 Kiyoshi Egawa
Kiyoshi Egawa Atsuo Fukuda
Atsuo Fukuda
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neural Circuits , 24 October 2013
Volume 7 - 2013 | https://doi.org/10.3389/fncir.2013.00170
This article is part of the Research Topic Pharmacology, physiology and regulation of tonic GABAA conductances View all 17 articles
High-affinity extrasynaptic gamma-aminobutyric acid A (GABAA) receptors are tonically activated by low and consistent levels of ambient GABA, mediating chronic inhibition against neuronal excitability (tonic inhibition) and the modulation of neural development. Synaptic (phasic) inhibition is spatially and temporally precise compared with tonic inhibition, which provides blunt yet strong integral inhibitory force by shunting electrical signaling. Although effects of acute modification of tonic inhibition are known, its pathophysiological significance remains unclear because homeostatic regulation of neuronal excitability can compensate for long-term deficit of extrasynaptic GABAA receptor activation. Nevertheless, tonic inhibition is of great interest for its pathophysiological involvement in central nervous system (CNS) diseases and thus as a therapeutic target. Together with the development of experimental models for various pathological states, recent evidence demonstrates such pathological involvements of tonic inhibition in neuronal dysfunction. This review focuses on the recent progress of tonic activation of GABAA conductance on the development and pathology of the CNS. Findings indicate that neuronal function in various brain regions are exacerbated with a gain or loss of function of tonic inhibition by GABA spillover. Disturbance of tonic GABAA conductance mediated by non-synaptic ambient GABA may result in brain mal-development. Therefore, various pathological states (epilepsy, motor dysfunctions, psychiatric disorders, and neurodevelopmental disorders) may be partly attributable to abnormal tonic GABAA conductances. Thus, the tone of tonic conductance and level of ambient GABA may be precisely tuned to maintain the regular function and development of the CNS. Therefore, receptor expression and factors for regulating the ambient GABA concentration are highlighted to gain a deeper understanding of pathology and therapeutic strategy for CNS diseases.
Neurotransmission comprises both excitatory and inhibitory signals. Therefore, understanding the mechanisms underlying the imbalance of these signals that are found in pathologies of the central nervous systems (CNSs) are vital in pursuing specific therapeutic strategies. In inhibitory neurotransmitter systems, gamma-aminobutyric acid (GABA) A receptor (GABAA)-mediated synaptic transmission is particularly important because its fast, ligand-gated inhibitory conductance allows fine homeostatic tuning of excitation-inhibition balance (see review Farrant and Nusser, 2005; Olsen and Sieghart, 2008).
Accumulating evidence has revealed that the function and distribution of the GABAA receptor are remarkably diverse because of their subunit assembly. Some GABAA receptor isoforms have been shown to be expressed outside synapses, and tonically activated by low concentrations of GABA (i.e., tonic inhibition) existing in the extrasynaptic space (Mody and Pearce, 2004; Farrant and Nusser, 2005). Although the proportion of GABAA receptor subtypes mediating tonic inhibition is estimated to be of minor abundance as compared with that mediating synaptic inhibition (Mohler et al., 2002), this effect nevertheless provides strong inhibitory force by shunting the electrical signal transmission. With increased knowledge about its functional significance, an alteration in tonic inhibition has received great attention as a mechanism underlying the pathophysiology of various CNS disorders. Its distinct properties and pharmacology may thus provide new therapeutic strategies for overcoming limitations of conventional GABAA receptor modulators. Thus far, deregulation of tonic inhibition has been shown to be a mechanism responsible for underlying a variety of CNS pathologies, including epilepsy, neurodevelopmental disorders, cognitive dysfunctions, and psychiatric disorders (see review, Brickley and Mody, 2012; Hines et al., 2012).
In addition to tonic inhibition induced by GABA generated by synaptic spillover, another form of tonic conductance occurs before synaptic formation. This tonic conductance is depolarizing because the intracellular chloride (Cl-) concentration is high in immature neurons due to the balance of Cl- transporters (Owens et al., 1996; Yamada et al., 2004; for review, see Ben-Ari, 2002). The release of GABA in the milieu occurs via non-vesicular mechanisms (Demarque et al., 2002; Manent et al., 2005; for review, see Owens and Kriegstein, 2002). This tonic GABAA receptor-mediated conductance is considered to be involved in a variety of developmental events, such as neurogenesis (LoTurco et al., 1995; Haydar et al., 2000; Andäng et al., 2008), migration (Behar et al., 1996, 1998, 2000, 2001; López-Bendito et al., 2003; Cuzon et al., 2006; Heck et al., 2007; Bortone and Polleux, 2009; Denter et al., 2010; Inada et al., 2011; Inoue et al., 2012) and synaptogenesis (Nakanishi et al., 2007; Wang and Kriegstein, 2008). Based on these findings, perturbation of tonic depolarization could also result in brain mal-development.
In this review, we provide an overview on the advancement in knowledge about the role of dysregulation of tonic inhibition in the pathophysiology of various CNS diseases (with the exception of epilepsy, which will be reviewed independently in this issue), and its involvement in their therapeutic strategies. Furthermore, we review the latest findings of tonic depolarization underlying brain mal-development (with the exception of the hippocampus, which will be reviewed independently in this issue).
Gamma-aminobutyric acid A receptors are assembled from a family of 19 homologous subunit gene products (six α subunits, three β subunits, three γ subunits, three ρ subunits, and one each of the ε, δ, θ, and π subunits) and form mostly hetero-oligomeric pentamers (for review, see Olsen and Sieghart, 2008). Most GABAA receptor subtypes are formed from two copies of a single α, two copies of a single β, and one copy of another subunit (γ, δ, or ε). Each subunit combination has a distinct distribution pattern in terms of subcellular domains. The δ subunit, generally partnered with the α4 (forebrain predominant) and α6 (cerebellum predominant) subunits, are shown to be exclusively localized in extrasynaptic membranes in various neuronal cells, including cerebellar granule cells (CGCs), dentate gyrus granule cells, neocortical layer 2/3 pyramidal cells (Nusser et al., 1998; Nusser and Mody, 2002; Wei et al., 2003), interneurons in the neocortex and hippocampus (Semyanov et al., 2003; Krook-Magnuson et al., 2008), thalamic relay neurons (Cope et al., 2005), medium spiny neurons of the striatum (Ade et al., 2008), and dorsal horn spinal neurons (Bonin et al., 2011). The δ subunit-containing GABAA (δ-GABAA) receptor shows high affinity and slow desensitization to GABA (Saxena and MacDonald, 1996), which allows tonic activation in response to low concentration of ambient GABA in extrasynaptic space (though see Bright et al., 2011).
The α5 subunit-containing GABAA receptors, which presumably do not include the δ subunit, are also expressed in extrasynaptic membranes. The α5 subunit is predominantly detected in hippocampal pyramidal neurons (Sperk et al., 1997), showing higher GABA sensitivity compared with α1 GABAA receptors, which allows tonic inhibition in hippocampal pyramidal neurons (Caraiscos et al., 2004; Glykys and Mody, 2006). The α1 and α5 subunit-mediated tonic inhibition is also shown in cortical principal neurons in layer 2/3 and layer 5, respectively (Yamada et al., 2007). In line with the predominant distribution of extrasynaptic α5 subunit-containing GABAA receptors (Brunig et al., 2002; Crestani et al., 2002; Caraiscos et al., 2004), perisynaptic α5 subunits also mediate the GABA spillover component of slow phasic inhibitory currents (Prenosil et al., 2006). The contribution of such a slow inhibitory synapse transmission to the signal computation may be a future issue to explore with further studies.
The other assemblies (e.g., α1γ2 subunit-containing receptor), which mainly mediate synaptic inhibition, also exist in the extrasynaptic membrane (Kasugai et al., 2010) and can contribute to tonic inhibition because their specific agonist, zolpidem, amplifies the tonic currents (Semyanov et al., 2003; Yamada et al., 2007). High affinity αβ subunit-containing GABAA receptors have also been shown to mediate tonic inhibition in rat hippocampal pyramidal neurons (Mortensen and Smart, 2006). Nevertheless, it is now clear that the GABAA receptor subtypes having the δ or α5 subunit are the dominant forms which are responsible for mediating tonic inhibition in the mammalian brain, including humans (Scimemi et al., 2006).
After the finding of tonic inhibition in the mature brain (Kaneda et al., 1995), its functional significance for regulating network excitability was subsequently revealed. In CGCs, blocking tonic inhibition decreases membrane shunting (Brickley et al., 1996) and increases information flow from granule cells to Purkinje cells (Hamann et al., 2002). In thalamic relay neurons or hippocampal interneurons, baseline membrane potentials and network oscillation are modulated (Cope et al., 2005; Song et al., 2011). Deregulation of tonic inhibition caused by a lack of synaptic plasticity-associated proteins has been shown in several mice models of neurological disorders (Curia et al., 2009; Olmos-Serrano et al., 2010; Egawa et al., 2012). However, the pathophysiological significance of long-term modification of extrasynaptic GABAA receptor activation can be masked by compensatory mechanisms (Brickley et al., 2001; Wisden et al., 2002). Some adaptive regulation responses could be attributed to the modification of GABAA receptor trafficking and/or clustering on the membrane surface (Saliba et al., 2012; see review Hines et al., 2012).
Progress in the development of pharmacological agents that predominantly modulate extrasynaptic GABAA receptors (summarized in Table 1) has contributed to the uncovering of the pathological significance of dysregulated tonic inhibition. Consequently, large efforts have been made to use these agents for clinical use. The negative allosteric modulator of the α5 GABAA receptor, α5IA, has recently been shown to improve ethanol-induced impaired performance in healthy subjects (Nutt et al., 2007). However, its use in a clinical trial was stopped due to renal toxicity (Atack, 2010). Another negative allosteric modulator of the α5 subunit, RO4938581, is presently under a phase 1 clinical trial for Down syndrome (the study is sponsored by Roche, and its outcomes have been made publicly available). The selective orthosteric agonist of the δ-GABAA receptor, 4,5,6,7-tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP, or also known as gaboxadol) was used as an hypnotic drug, reaching a phase 3 trial before its cessation because of side-effects, such as hallucination and disorientation (the study is sponsored by Merck, and its outcomes have been made publicly available). However, THIP has been recently shown to be effective against behavioral and motor dysfunction in mouse models of autism spectrum disorders (Olmos-Serrano et al., 2011; Egawa et al., 2012). Therefore, further cumulative evidence may hopefully renew an interest in using THIP for these diseases, which have no effective therapeutic strategies.
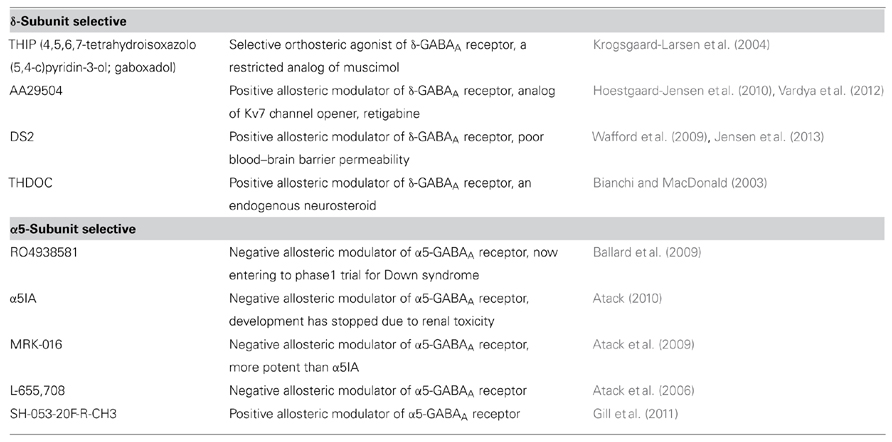
TABLE 1. Selective modulators for extrasynaptic GABAA receptor.
