; Dang et al., 2020; Eichmüller et al., 2020). Both transgenic and IUE-based animal models as well as in vitro models have been vital for mechanistic and preclinical drug studies.
The MCD pathology in TSC, FCDII, and HME are thought to be caused by constitutive mTORC1 hyperactivation in early neuroglial progenitor cells during embryonic development, leading to a subset of affected neurons within a diffuse or localized area of the cortex (Poduri et al., 2013; Prabowo et al., 2013; Tsai et al., 2014; D’Gama et al., 2017). It is thought that the timing of when the somatic mutations occur determines the extent of the affected brain region, such that mutations leading to broader MCDs in HME occur earlier in development while those leading to focal MCDs in TSC and FCDII occur later in development (D’Gama et al., 2017). Additionally, it has been proposed that somatic mutations occurring early on, before hemispheric cleavage, can lead to bilateral asymmetric FCDII, where one hemisphere display apparent focal MCDs whereas the other side does not have visible malformations despite containing mutated cells, due to an uneven hemispheric distribution of mutation load (Guerrini et al., 2021). While transgenic mouse models have been important in studies of global MCDs, models that more precisely recapitulate focal lesions in TSC and FCDII provide additional advantages. One such TSC mouse model combined exogenous injection of a Cre-expressing viral vector in Tsc1floxed/floxed pups. By controlling the dose of the viral vector, investigators were able to achieve mosaic Tsc1 loss in a localized region similar to those observed in human TSC (Prabhakar et al., 2013). Another approach that has emerged within the last decade is based on IUE. This technique allows for in vivo manipulation of specific cell types in select brain regions (LoTurco et al., 2009). Using this technique, plasmid DNA targeting PI3K-mTOR or GATOR1 components are microinjected into the ventricles of embryonic rodent brains and transfected via electrical pulses into neural progenitor cells (i.e., radial glia) that line the ventricles. The position of the electrodes that generate the electrical pulses, which is manually determined by the investigator, directs plasmid expression into the brain region of interest (Figure 2). Current IUE-based models of mTORopathies include expression of Pi3k, Akt, Rheb, and mTOR gain-of-function variants (mTORC1 activators) or suppression of Pten, Tsc1, Tsc2, Stradα, Depdc5, and Nprl3 (mTORC1 negative regulators) expression (Table 1 and Figure 1). Some of the gain-of-function variants are mutations that have been identified in patients with MCD and epilepsy while others are experimental mutations predicted to be pathogenic. Suppression of the negative regulators is achieved by CRISPR/Cas9 gene editing, which leads to gene knockout, or short hairpin RNA (shRNA)-mediated methods, which leads to gene knockdown. Additionally, two studies used a 2-hit model where a Cre plasmid was expressed by IUE in Tsc1floxedmutant or Depdc5floxed/mutant mice to drive cell-type and -region specificity. The studies reviewed here all targeted cortical pyramidal neurons of layer (L) 2/3 of either the somatosensory cortex (SSC) or medial prefrontal cortex (mPFC), as these principal cells are proposed to be most severely affected in MCDs (Blumcke et al., 2009; Hadjivassiliou et al., 2010; D’Gama et al., 2017). Expression in L2/3 pyramidal neurons is achieved by performing IUE at embryonic day (E) 14–15 (±0.5), which targets radial glia generating upper layer cortical neurons. It is important to note that although IUE targets radial glia that give rise to both neurons and astrocytes, episomal plasmids that are used in standard IUE will only be expressed in neurons due to a plasmid dilution effect that occurs in dividing cells (Feliciano et al., 2011; Chen and LoTurco, 2012). In contrast, CRISPR/Cas9 plasmids integrate into the genome of the electroporated radial glia and are expected to label all cell types in the radial glial cell lineage (i.e., neurons and astrocytes). Overall, in addition to providing region- and cell-specificity, IUE offers the flexibility to vary the type of gene variants expressed, the timing of plasmid expression, and the affected region in a relatively simple manner.
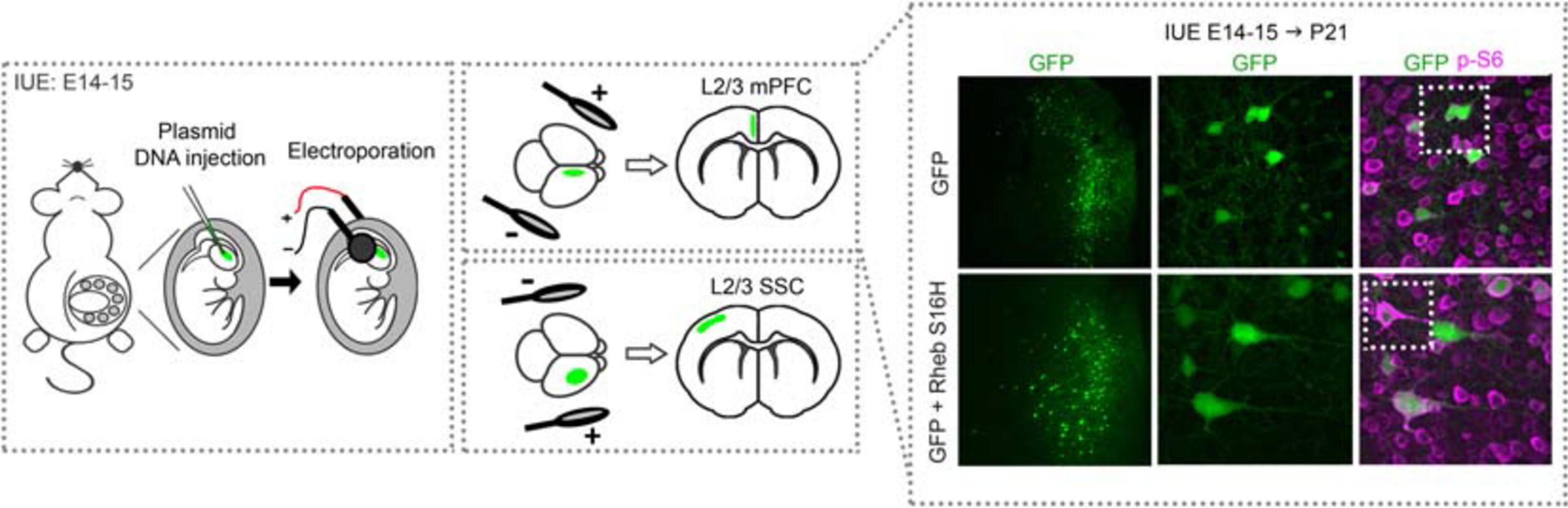
Figure 2. Schematic diagram of in utero electroporation and example of electroporated cortices. Rodents are electroporated at E14–15 (±0.5), which targets L2/3 cortical pyramidal neurons. Briefly, plasmid DNA is injected into the lateral ventricles and electrical pulses are passed through to electroporate the plasmid into neural progenitor cells (i.e., radial glia). The position of the electrodes that generate the pulses direct the plasmid into the specific brain region of interest, e.g., mPFC or SSC. An example of cortices from mice electroporated with GFP or GFP + Rheb S16H plasmids in the mPFC are shown. Note that GFP-expressing cells are positioned in L2/3 while GFP + Rheb S16H-expressing cells are misplaced across the cortical layers. GFP + Rheb S16H-expressing cells are also larger and exhibit increased phospho (p)-S6 levels, a marker of mTORC1 activity. E, embryonic day; L, layer; mPFC, medial prefrontal cortex; SSC, somatosensory cortex.
Conserved mTorc1-Dependent Phenotypes Across Pi3K-mTor and Gator1 Gene Variants
Ectopic neuronal placement and the presence of dysmorphic, cytomegalic neurons are major histopathological findings in TSC and FCDII (Wong, 2008; Blumcke et al., 2011). All gene variants reviewed here (see Table 1) consistently reproduce these phenotypes, supporting the notion that the underlying mechanisms for these alterations involve the mTORC1 pathway and its downstream processes. Additional histopathological phenotypes, such as the presence of balloon cells, have not been reported in these studies. Gliosis, including astrocyte and microglia reactivity, is regularly observed but seems to track with seizure activity (Feliciano et al., 2011; Nguyen et al., 2019), and is not discussed here.
Cortical pyramidal neurons destined to L2/3 are generated from radial glia in the ventricular zone at E14–15 in mice. Newborn neurons migrate from the ventricular zone shortly after and reach their final position in the cortex by postnatal day (P) 7 (Greig et al., 2013; Kast and Levitt, 2019). Disruption in this process leads to ectopic neuronal placement and cortical mislamination. In line with this timeframe, IUE at E13.5–15.5 resulted in neuronal migration defects and misplacement in the SSC and mPFC for all evaluated gene variants (Table 1). Despite being misplaced in other cortical layers, these neurons preserved the molecular identity of L2/3 pyramidal neurons, as evidenced by positive staining for the upper layer markers Cux1 or Satb2 and negative staining for the deeper layer marker Ctip2 (Tsai et al., 2014; Baek et al., 2015; Moon et al., 2015; Lin et al., 2016; Park et al., 2018; Tarkowski et al., 2019; Iffland et al., 2020; Onori et al., 2020; Zhong et al., 2021). Prenatal treatment with rapamycin to reduce mTORC1 activity prevented the migration defects and led to correct placement of the neurons in the cortex, supporting that mTORC1 hyperactivity contributes to these defects (Parker et al., 2013; Kassai et al., 2014; Tsai et al., 2014; Baek et al., 2015; Ribierre et al., 2018; Kim et al., 2019; Iffland et al., 2020; Onori et al., 2020). Postnatal rapamycin treatment after P7, when cortical neuronal migration is complete, is not expected to rescue neuronal misplacement. Indeed, this expectation was confirmed in the Pi3k E545 variant (Zhong et al., 2021). Additionally, two studies showed that mTORC1 hyperactivation once migration is complete, by using a conditional Rheb S16H or Rheb P37L plasmid with a tamoxifen-inducible Cre plasmid, had no effect on neuronal placement (Hsieh et al., 2016; Onori et al., 2020). Thus, the impact of mTORC1 hyperactivation on neuronal placement occurs during a specific developmental timeframe, and cortical mislamination can be prevented in a limited effectiveness window.
