 Cristina Colangelo
Cristina Colangelo Polina Shichkova
Polina Shichkova Daniel Keller
Daniel Keller Henry Markram
Henry Markram Srikanth Ramaswamy
Srikanth Ramaswamy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neural Circuits, 12 April 2019
Volume 13 - 2019 | https://doi.org/10.3389/fncir.2019.00024
This article is part of the Research TopicFrontiers in Neural Circuits - Editors' Pick 2021View all 10 articles
The neocortex is densely innervated by basal forebrain (BF) cholinergic neurons. Long-range axons of cholinergic neurons regulate higher-order cognitive function and dysfunction in the neocortex by releasing acetylcholine (ACh). ACh release dynamically reconfigures neocortical microcircuitry through differential spatiotemporal actions on cell-types and their synaptic connections. At the cellular level, ACh release controls neuronal excitability and firing rate, by hyperpolarizing or depolarizing target neurons. At the synaptic level, ACh impacts transmission dynamics not only by altering the presynaptic probability of release, but also the magnitude of the postsynaptic response. Despite the crucial role of ACh release in physiology and pathophysiology, a comprehensive understanding of the way it regulates the activity of diverse neocortical cell-types and synaptic connections has remained elusive. This review aims to summarize the state-of-the-art anatomical and physiological data to develop a functional map of the cellular, synaptic and microcircuit effects of ACh in the neocortex of rodents and non-human primates, and to serve as a quantitative reference for those intending to build data-driven computational models on the role of ACh in governing brain states.
The cholinergic system is one of the most well-studied neuromodulatory systems, and perhaps phylogenetically the oldest. Acetylcholine (ACh) is found in both vertebrates and invertebrates and together with adrenaline and noradrenaline (NA), it acts as one of the main effectors of the autonomic nervous system. In the central nervous system (CNS), ACh impacts cellular and synaptic physiology and may switch network dynamics resulting in behavioral transitions such as from sleep to wakefulness, distraction to attention, and learning and recall (Hasselmo and Sarter, 2011; Lee and Dan, 2012).
Cholinergic effects have been studied for more than a century. In 1869, Schmiedeberg and Koppe (1869) demonstrated how extracts of a common mushroom, Amanita muscaria, could slow, and at a higher concentration arrest the beat of the frog heart. They purified the extract and named it muscarine. This substance, when applied to the brain and spinal cord was able to produce flaccidity and weaken the peripheral reflexes. However, the pharmacology of the nitrite ester of choline was different in that it had considerable nicotinic activity (nicotine is the major alkaloid of tobacco, first isolated by Posselt and Reiman from Nicotiniana tabacum leaves in 1828; Koukouli et al., 2017). In 1921 experimental proof was obtained for ACh’s role as a chemical transmitter at the cardiac vagal endings. The active substance was initially named “vagusstoff” by Otto Loewi in 1921 (Loewi, 1924). Sir Henry Dale further described that muscarinic responses were antagonized by atropine, whereas the nicotine actions were antagonized by curare (Dale, 1914).
It has long been known that ACh is also present at the level of the CNS, however, it was not until 1953 that evidence of the release of ACh in the brain was provided (Eccles et al., 1953). Prior to this discovery, it was known that anti-cholinergic drugs could influence learning and memory—pharmacological activation of muscarinic ACh receptors (mAChRs) was known to produce delirium symptoms, while receptor blockade generates severe anterograde amnesia. Moreover, the dementia of Alzheimer’s and Parkinson’s diseases has been associated with the loss of cortical cholinergic innervation (Little et al., 1998; Giacobini, 2003; Sabri et al., 2008; Hasselmo and Sarter, 2011), and chronic administration of nicotine reverses hypofrontality in animal models of addiction and schizophrenia (Koukouli et al., 2017).
Classical notions sustain the view that the central cholinergic system works by a diffuse release of ACh across the cortex, activating its receptors globally and producing slow responses. While this view might be applicable to long-lasting behavioral phenomena, such as cortical arousal, it does not explain the modulation of processes that happen on a much faster scale, such as sensory gating, or plasticity (Muñoz and Rudy, 2014). ACh release in the neocortex originates from neurons distributed within the basal forebrain (BF) nuclei, including the medial septum, the vertical and horizontal diagonal band of Broca, the substantia innominata, and the nucleus basalis of Meynert (NBM). Release occurs through topographical projections, and all the projections arise from six groups of choline acetyl-transferase (ChAT)-positive neurons in the BF (Ch1–Ch4) and brainstem (Ch5–Ch6; Wevers, 2011). The innervation sparsely reaches all cortical layers, but layer 5 is more heavily innervated, particularly in the motor and sensory areas; cholinergic pathways often provide en passant innervation (Dani and Bertrand, 2007) to the neocortex. Additionally, ACh-releasing cells are found in cortical layer 2/3. These cells exhibit a bipolar morphology, stain positive for calretinin (CR) and vasoactive intestinal peptide (VIP), and are GABAergic (von Engelhardt et al., 2007; Granger et al., 2018).
The function of a neuromodulatory system is largely defined by the anatomy of its projections. Projections from the BF selectively control cortical activity and target neocortical regions more specifically than previously assumed (Hasselmo and Sarter, 2011; Muñoz and Rudy, 2014; Obermayer et al., 2017). Recent evidence suggests that a roughly topographical organizational scheme exists in the rostro-caudal sequence of neurons of the BF (Zaborszky et al., 2015) and that specific BF nuclei innervate specific cortical areas, as opposed to what happens with noradrenergic fibers originating from the locus coeruleus (Chaves-Coira et al., 2016; Kim et al., 2016). Cholinergic fibers can take one of four different routes to cortical structures: the septal pathway (which projects mainly to the hippocampal cortex) the medial pathway, the lateral pathway, or the internal capsule projection (which preferentially project to the neocortex; Poorthuis et al., 2014). Cholinergic terminals that reach the neocortex, mainly via layer 1 or layer 6 (Obermayer et al., 2017), can either exert a spread out control of cortical activity and regulate processes such as the transition from sleep to wakefulness and arousal, or contact a restricted number of cortical elements and have cell-type specific effects; here contextual cholinergic signals act in concert with local processing of sensory inputs in order to guide behavior.
The aim of this review is to bring together current knowledge of cholinergic modulation in the neocortex and to identify the gaps to propose future directions to advance the field of neuromodulation. Here, we summarize existing literature on ACh release in the neocortex of rodents and non-human primates, specifically focusing on how ACh-induced effects on the diversity of cell-types and synapses shape the emergence of network states and review theories that bridge the modulation of local circuit properties and the consequent reconfiguration of cortical states. Data-driven computational models allow predictions on the potential role of ACh in reconfiguring neocortical states (Ramaswamy et al., 2018). Therefore, this review reconciles the minimal, although sparse, datasets required to build a multi-scale computational model of the neocortical cholinergic system.
A major factor that determines the spatiotemporal precision of ACh action is the transmission mode at cholinergic terminals. Cholinergic cortical signaling has historically been considered a slow and diffuse process, which was established upon examination of the functional organization of cholinergic projections and was mainly based on reports indicating a nearly complete absence of classical synapses at the level of cholinergic terminals (Muñoz and Rudy, 2014). Before optogenetic techniques were available, cholinergic pathways could not be activated in a selective manner, and thus evidence of the existence of fast cholinergic synaptic transmission was lacking, although some proof of fast nicotinic responses was already available from hippocampal recordings (Kalmbach et al., 2012; Obermayer et al., 2017).
In the cerebral cortex, cholinergic fibers are distributed in an intricate network with a characteristic laminar pattern. They have a higher density in the deeper layers. Cholinergic innervation reflects the classic organizational scheme of information processing systems (Kennedy and Bullier, 1985), with a higher number of projections being present in higher-order areas. Presumed cholinergic release sites have been ultra-structurally inspected and the subtle presence of synapse-like contacts has indeed been revealed; however, a relatively large number of these small varicosities, which are often associated with accumulated synaptic vesicles, do not seem to effectively establish synaptic contact with neighboring neurons, or exhibit only a few morphologically identifiable synapses Furthermore, the scarceness of astrocytic processes in the immediate vicinity of ChAT-immuno-reactive axons (when compared to glutamatergic terminals) may also allow greater diffusion of ACh within the extracellular space (Aoki and Kabak, 1992). Thus, relatively low concentrations of ACh will reach locations that are distant from the release site. This produces volume transmission or bulk release: neuromodulators slowly diffuse in a wide cortical area and bind to a large pool of extra-synaptic receptors (Dani and Bertrand, 2007).
Many studies (Umbriaco et al., 1994; Descarries and Mechawar, 2000; Sarter et al., 2009; Yamasaki et al., 2010) conducted in the neocortex have suggested that ACh acts preferentially non-synaptically; however, central cholinergic synapses had already been observed in the early ‘90s. Actual synapses were found on cholinergic varicosities in the cingulate cortex of the rat (Umbriaco et al., 1994), and in macaque more than 40 percent of cholinergic varicosities contained synaptic specializations (Mrzijak et al., 1995). Later, Turrini et al. (2001) provide definitive evidence that suggests that synaptic mechanisms of cholinergic transmission not only exist but prevail in the rat neocortex. Ultrastructural observations that most (66%) cholinergic boutons—as revealed by IR assays for the specific cholinergic marker, vesicular ACh transporter (vAChT)—establish classical synapses in layer 5 of the rat parietal cortex. By applying an improved fixation protocol and by using an antibody for vAChT, Turrini et al. (2001) demonstrated that cholinergic boutons predominantly established symmetric synapses on layer 5 dendritic shafts. The authors also found that immuno-stained varicosities occasionally established asymmetric contacts, but always on dendritic spines.
Another study probed the molecular-anatomical relationship between detectable cholinergic varicosities and the most abundant receptor subtype in the cortex—the muscarinic receptor subtype M1 (Yamasaki et al., 2010). This study established that in the mouse neocortex M1 can be found almost exclusively on the extra-synaptic membrane of pyramidal cells (PCs). Here, they observed that M1 distribution is far denser than the putative cholinergic release sites and that it does not show any apposition pattern to the varicosities, nor to the cytomatrix active zone proteins that are normally found at glutamatergic terminals. Hence, M1’s function in cortical PCs may be to sense ambient ACh released from cholinergic terminals at variable distances, and the main modality through which it is recruited is likely to be volume transmission. These approaches not only contribute to building a more refined knowledge of the subcellular localization of receptor subtypes but also provide a method to qualitatively discriminate between two major modes of transmission. Because of a substantial difference in the distribution pattern of cholinergic receptors across species, it is very likely that experiments performed in different species will yield conflicting results. For instance, even though a low incidence of classical synapses was reported for the rodent brain, a much higher proportion of cholinergic synapses was found in primates (Smiley et al., 1997). In the human cerebral cortex, the same authors found that up to 67% of all cholinergic varicosities established synaptic contacts, suggesting that ACh signaling in humans is mostly mediated by point-to-point synaptic transmission; this mechanism appears to prevail in the primate brain, but whether the same can be said for rodents is still a matter of open debate.
Cholinergic innervation from the BF is more specific than previously considered; ACh can control cortical activity on a fine spatial scale as well. Indeed, these findings agree with the evidence of ACh signaling occurring through direct fast point-to-point synaptic transmission brought about by the application of optogenetic tools (Kalmbach et al., 2012). Overall, it is not completely clear yet whether one mode of cholinergic transmission prevails over the other. Instead, a growing body of evidence suggests that volume and synaptic transmission may be complementary mechanisms by which ACh modulates cortical function (Sarter et al., 2009). While bulk release is thought to cause a more tonic change in extracellular ACh concentration, in the scale of seconds and minutes, and is mainly mediated by activation of extra-synaptic receptors, ACh release occurring at junctional sites would have a more circumscribed influence, with the modulation of circuit activity being restricted to the contacted cortical elements and to a much more delimited spatiotemporal scale (Muñoz and Rudy, 2014). Taken together, evidence shows that ACh modulates microcircuit activity with different modalities, ranging from synaptic release to volume transmission, and exerts its effects by modifying membrane excitability or synaptic activity.
Instead of trying to proclaim one modality over the other, future research should address the issue of whether they can occur simultaneously and have a differential impact on the temporal aspects of the response. Traditional bath application of agonists results in broad spatial and temporal activation that might not reflect the accuracy of endogenous ACh release (Urban-Ciecko et al., 2018). It is thus of crucial importance to determine whether the different ways in which cholinergic agonists are experimentally applied reflect different transmission modalities, and how faithfully stimulation protocols replicate physiological conditions. In the future, ACh application should be standardized according to precisely obtained dose-response and sensitization kinetics curves, and ascending concentrations should be used in order to detect eventual dose-dependent responses. Furthermore, it would be of outstanding interest to better understand how ACh release obtained by optogenetic stimulation of cholinergic afferents compares against bath application of cholinergic agonists. In a recent study, optogenetic recruitment of cholinergic fibers was performed in parallel with 1 mM ACh bath-application to detect changes in Martinotti cells (MCs) activity: the two techniques yielded very similar results (Obermayer et al., 2018). Perhaps the high concentration of ACh used in this case is comparable with a more physiological activation of the cholinergic system. Further clarification is required on the matter, and future studies should, therefore, consider this issue and design their experiments accordingly.
Cholinergic projections are likely to be arranged according to a modular pattern, with isolated bands of neighboring ChAT+ cells in the BF having defined cortical targets that are, in turn, functionally associated. When retrograde dyes are injected in distant cortical areas, labeled regions of cholinergic cells in the BF still largely overlap, even though the innervated cortical space is quite restricted (Muñoz and Rudy, 2014). Furthermore, Zaborszky et al. (2015) assert that the degree of overlap of labeled neuronal locations within the BF is positively correlated to the connection strength between the different injected cortical regions. Such an organization could induce widespread modulation even when the system is only focally activated (Muñoz and Rudy, 2014). Nevertheless, the response to neuromodulatory inputs is determined by the interplay of multiple factors, such as post-synaptic target, receptor type and subunit composition, subcellular localization of the receptors and their sensitivity. This way, a diffusely-organized projection system can fine-tune microcircuit activity. The cholinergic projection system should be viewed as a highly dynamic structure, able to propagate inputs either selectively or diffusely, switching from one modality to another, depending on the needs.
The next section aims to unravel the contribution of each subtype of cholinergic receptor to microcircuit modulation and attempts to determine the physiological relevance of their compartmentalized distribution and differential activation.
Even though the differential pharmacological effects had already been characterized, it was not until the early 1950s that the idea of “receptors” as the binding site for ACh was firmly established by Eccles et al. (1953). Cholinergic receptors are composed of two classes of transmembrane macromolecular complexes, the muscarinic and the nicotinic receptor families, each of which is further divided into subclasses. The occurrence of many ACh receptor subtypes and their differential dendritic, somatic, axonal, and synaptic localization contribute to the varied roles that these receptors play in the CNS. Cholinergic receptors have been found on axons originating from thalamic, cortical or basalo-cortical fibers as well as on cortical pyramidal excitatory neurons and inhibitory GABAergic interneurons (Groleau et al., 2015). The precise layer-wise distribution of cholinergic terminals, the identification of cell-types that actually express cholinergic receptors, and the subcellular localization of these receptors are described in the following sections.
Cholinergic synapses throughout the CNS are composed of muscarinic receptors (mAChRs), which can be further differentiated into subtypes that are encoded by a single gene (Venter et al., 1988; Van der Zee and Luiten, 1999). Five genetically defined and pharmacologically characterized (M1 to M5) mAChR subtypes have been identified in the CNS with high levels of expression in subcortical structures and the cerebral cortex (Wevers, 2011). Immunocytochemical approaches have identified different levels of expression of mAChRs throughout the cerebral cortex. These studies have detected moderate levels of mAChRs in the frontal cortex, parietal cortex, temporal cortex, entorhinal cortex, occipital cortex, insular and cingulate cortex, with the highest values for the temporal and occipital cortex. M1 receptors are the most abundantly expressed among all subtypes of mAChRs (Wevers, 2011). The density of cholinergic terminals in the rat neocortex differs between the six layers and depends on the cortical region studied (Eckenstein et al., 1988; Lysakowski et al., 1989). The pattern of cellular staining for mAChRs in the neocortex is characterized by a clear laminar distribution: in most of the cortical mantle, especially in neocortical areas, predominantly layer 5 PCs (L5PCs) show strong immunoreactivity across mammals such as the mouse, golden hamster, rat, cat, and human (Van der Zee and Luiten, 1999).
The density of each mAChR subtype differs throughout the brain with M1 being the most abundantly expressed and M5 the least (Alger et al., 2014). In the hippocampus and neocortex, M1 is present at high levels, M3 is moderately represented (though generally low elsewhere) and M4 is present in high density, as almost anywhere else in the brain, even though its concentration is considerably lower than M1. M2 instead, is found at very low densities, and this class of receptors seems to be distributed according to a precise pattern. M2 receptors frequently reside on presynaptic axonal terminals, whereas M1 receptors are often located on somato-dendritic regions of neurons. The M5 subtype is believed to play an important role in cortical perfusion, and it is mainly expressed on endothelial cells of the cerebral vascular system (Elhusseiny and Hamel, 2000; Gericke et al., 2011) even though recent evidence suggests that the M3 subtype is also involved in this kind of process (Zuccolo et al., 2017). In the rodent visual cortex, the subtypes M1 and M2 predominate, while in primates the subtypes M1, M2 and M4 prevail. Besides a few regional variations, highest labeling densities have been observed in the superficial layers of most cortical areas for both M1 and M2 (Wevers, 2011).
Most cholinergic receptors are metabotropic and mediate slow responses, which are typically associated with volume transmission. In the neonatal and adult cortices of rodents and primates, M1–M5 subtypes of mAChRs occur in both pre-synaptic and post-synaptic positions (Mrzljak et al., 1993; Groleau et al., 2015). All mAChRs are transmembrane macromolecular complexes that are coupled to membrane-embedded G-proteins of different kinds; g-proteins act as intracellular effectors and initiate signaling cascades that ultimately have an effect on intracellular processes, leading to the opening or closing of some ion channel, or to the production of long-term modifications of genetic activity and protein expression. Different mAChRs are coupled to specific G-proteins. The pre-synaptic mAChRs M2 and M4 preferentially couple to Gi and Go proteins that generally have inhibitory effects on voltage-activated calcium channels or extend the opening of potassium channels. The resulting decrease in c-AMP signaling suppresses neurotransmitter release (Groleau et al., 2015). M1, M3 and M5 subtypes are preferentially coupled to Gq and G11 proteins and are mainly located post-synaptically. Their activation seems to trigger membrane depolarization and increases the input-resistance of the cell membrane. M1-like (M1-M3-M5) receptors are known to potentiate NMDA currents and also influence and modulate voltage-dependent calcium currents, mostly by upregulating phospholipase C (PLC) signaling and inositol triphosphate (IP3) turnover. One major effect that can be attributed to M1-type receptors is the inhibition of potassium currents, including the Im and the IAHP (both medium and slow rate). However, M1-type receptors can also potentiate cationic currents like the Ih and the TRP currents, and the Icat (Teles-Grilo Ruivo and Mellor, 2013). For a more detailed description of the effects of ACh on various currents and their associated intracellular signaling pathways, we direct the reader to the section “Subcellular Nicotinic and Muscarinic Pathways” of this review.
