 Dina Greenberg1,2Robert D’Cruz1,2†Jon L. Lacanlale1,2†Christopher J. Rowan1
Dina Greenberg1,2Robert D’Cruz1,2†Jon L. Lacanlale1,2†Christopher J. Rowan1 Norman D. Rosenblum1,2,3,4*
Norman D. Rosenblum1,2,3,4*- 1Program in Developmental and Stem Cell Biology, Hospital for Sick Children, Toronto, ON, Canada
- 2Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 3Division of Nephrology, Hospital for Sick Children, Toronto, ON, Canada
- 4Department of Pediatrics, University of Toronto, Toronto, ON, Canada
CAKUT is the leading cause of end-stage kidney disease in children and comprises a broad spectrum of phenotypic abnormalities in kidney and ureter development. Molecular mechanisms underlying the pathogenesis of CAKUT have been elucidated in genetic models, predominantly in the mouse, a paradigm for human renal development. Hedgehog (Hh) signaling is critical to normal embryogenesis, including kidney development. Hh signaling mediates the physiological development of the ureter and stroma and has adverse pathophysiological effects on the metanephric mesenchyme, ureteric, and nephrogenic lineages. Further, disruption of Hh signaling is causative of numerous human developmental disorders associated with renal malformation; Pallister-Hall Syndrome (PHS) is characterized by a diverse spectrum of malformations including CAKUT and caused by truncating variants in the middle-third of the Hh signaling effector GLI3. Here, we outline the roles of Hh signaling in regulating murine kidney development, and review human variants in Hh signaling genes in patients with renal malformation.
Introduction
Congenital Anomalies of the Kidney and Urinary Tract (CAKUT) constitute 20% of all congenital malformations and are the leading cause of end-stage kidney disease in children (1, 2). CAKUT comprises a broad phenotypic spectrum consisting of a range of kidney (renal agenesis, renal aplasia, renal dysplasia, hydronephrosis, multicystic dysplastic kidney, ectopic kidney, horseshoe kidney, renal tubular dysgenesis) and ureter (ureteropelvic junction obstruction (UPJO), vesicoureteral reflux (VUR), duplex collecting system, megaureter) anomalies (3, 4). While some forms of CAKUT can be mild with little or no functional impact, severe CAKUT at birth is associated with greater risk for chronic kidney disease and kidney replacement therapy (KRT), as well as increased risk of death in preterm infants (5, 6). CAKUT can appear as an isolated condition, and less commonly in syndromic form in concert with other extrarenal manifestations (i.e., defects in other organ systems) (7).
CAKUT is caused by disruption of the processes that regulate renal morphogenesis; this can be a result of in utero environmental, epigenetic, or genetic factors (3). To date, most investigations have focused on monogenic causes of CAKUT, with causative genes identified in both isolated and syndromic forms. However, monogenic CAKUT only accounts for 15-20% of all CAKUT cases, and poses a diagnostic challenge owing to incomplete penetrance and variable phenotypic expression (8–11). Other genetic causes of CAKUT include copy number variations (CNVs), chromosomal abnormalities, and, less commonly, oligogenic or polygenic CAKUT (7, 12–16).
Understanding the process of renal formation is key to elucidating the mechanisms which give rise to CAKUT. Human kidney development begins at week 3 of embryonic development with the formation of the pronephros, which regresses and is followed by formation of the mesonephros at 4 weeks and the metanephros at 5 weeks gestational age. Murine kidney development begins with reciprocal inductive interactions between the nephric duct (ND) and the metanephric mesenchyme (MM), which originate at embryonic day 8 (E8.0) from the anterior and posterior Osr1-expressing intermediate mesoderm (IM), respectively (17–19). At E10.5, inductive signaling from the MM stimulates the outgrowth of the ureteric bud (UB) from the ND. In the developing human embryo, this correlates with metanephros development at 5 weeks gestational age (20). The UB bifurcates into a T-shaped structure at E11.5 and undergoes iterative rounds of branching morphogenesis up until post-natal day 2 (P2) (or 34 weeks of human gestation); a process that ultimately gives rise to the mature renal collecting system (21–24). In parallel, inductive signaling from the UB signals the MM to differentiate into two distinct multipotent self-renewing progenitor populations: the Foxd1-expressing stromal and Six2 -expressing nephron progenitor lineages (18, 25, 26). The Foxd1-expressing stromal lineage subsequently gives rise to endothelial cells, vascular smooth muscle cells, pericytes, mesangial cells, and fibroblasts (25, 27). Six2-expressing nephron progenitors undergo mesenchymal-to-epithelial transition (MET) to form renal vesicles, wherein proximal/distal polarity is established. Polarized renal vesicles subsequently develop into comma-shaped bodies, S-shaped bodies, and finally, into mature nephrons (28). Murine nephrogenesis continues up until P4, while human embryonic nephrogenesis terminates at week 36 of gestation (29, 30). Overall, these processes are tightly regulated by a number of signaling pathways and factors, disruption of which has been shown to contribute to the phenotypic spectrum of CAKUT.
Hedgehog signaling
Hedgehog (Hh) signaling controls mammalian organogenesis, including that of the kidney (31). Dysregulation of Hh signaling is associated with various types of cancers, such as medullablastoma and basal cell carcinoma (32, 33). Activation of the Hh pathway can occur through a canonical (Glioma-Associated Oncogene (GLI)-dependent) or non-canonical (GLI-independent) mechanism. While non-canonical activation of the Hh pathway has been implicated in cancer, its role in mammalian kidney development has not yet been interrogated (34–36). Mammalian canonical Hh signaling is initiated when Hh ligand binds and inactivates the transmembrane protein receptor Patched1 (PTCH1), which results in PTCH1 being internalized and degraded. Degradation of PTCH1 results in accumulation of Smoothened (SMO), an intracellular receptor, at the primary cilium and activation of downstream cascades resulting in the processing and translocation of GLI transcription factors into the nucleus (32, 37). Absence of Hh ligand causes PTCH1 binding and inhibition of SMO activity. This results in phosphorylation of downstream GLI proteins, which are then targeted for processing. Phosphorylation of GLI proteins by combinations of Protein Kinase A and C (PKA/C), Casein Kinase 1 (CK1), Glycogen Synthase Kinase 3β (GSK3β), and Dual-Specificity Yak1-Related Kinase (DYRK1) targets GLI proteins for proteasome-dependent processing, giving rise to a truncated repressor form that translocates to the nucleus to repress Hh target gene transcription. In vertebrates, there are three GLI family members: GLI1, GLI2, and GLI3. GLI1 functions as a transcriptional activator, while GLI2 and GLI3 can function as both activators or repressors depending on their truncation. The major determinant of a cellular response to Hh ligand is the ratio of GLI activator to GLI repressor (GLIA/GLIR), as this ratio has been shown to control processes of organogenesis (38, 39).
Hedgehog signaling in kidney development
In mammals, there are three Hh genes, Sonic Hedgehog (Shh), Indian Hedgehog (Ihh), and Desert Hedgehog (Dhh). Of these, only Shh and Ihh expression is detectable in the developing mammalian kidney; Shh is expressed in the ureteric lineage and localizes to the epithelium of the presumptive ureter and medullary collecting ducts, while Ihh is localized to the nephrogenic tubules (40–43).
Previous studies have shown that the GLI family of transcription factors control the expression of cell cycle regulators N-myc and CyclinD1 (44), as well as genes critical to renal patterning such as Foxd1, Pax2, Sall1, and Tgfb2 (44, 45). Studies in multiple organ systems and cancers have also demonstrated that GLI transcription factors activate other Hh pathway components such as Ptch1, Smo, Gli1, Gli2, and Gli3, leading to positive and negative feedback of the Hh signaling pathway. Analysis of the temporospatial localization of these Hh signaling factors has therefore provided evidence of important roles for Hh signaling across murine and human kidney development. These studies are outlined below and are further supported by data drawn from published single-cell datasets of the developing mouse kidney (Figure 1).