Neuronal cytomegaly was also consistently reported for the evaluated gene variants. Increased neuron soma size was reported as early as E17.5 (Tarkowski et al., 2019) and persisted into adulthood (Lim et al., 2015, 2017; Park et al., 2018; Ribierre et al., 2018; Nguyen et al., 2019; Zhang et al., 2019; Dawson et al., 2020; Hsieh et al., 2020; Zhong et al., 2021). Furthermore, increasing mTORC1 activity levels by varying concentration of the Rheb S16H plasmid resulted in dose-dependent increases in neuron soma size, supporting a direct relationship between mTORC1 activity level and soma size (Nguyen et al., 2019). Unlike with the migration defects, mTORC1-induced neuronal dysmorphogenesis is not restricted to an early developmental window, as hyperactivating mTORC1 signaling via conditional Rheb S16H expression at P7 resulted in enlarged neurons (Hsieh et al., 2016). Neuron soma size was reduced with both pre- and postnatal rapamycin (or everolimus) treatment (Kassai et al., 2014; Tsai et al., 2014; Baek et al., 2015; Lim et al., 2015, 2017; Hsieh et al., 2016; Hu et al., 2018; Park et al., 2018; Zhang et al., 2019; Iffland et al., 2020; Zhong et al., 2021). Moreover, postnatal rapamycin treatment reduced soma size regardless of whether treatment began in neonatal or adult ages, suggesting mTORC1 hyperactivation impact neuron size via a dynamic, modifiable process. Dendrite morphology was assessed in the IUE studies targeting Pi3k, Pten, Rheb, and Depdc5, and all revealed dendritic overgrowth involving (for some) increased dendrite thickness and complexity (Chen et al., 2015; Hsieh et al., 2016; Lin et al., 2016; Ribierre et al., 2018; Sokolov et al., 2018; Zhang et al., 2019, 2020; Dawson et al., 2020; Onori et al., 2020; Zhong et al., 2021). Rescue of dendrite morphology by postnatal rapamycin treatment was reported in the Pi3k E545 and Rheb S16H variants, and rapamycin likely has the same effect for the other variants (Zhang et al., 2019; Zhong et al., 2021). Data on axons in the IUE models are more limited. One study reported accelerated axon growth in mPFC L2/3 neurons at P0 following Rheb S16H expression (Gong et al., 2015). Axon overgrowth to the contralateral cortex in SSC L2/3 neurons at P45 has also more recently been reported in mice expressing the Rheb P37L variant (Onori et al., 2020).
The effectiveness of rapamycin in restoring neuronal migration and morphology defects supports that these phenotypes are mTORC1-dependent processes. mTORC1 regulates several downstream intracellular pathways, which could contribute to these cytoarchitectural defects. We will discuss the following mTORC1-dependent mechanisms here: altered translational control via 4E-BP1/2 and S6K1/2, and defective neuronal ciliogenesis in the context of autophagy. We also briefly discuss mTORC1-mediated regulation of the reelin-disabled 1 (DAB1) signaling.
The best characterized function of mTORC1 is regulation of mRNA translation via direct phosphorylation of two major translational regulators, S6K1/2 and 4E-BP1/2 (Ma and Blenis, 2009). S6K1/2 controls translation initiation as well as other functions via a negative feedback loop through AKT (Manning, 2004). The only known function for 4E-BP1/2 so far is regulation of cap-dependent translation, whereby ribosome recruitment to mRNA is initiated via binding of the eukaryotic translation initiation factor 4F (eIF4F) complex to the mRNA 5′ cap structure (Richter and Sonenberg, 2005). 4E-BP1/2 serves as a translational repressor that inhibits eIF4F function. mTORC1 promotes translation by inhibiting 4E-BP1/2 activity, which disinhibits eIF4F (Figure 1). Studies by Lin et al. (2016) demonstrated that targeting 4E-BP1/2 via expression of a constitutive active 4E-BP1 mutant (4E-BP1 F113A), which resists inactivation by mTORC1, prevented Rheb S16H-induced neuronal misplacement. Furthermore, knockdown of 4E-BP2 alone, which leads to increased cap-dependent translation, led to ectopic neuronal placement (Lin et al., 2016). These findings support that increased cap-dependent translation is necessary and sufficient to induce neuronal misplacement. By contrast, targeting S6K1/2 did not rescue neuronal misplacement (Lin et al., 2016). The relevant role of cap-dependent translation in neuronal migration is corroborated by Kim et al. (2019) who demonstrated that knockdown of eIF4E, a component of the eIF4F complex, rescued misplacement in both the mTOR L2427P and C1483Y variants. With regards to neuronal morphology, expression of constitutive active 4E-BP1 or knockdown of eIF4E reduced soma enlargement caused by Rheb S16H and mTOR L2427P and C1483Y, respectively. Metformin, an inhibitor of cap-dependent translation, also reduced cell size in the two mTOR variants (Kim et al., 2019). Additionally, knockdown of S6K1/2 reduced the increased soma size associated mTOR SL1 + IT, an experimental mTOR variant that harbors four point mutations, V2198A, L2216H, L2260P, and I2017T (Kassai et al., 2014). However, one study showed that expressing a wildtype or a gain-of-function mutant of RPS6, the canonical S6K1/2 effector, did not lead to migration defects or altered neuron soma size in rats, suggesting that dysregulation in this axis alone is not sufficient to induce these phenotypes (Pelorosso et al., 2019). In terms of axons, studies by Gong et al. (2015) showed that enhancing 4E-BP1 or reducing S6K1/2 activity prevented Rheb S16H-induced axon overgrowth in vivo, supporting in vitro studies demonstrating both 4E-BP1/2 and S6K1/2 involvements in axon development (Choi et al., 2008; Li et al., 2008; Morita and Sobue, 2009).
Since newborn neurons migrate along radial glia scaffolds to their final position in the cortex and IUE targets radial glia, failure of neurons to migrate properly in these IUE models could be due to defects in the radial glia scaffolding rather than in the neurons. To address this question, Lin et al. (2016) co-electroporated a conditional Rheb S16H plasmid with a doublecortin (DCX)-Cre plasmid to selectively express Rheb S16H in DCX-expressing migrating neurons. They found that mTORC1 hyperactivation in migrating neurons alone was sufficient to cause neuronal misplacement, supporting that mTORC1-induced neuronal migration defects can occur independently of radial glia (Lin et al., 2016). Nevertheless, it is conceivable that radial glia defects can aggravate this phenotype. Neuronal-based mechanisms for cell misplacement is further supported by work from Park et al. (2018) who demonstrated that neuronal autophagy-mediated primary cilia defects cause mTORC1-induced neuronal misplacement. The authors found that impaired autophagy following mTOR L2427P and C1483Y expression led to an accumulation of the OFD1 protein, a regulator of neuronal ciliogenesis, and disrupted cilia formation. Neuron cilia are essential organelles that integrate many important developmental signaling pathways, such as Wnt and Sonic hedgehog, and defective neuron cilia-mediated signaling can lead to abnormal migration and ectopic neuronal placement (Guemez-Gamboa et al., 2014). The authors showed that knockdown of Ofd1 or expression of Wnt5a restored neuronal placement in the mTOR C1483Y condition, supporting that disrupted ciliogenesis underlies these alterations. Importantly, defective ciliogenesis was sensitive to rapamycin treatment in both the mTOR C1483Y and mTOR C1483Y conditions, verifying that this is an mTORC1-dependent mechanism that is potentially shared across the other gene variants. Another proposed mechanism for mTORC1-mediated migration defects is via the reelin-DAB1 signaling pathway. The reelin-DAB1 pathway controls cytoskeletal changes required for neuronal migration and is a well-known regulator of cortical lamination and cell positioning in the cortex (Forster et al., 2010; Frotscher, 2010). Reelin-DAB1 signaling is regulated by the ubiquitin E3 ligase, Cullin 5 (CUL5). Interestingly, studies by Moon et al. (2015) found that Tsc2 knockdown or wildtype Rheb overexpression resulted in increased CUL5 expression, and reducing CUL5 abrogated the migration defects resulting from these variants. They further showed that rapamycin decreased CUL5 levels in a conditional Tsc2–/– mouse model, suggesting that aberrant reelin- DAB1 signaling via CUL5 is an mTORC1-dependent mechanism for neuronal msiplacement observed in mTORopathies.
