 Eleftherios Gkekas1†Tsz Yau Tiffany Tang1†Alan Green1Han Davidson2Rachel Fraser2John A. Sayer1,2,3*Shalabh Srivastava1,4
Eleftherios Gkekas1†Tsz Yau Tiffany Tang1†Alan Green1Han Davidson2Rachel Fraser2John A. Sayer1,2,3*Shalabh Srivastava1,4- 1Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, United Kingdom
- 2Renal Services, The Newcastle upon Tyne Hospitals National Health Service (NHS) Foundation Trust, Newcastle, United Kingdom
- 3National Institute for Health and Care Research (NIHR) Bioresource Centre, Newcastle upon Tyne, United Kingdom
- 4Nephrology Department, South Tyneside and Sunderland National Health Service (NHS) Foundation Trust, Sunderland, United Kingdom
Autosomal dominant polycystic kidney disease (ADPKD) is a cause of end-stage kidney disease (ESKD). The vasopressin V2-receptor antagonist tolvaptan has been shown within randomized clinical trials to slow down decline of kidney function in patients with ADPKD at risk of rapid progression. We performed a retrospective review of a Northeast England cohort of adult ADPKD patients who had been established on tolvaptan therapy to determine its efficacy in a real-world clinic setting. Other inclusion criteria involved a pre-treatment decline in greater than 2.5 ml/min/1.73m2/year based on readings for a 3 year period, and ability to tolerate and maintain tolvaptan treatment for at least 12 months. We calculated based on eGFR slopes, predicted time to reach ESKD with and without tolvaptan therapy. The cohort of patients included 21 from the Northeast of England. The mean rate of eGFR decline prior to treatment was -6.02 ml/min/1.73m2/year for the cohort. Following tolvaptan treatment, the average decline in eGFR was reduced to -2.47 ml/min/1.73m2/year, gaining a mean 8 years and 4 months delay to reach ESKD. The majority of patients (n=19) received and tolerated full dose tolvaptan (90 mg/30 mg). The real-life use of tolvaptan gave a dramatic improvement in eGFR slopes, much more than previously reported in clinical studies. These effects may be in part due to careful patient identification, selection and inclusion of patients who were able to tolerate tolvaptan therapy, excellent compliance with medication and a “tolvaptan clinic” effect where great personal care was given to these patients.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited kidney disorder leading to end stage kidney disease (ESKD) (1). The cystic change within the kidneys causes enlargement of the kidneys, leading to increases in total kidney volume (TKV), elevated blood pressure and a progressive decline in kidney function resulting in ESKD (2). Cysts may form in other organs including the liver, spleen and pancreas and there may be vascular abnormalities including cerebral aneurysms that may lead to significant morbidity and mortality (3).
ADPKD will most often present clinically in the 3rd or 4th decade of life with symptoms that include abdominal pain and fullness, infection of cysts, cyst hemorrhage with hematuria and hypertension (2).
ADPKD phenotypes are associated with a single heterozygous mutation in one of two main genes, PKD1 and PKD2 and a growing number of minor genes, including IFT140 (4). Mutations in PKD1 account for 85% of cases and are associated with more rapid disease progression (5).
The treatment of ADPKD patients traditionally relied upon supportive measures including blood pressure control, pain control, and prompt treatment of infected cysts, urinary tract infections and renal stones. Systemic blood pressure management was previously the only pharmacologically modifiable target based on evidence from clinical trials (6). In 2012, a treatment called tolvaptan, a vasopressin V2 receptor antagonist was shown to reduce both kidney growth and loss of estimated glomerular filtration rate (eGFR). This was based on the results of the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes 3:4 Trial [TEMPO 3:4], a 3-year randomized double-blinded placebo controlled trial involving 1445 ADPKD patients (7). The effect of tolvaptan on eGFR was a slower decline in renal function as measured by reciprocal of serum creatinine level, compared with placebo. The decline in eGFR per year was -0.71 ml/min/1.73m2 per year in tolvaptan treated compared to -2.1 in historical controls (7).