What anatomical and functional evidence exists on the distribution of mAChRs in the neocortex? Muscarinic cholinergic activity influences sensory processing by facilitating or depressing neuronal responses to specific stimuli, and by modulating connections strength and neural synchronization: this results in the fine-tuning of cellular and network properties during developmental processes, the execution of attention tasks and perceptual learning (Groleau et al., 2015). These effects can largely be attributed to M1 and M2 subtypes, which appear to be highly prevalent in the neocortex. The presence of M1 and M2 mAChRs on PC somata and apical dendrites in non-human primates is well established, but M2 receptors are also found on excitatory and inhibitory axons in the primate neocortex (Mrzljak et al., 1993). Disney et al. (2006) report that M1 and M2 receptor labeling can be observed, but is quite weak in axons and terminals in the macaque visual cortex, whereas mAChRs are mostly expressed at the level of the soma of GABAergic neurons and in the dendritic compartments of glutamatergic cells.
Among the presynaptic receptors in the rodent and human visual cortex, M2 is very abundant while M4 is less prevalent (Groleau et al., 2015). M2 and M4 are mostly found at the presynaptic terminals; activation of these receptor subtypes causes membrane hyperpolarization and conveys a self-inhibitory signal. Thus, extracellular levels of ACh are regulated by means of negative feedback. In the rat’s primary visual cortex (V1) M2 is mainly found at the level of cholinergic terminals in layer 4 and layer 5. Being the main inhibitory auto-receptor, it contributes to the suppression of presynaptic ACh release (Mrzljak et al., 1993).
It is not yet clear whether the presence of M2-like subtypes at the level of the presynaptic terminal is a distinctive feature of cholinergic axons innervating the neocortex. Conflicting results emerge when looking at rodent studies, while experiments done on non-human primates and cats corroborate M2 receptors as the main auto-receptors localized on BF cholinergic axons. Subsequent research should, therefore, address this issue and determine the extent to which presynaptic M2-like receptors account for negative feedback via auto-inhibition, since this type of self-regulatory process is crucial for the fine-tuning of the response. Moreover, given that BF fibers originating from distinct neuron clusters differentially innervate separate cortical areas (Zaborszky et al., 2015; Chaves-Coira et al., 2016; Kim et al., 2016), discrepancies should be expected when assessing receptor subtype distributions across neocortical regions. Estimation of the physiological presynaptic distribution profile of inhibitory auto-receptors in the rodent sensory cortex is of key importance to understanding the system’s self-calibrating features. A systematic anatomical profiling of receptor expression should be performed in the rodent models, and quantitative comparisons should be made across sensory areas.
Neocortical PCs and inhibitory interneurons are strongly innervated by cholinergic axons, with L5PCs being the most densely innervated cells; however, numerous immuno-reactive interneurons can be found in all layers, but most frequently in layer 2/3 and layer 5. Here, the mAChR positive interneurons are intermingled with labeled PCs, but in general, the immunostaining of interneurons is less dense than that of the PCs (Van der Zee and Luiten, 1999). While mAChRs are more easily found in the dendritic compartments of PCs, their expression profile throughout the diversity of inhibitory interneurons is quite homogeneous, as these receptors are detected in proximity of the somatic compartment (Disney et al., 2006). mAChRs are expressed by different types of interneurons. In macaque, M2 receptors are found in 31% of PV neurons, 23% of CB neurons, and 25% of CR neurons. 87% of PV+ neurons, 60% of CB+ neurons and 40% of CR+ neurons however, express M1-type mAChRs. The M1 subtype is found across the cortical mantle on the cell bodies and dendrites of post-synaptic PCs, and it appears to be present mainly in layers 2/3 and 6, but it can be found across all cortical layers. In macaque V1, M1 is mostly expressed on GABAergic interneurons, but it is also found on cortico-cortical fibers (Mrzljak et al., 1993; Groleau et al., 2015). M1 immuno-reactivity is also observable in interneurons of the rat neocortex (Levey et al., 1991), although other studies have pointed to a low expression of M1 in primary sensory cortices of rats, such as S1 and V1. Some found M1 expression on PV+ neurons to be low or even undetectable in mice neocortex (Yamasaki et al., 2010). The significant difference in expression between rodents and primates could be explained by the fact that M1 receptors are much more associated to the extra-synaptic membrane compartments and are usually activated by volume transmission. Given that the BF cholinergic projection system is scaled-up in primates relative to rodents, there could be a more widespread distribution of M1 receptors throughout cortical interneurons. M1 immuno-reactivity is also detected at the synaptic level, in both inhibitory and excitatory synapses across cortical layers, but more frequently on asymmetric synapses, and here, preferentially on dendritic spines, as opposed to symmetric synapses where M1 is found mostly on dendritic shafts (Mrzljak et al., 1993). This preferential distribution perspective is challenged though, by experimental evidence that cholinergic boutons form synapses mainly with dendritic shafts, much fewer with dendritic spines and only occasionally on neuronal somata (Beaulieu and Somogyi, 1991; Mrzljak et al., 1993; Umbriaco et al., 1994). However, in mice, the highest density of M1 immuno-particles is observed in small-caliber oblique dendrites (smaller than 0.66 μm in diameter) of PCs (Yamasaki et al., 2010).
In L5PCs, M2 mAChRs are mainly localized postsynaptically, where they bring about a decrease in excitatory conductances, but M2 and M4 receptors are also present on the cell bodies of GABAergic interneurons in layers 2/3 and 4; here, M2 activation inhibits GABA release. The M3 subtype is localized postsynaptically in rodent inhibitory neurons and dendrites, where it enhances inhibitory transmission (Mrzljak et al., 1993; Groleau et al., 2015). Finally, M4 mAChRs are expressed in cortical excitatory neurons, in particular, in layer 4 spiny stellate neurons (L4SS) across different neocortical regions—S1, V1, and prefrontal cortex (PFC)—where they generate a persistent hyperpolarizing response (Radnikow and Feldmeyer, 2018). Perhaps the presence of M4 mAChRs is a marker to tell apart layer 4 from other layers.
Cholinergic inputs to the cortex generate different responses depending on which receptor is recruited: while M1-like (M1-M3-M5) receptors activation generally leads to an increase in postsynaptic conductance, M2-like receptors (M2-M4) have the opposite tendency to decrease synaptic transmission, by regulating presynaptic ACh release or by directly hyperpolarizing the post-synaptic membrane. mAChRs thus seem to be distributed both at the presynaptic and the postsynaptic level, and the resulting effect depends mostly on which subtype is activated. A detailed understanding of the cellular localization of each receptor subtype for every cell-type is still lacking; some generalizations can be made (as can be seen in Figure 3), but in order to precisely understand how neuromodulatory signals affect neural computation, a detailed knowledge of the amount and distribution of receptor subtypes at the level of each compartment is essential. Furthermore, it is of vital importance to gather this information for each neocortical cell-type. Neuromodulatory inputs very likely affect each cell-type differently, unlocking the possibility of fine-tuning the response and allowing delicate recalibration based on contextual information processing. This is most likely achieved by differentially distributing receptors along cellular compartments, thus creating modulatory micro-domains.
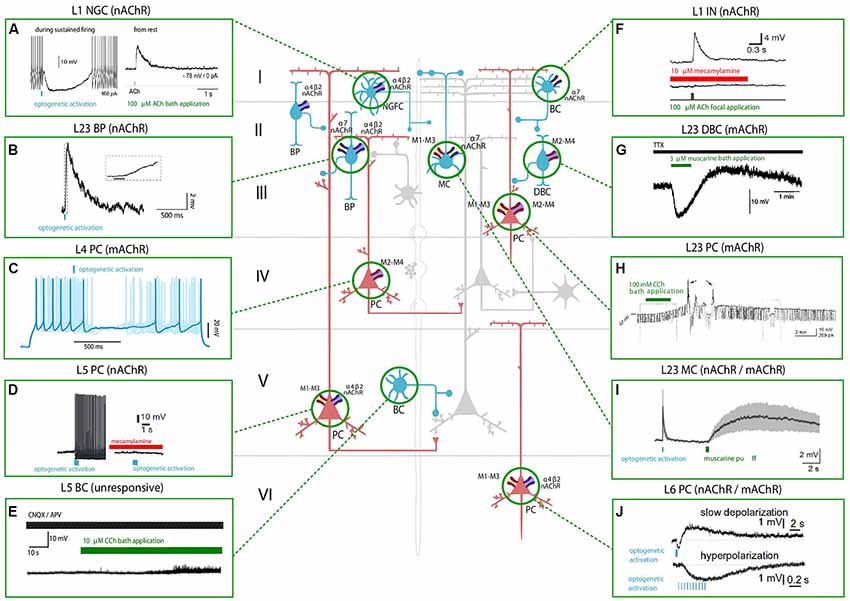
Figure 1. Effect of nicotinic acetylcholine receptors (nAChRs) and muscarinic ACh receptors (mAChRs) activation on the membrane potential of various neocortical cell types. The central schema represents the main cell types in the neocortex. Excitatory neurons are shown in red and inhibitory GABAergic neurons are shown in blue. The electrophysiological responses to the optogenetic activation of cholinergic fibers (in light blue) or the application of a cholinergic agonist (shown in green) or antagonist (shown in red) of each cell type are depicted in the inserts. Timing of cholinergic manipulation is shown as a vertical or horizontal bar. Muscarinic and nicotinic cholinergic receptors associated with the observed response, when known, are shown as four main subtypes: M1-M3-M5 like receptors (yellow and red), M2-M4 like receptors (violet and red), α4β2 heteromeric nAChRs (violet and blue) and α7 homomeric nAChRs (yellow and blue). All shown experimental traces reflect studies listed in Tables 1, 2. Selected traces were recorded in sensory areas of the rodent neocortex. Inclusion criteria for the experimental traces comprise knowledge of the cell-types and the receptor subtype (nicotinic or muscarinic) involved in the electrophysiological response. Abbreviations: PC, pyramidal cell; SS, spiny-stellate cell; IN, interneuron; MC, Martinotti cell; BC, basket cell; DBC, double-bouquet cell; NGFC, neurogliaform cell; BPC, bipolar cell. Reproduced and adapted from: (left, top to bottom): (A). Brombas et al., 2014; (B) Arroyo et al., 2012; (C) Dasgupta et al., 2018; (D) Hedrick and Waters, 2015; (E) Kawaguchi, 1997 (Right, top to bottom): (F) Gulledge et al., 2007; (G) Kawaguchi, 1997; (H) Shalinsky et al., 2002; (I) Dasgupta et al., 2018; (J) Hedrick and Waters, 2015. For more exhaustive information on agonist concentration, species and cortical area examined, see Tables 1, 2.
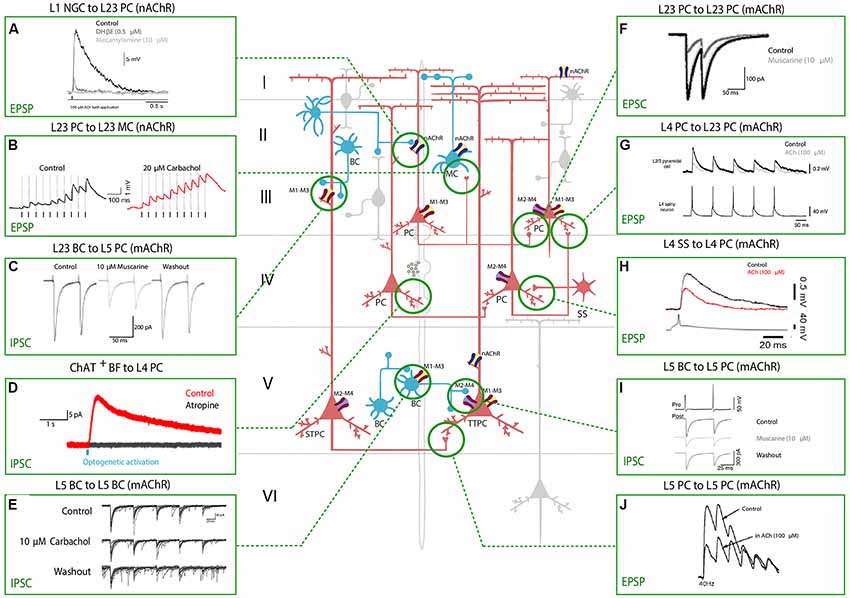
Figure 2. Effect of nAChRs and mAChRs activation on neocortical synaptic dynamics. The central schema represents the main neocortical cell types and their synaptic connections. A fiber of subcortical provenance associated with cholinergic boutons is also shown. Excitatory neurons are shown in red and inhibitory GABAergic neurons are shown in blue. The electrophysiological responses to the application of a cholinergic agonist or antagonist or to basal forebrain (BF) optical stimulation are depicted in the inserts. Panels show the modulation of synaptic dynamics in terms of increase or decrease in PSP/PSC size. Muscarinic and nicotinic cholinergic receptors associated with the observed response, when known, are shown as four main subtypes: M1-M3-M5 like receptors (yellow and red), M2-M4 like receptors (violet and red), α4β2 heteromeric nAChRs (violet and blue) and α7 homomeric nAChRs (yellow and blue). All shown experimental traces reflect studies listed in Table 3. Selected traces were recorded in sensory areas of the rodent neocortex. Inclusion criteria for the experimental traces comprise knowledge of the pre and postsynaptic cell-types and the receptor subtype (nicotinic or muscarinic) involved in the response. Abbreviations: PC, pyramidal cell; TTPC, thick tufted pyramidal cell; STPC, slender tufted pyramidal cell; SS, spiny-stellate cell; MC, Martinotti cell; BC, basket cell; NGFC, neurogliaform cell; BPC, bipolar pyramidal cell; IPC, inverted pyramidal cell. Reproduced and adapted from: (left, top to bottom): (A) Brombas et al., 2014; (B) Urban-Ciecko et al., 2018; (C) Kruglikov and Rudy, 2008; (D) Dasgupta et al., 2018; (E) Yamamoto et al., 2010; (F) Salgado et al., 2007; (G,H) Eggermann and Feldmeyer, 2009; (I) Kruglikov and Rudy, 2008; (J) Markram et al., 1997. For more exhaustive information on technique, species and cortical area examined, see Table 3.
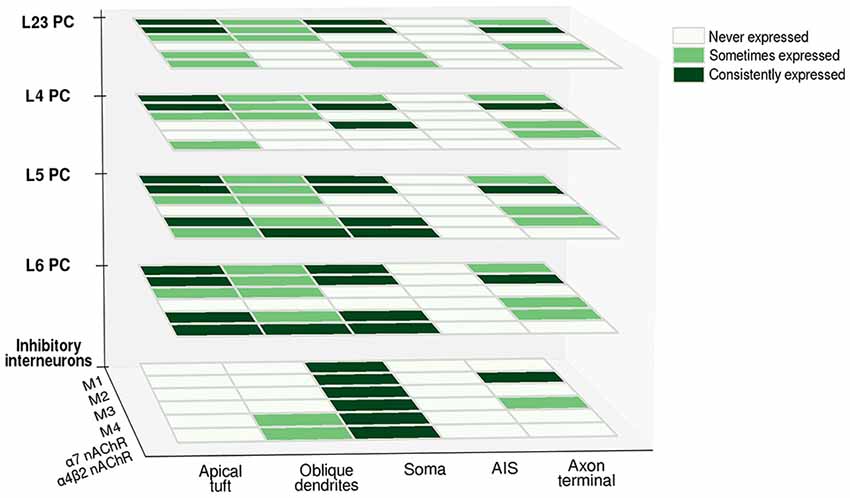
Figure 3. Differential expression of cholinergic receptors in various neuronal compartments across cell-types. Heatmap matrices show the occurrence of cholinergic receptor subtypes at the level of different cell-types. The presence of a given subtype in a cellular compartment is classified as consistently expressed (consistent findings across experimental studies), sometimes expressed (evidence of its presence is only partial) and never expressed (presence of a given subtype is undetectable). Abbreviations: PC, pyramidal cell; M1, M2, M3, M4, muscarinic cholinergic receptors 1–4; nAChR, nicotinic acetylcholine (ACh) receptor.
ACh can either increase or decrease neurotransmitter release probability, consistent with its role as a neuromodulator rather than a transmitter, and the effect on synaptic release probability depends on the identity of the pre and postsynaptic partners. Cell-types in the neocortex are differentially regulated by ACh, and the effects of cholinergic release include modulation of membrane properties (Figure 1) and synaptic dynamics (Figure 2).
The effects of ACh on neocortical PCs have been thoroughly investigated, and many studies (Gil et al., 1997; Disney et al., 2007) have come to the conclusion that besides generating direct PC depolarization, cholinergic modulation has an overall effect of increasing the signal to noise ratio (SNR) of incoming thalamic inputs. ACh seems to plays a role in enhancing circuit responses to relevant stimuli, providing a mechanism to regulate sensory processing during learning and attention.
The involvement of mAChRs in the depolarizing response of PCs to BF cholinergic inputs has been established by numerous studies (McCormick and Prince, 1985; Delmas and Brown, 2005; Gulledge and Stuart, 2005; Carr and Surmeier, 2007; Zhang and Séguéla, 2010), which report that muscarinic activation in PCs leads to an initial SK-mediated hyperpolarization, followed by a more sustained and slow depolarization (Table 1, Figure 1). Interestingly, the same biphasic response can be induced by bath perfusion of muscarinic agonists in hippocampal interneurons (Heys and Hasselmo, 2012; Heys et al., 2012). The mechanism by which this depolarization emerges has not been fully clarified yet, but some authors suggest the suppression of muscarinic-sensitive and voltage-dependent K+ conductance termed the M current (Im) or the activation of a non-specific cationic current both support the observed depolarization (McCormick and Prince, 1985; Krnjević, 2004).
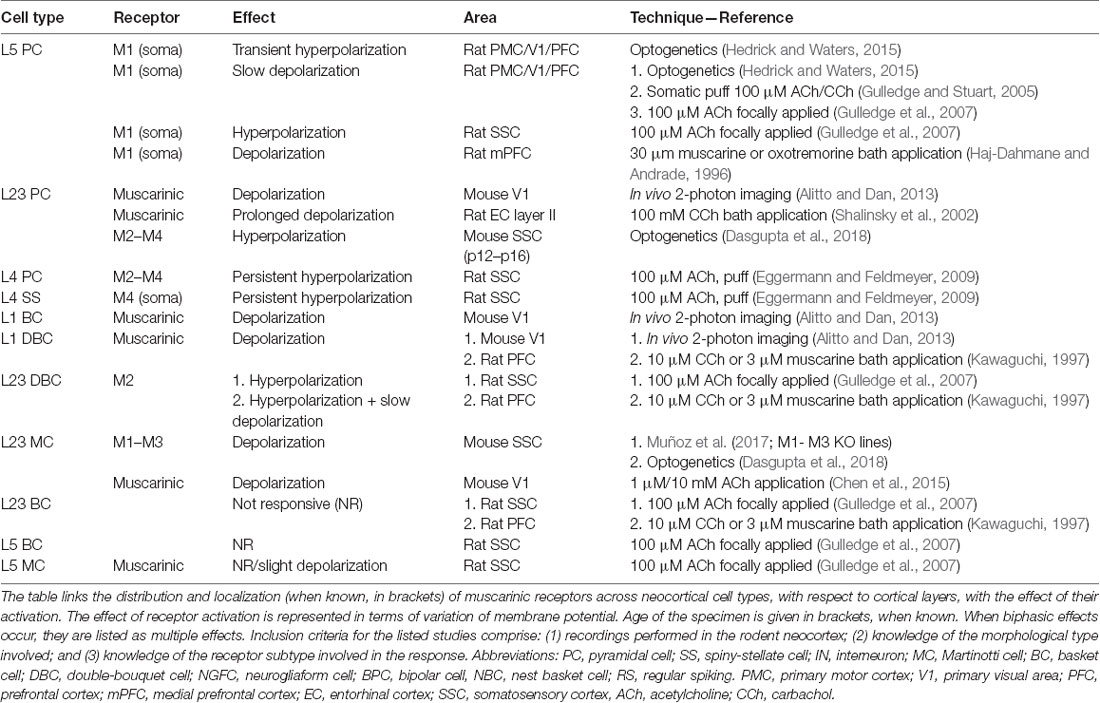
Table 1. Effect of muscarinic acetylcholine receptors (mAChRs) activation on membrane potential in various neocortical cell types.