Overall, these IUE studies demonstrate that defects in neuronal migration and morphology are conserved phenotypes across distinct PI3K-mTOR and GATOR1 gene variants, and the mechanisms that underlie these alterations likely converge on mTORC1 and its downstream effectors. For example, cap-dependent translation was shown to be an important regulator of the migration and morphological phenotypes observed in the Rheb and mTOR variants, and it is likely that the other variants such as Tsc1, Tsc2, Depdc5, etc. also share these mechanisms. However, it is important to emphasize that while 4E-BP1/2 and eIF4E contribute to neuron size, other molecular players such as S6K1/2 may also be involved (Fingar et al., 2002). It is also important to recognize that distinct PI3K-mTOR and GATOR1 genes differentially activate other intracellular molecules, which can also contribute to the observed phenotypes independently of mTORC1. Some of these mTORC1-independent pathways are discussed more in the later sections. Lastly, while some targets are important for multiple phenotypes (e.g., 4E-BP1/2 on neuronal migration and morphology), other targets regulate specific pathological features. For example, targeting OFD1 rescued migration defects but not neuron size in the mTOR variants.
In the context of epilepsy, spontaneous seizures were reported in all studies that evaluated this phenotype. Postnatal rapamycin treatment, starting before or after seizure onset, sufficiently suppresses seizures in multiple models (Lim et al., 2015, 2017; Hsieh et al., 2016; Zhao et al., 2019; Onori et al., 2020). Interestingly, these studies consistently reported that mTORC1-induced seizures occur independently of neuronal placement, and preventing this alteration did not prevent seizures. Hsieh et al. (2016) and Onori et al. (2020) used a conditional Rheb S16H and Rheb P37L plasmid, respectively, to bypass the developmental window for neuronal migration and found that seizures still developed despite proper placement of the mutant neurons. Similarly, Park et al. (2018) showed that targeting the Ofd1 gene responsible for disrupting neuronal ciliogenesis rescued cilia defects and neuronal misplacement but not seizures in the mTOR C1483Y condition. These findings support neuronal misplacement of mutant neurons is not necessary for seizure generation. It is nonetheless conceivable that neuronal misplacement renders seizures worse, but this has not been examined. In terms of neuronal morphology, increased neuron soma size, as well as altered dendritic complexity and axon length, are thought to impact neuronal integration into cortical circuits and thereby contribute to cortical hyperexcitability and network dysfunction that can lead to seizures and epilepsy (Lasarge and Danzer, 2014; Represa, 2019).
Potential Shared and Divergent Sources of Cell-Autonomous Hyperexcitability
There is a large number of electrophysiological studies in in vitro (primarily in hippocampal neurons and some in cortical neurons) and transgenic mouse models of mTORopathies [for some examples, see: (Tavazoie et al., 2005; Wang et al., 2007; Bateup et al., 2011, 2013; Luikart et al., 2011; Weston et al., 2012; Xiong et al., 2012; De Fusco et al., 2020)]. These studies are not reviewed here, but they highlight gene-specific differences in neurons and tissue hyperexcitability. Findings of cell-autonomous changes in excitability came from original studies in human TSC and FCDII tissue samples. These studies reported that dysmorphic, cytomegalic neurons within the tissue samples exhibit altered intrinsic excitability that may contribute to the generation of epileptic activity (Cepeda et al., 2003, 2005, 2012; Wang et al., 2007). In addition, a recent IUE study in Rheb S16H mutant pyramidal neurons reported that decreasing neuronal excitability by expressing an inwardly-rectifying potassium channel (Kir2.1) significantly reduced seizures, supporting that mutant neurons are directly involved in seizure generation (Hsieh et al., 2020). In light of these findings supporting a cell autonomous mechanism of hyperexcitability, we review the electrophysiological data on neurons expressing the different PI3K-mTOR and GATOR1 gene variants. We note that non-cell autonomous mechanisms of hyperexcitability in the surrounding environment have been described in some of the reviewed studies (Zhang et al., 2019; Onori et al., 2020; Zhong et al., 2021), but there are not enough data to compare across gene variants and they will thus not be discussed.
Two main groups of functional parameters account for neuronal excitability: intrinsic biophysical properties [i.e., passive membrane properties and ion channel complements important for the generation of action potentials (AP)] and synaptic properties [i.e., dendritic spine properties and excitatory and inhibitory postsynaptic currents (EPSC and IPSC)]. These parameters have been examined by patch clamp electrophysiology recordings in acute brain slices in 9 IUE studies targeting Pi3k, Pten, Tsc1, Rheb, mTOR, and Depdc5 (Table 1). While these variants display strikingly similar morphological phenotypes (as described above), the functional phenotypes are more variable. Most recordings were performed from L2/3 cortical pyramidal neurons in the SSC except for the Rheb S16H condition, which was performed in the mPFC. Of note, no single study examined all the above parameters at once and the age of recording was different for each study. The latter variable is particularly important for evaluating synaptic activity because under normal conditions, spine growth is complete by P21 and a period of autophagy-mediated spine elimination follows until P28 (White et al., 1997). A study in conditional Tsc2± mice reported no changes in dendritic spine properties at P21 but found an increase in spine density at P28 (Tang et al., 2014). This was accounted for by the observation that Tsc2± mice have decreased mTORC1-regulated autophagy, which reduced spine elimination but did not affect spine formation. Thus, age needs to be carefully considered when assessing synaptic activity.
Regardless of the differences in age of recording and cortical area (i.e., mPFC vs. SSC), a few biophysical changes were conserved across the gene variants. In particular, increased capacitance was found in mutant neurons expressing Rheb P37L (Onori et al., 2020) and Depdc5 (Ribierre et al., 2018) variants, which is consistent with increased cell size. Additionally, Pten, Tsc1, Rheb S16H, Rheb P37L, and Depdc5 mutant neurons all displayed decreased input resistance (Rin) (Chen et al., 2015; Hu et al., 2018; Ribierre et al., 2018; Goz et al., 2020; Hsieh et al., 2020; Onori et al., 2020). As a result of decreased Rin, mutant neurons required a larger current injection to reach AP threshold [i.e., decreased input/output (I/O) or increased rheobase]. These findings support a decreased firing responsiveness to excitatory inputs for these gene variants. Nevertheless, above the firing threshold, Depdc5 mutant neurons were reported to fire an initial AP doublet (Hu et al., 2018) or displayed increased gain of firing frequency (i.e., generate more APs) (Ribierre et al., 2018), suggesting that once neurons reach their firing threshold, they are more excitable. These findings also suggest an alteration in the ion channel complement of the Depdc5 mutant neurons. Interestingly, no changes in Rin or I/O curve were observed for neurons expressing the Pi3k E545K variant (Goz et al., 2020). Regarding the resting membrane potential (RMP), Rheb S16H mutant neurons in the mPFC displayed a depolarized RMP (Lin et al., 2016; Hsieh et al., 2020). In contrast, Pi3k E545K, Pten, Rheb P37L, and Depdc5 mutant neurons in the SSC displayed no changes in RMP (Chen et al., 2015; Hu et al., 2018; Goz et al., 2020; Onori et al., 2020). Tsc1 mutant neurons (by the CRISPR/Cas9 system) had hyperpolarized RMP when compared to control eGFP/mRFP-expressing neurons, although it is unknown whether this would also be the case when compared to control CRISPR/Cas9 neurons (Goz et al., 2020). It remains unclear why Rheb S16H and Rheb P37L variants have different effects on RMP. One possible reason is that Rheb S16H was expressed in the mPFC whereas Rheb P37L was expressed in the SSC. Since a neuron’s RMP is set by its ion channel complement, differentially expressed ion channels in mPFC vs. SSC Rheb mutant neurons may contribute to this difference, but this needs to be further explored. Although the complement of ion channels has not been thoroughly examined in these studies, one study has reported abnormal expression of an HCN channel isoform, HCN4 (discussed below) as a source of hyperexcitability (Hsieh et al., 2020).
With regards to synaptic function, the reported findings are highly variable and depend on the gene variant. Pten mutant neurons (at P21-P30) displayed increased miniature and spontaneous excitatory postsynaptic current (mEPSC and sEPSC, respectively) frequency (Chen et al., 2015), consistent with previous findings in conditional Pten–/– mice (Williams et al., 2015). In contrast, Rheb S16H and mTOR L2427P and C1483Y mutant neurons (at P28–42 and P21, respectively) all displayed decreased spine density (Lin et al., 2016; Kim et al., 2019). Rheb S16H mutant neurons also had decreased sEPSC frequency. Similar decreases in cortical neuron spine density were observed in conditional Tsc1–/+ mice (Meikle et al., 2008). Depdc5 mutant neurons (at P20–24) displayed no changes in spine density, but the size of the spine heads was increased, which was accompanied by an increase in sEPSC amplitude and no change in sEPSC frequency (Ribierre et al., 2018). This would likely result in an increased total excitatory charge. Only one study examined inhibitory postsynaptic currents (IPSCs), and it was reported that the frequency and amplitude of miniature IPSC (mIPSC) were decreased in Pi3k E545K mutant neurons (at P24–P28) (Zhong et al., 2021). These data suggest decreased inhibition onto the mutant neurons, assuming GABAA receptor activation remains hyperpolarizing (Talos et al., 2012).