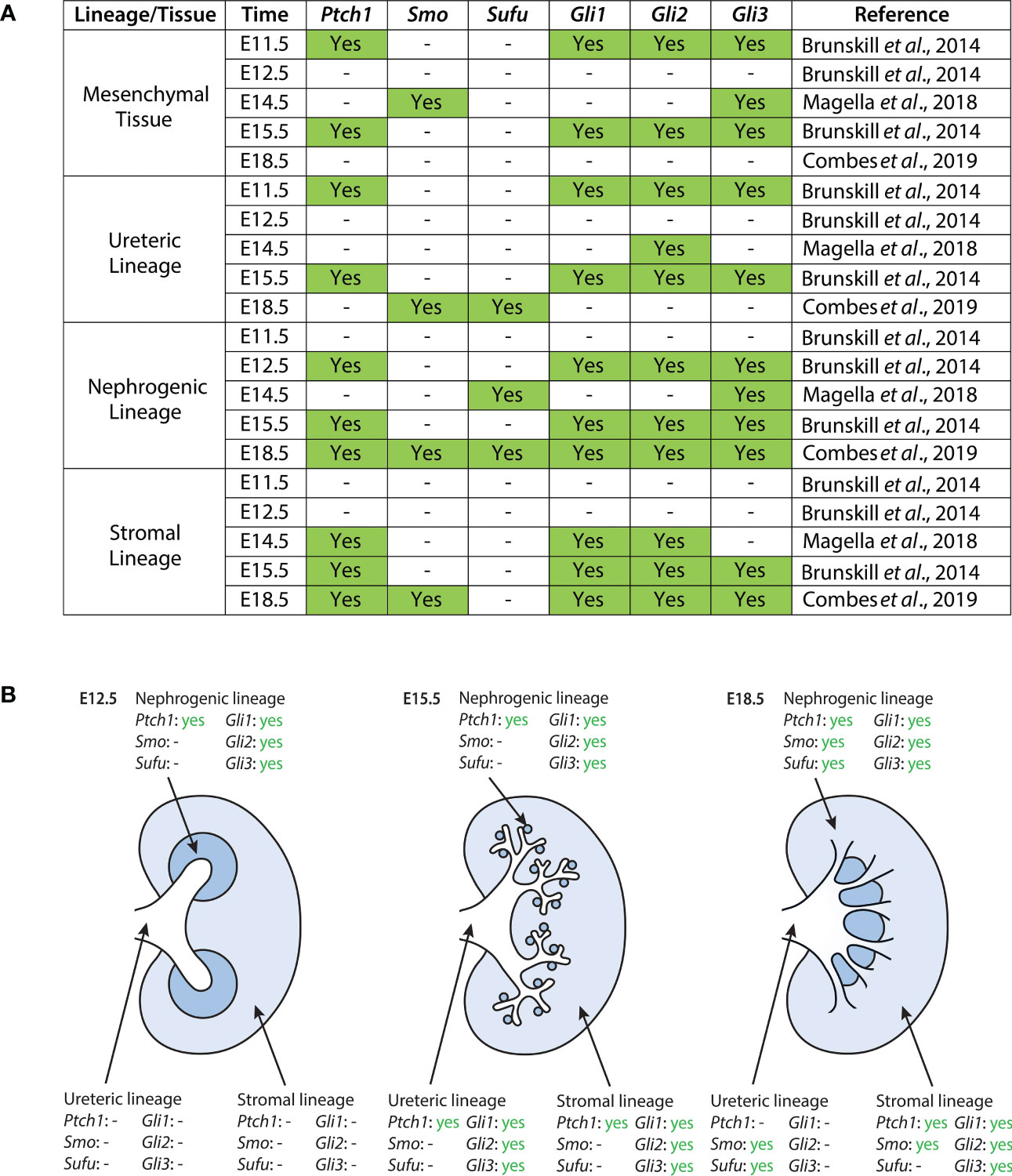
Figure 1 Expression of Hh components in the developing mouse kidney as reported in published single-cell RNA sequencing datasets (40, 41, 46). (A) Datasets were screened for expression of Ptch1, Smo, Sufu, Gli1, Gli2, and Gli3. Expression here is reported as binary; “yes” indicates statistically significant expression of a gene, as determined by the study. The exception to this is the dataset from Magella et al. (E14.5), which reports a cluster of greatest correlation for each gene; here, we include correlations based on both the Fluidigm and Chromium platforms, as they are the ones used by Brunskill et al. and Combes et al., respectively. A dash indicates either no statistically significant expression of a gene within a lineage cluster, or the absence of expression data reported altogether. (B) Expression of Hh pathway components in the developing mouse kidney at E12.5, E15.5, and E18.5. Data shown here is provided in greater detail in (A).
Ptch1
During murine kidney development, Ptch1 expression is weakly localized to the epithelium of the presumptive ureter and medullary collecting ducts, and strongly expressed in the surrounding stromal mesenchyme (47). In the adult mouse kidney, Ptch1 expression is localized at the corticomedullary junction (42). Single-cell data suggests that between E11.5 and E18.5, Ptch1 is most strongly expressed in the mesenchymal and stromal clusters, with additional expression in the nephrogenic and ureteric clusters (40, 41, 46).
Smo
Single-cell data suggests that in the E14.5 mouse kidney, Smo is expressed most strongly in the mesenchymal and endothelial cell lineages (46). Further, Combes et al. detected expression of Smo in the stromal, nephrogenic, and endothelial lineages of the E18.5 murine kidney (41).
Gli1
Expression of Gli1 in the murine embryonic kidney is localized to the interstitial stromal cells (42, 44, 45). Single-cell data shown by Brunskill et al. further demonstrated low levels of Gli1 expression in the ureteric lineage at E11.5 and E15.5, and Gli1 expression in developing mesenchymal and stromal lineages (40, 41, 46).
Gli2
Gli2 is highly expressed within the medulla of the adult murine kidney but localized to the cortex at E14.5 (42, 44). Single-cell expression data suggests Gli2 is largely expressed in the mesenchymal and stromal lineages, with additional expression in the nephrogenic and ureteric lineages (40, 41, 46).
Gli3
Gli3 protein expression is ubiquitous at E14.5 (44). Single-cell expression data demonstrates that Gli3 is highly expressed in mesenchymal clusters at E11.5 and E14.5, with subsequent expression in stromal and nephrogenic clusters. Gli3 expression is also detected in the ureteric lineage at E11.5 and E15.5 (40, 41, 46).
Physiological roles for hedgehog signaling in murine kidney development
Previous studies using genetic mouse models have demonstrated important physiological roles for Hh signaling during murine kidney development. Mice with global deficiency of Shh ligand display a spectrum of renal abnormalities including bilateral renal aplasia and ectopic dysplastic kidneys (44, 48). Subsequent studies have identified key physiological roles for Hh signaling in controlling ureter and stromal development. These studies are outlined below and summarized in Table 1 and Figure 2.