In L5PCs, transient activation of M1-type mAChRs induces calcium release from IP3-sensitive intracellular calcium stores and subsequent activation of an apamin-sensitive, SK-type calcium-activated potassium conductance (Gulledge et al., 2007). Conversely, M4-mediated activation of a potassium conductance (Kir3) in L4SS generates a persistent membrane hyperpolarization and induces suppression of neurotransmitter release (Table 1, Figure 1). The observed hyperpolarizing response is supported by a decrease in presynaptic calcium conductance, at synapses between L4PCs and also at synapses between L4PCs and L23PCs (see Table 3, Figure 2; Eggermann and Feldmeyer, 2009). Focal application of ACh onto the soma of L5PCs evokes a biphasic response in which a transient membrane hyperpolarization precedes a slower and longer-lasting depolarization. Pharmacological evidence suggests that this effect is mediated by M1 receptors. Compared with the pressure application of ACh, activation of cholinergic synapses with brief bursts provides relatively weak activation of mAChRs that often fails to affect the somatic membrane potential at rest (Hedrick and Waters, 2015). One possible interpretation of these results might be that synaptically released ACh activates first nAChRs and usually fails to activate mAChRs, whereas pressure ejection onto the soma recruits primarily mAChRs.
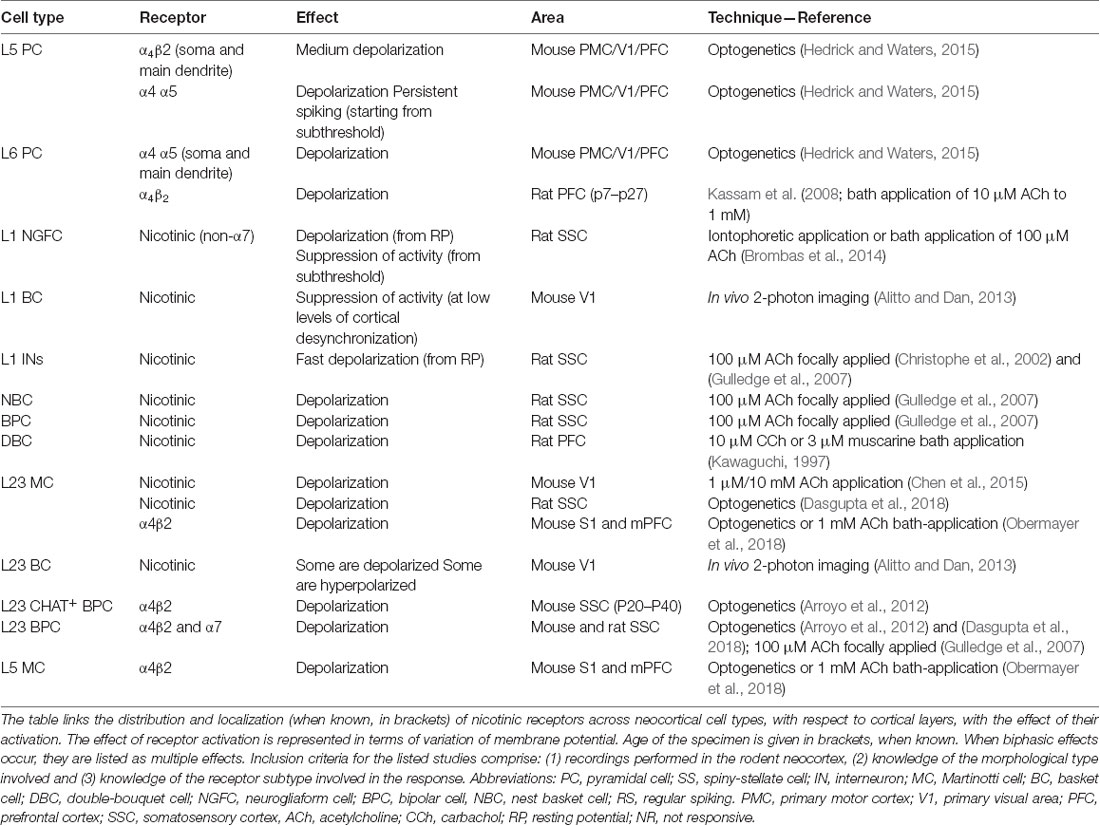
Table 2. Effect of nAChRs activation on membrane potential in various neocortical cell types.
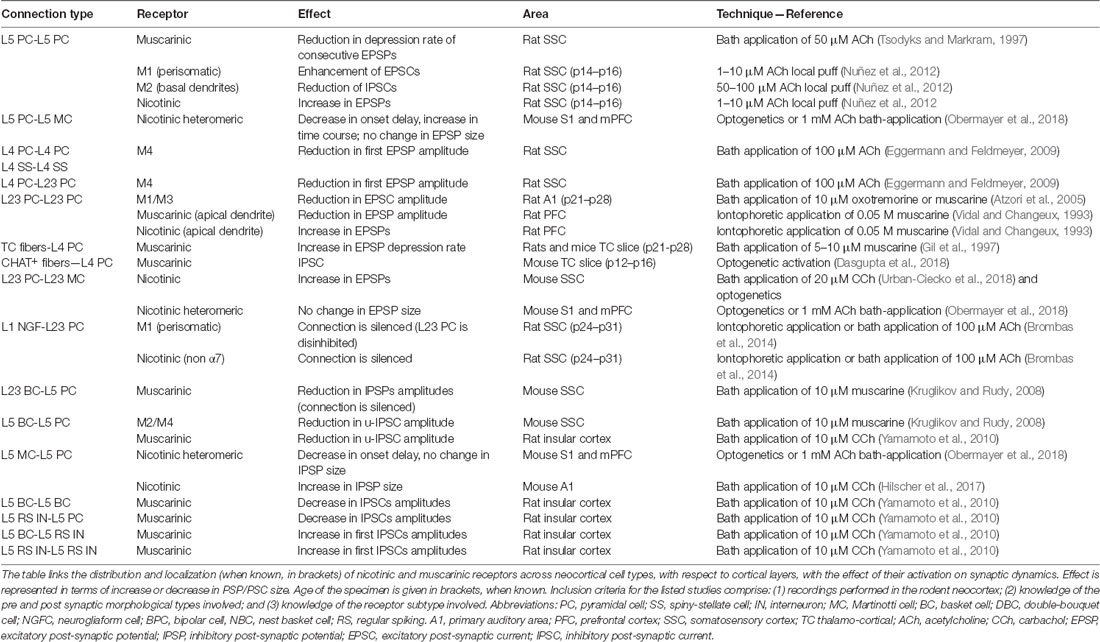
Table 3. Cholinergic mediated modulation of neocortical synaptic dynamics.
Muscarinic activation modulates K+ conductances (McCormick, 1992), but the reversal potential for K+ is approximately −90 mV: mAChR activation, therefore, exerts a little effect at resting membrane potential. However, when a neuron is depolarized, the observable mAChR-mediated hyperpolarization and subsequent depolarization are larger. The reported biphasic effect affects both cortico-pontine (CPn) and commissural (COM) pyramidal neurons; however, COM neurons show a more pronounced inhibitory phase, while CPn neurons have a larger and longer-lasting depolarizing phase (Baker et al., 2018). While these effects have been characterized thoroughly in deep-layers PCs, others report that ACh has limited ability to inhibit superficial PCs via changes in membrane potential (Gulledge et al., 2007).
Cortical inhibitory interneurons are, as well as PCs, a prominent target of cholinergic neuromodulation. The ways in which ACh modulates the dynamics of local interneurons have not been completely clarified yet, because the effects of BF cholinergic stimulation and bath application of cholinergic agonists (Table 1) strongly depend on the inhibitory cell-type.
Exogenous application is unlikely to mimic accurately the spatiotemporal profile of ACh release from cholinergic axons, and furthermore, there seems to be no agreement within the neuroscientific community on which concentration of cholinergic agonists should be used to promote activation of the cholinergic receptors. The applied dose ranges from 10 to 100 micromolar across different experimental groups, and in other cases, it even spans the millimolar range. These discrepancies arise from the fact that to measure the physiological extracellular concentration of ACh is experimentally challenging, because of the prompt intervention of hydrolases in the synaptic cleft. Application of acetylcholinesterase inhibitors cannot be avoided, making it extremely difficult to detect physiological levels of ACh in the extracellular space. Moreover, while mAChR agonists have been extensively used and are known to generate a multitude of responses in cortical neurons, much fewer studies (Hedrick and Waters, 2015; Dasgupta et al., 2018) have discerned muscarinic responses evoked by endogenous ACh release (see Figures 1, 2).
Cholecystokinin-immunoreactive (CCK) cells are affected heterogeneously by cholinergic agonists depending on their sizes. For example, small CCK cells are promptly depolarized by cholinergic inputs, while bigger CCK cells show a biphasic response comprising an initial hyperpolarization and a subsequent depolarization similarly to PCs (Kawaguchi, 1997). There is a general consensus (Gulledge et al., 2007; Kruglikov and Rudy, 2008; Poorthuis et al., 2013) that cholinergic modulation of fast-spiking PV positive (PV+) interneurons does not produce any effect on membrane excitability (Table 1). However, evidence also shows the opposite. For example, Alitto and Dan (2013) report in their review that PV+ interneurons are depolarized via muscarinic activation, but when mAChRs are blocked by antagonist application, the excitation is converted to inhibition; in turn inhibition of PV+ cells is converted to excitation when nAChRs are blocked, suggesting that excitation and inhibition compete in the same population of PV+ interneurons through the activity of the different receptors.
The subpopulation of dendrite-targeting interneurons, that is identified as somatostatin (Sst) expressing (Sst+) interneurons (MCs), can be depolarized by activation of mAChRs (Fanselow et al., 2008). However, some studies report that only very few Sst+ interneurons display excitation or inhibition in response to BF stimulation and that the inhibitory cells displaying the strongest excitation by ACh are L1 and VIP+ interneurons). Recent findings outlined by Muñoz et al. (2017) challenge these results. In their study, they claim that cholinergic modulation of Sst+ interneurons via M1 and/or M3 mAChRs provides a major excitatory drive to these cells during whisking activity.
VIP expressing interneurons are highly responsive to cholinergic inputs and show a mixed activation profile that is partially blocked by both nicotinic and muscarinic receptor antagonists (Kawaguchi and Kubota, 1997).
In summary, muscarinic activation has differential effects on membrane potential, based on which subtypes are expressed in a specific cell-type and in cellular compartments. These heterogeneous responses might play different roles in neocortical information processing: the initial hyperpolarizing phase observed in PCs and some CCK+ cells could be used to push the cell away from threshold, while the subsequent depolarization selectively augments inputs that are strong enough to reach threshold, therefore increasing the SNR, and at the same time promoting synchronization of neural activity. At the same time, the presynaptic inhibition of excitatory feedback could serve as a mechanism to prevent interference during the encoding of new stimuli and reduce top-down influences on perceptive processes. In addition, muscarinic receptors contribute to the generation of the gamma rhythm by inducing synchronized oscillations in both excitatory and inhibitory neurons (Heys et al., 2012).
Another class of receptors contributes to cholinergic signaling in the neocortex. Nicotinic receptors exert fast cortical actions, playing a key role in many cognitive processes (Dani and Bertrand, 2007), as described in the following section (Dani and Bertrand, 2007).
ACh is primarily regarded as a neuromodulator rather than a neurotransmitter in the CNS because its physiological effects have a latency of onset of tens of milliseconds to minutes (Van der Zee and Luiten, 1999). This great variability in the response of cortical neurons to ACh stimulation originates from the fact that there are two main types of ACh receptor proteins. Neuronal nicotinic receptors (nAChRs) are ionotropic receptors which are composed of combinations of twelve different nAChR subunits: α2 to α10 and β2, β3, β4. Each receptor is made of five subunits. It is generally assumed that nicotinic actions are fast and precise; however, the depolarization rate produced by the opening of the nicotinic channel can vary depending on the specific subunit composition. Because mAChR signaling acts through G-proteins, mAChR signaling might be expected to be slower than ionic nAChR signaling. However, homomeric (α7) nAChRs can also mediate slow responses, and the time course of muscarinic action may also vary widely, depending on the signal pathways involved (Muñoz and Rudy, 2014).
The nicotinic branch of the AChR family can be further divided into at least two classes, based on the affinity that their binding sites have for nicotine itself or the snake toxin α-bungarotoxin. At their simplest neuronal nAChRs are homomeric (constituted from five identical subunits) while the more complex forms are heteromeric, composed of at least one α and one β subtype. Binding studies using [3H]-nicotine have shown that high-affinity nAChR binding sites are very common for the human cerebral cortex, while autoradiographic labeling of nAChRs shows an inhomogeneous distribution over architectonically identified cortical areas of the rat brain, with highest concentrations in the medial PFC (mPFC) and generally frontal areas.
As for mAChRs, the expression of different subunit combinations varies across layers and across cortical areas. Given the involvement of the nicotinergic system in the treatment of tobacco addiction, many studies have been performed in the human brain. Most data on the distribution of nAChRs has been obtained from human autopsy tissue homogenates using techniques such as ligand binding, RT-PCR, immunoprecipitation, and Western blot.
Currently-available nAChR agonists and antagonists used for receptor auto-radiography are not subtype specific, although they act on nAChR subtypes with a distinct profile: labeling experiments carried out with different probes revealed that nAChRs are widely expressed in the cortex, both at the level of gray and white matter; many fibers show immunoreactivity at the neuropil level (Schröder, 1992). Five α subunits (3–7) and three β subunits (2–4) are expressed in the human brain. The expression of α4 and β2 subunits in the frontal cortex, parietal cortex, and temporal cortex shows a characteristic laminar distribution. Higher receptor binding is observed in layers 1, 3 and 5. These results are in agreement with the observed distribution of α3 and α4 mRNAs that are mostly found in PCs of layer 2/3 and layer 5 of the frontal cortex (Wevers, 2011). However, other studies report that the α3 mRNA is exclusively expressed in layer 4, while α4 subunit is moderately expressed in all layers (Radnikow and Feldmeyer, 2018). The α7 subunit is found mostly in layer 1–3 and 5 and is virtually absent in layer 4, while α4 and β2 immunoreactive fibers were observed in layer 4 of the PFC (Sparks et al., 2018). The α2 subunit is a characteristic feature of L5MCs that project to layer 1 and specifically target L5TTPCs (Hilscher et al., 2017). The detection of nicotinic subunits is possible because of the existence of specific antisubunit-antibodies and the introduction of nAChR subunit-Cre mouse lines. Nevertheless, nicotinic receptors are made up of multiple subunits and are either homomeric or heteromeric. The most abundant receptor subtypes in the neocortex are the homomeric receptor α7 and the heteromeric α4β2 channel (which is often associated with the regulatory subunit α5; Radnikow and Feldmeyer, 2018). Nicotinic receptors can be activated both via volume transmission and fast synaptic activity (Dani and Bertrand, 2007; Hedrick and Waters, 2015; Hay et al., 2016).
None of the studies mentioned above investigates the precise cellular localization of cholinergic receptors, which is crucial in determining the outcome of the response. This is especially true for nAChRs, because their activation directly leads to a cation influx into the cell, and immediately results in a voltage change in the underlying compartment.
nAChRs are expressed on glutamatergic inputs to layer 5, mostly contacting layer 5 interneurons and L5/L6 PCs. L5PCs and L6PCs are modulated by α7 and β2 nAChRs, respectively, while L23PCs and glutamatergic inputs to these cells do not contain nAChRs. Interneurons across layers contain mixed combinations of nAChRs (Poorthuis et al., 2013). Some subtypes, such as α7 homomeric receptors, are preponderantly expressed in presynaptic areas, whereas heteromeric receptors are more expressed on cell bodies and main dendrites (Bertrand, 2010). Cholinergic axons that diffusely innervate the cortex are thought to make en passant connections in the area of the main dendrite of the PCs from layer 5 and VI, therefore causing a volume release of ACh. Pre-synaptically, nAChRs generally increase the release of GABA and glutamate (Dani and Bertrand, 2007). However, both nAChR and mAChRs can reduce EPSPs by acting pre-synaptically (Levy et al., 2006).
The distribution of nAChRs at the light and electron microscopic level was studied in the human cerebral cortex using anti-nAChR monoclonal antibody (mAb) WF-6, which is not subunit selective (Schröder et al., 1990): nAChR immunoreactivity revealed a pattern for the frontal and temporal cortex that was very similar to that obtained with the auto-radiography. In the frontal cortex, in situ hybridization techniques display numerous labeled neurons, mostly PCs bearing the α7 mRNA in the cell body and in the apical dendrite. In the motor cortex, many PCs showed signals in the proximal part of their apical dendrite.
As reported by Schröder et al. (1989) and Schröder (1992) nAChR localization is predominant in L23 and L5 PCs; a few nAChR-expressing fusiform cells can be detected in layer 4 and VI. Many PCs show nAChRs on basal dendrites that originate in layer 5, cross the superficial layers of the cortex perpendicular to the pial surface, and branch between layers 1 and 2. Immuno-precipitate is detectable both in cell bodies and in their apical dendrites, in branches of various diameters, and in the PSD of synaptic junctions. In a double-labeling approach conducted in the temporal cortex, it was further demonstrated that PV+ interneurons express α4 and α7 subunit protein (Wevers, 2011). Double-labeling studies have shown that at least 30% of cortical neurons contain both nAChR and mAChR proteins, the majority of these being PCs. In the human cortex, nicotinic immuno-staining in individual neurons appears generally comparable to that seen in the rodent model (Schröder et al., 1989; Schröder, 1992): as in the rat occipital cortex, nAChRs can be detected on the cell bodies and dendrites of L23 and L5 PCs.