The diverging effects of Pi3k, Pten, Tsc1, Rheb, mTOR, and Depdc5 variants on neuronal excitability, particularly synaptic activity, emphasize a critical need to systematically investigate the functional effects of these gene variants side-by-side in the same system (i.e., IUE), cell type, cortical region, and age to parse out the source of the differences (i.e., whether the observed effects are due to variant-specific functional differences giving rise to distinct effects or variable experimental conditions). Such side-by-side comparisons to investigate the effects of Pten and Tsc1 knockout on hippocampal neurons have been performed in vitro by Weston et al. (2014). The authors found that Pten deletion led to an increase in both excitatory and inhibitory synaptic transmission, while Tsc1 deletion reduced inhibitory but did not affect excitatory synaptic transmission within the same culture system. These findings support diverging effects of Pten and Tsc1 on synaptic transmission, which may be accounted for by the differential impact of Pten and Tsc1 deletion on AKT activation (discussed more in the next section). One consideration for the apparent differences in the functional phenotypes in the IUE studies is that different PI3K-mTOR and GATOR1 gene variants can lead to different levels of mTORC1 activation, which could contribute to variable phenotypes. Studies by Nguyen et al. (2019) showed that increasing concentration of the Rheb S16H plasmid during IUE led to dose-dependent increases in mTORC1 activation. Importantly, low concentrations of Rheb S16H did not result in seizures whereas higher concentrations of Rheb S16H resulted in robust seizure activity, emphasizing an mTORC1 activity level-dependent effect on these phenotypes. Thus, it will be important to carefully assess and compare the degree of mTORC1 activation with these variants. Finally, only a few studies have directly examined the dependence of mTORC1 activity on the above electrophysiological findings so far. In the Pi3k E545K mutant neurons, reducing mTORC1 activity with rapamycin treatment restored mIPSC frequency but not amplitude (Zhong et al., 2021). In Rheb S16H mutant neurons, neonatal rapamycin treatment eliminated aberrant HCN4 channel expression (Hsieh et al., 2020). Additionally, targeting mTORC1-regulated cap-dependent translation via expression of constitutive active 4E-BP1 restored RMP and sEPSC frequency, but it not did rescue spine density in these neurons (Lin et al., 2016). The reason for the discrepancy between sEPSC and spines data in Rheb S16H mutant neurons remains unclear. In contrast, knockdown of eIF4E to reduce cap-dependent translation rescued spine density in both mTOR L2427P and C1483Y mutant neurons. Given these few studies, the extent to which the observed functional phenotypes in the PI3K-mTOR and GATOR1 gene variants converge on mTORC1 and its downstream effectors remains unclear.
With regards to the source of epileptogenicity, all evaluated mutant neurons required higher depolarizations to generate APs as discussed above. How intrinsically more hypoexcitable neurons lead to cortical hyperexcitability and epilepsy remains unclear and is an active area of research. In Pten mutant neurons, increased excitatory neurotransmission might induce greater depolarization locally compared to control neurons, but it is unclear whether this is sufficient to counterbalance the low Rin to trigger firing (Chen et al., 2015). In Depdc5 mutant neurons, once the AP threshold was reached, neurons displayed a higher firing gain (i.e., fire more APs) (Ribierre et al., 2018). Whether the combined increase in total excitatory charge and increased firing grain is sufficient to trigger the seizures observed in this model remains unknown. In Rheb S16H mutant neurons, abnormal expression of an HCN4 channel was found to be responsible for depolarized RMPs and repetitive firing in these neurons (Hsieh et al., 2020). Silencing HCN4 channel activity in Rheb S16H mutant neurons decreased RMPs and reduced the seizure frequency, thus providing a novel mechanism of mTORC1-induced hyperexcitability. HCN4 channels are highly sensitive to intracellular cAMP levels and do not depend on membrane depolarization for gating (Robinson and Siegelbaum, 2003; Brennan et al., 2016). Indeed, increasing intracellular cAMP levels by forskolin application depolarized Rheb S16H mutant neurons and led to repetitive AP firing (Hsieh et al., 2020), but how HCN4 channels are activated in vivo leading to seizures in Rheb S16H mice is unknown. Cortical neurons, including mPFC pyramidal neurons, receive dense noradrenergic projections from the locus coeruleus and dopaminergic projections from the ventral tegmental area (Chandler et al., 2014; Radnikow and Feldmeyer, 2018). Noradrenaline and dopamine released from these projections activate several receptors, including Gs-coupled β-adrenergic and D1/D5 dopaminergic receptors, which could contribute to cAMP increase and HCN4 channel activation in Rheb S16H mutant neurons (Grzelka et al., 2017; Radnikow and Feldmeyer, 2018). Nevertheless, this needs to be further investigated. Given that the abnormal expression of HCN4 in Rheb S16H mutant neurons is an mTORC1-dependent process (rapamycin treatment eliminated aberrant HCN4 expression) that can modulate epilepsy, it would be important to examine whether this phenotype is conserved across the different gene variants.
Differential Activation of Intracellular Pathways in mTORopathies
While pathological variants in the PI3K-mTOR pathway and GATOR1 complex all lead to mTORC1 hyperactivity, they can also differentially modulate other intracellular signaling pathways independently of mTORC1. Here, we discuss several findings from the IUE studies reporting activation of parallel pathways by specific proteins in the PI3K-mTOR pathway: AKT-GSK3β, AKT- FOXG1-reelin, and TSC/RHEB-MAPK/ERK-FLNA (Figure 1). Other examples, such as the non-canonical function of TSC/RHEB on the notch signaling pathway, have been reviewed elsewhere (Neuman and Henske, 2011), although the function of mTORC1-independent pathways in mTORopathies overall remains understudied.
One of the best-established function of AKT, aside from regulation of cell growth via TSC1/2-RHEB-mTORC1 signaling, is phosphorylation and inhibition of the glycogen synthase GSK3β (Bellacosa et al., 2004). GSK3β regulates many neuronal processes, including neuronal polarization, axon growth, and axon branching (Kim Y. T. et al., 2011). Studies in the cancer field have shown that gain-of-function mutations in Pi3k and Akt, as well as Pten loss-of-function, upregulate AKT and lead to decreased GSK3β activity (Duda et al., 2020). By contrast, Tsc1 loss-of-function or Rheb gain-of-function (genes downstream of AKT) indirectly downregulates AKT via a negative homeostatic feedback loop through mTORC1-S6K1/2, thereby increasing GSK3β activity (Shah et al., 2004; Meikle et al., 2008; Gong et al., 2015). It is expected that variants of the GATOR1 complex would lead to decreased AKT activity through the same feedback mechanism, but this needs to be examined. IUE-based studies by Gong et al. (2015) confirmed increased S6K1/2 activity and decreased GSK3β activity in mice expressing the Rheb S16H variant. Moreover, they found that decreasing GSK3β activity by expression of a dominant-negative GSK3β mutant prevented Rheb S16H-induced axon overgrowth in vivo. This effect was also observed when using the pharmacological GSK3β inhibitor, lithium chloride. Thus, one mechanism by which RHEB-induced mTORC1 hyperactivation causes axon dysmorphogenesis is via S6K1/2 feedback inhibition on AKT-GSK3β signaling. It remains unknown whether inhibiting GSK3β activity would also reduce axon overgrowth in neurons expressing other discussed variants downstream of AKT (e.g., Tsc1, Tsc2, and mTOR). Interestingly, mutant hippocampal neurons in conditional Pten–/– mice with reduced GSK3β activity also displayed increased axon growth (Kwon et al., 2006), suggesting that too little or too much GSK3β activity leads to a similar axonal phenotype in hyperactive mTORC1 conditions.
Another described function of AKT is the regulation of the FOXG1 transcriptional repressor (Datta et al., 1999; Regad et al., 2007; Dastidar et al., 2011). FOXG1 plays crucial roles in early brain development, and loss of FOXG1 leads to microcephaly, developmental delay, and cerebral atrophy (Hettige and Ernst, 2019). Studies by Baek et al. (2015) showed that expression of the Akt E17K variant by IUE altered AKT-FOXG1 signaling in mice, resulting in increased reelin expression during development. They further showed that expressing a Foxg1 T271A mutant that resists modulation by AKT or reducing reelin via siRNA partially restored the migration deficits caused by Akt3 E17K expression. These findings suggest a role for AKT-FOXG1-reelin signaling in AKT-induced cortical mislamination, but it is unclear whether this is conserved across the other PI3K-mTOR or GATOR1 gene variants.