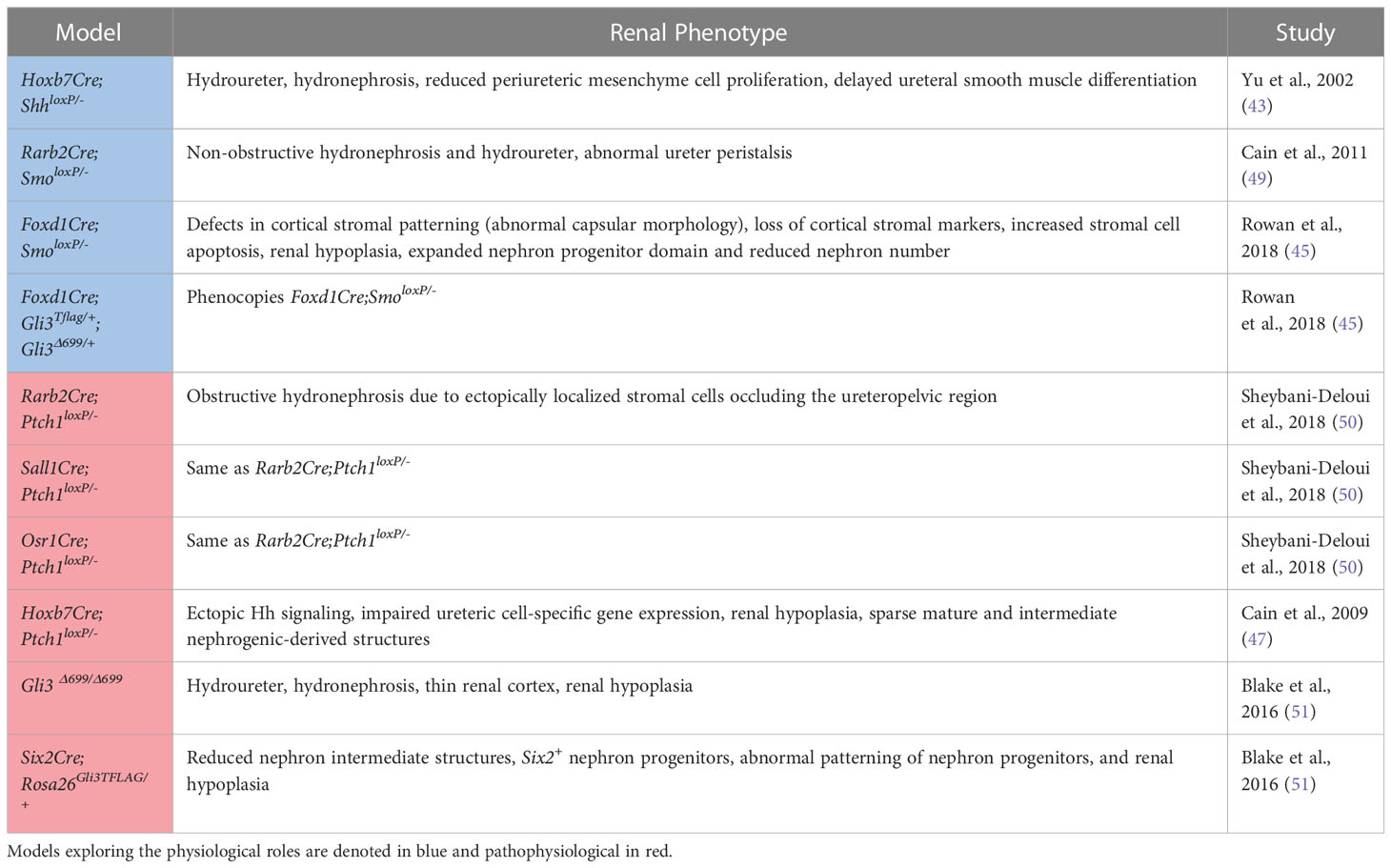
Table 1 A summary of the mouse models used to investigate the physiological and pathophysiological roles of Hedgehog signaling in the developing kidney along with the associated phenotypes.
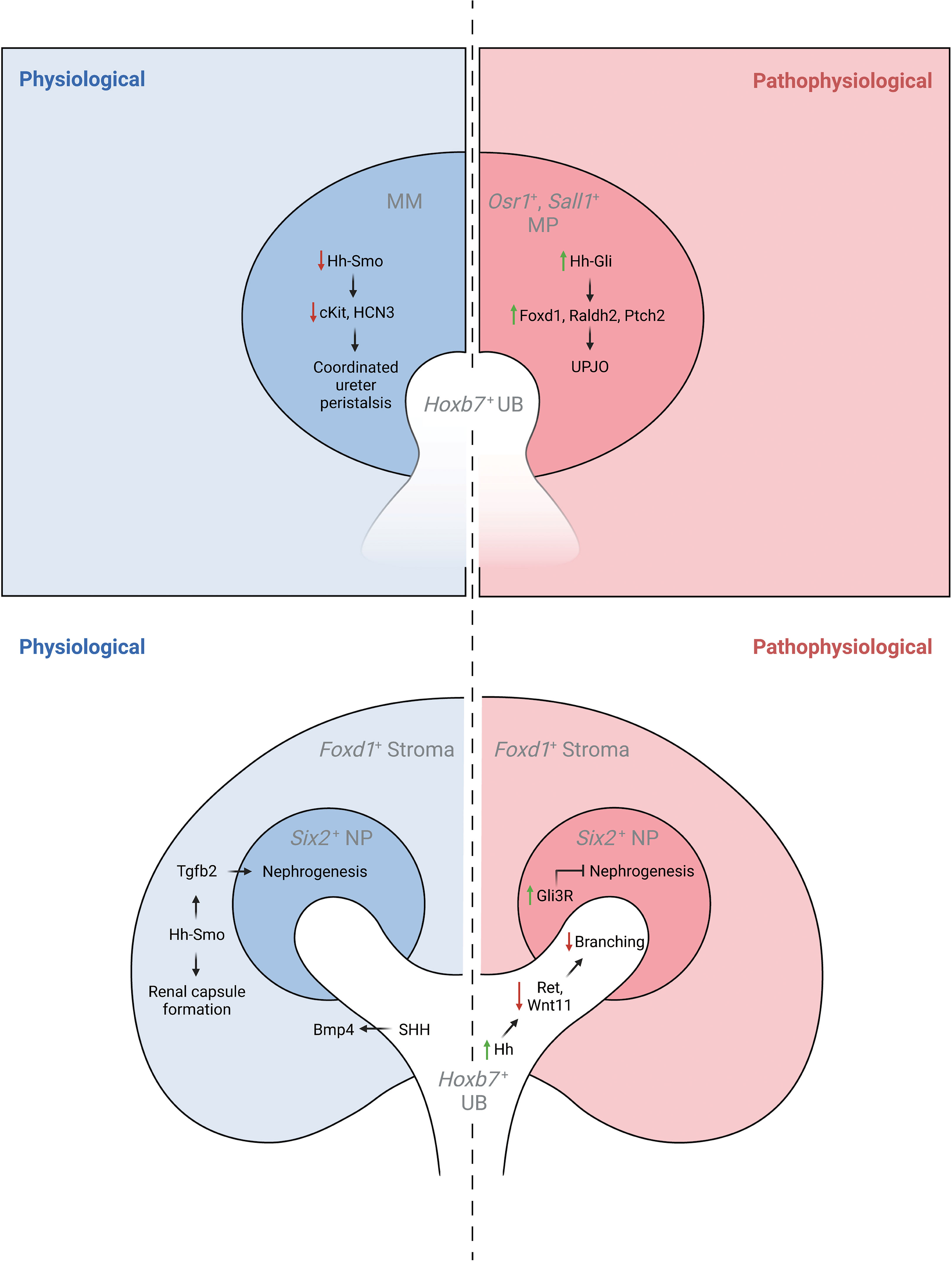
Figure 2 The physiological and pathophysiological roles of Hh signaling in specific lineages of the developing kidney. MM, metanephric mesenchyme; MP, mesenchymal progenitors; UB, ureteric bud.
Ureter development
Genetic inactivation of Shh in the ureteric lineage results in hydroureter and hydronephrosis, a reduction in periureteric mesenchyme cell proliferation, and delayed ureteral smooth muscle differentiation associated with reduced expression of Bmp4 and Ptch1 (43). Further, targeted inactivation of Smo in intermediate mesenchyme that gives rise to the metanephros (Rarb2Cre;Smolox/P-) results in non-obstructive hydronephrosis and hydroureter. In contrast to mice with genetic deficiency of Shh in the ureteric lineage, Rarb2Cre;SmoloxP/- mice exhibit normal ureteric smooth muscle but abnormal ureter peristalsis and a reduction in expression of urinary tract pacemaker cell (utPMC) markers cKit and Hcn3 (49, 52). Together, these findings demonstrate a physiologic role for Hh signaling in regulating urinary tract pacemaker activity and development of urinary pacemaker cells.
Stromal development
Foxd1+ stromal cells play important roles in regulating renal development (53). Expression data demonstrates that Gli1 is present in the developing cortical stroma, suggesting a role for Hh signaling in cortical stromal function (42, 45) and single-cell RNA sequencing demonstrates expression of Gli1 in stromal clusters starting at E14.5 (40, 41, 46). Recent studies have demonstrated that genetic deletion of Smo in the stromal lineage (Foxd1Cre;SmoloxP/-) causes loss of the cortical stroma after Foxd1+ cells initially migrate to take up residence in the renal cortex, resulting in loss of cortical stromal markers such as Foxd1 and Raldh2, and near-absence of the renal capsule (Rowan, 2018). Inactivation of stromal Hh signaling also negatively regulates the nephrogenic domain, as Foxd1Cre;SmoloxP/- mice exhibit renal hypoplasia with an expanded nephron progenitor domain and fewer nephrons (45). The contribution of GLI3R to these defects was demonstrated in mice with obligate GLI3R expression targeted to the stroma (Foxd1Cre;Gli3Tflag/+;Gli3Δ699/+), in which these defects were replicated, and in mice with genetic deletion of Gli3 in a Foxd1Cre;SmoloxP/- background, in which these abnormalities were rescued (45). While the downstream mediators of these Hh-dependent effects are only beginning to be elucidated, it has been demonstrated that Tgfb signaling acts downstream of Hh signaling to exert cell autonomous and non-cell autonomous functions (45).