Most studies agree that nAChRs are preferentially found in infragranular layers, mostly at the level of L5 and L6PCs, but also at the level of inhibitory interneurons; CB-immunoreactive neurons, as well as PV+ neurons all express nAChRs, while that is not true for CR-ir neurons (Coppola and Disney, 2018); furthermore, nAChRs are expressed at the level of layer 2/3 as well, both in PC bodies and in the apical dendrites of deeper-layer placed cells. However, only a small subset of layer 2/3 excitatory neurons and no layer 4 neurons express nAChRs; layer 6 expression profile can be set apart from the rest, given that these neurons predominantly express the slowly desensitizing heteromeric α4β2 channel (Radnikow and Feldmeyer, 2018).
The distribution of nAChRs and the subunits combination, therefore, depends on cell-types, laminar position and on the cortical area studied, similarly to mAChRs; nowadays the possibility of systematically studying the distribution profile of cholinergic receptors has greatly increased, due to the advancement in the production of anti-subunit-specific-antisera and to the development of better immunoprecipitation and ligand binding techniques. Such studies exist and are quite informative as regards, for instance, the striatum (Zoli et al., 2002), but a comprehensive and detailed investigation of the expression of subunits in the neocortex is still lacking. Nicotinic activation prevalently modulates the excitability of deep cortical layers: in the next section, we move on and explore the contribution of nicotinic stimulation to local circuit properties and examine studies that investigated the involvement of the nicotinergic system in the modulation of neocortical activity.
Even though nAChRs are predominantly expressed pre-synaptically, where their activation modulates neurotransmitter release through calcium influx or terminal depolarization (Nashmi and Lester, 2006), there is evidence that nAChRs may also influence post-synaptic signaling and that these effects vary based on the subcellular localization of the receptor (Tables 2, 3). nAChRs expressed on distal dendrites are thought to cause the generation of fast excitatory post-synaptic potentials since activation of nAChRs on distal apical dendrites promotes PC depolarization and leads to an increase in action potential firing. On the contrary, activation of nAChRs on the proximal apical dendrites (closer to the cell body) reduces membrane impedance and shunts signal incoming from the apical tuft: when the nAChRs opens, the membrane resistance of the PC decreases and signals incoming from the apical dendrites get attenuated (Dani and Bertrand, 2007).
Optogenetic activation of cortical cholinergic input generates an increase in membrane excitability (Table 2) mediated by nAChRs and promotes spiking in L5PCs (Hedrick and Waters, 2015). When the stimulation is paired with additional depolarization, spiking activity becomes persistent and can be blocked by BAPTA application, suggesting that the observed depolarization is mediated by intracellular Ca++ transients. As suggested by kinetic analysis it is likely that non-α7 nAChRs determine this response. The depolarizing response spans all layers, but occurs with laminar and regional differences; additionally, the effect of the depolarization can be moderate and transitory or pronounced and persistent depending on the cell membrane potential. Although the modulatory effect was found to be stronger in deeper layers, the authors report that it was similar in M1, V1 and prefrontal (PF) cortices. The preferential modulation of deep neocortical layers is likely to influence the flow of excitation occurring throughout the neocortex that originates in layer 4 and then propagates to the superficial layers, whose role is to modify the output of layer 5. Altogether this study showed that nAChR activation increases the excitability of neocortical PCs; in the light of previous evidence that α4 and α5 subunits are highly expressed in layer 6 (Tribollet et al., 2004), and nAChR-mediated responses in layer 6 of the PFC have already been reported by many studies (Kassam et al., 2008; Bailey et al., 2010; Poorthuis et al., 2013), the authors suggest that the presence of α4 and α5-mediated PSPs could be a characteristic feature of L6PCs across neocortical regions (see Table 2, Figure 1).
Pyramidal-to-PCs connections in layer 5 can be potentiated by using an spike-timing-dependent-plasticity (STDP) protocol. Bath-application of 10 μM (or 300 nM) nicotine impairs L5PC to L5PC potentiation and favors the induction of LTD. When monitoring spontaneous synaptic events, application of nicotine increases the frequency and amplitude of sEPSCs. Evoked excitatory post-synaptic currents (EPSCs) behave differently and are reduced in amplitude by nicotine. However, puffing nicotine directly on PCs fails to elicit an inward current, and application of gabazine prevents the de-potentiation. Therefore, the effects of nicotine on L5PC to L5PC synapses should be attributed to an enhancement of GABAergic transmission, rather than the direct activation of a PCs (Couey et al., 2007).
nAChRs are known to be distributed throughout the dendritic trees of cortical PCs (van der Zee et al., 1992), but a comprehensive mapping of cholinergic synapses apposition remains elusive. To provide concomitant information on receptor localization while recording electrical responses more researchers should apply the strategy used by Hedrick and Waters (2015), who measured nicotinic PSPs during restricted illumination of the slice: illumination of the tuft dendrites failed to evoke a nicotinic PSP at the soma and therefore the authors concluded that nAChRs that contribute to the somatic depolarization are likely to be within 300 μm of the soma and many are probably located in the proximal 50 μm of the apical and basal arbor. This technique sheds light on the compartmental origin of the observed response and it is immensely useful to causally link the distribution of cholinergic receptors and their physiological role. A subsequent investigation should combine this strategy with pharmacological inactivation of specific receptor subunits and provide further proof that PCs responses to cholinergic inputs in different layers are mediated by specific receptor subunits and that their distribution profile is greatly involved in determining the outcome of neural computations.
Although nAChRs are mainly found on PCs, there is extensive evidence that nAChRs are expressed on the membrane of cortical interneurons (Table 2), such as MC, chandelier cells (ChCs) and basket cells (BCs), where they contribute to the modulation of GABAergic signaling (Couey et al., 2007; Wevers, 2011). The subpopulation of serotonin receptor 5-HT3aR expressing GABAergic interneurons is depolarized by ACh via nAChRs (Gulledge et al., 2007; Poorthuis et al., 2013); this embryologically distinguished subpopulation, that accounts for about 30% of the total number of cortical inhibitory interneurons, is heterogeneous and includes all the VIP+ interneurons, as well as the VIP− neurogliaform cells (NGCs; Rudy et al., 2011). VIP+ interneurons show a mixed activation profile in which both nicotinic and muscarinic receptors are involved (Figure 1; Kawaguchi, 1997).
Prominent nAChRs expression is a hallmark of layer 1 inhibitory interneurons both in rodents and humans (Letzkus et al., 2011; Alitto and Dan, 2013) and endogenous cholinergic release is known to rapidly recruit this receptor subpopulation during locomotion and attentive processes. These fast, nicotinic responses are mediated by α7 and β2 containing receptors (Poorthuis et al., 2018). When at rest, all layer 1 interneurons are depolarized via nicotinic activation (Figure 1, Table 2); however, when these interneurons are engaged in repetitive firing, ACh inhibits the activity of L1 NGCs (Brombas et al., 2014). Conversely, single bouquet cells (SBCs) are activated by ACh in the regime of repetitive firing (Jiang et al., 2013). Layer 1 interneurons responses are abolished by application of nAChR antagonists (Figure 1; Christophe et al., 2002).
ACh enhances the activation of neocortical deep-layers PCs by ascending thalamic inputs via mAChR-mediated depolarization and subsequent enhanced glutamate release from thalamocortical terminals in layer 4 (Gil et al., 1997; Metherate and Hsieh, 2004; Disney et al., 2007), but it also releases inhibition on superficial layers PCs. There is extensive evidence that ACh mediates activation of layer 1 and layer 2/3 non-fast spiking PV− cortical interneurons via non-α7 nAChRs. These interneurons, in turn, inhibit MCs and BCs that directly target PCs: nAChR-mediated inhibition of superficial interneurons reduces inhibition of superficial PCs (Gulledge et al., 2007; Arroyo et al., 2012; Brombas et al., 2014). Photostimulation of ChAT+ neurons in the BF evokes a prolonged disynaptic inhibition in PCs; pharmacological manipulation of the response suggests that it is supported by non-α7 mediated excitation of specific interneurons subtypes. This finding indicates that nicotinic cholinergic input originating from BF fibers is also comprised of a slow component. The observed delayed barrage of inhibitory post-synaptic current (IPSC) in L23PCs exhibits a long latency (of about 26 ms) characteristic of dysynaptic inhibition. Layer 1 and layer 2/3 inhibitory interneurons, and in particular in late-spiking cells and L23 ChAT+ bipolar cells are responsible for this phenomenon (Arroyo et al., 2012). In agreement with previous reports (Poorthuis et al., 2014) fast-spiking cells such as BCs and ChCs do not exhibit EPSPs in response to optogenetic stimulation of ChAT+ BF neurons, but rather respond similarly to PCs and are swamped by an IPSC barrage as well. While layer 1 and layer 2/3 late spiking cells (LS) exhibit both a fast and a slow response, L23 ChAT bipolar cells display only a slow response. This study demonstrates that the fast and slow components are mediated by α7 receptors and non-α7 receptors, respectively, and that non-α7 receptor-mediated excitation elicits action potentials in cortical interneurons that in turn produce a delayed and prolonged wave of inhibition in L23PCs and FS cells. One proposed explanation for the slow response is that it may arise from a cholinergic bulk transmission and that it may sustain the high metabolic demand of processes such as attention and memory (Cauli et al., 2004). Cortical ChAT+/VIP+ interneurons have been shown to dilate local microvasculature to increase blood supply during periods of elevated neuronal activity (Kocharyan et al., 2008) during the execution of memory and attention tasks, following electrical BF stimulation. The fast component of the cholinergic response may also be implicated in the emergence of a broader phenomenon like synchronized neuronal activity; it has been shown that LS cells are connected via gap junctions, and this fast response may thus play a fundamental role in the emergence of network oscillations that sustain plasticity and attention mechanisms.
Couey et al. (2007) realized that the effect of nicotine on L5PC to L5PC connections is mostly due to an enhancement of GABAergic transmission, and they decided to dissect the effects of nicotine on three different interneurons types. First, they looked at the activity of FS cells in layer 5, and observed no effect when adding nicotine to the bath; later they stained the cells for certain neuropeptides and several nAChR subunits and found an extremely low amount of mRNA coding for nicotinic subunits in FS cells, which might explain their unresponsiveness. Once again, another piece of evidence emerges confirming that (putative) BCs have a tendency not to respond to the application of cholinergic agonists. The authors identified another type of interneuron as a regular-spiking-non-PC (RSNPC), and observed a fast inward current after application of nicotine. LTS cells (putative MC) showed an even bigger inward current response; in both cell-types the most abundantly stained nicotinic subunit was α4, but β2 and α7 were also present. In this study, nicotine application increases the frequency and amplitude of spontaneous EPSCs in putative BCs and MCs; as for putative ChC (RSNP) a decrease in the frequency, but not the amplitude of sEPSCs can be observed (Couey et al., 2007).
Pyramidal to SST+ interneurons neocortical connections are relatively weak, but local excitatory input to SST neurons is selectively enhanced during cholinergic modulation of network activity. In a recent 2018 study, it was shown that endogenous ACh release activates presynaptic nAChRs and boosts glutamatergic input in a target-cell specific manner (Urban-Ciecko et al., 2018). Thus, there is evidence that local excitatory input to SST neurons is selectively enhanced during nicotinic modulation of network activity (Table 2, Figure 2). In a recent study by Obermayer et al. (2018) examined PC-MC-PC disynaptic connections in both layer 2/3 and layer 5 and found that the typical delayed disynaptic inhibitory response in the post-synaptic PC is faster and stronger when cholinergic inputs are activated optogenetically, or by means of 1 mM ACh bath application. When looking at the activity of a single MC, they observed that ACh inputs lead to a significant decrease of the onset delay of AP firing and increases the number of APs fired in MCs, which can account for the earlier onset and prolonged duration of disynaptic inhibition. This effect was abolished by application of 10 μM DHβE demonstrating that it is mediated by heteromeric nicotinic receptors (Table 2, Figure 2). However, when they recorded from synaptically connected PC-MC pairs during concurrent activation of cholinergic fibers, they could only observe an increase in the membrane depolarization level, but not in EPSP sizes. The same effect was found in MC-PC connections, and this as well was confirmed to be nicotinic in nature, contradicting the result obtained by Urban-Ciecko and others and others. The setups of the two experiments are comparable: both studies were performed in the adult mouse somatosensory cortex. However, the first remarkable difference lies in the nature of the cholinergic input used in the two experiments: while Obermayer et al. (2018) used bath-application of 1 mM ACh and optogenetic activation, Urban-Ciecko et al. (2018) decided to record activity in the presence of 20 μM CCh, a non-hydrolyzable analogue of ACh. Not only the two concentrations differ by two orders of magnitude, but the two cholinergic agonists work in fundamentally different ways. While ACh is almost immediately hydrolyzed by the cholinesterase in the synaptic cleft (within a few milliseconds), carbachol has a much more prolonged effect (Katz and Miledi, 1973). Nevertheless, the results obtained by bath-application of ACh are in agreement with the results achieved by optogenetic activation of the cholinergic system, which is supposed to be a more physiological way of stimulating cholinergic release (Obermayer et al., 2018).
Interestingly, optogenetic activation of cholinergic inputs did not affect the typical fast disynaptic post-PC response mediated by BCs, which provides yet another example of how BCs tend to be unresponsive to cholinergic release in both layer 2/3 and layer 5, or more generally show a more heterogeneous response profile to ACh inputs (Obermayer et al., 2018). This could be explained by the lack of a precise morphological identification of various subtypes of BCs, which could express cholinergic receptors in different subcellular locations or in a different amount, and therefore show differential responses to ACh inputs. These findings indicate that subcortical neuromodulatory projections recruit nicotinic receptors to alter network function through increased inhibition and provide a potential mechanism by which attention controls the gain of local circuits.
What are the receptor affinities to various agonists and can this be related to the actual amount of nicotinergic modulation? The relative activation of receptors vs. the concentration of agonist has been measured (Table 4).
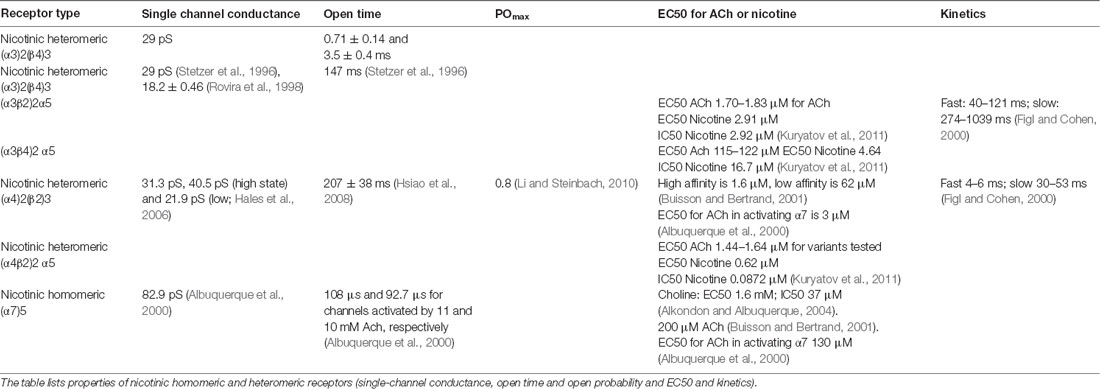
Table 4. Nicotinic homomeric and heteromeric receptors kinetics.
Muscarine reversibly reduces Ca2+ currents in a dose-dependent manner. The modulation is rapid, with an onset time constant of 1.2 s. A slowly developing component of the modulation also is observed, with a time constant of 17 s. Under elevated Ca2+ conditions, the fast component is due to a reduction in both N- and P-type calcium currents, whereas the slow component involves L-type current (Stewart et al., 1999). Receptor properties such as conductance, open time, and sensitivity to ACh depend on the nicotinic subunit composition (Table 4). (α4)2(β2)3 nAChRs are sensitive to micromolar scale changes, while (α7)5 receptors have a half-maximal sensitivity of more than a hundred micromolar. Extracellular choline is normally 3–5 μM but can attain 20 μM in some pathological cases. However, ACh reaches the millimolar range at the site of release (Alkondon and Albuquerque, 2004). Responses mediated by α7 nAChRs are short-lasting, whereas those mediated by α4β2 nAChRs are long-lasting. This is because the mean open time of α7 nAChRs is shorter than that of α4β2 nAChRs. Also, α7 nAChRs desensitize much faster than α4β2 nAChRs (Alkondon et al., 1999).
An interesting hypothesis was put forward by Albuquerque et al. (2000). α7 but not α4β2 nAChRs can be fully activated by choline (Nguyen et al., 1995; Alkondon et al., 1999). Choline and acetate are the products of hydrolyzation of synaptically released ACh by ACh-esterase in the synaptic cleft. This process occurs quickly, but reuptake of choline into presynaptic terminals is slow. Therefore, the ACh concentration in the synaptic cleft should decay rapidly, with only low levels of diffusing ACh reaching peri-synaptic sites. But choline levels should rapidly rise in the synaptic cleft with high levels of diffusing choline reaching peri-synaptic sites. This implies that extrasynaptically located α4β2 nAChRs (i.e., the high affinity nAChRs) could be activated by diffusing, low levels of ACh, extrasynaptically located while low-affinity α7 nAChRs may be activated by diffusing choline. Thus, α7 and α4β2 nAChRs might exhibit differential control (Albuquerque et al., 2000).
ACh affects membrane conductance through several subcellular pathways, as illustrated in Figure 4, leading to both hyperpolarizing and depolarizing effects (Tables 1, 2). ACh can act on both pre and post-synaptic membranes, binding to muscarinic and nicotinic receptors. The interplay among intracellular pathways leads to a dynamically changing outcome, such as the transient hyperpolarization and following long-term depolarization resulting from the binding of ACh to M1 mAChR (Dasari et al., 2017). When ACh interacts with M1, the exchange of coupled GDP for GTP produces the dissociation of the G-protein complex from the receptor. The released α subunit of the Gq protein then activates the enzyme phospholipase C (PLC β) which hydrolyzes phosphatidyl-inositol 4,5 bisphosphate (PIP2), leading to its dissociation from the membrane and the subsequent formation of diacylglycerol (DAG) and IP3. IP3 initiates calcium ions release from the endoplasmic reticulum (ER), serving as a trigger for this process. Refilling of the ER with Ca2+ions is then obtained by the activity of the sarco-ER Ca2+-ATPase (SERCA). Extracellular calcium ions are therefore crucial for the maintenance of calcium cycling. M1 activation facilitates voltage-dependent refilling of calcium stores by promoting excitation. Thus, fine-tuned calcium dynamics govern complex reciprocal relations among many different proteins contributing to changes in membrane potential. Ultimately, changes in K+, Ca2+-activated K+-currents and non-specific cationic currents support a shift from transient hyperpolarization to a sustained excitation.