More recently, studies by Zhang et al. (2020) demonstrated increased levels of an actin cross-linking molecule, Filamin A (FLNA) following expression of Rheb S16H by IUE in mice. Histological analysis in resected human cortical tissue from individuals with FCDII confirmed increased FLNA expression in dysmorphic pyramidal neurons and balloon cells (Zhang et al., 2020). FLNA regulates cytoskeletal dynamics through interaction with multiple membrane proteins and plays important functions in neuronal migration (Feng and Walsh, 2004). Mutations in FLNA cause a neurodevelopmental disorder known as X-linked periventricular nodular heterotopia, which is associated with cortical malformation and seizures (Parrini et al., 2006). The authors showed that knockdown of FLNA prevented neuronal misplacement and reduced soma and dendrite hypertrophy in Rheb S16H-expressing mice, supporting a role for FLNA in cortical mislamination and neuronal dysmorphogenesis in mTORopathies. Aberrant neuronal morphology was also rescued using a pharmacological modulator of FLNA, PTI-125. More importantly, both FLNA knockdown and PTI-125 treatment reduced seizure activity in Rheb S16H mice, demonstrating a direct contribution of FLNA to epilepsy (Zhang et al., 2020). Interestingly, Zhang et al. (2020) showed that the Rheb S16H-induced FLNA increase in mice was insensitive to rapamycin treatment. Instead, inhibiting MAPK/ERK pathway, a parallel regulator of cell growth and development that is dysregulated in TSC (Maldonado et al., 2003; Magri et al., 2011; Chevere-Torres et al., 2012; Zhang et al., 2014), with PD0325901 treatment successfully reduced FLNA levels (Zhang et al., 2020). These findings revealed an mTORC1-independent process for Rheb-induced FLNA changes. Indeed, expressing the gain-of-function mTOR variants, mTOR L1460P and S2215Y, in cortical neurons in vitro did not lead to an increase in FLNA level (Zhang et al., 2020). Dysregulation of MAPK/ERK signaling and increased FLNA levels have also been reported in conditional Tsc1–/– and Tsc1± mice (Zhang et al., 2014, 2016). Thus, altered MAPK/ERK-FLNA function represents an mTORC1-independent mechanism that contributes to cortical malformations and epilepsy in TSC- and RHEB-related mTORopathies. It remains to be examined whether FLNA levels are increased in the other variants.
Treatment for Human mTORopathies Beyond mTorc1 Inhibition
The crucial role of aberrant mTORC1 signaling in epilepsy is undebatable. The efficacy of rapamycin treatment in suppressing seizures has been demonstrated in numerous animal models of mTORopathies (Ostendorf and Wong, 2015), and the rapamycin analog, everolimus, has been approved for treating seizures in human TSC (French et al., 2016). The breakthrough usage of everolimus in clinics provides a new treatment option for intractable seizures in patients with TSC, however, everolimus is and remains the only mTORC1-targeting drug approved for epilepsy treatment. Given the growing body of genetic and molecular evidence for mTORC1 dysregulation in TSC, FCDII, and HME, there is a crucial need for novel treatment options that directly target the underlying molecular mechanisms.
Studies in the IUE-based models of mTORopathies that we review here provide some promising, potential molecular candidates for therapeutic intervention. Specifically, effective reduction of seizures was observed by modulating the activity of FLNA, HCN4, eIF4E, and adenosine kinase (ADK) in mice with mTORC1 hyperactivation. Work by Zhang et al. (2020) demonstrated that FLNA levels were aberrantly upregulated in dysmorphic neurons in patients with FCDII and mice expressing Rheb S16H. They further showed that knockdown of FLNA or treatment with an FLNA modulator, PTI-125, significantly reduced seizure activity by >60% in these mice (Zhang et al., 2020). Work by Hsieh et al. (2020) demonstrated that HCN4 was ectopically expressed in dysmorphicr neurons in patients in FCDII and TSC as well as mice expressing Rheb S16H. Silencing HCN4 activity by expression of a non-functional mutant form of HCN4 prevented epilepsy in these mice (Hsieh et al., 2020). Increased FLNA and HCN4 were found in the same Rheb S16H model and are mTORC1-independent and -dependent, respectively. Normalizing FLNA levels or blocking HCN4 activity reduced seizures, suggesting that both alterations contribute to seizures through different mechanisms and targeting one of these mechanisms is sufficient to reduce seizures. Work by Kim et al. (2019) provided evidence for aberrant translational regulation in patients with FCDII and mice expressing the patient variants mTOR L2427P and C1483Y. They showed that knockdown of the mTORC1-mediated translational regulator eIF4E or treatment with the eIF4E inhibitor, metformin, significantly decreased seizures in these mice. Kim et al. (2019) also showed that genetic or pharmacological inhibition of ADK, a novel downstream target of the mTORC1/eIF4F axis that was shown to be upregulated in patients with TSC, FCDII, and HME, alleviated seizures.
These studies are important because they show that while a combination of molecular and functional alterations is required for epileptogenesis, targeting a subset of these abnormalities is sufficient to reduce seizures. The strength of these studies is that they also reported abnormal expression of these potential targets in patients, establishing a clinical relevance for these molecules. FLNA and eIF4E can be pharmacologically targeted with available drugs, and therefore represent attractive targets. Both studies by Kim et al. (2019) and Zhang et al. (2020) showed that targeting FLNA or eIF4E, respectively, via drug treatment after seizure onset alleviated seizures, which is clinically important since seizures are often well established by the time patients are diagnosed with epilepsy and considered for treatment. Moreover, metformin is an FDA-approved drug to treat type 2 diabetes that could potentially be rapidly translated into clinical use for epilepsy. One limitation with targeting FLNA is that this is an mTORC1-independent mechanism (as discussed above), and thus may only apply for TSC and a subset of FCDII. As expected, PTI-125 treatment did not affect mTORC1 activity, suggesting that combination therapy with rapamycin may have added therapeutic benefits. Regarding HCN4, specific pharmacological inhibitors for these channels are not available and systematic inhibition of HCN4 function via a drug treatment would be unsafe given that HCN4 channels are responsible for the pacemaking activity of the heart (DiFrancesco, 2010). However, HCN4 could represent a potential molecular target for targeted gene therapies. Indeed, since focal MCDs, such as those observed in TSC and FCDII, are localized to specific areas in the brain, targeted gene delivery via direct brain injection could be an innovative treatment avenue for these mTORopathies.
Conclusion
Studies using IUE-based models and others demonstrate that gene variants along the PI3K-mTOR signaling pathway and GATOR1 complex produce both shared and unshared phenotypes. These findings have significant clinical implications as they underscore a need to consider patient-specific variants when evaluating treatment strategies for intractable epilepsy. Functional (e.g., electrophysiology) and non-cell autonomous effects of these variants have been under-investigated and should be addressed in future studies as they will provide valuable insights into mechanisms of human pathology and disease etiology. As healthcare is moving toward precision medicine, we propose the idea of a targeted therapeutic strategy for epilepsy based on individual patient variants on a case-by-case basis. This would, however, require new diagnostic tools beyond neuroimaging and pathological examination of brain specimen to systematically detect the underlying gene variants and to visualize associated molecular changes.
Author Contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH-NINDS R01 NS086329 (AB), NIH-NICHD F32 HD095567 (LN), American Epilepsy Society Postdoctoral Fellowship (LN), and Yale Swebilius Foundation Grant (AB and LN).
Conflict of Interest
LN and AB are co-inventors on one patent application, PCT/US2020/054007 entitled “Targeting Cap-Dependent Translation to Reduce Seizures in mTOR disorders.” AB is an inventor on two patent applications, PCT/US2020/020994 entitled “Methods of Treating and Diagnosing Epilepsy” and PCT/US2020/018136 entitled “Methods of Treating Epilepsy.”