Because extrasynaptic GABAA receptors are activated by ambient GABA in the extrasynaptic space, regulation of ambient GABA concentrations must therefore be an important factor for determining the degree of tonic inhibition. The concentration of ambient GABA has been estimated to be very low, less than 300 nM in both in vivo (Kennedy et al., 2002) and in vitro (Wu et al., 2003; Santhakumar et al., 2006) conditions. A considerable part of ambient GABA may be derived from spillover from the synaptic cleft (Glykys and Mody, 2007). Thus, presynaptic functions, such as GABA synthesis and GABA release, should also regulate tonic GABA conductance. To date, reverse mode operation of GABA transporters (Wu et al., 2003; Heja et al., 2012) and GABA release via bestrophin1 anion channel (Lee et al., 2010) have been proposed as mechanisms underlying the non-vesicular release of GABA, both of which are currently under debate (Diaz et al., 2011; Kersante et al., 2013).
Regardless of the source of ambient GABA, GABA transporters play a pivotal role in regulating tonic inhibition. In the rat hippocampus, presynaptically located GABA transporter 1 (GAT1) and astrocytic GAT3 (corresponding to GAT4 in mice) synergistically modulate the ambient concentration of GABA via its uptake from the extrasynaptic space (Egawa et al., 2013; Kersante et al., 2013; Song et al., 2013). In vivo analysis has shown that neuronal GAT1 plays a predominant role under resting conditions and that the uptake of GABA by astrocytic GAT3 occurs during membrane depolarization (Kersante et al., 2013). In general, GAT1 and GAT3 is expressed in presynaptic neurons and astrocytes, respectively. However, this expression pattern is not consistent throughout the brain regions. For example, thalamic GAT1 and GAT3 are exclusively expressed in astrocytes, but not in presynaptic neurons (De Biasi et al., 1998), and thus the regulatory mechanisms for ambient GABA in the brain can differ depending on the region. Recently, compromised function of glial GATs (associated with increased tonic inhibition), has been shown to underlie the pathophysiology of absence epilepsy (Cope et al., 2009) and prevent recovery after stroke (Clarkson et al., 2010). Insufficient (Chiu et al., 2005) or excessive (Egawa et al., 2012) amounts of GAT1 can cause cerebellar dysfunction, at least in part, to deregulation of tonic inhibition.
A Schematic drawing of the regulation of tonic inhibition with some implications for CNS disorders possibly associated with dysfunction of tonic inhibition (details will be shown below) is shown as Figure 1.
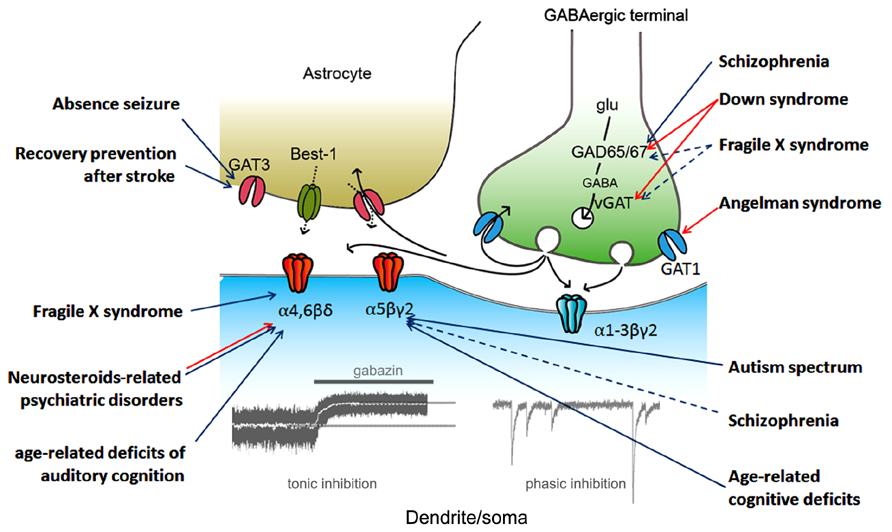
FIGURE 1. Regulation of tonic inhibition and CNS disorders. Aberrant tonic inhibition is suspected to be a mechanism underlying the neuronal dysfunctions in the listed disorders based on at least one of the following: (1) a direct measurement of tonic inhibition in model animals, (2) the use of extrasynaptic gamma-aminobutyric acid A (GABAA) receptor selective modulators in model animals, and (3) evaluation of α5 subunit expression in living human subjects (via positron emission tomography). Colored arrows indicate the proposed protein/subunit responsible for aberrant tonic inhibition (blue, down-regulation; red, up-regulation; black, GABA signaling during synaptic transmission). Dashed arrows indicate pathways still under debate. vGAT, vesicular GABA transporter; Best-1, bestrophin-1; glu, glutamate.
Tonic GABA conductances exist within ascending sensory pathways on each level, including spiny neurons of the dorsal horn in the spinal cord (Bonin et al., 2011), thalamic relay neurons (Cope et al., 2005; Richardson et al., 2011), granule cells of the olfactory bulb (Labarrera et al., 2013), the brain stem auditory pathway (Campos et al., 2001), and the primary sensory cortex (Yamada et al., 2007; Krook-Magnuson et al., 2008; Imbrosci et al., 2012). Tonic inhibition is thought to be important for sensory processing because it reduces the impact of low frequency excitation, thereby increasing its signal-to-noise ratio.
4,5,6,7-Tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP) acts as an anti-nociceptive in rodent models of acute pain (Enna and McCarson, 2006; Munro et al., 2008; Bonin et al., 2011). The anti-nociceptive effect is absent from δ- or α4-GABAA receptor null mutant mice (Chandra et al., 2006; Bonin et al., 2011), indicating that tonic inhibition plays major roles in the regulation of acute nociception. The effects of THIP have been attributed to actions at supraspinal targets, such as thalamic relay neurons (Zorn and Enna, 1985; Rode et al., 2005; Chandra et al., 2006). Tonic GABA currents mediated by the δ subunit also exist in the spinal cord dorsal horn (Bonin et al., 2011). Studies on δ subunit knock-out (KO) mice indicate that tonic inhibition likely regulates the late phase of nociceptor activity, thus corresponding to central sensitization (Bonin et al., 2011). This response may result from amplification of tonic inhibition caused by activity-dependent GABA spillover or induction of neurosteroids during early phase of nociception. Nociceptor activity in early phase was shown to be comparable between wild type (WT) and δ-GABAA receptor KO mice (Bonin et al., 2011). Therefore, a tonic inhibition in spinal cord neurons may also contribute to regulating central sensitization and acute nociception.
In the auditory thalamus, the medial geniculate body is essential for processing acoustic information. A recent study has demonstrated that age-related loss of inhibition in this area is more dominant in tonic rather than phasic inhibition (Richardson et al., 2013). Decreased expression of α4δ-GABAA receptors and down-regulation of glutamic acid decarboxylase (GAD) 67 have been found to be possible mechanisms underlying this difference (Richardson et al., 2013). Because the efficacy of auditory coding is directly correlated with the strength of GABAergic inhibition (Gleich et al., 2003; de Villers-Sidani et al., 2010), enhancement of tonic inhibition was proposed to be a potential strategy against age-related decline of auditory cognition. A reduction of tonic inhibition has been reported in the auditory cortex of hearing-lesioned rat (Yang et al., 2011). In this model, GAD67 was reduced in the affected region, resulting in down-regulation of both phasic and tonic inhibition. In addition, hearing-lesioned animals displayed tinnitus with a pitch in the hearing loss range, which was eliminated by the GABA transaminase inhibitor, vigabatrin, or by reducing the uptake of GABA by the GAT1 inhibitor, NO711.
Because cognition involves a group of mental processes such as memory, attention, understanding language, and learning, numerous brain regions are thus responsible for cognitive dysfunctions. The hippocampus plays a critical role in learning and memory, and recent evidence suggests that the α5-GABAA receptor is abundantly expressed in this brain region, and thus may be a therapeutic target for cognitive dysfunction (Sperk et al., 1997).
Down syndrome is caused by the trisomy for chromosome 21, and is the most frequent neurodevelopmental disorder of chromosomal origin. Almost all affected individuals exhibit cognitive impairment, with specific deficits in learning and memory mediated by hippocampal dysfunctions (Carlesimo et al., 1997). This phenotype is well replicated in the Ts65Dn mouse model of Down syndrome. The finding that low-dose picrotoxin restored hippocampal long-term potentiation in this model (Kleschevnikov et al., 2004) subsequently led to the hypothesis of “excessive inhibition,” and was strongly recognized to play a pathological role in this syndrome. In support of this hypothesis, Ts65Dn mice were found to exhibit increased immunoreactivity of GAD65/67 and vesicular GABA transporter (vGAT; Belichenko et al., 2009; Perez-Cremades et al., 2010; Martinez-Cue et al., 2013). Numerous studies indicate that α5-GABAA receptor-mediated inhibition (mainly in the tonic form) reduces learning and memory. Mice deficient in the α5 subunit show better performance of cognitive functions compared with wild-type (Collinson et al., 2002; Crestani et al., 2002; Yee et al., 2004). Similarly, rodents systemically administrated with various α5 subunit-selective negative allosteric modulators displayed a facilitation in learning and memory tasks, associated with increased long-term potentiation (Navarro et al., 2002; Atack et al., 2006, 2009; Dawson et al., 2006; Ballard et al., 2009). Therefore, specific antagonism of the α5 subunit may be a potential therapeutic strategy against cognitive impairment in Down syndrome. Indeed, recent studies have revealed that cognitive function is ameliorated with the α5 subunit-selective negative allosteric modulators, α5IA, or RO4938581, in Ts65Dn mice without any side effects (Braudeau et al., 2011; Martinez-Cue et al., 2013). RO4938581 is currently in clinical trials for adults with Down syndrome (the study is sponsored by Roche, and its outcomes have been made publicly available). The α5-selective negative allosteric modulators have also been shown as effective enhancers for learning and memory in other cognitive deficits induced by acute inflammation (Wang et al., 2012) or general anesthesia (Saab et al., 2010; Zurek et al., 2012) in mice, and by alcoholism in humans (Nutt et al., 2007; Atack, 2010).
In the aged brain, hippocampal tonic inhibition can regulate cognitive functions. Recent animal studies and human brain imaging of elderly individuals have indicated a strong positive correlation between hippocampal hyperactivity and cognitive impairment (Wilson et al., 2005; Ewers et al., 2011). In support of the “excessive excitation” hypothesis, the positive (and not negative) allosteric modulator for the α5 subunit improves memory tasks in aged rats with cognitive deficits (Wilson et al., 2005; Koh et al., 2013). This hypothesis may be applied to autism spectrum disorders because antiepileptic drugs sometimes improve cognitive functions in these affected individuals (Di Martino and Tuchman, 2001). α5 subunit selective allosteric modulators currently in clinical trials have shown promise as a potential therapeutic strategy for cognitive deficits in various CNS diseases.
Several reports have demonstrated that the inhibitory balance between phasic and tonic inhibition shifts to tonic after cortical lesion (Clarkson et al., 2010; Imbrosci et al., 2012). This shift may provide protection against cell death in the acute stage, and also prevent homeostatic plasticity during the recovery period. In a cortical stroke model, excessive tonic inhibition has been proposed as a novel pharmacological target for promoting recovery after stroke (Clarkson et al., 2010). Therefore, negative allosteric modulators for the α5 subunit may be effective against learning and memory deficits as well as for dysfunctions after brain injury.