Pathophysiological roles for hedgehog signaling in murine kidney development
Increased Hh activity in non-renal tissue causes cancer of different types and has been shown to be pathogenic during lung and limb development (54–56). Here, we review how Hh signaling targeted to specific embryonic kidney cell lineages is detrimental to renal morphogenesis. These studies are reviewed below and summarized in Table 1 and Figure 2.
Metanephric mesenchyme
Sheybani-Deloui et al. investigated the role of Hh constitutive activation in cells giving rise to the metanephric mesenchyme using genetic mouse models with Cre-mediated excision of Ptch1. Tamoxifen-induced Cre-mediated Ptch1 deficiency in Osr1+ cells, the progenitors to both nephrogenic and stromal cells, starting at E9.5 caused obstructive hydronephrosis and ureteropelvic junction (UPJ) obstruction in embryonic kidneys. Investigation of underlying cellular and molecular mechanisms demonstrated ectopic localization of Foxd1+ and Raldh2+ cells at the UPJ with resulting blockage of urine outflow. Lineage tracing experiments confirmed that these blocking cells are, indeed, derived from metanephric mesenchyme. Analysis of RNA expressed by these cells revealed other markers of the cortical stroma and Hh-targets. Analysis of UPJ tissue in children with UPJO demonstrated expression of both stromal and Hh-target genes in a subset of affected patients (50). Together, these findings suggest that increased Hh signaling in Osr1+ progenitor cells during a narrow time window causes abnormal stromal cell localization to the UPJ with resultant obstruction.
Ureteric lineage
Increased Hh signaling activity in the Hoxb7+ ureteric lineage via deletion of Ptch1 (Hoxb7Cre;Ptch1loxP/-) results in ectopic Hh signaling in ureteric branch tips, impaired expression of genes specific to ureteric tip cells, and renal hypoplasia. Overexpression of GLI3R in mice with Ptch1-deficiency in the ureteric lineage rescued the normal pattern of Hh signaling and expression of ureteric genes as well as renal hypoplasia (47).
Germline expression of GLI3R (Gli3D699/D699) results in a complex CAKUT phenotype consisting of hydroureter, hydronephrosis, a thin renal cortex, and renal hypoplasia. Among the mechanisms underlying these phenotypes is impairment of ureteric bud lengthening (51). Collectively, these studies demonstrate that regulation of Hh activity and GLI3R expression is critical to development of the ureteric bud and its branches.
Nephrogenic lineage
Obligate expression of GLI3R (Six2Cre;Rosa26Gli3TFLAG/+) targeted to the Six2+ nephrogenic lineage inhibits nephrogenesis via inhibition of Six2+ progenitor cell self-renewal resulting in renal hypoplasia (51).
Hh signaling in human kidney development
Below, we outline human renal malformations characterized by disruption of genes required for Hh signaling.
SHH
Numerous cases of CAKUT in patients carrying terminal deletions in the long arm of chromosome 7q32-36 (encompassing the SHH locus) have been reported with phenotypes including renal hypoplasia, renal dysplasia, ectopic kidney, duplex collecting system and VUR (Table 2). Patients had other extrarenal manifestations including, but not limited to, holoprosencephaly, facial dimorphisms, and developmental delay (57–60). Several SHH variants have further been identified as resulting in CAKUT; a nonsense mutation (c.388G>T) resulting in truncation at exon 2 of SHH caused renal hypoplasia along with holoprosencephaly and facial dimorphisms in a male patient (61). A female patient carrying the c.1061T>C missense mutation in exon 3 of SHH had renal hypoplasia, holoprosencephaly, cerebellar, and retinal defects (61). SHH encompasses 3 exons; nascent SHH undergoes proteolysis to generate an N-terminal (N-SHH; comprising of exons 1 and 2) or C-terminal (C-SHH; comprising of exons 1 and 3) form (Figure 3). Although it is N-SHH that ultimately binds PTCH1 to initiate the Hh signaling cascade, C-SHH is critical to the processing of N-SHH (62, 63). Thus, disruption to SHH may be detrimental to human renal morphogenesis.
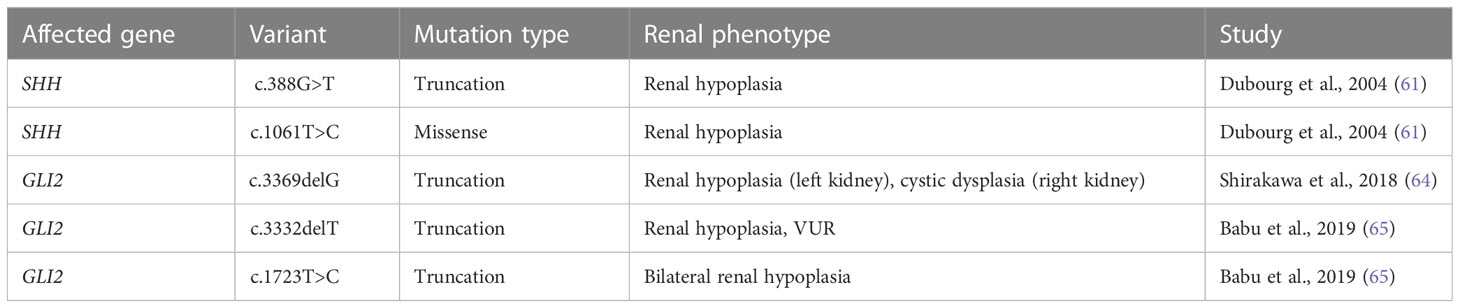
Table 2 SHH and GLI2 variants identified in humans and their associated renal phenotypes.
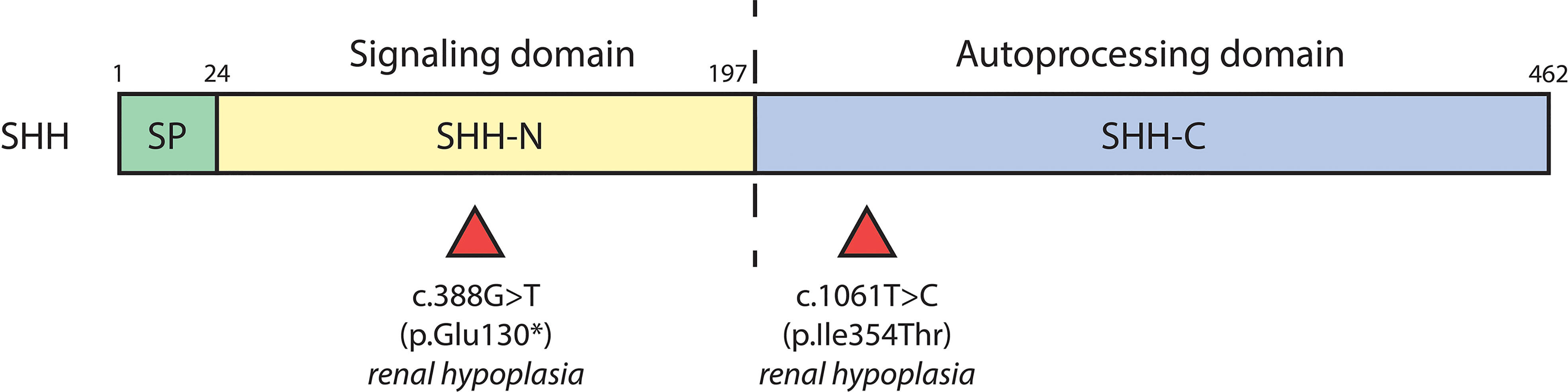
Figure 3 SHH variants associated with CAKUT. Schematic diagram of the biochemical domains of human SHH shows the signal peptide (SP, a.a. 1-23), signaling domain (SHH-N, a.a. 24-197), and autoprocessing domain (SHH-C, a.a. 198-462). The dashed line indicates the site of autocatalytic cleavage. Red triangles underneath denote location of SHH variants and their associated renal phenotypes.