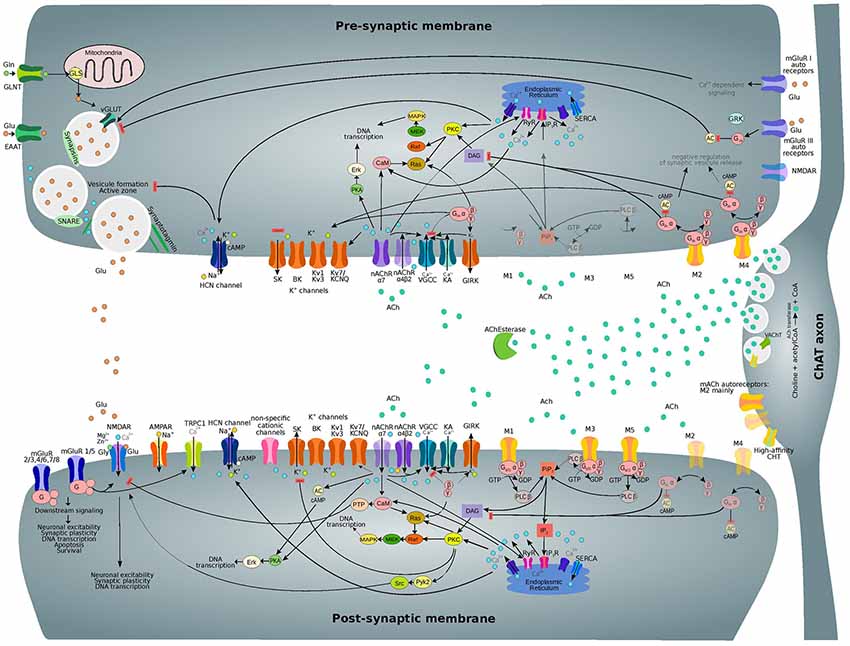
Figure 4. Subcellular nicotinic and muscarinic signaling processes at the glutamatergic synapse being modulated by ACh. Only the main relevant pathways and components are shown. Receptor subtypes which are less expressed on pre and post-synaptic membranes and related downstream processes are shown in semi-transparent colors. Abbreviations: ACh, acetylcholine; ACh Esterase, acetylcholinesterase; M1-M5, muscarinic acetylcholine receptor types 1–5; nAChR (α7, α4β2), nicotinic acetylcholine receptor (types α7, α4β2); VGCC, voltage-gated calcium channel; KA, kainate receptor; GIRK, G-protein activated inward rectifier K+ channel; PKA, protein kinase A; CaM, calmodulin; AC, adenylyl cyclase; DAG, diacylglycerol; PKC, protein kinase C; NOS, NO-synthase; HO-2, heme oxygenase 2; sGC, soluble guanylyl cyclase; PKG, cGMP-dependent protein kinase; HCN, hyperpolarization-activated cyclic nucleotide-gated channel; TRPC1, transient receptor potential cation channel 1; mGluR, metabotropic glutamate receptor; Pyk2, protein-tyrosine kinase 2; PiP2, phosphoinositol-1,4,5-biphosphate; PLC β, phospholipase C β; IP3, inositol triphosphate; IP3R, IP3 receptor; RyR, ryanodine receptor; SERCA, sarco-endoplasmic reticulum Ca2+-ATPase.
Meanwhile, DAG together with Ca2+ ions activate kinases such as protein kinase C (PKC), causing multiple downstream effects. PKC controls the function of many proteins including members of both pre and post-synaptic membranes. PKC is also involved in synaptic plasticity regulation and causes the internalization of AMPARs and NMDARs, leading to LTD phenomena (Callender and Newton, 2017).
PKC can also phosphorylate metabotropic glutamate receptor 5 (mGluR5; Hwang et al., 2005) as well as many other proteins. Moreover, PKC activates heme-oxygenase 2 (HO-2; Artinian et al., 2001) and inhibits NO-synthase (NOS), interfering with the calcium/calmodulin activation of NOS enzyme (Borda et al., 1998). These effects contribute to the downstream processes involving carbon monoxide (CO) and nitric oxide (NO) as interacting messengers (Mathes and Thompson, 1996; Artinian et al., 2001). Long-term effects of PKC activation include changes in DNA transcription that are mediated by MAPK/Erk signaling. Furthermore, there is recent evidence for the direct interaction of M3 mAChR with PLC β, which increases signaling efficiency (Kan et al., 2014).
The downstream signaling pathways of M3 and M5 receptors overlap with that of M1, and therefore they are grouped as M1-like receptors; similarly, M2-type mAChRs comprise both M2 and M4 receptors. Binding of ACh to M2-type mAChRs results in the inhibition of adenylyl cyclase (AC) by the α subunit of Gi/o protein and in the subsequent reduction of cAMP levels (Muñoz and Rudy, 2014). However, there are some differences between the Gi and Go mechanisms of AC regulation (Jiang and Bajpayee, 2009). The βγ-complex of the dissociated G-protein can activate the G-protein activated inward rectifier K+ channels (GIRK) and inhibit voltage-gated calcium channels (VGCCs). Moreover, Go proteins can also regulate Na+ channels (Jiang and Bajpayee, 2009). Particular effects of M1 and M2 receptors on different ion channels have been already summarized by Thiele et al. (2012).
A significant increase in intracellular calcium concentration comes from the direct flow of ions due to the permeability of nAChRs to Ca2+. However, nAChR activation also leads to the activation of VGCC and subsequent Ca2+ influx. (Dajas-Bailador and Wonnacott, 2004; Shen and Yakel, 2009). Moreover, functional cross-talk among presynaptic nAChRs has been shown to affect signal transduction (Marchi and Grilli, 2010). Therefore, the action of one receptor might depend on the function of co-existing receptor subtypes in the same cell. The interaction between presynaptic nicotinic receptors with other ionotropic or metabotropic receptors serves the purpose of producing an integrated response.
In recent years, a wealth of transcriptomic data from the mouse brain has become available (Saunders et al., 2018; Zeisel et al., 2018). Many different cell types may exist; one study found 565 different cell groups, for example (Saunders et al., 2018). Since a standard classification of cortical cell types is still emerging, most articles employ different approaches to arrive at cell type specific transcriptomes.
We examined a representative data set from the somatosensory cortex in order to interpret possible cell-specific differences in cholinergic receptor expression (Figure 5). We chose this data set since excitatory cell types are mapped to layer-specific types, allowing the easiest comparison with the types referenced in this review. In this dataset, normalized expression of M1 receptors is highest in L4 PCs. There is a strong expression of M2 in deep layer neurons, particularly in layer 5a. M3 is highly expressed in layer 2/3 and layer 5a, while M4 is highest in layer 4. α3 nAChR subunits are highest in layer 4, but also in the deep layers. β subunit expression is highest in layer 6 and layer 6a neurons. Inhibitory interneuron expression of cholinergic receptors is definitely cell-type specific, though heterologous. PV cells express more nAchRα3 than do somatostatin-expressing interneurons (Figure 5B). Somatostatin expression is best correlated with M2 expression and nicotinic β subunit expression and negatively correlated with M1 expression (Figure 5C). VIP and Htr3a expression is correlated with nAchRα3, nAchRα4, and nAchRα5. Furthermore, ChAT expression is correlated with M1 expression. In layer 5a, the effects of the predominantly-expressed nAChR and mAChRs seemed to be synergistic.
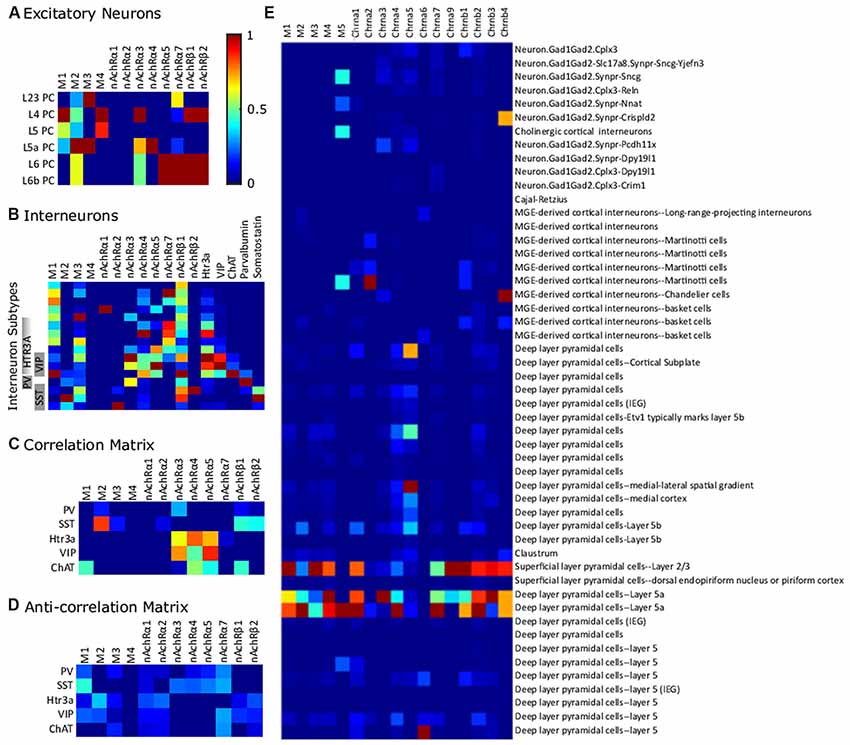
Figure 5. Differential expression of cholinergic receptors in transcriptome-derived cell types. (A) Excitatory cell types. (B) Interneurons in somatosensory cortex. Gene expression is normalized to a maximum of 1 on a gene-by-gene basis. (C) Correlation matrix (positive values of correlation matrix Pearson correlation coefficient matrix). (D) Anti-correlation matrix (negative values of correlation matrix). The data is from Zeisel et al. (2018) and was collected with high-throughput single-cell RNA sequencing, a method which counts individual RNA molecules. Abbreviations: PV, parvalbumin; SST, somatostatin; VIP, vasointestinal peptide; ChAT, choline acetyltransferase. (E) Expression of ACh receptor genes across the Frontal cortex cell-clusters identified in Saunders et al. (2018). The data was collected using Drop-seq (a method which allows the use of older animals and elimination of certain technical artifacts) to profile the RNA expression of individual cells. Semi-supervised independent component analysis was used to group cells into the sub-clusters using network-based clustering (ibid). Expression levels were normalized to the highest expression across all the selected genes. In this data set, receptor expression was particularly high in L23 and L5a PCs.
We also examined an additional dataset for frontal cortex (Figure 5E; Saunders et al., 2018). M5 is expressed in a subset of interneurons, including some cholinergic and MCs. The nicotinic receptor Chrna5 is expressed in a subset of deep PCs. Chrna6 is most expressed in a particular type of layer 5 PC. This dataset illustrates that the degree of sub-classification of PCs is likely to be important. For example, there are many subtypes of L5PCs, which have different cholinergic receptor expression. Both datasets showed consistency in M3 expression in L2/3 and L5a PCs but not L4 and L5 PCs.
In addition to cell-type specific correlation, nAChR genes that encode heteromeric α/β subunits are well correlated among themselves (Zoli et al., 2015; Saunders et al., 2018). The genes encoding the α subunits correlate well with the corresponding β subunit.
Cholinergic neurons can be identified by cluster analysis (Zeisel et al., 2018). In particular, separate types have been identified in the red nucleus and habenular nucleus of the thalamus (ibid). ACh often is released in neurons releasing other neurotransmitters (Zeisel et al., 2018). In the habenular nucleus, the glutamate transporter Slc17a6, in cholinergic cells, suggesting co-release of glutamate and ACh (Mancarci et al., 2017). In the ventral midbrain, a neuron type that was both dopaminergic and cholinergic was identified (Zeisel et al., 2018). Many forebrain cholinergic neurons also are GABAergic (Mancarci et al., 2017), consistent with the co-release of these two substances (Saunders et al., 2015).
The transition between different brain states that occurs whenever an organism switches from one behavioral state to another is associated with changes in the overall pattern of neural activity, which can be captured with EEG or LFP recordings. The pattern of EEG activity can change dramatically with the behavioral state of the animal (Lee and Dan, 2012), as can be seen in the transition from slow-wave sleep to wakefulness (or from deep sleep to REM sleep), when the EEG pattern shifts from large and synchronous waves of neural activity to a more desynchronized and short-amplitude wave pattern (Berger, 1929). Ensemble neuronal activity undergoes impressive changes during behavioral state transitions, and different brain states have been associated with different brain functions; definitive evidence for these functions although, is still lacking, and the mechanism by which these transitions are achieved in the cortical network is not yet understood. Many authors have proposed that the switch between cortical states may be driven by the action of neuromodulators like ACh (Lee and Dan, 2012). However, precisely how these neuromodulators influence global cortical processing by locally targeting specific cells is largely an unsolved mystery.
A large body of evidence suggests that the BF, a complex and heterogeneous structure classically defined by the presence of clusters of cholinergic neurons, is crucial for the maintenance of the sleep/wake cycle and for processes that underlie arousal and attentional modulation, but it is unclear which BF neurons promote each brain state and how they interact with each other to regulate transitions between states (Anaclet et al., 2015). Already since 1930, it was known that BF lesions could cause severe insomnia (Saper et al., 2001); however, this evidence has been an object of constant challenge over the years, and the attempts to replicate this experiment would yield different results. Finally, Szymusiak and McGinty (1986) observed that sleep-active cells were confined to the ventral BF in the cat (the horizontal limb of the diagonal bands of Broca, substantia innominata, entopeduncular nucleus and ventral globus pallidus) and that these areas partially overlap with those where chemical and electrical stimulations evoke sleep, and where lesions suppress sleep. The sleep-active cells were thus considered optimal candidates for mediating some of the sleep-promoting functions attributed to the BF (Szymusiak and McGinty, 1986).
Many BF neurons are active during wake and during REM sleep (Lee and Dan, 2012), and specific lesions reduce wakefulness, in agreement with the finding that BF lesions cause significant increases in delta waves occurrence during wakefulness, and that BF stimulation induces cortical desynchronization of EEG or LFP signals, accompanied by a decrease in correlated spiking. Furthermore, the BF receives inputs from the LDT and PPT pontine nuclei; cholinergic neurons that can be found at the level of the LDT nucleus exhibit an increase in firing rate during cortical activation, just before the transition from slow-wave sleep frequencies to faster frequencies (Saper et al., 2010).
Therefore, it seems reasonable to hypothesize the existence of functionally diverse neurons in the BF: according to Duque et al. (2000), BF cells that exhibit different wake/sleep activity pattern, also express different molecular markers (Zaborszky and Duque, 2000). There are three major neuronal types in the BF: cholinergic, glutamatergic and GABAergic cells (Anaclet et al., 2015; Xu et al., 2015). There might be extensive local synaptic interactions among BF neurons mediating local reciprocal inhibition between GABAergic neurons and sleep-active and wake-active cholinergic neurons. The well-known flip-flop circuit for sleep/wake cycle control (Saper et al., 2010) could, therefore, comprise multiple loops and switches. However, some findings suggest that BF GABAergic neurons provide major contributions to wakefulness, while cholinergic and glutamatergic neurons appear to play a lesser role; chemogenetic activation of GABAergic neurons promotes wake and high-frequency EEG activity, whereas cholinergic or glutamatergic activation have a destabilizing effect on slow-wave-sleep (SWS), but has no effect on total wake (Anaclet et al., 2015).
Cholinergic neurons residing in the BF can be divided into two subpopulations, that might be involved in different functions: an early-spiking population may reflect phasic changes in cortical ACh release associated with attention, while the late-spiking group could be more suited for the maintenance of the cholinergic tone during general cortical arousal (Unal et al., 2012).
Nevertheless, functional co-transmission of ACh and GABA seems to be a common feature of nearly all forebrain ACh-producing neurons (Henny and Jones, 2008; Granger et al., 2016). BF inputs to the neocortex are therefore not only constituted of different fibers, but also use a mixture of functionally diverse neurotransmitters (Kalmbach et al., 2012). This opens the question of whether there is a substantial difference between the cholinergic modulation and the BF modulation of neocortical activity. The contribution of GABA needs to be considered when studying the functional impact of ACh-producing neurons: electrical stimulation of BF fibers might evoke markedly different responses than optogenetically-evoked selective cholinergic release.
Does the co-release happen in a target-specific modality, at different terminals branching from the same axon, or is the release site the same for both transmitters? And if so, how does GABA affect the ongoing cholinergic modulation? Release of an excitatory (ACh) and inhibitory (GABA) neurotransmitter by the same axons seems to be functionally antagonistic. However, both transmitters could act in parallel, depending on the mode of co-transmission (Granger et al., 2016). If both ACh and GABA are released simultaneously onto the same post-synaptic cells, then GABA may act to shunt the (supposed) excitation generated by ACh. Otherwise, they could target different postsynaptic cells, such that GABA inhibits one cell population while ACh excites another. Given previous experimental results showing that GABA release from VIP interneurons shunts activity of Sst+ interneurons, but not other VIP interneurons, it is thought that VIP/ChAT cortical interneurons may release ACh and GABA onto different post-synaptic targets, perhaps from separate synaptic vesicle populations (Granger et al., 2016). Indeed, a recent analysis of the molecular composition of the pre-synaptic terminals of cortical VIP/ChAT interneurons revealed that ACh and GABA vesicles are confined to separate boutons. At the post-synaptic level, the subset of GABAergic boutons seems to contact prevalently other inhibitory interneurons, while ACh boutons target mostly L1 interneurons and other VIP/ChAT cortical interneurons. Here, ACh evokes EPSCs that are mediated by nicotinic receptors (Granger et al., 2018). Another recent study conducted in the mPFC confirms that only 10%–20% of post-synaptic targets of VIP/ChAT cortical interneurons are contacted by both cholinergic and GABAergic inputs (Obermayer et al., 2018); here they report that VIP/ChAT neurons directly excite interneurons in layers 1–3 as well as PCs in L2/3 and L6 by fast nicotinic transmission.
Immunolabeling studies (Beaulieu and Somogyi, 1991) have shown substantial co-labeling of presynaptic cholinergic terminals for both GABA and ChAT in the neocortex, but more studies should address the functional consequences of the synaptic co-release of these neurotransmitters and try to dissect the differential impact of each transmitter on postsynaptic cells excitability. Analysing the co-localization of post-synaptic receptors or scaffolding proteins could also allow the identification of individual synapses that are sensitive to both ACh and GABA. These possibilities should be addressed systematically in order to precisely understand the contribution of each neurotransmitter to cortical processing.
Apart from the fine-tuning of sleep/wake transitions, cholinergic neuromodulation is tightly implicated in regulating selective attention to a given sensory stimulus by altering the activity of the sensory cortex that perceives that modality (Kim et al., 2016). ACh is known to be especially involved in cortical arousal (Saper et al., 2010) and in the state-dependent modulation of cortical activity; cholinergic neurons are active during locomotion (Buzsaki et al., 1988) and during transition to the attentive state (Kim et al., 2016). Studies have shown that the occurrence of relevant sensory events evokes a transient increase in ACh concentration in the rat PFC (Hasselmo and Sarter, 2011). Conversely, activating cholinergic transmission in the PFC determines an improvement in subject’s performance during sustained attention tasks (Saper et al., 2010). It is, therefore, reasonable to hypothesize that ACh can induce long-lasting changes in neuronal excitability, and indeed this was demonstrated. Pioneering experiments showing that ablation of noradrenergic and cholinergic innervation in the striate cortex substantially impairs ocular dominance plasticity in kittens (Bear and Singer, 1986) opened the way for subsequent studies on the involvement of ACh in cortical plasticity. Some showed that when a tone is paired with NBM stimulation or ACh application, auditory cortex receptive fields change and prolonged enhanced responses to the paired frequency can be observed (Metherate and Weinberger, 1990; Rasmusson, 2000). Others discovered that co-application of muscarinic agonists with glutamate induces a prolonged increase in response to glutamate in somatosensory cortical neurons (Sugihara et al., 2016), and that these effects concern as well the somatosensory cortex and the primary visual area V1. According to Metherate and Weinberger (1990), the potentiation can be blocked by cortical application of atropine, but others (Sugihara et al., 2016) report that cholinergic antagonists cannot reverse the prolonged changes, thereby confirming that ACh is necessary for the induction, but not the maintenance of these modifications. ACh seems to act more as an instructive, rather than a permissive signal (Lin et al., 2015).