Schizophrenia is generally recognized to develop from the disruption of various kinds of neurotransmission. Dysfunction in GABAergic transmission has drawn significant attention as a mechanism underlying cognitive deficits in schizophrenia (Guidotti et al., 2005; Lewis et al., 2005). Postmortem analyses and brain imaging of living schizophrenic subjects have revealed a deficiency in the synthesis of GABA resulting from reduced transcription of GAD67 (Akbarian et al., 1995b; Volk et al., 2000; Hashimoto et al., 2003; Yoon et al., 2010). Results of studies investigating the expression of GABAA receptor subunits in postmortem tissue have been inconsistent. The expression of α1, α2, and α5 subunits have been reported to be increased (Impagnatiello et al., 1998; Ishikawa et al., 2004; Lewis et al., 2004), decreased (Hashimoto et al., 2003), or unchanged (Akbarian et al., 1995a). Despite the limited number of reports investigating the expression of the δ subunit, several reports have shown decreased expression of this subunit in the prefrontal cortex (Hashimoto et al., 2003; Maldonado-Aviles et al., 2009). A considerable limitation of the postmortem tissue analyses in these studies is the chronic consumption of antipsychotics and/or benzodiazepines in the subjects. Furthermore, symptoms of schizophrenia dynamically change with the clinical stage. A recent study involving positron emission tomography (PET) using the highly selective α5 subunit ligand, [11C]Ro15-4513, revealed an inverse correlation between binding potential and severity of the negative symptoms in medication-free subjects (Asai et al., 2008). Therefore, this result suggests that decreased expression of the α5 subunit may be involved in the pathophysiology of schizophrenia during the negative stage. Supporting this hypothesis, use of the positive allosteric modulator for the α5 subunit has been shown to improve the hyperactivity and cognitive dysfunctions in rat models of schizophrenia (Damgaard et al., 2011; Gill et al., 2011). However, Redrobe et al. (2012) have shown that a negative allosteric modulator of the α5 subunit also attenuates cognitive deficits, using a different mouse model of schizophrenia. Interestingly, the authors also showed that hyperactivity was ameliorated by both positive and negative modulators for the α5 subunit in these mice. Excitatory and inhibitory biphasic changes induced by tonic conductance have been recently shown in single neurons with depolarizing reversal potential for GABA currents (Song et al., 2011). Therefore, an increase or decrease of tonic inhibition may result in net inhibitory effects in relevant neuronal circuits in this model. Further studies are required to validate the usefulness of α5 subunit selective allosteric modulators for the treatment of schizophrenia.
Neurosteroids, metabolites of progesterone, other sex hormones, and several stress-induced steroids are potent modulators of GABAA receptors (Lambert et al., 2003). In particular, the extrasynaptic δ-GABAA receptor shows higher sensitivity for neurosteroids compared with synaptic GABAA receptors (Davies et al., 1997; Stell et al., 2003; see review Brickley and Mody, 2012). Therefore, dysregulation of tonic inhibition may play a pivotal role in psychiatric disorders, such as anxiety and mood disorders, which are often associated with sex hormone alterations or stress. For example, expression of the δ subunit has been shown to decrease during pregnancy due to adaptive mechanisms against large elevation of progesterone metabolites (Maguire and Mody, 2008). The rapid decrease of neurosteroids after birth can thus induce the down-regulation of tonic inhibition, resulting in postpartum depression (Maguire and Mody, 2008).
In a rat model of premenstrual dystrophic disorders, withdrawal of progesterone leads to anxiety and increased seizure susceptibility, associated with up-regulation of the α4 subunit (Smith et al., 1998). In contrast, increased levels of neurosteroids during physiological ovarian cycles leads to reduction of anxiety and seizure susceptibility caused by up-regulation of the δ subunit (Maguire et al., 2005). This difference may thus suggest that neurosteroids can alter the effects of tonic GABA conductance by dose and/or exposure time-dependent mechanisms, thereby regulating the expression of receptor subunits. In accordance with this speculation, evaluating anxiety at puberty revealed that neurosteroids negatively modulated tonic inhibition by decreasing outward currents, via α4δ-GABAA receptors (Shen et al., 2007). This regulation was shown to contribute to psychiatric disorders during puberty, such as mood swings, anxiety, and anorexia (Aoki et al., 2012).
Excessive stress leads to physiological and behavioral responses resulting in numerous psychiatric disorders. The hypothalamic-pituitary-adrenal (HPA) axis plays an important role for mediating physiological responses by promoting stress-induced steroid synthesis (see review Kudielka and Kirschbaum, 2005). A recent study has demonstrated that stress-derived neurosteroid acts on δ-GABAA receptors of corticotrophin releasing hormone (CRH) neurons, biphasically modulating their excitability depending on the presence or absence of stress (Hewitt et al., 2009). The study showed that stress decreased the activity of the outward-directed K+, Cl- co-transporter, potassium-chloride transporter 2 (KCC2), in CRH neurons, switching tonic GABA conductance from inhibitory to excitatory and resulting in increased activity of the HPA axis and elevated levels of corticosterone (Hewitt et al., 2009). Because blockage of corticosterone synthesis prevents stress-induced anxiety, this positive feedback loop may be one of the mechanisms underlying stress-induced psychiatric disorders (Sarkar et al., 2011). Overall, emerging evidence has uncovered a bidirectional modification of tonic inhibition by neurosteroids, and thus may explain why increases in neurosteroids can lead to various types of psychiatric disorders, including depression and anxiety.
“Autism spectrum” encompasses a wide range of disorders, which can be divided into two groups with respect to their etiology: (1) recognizably distinct syndromes caused by mutations in specific genes or chromosomal loci, and (2) more common, genetically heterogeneous subjects, referred to as idiopathic autism (Coghlan et al., 2012). The number of subjects in the first group is much lower than idiopathic autisms (Coghlan et al., 2012), however their pathophysiology can be rigorously analyzed by using a genetic mouse model.
Fragile X syndrome (FXS) is a neurodevelopmental disorder caused by malfunction of the fragile X mental retardation 1 (FMR1) gene on the X chromosome, which encodes the FMR protein (FMRP; Verkerk et al., 1991). Common symptoms include intellectual disability, a distinctive physical phenotype, and autism-like behavior often associated with epilepsy. FMRP is known to regulate the mRNA transcription of various synaptic plasticity-associated proteins in concert with activation of metabotropic glutamate receptors (mGluR; Bassell and Warren, 2008). mRNA of some GABAA receptor subunits, particularly the δ subunit, are targeted substrates of FMRP (Miyashiro et al., 2003; Dictenberg et al., 2008). Dendritic localization of δ subunit mRNA has been shown to be regulated by mGluR activation under the presence of FMRP (Dictenberg et al., 2008), suggesting that tonic inhibition plays an important role for the homeostatic regulation of excitation-inhibition during development. Indeed, electrophysiological analysis in FMR1 KO mice revealed that tonic, but not phasic, inhibition in subicular pyramidal neurons is significantly decreased, with decreased expression of δ and α5 subunits in this region (Curia et al., 2009). Decreased expression of the δ subunit protein has also been shown in the cortex and hippocampus of this mouse model (El Idrissi et al., 2005). Olmos-Serrano et al. (2010) demonstrated that FMR1 KO mice exhibit remarkably reduced tonic and phasic inhibitory currents in the amygdala principal neurons, with reduced GAD65 and GAD67 expression. This group also showed that THIP rescued neuronal hyperexcitability, in vitro, and ameliorated hyperexcitability, in vivo (Olmos-Serrano et al., 2010, 2011; Figures 2A–C), indicating that tonic inhibition is a potent therapeutic target for FXS. In contrast, increased GAD65 and GAD67 expression was also observed in the cortex and hippocampus of FMR1 KO mice (El Idrissi et al., 2005; Adusei et al., 2010), thus indicating regional differentiation of deregulation mechanisms of inhibition.
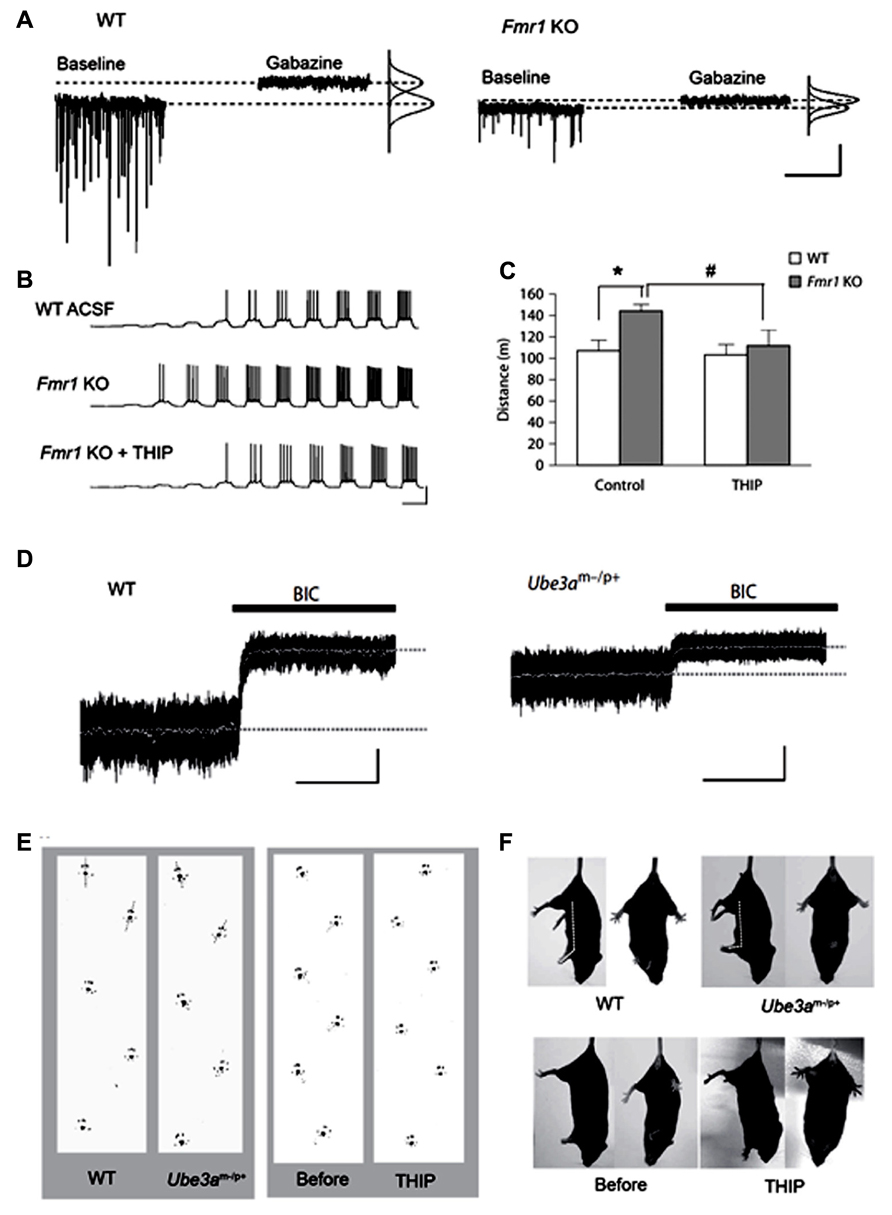
FIGURE 2. Decreased tonic inhibition in mouse models of the neurodevelopmental disorders, fragile X syndrome (FXS) and Angelman syndrome. (A–C) Analyses in fragile X mental retardation 1 (Fmr1) knock out (KO) mice (FXS model). (A) Voltage clamp traces from principal neurons of amygdala in wild type (WT) and Fmr1 KO mice, showing impaired phasic and tonic inhibition in KO mice. (B) Fmr1 KO mice have higher action potential (AP) firing rates for a given depolarizing current step and a lower threshold for AP generation (middle panel), which is reverted to WT levels with THIP (lower panel). (C) In the open field test, THIP prevents hyper locomotor activity in Fmr1 KO mice. *p < 0.05, WT control vs. Fmr KO control; #p < 0.05, Fmr1 KO control vs. Fmr KO THIP. (D,E) Analyses in ubiquitin-protein ligase E3A (Ube3a) KO mice (Angelman syndrome model). (D) Decreased cerebellar tonic inhibition in Ube3a KO mice [black bars: 20 μM bicuculline (BIC)]. (E) Ataxic gait rescued by administration of THIP (right panel) in Ube3a KO mice. (F) THIP (lower panels) improves clasping reflex (via the tail-suspension test) in Ube3a KO mice. (A–C) Adapted from Olmos-Serrano et al. (2010), 2011; (D–F) adapted from Egawa et al. (2012).