GLI2
GLI2 variants have been linked to polydactyly and dysmorphic facial features. There have only been three recorded cases of GLI2 variants resulting in CAKUT, outlined in Table 2 and Figure 4. One study identified a male carrying one copy of a c.3369delG GLI2 allele, which was predicted to generate a truncation in the C-terminal GLI2 transactivation domain. The patient presented with a left hypoplastic kidney, a right cystic dysplastic kidney, as well as urinary tract obstruction, eventually progressing to end-stage-renal failure. Other features included postaxial polydactyly and cognitive developmental delay (64). Another study identified a female with a heterozygous c.3332delT GLI2 variant, similarly predicted to cause C-terminal truncation. The patient had renal hypoplasia and VUR, as well as congenital heart disease, cleft palate, deafness, ocular, and cognitive impairment. The same study identified another female patient with a heterozygous c.1723T>C GLI2 variant with bilateral renal hypoplasia, polydactyly, cleft palate, craniofacial abnormalities, and pituitary hypoplasia. The c.1723C>T variant was predicted to cause truncation in the C-terminal portion of the DNA binding domain of GLI2. In vitro co-transfection of a truncated c.1723C>T GLI2 plasmid with a full length GLI2 plasmid showed a 55% reduction in transcriptional activation (65). GLI2 is regarded as a potent activator of Hh signaling, but owing to inefficient processing, is a weak transcriptional repressor, suggesting a mechanism of loss of function of GLI2A that results in decreased Hh activity, rather than constitutive repression (66, 67).
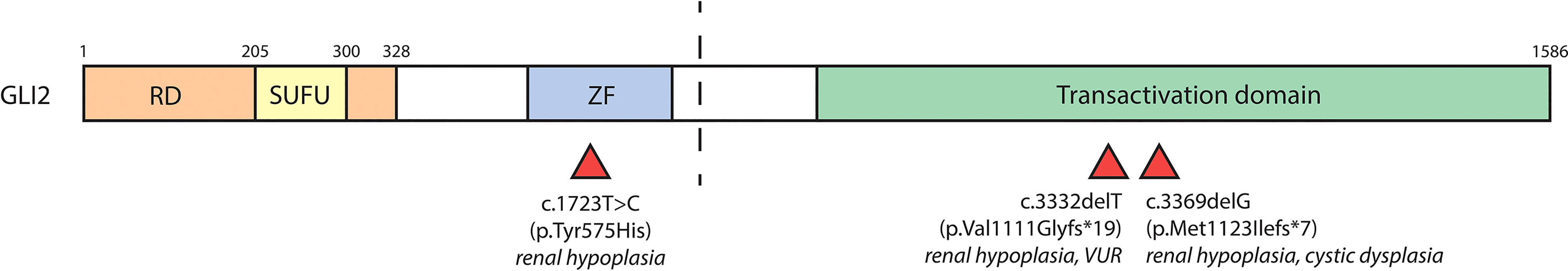
Figure 4 GLI2 variants associated with CAKUT. The human GLI2 protein is comprised of a repressor domain (RD, a.a. 1-328), SUFU-binding domain (SUFU, a.a. 205-300), zinc finger DNA-binding domain (ZF) and C-terminal transactivation domain. The zinc finger and transactivation domains of GLI2 have not been well positionally defined. The dashed line indicates the site of proteolytic cleavage that results in formation of GLI2R. Red triangles underneath denote location of GLI2 variants and their associated renal phenotypes.
GLI3
Human variants in the GLI3 gene are known to cause Pallister-Hall Syndrome (PHS; OMIM: 146510) and Greig cephalopolysyndactyly syndrome (GCPS; OMIM: 175700), and non-syndromic postaxial polydactyly types A1 and B (PAPA1/PAPB; OMIM: 174200) and preaxial polydactyly type IV (PPD-IV; OMIM: 174700). Congenital diseases driven by mutations in GLI3 are inherited in an autosomal dominant fashion and give rise to a broad spectrum of clinical phenotypes. A diagnosis of Pallister-Hall Syndrome is met upon the criteria of hypothalamic hamartoma, mesoaxial polydactyly, and a confirmed mutation in the central-third of the GLI3 gene (68–71). PHS is characterized by a further spectrum of multi-organ malformations, including CAKUT (71, 72). An estimated 27% of PHS patients present with CAKUT, with hypoplasia (with or without dysplasia) and agenesis being most common (accounting for just under one third of PHS cases with CAKUT, each). Other types of CAKUT found in PHS include dysplasia, ectopic kidney, single kidney, and VUR, all of which are reported to occur unilaterally and bilaterally at equal frequency. PHS patients with CAKUT are more likely to experience craniofacial defects, bifid epiglottis, and genital hypoplasia (72).
Several large patient cohort studies have allowed for the identification of distinct genotype-phenotype correlations of GLI3 variants (71–76). Patients with PHS have truncating variants (frameshift or nonsense) almost exclusively within the central-third (amino acids 661-1159) of the GLI3 gene. As these truncations take place C-terminally to the zinc finger DNA-binding domain of GLI3 and cause loss of the N-terminal transactivation domain, it is believed that the resulting protein mimics a constitutively active GLI3R (74, 77, 78). It has been proposed that constitutive GLI3R activity is the driver of CAKUT phenotypes in PHS; this notion is supported by the exclusive association of CAKUT with PHS-causing GLI3 variants, as well as the requirement for balanced GLI3A and GLI3R activities in normal kidney formation (31, 44, 45, 51, 79). Indeed, GLI3 variants found in GCPS or isolated polydactylies, which have no renal manifestations, are proposed to cause functional haploinsufficiency of the GLI3 protein (74–76).
Although all PHS-causing GLI3 variants are predicted to generate GLI3R-like transcripts, only a quarter of PHS patients present with CAKUT. A recent patient cohort study by McClelland et al. investigated the link between GLI3 variants and renal phenotype in PHS patients; the study demonstrated that PHS-causing GLI3 variants are evenly distributed within the central third region of the GLI3 gene, and found no correlation between locus of mutagenesis and prevalence of CAKUT (72). It may be, however, that the spectrum of renal phenotypes found in PHS patients is mediated by GLI3 variant-specific mechanisms that are difficult to elucidate from genotype alone.
GLI3 mRNA stability may be an important mediator of GLI3 variant induced renal phenotype. One study identified three cases of GLI3 variants (c.1320_1321insT, c.2372delC, and c.2374C>T) in patients with isolated polydactyly that were subject to nonsense-mediated mRNA decay (NMD). C.2372delC and c.2374C>T fall within the central region of GLI3 that is typically associated with PHS; interestingly, the c.2374C>T variant has been identified in six other cases of GCPS (74, 80). Given that GCPS is characterized by loss of GLI3 function, whereas PHS is believed to arise from constitutive GLI3R activity, it is possible that PHS-causing GLI3 variants are less likely be subject to NMD. Further investigation is required to elucidate the contribution of NMD to CAKUT within the PHS phenotypic spectrum.
Renal phenotype in PHS may also be a consequence of GLI3 variant structure and function. Physiological processing of nascent GLI3 into GLI3A or GLI3R requires interaction with multiple binding partners at domains spanning the full length of GLI3, outlined in Figure 5. PHS-causing variants fall within the central region of GLI3 (amino acids 661-1159), and without exception truncate the TA1 and TA2 domains, thus disrupting GLI3A activity. It is possible that variant-specific positional truncation of GLI3 is determinant of the success of posttranslational modification, particularly within regions critical to GLI3R processing, and subsequent proteolytic cleavage (67, 81). Alternatively, it may be that truncated GLI3 variants are of sufficient structural similarity to wildtype proteosomally cleaved GLI3R and are thus able to emulate GLI3R activity. Future studies of patient GLI3 variants would be beneficial to our understanding of the molecular mechanisms underlying phenotypic variation in PHS.