References
Andrews, M. G., Subramanian, L., and Kriegstein, A. R. (2020). mTOR signaling regulates the morphology and migration of outer radial glia in developing human cortex. eLife 9:e58737. doi: 10.7554/eLife.58737
Aronica, E., Boer, K., Baybis, M., Yu, J., and Crino, P. (2007). Co-expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol. 114, 287–293. doi: 10.1007/s00401-007-0225-6
Baek, S. T., Copeland, B., Yun, E. J., Kwon, S. K., Guemez-Gamboa, A., Schaffer, A. E., et al. (2015). An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat. Med. 21, 1445–1454. doi: 10.1038/nm.3982
Baldassari, S., Ribierre, T., Marsan, E., Adle-Biassette, H., Ferrand-Sorbets, S., Bulteau, C., et al. (2019). Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 138, 885–900. doi: 10.1007/s00401-019-02061-5
Bar-Peled, L., and Sabatini, D. M. (2014). Regulation of mTORC1 by amino acids. Trends Cell Biol. 24, 400–406. doi: 10.1016/j.tcb.2014.03.003
Bateup, H. S., Johnson, C. A., Denefrio, C. L., Saulnier, J. L., Kornacker, K., and Sabatini, B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. doi: 10.1016/j.neuron.2013.03.017
Bateup, H. S., Takasaki, K. T., Saulnier, J. L., Denefrio, C. L., and Sabatini, B. L. (2011). Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J. Neurosci. 31, 8862–8869. doi: 10.1523/jneurosci.1617-11.2011
Baybis, M., Yu, J., Lee, A., Golden, J. A., Weiner, H., and McKhann, G. II, et al. (2004). mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann. Neurol. 56, 478–487. doi: 10.1002/ana.20211
Bellacosa, A., Testa, J. R., Moore, R., and Larue, L. (2004). A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther 3, 268–275. doi: 10.4161/cbt.3.3.703
Blair, J. D., Hockemeyer, D., and Bateup, H. S. (2018). Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 24, 1568–1578. doi: 10.1038/s41591-018-0139-y
Blumcke, I., Thom, M., Aronica, E., Armstrong, D. D., Vinters, H. V., Palmini, A., et al. (2011). The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia 52, 158–174. doi: 10.1111/j.1528-1167.2010.02777.x
Blumcke, I., Vinters, H. V., Armstrong, D., Aronica, E., Thom, M., and Spreafico, R. (2009). Malformations of cortical development and epilepsies: neuropathological findings with emphasis on focal cortical dysplasia. Epileptic Disord. 11, 181–193. doi: 10.1684/epd.2009.0261
Brennan, G. P., Baram, T. Z., and Poolos, N. P. (2016). Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) channels in epilepsy. Cold Spring Harb. Perspect. Med. 6:a022384. doi: 10.1101/cshperspect.a022384
Cepeda, C., Andre, V. M., Hauptman, J. S., Yamazaki, I., Huynh, M. N., Chang, J. W., et al. (2012). Enhanced GABAergic network and receptor function in pediatric cortical dysplasia Type IIB compared with Tuberous Sclerosis complex. Neurobiol. Dis. 45, 310–321. doi: 10.1016/j.nbd.2011.08.015
Cepeda, C., Andre, V. M., Vinters, H. V., Levine, M. S., and Mathern, G. W. (2005). Are cytomegalic neurons and balloon cells generators of epileptic activity in pediatric cortical dysplasia? Epilepsia 46(Suppl. 5), 82–88. doi: 10.1111/j.1528-1167.2005.01013.x
Cepeda, C., Hurst, R. S., Flores-Hernandez, J., Hernandez-Echeagaray, E., Klapstein, G. J., Boylan, M. K., et al. (2003). Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J. Neurosci. Res. 72, 472–486. doi: 10.1002/jnr.10604
Chandler, D. J., Gao, W. J., and Waterhouse, B. D. (2014). Heterogeneous organization of the locus coeruleus projections to prefrontal and motor cortices. Proc. Natl. Acad. Sci. U.S.A. 111, 6816–6821. doi: 10.1073/pnas.1320827111
Chen, F., and LoTurco, J. (2012). A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J. Neurosci. Methods 207, 172–180. doi: 10.1016/j.jneumeth.2012.03.016
Chen, F., Rosiene, J., Che, A., Becker, A., and LoTurco, J. (2015). Tracking and transforming neocortical progenitors by CRISPR/Cas9 gene targeting and piggyBac transposase lineage labeling. Development 142, 3601–3611. doi: 10.1242/dev.118836
Chevere-Torres, I., Kaphzan, H., Bhattacharya, A., Kang, A., Maki, J. M., Gambello, M. J., et al. (2012). Metabotropic glutamate receptor-dependent long-term depression is impaired due to elevated ERK signaling in the DeltaRG mouse model of tuberous sclerosis complex. Neurobiol. Dis. 45, 1101–1110. doi: 10.1016/j.nbd.2011.12.028
Choi, Y. J., Di Nardo, A., Kramvis, I., Meikle, L., Kwiatkowski, D. J., Sahin, M., et al. (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev. 22, 2485–2495. doi: 10.1101/gad.1685008
Costa-Mattioli, M., and Monteggia, L. M. (2013). mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 16, 1537–1543. doi: 10.1038/nn.3546
Crino, P. B. (2015). mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb. Perspect. Med. 5:a022442. doi: 10.1101/cshperspect.a022442
Crino, P. B. (2020). mTORopathies: a road well-traveled. Epilepsy Curr. 20, 64S–66S. doi: 10.1177/1535759720959320
Dang, L. T., Glanowska, K. M., Iffland, Ii, P. H., Barnes, A. E., Baybis, M., et al. (2020). Multimodal analysis of STRADA function in brain development. Front. Cell. Neurosci. 14:122. doi: 10.3389/fncel.2020.00122
Dastidar, S. G., Landrieu, P. M., and D’Mello, S. R. (2011). FoxG1 promotes the survival of postmitotic neurons. J. Neurosci. 31, 402–413. doi: 10.1523/jneurosci.2897-10.2011
Datta, S. R., Brunet, A., and Greenberg, M. E. (1999). Cellular survival: a play in three Akts. Genes Dev. 13, 2905–2927. doi: 10.1101/gad.13.22.2905
Dawson, R. E., Nieto Guil, A. F., Robertson, L. J., Piltz, S. G., Hughes, J. N., and Thomas, P. Q. (2020). Functional screening of GATOR1 complex variants reveals a role for mTORC1 deregulation in FCD and focal epilepsy. Neurobiol. Dis. 134:104640. doi: 10.1016/j.nbd.2019.104640
de Calbiac, H., Dabacan, A., Marsan, E., Tostivint, H., Devienne, G., Ishida, S., et al. (2018). Depdc5 knockdown causes mTOR-dependent motor hyperactivity in zebrafish. Ann. Clin. Transl. Neurol. 5, 510–523. doi: 10.1002/acn3.542
De Fusco, A., Cerullo, M. S., Marte, A., Michetti, C., Romei, A., Castroflorio, E., et al. (2020). Acute knockdown of Depdc5 leads to synaptic defects in mTOR-related epileptogenesis. Neurobiol. Dis. 139:104822. doi: 10.1016/j.nbd.2020.104822
D’Gama, A. M., Woodworth, M. B., Hossain, A. A., Bizzotto, S., Hatem, N. E., LaCoursiere, C. M., et al. (2017). Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 21, 3754–3766. doi: 10.1016/j.celrep.2017.11.106
Dibble, C. C., and Cantley, L. C. (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555. doi: 10.1016/j.tcb.2015.06.002
DiFrancesco, D. (2010). The role of the funny current in pacemaker activity. Circ. Res. 106, 434–446. doi: 10.1161/circresaha.109.208041
Duda, P., Akula, S. M., Abrams, S. L., Steelman, L. S., Martelli, A. M., Cocco, L., et al. (2020). Targeting GSK3 and associated signaling pathways involved in cancer. Cells 9:1110. doi: 10.3390/cells9051110
Eichmüller, O. L., Corsini, N. S., Vértesy, Á, Scholl, T., Gruber, V.-E., Peer, A. M., et al. (2020). Cerebral organoid model reveals excessive proliferation of human caudal late interneuron progenitors in Tuberous Sclerosis complex. bioRxiv doi: 10.1101/2020.02.27.967802
Feliciano, D. M., Su, T., Lopez, J., Platel, J. C., and Bordey, A. (2011). Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Invest. 121, 1596–1607. doi: 10.1172/jci44909
Feng, Y., and Walsh, C. A. (2004). The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 6, 1034–1038. doi: 10.1038/ncb1104-1034
Fingar, D. C., Salama, S., Tsou, C., Harlow, E., and Blenis, J. (2002). Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16, 1472–1487. doi: 10.1101/gad.995802
Forster, E., Bock, H. H., Herz, J., Chai, X., Frotscher, M., and Zhao, S. (2010). Emerging topics in Reelin function. Eur. J. Neurosci. 31, 1511–1518. doi: 10.1111/j.1460-9568.2010.07222.x
French, J. A., Lawson, J. A., Yapici, Z., Ikeda, H., Polster, T., Nabbout, R., et al. (2016). Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388, 2153–2163. doi: 10.1016/s0140-6736(16)31419-2
Frotscher, M. (2010). Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 33, 407–414. doi: 10.1016/j.tins.2010.06.001
Gong, X., Zhang, L., Huang, T., Lin, T. V., Miyares, L., Wen, J., et al. (2015). Activating the translational repressor 4E-BP or reducing S6K-GSK3beta activity prevents accelerated axon growth induced by hyperactive mTOR in vivo. Hum. Mol. Genet. 24, 5746–5758. doi: 10.1093/hmg/ddv295
Goz, R. U., Akgul, G., and LoTurco, J. J. (2020). BRAFV600E expression in neural progenitors results in a hyperexcitable phenotype in neocortical pyramidal neurons. J. Neurophysiol. 123, 2449–2464. doi: 10.1152/jn.00523.2019
Greig, L. C., Woodworth, M. B., Galazo, M. J., Padmanabhan, H., and Macklis, J. D. (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 14, 755–769. doi: 10.1038/nrn3586