ACh is as well involved in the generation of LTD at synapses between cortical pyramidal neurons and striatal medium spiny neurons through disinhibition of Cav channels. Here, the activation of D2 receptors reduces basal ACh release from cholinergic striatal interneurons and lowers M1 receptor tone in medium spiny neurons, which leads to enhanced opening of intraspine Cav1.3 Ca2+ channels in response to synaptic depolarization. The calcium transient results in enhanced production of endocannabinoids (ECs) such as 2-arachidonoylglycerol, and activation of presynaptic CB1 receptors that reduce glutamate release (Wang et al., 2006).
Furthermore, the role of several neuromodulatory systems in STDP induction (Pawlak et al., 2010) has been studied across multiple brain areas. While dopamine (DA) and NA modulation of STDP has been mostly investigated in subcortical areas, ACh’s role in STDP induction has been extensively researched in neocortical sensory areas and in the PFC. In mouse mPFC, nicotine application increases the threshold for STDP in L5PCs by reducing their dendritic calcium signals. This effect, however, is due to an enhancement in GABAergic transmission in various types of interneurons in the PFC network, that express multiple types of nAChRs (Couey et al., 2007), and not to a direct nicotinic action on PCs.
Taken together, evidence suggests that cholinergic inputs to the cortex incoming from the BF should be viewed more as teaching, rather than motivational signals. Overall, activation of the cholinergic system controls the shift from a correlated or synchronized state, to a decorrelated or desynchronized state and results in an enhancement of cortical information processing (Lee and Dan, 2012). However, exactly how the detection of relevant stimuli is enhanced and which are the mechanisms at the basis of this ACh-induced desynchronization are still a matter of open debate.
NBM stimulation has a differential effect on spontaneous and sensory-evoked activity. In a recent study, Meir et al. (2018) showed that NBM stimulation desynchronizes cortical LFP and increases the SNR of sensory-evoked responses while suppressing ongoing spontaneous synaptic activity. The authors recorded spontaneous PSPs occurring in L4 and showed that following NBM stimulation the frequency and amplitude of sPSPs were decreased. Moreover, the mean membrane voltage of the response became more hyperpolarized, and trial-to-trial variability was decreased, both during spontaneous and evoked activity. However, sensory stimulation did not change the amplitude of the response, whereas it caused a prominent reduction in the noise amplitude, therefore changing the SNR of the sensory response. By analyzing the coupling of Vm and LFP signals, they also showed that cholinergic activation largely reduced fluctuations in the membrane potential and caused a decorrelation in network activity.
Chen et al. (2015) were able to identify a defined microcircuit in the superficial layers of mouse V1 that supports ACh driven desynchronization. The authors measured the activity of different inhibitory interneurons while optogenetically stimulating superficial cholinergic axons, and found that cholinergic inputs facilitate Sst+ interneurons, which in turn inhibit PV+ interneurons and PCs. Optogenetic inhibition of Sst+ neurons blocks desynchronization, whereas direct activation of Sst+ neurons is sufficient to induce desynchronization (Chen et al., 2015). The observed desynchronization in cortical activity may explain the role of ACh in mediating transitions between phases of the sleep-wake cycle, but it fails to explain how ACh enhances sensory processing. A large body of evidence suggests that ACh enhances sensory inputs while simultaneously suppressing intrinsic cortical activation (Kimura et al., 1999; Disney et al., 2007; Newman et al., 2012), but a detailed understanding of this process is currently lacking. ACh’s role may substantially differ across sensory areas and affect different tuning properties.
Nucleus basalis activation affects sensory responses to natural stimuli of a population of cortical neurons. Before BF stimulation, multi-unit activity (MUA) in the rat’s V1 is highly correlated but poorly time-locked to the stimulus; after BF stimulation it becomes less correlated but more time-locked to the sensory event. NBM stimulation also decreases single-unit activity (SU) correlation (between cells correlation) and increases response reliability (between trials correlation coefficient) but does not induce any significant change in receptive field size, orientation tuning nor direction selectivity. Atropine application decreases NBM induced decorrelation, indicating that mAChRs support this effect (Goard and Dan, 2009). After NBM stimulation a shift in the firing modality of the LGN resembling that found at the level of the thalamus can be observed, namely a transition from burst to tonic mode (Bazhenov et al., 2002; Castro-Alamancos and Gulati, 2014). A similar study (Thiele et al., 2012) was conducted in the extrastriate cortex of the macaque and yielded opposing results: at the level of the middle temporal (MT) area it revealed how other tuning properties, like orientation and direction discriminability, are also affected by cholinergic modulation; in this case, ACh had little effect on response reliability, though it is still not clear whether these differences are attributable to differences existing between rodents and primates or to functional differences between sensory areas. In an effort to clarify the precise role of neocortical cholinergic modulation, Disney et al. (2007) concentrated on the role of nAChRs in a well-studied cortical model system, the V1 of the macaque monkey. Here they showed in vivo that nicotine reliably enhances the gain of responses to visual stimuli in layer 4c, but not in other layers. Having found β2-nAChR in a pre-synaptic position at the level of thalamo-cortical synapses on PV+ interneurons, they prove that nicotine enhances detection of visual stimuli through enhanced TC transmission. These findings confirm that cholinergic activation causes an increase in cortical sensory responses through enhancement of thalamic synaptic transmission and suppression of intracortical inputs. A systematic effort to extend these results to other sensory areas is therefore needed in order to decipher whether the mechanism supporting cholinergic modulation is common throughout all cortical areas or if different tuning properties are affected each time.
Castro-Alamanco and Gulati recorded, multi-electrode activity (MUA) and field potential from adult rat barrel cortex following multi-whisker stimulation at 0.2 Hz, while increasing concentrations of carbachol or other drugs were applied by means of micro-dialysis. The authors found that the application of 50 μM carbachol, but not norepinephrine, can stop the emergence of the 10–15 Hz oscillations that are observed during baseline recordings and that in the presence of atropine these oscillations are even enhanced (Castro-Alamancos and Gulati, 2014). The effect of carbachol on barrel cortex LFP is thus congruent with the traditionally termed desynchronization for doses higher than 50 μM (Moruzzi and Magoun, 1949; Steriade et al., 1993). A low tone of cholinergic activation (0.5–1 μM) however, reinforces the deactivated cortical state by enhancing synchronous slow oscillations. A very high tone of cholinergic activation (250–2,500 μM) leads to a significant increase in tonic firing, without altering the overall firing rate. An interesting follow-up to this experiment would be to check whether the same effect can be observed in the whole somatosensory region, and across other sensory cortices. The group then tried to decipher whether cholinergic activation would also modulate thalamocortical activity: by recording from the VPM, they found that cholinergic cortical activation suppresses burst-firing in the thalamus and changes neuronal firing to a tonic mode. This result is fairly consistent with the outcome predicted by the model of thalamo-cortical slow-wave sleep oscillations and transition to activated states generated by Bazhenov et al. (2002). Here, the increase in ACh activity was modeled by the reduction of a K+ leak current in pyramidal and thalamo-cortical cells and resulted in the abolishment of the hyperpolarizing phase of network activity and a consequent increase in the input/resistance relationship, accompanied by a switch to the tonic firing (15–20 Hz) modality. The transition from bursting to tonic firing thus seems to be a characteristic feature of relay diencephalic structures like the thalamus and the meta-thalamus.
Enhanced thalamo-cortical transmission seems to be a constant finding across a vast number of articles and reviews (Bazhenov et al., 2002; Disney et al., 2007; Hasselmo and Sarter, 2011) with the aim of revealing the mechanisms by which cholinergic neuromodulation operates. Next studies in this field should, therefore, consider the possibility that cholinergic inputs reach the cortex not only through direct BF projections but also exploiting the thalamo-cortical loop.
Voltage-sensitive dye imaging revealed that ACh application to the neocortex, upon stimulation of layer 2/3, suppresses the spread of excitation to nearby areas. Thus, ACh seems to play an important role in coding sensory stimuli by enhancing thalamocortical inputs, but at the same time, by suppressing intracortical interactions (Kimura et al., 1999).
One of the proposed models for the cholinergic mediated shift from default mode to detection mode suggests that ACh acts to enhance the glutamatergic representation of thalamic input through stimulation of nAChRs, while suppressing the cortical spread of associational input through activation of mAChRs (Hasselmo and Sarter, 2011). Minces et al. (2017) recently evaluated the effect of increases in cortical ACh following optogenetic BF stimulation on the correlation structure of the visual network and found that transient cholinergic release in the cortex decreases the slope between signal and noise correlations. The authors propose that this mechanism acts to increase the encoding capacity of the network.
Another article evaluated the impact of ACh on local circuit activation and found that cholinergic inputs exclude unreliable neurons from contributing to circuit activity while conserving neurons that were active in response to thalamic activity and showed strong correlations. Moreover, weak functional connections were pruned, thus yielding a more modular and hierarchical circuit structure. Once again, these results highlight how ACh is able to reorganize the circuit function in a way that promotes the discriminability of thalamic inputs at the expense of weak pairwise relationships (Runfeldt et al., 2014).
Many studies (Disney et al., 2007; Minces et al., 2017) have focused on trying to understand the role played by ACh in improving stimuli detection or modifying receptor fields size in the visual cortex. While many of them have been done in primates, others have privileged the somatosensory areas and highlight the involvement of the cholinergic system in the regulation of sensory cortical processing in rodents as well, supporting the idea that cholinergic modulation of cortical microcircuits is functionally equivalent across brain areas and model organisms, even though a canonical and anatomically equivalent system is not strictly identifiable (Coppola and Disney, 2018).
The finding that distinct neuronal clusters in the BF project selectively to specific sensory areas (Kim et al., 2016) and that cholinergic inputs to sensory cortices are spatially segregated supports the idea that cholinergic release improves sensory discrimination in a modality-selective manner and with a high degree of specificity. The authors mapped BF projections to different sensory areas and found retrobead-labeled neurons from three different sensory cortices within the BF, with a clear distinction between the clusters of cells: neurons in the HDB project preferentially to V1, the posterior part of NBM projects to A1, while the aNBM preferentially projects to S1. These results were further confirmed by another experiment in which the authors optogenetically activated cholinergic neurons in the BF subnuclei and successfully induced modality-selective desynchronization in specific sensory cortices.
A similar experiment was performed by Chaves-Coira et al. (2016), who also used retrograde anatomical procedures to demonstrate the existence of specific neuronal groups in the BF implicated in the modulation of specific sensory cortices. However, here the authors found that most of the neurons located in the HDB projected to the S1 cortex, suggesting that this area is specialized in the sensory processing of tactile stimuli, and the NBM was found to have a similar number of cells projecting to S1 as to A1. Furthermore, optogenetic HDB stimulation induced a larger facilitation of tactile evoked potentials in S1 than auditory evoked potentials in A1, while optogenetic stimulation of the NBM facilitated either tactile or auditory evoked potentials equally. These results suggest that cholinergic projections to the cortex are organized into spatially segregated pools of neurons that modulate specific cortical areas; although, additional research will be needed in order to provide a clear and definitive picture of the topographical organization of the projections arising from the BF region and innervating the cortex. Despite the many attempts to clarify this issue, it remains unclear whether there exist distinct neuronal populations in the HDB, or whether the differences observed in the outcomes of the experiments mentioned above are due to discrepancies existing in the transgenic mouse lines used or to the slightly different techniques that were employed.
ACh is thus involved both in the bottom-up attentional process that leads to a general and whole-state arousal of the cortex and in the top-down modifications of circuit activity that occur during detection of behaviorally relevant sensory stimuli. Cognitive functions of cholinergic projection systems vary according to the brain area that is being modulated. Cholinergic modulation may act as a common mechanism to improve sensory encoding in several brain areas.
ACh release in the neocortex controls transitions between brain states, such as attention, memory and wakefulness, and can occur through volume or synaptic transmission. However, it is not clear yet whether one modality prevails upon the other or if they are complementary mechanisms. Further studies are needed to establish correlations between the distribution profile of the receptor subtypes, the relative proximity and density of cholinergic varicosities to assess differences between the two modalities. Moreover, as results could vastly vary across species, a systematic effort is crucial to be able to compare quantitative measurements.
The expression of muscarinic and nicotinic cholinergic receptors—the two main types—varies according to the cell-type and the pattern of receptor localization varies across cortical layers. A detailed knowledge of the subcellular localization of cholinergic receptors is, however, currently lacking. The detection of cholinoceptive structures such as the receptor protein has become easier with the advent of polyclonal antibodies targeting different subtypes. Future investigations should, therefore, converge on systematically measuring the amount of each receptor subtype across cellular compartments.
In this review, we have endeavored to determine, in a quantitative manner, the cellular and synaptic effects of ACh release in the neocortex. While the cholinergic modulation of excitatory PCs has been extensively researched, its effect on inhibitory interneurons is still largely unknown. For example, the effect of ACh on BCs (fast-spiking, PV+ interneurons) remains unclear. This could be due to the lack of a thorough classification of diverse morphological types of BCs where a differential distribution of cholinergic receptors could modulate divergent cellular and synaptic effects. Furthermore, it is not clear whether bath-application of cholinergic agonists is comparable to a physiological activation of the cholinergic system. Applied concentrations of cholinergic agonists vary substantially (up to three orders of magnitude) across electrophysiological studies, which seldom use more than one concentration. To obtain carefully designed dose-response curves of the effects of cholinergic agonists is paramount to dissect the consequences of physiological ACh release in the neocortex. The advent of optogenetics holds promise in designing physiological protocols of ACh release. Future experiments should not only merely employ traditional bath-application of cholinergic agonists but also exploit optogenetics to reconcile how doses of agonists directly map to effects of endogenous, physiological release of ACh.
The effects of ACh on synaptic connections can vary drastically according to the identity of the presynaptic terminal and its postsynaptic partner. Additionally, the magnitude of the postsynaptic response also depends on the receptor subtype being activated. Therefore, there is a clear requirement for systematic investigations of the effects of ACh on different synapse-types, combined with knowledge of implicated cell-types and receptor subtypes to unravel the effects of ACh release on necortical synaptic transmission.
ACh is involved in the induction of synaptic plasticity mechanisms, which could support its role in cortical learning and memory. In addition, ACh enhances sensory processing by affecting receptor fields size and tuning properties. It is not clear, however, if the effects of ACh are modality-specific or can be generalized to all sensory processing, nor exactly which tuning properties are affected. Many studies point to a role of ACh in increasing the SNR of a sensory response, and others describe how ACh suppresses cortico-cortical interactions in favor of thalamic transmission. Therefore, further clarification is required on the matter. Moreover, special attention must be paid in integrating data from primates and rodents: neuromodulatory systems are commonly the object of evolutionary modifications, even though they might maintain some functional similarity throughout species.
The mechanisms of ACh-induced changes in the physiology of neocortical neurons and their synapses, and how these changes shape the emergence of global network states still remains elusive. The impact of ACh on global cortical computations sustains cognitive functions such as attention, learning and memory, which are characterized by desynchronized network activity. Cholinergic inputs mainly originate in the BF, a structure comprising distinct multi-transmitter neuronal populations. The functional relevance of neuronal subpopulations in the BF and the co-release of two potentially antagonistic transmitters to the desynchronization of cortical activity is unknown. Furthermore, recent work identifies that a sub-population of VIP+ cortical interneurons co-release ACh and GABA with potentially differing functions across species. Future research should, therefore, focus on dissecting the impact of each transmitter on cellular excitability. In addition, analyzing the co-localization of post-synaptic receptors could also allow the identification of individual synapses that are sensitive to multiple neurotransmitters. All these possibilities should be addressed systematically in order to precisely understand the contribution of each neurotransmitter to ACh-induced effects on the emergence of cortical network states in health and disease.
CC, DK, PS and SR wrote the manuscript and drafted the figures and tables. SR, DK and HM reviewed and edited the manuscript and the figures. SR conceived the idea and supervised the study.
This work was supported by funding from the ETH Domain for the Blue Brain Project (BBP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr Natali Barros-Zulaica and Taylor H. Newton for useful discussions.
Albuquerque, E. X., Pereira, E. F., Mike, A., Eisenberg, H. M., Maelicke, A., and Alkondon, M. (2000). Neuronal nicotinic receptors in synaptic functions in humans and rats: physiological and clinical relevance. Behav. Brain Res. 113, 131–141. doi: 10.1016/s0166-4328(00)00208-4
Alger, B. E., Nagode, D. A., and Tang, A.-H. (2014). Muscarinic cholinergic receptors modulate inhibitory synaptic rhythms in hippocampus and neocortex. Front. Synaptic Neurosci. 6:18. doi: 10.3389/fnsyn.2014.00018
Alitto, H. J., and Dan, Y. (2013). Cell-type-specific modulation of neocortical activity by basal forebrain input. Front. Syst. Neurosci. 6:79. doi: 10.3389/fnsys.2012.00079
Alkondon, M., and Albuquerque, E. X. (2004). The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog. Brain Res. 145, 109–120. doi: 10.1016/s0079-6123(03)45007-3
Alkondon, M., Pereira, E. F., Eisenberg, H. M., and Albuquerque, E. X. (1999). Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J. Neurosci. 19, 2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999
Anaclet, C., Pedersen, N. P., Ferrari, L. L., Venner, A., Bass, C. E., Arrigoni, E., et al. (2015). Basal forebrain control of wakefulness and cortical rhythms. Nat. Commun. 6:8744. doi: 10.1038/ncomms9744
Aoki, C., and Kabak, S. (1992). Cholinergic terminals in the cat visual cortex: ultrastructural basis for interaction with glutamate-immunoreactive neurons and other cells. Vis. Neurosci. 8, 177–191. doi: 10.1017/s0952523800002832
Arroyo, S., Bennett, C., Aziz, D., Brown, S. P., and Hestrin, S. (2012). Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J. Neurosci. 32, 3859–3864. doi: 10.1523/JNEUROSCI.0115-12.2012
Artinian, L. R., Ding, J. M., and Gillette, M. U. (2001). Carbon monoxide and nitric oxide: interacting messengers in muscarinic signaling to the brain’s circadian clock. Exp. Neurol. 171, 293–300. doi: 10.1006/exnr.2001.7781
Atzori, M., Kanold, P. O., Pineda, J. C., Flores-Hernandez, J., and Paz, R. D. (2005). Dopamine prevents muscarinic-induced decrease of glutamate release in the auditory cortex. Neuroscience 134, 1153–1165. doi: 10.1016/j.neuroscience.2005.05.005
Bailey, C. D. C., De Biasi, M., Fletcher, P. J., and Lambe, E. K. (2010). The nicotinic acetylcholine receptor α5 subunit plays a key role in attention circuitry and accuracy. J. Neurosci. 30, 9241–9252. doi: 10.1523/JNEUROSCI.2258-10.2010
Baker, A. L., O’Toole, R. J., and Gulledge, A. T. (2018). Preferential cholinergic excitation of corticopontine neurons. J. Physiol. 596, 1659–1679. doi: 10.1113/jp275194
Bazhenov, M., Timofeev, I., Steriade, M., and Sejnowski, T. J. (2002). Model of thalamocortical slow-wave sleep oscillations and transitions to activated states. J. Neurosci. 22, 8691–8704. doi: 10.1523/JNEUROSCI.22-19-08691.2002
Bear, M. F., and Singer, W. (1986). Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature 320, 172–176. doi: 10.1038/320172a0
Beaulieu, C., and Somogyi, P. (1991). Enrichment of cholinergic synaptic terminals on GABAergic neurons and coexistence of immunoreactive GABA and choline acetyltransferase in the same synaptic terminals in the striate cortex of the cat. J. Comp. Neurol. 304, 666–680. doi: 10.1002/cne.903040412
Berger, H. (1929). Über das elektrenkephalogramm des menschen. Arch. Für Psychiatr. Nervenkrankh. 87, 527–570. doi: 10.1007/BF01797193
Bertrand, D. (2010). Neurocircuitry of the nicotinic cholinergic system. Dialogues Clin. Neurosci. 12, 463–470.