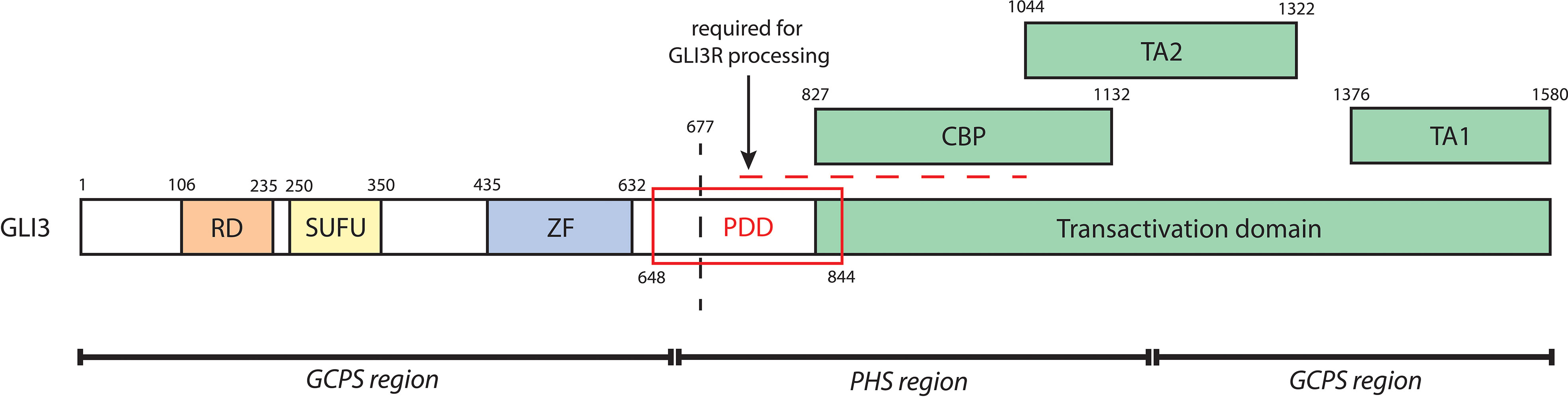
Figure 5 A schematic of biochemical domains of human GLI3 containing the repressor domain (RD, a.a. 106-235), SUFU-binding domain (SUFU, 250-350), zinc finger DNA-binding domain (ZF, a.a. 435-632), and transactivation domain (a.a. 827-1580). The transactivation domain, which is critical to GLI3A, contains a CREB-binding protein domain (CBP, a.a. 827-1132), transactivation domain 2 (TA2, a.a. 1044-1322), and transactivation domain 1 (TA1, a.a. 1376-1580). The processing determinant domain (PDD, a.a. 648-844) is critical to the proteolytic cleavage of GLI3. The horizontal dashed line further denotes a series of sites required for GLI3R processing; GLI3 is phosphorylated at its PKA sites (a.a. 849, 869, 877, 907, 980, 1006) and GSK3β sites (a.a. 861, 873, 903). This leads to binding of βTrCP (at GLI3 a.a. 856, 864, 855, 894) and subsequent ubiquitination by SCFTrCP (at GLI3 a.a. 773, 779, 784, 800), causing proteolytic cleavage (indicated by the vertical dashed line) and thus generation of GLI3R. Regions of mutagenesis of GLI3 associated with either GCPS (a.a. 1-660, 1160-1580) or PHS (a.a. 661-1159) are indicated at the bottom.
Lastly, given the extensive network of genes that govern kidney development, it is also possible that the variable penetrance of CAKUT in PHS may be a factor of not only specific GLI3 variants, but also secondary genetic background factors (i.e., modifier gene interactions, stochastic effects, epigenetic mechanisms), as has been demonstrated in other human diseases (3, 82).
Further investigation is required to elucidate the molecular mechanism by which GLI3 variants associated with PHS give rise to variable penetrance of CAKUT. While studies in mouse models have shed light on the effects of constitutive GLI3R activity on kidney development, the hemizygous Gli3Δ699/+ mouse model is phenotypically normal, indicating that there are likely differences in Hh-mediated regulation between murine and human kidney development. For example, there may be variability in Hh sensitivity across human and mouse kidney cell lineages. Similarly, it appears that GLI3R exerts its effects on mouse and human tissue at different dosages. Use of a human kidney organoid model would permit the interrogation of these key differences. Kidney organoids could be used to assay the impacts of Hh signaling on distinct kidney cell lineages, as well as the effects of gain of function or loss of function of Hh signaling. Changes to Hh signaling and kidney development driven by specific GLI3 variants can be similarly investigated in kidney organoids. Further, the use of an isogenic kidney organoid model, where possible, has the added benefit of preservation of genetic background, which may be an important contributor to the phenotypic variance seen in PHS, as well as other forms of CAKUT (83, 84).
Conclusion
CAKUT represents a broad spectrum of kidney and urinary tract malformations that are a significant source of morbidity in the pediatric population. Dysregulation of genes associated with normal kidney and ureter development lead to the development of CAKUT. Previous work using genetic mouse models has demonstrated that Hh signaling plays important physiological and pathophysiological roles in kidney and ureter morphogenesis, and mutations in Hh genes have been shown to cause human renal malformation. Future studies using a combination of genetic mouse models and human kidney organoids can help elucidate the precise variant-specific molecular mechanism underlying Hh-mediated renal malformation.
Author contributions
All authors contributed to conception and design of the review. DG, RD, and JL wrote the manuscript. All authors contributed to manuscript revision and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Canadian Institute for Health Research and a Tier I Canada Research Chair to NR; MD/PhD funding from the University of Toronto to RD.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chesnaye N, Bonthuis M, Schaefer F, Groothoff JW, Verrina E, Heaf JG, et al. Demographics of paediatric renal replacement therapy in Europe: a report of the ESPN/ERA–EDTA registry. Pediatr Nephrol (2014) 29:2403–10. doi: 10.1007/s00467-014-2884-6
2. Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat Diagn (2019) 39:679–92. doi: 10.1002/pd.5536
3. Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol (2015) 11:720–31. doi: 10.1038/nrneph.2015.140
4. Schedl A. Renal abnormalities and their developmental origin. Nat Rev Genet (2007) 8:791–802. doi: 10.1038/nrg2205
5. Hays T, Thompson MV, Bateman DA, Sahni R, Tolia VN, Clark RH, et al. The prevalence and clinical significance of congenital anomalies of the kidney and urinary tract in preterm infants. JAMA Network Open (2022) 5:e2231626. doi: 10.1001/jamanetworkopen.2022.31626
6. Nishi K, Uemura O, Harada R, Yamamoto M, Okuda Y, Miura K, et al. Early predictive factors for progression to kidney failure in infants with severe congenital anomalies of the kidney and urinary tract. Pediatr Nephrol (2023) 38:1057–66. doi: 10.1007/s00467-022-05703-1
7. Stoll C, Dott B, Alembik Y, Roth M-P. Associated nonurinary congenital anomalies among infants with congenital anomalies of kidney and urinary tract (CAKUT). Eur J Med Genet (2014) 57:322–8. doi: 10.1016/j.ejmg.2014.04.014
8. Connaughton DM, Hildebrandt F. Disease mechanisms of monogenic congenital anomalies of the kidney and urinary tract American journal of medical genetics part c. Am J Med Genet C Semin Med Genet (2022) 190:325–43. doi: 10.1002/ajmg.c.32006
9. Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al. Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol (2017) 28:2901–14. doi: 10.1681/ASN.2017010043
10. Hwang D-Y, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int (2014) 85:1429–33. doi: 10.1038/ki.2013.508
11. van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol (2018) 29:2348. doi: 10.1681/ASN.2017121265
12. Ahn YH, Lee C, Kim NKD, Park E, Kang HG, Ha I-S, et al. Targeted exome sequencing provided comprehensive genetic diagnosis of congenital anomalies of the kidney and urinary tract. J Clin Med (2020) 9:751. doi: 10.3390/jcm9030751
13. Caruana G, Wong MN, Walker A, Heloury Y, Webb N, Johnstone L, et al. Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol (2015) 30:487–95. doi: 10.1007/s00467-014-2962-9
14. Schild R, Knüppel T, Konrad M, Bergmann C, Trautmann A, Kemper MJ, et al. Double homozygous missense mutations in DACH1 and BMP4 in a patient with bilateral cystic renal dysplasia. Nephrol Dialysis Transplant (2013) 28:227–32. doi: 10.1093/ndt/gfs539
15. Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, et al. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet (2019) 51:117–27. doi: 10.1038/s41588-018-0281-y