Similar presynaptic dysfunction in GABAergic neurotransmission has been proposed as a mechanism underlying the autism spectrum-related disorder, Rett syndrome. Mice lacking the causal gene, methyl CpG binding protein 2, from GABAergic neurons recapitulate phenotypes as those seen in Rett syndrome, with decreased GABA levels, reduced expression of GAD65/67, and miniature inhibitory postsynaptic current amplitudes in cortical pyramidal neurons (Chao et al., 2010). Therefore, reduction of tonic inhibition may also contribute to the pathophysiology of Rett syndrome.
Findings from our recent study have shown decreased tonic inhibition as a mechanism underlying cerebellar ataxia in the autism spectrum-related disorder, Angelman syndrome (Egawa et al., 2012). This syndrome is one of the ubiquitin-proteasome pathway-associated diseases with the causal gene, ubiquitin-protein ligase E3A (UBE3A), encoding the Ube3a protein (Kishino et al., 1997). We found that Ube3a binds to GAT1 and directly controls its degradation in the cerebellum (Egawa et al., 2012). Furthermore, a surplus of GAT1 was found to decrease tonic currents of CGCs in Ube3a KO mice. Because THIP can improve cerebellar dysfunctions in this mouse model under in vitro and in vivo conditions (Figures 2D–F), the reduction of tonic inhibition could thus underlie cerebellar ataxia in Angelman syndrome. Expression of GAT1 and Ube3a is increased during development (Takayama and Inoue, 2005; Sato and Stryker, 2010). Therefore, our findings suggest that Ube3a-dependent degradation of GAT1 plays an important role for neuronal plasticity of inhibitory systems by regulating tonic inhibition. Disruption of CaMK II-mediated regulation of membrane trafficking of extrasynaptic GABAA receptors may also result in decreased tonic inhibition, because deactivation of CaMK II is known to play a role in the pathophysiology of this syndrome (Weeber et al., 2003). Ube3a is widely expressed in the CNS (Jiang et al., 1998), thus decreased tonic inhibition may also be responsible for other symptoms, such as autistic behavior, mental retardation, and seizures. Notably, Angelman syndrome is predominantly caused by the large deletion of the relevant chromosome, 15q11-13, containing genes that encode the GABAA receptor α5, β3, and γ3 subunits. Therefore, these individuals show a greater severity in phenotypes compared with individuals without the deletion (Lossie et al., 2001; Egawa et al., 2008). The relative expression of β3 and α5 was shown to be reduced in postmortem brain tissue from patients lacking these genes (Roden et al., 2010). Therefore, tonic inhibition may be lower in typical Angelman syndrome individuals with the large deletion of 15q11-13, which could result in the phenotypic difference in patients with or without the deletion.
Accumulating evidence has indicated that the chromosome 15q11-13 abnormalities are highly correlated with the prevalence of idiopathic autisms. Genetic multi-linkage analysis has identified GABAA receptor α5 and β3 subunit genes on 15q11-13 that contribute to autism susceptibility (McCauley et al., 2004). Furthermore, this region is one of the most common loci where copy number variants have been observed in idiopathic autisms. The prevalence of 15q11–13 duplications in idiopathic autisms has been estimated to be at 3% (Coghlan et al., 2012). Although an increased copy number of 15q11–q13 has been predicted to elevate the gene expression of GABAA receptor subunits, recent in vitro studies using human neuronal cell lines have shown contradictory results, found to be due to impaired homologous pairing (Meguro-Horike et al., 2011). Therefore, expression of GABAA receptor α5 and β3 subunits is likely to be reduced in individuals with duplicated 15q11-13. Furthermore, attenuated expression of α5 and β3 subunits may be a common pathophysiological feature in idiopathic autisms without the 15q11-13 mutation. In support of this hypothesis, postmortem brain tissue analyses have reported that the expression of α5 and β3 subunits are significantly decreased (in four out of eight cases) in idiopathic autism spectrum disorders (Hogart et al., 2007). Further support was shown in a PET study in which binding of [11C]Ro15-4513 was significantly lower throughout the brain, particularly in the amygdala, in adult idiopathic autism patients who did not take any neuro-modulative medications (Momosaki et al., 2010). However, compensatory regulation that may reduce the expression of the α5 receptor in idiopathic autism subjects cannot be excluded. Nevertheless, findings from mouse models of specific gene-associated autism spectrum disorders and brain imaging of human subjects, suggest that reduced tonic inhibition may be one of the common mechanisms underlying neuronal dysfunctions in autism spectrum disorders.
Gamma-aminobutyric acid induces depolarization and hyperpolarization in the immature and adult brain, respectively. The GABAA receptor is a Cl- channel (also permeable for HCO3-), and thus the developmental switch of GABA-mediated depolarization to hyperpolarization is induced by changes in the transmembranous Cl- gradient, from its efflux to influx, respectively. This change is regulated by cation-Cl- cotransporters [Na–K–Cl cotransporter 1 (NKCC1), Cl- uptake; and KCC2, Cl- extrusion]. Furthermore, in immature neurons the release mechanism of GABA is non-vesicular and non-synaptic, and therefore GABA-mediated activity is generally tonic. These tonic depolarizing (and excitatory on occasion) GABA actions are necessary for neurogenesis, differentiation, migration, and synaptogenesis (Figure 3).
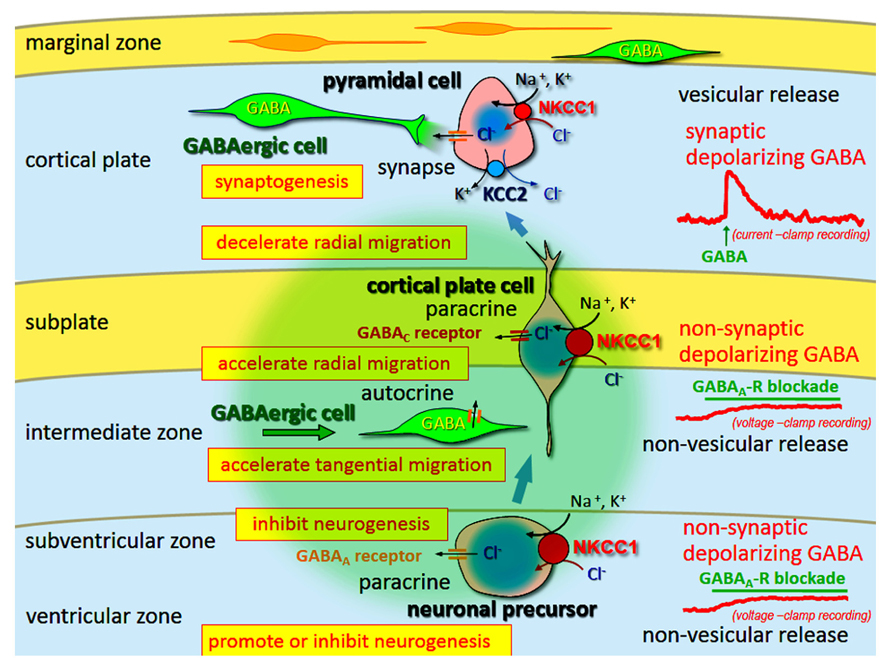
FIGURE 3. Multi-modal actions of tonic modulation of the GABAA receptor during corticogenesis. In the ventricular zone (VZ) and subventricular zone (SVZ), ambient GABA affects neurogenesis. Post-mitotic neurons migrating to the cortical plate are tonically depolarized by non-synaptic and non-vesicular ambient GABA, released from tangentially migrating GABA neurons prior to forming synapses, and changing migratory pace. GABA also acts in an autocrine manner to accelerate tangential migration. Vesicular release of GABA, which remains depolarizing, may contribute to synapse formation. Following the establishment of GABAergic synapses (and prior to hyperpolarization), GABA acts as an excitatory neurotransmitter due to high intracellular Cl- concentration levels, predominantly resulting from Na–K–Cl cotransporter 1 (NCC1) followed by K–Cl cotransporter 2 (KCC2). Green circle: ambient GABA. Adapted from Fukuda and Wang (2013).
During development, GABAA receptor subunits exhibit a completely different expression pattern (Laurie et al., 1992; Fritschy et al., 1994) compared with that in the adult. At the prenatal stage and during early postnatal days, cortical neurons predominantly express α2, α3, α4, α5, β2, β3, γ1, and γ2 subunits (Laurie et al., 1992). Gene transcripts of α2, α3, and α5 subunits have been detected in the developing CNS (Laurie et al., 1992). At rat embryonic day 17 and 20, α4, β1, and γ1 subunit transcripts have been found to be abundant in the inner half of the germinal matrix corresponding to the ventricular zone (VZ), [although undetectable in the intermediate zone (IZ)], thus implying that proliferating cells may express these subunits (Ma and Barker, 1995).
GABAA receptor-mediated signaling affects neurogenesis during development. A pioneering study by LoTurco et al. (1995) provided evidence that GABA can affect the proliferation of progenitor cells. They showed that depolarizing GABA actions led to a decrease in both DNA synthesis and the number of bromodeoxyuridine (BrdU)-labeled cells in the rat embryonic neocortex. However, later studies have revealed that GABA also has varying effects on neurogenesis in different cell-types and/or brain regions. Using organotypic slices of mouse embryonic neocortex, Haydar et al. (2000) revealed that differential regulation of cell production by depolarizing GABA activity occurs in cortical progenitor cells located in different regions [i.e., promotion and inhibition of cell division in the VZ and subventricular zone (SVZ), respectively]. These opposing effects may be attributed to the difference in the subunits of GABAA receptors or in the regional concentration of ambient GABA (Morishima et al., 2010).