Borda, T., Genaro, A., Sterin-Borda, L., and Cremaschi, G. (1998). Involvement of endogenous nitric oxide signalling system in brain muscarinic acetylcholine receptor activation. J. Neural Transm. 105, 193–204. doi: 10.1007/s007020050048
Brombas, A., Fletcher, L. N., and Williams, S. R. (2014). Activity-dependent modulation of layer 1 inhibitory neocortical circuits by acetylcholine. J. Neurosci. 34, 1932–1941. doi: 10.1523/JNEUROSCI.4470-13.2014
Buisson, B., and Bertrand, D. (2001). Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J. Neurosci. 21, 1819–1829. doi: 10.1523/JNEUROSCI.21-06-01819.2001
Buzsaki, G., Bickford, R. G., Ponomareff, G., Thal, L. J., Mandel, R., and Gage, F. H. (1988). Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J. Neurosci. 8, 4007–4026. doi: 10.1523/JNEUROSCI.08-11-04007.1988
Callender, J. A., and Newton, A. C. (2017). Conventional protein kinase C in the brain: 40 years later. Neuronal Signal. 1:NS20160005. doi: 10.1042/ns20160005
Carr, D. B., and Surmeier, D. J. (2007). M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J. Neurophysiol. 97, 3432–3438. doi: 10.1152/jn.00828.2006
Castro-Alamancos, M. A., and Gulati, T. (2014). Neuromodulators produce distinct activated states in neocortex. J. Neurosci. 34, 12353–12367. doi: 10.1523/JNEUROSCI.1858-14.2014
Cauli, B., Tong, X.-K., Rancillac, A., Serluca, N., Lambolez, B., Rossier, J., et al. (2004). Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J. Neurosci. 24, 8940–8949. doi: 10.1523/JNEUROSCI.3065-04.2004
Chaves-Coira, I., Barros-Zulaica, N., Rodrigo-Angulo, M., and Núñez, Á. (2016). Modulation of specific sensory cortical areas by segregated basal forebrain cholinergic neurons demonstrated by neuronal tracing and optogenetic stimulation in mice. Front. Neural Circuits 10:28. doi: 10.3389/fncir.2016.00028
Chen, N., Sugihara, H., and Sur, M. (2015). An acetylcholine-activated microcircuit drives temporal dynamics of cortical activity. Nat. Neurosci. 18, 892–902. doi: 10.1038/nn.4002
Christophe, E., Roebuck, A., Staiger, J. F., Lavery, D. J., Charpak, S., and Audinat, E. (2002). Two types of nicotinic receptors mediate an excitation of neocortical layer I interneurons. J. Neurophysiol. 88, 1318–1327. doi: 10.1152/jn.2002.88.3.1318
Coppola, J. J., and Disney, A. A. (2018). Is there a canonical cortical circuit for the cholinergic system? Anatomical differences across common model systems. Front. Neural Circuits 12:8. doi: 10.3389/fncir.2018.00008
Couey, J. J., Meredith, R. M., Spijker, S., Poorthuis, R. B., Smit, A. B., Brussaard, A. B., et al. (2007). Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron 54, 73–87. doi: 10.1016/j.neuron.2007.03.006
Dajas-Bailador, F., and Wonnacott, S. (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 25, 317–324. doi: 10.1016/s0165-6147(04)00118-x
Dale, H. H. (1914). The action of certain esters and ethers of choline and their relation to muscarine. J. Pharmacol. Exp. Ther. 6, 147–190.
Dani, J. A., and Bertrand, D. (2007). Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 47, 699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214
Dasari, S., Hill, C., and Gulledge, A. T. (2017). A unifying hypothesis for M1 muscarinic receptor signalling in pyramidal neurons. J. Physiol. 595, 1711–1723. doi: 10.1113/jp273627
Dasgupta, R., Seibt, F., and Beierlein, M. (2018). Synaptic release of acetylcholine rapidly suppresses cortical activity by recruiting muscarinic receptors in layer 4. J. Neurosci. 38, 5338–5350. doi: 10.1523/JNEUROSCI.0566-18.2018
Delmas, P., and Brown, D. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862. doi: 10.1038/nrn1785
Descarries, L., and Mechawar, N. (2000). “Ultrastructural evidence for diffuse transmission by monoamine and acetylcholine neurons of the central nervous system,” in Progress in Brain Research, eds L. F. Agnati, K. Fuxe, C. Nicholson and E. Syková (Amsterdam: Elsevier), 27–47.
Disney, A. A., Aoki, C., and Hawken, M. J. (2007). Gain modulation by nicotine in macaque V1. Neuron 56, 701–713. doi: 10.1016/j.neuron.2007.09.034
Disney, A. A., Domakonda, K. V., and Aoki, C. (2006). Differential expression of muscarinic acetylcholine receptors across excitatory and inhibitory cells in visual cortical areas V1 and V2 of the macaque monkey. J. Comp. Neurol. 499, 49–63. doi: 10.1002/cne.21096
Duque, A., Balatoni, B., Detari, L., and Zaborszky, L. (2000). EEG correlation of the discharge properties of identified neurons in the basal forebrain. J. Neurophysiol. 84, 1627–1635. doi: 10.1152/jn.2000.84.3.1627
Eccles, J. C., Fatt, P., and Koketsu, K. (1953). Cholinergic and inhibitory synapses in a central nervous pathway. Aust. J. Sci. 16, 50–54.
Eckenstein, F. P., Baughman, R. W., and Quinn, J. (1988). An anatomical study of cholinergic innervation in rat cerebral cortex. Neuroscience 25, 457–474. doi: 10.1016/0306-4522(88)90251-5
Eggermann, E., and Feldmeyer, D. (2009). Cholinergic filtering in the recurrent excitatory microcircuit of cortical layer 4. Proc. Natl. Acad. Sci. U S A 106, 11753–11758. doi: 10.1073/pnas.0810062106
Elhusseiny, A., and Hamel, E. (2000). Muscarinic—but not nicotinic—acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J. Cereb. Blood Flow Metab. 20, 298–305. doi: 10.1097/00004647-200002000-00011
Fanselow, E. E., Richardson, K. A., and Connors, B. W. (2008). Selective, state-dependent activation of somatostatin-expressing inhibitory interneurons in mouse neocortex. J. Neurophysiol. 100, 2640–2652. doi: 10.1152/jn.90691.2008
Figl, A., and Cohen, B. N. (2000). The subunit dominates the relaxation kinetics of heteromeric neuronal nicotinic receptors. J. Physiol. 524, 685–699. doi: 10.1111/j.1469-7793.2000.00685.x
Gericke, A., Sniatecki, J. J., Mayer, V. G. A., Goloborodko, E., Patzak, A., Wess, J., et al. (2011). Role of M1, M3, and M5 muscarinic acetylcholine receptors in cholinergic dilation of small arteries studied with gene-targeted mice. Am. J. Physiol. Heart Circ. Physiol. 300, H1602–H1608. doi: 10.1152/ajpheart.00982.2010
Giacobini, E. (2003). Cholinergic function and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 18, S1–S5. doi: 10.1002/gps.935
Gil, Z., Connors, B. W., and Amitai, Y. (1997). Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19, 679–686. doi: 10.1016/s0896-6273(00)80380-3
Goard, M., and Dan, Y. (2009). Basal forebrain activation enhances cortical coding of natural scenes. Nat. Neurosci. 12, 1444–1449. doi: 10.1038/nn.2402
Granger, A. J., Mulder, N., Saunders, A., and Sabatini, B. L. (2016). Cotransmission of acetylcholine and GABA. Neuropharmacology 100, 40–46. doi: 10.1016/j.neuropharm.2015.07.031
Granger, A. J., Wang, W., Robertson, K., El-Rifai, M., Zanello, A., Bistrong, K., et al. (2018). Target-specific co-transmission of acetylcholine and GABA from a subset of cortical VIP+ interneurons. BioRxiv:69064 [preprint]. doi: 10.1101/469064
Groleau, M., Kang, J. I., Huppé-Gourgues, F., and Vaucher, E. (2015). Distribution and effects of the muscarinic receptor subtypes in the primary visual cortex. Front. Synaptic Neurosci. 7:10. doi: 10.3389/fnsyn.2015.00010
Gulledge, A. T., Park, S. B., Kawaguchi, Y., and Stuart, G. J. (2007). Heterogeneity of phasic cholinergic signaling in neocortical neurons. J. Neurophysiol. 97, 2215–2229. doi: 10.1152/jn.00493.2006
Gulledge, A. T., and Stuart, G. J. (2005). Cholinergic inhibition of neocortical pyramidal neurons. J. Neurosci. 25, 10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005
Haj-Dahmane, S., and Andrade, R. (1996). Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J. Neurosci. 16, 3848–3861. doi: 10.1523/JNEUROSCI.16-12-03848.1996
Hales, T. G., Dunlop, J. I., Deeb, T. Z., Carland, J. E., Kelley, S. P., Lambert, J. J., et al. (2006). Common determinants of single channel conductance within the large cytoplasmic loop of 5-hydroxytryptamine type 3 and alpha4beta2 nicotinic acetylcholine receptors. J. Biol. Chem. 281, 8062–8071. doi: 10.1074/jbc.M513222200
Hasselmo, M. E., and Sarter, M. (2011). Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology 36, 52–73. doi: 10.1038/npp.2010.104
Hay, Y. A., Lambolez, B., and Tricoire, L. (2016). Nicotinic transmission onto layer 6 cortical neurons relies on synaptic activation of non-α7 receptors. Cereb. Cortex 26, 2549–2562. doi: 10.1093/cercor/bhv085
Hedrick, T., and Waters, J. (2015). Acetylcholine excites neocortical pyramidal neurons via nicotinic receptors. J. Neurophysiol. 113, 2195–2209. doi: 10.1152/jn.00716.2014
Henny, P., and Jones, B. E. (2008). Projections from basal forebrain to prefrontal cortex comprise cholinergic, GABAergic and glutamatergic inputs to pyramidal cells or interneurons. Eur. J. Neurosci. 27, 654–670. doi: 10.1111/j.1460-9568.2008.06029.x
Heys, J. G., and Hasselmo, M. E. (2012). Neuromodulation of Ih in layer II medial entorhinal cortex stellate cells: a voltage-clamp study. J. Neurosci. 32, 9066–9072. doi: 10.1523/JNEUROSCI.0868-12.2012
Heys, J. G., Schultheiss, N. W., Shay, C. F., Tsuno, Y., and Hasselmo, M. E. (2012). Effects of acetylcholine on neuronal properties in entorhinal cortex. Front. Behav. Neurosci. 6:32. doi: 10.3389/fnbeh.2012.00032
Hilscher, M. M., Leão, R. N., Edwards, S. J., Leão, K. E., and Kullander, K. (2017). Chrna2-martinotti cells synchronize layer 5 type a pyramidal cells via rebound excitation. PLoS Biol. 15:e2001392. doi: 10.1371/journal.pbio.2001392
Hsiao, B., Mihalak, K. B., Magleby, K. L., and Luetje, C. W. (2008). Zinc potentiates neuronal nicotinic receptors by increasing burst duration. J. Neurophysiol. 99, 999–1007. doi: 10.1152/jn.01040.2007
Hwang, J.-I., Kim, H. S., Lee, J. R., Kim, E., Ryu, S. H., and Suh, P.-G. (2005). The interaction of phospholipase C-β3 with Shank2 regulates mGluR-mediated calcium signal. J. Biol. Chem. 280, 12467–12473. doi: 10.1074/jbc.M410740200
Jiang, M., and Bajpayee, N. S. (2009). Molecular mechanisms of go signaling. Neurosignals 17, 23–41. doi: 10.1159/000186688
Jiang, X., Wang, G., Lee, A. J., Stornetta, R. L., and Zhu, J. J. (2013). The organization of two new cortical interneuronal circuits. Nat. Neurosci. 16, 210–218. doi: 10.1038/nn.3305
Kalmbach, A., Hedrick, T., and Waters, J. (2012). Selective optogenetic stimulation of cholinergic axons in neocortex. J. Neurophysiol. 107, 2008–2019. doi: 10.1152/jn.00870.2011
Kan, W., Adjobo-Hermans, M., Burroughs, M., Faibis, G., Malik, S., Tall, G. G., et al. (2014). M3 muscarinic receptor interaction with phospholipase C β3 determines its signaling efficiency. J. Biol. Chem. 289, 11206–11218. doi: 10.1074/jbc.M113.538546
Kassam, S. M., Herman, P. M., Goodfellow, N. M., Alves, N. C., and Lambe, E. K. (2008). Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J. Neurosci. 28, 8756–8764. doi: 10.1523/JNEUROSCI.2645-08.2008
Katz, B., and Miledi, R. (1973). The binding of acetylcholine to receptors and its removal from the synaptic cleft. J. Physiol. 231, 549–574. doi: 10.1113/jphysiol.1973.sp010248
Kawaguchi, Y. (1997). Selective cholinergic modulation of cortical GABAergic cell subtypes. J. Neurophysiol. 78, 1743–1747. doi: 10.1152/jn.1997.78.3.1743
Kawaguchi, Y., and Kubota, Y. (1997). GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb. Cortex 7, 476–486. doi: 10.1093/cercor/7.6.476
Kennedy, H., and Bullier, J. (1985). A double-labeling investigation of the afferent connectivity to cortical areas V1 and V2 of the macaque monkey. J. Neurosci. 5, 2815–2830. doi: 10.1523/JNEUROSCI.05-10-02815.1985
Kim, J.-H., Jung, A.-H., Jeong, D., Choi, I., Kim, K., Shin, S., et al. (2016). Selectivity of neuromodulatory projections from the basal forebrain and locus ceruleus to primary sensory cortices. J. Neurosci. 36, 5314–5327. doi: 10.1523/JNEUROSCI.4333-15.2016
Kimura, F., Fukuda, M., and Tsumoto, T. (1999). Acetylcholine suppresses the spread of excitation in the visual cortex revealed by optical recording: possible differential effect depending on the source of input. Eur. J. Neurosci. 11, 3597–3609. doi: 10.1046/j.1460-9568.1999.00779.x
Kocharyan, A., Fernandes, P., Tong, X.-K., Vaucher, E., and Hamel, E. (2008). Specific subtypes of cortical GABA interneurons contribute to the neurovascular coupling response to basal forebrain stimulation. J. Cereb. Blood Flow Metab. 28, 221–231. doi: 10.1038/sj.jcbfm.9600558
Koukouli, F., Rooy, M., Tziotis, D., Sailor, K. A., O’Neill, H. C., Levenga, J., et al. (2017). Nicotine reverses hypofrontality in animal models of addiction and schizophrenia. Nat. Med. 23, 347–354. doi: 10.1038/nm.4274
Krnjević, K. (2004). Synaptic mechanisms modulated by acetylcholine in cerebral cortex. Prog. Brain Res. 145, 81–93. doi: 10.1016/s0079-6123(03)45005-x
Kruglikov, I., and Rudy, B. (2008). Perisomatic GABA release and thalamocortical integration onto neocortical excitatory cells are regulated by neuromodulators. Neuron 58, 911–924. doi: 10.1016/j.neuron.2008.04.024
Kuryatov, A., Berrettini, W., and Lindstrom, J. (2011). Acetylcholine receptor (AChR) a5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (a4β2)2a5 AChR function. Mol. Pharmacol. 79, 119–125. doi: 10.1124/mol.110.066357
Lee, S.-H., and Dan, Y. (2012). Neuromodulation of brain states. Neuron 76, 209–222. doi: 10.1016/j.neuron.2012.09.012
Letzkus, J. J., Wolff, S. B. E., Meyer, E. M. M., Tovote, P., Courtin, J., Herry, C., et al. (2011). A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480, 331–335. doi: 10.1038/nature10674
Levey, A. I., Kitt, C. A., Simonds, W. F., Price, D. L., and Brann, M. R. (1991). Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 11, 3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991
Levy, R. B., Reyes, A. D., and Aoki, C. (2006). Nicotinic and muscarinic reduction of unitary excitatory postsynaptic potentials in sensory cortex; dual intracellular recording in vitro. J. Neurophysiol. 95, 2155–2166. doi: 10.1152/jn.00603.2005
Li, P., and Steinbach, J. H. (2010). The neuronal nicotinic alpha4beta2 receptor has a high maximal probability of being open. Br. J. Pharmacol. 160, 1906–1915. doi: 10.1111/j.1476-5381.2010.00761.x
Lin, S.-C., Brown, R. E., Shuler, M. G. H., Petersen, C. C. H., and Kepecs, A. (2015). Optogenetic dissection of the basal forebrain neuromodulatory control of cortical activation, plasticity, and cognition. J. Neurosci. 35, 13896–13903. doi: 10.1523/JNEUROSCI.2590-15.2015
Little, J. T., Johnson, D. N., Minichiello, M., Weingartner, H., and Sunderland, T. (1998). Combined nicotinic and muscarinic blockade in elderly normal volunteers: cognitive, behavioral, and physiologic responses. Neuropsychopharmacology 19, 60–69. doi: 10.1016/s0893-133x(98)00002-5
Loewi, O. (1924). Über humorale Übertragbarkeit der Herznervenwirkung: V. Mitteilung. Die Übertragbarkeit der negativ chrono- und dromotropen Vaguswirkung. Pflüg. Arch. Für Gesamte Physiol. Menschen Tiere 204, 629–640. doi: 10.1007/BF01731235
Lysakowski, A., Wainer, B. H., Bruce, G., and Hersh, L. B. (1989). An atlas of the regional and laminar distribution of choline acetyltransferase immunoreactivity in rat cerebral cortex. Neuroscience 28, 291–336. doi: 10.1016/0306-4522(89)90180-2
Mancarci, B. O., Toker, L., Tripathy, S. J., Li, B., Rocco, B., Sibille, E., et al. (2017). Cross-laboratory analysis of brain cell type transcriptomes with applications to interpretation of bulk tissue data. eNeuro 4:ENEURO.0212–17.2017. doi: 10.1523/eneuro.0212-17.2017
Marchi, M., and Grilli, M. (2010). Presynaptic nicotinic receptors modulating neurotransmitter release in the central nervous system: functional interactions with other coexisting receptors. Prog. Neurobiol. 92, 105–111. doi: 10.1016/j.pneurobio.2010.06.004
Markram, H., Lübke, J., Frotscher, M., Roth, A., and Sakmann, B. (1997). Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J. Physiol. 500, 409–440. doi: 10.1113/jphysiol.1997.sp022031