16. Yoshimura-Furuhata M, Nishimura-Tadaki A, Amano Y, Ehara T, Hamasaki Y, Muramatsu M, et al. Renal complications in 6p duplication syndrome: microarray-based investigation of the candidate gene(s) for the development of congenital anomalies of the kidney and urinary tract (CAKUT) and focal segmental glomerular sclerosis (FSGS). Am J Med Genet Part A (2015) 167:592–601. doi: 10.1002/ajmg.a.36942
17. Little MH, Combes AN, Takasato M. Understanding kidney morphogenesis to guide renal tissue regeneration. Nat Rev Nephrol (2016) 12:624–35. doi: 10.1038/nrneph.2016.126
18. Mugford JW, Sipilä P, McMahon JA, McMahon AP. Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev Biol (2008) 324:88–98. doi: 10.1016/j.ydbio.2008.09.010
19. Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell (2014) 14:53–67. doi: 10.1016/j.stem.2013.11.010
20. Ludwig KS, Landmann L. Early development of the human mesonephros. Anat Embryol (Berl) (2005) 209:439–47. doi: 10.1007/s00429-005-0460-3
21. Hartman HA, Lai HL, Patterson LT. Cessation of renal morphogenesis in mice. Dev Biol (2007) 310:379–87. doi: 10.1016/j.ydbio.2007.08.021
22. Schreuder MF. Safety in glomerular numbers. Pediatr Nephrol (2012) 27:1881–7. doi: 10.1007/s00467-012-2169-x
23. Blake J, Rosenblum ND. Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin Cell Dev Biol (2014) 36:2–12. doi: 10.1016/j.semcdb.2014.07.011
24. Short KM, Combes AN, Lefevre J, Ju AL, Georgas KM, Lamberton T, et al. Global quantification of tissue dynamics in the developing mouse kidney. Dev Cell (2014) 29:188–202. doi: 10.1016/j.devcel.2014.02.017
25. Kobayashi A, Mugford JW, Krautzberger AM, Naiman N, Liao J, McMahon AP. Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Rep (2014) 3:650–62. doi: 10.1016/j.stemcr.2014.08.008
26. Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell (2008) 3:169–81. doi: 10.1016/j.stem.2008.05.020
27. Li W, Hartwig S, Rosenblum ND. Developmental origins and functions of stromal cells in the normal and diseased mammalian kidney. Dev Dynamics (2014) 243:853–63. doi: 10.1002/dvdy.24134
28. O’Brien LL, McMahon AP. Induction and patterning of the metanephric nephron. Semin Cell Dev Biol (2014) 36:31–8. doi: 10.1016/j.semcdb.2014.08.014
29. Elmore SA, Kavari SL, Hoenerhoff MJ, Mahler B, Scott BE, Yabe K, et al. Histology atlas of the developing mouse urinary system with emphasis on prenatal days E10.5-E18.5. Toxicol Pathol (2019) 47:865–86. doi: 10.1177/0192623319873871
30. Connolly JO, Chan MMY, Neild GH. Congenital anomalies of the kidney and urinary tract. In: Comprehensive clinical nephrology. (Philadephia, PA: Elsevier Inc) (2019). p. 607–25.
31. Cain JE, Rosenblum ND. Control of mammalian kidney development by the hedgehog signaling pathway. Pediatr Nephrol (2011) 26:1365–71. doi: 10.1007/s00467-010-1704-x
32. Carballo GB, Honorato JR, de Lopes GPF, de Spohr TCLSE. A highlight on sonic hedgehog pathway. Cell Commun Signal (2018) 16:11. doi: 10.1186/s12964-018-0220-7
33. Jiang J, Hui C-C. Hedgehog signaling in development and cancer. Dev Cell (2008) 15:801–12. doi: 10.1016/j.devcel.2008.11.010
34. Colavito SA, Zou MR, Yan Q, Nguyen DX, Stern DF. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated b cells (NFκB) pathway. Breast Cancer Res (2014) 16:444. doi: 10.1186/s13058-014-0444-4
35. Singh R, Dhanyamraju PK, Lauth M. DYRK1B blocks canonical and promotes non-canonical hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget (2017) 8:833–45. doi: 10.18632/oncotarget.13662
36. Wang Y, Jin G, Li Q, Wang Z, Hu W, Li P, et al. Hedgehog signaling non-canonical activated by pro-inflammatory cytokines in pancreatic ductal adenocarcinoma. J Cancer (2016) 7:2067–76. doi: 10.7150/jca.15786
37. Briscoe J, Thérond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol (2013) 14:416–29. doi: 10.1038/nrm3598
38. Hui C-C, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol (2011) 27:513–37. doi: 10.1146/annurev-cellbio-092910-154048
39. Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature (2002) 418:979–83. doi: 10.1038/nature01033
40. Brunskill EW, Park J-S, Chung E, Chen F, Magella B, Potter SS. Single cell dissection of early kidney development: multilineage priming. Development (2014) 141:3093–101. doi: 10.1242/dev.110601
41. Combes AN, Phipson B, Lawlor KT, Dorison A, Patrick R, Zappia L, et al. Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk. Development (2019) 146:dev178673. doi: 10.1242/dev.178673
42. Fabian SL, Penchev RR, St-Jacques B, Rao AN, Sipilä P, West KA, et al. Hedgehog-gli pathway activation during kidney fibrosis. Am J Pathol (2012) 180:1441–53. doi: 10.1016/j.ajpath.2011.12.039
43. Yu J, Carroll TJ, McMahon AP. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development (2002) 129:5301–12. doi: 10.1242/dev.129.22.5301
44. Hu MC, Mo R, Bhella S, Wilson CW, Chuang P-T, Hui C-C, et al. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development (2006) 133:569–78. doi: 10.1242/dev.02220
45. Rowan CJ, Li W, Martirosyan H, Erwood S, Hu D, Kim Y-K, et al. Hedgehog-GLI signaling in Foxd1-positive stromal cells promotes murine nephrogenesis via TGFβ signaling. Development (2018) 145:dev159947. doi: 10.1242/dev.159947
46. Magella B, Adam M, Potter AS, Venkatasubramanian M, ChEtal K, Hay SB, et al. Cross-platform single cell analysis of kidney development shows stromal cells express gdnf. Dev Biol (2018) 434:36–47. doi: 10.1016/j.ydbio.2017.11.006
47. Cain JE, Islam E, Haxho F, Chen L, Bridgewater D, Nieuwenhuis E, et al. GLI3 repressor controls nephron number via regulation of Wnt11 and ret in ureteric tip cells. PLoS One (2009) 4:e7313. doi: 10.1371/journal.pone.0007313
48. Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, et al. Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature (1996) 383:407–13. doi: 10.1038/383407a0
49. Cain JE, Islam E, Haxho F, Blake J, Rosenblum ND. GLI3 repressor controls functional development of the mouse ureter. J Clin Invest (2011) 121:1199–206. doi: 10.1172/JCI45523
50. Sheybani-Deloui S, Chi L, Staite MV, Cain JE, Nieman BJ, Henkelman RM, et al. Activated hedgehog-GLI signaling causes congenital ureteropelvic junction obstruction. J Am Soc Nephrol (2018) 29:532–44. doi: 10.1681/ASN.2017050482
51. Blake J, Hu D, Cain JE, Rosenblum ND. Urogenital development in pallister–hall syndrome is disrupted in a cell-lineage-specific manner by constitutive expression of GLI3 repressor. Hum Mol Genet (2016) 25:437–47. doi: 10.1093/hmg/ddv483
52. Iskander SM, Feeney MM, Yee K, Rosenblum ND. Protein kinase 2β is expressed in neural crest-derived urinary pacemaker cells and required for pyeloureteric contraction. J Am Soc Nephrol (2018) 29:1198–209. doi: 10.1681/ASN.2017090951
53. Rowan CJ, Sheybani-Deloui S, Rosenblum ND. Origin and function of the renal stroma in health and disease. In: Miller RK, editor. Kidney development and disease. results and problems in cell differentiation. Cham: Springer International Publishing (2017). p. 205–29. doi: 10.1007/978-3-319-51436-9_8