In the neocortex, different classes of neurons, pyramidal cortical neurons, and GABAergic interneurons, originate from different sources and have distinct migration routes. Glutamatergic pyramidal neurons radially migrate from the dorsal telencephalic VZ where they are generated via radial glial scaffolding toward the cortical plate. In contrast, GABAergic interneurons arising from the medial ganglionic eminence (MGE) tangentially migrate into the cerebral wall (for review, see Marïn and Rubenstein, 2001). GABAA receptor signaling is well known to have distinct effects on radially or tangentially migrating neurons. Behar et al. (1996) demonstrated that the GABAergic modulation of neuronal migration was strongly concentration-dependent, in which femtomolar concentrations of GABA stimulated the migration along a chemical gradient (i.e., chemotaxis) while micromolar concentrations increased random migratory movement (i.e., chemokinesis). Furthermore, both processes were shown to be mediated by an increase in intracellular calcium, suggesting that GABA mediates chemotactic as well as chemokinetic migratory responses in embryonic neocortical neurons. The effect of GABA on migration is dependent on its location and receptor. Activation of neuroblastic GABAB or ρ-subunit-containing GABAA receptors promotes GABA migration from the SVZ and IZ, and the activation of GABAA receptors in cortical plate cells induces a stop signal (Behar et al., 2000; Heck et al., 2007; Denter et al., 2010). GABA also regulates the tangential migration of immature GABAergic cortical interneurons. After generation in the MGE, GABAergic interneurons migrate to the cortex via the corticostriatal junction (CSJ), avoiding the striatum (Cuzon et al., 2006). Although ambient GABA levels in the CSJ are similar to those in the MGE, the response of tonic GABAA receptor to ambient GABA is likely to be enhanced in the CSJ region, suggesting that a dynamic expression of GABAA receptor isoforms on MGE-derived cells promotes the cortical entry of tangentially migrating MGE-derived cells (Cuzon and Yeh, 2011). When GABAergic interneurons terminate their migration in the cortex their response to ambient GABA changes to a stop signal via up-regulation of KCC2 (Bortone and Polleux, 2009).
Tonic activation of GABAA receptors precedes phasic activation in early development (for review, see Owens and Kriegstein, 2002), hence its possible requirement for synaptogenesis during this period (Demarque et al., 2002; Liu et al., 2006). Spontaneous depolarizations of GABA would be the first excitatory drive required for activity-dependent synaptogenesis (Nakanishi et al., 2007; Wang and Kriegstein, 2008; for review, see Ben-Ari et al., 2007).
As mentioned previously in this review, tonic depolarizing responses to ambient GABA is locally controlled by uptake and/or release mechanisms allowing GABAA receptor-mediated actions to control a variety of developmental events in a time- and region-specific fashion. Thus, a defect in GABAA receptor-mediated multimodal functions during development can cause neuronal circuit dysfunctions hence neurological disorders. Therefore, use of GABAA receptor activating drugs to enhance inhibition in the immature brain may result in unexpected deleterious effects. A growing number of studies in immature animal models have demonstrated degenerative effects of several anesthetics on neuronal structure and brain functions, such as social behaviors, fear conditioning, and spatial reference memory tasks (Jevtovic-Todorovic et al., 2003; Loepke et al., 2009; Satomoto et al., 2009). Several animal studies have demonstrated that sevoflurane (Satomoto et al., 2009) and isoflurane (Sall et al., 2009; Stratmann et al., 2009), which bind to GABAA receptors, exhibit deleterious effects on neuronal survival and neurogenesis when exposed at early periods of life.
Neocortical malformations, such as polymicrogyria show the presence of cortical cells in heterotopic positions; often caused by a disruption in neuronal migration (Walsh, 1999; Francis et al., 2006). Therefore, perturbation of the numerous functions of GABA during development may underlie the etiology of cortical malformations. Focal freeze lesion in the cerebral cortex of newborn (P0) rats was shown to produce microgyrus, a focal cortical malformation with a small sulcus and a 3- or 4-layered microgyric cortex. This defect resembles human 4-layered polymicrogyria (Dvorák and Feit, 1977), a clinical condition that results from abnormal neuronal migration. Wang et al. (2013) have reported that temporally increased ambient GABA causes tonic activation of neuronal GABAA receptors (possibly due to depolarization because NKCC1 is up-regulated and KCC2 is down-regulated), resulting in the modulation of intracellular Ca2+ oscillations. These oscillations could differentially affect the migratory status of GABAergic cells (“go”) and cortical plate cells (“stop”). Therefore, immature cortical plate neurons and GABAergic neurons migrate in distinct patterns to form the microgyrus and thus, abnormally induced tonic functions of GABA could be involved in migration disorders that ultimately result in cortical malformations.
This review has focused on the recent progress in understanding the impact of dysregulated tonic inhibition in CNS diseases. Accumulating evidence highlights the pathophysiological significance of dysregulated tonic inhibition in a number of neurodevelopmental disorders, psychiatric disorders, and cognitive dysfunctions (see Figure 1). In addition to pharmacological progress, animal model studies are presently opening the door for practical medicine. However, an overview of these studies also presents problems for clinical application. For example, the same phenotype may result from a decrease or increase of tonic inhibition. Therefore, the net shift from excitation to inhibition may not always occur with a modulation of tonic inhibition. This paradox is possibly due to the complexity of network regulation by tonic conductance. For example, simultaneous receptor activation in either inhibitory or excitatory neurons, or multiple mechanisms can be responsible for changing membrane properties, such as shunting inhibition and a shift in intracellular Cl- concentration. Although temporal characteristics of tonic inhibition are weak, maintaining network functions can possibly regulate its strength, thus suggesting a narrow therapeutic range for such modulators. A detailed assessment of these mechanisms would therefore be needed to develop a therapeutic strategy.
This review also discussed tonic depolarization induced by ambient GABA during early development, where the release of GABA is possibly non-vesicular and occurring before synaptic organization (see Figure 3). Thus far, very few studies have addressed this series of events as a potential risk factor for CNS diseases. Therefore, research focused on the pathological effects of multimodal functions of GABA during neurodevelopment may shed light on understanding the pathology of neurodevelopmental disorders in which GABA is known to play a role, for example in schizophrenia and autism spectrum disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. S. Saito for comments on the manuscript.
Ade, K. K., Janssen, M. J., Ortinski, P. I., and Vicini, S. (2008). Differential tonic GABA conductances in striatal medium spiny neurons. J. Neurosci. 28, 1185–1197. doi: 10.1523/JNEUROSCI.3908-07.2008
Adusei, D. C., Pacey, L. K., Chen, D., and Hampson, D. R. (2010). Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology 59, 167–171. doi: 10.1016/j.neuropharm.2010.05.002
Akbarian, S., Huntsman, M. M., Kim, J. J., Tafazzoli, A., Potkin, S. G., Bunney, W. E., et al. (1995a). GABAA receptor subunit gene expression in human prefrontal cortex: comparison of schizophrenics and controls. Cereb. Cortex 5, 550–560. doi: 10.1093/cercor/5.6.550
Akbarian, S., Kim, J. J., Potkin, S. G., Hagman, J. O., Tafazzoli, A., Bunney, W. E., et al. (1995b). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 52, 258–266. doi: 10.1001/archpsyc.1995.03950160008002
Andäng, M., Hjerling-Leffler, J., Moliner, A., Lundgren, T. K., Castelo-Branco, G., Nanou, E., et al. (2008). Histone H2AX-dependent GABAA receptor regulation of stem cell proliferation. Nature 451, 460–464. doi: 10.1038/nature06488
Aoki, C., Sabaliauskas, N., Chowdhury, T., Min, J. Y., Colacino, A. R., Laurino, K., et al. (2012). Adolescent female rats exhibiting activity-based anorexia express elevated levels of GABAA receptor α4 and δ subunits at the plasma membrane of hippocampal CA1 spines. Synapse 66, 391–407. doi: 10.1002/syn.21528
Asai, Y., Takano, A., Ito, H., Okubo, Y., Matsuura, M., Otsuka, A., et al. (2008). GABAA/Benzodiazepine receptor binding in patients with schizophrenia using [11C]Ro15-4513, a radioligand with relatively high affinity for α5 subunit. Schizophr. Res. 99, 333–340. doi: 10.1016/j.schres.2007.10.014
Atack, J. R. (2010). Preclinical and clinical pharmacology of the GABAA receptor α5 subtype-selective inverse agonist α5IA. Pharmacol. Ther. 125, 11–26. doi: 10.1016/j.pharmthera.2009.09.001
Atack, J. R., Bayley, P. J., Seabrook, G. R., Wafford, K. A., Mckernan, R. M., and Dawson, G. R. (2006). L-655,708 enhances cognition in rats but is not proconvulsant at a dose selective for α5-containing GABAA receptors. Neuropharmacology 51, 1023–1029. doi: 10.1016/j.neuropharm.2006.04.018
Atack, J. R., Maubach, K. A., Wafford, K. A., O’Connor, D., Rodrigues, A. D., Evans, D. C., et al. (2009). In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5- ylmethoxy)-pyrazolo[1,5-d]-[1,2,4] triazine (MRK-016), a GABAA receptor α5 subtype-selective inverse agonist. J. Pharmacol. Exp. Ther. 331, 470–484. doi: 10.1124/jpet.109.157636
Ballard, T. M., Knoflach, F., Prinssen, E., Borroni, E., Vivian, J. A., Basile, J., et al. (2009). RO4938581, a novel cognitive enhancer acting at GABAA α5 subunit-containing receptors. Psychopharmacology (Berl.) 202, 207–223. doi: 10.1007/s00213-008-1357-7
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Behar, T. N., Li, Y. X., Tran, H. T., Ma, W., Dunlap, V., Scott, C., et al. (1996). GABA stimulates chemotaxis, and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J. Neurosci. 16, 1808–1818.
Behar, T. N., Schaffner, A. E., Scott, C. A., Greene, C. L., and Barker, J. L. (2000). GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb. Cortex 10, 899–909. doi: 10.1093/cercor/10.9.899
Behar, T. N., Schaffner, A. E., Scott, C. A., O’Connell, C., and Barker, J. L. (1998). Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J. Neurosci. 18, 6378–6387.
Behar, T. N., Smith, S. V., Kennedy, R. T., McKenzie, J. M., Maric, I., and Barker, J. L. (2001). GABAB receptors mediate motility signals for migrating embryonic cortical cells. Cereb. Cortex 11, 744–753. doi: 10.1093/cercor/11.8.744
Belichenko, P. V., Kleschevnikov, A. M., Masliah, E., Wu, C., Takimoto-Kimura, R., Salehi, A., et al. (2009). Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 512, 453–466. doi: 10.1002/cne.21895
Ben-Ari, Y. (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Bianchi, M. T., and MacDonald, R. L. (2003). Neurosteroids shift partial agonist activation of GABAA receptor channels from low- to high-efficacy gating patterns. J. Neurosci. 23, 10934–10943.
Bonin, R. P., Labrakakis, C., Eng, D. G., Whissell, P. D., De Koninck, Y., and Orser, B. A. (2011). Pharmacological enhancement of δ-subunit-containing GABAA receptors that generate a tonic inhibitory conductance in spinal neurons attenuates acute nociception in mice. Pain 152, 1317–1326. doi: 10.1016/j.pain.2011.02.011
Bortone, D., and Polleux, F. (2009). KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62, 53–71. doi: 10.1016/j.neuron.2009.01.034
Braudeau, J., Delatour, B., Duchon, A., Pereira, P. L., Dauphinot, L., De Chaumont, F., et al. (2011). Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J. Psychopharmacol. 25, 1030–1042. doi: 10.1177/0269881111405366
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. 497, 753–759.