Grzelka, K., Kurowski, P., Gawlak, M., and Szulczyk, P. (2017). Noradrenaline modulates the membrane potential and holding current of medial prefrontal cortex pyramidal neurons via beta1-adrenergic receptors and HCN channels. Front. Cell. Neurosci. 11:341. doi: 10.3389/fncel.2017.00341
Guemez-Gamboa, A., Coufal, N. G., and Gleeson, J. G. (2014). Primary cilia in the developing and mature brain. Neuron 82, 511–521. doi: 10.1016/j.neuron.2014.04.024
Guerrini, R., Cavallin, M., Pippucci, T., Rosati, A., Bisulli, F., Dimartino, P., et al. (2021). Is focal cortical dysplasia/epilepsy caused by somatic MTOR mutations always a unilateral disorder? Neurol. Genet. 7:e540. doi: 10.1212/nxg.0000000000000540
Hadjivassiliou, G., Martinian, L., Squier, W., Blumcke, I., Aronica, E., Sisodiya, S. M., et al. (2010). The application of cortical layer markers in the evaluation of cortical dysplasias in epilepsy. Acta Neuropathol. 120, 517–528. doi: 10.1007/s00401-010-0686-x
Hanai, S., Sukigara, S., Dai, H., Owa, T., Horike, S. I., Otsuki, T., et al. (2017). Pathologic active mTOR mutation in brain malformation with intractable epilepsy leads to cell-autonomous migration delay. Am. J. Pathol. 187, 1177–1185. doi: 10.1016/j.ajpath.2017.01.015
Harvey, A. S., Cross, J. H., Shinnar, S., Mathern, G. W., and Taskforce, I. P. E. S. S. (2008). Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia 49, 146–155. doi: 10.1111/j.1528-1167.2007.01421.x
Hettige, N. C., and Ernst, C. (2019). FOXG1 dose in brain development. Front. Pediatr. 7:482. doi: 10.3389/fped.2019.00482
Hsieh, L. S., Wen, J. H., Claycomb, K., Huang, Y., Harrsch, F. A., Naegele, J. R., et al. (2016). Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat. Commun. 7:11753. doi: 10.1038/ncomms11753
Hsieh, L. S., Wen, J. H., Nguyen, L. H., Zhang, L., Getz, S. A., Torres-Reveron, J., et al. (2020). Ectopic HCN4 expression drives mTOR-dependent epilepsy in mice. Sci. Transl. Med. 12:eabc1492. doi: 10.1126/scitranslmed.abc1492
Hu, S., Knowlton, R. C., Watson, B. O., Glanowska, K. M., Murphy, G. G., Parent, J. M., et al. (2018). Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann. Neurol. 84, 140–146. doi: 10.1002/ana.25272
Iffland, P. H., Everett, M. E., Cobb-Pitstick, K. M., Bowser, L. E., Barnes, A. E., Babus, J. K., et al. (2020). NPRL3: direct effects on human phenotypic variability, mTOR signaling, subcellular mTOR localization, cortical lamination, and seizure susceptibility. bioRxiv doi: 10.1101/2020.12.11.421214
Itoh, Y., Higuchi, M., Oishi, K., Kishi, Y., Okazaki, T., Sakai, H., et al. (2016). PDK1–Akt pathway regulates radial neuronal migration and microtubules in the developing mouse neocortex. Proc. Natl. Acad. Sci. U.S.A. 113, E2955–E2964. doi: 10.1073/pnas.1516321113
Kassai, H., Sugaya, Y., Noda, S., Nakao, K., Maeda, T., Kano, M., et al. (2014). Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 7, 1626–1639. doi: 10.1016/j.celrep.2014.04.048
Kast, R. J., and Levitt, P. (2019). Precision in the development of neocortical architecture: from progenitors to cortical networks. Prog. Neurobiol. 175, 77–95. doi: 10.1016/j.pneurobio.2019.01.003
Kim, J. K., Cho, J., Kim, S. H., Kang, H. C., Kim, D. S., Kim, V. N., et al. (2019). Brain somatic mutations in MTOR reveal translational dysregulations underlying intractable focal epilepsy. J. Clin. Invest. 129, 4207–4223. doi: 10.1172/jci127032
Kim, J. K., and Lee, J. H. (2019). Mechanistic target of rapamycin pathway in epileptic disorders. J. Korean Neurosurg. Soc. 62, 272–287. doi: 10.3340/jkns.2019.0027
Kim, S. H., Speirs, C. K., Solnica-Krezel, L., and Ess, K. C. (2011). Zebrafish model of tuberous sclerosis complex reveals cell-autonomous and non-cell-autonomous functions of mutant tuberin. Dis. Model. Mech. 4, 255–267. doi: 10.1242/dmm.005587
Kim, Y. T., Hur, E. M., Snider, W. D., and Zhou, F. Q. (2011). Role of GSK3 signaling in neuronal morphogenesis. Front. Mol. Neurosci. 4:48. doi: 10.3389/fnmol.2011.00048
Konno, D., Yoshimura, S., Hori, K., Maruoka, H., and Sobue, K. (2005). Involvement of the phosphatidylinositol 3-kinase/rac1 and cdc42 pathways in radial migration of cortical neurons. J. Biol. Chem. 280, 5082–5088. doi: 10.1074/jbc.m408251200
Kwiatkowski, D. J. (2003). Tuberous sclerosis: from tubers to mTOR. Ann. Hum. Genet. 67, 87–96. doi: 10.1046/j.1469-1809.2003.00012.x
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Lasarge, C. L., and Danzer, S. C. (2014). Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front. Mol. Neurosci. 7:18. doi: 10.3389/fnmol.2014.00018
Lee, J. H., Huynh, M., Silhavy, J. L., Kim, S., Dixon-Salazar, T., Heiberg, A., et al. (2012). De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 44, 941–945. doi: 10.1038/ng.2329
Li, Y. H., Werner, H., and Puschel, A. W. (2008). Rheb and mTOR regulate neuronal polarity through Rap1B. J. Biol. Chem. 283, 33784–33792. doi: 10.1074/jbc.M802431200
Lim, J. S., Gopalappa, R., Kim, S. H., Ramakrishna, S., Lee, M., Kim, W. I., et al. (2017). Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am. J. Hum. Genet. 100, 454–472. doi: 10.1016/j.ajhg.2017.01.030
Lim, J. S., Kim, W. I., Kang, H. C., Kim, S. H., Park, A. H., Park, E. K., et al. (2015). Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 21, 395–400. doi: 10.1038/nm.3824
Lin, T. V., Hsieh, L., Kimura, T., Malone, T. J., and Bordey, A. (2016). Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc. Natl. Acad. Sci. U.S.A. 113, 11330–11335. doi: 10.1073/pnas.1605740113
Lipton, J. O., and Sahin, M. (2014). The neurology of mTOR. Neuron 84, 275–291. doi: 10.1016/j.neuron.2014.09.034
Ljungberg, M. C., Bhattacharjee, M. B., Lu, Y., Armstrong, D. L., Yoshor, D., Swann, J. W., et al. (2006). Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann. Neurol. 60, 420–429. doi: 10.1002/ana.20949
LoTurco, J., Manent, J. B., and Sidiqi, F. (2009). New and improved tools for in utero electroporation studies of developing cerebral cortex. Cereb. Cortex 19(Suppl. 1), i120–i125. doi: 10.1093/cercor/bhp033
Luikart, B. W., Schnell, E., Washburn, E. K., Bensen, A. L., Tovar, K. R., and Westbrook, G. L. (2011). Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J. Neurosci. 31, 4345–4354. doi: 10.1523/jneurosci.0061-11.2011
Ma, X. M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318. doi: 10.1038/nrm2672
Magri, L., Cambiaghi, M., Cominelli, M., Alfaro-Cervello, C., Cursi, M., Pala, M., et al. (2011). Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9, 447–462. doi: 10.1016/j.stem.2011.09.008
Maldonado, M., Baybis, M., Newman, D., Kolson, D. L., Chen, W., and McKhann, G. II, et al. (2003). Expression of ICAM-1, TNF-alpha, NF kappa B, and MAP kinase in tubers of the tuberous sclerosis complex. Neurobiol. Dis. 14, 279–290. doi: 10.1016/s0969-9961(03)00127-x
Manning, B. D. (2004). Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J. Cell Biol. 167, 399–403. doi: 10.1083/jcb.200408161
Marsan, E., and Baulac, S. (2018). Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 44, 6–17. doi: 10.1111/nan.12463
Meikle, L., Pollizzi, K., Egnor, A., Kramvis, I., Lane, H., Sahin, M., et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 28, 5422–5432. doi: 10.1523/jneurosci.0955-08.2008
Miyata, H., Chiang, A. C., and Vinters, H. V. (2004). Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann. Neurol. 56, 510–519. doi: 10.1002/ana.20234
Moon, U. Y., Park, J. Y., Park, R., Cho, J. Y., Hughes, L. J., and McKenna, J. III, et al. (2015). Impaired Reelin-Dab1 signaling contributes to neuronal migration deficits of tuberous sclerosis complex. Cell Rep. 12, 965–978. doi: 10.1016/j.celrep.2015.07.013
Morita, T., and Sobue, K. (2009). Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J. Biol. Chem. 284, 27734–27745. doi: 10.1074/jbc.m109.008177
Muhlebner, A., Bongaarts, A., Sarnat, H. B., Scholl, T., and Aronica, E. (2019). New insights into a spectrum of developmental malformations related to mTOR dysregulations: challenges and perspectives. J. Anat. 235, 521–542. doi: 10.1111/joa.12956
Nakashima, M., Saitsu, H., Takei, N., Tohyama, J., Kato, M., Kitaura, H., et al. (2015). Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann. Neurol. 78, 375–386. doi: 10.1002/ana.24444
Neuman, N. A., and Henske, E. P. (2011). Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol. Med. 3, 189–200. doi: 10.1002/emmm.201100131
Nguyen, L. H., Mahadeo, T., and Bordey, A. (2019). mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J. Neurosci. 39, 2762–2773. doi: 10.1523/jneurosci.2260-18.2019
Onori, M. P., Koene, L. M. C., Schafer, C. B., Nellist, M., de Brito, van Velze, M., et al. (2020). RHEB/mTOR-hyperactivity causing cortical malformations drives seizures through increased axonal connectivity. bioRxiv doi: 10.1101/2020.07.08.189399
Orlova, K. A., and Crino, P. B. (2010). The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 1184, 87–105. doi: 10.1111/j.1749-6632.2009.05117.x