54. Chuang P-T, Kawcak T, McMahon AP. Feedback control of mammalian hedgehog signaling by the hedgehog-binding protein, Hip1, modulates fgf signaling during branching morphogenesis of the lung. Genes Dev (2003) 17:342–7. doi: 10.1101/gad.1026303
55. Zhulyn O, Nieuwenhuis E, Liu YC, Angers S, Hui C. Ptch2 shares overlapping functions with Ptch1 in smo regulation and limb development. Dev Biol (2015) 397:191–202. doi: 10.1016/j.ydbio.2014.10.023
56. Raleigh DR, Reiter JF. Misactivation of hedgehog signaling causes inherited and sporadic cancers. J Clin Invest (2019) 129:465–75. doi: 10.1172/JCI120850
57. Masuno M, Fukushima Y, Sugio Y, Ikeda M, Kuroki Y. Two unrelated cases of single maxillary central incisor with 7q terminal deletion. Jinrui Idengaku Zasshi (1990) 35:311–7. doi: 10.1007/BF01883753
58. Benzacken B, Siffroi JP, Bourhis CL, Krabchi K, Joyé N, Maschino F, et al. Different proximal and distal rearrangements of chromosome 7q associated with holoprosencephaly. J Med Genet (1997) 34:899–903. doi: 10.1136/jmg.34.11.899
59. Wang J, Spitz L, Hayward R, Kiely E, Hall CM, O’Donoghue DP, et al. Sacral dysgenesis associated with terminal deletion of chromosome 7q: a report of two families. Eur J Pediatr (1999) 158:902–5. doi: 10.1007/s004310051238
60. Zen PRG, Riegel M, Rosa RFM, Pinto LLC, Graziadio C, Schwartz IVD, et al. Esophageal stenosis in a child presenting a de novo 7q terminal deletion. Eur J Med Genet (2010) 53:333–6. doi: 10.1016/j.ejmg.2010.06.008
61. Dubourg C, Lazaro L, Pasquier L, Bendavid C, Blayau M, Le Duff F, et al. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: mutation review and genotype-phenotype correlations. Hum Mutat (2004) 24:43–51. doi: 10.1002/humu.20056
62. Porter JA, von Kessler DP, Ekker SC, Young KE, Lee JJ, Moses K, et al. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature (1995) 374:363–6. doi: 10.1038/374363a0
63. Qi X, Schmiege P, Coutavas E, Wang J, Li X. Structures of human patched and its complex with native palmitoylated sonic hedgehog. Nature (2018) 560:128–32. doi: 10.1038/s41586-018-0308-7
64. Shirakawa T, Nakashima Y, Watanabe S, Harada S, Kinoshita M, Kihara T, et al. A novel heterozygous GLI2 mutation in a patient with congenital urethral stricture and renal hypoplasia/dysplasia leading to end-stage renal failure. CEN Case Rep (2018) 7:94–7. doi: 10.1007/s13730-018-0302-9
65. Babu D, Fanelli A, Mellone S, Muniswamy R, Wasniewska M, Prodam F, et al. Novel GLI2 mutations identified in patients with combined pituitary hormone deficiency (CPHD): evidence for a pathogenic effect by functional characterization. Clin Endocrinol (Oxf) (2019) 90:449–56. doi: 10.1111/cen.13914
66. Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of shh signaling. Development (1999) 126:3915–24. doi: 10.1242/dev.126.17.3915
67. Pan Y, Wang B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J Biol Chem (2007) 282:10846–52. doi: 10.1074/jbc.M608599200
68. Clarren SK, Alvord EC, Hall JG. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly–a new syndrome? part II: neuropathological considerations. Am J Med Genet (1980) 7:75–83. doi: 10.1002/ajmg.1320070111
69. Hall JG, Pallister PD, Clarren SK, Beckwith JB, Wiglesworth FW, Fraser FC, et al. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly–a new syndrome? part I: clinical, causal, and pathogenetic considerations. Am J Med Genet (1980) 7:47–74. doi: 10.1002/ajmg.1320070110
70. Kang S, Graham JM, Olney AH, Biesecker LG. GLI3 frameshift mutations cause autosomal dominant pallister-hall syndrome. Nat Genet (1997) 15:266–8. doi: 10.1038/ng0397-266
71. Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat (2010) 31:1142–54. doi: 10.1002/humu.21328
72. McClelland K, Li W, Rosenblum ND. Pallister-hall syndrome, GLI3, and kidney malformation. Am J Med Genet C Semin Med Genet (2022) 190(3):264–78. doi: 10.1002/ajmg.c.31999
73. Radhakrishna U, Bornholdt D, Scott HS, Patel UC, Rossier C, Engel H, et al. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; no phenotype prediction from the position of GLI3 mutations. Am J Hum Genet (1999) 65:645–55. doi: 10.1086/302557
74. Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and pallister-hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet (2005) 76:609–22. doi: 10.1086/429346
75. Démurger F, Ichkou A, Mougou-Zerelli S, Le Merrer M, Goudefroye G, Delezoide A-L, et al. New insights into genotype–phenotype correlation for GLI3 mutations. Eur J Hum Genet (2015) 23:92–102. doi: 10.1038/ejhg.2014.62
76. Sczakiel HL, Hülsemann W, Holtgrewe M, Abad-Perez AT, Elsner J, Schwartzmann S, et al. GLI3 variants causing isolated polysyndactyly are not restricted to the protein’s c-terminal third. Clin Genet (2021) 100:758–65. doi: 10.1111/cge.14059
77. Ruppert JM, Vogelstein B, Arheden K, Kinzler KW. GLI3 encodes a 190-kilodalton protein with multiple regions of GLI similarity. Mol Cell Biol (1990) 10:5408–15. doi: 10.1128/mcb.10.10.5408-5415.1990
78. Shin SH, Kogerman P, Lindström E, Toftgárd R, Biesecker LG. GLI3 mutations in human disorders mimic drosophila cubitus interruptus protein functions and localization. Proc Natl Acad Sci U.S.A. (1999) 96:2880–4. doi: 10.1073/pnas.96.6.2880
79. D’Cruz R, Stronks K, Rowan CJ, Rosenblum ND. Lineage-specific roles of hedgehog-GLI signaling during mammalian kidney development. Pediatr Nephrol (2020) 35:725–31. doi: 10.1007/s00467-019-04240-8
80. Furniss D, Critchley P, Giele H, Wilkie AOM. Nonsense-mediated decay and the molecular pathogenesis of mutations in SALL1 and GLI3. Am J Med Genet Part A (2007) 143A:3150–60. doi: 10.1002/ajmg.a.32097
81. Matissek SJ, Elsawa SF. GLI3: a mediator of genetic diseases, development and cancer. Cell Commun Signal (2020) 18:1–20. doi: 10.1186/s12964-020-00540-x
82. Kammenga JE. The background puzzle: how identical mutations in the same gene lead to different disease symptoms. FEBS J (2017) 284:3362–73. doi: 10.1111/febs.14080
83. Jansen J, van den Berge BT, van den Broek M, Maas RJ, Daviran D, Willemsen B, et al. Human pluripotent stem cell-derived kidney organoids for personalized congenital and idiopathic nephrotic syndrome modeling. Development (2022) 149:dev200198. doi: 10.1242/dev.200198
Keywords: kidney, CAKUT, hedgehog signaling, stromal, ureteric, nephrogenic, Pallister-Hall syndrome
Citation: Greenberg D, D’Cruz R, Lacanlale JL, Rowan CJ and Rosenblum ND (2023) Hedgehog-GLI mediated control of renal formation and malformation. Front. Nephrol. 3:1176347. doi: 10.3389/fneph.2023.1176347
Received: 28 February 2023; Accepted: 31 March 2023;
Published: 20 April 2023.
Edited by:
Giorgia Schena, Yale University, United StatesReviewed by:
Michael Caplan, Yale University, United StatesMara Medeiros, Federico Gómez Children’s Hospital, Mexico
Copyright © 2023 Greenberg, D’Cruz, Lacanlale, Rowan and Rosenblum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norman D. Rosenblum, bm9ybWFuLnJvc2VuYmx1bUBzaWNra2lkcy5jYQ==
†These authors have contributed equally to this work