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Brickley, S. G., Revilla, V., Cull-Candy, S. G., Wisden, W., and Farrant, M. (2001). Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature 409, 88–92. doi: 10.1038/35051086
Bright, D. P., Renzi, M., Bartram, J., McGee, T. P., MacKenzie, G., Hosie, A. M., et al. (2011). Profound desensitization by ambient GABA limits activation of δ-containing GABAA receptors during spillover. J. Neurosci. 31, 753–763. doi: 10.1523/JNEUROSCI.2996-10.2011
Brunig, I., Scotti, E., Sidler, C., and Fritschy, J. M. (2002). Intact sorting, targeting, and clustering of γ-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J. Comp. Neurol. 443, 43–55. doi: 10.1002/cne.10102
Campos, M. L., De Cabo, C., Wisden, W., Juiz, J. M., and Merlo, D. (2001). Expression of GABAA receptor subunits in rat brainstem auditory pathways: cochlear nuclei, superior olivary complex and nucleus of the lateral lemniscus. Neuroscience 102, 625–638. doi: 10.1016/S0306-4522(00)00525-X
Caraiscos, V. B., Elliott, E. M., You-Ten, K. E., Cheng, V. Y., Belelli, D., Newell, J. G., et al. (2004). Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 3662–3667. doi: 10.1073/pnas.0307231101
Carlesimo, G. A., Marotta, L., and Vicari, S. (1997). Long-term memory in mental retardation: evidence for a specific impairment in subjects with Down’s syndrome. Neuropsychologia 35, 71–79. doi: 10.1016/S0028-3932(96)00055-3
Chandra, D., Jia, F., Liang, J., Peng, Z., Suryanarayanan, A., Werner, D. F., et al. (2006). GABAA receptor α 4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc. Natl. Acad. Sci. U.S.A. 103, 15230–15235. doi: 10.1073/pnas.0604304103
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chiu, C. S., Brickley, S., Jensen, K., Southwell, A., McKinney, S., Cull-Candy, S., et al. (2005). GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J. Neurosci. 25, 3234–3245. doi: 10.1523/JNEUROSCI.3364-04.2005
Clarkson, A. N., Huang, B. S., MacIsaac, S. E., Mody, I., and Carmichael, S. T. (2010). Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. doi: 10.1038/nature09511
Coghlan, S., Horder, J., Inkster, B., Mendez, M. A., Murphy, D. G., and Nutt, D. J. (2012). GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci. Biobehav. Rev. 36, 2044–2055. doi: 10.1016/j.neubiorev.2012.07.005
Collinson, N., Kuenzi, F. M., Jarolimek, W., Maubach, K. A., Cothliff, R., Sur, C., et al. (2002). Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α 5 subunit of the GABAA receptor. J. Neurosci. 22, 5572–5580.
Cope, D. W., Di Giovanni, G., Fyson, S. J., Orban, G., Errington, A. C., Lorincz, M. L., et al. (2009). Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 15, 1392–1398. doi: 10.1038/nm.2058
Cope, D. W., Hughes, S. W., and Crunelli, V. (2005). GABAA receptor-mediated tonic inhibition in thalamic neurons. J. Neurosci. 25, 11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005
Crestani, F., Keist, R., Fritschy, J. M., Benke, D., Vogt, K., Prut, L., et al. (2002). Trace fear conditioning involves hippocampal α5 GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 99, 8980–8985. doi: 10.1073/pnas.142288699
Curia, G., Papouin, T., Seguela, P., and Avoli, M. (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1515–1520. doi: 10.1093/cercor/bhn159
Cuzon, V. C., and Yeh, H. H. (2011). GABAA receptor subunit profiles of tangentially migrating neurons derived from the medial ganglionic eminence. Cereb. Cortex 21, 1792–1802. doi: 10.1093/cercor/bhq247
Cuzon, V. C., Yeh, P. W., Cheng, Q., and Yeh, H. H. (2006). Ambient GABA promotes cortical entry of tangentially migrating cells derived from the medial ganglionic eminence. Cereb. Cortex 16, 1377–1388. doi: 10.1093/cercor/bhj084
Damgaard, T., Plath, N., Neill, J. C., and Hansen, S. L. (2011). Extrasynaptic GABAA receptor activation reverses recognition memory deficits in an animal model of schizophrenia. Psychopharmacology 214, 403–413. doi: 10.1007/s00213-010-2039-9
Davies, P. A., Hanna, M. C., Hales, T. G., and Kirkness, E. F. (1997). Insensitivity to anaesthetic agents conferred by a class of GABAA receptor subunit. Nature 385, 820–823. doi: 10.1038/385820a0
Dawson, G. R., Maubach, K. A., Collinson, N., Cobain, M., Everitt, B. J., MacLeod, A. M., et al. (2006). An inverse agonist selective for α5 subunit-containing GABAA receptors enhances cognition. J. Pharmacol. Exp. Ther. 316, 1335–1345. doi: 10.1124/jpet.105.092320
De Biasi, S., Vitellaro-Zuccarello, L., and Brecha, N. C. (1998). Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience 83, 815–828. doi: 10.1016/S0306-4522(97)00414-4
Demarque, M., Represa, A., Becq, H., Khalilov, I., Ben-Ari, Y., and Aniksztejn, L. (2002). Paracrine intercellular communication by a Ca2+ and SNARE-independent release of GABA and glutamate prior to synapse formation. Neuron 36, 1051–1061. doi: 10.1016/S0896-6273(02)01053-X
Denter, D. G., Heck, N., Riedemann, T., White, R., Kilb, W., and Luhmann, H. J. (2010). GABAC receptors are functionally expressed in the intermediate zone and regulate radial migration in the embryonic mouse neocortex. Neuroscience 167, 124–134. doi: 10.1016/j.neuroscience.2010.01.049
de Villers-Sidani, E., Alzghoul, L., Zhou, X., Simpson, K. L., Lin, R. C., and Merzenich, M. M. (2010). Recovery of functional and structural age-related changes in the rat primary auditory cortex with operant training. Proc. Natl. Acad. Sci. U.S.A. 107, 13900–13905. doi: 10.1073/pnas.1007885107
Diaz, M. R., Wadleigh, A., Hughes, B. A., Woodward, J. J., and Valenzuela, C. F. (2011). Bestrophin1 channels are insensitive to ethanol and do not mediate tonic GABAergic currents in cerebellar granule cells. Front. Neurosci. 5:148. doi: 10.3389/fnins.2011.00148
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. doi: 10.1016/j.devcel.2008.04.003
Di Martino, A., and Tuchman, R. F. (2001). Antiepileptic drugs: affective use in autism spectrum disorders. Pediatr. Neurol. 25, 199–207. doi: 10.1016/S0887-8994(01)00276-4
Dvorák, K., and Feit, J. (1977). Migration of neuroblasts through partial necrosis of the cerebral cortex in newborn rats-contribution to the problems of morphological development and developmental period of cerebral microgyria. Histological and autoradiographical study. Acta Neuropathol. 38, 203–212. doi: 10.1007/BF00688066
Egawa, K., Asahina, N., Shiraishi, H., Kamada, K., Takeuchi, F., Nakane, S., et al. (2008). Aberrant somatosensory-evoked responses imply GABAergic dysfunction in Angelman syndrome. Neuroimage 39, 593–599. doi: 10.1016/j.neuroimage.2007.09.006
Egawa, K., Kitagawa, K., Inoue, K., Takayama, M., Takayama, C., Saitoh, S., et al. (2012). Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci. Transl. Med. 4, 163ra157.
Egawa, K., Yamada, J., Furukawa, T., Yanagawa, Y., and Fukuda, A. (2013). Cl- homeodynamics in gap-junction-coupled astrocytic networks on activation of GABAergic synapses. J. Physiol. 591(Pt. 16), 3901–3917. doi: 10.1113/jphysiol.2013.257162
El Idrissi, A., Ding, X. H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABAA receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146. doi: 10.1016/j.neulet.2004.11.087
Enna, S. J., and McCarson, K. E. (2006). The role of GABA in the mediation and perception of pain. Adv. Pharmacol. 54, 1–27. doi: 10.1016/S1054-3589(06)54001-3
Ewers, M., Sperling, R. A., Klunk, W. E., Weiner, M. W., and Hampel, H. (2011). Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 34, 430–442. doi: 10.1016/j.tins.2011.05.005
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Francis, F., Meyer, G., Fallet-Bianco, C., Moreno, S., Kappeler, C., Socorro, A. C., et al. (2006). Human disorders of cortical development: from past to present. Eur. J. Neurosci. 23, 877–893. doi: 10.1111/j.1460-9568.2006.04649.x
Fritschy, J. M., Paysan, J., Enna, A., and Mohler, H. (1994). Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J. Neurosci. 14, 5302–5324.
Fukuda, A., and Wang, T. (2013). A perturbation of multimodal GABA functions underlying the formation of focal cortical malformations: assessments by using animal models. Neuropathology 33, 480–486. doi: 10.1111/neup.12021
Gill, K. M., Lodge, D. J., Cook, J. M., Aras, S., and Grace, A. A. (2011). A novel α5 GABAAR-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology 36, 1903–1911. doi: 10.1038/npp.2011.76
Gleich, O., Hamann, I., Klump, G. M., Kittel, M., and Strutz, J. (2003). Boosting GABA improves impaired auditory temporal resolution in the gerbil. Neuroreport 14, 1877–1880. doi: 10.1097/01.wnr.0000089569.45990.74
Glykys, J., and Mody, I. (2006). Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABAA receptor α5 subunit-deficient mice. J. Neurophysiol. 95, 2796–2807. doi: 10.1152/jn.01122.2005
Glykys, J., and Mody, I. (2007). The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 582, 1163–1178. doi: 10.1113/jphysiol.2007.134460
Guidotti, A., Auta, J., Davis, J. M., Dong, E., Grayson, D. R., Veldic, M., et al. (2005). GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology 180, 191–205. doi: 10.1007/s00213-005-2212-8
Hamann, M., Rossi, D. J., and Attwell, D. (2002). Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron 33, 625–633. doi: 10.1016/S0896-6273(02)00593-7
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., et al. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326.
Haydar, T. F., Wang, F., Schwartz, M. L., and Rakic, P. (2000). Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J. Neurosci. 20, 5764–5774.
Heck, N., Kilb, W., Reiprich, P., Kubota, H., Furukawa, T., Fukuda, A., et al. (2007). GABA-A receptors regulate neocortical neuronal migration in vitro and in vivo. Cereb. Cortex 17, 138–148. doi: 10.1093/cercor/bhj135
Heja, L., Nyitrai, G., Kekesi, O., Dobolyi, A., Szabo, P., Fiath, R., et al. (2012). Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 10:26. doi: 10.1186/1741-7007-10-26
Hewitt, S. A., Wamsteeker, J. I., Kurz, E. U., and Bains, J. S. (2009). Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 12, 438–443. doi: 10.1038/nn.2274
Hines, R. M., Davies, P. A., Moss, S. J., and Maguire, J. (2012). Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 22, 552–558. doi: 10.1016/j.conb.2011.10.007
Hoestgaard-Jensen, K., Dalby, N. O., Wolinsky, T. D., Murphey, C., Jones, K. A., Rottlander, M., et al. (2010). Pharmacological characterization of a novel positive modulator at α4β3δ-containing extrasynaptic GABAA receptors. Neuropharmacology 58, 702–711. doi: 10.1016/j.neuropharm.2009.12.023
Hogart, A., Nagarajan, R. P., Patzel, K. A., Yasui, D. H., and Lasalle, J. M. (2007). 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 16, 691–703. doi: 10.1093/hmg/ddm014
Imbrosci, B., Neubacher, U., White, R., Eysel, U. T., and Mittmann, T. (2012). Shift from phasic to tonic GABAergic transmission following laser-lesions in the rat visual cortex. Pflugers Arch. 465, 879–893. doi: 10.1007/s00424-012-1191-y
Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95, 15718–15723. doi: 10.1073/pnas.95.26.15718
Inada, H., Watanabe, M., Uchida, T., Ishibashi, H., Wake, H., Nemoto, T., et al. (2011). GABA regulates the multidirectional tangential migration of GABAergic interneurons in living neonatal mice. PLoS ONE 6:e27048. doi: 10.1371/journal.pone.0027048
Inoue, K., Furukawa, T., Kumada, T., Yamada, J., Wang, T., Inoue, R., et al. (2012). Taurine inhibits the K+–Cl--cotransporter KCC2 to regulate embryonic Cl- homeostasis via the with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem. 287, 20839–20850. doi: 10.1074/jbc.M111.319418
Ishikawa, M., Mizukami, K., Iwakiri, M., Hidaka, S., and Asada, T. (2004). Immunohistochemical and immunoblot study of GABAA α1 and β2/3 subunits in the prefrontal cortex of subjects with schizophrenia and bipolar disorder. Neurosci. Res. 50, 77–84. doi: 10.1016/j.neures.2004.06.006
Jensen, M. L., Wafford, K. A., Brown, A. R., Belelli, D., Lambert, J. J., and Mirza, N. R. (2013). A study of subunit selectivity, mechanism and site of action of the δ selective compound 2 (DS2) at human recombinant and rodent native GABAA receptors. Br. J. Pharmacol. 168, 1118–1132. doi: 10.1111/bph.12001
Jevtovic-Todorovic, V., Hartman, R. E., Izumi, Y., Benshoff, N. D., Dikranian, K., Zorumski, C. F., et al. (2003). Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 23, 876–882.