Orlova, K. A., Parker, W. E., Heuer, G. G., Tsai, V., Yoon, J., Baybis, M., et al. (2010). STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J. Clin. Invest. 120, 1591–1602. doi: 10.1172/jci41592
Ostendorf, A. P., and Wong, M. (2015). mTOR inhibition in epilepsy: rationale and clinical perspectives. CNS Drugs 29, 91–99. doi: 10.1007/s40263-014-0223-x
Park, S. M., Lim, J. S., Ramakrishina, S., Kim, S. H., Kim, W. K., Lee, J., et al. (2018). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron 99, 83–97e87. doi: 10.1016/j.neuron.2018.05.039
Parker, W. E., Orlova, K. A., Parker, W. H., Birnbaum, J. F., Krymskaya, V. P., Goncharov, D. A., et al. (2013). Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci. Transl. Med. 5:182ra153. doi: 10.1126/scitranslmed.3005271
Parrini, E., Ramazzotti, A., Dobyns, W. B., Mei, D., Moro, F., Veggiotti, P., et al. (2006). Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain 129, 1892–1906. doi: 10.1093/brain/awl125
Pelorosso, C., Watrin, F., Conti, V., Buhler, E., Gelot, A., Yang, X., et al. (2019). Somatic double-hit in MTOR and RPS6 in hemimegalencephaly with intractable epilepsy. Hum. Mol. Genet. 28, 3755–3765. doi: 10.1093/hmg/ddz194
Poduri, A., Evrony, G. D., Cai, X., Elhosary, P. C., Beroukhim, R., Lehtinen, M. K., et al. (2012). Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48. doi: 10.1016/j.neuron.2012.03.010
Poduri, A., Evrony, G. D., Cai, X., and Walsh, C. A. (2013). Somatic mutation, genomic variation, and neurological disease. Science 341:1237758. doi: 10.1126/science.1237758
Prabhakar, S., Goto, J., Zhang, X., Sena-Esteves, M., Bronson, R., Brockmann, J., et al. (2013). Stochastic model of Tsc1 lesions in mouse brain. PLoS One 8:e64224. doi: 10.1371/journal.pone.0064224
Prabowo, A. S., Anink, J. J., Lammens, M., Nellist, M., van den Ouweland, A. M., Adle-Biassette, H., et al. (2013). Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol. 23, 45–59. doi: 10.1111/j.1750-3639.2012.00616.x
Puffenberger, E. G., Strauss, K. A., Ramsey, K. E., Craig, D. W., Stephan, D. A., Robinson, D. L., et al. (2007). Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain 130, 1929–1941. doi: 10.1093/brain/awm100
Radnikow, G., and Feldmeyer, D. (2018). Layer- and cell type-specific modulation of excitatory neuronal activity in the neocortex. Front. Neuroanat. 12:1. doi: 10.3389/fnana.2018.00001
Regad, T., Roth, M., Bredenkamp, N., Illing, N., and Papalopulu, N. (2007). The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nat. Cell Biol. 9, 531–540. doi: 10.1038/ncb1573
Reijnders, M. R. F., Kousi, M., van Woerden, G. M., Klein, M., Bralten, J., Mancini, G. M. S., et al. (2017). Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 8:1052. doi: 10.1038/s41467-017-00933-6
Represa, A. (2019). Why malformations of cortical development cause epilepsy. Front. Neurosci. 13:250. doi: 10.3389/fnins.2019.00250
Ribierre, T., Deleuze, C., Bacq, A., Baldassari, S., Marsan, E., Chipaux, M., et al. (2018). Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest. 128, 2452–2458. doi: 10.1172/jci99384
Richter, J. D., and Sonenberg, N. (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. doi: 10.1038/nature03205
Robinson, R. B., and Siegelbaum, S. A. (2003). Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65, 453–480. doi: 10.1146/annurev.physiol.65.092101.142734
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. doi: 10.1016/j.cell.2017.03.035
Shah, O. J., Wang, Z., and Hunter, T. (2004). Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650–1656. doi: 10.1016/j.cub.2004.08.026
Sim, N. S., Ko, A., Kim, W. K., Kim, S. H., Kim, J. S., Shim, K. W., et al. (2019). Precise detection of low-level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol. 138, 901–912. doi: 10.1007/s00401-019-02052-6
Sokolov, A. M., Seluzicki, C. M., Morton, M. C., and Feliciano, D. M. (2018). Dendrite growth and the effect of ectopic Rheb expression on cortical neurons. Neurosci. Lett. 671, 140–147. doi: 10.1016/j.neulet.2018.02.021
Swaminathan, A., Hassan-Abdi, R., Renault, S., Siekierska, A., Riche, R., Liao, M., et al. (2018). Non-canonical mTOR-independent role of DEPDC5 in regulating GABAergic network development. Curr. Biol. 28, 1924.e5–1937.e5. doi: 10.1016/j.cub.2018.04.061
Switon, K., Kotulska, K., Janusz-Kaminska, A., Zmorzynska, J., and Jaworski, J. (2017). Molecular neurobiology of mTOR. Neuroscience 341, 112–153. doi: 10.1016/j.neuroscience.2016.11.017
Takei, N., and Nawa, H. (2014). mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 7:28. doi: 10.3389/fnmol.2014.00028
Talos, D. M., Sun, H., Kosaras, B., Joseph, A., Folkerth, R. D., Poduri, A., et al. (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. doi: 10.1002/ana.22696
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tarkowski, B., Kuchcinska, K., Blazejczyk, M., and Jaworski, J. (2019). Pathological mTOR mutations impact cortical development. Hum. Mol. Genet. 28, 2107–2119. doi: 10.1093/hmg/ddz042
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
Tsai, V., Parker, W. E., Orlova, K. A., Baybis, M., Chi, A. W., Berg, B. D., et al. (2014). Fetal brain mTOR signaling activation in tuberous sclerosis complex. Cereb. Cortex 24, 315–327. doi: 10.1093/cercor/bhs310
Wang, Y., Greenwood, J. S., Calcagnotto, M. E., Kirsch, H. E., Barbaro, N. M., and Baraban, S. C. (2007). Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann. Neurol. 61, 139–152. doi: 10.1002/ana.21058
Weston, M. C., Chen, H., and Swann, J. W. (2012). Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J. Neurosci. 32, 11441–11452. doi: 10.1523/jneurosci.1283-12.2012
Weston, M. C., Chen, H., and Swann, J. W. (2014). Loss of mTOR repressors Tsc1 or Pten has divergent effects on excitatory and inhibitory synaptic transmission in single hippocampal neuron cultures. Front. Mol. Neurosci. 7:1. doi: 10.3389/fnmol.2014.00001
White, E. L., Weinfeld, L., and Lev, D. L. (1997). A survey of morphogenesis during the early postnatal period in PMBSF barrels of mouse SmI cortex with emphasis on barrel D4. Somatosens. Mot. Res. 14, 34–55. doi: 10.1080/08990229771204
Williams, M. R., DeSpenza, T. Jr., Li, M., Gulledge, A. T., and Luikart, B. W. (2015). Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J. Neurosci. 35, 943–959. doi: 10.1523/jneurosci.3144-14.2015
Wong, M. (2008). Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia 49, 8–21. doi: 10.1111/j.1528-1167.2007.01270.x
Xiong, Q., Oviedo, H. V., Trotman, L. C., and Zador, A. M. (2012). PTEN regulation of local and long-range connections in mouse auditory cortex. J. Neurosci. 32, 1643–1652. doi: 10.1523/jneurosci.4480-11.2012
Yuskaitis, C. J., Jones, B. M., Wolfson, R. L., Super, C. E., Dhamne, S. C., Rotenberg, A., et al. (2018). A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 111, 91–101. doi: 10.1016/j.nbd.2017.12.010
Zhang, L., Bartley, C. M., Gong, X., Hsieh, L. S., Lin, T. V., Feliciano, D. M., et al. (2014). MEK-ERK1/2-dependent FLNA overexpression promotes abnormal dendritic patterning in tuberous sclerosis independent of mTOR. Neuron 84, 78–91.
Zhang, L., Huang, T., and Bordey, A. (2016). Tsc1 haploinsufficiency is sufficient to increase dendritic patterning and Filamin A levels. Neurosci. Lett. 629, 15–18. doi: 10.1016/j.neulet.2016.06.037
Zhang, L., Huang, T., Teaw, S., and Bordey, A. (2019). Hypervascularization in mTOR-dependent focal and global cortical malformations displays differential rapamycin sensitivity. Epilepsia 60, 1255–1265. doi: 10.1111/epi.15969
Zhang, L., Huang, T., Teaw, S., Nguyen, L. H., Hsieh, L. S., Gong, X., et al. (2020). Filamin A inhibition reduces seizure activity in a mouse model of focal cortical malformations. Sci. Transl. Med. 12:eaay0289. doi: 10.1126/scitranslmed.aay0289
Zhao, S., Li, Z., Zhang, M., Zhang, L., Zheng, H., Ning, J., et al. (2019). A brain somatic RHEB doublet mutation causes focal cortical dysplasia type II. Exp. Mol. Med. 51:84. doi: 10.1038/s12276-019-0277-4
Keywords: neuron migration, tuberous sclerosis complex, focal cortical dysplasia, GATOR1 complex, in utero electroporation, mTOR, cortical development, epilepsy
Citation: Nguyen LH and Bordey A (2021) Convergent and Divergent Mechanisms of Epileptogenesis in mTORopathies. Front. Neuroanat. 15:664695. doi: 10.3389/fnana.2021.664695
Received: 05 February 2021; Accepted: 17 March 2021;
Published: 09 April 2021.
Edited by:
Alfonso Represa, INSERM U901 Institut de Neurobiologie de la Méditerranée, FranceReviewed by:
Renzo Guerrini, University of Florence, ItalyStéphanie Baulac, INSERM U1127 Institut du Cerveau et de la Moelle Épinière (ICM), France
Copyright © 2021 Nguyen and Bordey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angélique Bordey, YW5nZWxpcXVlLmJvcmRleUB5YWxlLmVkdQ==