Mathes, C., and Thompson, S. H. (1996). The nitric oxide/cGMP pathway couples muscarinic receptors to the activation of Ca2+ influx. J. Neurosci. 16, 1702–1709. doi: 10.1523/JNEUROSCI.16-05-01702.1996
McCormick, D. A. (1992). Cellular mechanisms underlying cholinergic and noradrenergic modulation of neuronal firing mode in the cat and guinea pig dorsal lateral geniculate nucleus. J. Neurosci. 12, 278–289. doi: 10.1523/JNEUROSCI.12-01-00278.1992
McCormick, D. A., and Prince, D. A. (1985). Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc. Natl. Acad. Sci. U S A 82, 6344–6348. doi: 10.1073/pnas.82.18.6344
Meir, I., Katz, Y., and Lampl, I. (2018). Membrane potential correlates of network decorrelation and improved SNR by cholinergic activation in the somatosensory cortex. J. Neurosci. 38, 10692–10708. doi: 10.1523/JNEUROSCI.1159-18.2018
Metherate, R., and Hsieh, C. Y. (2004). Synaptic mechanisms and cholinergic regulation in auditory cortex. Prog. Brain Res. 145, 143–156. doi: 10.1016/s0079-6123(03)45010-3
Metherate, R., and Weinberger, N. M. (1990). Cholinergic modulation of responses to single tones produces tone-specific receptive field alterations in cat auditory cortex. Synapse 6, 133–145. doi: 10.1002/syn.890060204
Minces, V., Pinto, L., Dan, Y., and Chiba, A. A. (2017). Cholinergic shaping of neural correlations. Proc. Natl. Acad. Sci. U S A 114, 5725–5730. doi: 10.1073/pnas.1621493114
Moruzzi, G., and Magoun, H. W. (1949). Brain stem reticular formation and activation of the EEG. Electroencephalogr. Clin. Neurophysiol. 1, 455–473. doi: 10.1016/0013-4694(49)90066-8
Mrzijak, L., Pappy, M., Leranth, C., and Goldman-Rakic, P. S. (1995). Cholinergic synaptic circuitry in the macaque prefrontal cortex. J. Comp. Neurol. 357, 603–617. doi: 10.1002/cne.903570409
Mrzljak, L., Levey, A. I., and Goldman-Rakic, P. S. (1993). Association of m1 and m2 muscarinic receptor proteins with asymmetric synapses in the primate cerebral cortex: morphological evidence for cholinergic modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U S A 90, 5194–5198. doi: 10.1073/pnas.90.11.5194
Muñoz, W., and Rudy, B. (2014). Spatiotemporal specificity in cholinergic control of neocortical function. Curr. Opin. Neurobiol. 26, 149–160. doi: 10.1016/j.conb.2014.02.015
Muñoz, W., Tremblay, R., Levenstein, D., and Rudy, B. (2017). Layer-specific modulation of neocortical dendritic inhibition during active wakefulness. Science 355, 954–959. doi: 10.1126/science.aag2599
Nashmi, R., and Lester, H. A. (2006). CNS localization of neuronal nicotinic receptors. J. Mol. Neurosci. 30, 181–184. doi: 10.1385/jmn:30:1:181
Newman, E. L., Gupta, K., Climer, J. R., Monaghan, C. K., and Hasselmo, M. E. (2012). Cholinergic modulation of cognitive processing: insights drawn from computational models. Front. Behav. Neurosci. 6:24. doi: 10.3389/fnbeh.2012.00024
Nguyen, Q., Sapp, D. W., Van Ness, P. C., and Olsen, R. W. (1995). Modulation of GABAA receptor binding in human brain by neuroactive steroids: species and brain regional differences. Synapse 19, 77–87. doi: 10.1002/syn.890190203
Nuñez, A., Domínguez, S., Buño, W., and Fernández de Sevilla, D. (2012). Cholinergic-mediated response enhancement in barrel cortex layer V pyramidal neurons. J. Neurophysiol. 108, 1656–1668. doi: 10.1152/jn.00156.2012
Obermayer, J., Heistek, T. S., Kerkhofs, A., Goriounova, N. A., Kroon, T., Baayen, J. C., et al. (2018). Lateral inhibition by Martinotti interneurons is facilitated by cholinergic inputs in human and mouse neocortex. Nat. Commun. 9:4101. doi: 10.1038/s41467-018-06628-w
Obermayer, J., Verhoog, M. B., Luchicchi, A., and Mansvelder, H. D. (2017). Cholinergic modulation of cortical microcircuits is layer-specific: evidence from rodent, monkey and human brain. Front. Neural Circuits 11:100. doi: 10.3389/fncir.2017.00100
Pawlak, V., Wickens, J. R., Kirkwood, A., and Kerr, J. N. (2010). Timing is not everything: neuromodulation opens the STDP gate. Front. Synaptic Neurosci. 2:146. doi: 10.3389/fnsyn.2010.00146
Poorthuis, R. B., Bloem, B., Schak, B., Wester, J., de Kock, C. P. J., and Mansvelder, H. D. (2013). Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb. Cortex 23, 148–161. doi: 10.1093/cercor/bhr390
Poorthuis, R. B., Enke, L., and Letzkus, J. J. (2014). Cholinergic circuit modulation through differential recruitment of neocortical interneuron types during behaviour. J. Physiol. 592, 4155–4164. doi: 10.1113/jphysiol.2014.273862
Poorthuis, R. B., Muhammad, K., Wang, M., Verhoog, M. B., Junek, S., Wrana, A., et al. (2018). Rapid neuromodulation of layer 1 interneurons in human neocortex. Cell Rep. 23, 951–958. doi: 10.1016/j.celrep.2018.03.111
Radnikow, G., and Feldmeyer, D. (2018). Layer- and cell type-specific modulation of excitatory neuronal activity in the neocortex. Front. Neuroanat. 12:1. doi: 10.3389/fnana.2018.00001
Ramaswamy, S., Colangelo, C., and Markram, H. (2018). Data-driven modeling of cholinergic modulation of neural microcircuits: bridging neurons, synapses and network activity. Front. Neural Circuits 12:77. doi: 10.3389/fncir.2018.00077
Rasmusson, D. (2000). The role of acetylcholine in cortical synaptic plasticity. Behav. Brain Res. 115, 205–218. doi: 10.1016/s0166-4328(00)00259-x
Rovira, J. C., Ballesta, J. J., Vicente-Agulló, F., Campos-Caro, A., Criado, M., Sala, F., et al. (1998). A residue in the middle of the M2–M3 loop of the beta4 subunit specifically affects gating of neuronal nicotinic receptors. FEBS Lett. 433, 89–92. doi: 10.1016/s0014-5793(98)00889-8
Rudy, B., Fishell, G., Lee, S., and Hjerling-Leffler, J. (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61. doi: 10.1002/dneu.20853
Runfeldt, M. J., Sadovsky, A. J., and MacLean, J. N. (2014). Acetylcholine functionally reorganizes neocortical microcircuits. J. Neurophysiol. 112, 1205–1216. doi: 10.1152/jn.00071.2014
Sabri, O., Kendziorra, K., Wolf, H., Gertz, H.-J., and Brust, P. (2008). Acetylcholine receptors in dementia and mild cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging 35, S30–S45. doi: 10.1007/s00259-007-0701-1
Salgado, H., Bellay, T., Nichols, J. A., Bose, M., Martinolich, L., Perrotti, L., et al. (2007). Muscarinic M2 and M1 receptors reduce GABA release by Ca2+ channel modulation through activation of PI3K/Ca2+ -independent and PLC/Ca2+ -dependent PKC. J. Neurophysiol. 98, 952–965. doi: 10.1152/jn.00060.2007
Saper, C. B., Chou, T. C., and Scammell, T. E. (2001). The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 24, 726–731. doi: 10.1016/s0166-2236(00)02002-6
Saper, C. B., Fuller, P. M., Pedersen, N. P., Lu, J., and Scammell, T. E. (2010). Sleep state switching. Neuron 68, 1023–1042. doi: 10.1016/j.neuron.2010.11.032
Sarter, M., Parikh, V., and Howe, W. M. (2009). Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat. Rev. Neurosci. 10, 383–390. doi: 10.1038/nrn2635
Saunders, A., Granger, A. J., and Sabatini, B. L. (2015). Corelease of acetylcholine and GABA from cholinergic forebrain neurons. Elife 4:e06412. doi: 10.7554/elife.06412
Saunders, A., Macosko, E. Z., Wysoker, A., Goldman, M., Krienen, F. M., de Rivera, H., et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015.e16–1030.e16. doi: 10.1016/j.cell.2018.07.028
Schmiedeberg, O., and Koppe, R. (1869). Das Muscarin. Das Giftige Alkaloid des Fliegenpilzes. Leipzig: Vogel.
Schröder, H. (1992). Immunohistochemistry of cholinergic receptors. Anat. Embryol. 186, 407–429. doi: 10.1007/bf00185457
Schröder, H., Zilles, K., Luiten, P. G. M., and Strosberg, A. (1990). Immunocytochemical visualization of muscarinic cholinoceptors in the human cerebral cortex. Brain Res. 514, 249–258. doi: 10.1016/0006-8993(90)91420-l
Schröder, H., Zilles, K., Maelicke, A., and Hajós, F. (1989). Immunohisto- and cytochemical localization of cortical nicotinic cholinoceptors in rat and man. Brain Res. 502, 287–295. doi: 10.1016/0006-8993(89)90624-0
Shalinsky, M. H., Magistretti, J., Ma, L., and Alonso, A. A. (2002). Muscarinic activation of a cation current and associated current noise in entorhinal-cortex layer-II neurons. J. Neurophysiol. 88, 1197–1211. doi: 10.1152/jn.2002.88.3.1197
Shen, J., and Yakel, J. L. (2009). Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 30, 673–680. doi: 10.1038/aps.2009.64
Smiley, J. F., Morrell, F., and Mesulam, M. M. (1997). Cholinergic synapses in human cerebral cortex: an ultrastructural study in serial sections. Exp. Neurol. 144, 361–368. doi: 10.1006/exnr.1997.6413
Sparks, D. W., Tian, M. K., Sargin, D., Venkatesan, S., Intson, K., and Lambe, E. K. (2018). Opposing cholinergic and serotonergic modulation of layer 6 in prefrontal cortex. Front. Neural Circuits 11:107. doi: 10.3389/fncir.2017.00107
Steriade, M., Amzica, F., and Nunez, A. (1993). Cholinergic and noradrenergic modulation of the slow (approximately 0.3 Hz) oscillation in neocortical cells. J. Neurophysiol. 70, 1385–1400. doi: 10.1152/jn.1993.70.4.1385
Stetzer, E., Ebbinghaus, U., Storch, A., Poteur, L., Schrattenholz, A., Kramer, G., et al. (1996). Stable expression in HEK-293 cells of the rat alpha3/beta4 subtype of neuronal nicotinic acetylcholine receptor. FEBS Lett. 397, 39–44. doi: 10.1016/s0014-5793(96)01115-5
Stewart, A. E., Yan, Z., Surmeier, D. J., and Foehring, R. C. (1999). Muscarine modulates Ca2+ channel currents in rat sensorimotor pyramidal cells via two distinct pathways. J. Neurophysiol. 81, 72–84. doi: 10.1152/jn.1999.81.1.72
Sugihara, H., Chen, N., and Sur, M. (2016). Cell-specific modulation of plasticity and cortical state by cholinergic inputs to the visual cortex. J. Physiol. Paris 110, 37–43. doi: 10.1016/j.jphysparis.2016.11.004
Szymusiak, R., and McGinty, D. (1986). Sleep suppression following kainic acid-induced lesions of the basal forebrain. Exp. Neurol. 94, 598–614. doi: 10.1016/0014-4886(86)90240-2
Teles-Grilo Ruivo, L. M., and Mellor, J. R. (2013). Cholinergic modulation of hippocampal network function. Front. Synaptic Neurosci. 5:2. doi: 10.3389/fnsyn.2013.00002
Thiele, A., Herrero, J. L., Distler, C., and Hoffmann, K.-P. (2012). Contribution of cholinergic and GABAergic mechanisms to direction tuning, discriminability, response reliability and neuronal rate correlations in macaque middle temporal area. J. Neurosci. 32, 16602–16615. doi: 10.1523/JNEUROSCI.0554-12.2012
Tribollet, E., Bertrand, D., Marguerat, A., and Raggenbass, M. (2004). Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience 124, 405–420. doi: 10.1016/j.neuroscience.2003.09.028
Tsodyks, M. V., and Markram, H. (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. U S A 94, 719–723. doi: 10.1073/pnas.94.2.719
Turrini, P., Casu, M. A., Wong, T. P., De Koninck, Y., Ribeiro-da-Silva, A., and Cuello, A. C. (2001). Cholinergic nerve terminals establish classical synapses in the rat cerebral cortex: synaptic pattern and age-related atrophy. Neuroscience 105, 277–285. doi: 10.1016/s0306-4522(01)00172-5
Umbriaco, D., Watkins, K. C., Descarries, L., Cozzari, C., and Hartman, B. K. (1994). Ultrastructural and morphometric features of the acetylcholine innervation in adult rat parietal cortex: an electron microscopic study in serial sections. J. Comp. Neurol. 348, 351–373. doi: 10.1002/cne.903480304
Unal, C. T., Golowasch, J. P., and Zaborszky, L. (2012). Adult mouse basal forebrain harbors two distinct cholinergic populations defined by their electrophysiology. Front. Behav. Neurosci. 6:21. doi: 10.3389/fnbeh.2012.00021
Urban-Ciecko, J., Jouhanneau, J.-S., Myal, S. E., Poulet, J. F. A., and Barth, A. L. (2018). Precisely timed nicotinic activation drives SST inhibition in neocortical circuits. Neuron 97, 611–625.e5. doi: 10.1016/j.neuron.2018.01.037
Van der Zee, E. A., and Luiten, P. G. (1999). Muscarinic acetylcholine receptors in the hippocampus, neocortex and amygdala: a review of immunocytochemical localization in relation to learning and memory. Prog. Neurobiol. 58, 409–471. doi: 10.1016/s0301-0082(98)00092-6
van der Zee, E. A., Streefland, C., Strosberg, A. D., Schröder, H., and Luiten, P. G. (1992). Visualization of cholinoceptive neurons in the rat neocortex: colocalization of muscarinic and nicotinic acetylcholine receptors. Mol. Brain Res. 14, 326–336. doi: 10.1016/0169-328x(92)90100-p
Venter, J. C., di Porzio, U., Robinson, D. A., Shreeve, S. M., Lai, J., Kerlavage, A. R., et al. (1988). Evolution of neurotransmitter receptor systems. Prog. Neurobiol. 30, 105–169. doi: 10.1016/0301-0082(88)90004-4
Vidal, C., and Changeux, J.-P. (1993). Nicotinic and muscarinic modulations of excitatory synaptic transmission in the rat prefrontal cortexin vitro. Neuroscience 56, 23–32. doi: 10.1016/0306-4522(93)90558-w
von Engelhardt, J., Eliava, M., Meyer, A. H., Rozov, A., and Monyer, H. (2007). Functional characterization of intrinsic cholinergic interneurons in the cortex. J. Neurosci. 27, 5633–5642. doi: 10.1523/JNEUROSCI.4647-06.2007
Wang, Z., Kai, L., Day, M., Ronesi, J., Yin, H. H., Ding, J., et al. (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50, 443–452. doi: 10.1016/j.neuron.2006.04.010
Wevers, A. (2011). Localisation of pre- and postsynaptic cholinergic markers in the human brain. Behav. Brain Res. 221, 341–355. doi: 10.1016/j.bbr.2010.02.025
Xu, M., Chung, S., Zhang, S., Zhong, P., Ma, C., Chang, W.-C., et al. (2015). Basal forebrain circuit for sleep-wake control. Nat. Neurosci. 18, 1641–1647. doi: 10.1038/nn.4143
Yamamoto, K., Koyanagi, Y., Koshikawa, N., and Kobayashi, M. (2010). Postsynaptic cell type-dependent cholinergic regulation of GABAergic synaptic transmission in rat insular cortex. J. Neurophysiol. 104, 1933–1945. doi: 10.1152/jn.00438.2010
Yamasaki, M., Matsui, M., and Watanabe, M. (2010). Preferential localization of muscarinic M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J. Neurosci. 30, 4408–4418. doi: 10.1523/JNEUROSCI.5719-09.2010
Zaborszky, L., and Duque, A. (2000). Local synaptic connections of basal forebrain neurons. Behav. Brain Res. 115, 143–158. doi: 10.1016/s0166-4328(00)00255-2
Zaborszky, L., Duque, A., Gielow, M., Gombkoto, P., Nadasdy, Z., and Somogyi, J. (2015). “Organization of the basal forebrain cholinergic projection system,” in The Rat Nervous System, ed. G. Paxinos (Amsterdam: Elsevier Inc.), 491–507.
Zeisel, A., Hochgerner, H., Lönnerberg, P., Johnsson, A., Memic, F., van der Zwan, J., et al. (2018). Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22. doi: 10.1016/j.cell.2018.06.021
Zhang, Z., and Séguéla, P. (2010). Metabotropic induction of persistent activity in layers II/III of anterior cingulate cortex. Cereb. Cortex 20, 2948–2957. doi: 10.1093/cercor/bhq043
Zoli, M., Moretti, M., Zanardi, A., McIntosh, J. M., Clementi, F., and Gotti, C. (2002). Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J. Neurosci. 22, 8785–8789. doi: 10.1523/jneurosci.22-20-08785.2002
Zoli, M., Pistillo, F., and Gotti, C. (2015). Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96, 302–311. doi: 10.1016/j.neuropharm.2014.11.003
Keywords: neuromodulation, acetylcholine, Ach receptors, neocortex, cellular excitability, synaptic transmission, network activity
Citation: Colangelo C, Shichkova P, Keller D, Markram H and Ramaswamy S (2019) Cellular, Synaptic and Network Effects of Acetylcholine in the Neocortex. Front. Neural Circuits 13:24. doi: 10.3389/fncir.2019.00024
Received: 28 January 2019; Accepted: 22 March 2019;
Published: 12 April 2019.
Edited by:
Robert C. Froemke, New York University, United StatesReviewed by:
Heiko J. Luhmann, Johannes Gutenberg University Mainz, GermanyCopyright © 2019 Colangelo, Shichkova, Keller, Markram and Ramaswamy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Colangelo, Y3Jpc3RpbmEuY29sYW5nZWxvQGVwZmwuY2g=
Srikanth Ramaswamy, c3Jpa2FudGgucmFtYXN3YW15QGVwZmwuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.