Jiang, Y. H., Armstrong, D., Albrecht, U., Atkins, C. M., Noebels, J. L., Eichele, G., et al. (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811. doi: 10.1016/S0896-6273(00)80596-6
Kaneda, M., Farrant, M., and Cull-Candy, S. G. (1995). Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J. Physiol. 485(Pt 2), 419–435.
Kasugai, Y., Swinny, J. D., Roberts, J. D., Dalezios, Y., Fukazawa, Y., Sieghart, W., et al. (2010). Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur. J. Neurosci. 32, 1868–1888. doi: 10.1111/j.1460-9568.2010.07473.x
Kennedy, R. T., Thompson, J. E., and Vickroy, T. W. (2002). In vivo monitoring of amino acids by direct sampling of brain extracellular fluid at ultralow flow rates and capillary electrophoresis. J. Neurosci. Methods 114, 39–49. doi: 10.1016/S0165-0270(01)00506-4
Kersante, F., Rowley, S. C., Pavlov, I., Gutierrez-Mecinas, M., Semyanov, A., Reul, J. M., et al. (2013). A functional role for both γ-aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J. Physiol. 591, 2429–2441. doi: 10.1113/jphysiol.2012.246298
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004
Koh, M. T., Rosenzweig-Lipson, S., and Gallagher, M. (2013). Selective GABAA α5 positive allosteric modulators improve cognitive function in aged rats with memory impairment. Neuropharmacology 64, 145–152. doi: 10.1016/j.neuropharm.2012.06.023
Krogsgaard-Larsen, P., Frolund, B., Liljefors, T., and Ebert, B. (2004). GABAA agonists and partial agonists: THIP (gaboxadol) as a non-opioid analgesic and a novel type of hypnotic. Biochem. Pharmacol. 68, 1573–1580. doi: 10.1016/j.bcp.2004.06.040
Krook-Magnuson, E. I., Li, P., Paluszkiewicz, S. M., and Huntsman, M. M. (2008). Tonically active inhibition selectively controls feedforward circuits in mouse barrel cortex. J. Neurophysiol. 100, 932–944. doi: 10.1152/jn.01360.2007
Kudielka, B. M., and Kirschbaum, C. (2005). Sex differences in HPA axis responses to stress: a review. Biol. Psychol. 69, 113–132. doi: 10.1016/j.biopsycho.2004.11.009
Labarrera, C., London, M., and Angelo, K. (2013). Tonic inhibition sets the state of excitability in olfactory bulb granule cells. J. Physiol. 591, 1841–1850. doi: 10.1113/jphysiol.2012.241851
Lambert, J. J., Belelli, D., Peden, D. R., Vardy, A. W., and Peters, J. A. (2003). Neurosteroid modulation of GABAA receptors. Prog. Neurobiol. 71, 67–80. doi: 10.1016/j.pneurobio.2003.09.001
Laurie, D. J., Wisden, W., and Seeburg, P. H. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172.
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., et al. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796. doi 10.1126/science.1184334
Lewis, D. A., Hashimoto, T., and Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324. doi: 10.1038/nrn1648
Lewis, D. A., Volk, D. W., and Hashimoto, T. (2004). Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology 174, 143–150. doi: 10.1007/s00213-003-1673-x
Liu, Z., Neff, R. A., and Berg, D. K. (2006). Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science 314, 1610–1613. doi: 10.1126/science.1134246
Loepke, A. W., Istaphanous, G. K., McAuliffe, J. J. III, Miles, L., Hughes, E. A., McCann, J. C., et al. (2009). The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth. Analg. 108, 90–104. doi: 10.1213/ane.0b013e31818cdb29
López-Bendito, G., Luján, R., Shigemoto, R., Ganter, P., Paulsen, O., and Molnár, Z. (2003). Blockade of GABAB receptors alters the tangential migration of cortical neurons. Cereb. Cortex 13, 932–942. doi: 10.1093/cercor/13.9.932
Lossie, A. C., Whitney, M. M., Amidon, D., Dong, H. J., Chen, P., Theriaque, D., et al. (2001). Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 38, 834–845. doi: 10.1136/jmg.38.12.834
LoTurco, J. J., Owens, D. F., Heath, M. J., Davis, M. B., and Kriegstein, A. R. (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298. doi: 10.1016/0896-6273(95)90008-X
Ma, W., and Barker, J. L. (1995). Complementary expressions of transcripts encoding GAD67 and GABAA receptor α4, β1, and γ1 subunits in the proliferative zone of the embryonic rat central nervous system. J. Neurosci. 15, 2547–2560.
Maguire, J., and Mody, I. (2008). GABAAR plasticity during pregnancy: relevance to postpartum depression. Neuron 59, 207–213. doi: 10.1016/j.neuron.2008.06.019
Maguire, J. L., Stell, B. M., Rafizadeh, M., and Mody, I. (2005). Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 8, 797–804. doi: 10.1038/nn1469
Maldonado-Aviles, J. G., Curley, A. A., Hashimoto, T., Morrow, A. L., Ramsey, A. J., O’Donnell, P., et al. (2009). Altered markers of tonic inhibition in the dorsolateral prefrontal cortex of subjects with schizophrenia. Am. J. Psychiatry 166, 450–459. doi: 10.1176/appi.ajp.2008.08101484
Manent, J. B., Demarque, M., Jorquera, I., Pellegrino, C., Ben-Ari, Y., Aniksztejn, L., et al. (2005). A noncanonical release of GABA and glutamate modulates neuronal migration. J. Neurosci. 25, 4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005
Marïn, O., and Rubenstein, J. L. R. (2001). A long, remarkable journey: tangential migration in the telencephalon. Nat. Rev. Neurosci. 2, 780–790. doi: 10.1038/35097509
Martinez-Cue, C., Martinez, P., Rueda, N., Vidal, R., Garcia, S., Vidal, V., et al. (2013). Reducing GABAA α5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of Down syndrome. J. Neurosci. 33, 3953–3966. doi: 10.1523/JNEUROSCI.1203-12.2013
McCauley, J. L., Olson, L. M., Delahanty, R., Amin, T., Nurmi, E. L., Organ, E. L., et al. (2004). A linkage disequilibrium map of the 1-Mb 15q12 GABAA receptor subunit cluster and association to autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 131B, 51–59. doi: 10.1002/ajmg.b.30038
Meguro-Horike, M., Yasui, D. H., Powell, W., Schroeder, D. I., Oshimura, M., Lasalle, J. M., et al. (2011). Neuron-specific impairment of inter-chromosomal pairing and transcription in a novel model of human 15q-duplication syndrome. Hum. Mol. Genet. 20, 3798–3810. doi: 10.1093/hmg/ddr298
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L., et al. (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. doi: 10.1016/S0896-6273(03)00034-5
Mody, I., and Pearce, R. A. (2004). Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci. 27, 569–575. doi: 10.1016/j.tins.2004.07.002
Mohler, H., Fritschy, J. M., and Rudolph, U. (2002). A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 300, 2–8. doi: 10.1124/jpet.300.1.2
Momosaki, S., Hosoi, R., Abe, K., and Inoue, O. (2010). Remarkable selectivity of the in vivo binding of [3H]Ro15-4513 to α5 subtype of benzodiazepine receptor in the living mouse brain. Synapse 64, 928–936. doi: 10.1002/syn.20812
Morishima, T., Uematsu, M., Furukawa, T., Yanagawa, Y., Fukuda, A., and Yoshida, S. (2010). GABA imaging in brain slices using immobilized enzyme-linked photo analysis. Neurosci. Res. 67, 347–353. doi: 10.1016/j.neures.2010.04.005
Mortensen, M., and Smart, T. G. (2006). Extrasynaptic αβ subunit GABAA receptors on rat hippocampal pyramidal neurons. J. Physiol. 577, 841–856. doi: 10.1113/jphysiol.2006.117952
Munro, G., Lopez-Garcia, J. A., Rivera-Arconada, I., Erichsen, H. K., Nielsen, E. O., Larsen, J. S., et al. (2008). Comparison of the novel subtype-selective GABAA receptor-positive allosteric modulator NS11394 [3′-[5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile] with diazepam, zolpidem, bretazenil, and gaboxadol in rat models of inflammatory and neuropathic pain. J. Pharmacol. Exp. Ther. 327, 969–981. doi: 10.1124/jpet.108.144568
Nakanishi, K., Yamada, J., Takayama, C., Oohira, A., and Fukuda, A. (2007). NKCC1 activity modulates formation of functional inhibitory synapses in cultured neocortical neurons. Synapse 61, 138–149. doi: 10.1002/syn.20352
Navarro, J. F., Buron, E., and Martin-Lopez, M. (2002). Anxiogenic-like activity of L-655,708, a selective ligand for the benzodiazepine site of GABAA receptors which contain the α-5 subunit, in the elevated plus-maze test. Prog. Neuropsychopharmacol. Biol. Psychiatry 26, 1389–1392. doi: 10.1016/S0278-5846(02)00305-6
Nusser, Z., and Mody, I. (2002). Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 87, 2624–2628.
Nusser, Z., Sieghart, W., and Somogyi, P. (1998). Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J. Neurosci. 18, 1693–1703.
Nutt, D. J., Besson, M., Wilson, S. J., Dawson, G. R., and Lingford-Hughes, A. R. (2007). Blockade of alcohol’s amnestic activity in humans by an α5 subtype benzodiazepine receptor inverse agonist. Neuropharmacology 53, 810–820. doi: 10.1016/j.neuropharm.2007.08.008
Olmos-Serrano, J. L., Corbin, J. G., and Burns, M. P. (2011). The GABAA receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 33, 395–403. doi: 10.1159/000332884
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 30, 9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010
Olsen, R. W., and Sieghart, W. (2008). International union of pharmacology. LXX. Subtypes of γ-aminobutyric acid A receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260. doi: 10.1124/pr.108.00505
Owens, D. F., Boyce, L. H., Davis, M. B., and Kriegstein, A. R. (1996). Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 16, 6414–6423.
Owens, D. F., and Kriegstein, A. R. (2002). Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3, 715–727. doi: 10.1038/nrn919
Perez-Cremades, D., Hernandez, S., Blasco-Ibanez, J. M., Crespo, C., Nacher, J., and Varea, E. (2010). Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down’s syndrome. J. Neural. Transm. 117, 445–455. doi: 10.1007/s00702-010-0376-9
Prenosil, G. A., Schneider Gasser, E. M., Rudolph, U., Keist, R., Fritschy, J. M., and Vogt, K. E. (2006). Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J. Neurophysiol. 96, 846–857.
Redrobe, J. P., Elster, L., Frederiksen, K., Bundgaard, C., De Jong, I. E., Smith, G. P., et al. (2012). Negative modulation of GABAA α5 receptors by RO4938581 attenuates discrete sub-chronic and early postnatal phencyclidine (PCP)-induced cognitive deficits in rats. Psychopharmacology 221, 451–468. doi: 10.1007/s00213-011-2593-9
Richardson, B. D., Ling, L. L., Uteshev, V. V., and Caspary, D. M. (2011). Extrasynaptic GABAA receptors and tonic inhibition in rat auditory thalamus. PLoS ONE 6:e16508. doi: 10.1371/journal.pone.0016508
Richardson, B. D., Ling, L. L., Uteshev, V. V., and Caspary, D. M. (2013). Reduced GABAA receptor-mediated tonic inhibition in aged rat auditory thalamus. J. Neurosci. 33, 1218–1227. doi: 10.1523/JNEUROSCI.3277-12.2013
Rode, F., Jensen, D. G., Blackburn-Munro, G., and Bjerrum, O. J. (2005). Centrally-mediated antinociceptive actions of GABAA receptor agonists in the rat spared nerve injury model of neuropathic pain. Eur. J. Pharmacol. 516, 131–138. doi: 10.1016/j.ejphar.2005.04.034
Roden, W. H., Peugh, L. D., and Jansen, L. A. (2010). Altered GABAA receptor subunit expression and pharmacology in human Angelman syndrome cortex. Neurosci. Lett. 483, 167–172. doi: 10.1016/j.neulet.2010.08.001
Saab, B. J., MacLean, A. J., Kanisek, M., Zurek, A. A., Martin, L. J., Roder, J. C., et al. (2010). Short-term memory impairment after isoflurane in mice is prevented by the α5 γ-aminobutyric acid type A receptor inverse agonist L-655,708. Anesthesiology 113, 1061–1071. doi: 10.1097/ALN.0b013e3181f56228
Saliba, R. S., Kretschmannova, K., and Moss, S. J. (2012). Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 31, 2937–2951. doi: 10.1038/emboj.2012.109
Sall, J. W., Stratmann, G., Leong, J., McKleroy, W., Mason, D., Shenoy, S., et al. (2009). Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology 110, 826–833. doi: 10.1097/ALN.0b013e31819b62e2
Santhakumar, V., Hanchar, H. J., Wallner, M., Olsen, R. W., and Otis, T. S. (2006). Contributions of the GABAA receptor α6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the α6 gene. J. Neurosci. 26, 3357–3364. doi: 10.1523/JNEUROSCI.4799-05.2006
Sarkar, J., Wakefield, S., MacKenzie, G., Moss, S. J., and Maguire, J. (2011). Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J. Neurosci. 31, 18198–18210. doi: 10.1523/JNEUROSCI.2560-11.2011
Sato, M., and Stryker, M. P. (2010). Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. U.S.A. 107, 5611–5616. doi: 10.1073/pnas.1001281107
Satomoto, M., Satoh, Y., Terui, K., Miyao, H., Takishima, K., Ito, M., et al. (2009). Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 110, 628–637. doi: 10.1097/ALN.0b013e3181974fa2
Saxena, N. C., and MacDonald, R. L. (1996). Properties of putative cerebellar γ-aminobutyric acid A receptor isoforms. Mol. Pharmacol. 49, 567–579.
Scimemi, A., Andersson, A., Heeroma, J. H., Strandberg, J., Rydenhag, B., McEvoy, A. W., et al. (2006). Tonic GABAA receptor-mediated currents in human brain. Eur. J. Neurosci. 24, 1157–1160. doi: 10.1111/j.1460-9568.2006.04989.x
Semyanov, A., Walker, M. C., and Kullmann, D. M. (2003). GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat. Neurosci. 6, 484–490.
Shen, H., Gong, Q. H., Aoki, C., Yuan, M., Ruderman, Y., Dattilo, M., et al. (2007). Reversal of neurosteroid effects at α4β2δ GABAA receptors triggers anxiety at puberty. Nat. Neurosci. 10, 469–477.
Smith, S. S., Gong, Q. H., Hsu, F. C., Markowitz, R. S., ffrench-Mullen, J. M. H., and Li, X. (1998). GABAA receptor α4 subunit suppression prevents withdrawal properties of an endogenous steroid. Nature 392, 926–930. doi: 10.1038/31948
Song, I., Savtchenko, L., and Semyanov, A. (2011). Tonic excitation or inhibition is set by GABAA conductance in hippocampal interneurons. Nat. Commun. 2, 376. doi: 10.1038/ncomms1377
Song, I., Volynski, K., Brenner, T., Ushkaryov, Y., Walker, M., and Semyanov, A. (2013). Different transporter systems regulate extracellular GABA from vesicular and non-vesicular sources. Front. Cell. Neurosci. 7:23. doi: 10.3389/fncel.2013.00023
Sperk, G., Schwarzer, C., Tsunashima, K., Fuchs, K., and Sieghart, W. (1997). GABAA receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits. Neuroscience 80, 987–1000. doi: 10.1016/S0306-4522(97)00146-2
Stell, B. M., Brickley, S. G., Tang, C. Y., Farrant, M., and Mody, I. (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 14439–14444. doi: 10.1073/pnas.2435457100
Stratmann, G., Sall, J. W., May, L. D., Bell, J. S., Magnusson, K. R., Rau, V., et al. (2009). Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology 110, 834–848. doi: 10.1097/ALN.0b013e31819c463d
Takayama, C., and Inoue, Y. (2005). Developmental expression of GABA transporter-1 and 3 during formation of the GABAergic synapses in the mouse cerebellar cortex. Brain Res. Dev. Brain Res. 158, 41–49. doi: 10.1016/j.devbrainres.2005.05.007
Vardya, I., Hoestgaard-Jensen, K., Nieto-Gonzalez, J. L., Dosa, Z., Boddum, K., Holm, M. M., et al. (2012). Positive modulation of δ-subunit containing GABAA receptors in mouse neurons. Neuropharmacology 63, 469–479. doi: 10.1016/j.neuropharm.2012.04.023
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Volk, D. W., Austin, M. C., Pierri, J. N., Sampson, A. R., and Lewis, D. A. (2000). Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical γ-aminobutyric acid neurons in subjects with schizophrenia. Arch. Gen. Psychiatry 57, 237–245. doi: 10.1001/archpsyc.57.3.237
Wafford, K. A., Van Niel, M. B., Ma, Q. P., Horridge, E., Herd, M. B., Peden, D. R., et al. (2009). Novel compounds selectively enhance δ subunit containing GABAA receptors and increase tonic currents in thalamus. Neuropharmacology 56, 182–189. doi: 10.1016/j.neuropharm.2008.08.004
Walsh, C. A. (1999). Genetic malformations of the human cerebral cortex. Neuron 23, 19–29. doi: 10.1016/S0896-6273(00)80749-7
Wang, D. D., and Kriegstein, A. R. (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28, 5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008
Wang, D. S., Zurek, A. A., Lecker, I., Yu, J., Abramian, A. M., Avramescu, S., et al. (2012). Memory deficits induced by inflammation are regulated by α5-subunit-containing GABAA receptors. Cell Rep. 2, 488–496. doi: 10.1016/j.celrep.2012.08.022
Wang, T., Kumada, T., Morishima, T., Iwata, S., Kaneko, T., Yanagawa, Y., et al. (2013). Accumulation of GABAergic neurons, causing a focal ambient GABA gradient, and downregulation of KCC2 are induced during microgyrus formation in a mouse model of polymicrogyria. Cereb. Cortex. doi: 10.1093/cercor/bhs375 [Epub ahead of print].
Weeber, E. J., Jiang, Y. H., Elgersma, Y., Varga, A. W., Carrasquillo, Y., Brown, S. E., et al. (2003). Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 23, 2634–2644.
Wei, W., Zhang, N., Peng, Z., Houser, C. R., and Mody, I. (2003). Perisynaptic localization of δ subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J. Neurosci. 23, 10650–10661.
Wilson, I. A., Ikonen, S., Gallagher, M., Eichenbaum, H., and Tanila, H. (2005). Age-associated alterations of hippocampal place cells are subregion specific. J. Neurosci. 25, 6877–6886. doi: 10.1523/JNEUROSCI.1744-05.2005
Wisden, W., Cope, D., Klausberger, T., Hauer, B., Sinkkonen, S. T., Tretter, V., et al. (2002). Ectopic expression of the GABAA receptor α6 subunit in hippocampal pyramidal neurons produces extrasynaptic receptors and an increased tonic inhibition. Neuropharmacology 43, 530–549. doi: 10.1016/S0028-3908(02)00151-X
Wu, Y., Wang, W., and Richerson, G. B. (2003). Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J. Neurophysiol. 89, 2021–2034. doi: 10.1152/jn.00856.2002
Yamada, J., Furukawa, T., Ueno, S., Yamamoto, S., and Fukuda, A. (2007). Molecular basis for the GABAA receptor-mediated tonic inhibition in rat somatosensory cortex. Cereb. Cortex 17, 1782–1787. doi: 10.1093/cercor/bhl087
Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J., and Fukuda, A. (2004). Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 557,829–841. doi: 10.1113/jphysiol.2004.062471
Yang, S., Weiner, B. D., Zhang, L. S., Cho, S. J., and Bao, S. (2011). Homeostatic plasticity drives tinnitus perception in an animal model. Proc. Natl. Acad. Sci. U.S.A. 108, 14974–14979. doi: 10.1073/pnas.1107998108
Yee, B. K., Hauser, J., Dolgov, V. V., Keist, R., Mohler, H., Rudolph, U., et al. (2004). GABA receptors containing the α5 subunit mediate the trace effect in aversive and appetitive conditioning and extinction of conditioned fear. Eur. J. Neurosci. 20, 1928–1936. doi: 10.1111/j.1460-9568.2004.03642.x
Yoon, J. H., Maddock, R. J., Rokem, A., Silver, M. A., Minzenberg, M. J., Ragland, J. D., et al. (2010). GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J. Neurosci. 30, 3777–3781. doi: 10.1523/JNEUROSCI.6158-09.2010
Zorn, S. H., and Enna, S. J. (1985). The effect of mouse spinal cord transection on the antinociceptive response to the γ-aminobutyric acid agonists THIP (4,5,6,7-tetrahydroisoxazolo [5,4-c]pyridine-3-ol) and baclofen. Brain Res. 338, 380–383. doi: 10.1016/0006-8993(85)90173-8
Keywords: GABA, GAT, extrasynaptic, ambient, transporter, tonic inhibition, neurological disease, GABAA receptor
Citation: Egawa K and Fukuda A (2013) Pathophysiological power of improper tonic GABAA conductances in mature and immature models. Front. Neural Circuits 7:170. doi: 10.3389/fncir.2013.00170
Received: 02 August 2013; Paper pending published: 12 September 2013;
Accepted: 28 September 2013; Published online: 24 October 2013.
Edited by:
Alexey Semyanov, RIKEN Brain Science Institute, JapanReviewed by:
Vincenzo Crunelli, Cardiff University, UKCopyright © 2013 Egawa and Fukuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsuo Fukuda, Department of Neurophysiology, Hamamatsu University School of Medicine, 20-1 Handayama 1-chome, Higashi-ku, Hamamatsu, Shizuoka 431-3192, Japan e-mail:YXhmdWt1ZGFAaGFtYS1tZWQuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.