In the United Kingdom, tolvaptan was approved by the National Institute of Clinical Excellence (NICE) in 2015 as the first drug to be used in the treatment ADPKD) (8). Since then, tolvaptan use has been advocated for ADPKD patients with evidence of rapid progression. Defining the patient cohort of rapid progression is subjective but the UK Renal Association provided some guidelines for its use in clinical practice (https://ukkidney.org/sites/renal.org/files/tolvaptan-in-adpkd-nice-commentary.pdf) as did the European Renal Association (9). A second study called REPRISE confirmed that tolvaptan was able to slow decline in eGFR on patients with later-stage ADPKD (10), with treatment effects being a gain of 2.36. ml/min/1.73m2 in patients with baseline eGFR of 45 to 59 following the 1 year study. Real world experience with the use of tolvaptan is now growing and the definitions of rapid progression have been clarified (8) allowing cohorts of patients separate from any clinical trials to be initiated and established on this therapy. Studies assessing long-term administration of tolvaptan for ADPKD convincingly show a sustained reduction of annual rate of eGFR decline (11).
Here, we present the outcomes from a Northeast England cohort of patients with ADPKD who were identified as being at risk of rapid progression and were commenced on tolvaptan within the renal outpatient department settings from two dedicated renal genetics clinics.
Within local patient databases, patients with ADPKD, who had features of rapid progression were identified and offered tolvaptan, as recommended by the UK Renal Association. This included patients over the age of 18 years with and established diagnosis of ADPKD, stage 2-3 CKD (30-89ml.min/1,73m2), a documented sustained decline in eGFR of ≥2.5 ml/min/1.73m2. Following these guidelines, we have used tolvaptan to treat ADPKD since its approval by NICE within dedicated renal genetics clinics serving the population of the Northeast England.
Methods
This was a retrospective observational study to assess the impact of tolvaptan on the kidney function as measured by eGFR in patients with a clinical diagnosis of ADPKD. Patients underwent detailed phenotyping and long-term follow-up within the National Registry of Rare Kidney Diseases (RaDaR) (https://rarerenal.org/radar-registry/). We reviewed our cohort of patients on tolvaptan and included patients who met the following criteria:
1. Confirmed diagnosis of ADPKD (clinical/radiological findings consistent with the diagnosis and molecular genetic studies where available)
2. Age – 18-65 years
3. Estimated Glomerular filtration (eGFR) rate – 30-89 ml/min/1.73m2 as calculated by the CKD-EPI formula at the initiation of tolvaptan (12)
4. eGFR slope ≥-2.5 ml/min/1.73m2/year prior to initiation of tolvaptan using CKD-EPI formula and excluding values >90 ml/min/1.73m2. eGFR values at times of intercurrent illness and associated acute kidney injury were filtered out.
5. Tolvaptan therapy (at any dose) for greater than 18 months duration without treatment breaks/interruptions. The observation period for each patient was therefore at least 18 months. Throughout tolvaptan treatment patients would have an assessment of liver enzymes and eGFR on a 3 monthly basis.
We calculated the imaging classification of ADPKD patients using the Mayo Clinic algorithm (https://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754) by imputing total kidney volume (calculated by stereology measurements from kidney MRI or CT scan), patient height and patient age, resulting in Class 1A-1E. To plot eGFR slopes we retrospectively compared the slope of eGFR prior to tolvaptan initiation and following tolvaptan therapy. Specifically, prior to treatment, eGFR levels were collected from the first available levels up to three years before the date the patient was started on tolvaptan. The tolvaptan treatment eGFR levels were collected from the first available levels three months following the start date of tolvaptan therapy and up to four years after tolvaptan was started. eGFR levels taken within three months of starting tolvaptan were excluded to avoid the acute changes in eGFR upon drug commencement. The eGFR slopes were obtained using the function SLOPE() on Microsoft Excel on the raw data collected with the slope defined as the rate of change in eGFR over 1 year. We predicted time to ESKD (CKD stage 5, eGFR <15mls/min/1.73m2) for each patient based on the rate of eGFR decline. For graphical representation, we plotted the raw eGFR data in a scatter-plot graph for each patient, with the x-axis representing time (in years) and the y-axis representing eGFR level. A line of best fit was generated for pre-tolvaptan and tolvaptan eGFR values over time with confidence intervals of 95% added using a linear regression model (GraphPad Prism). Side effects including liver enzymes were assessed for each patient, overall bloods pressure control was reviewed and episodes of intercurrent illness noted.
Results
A total of 21 patients with ADPKD who had been treated with tolvaptan met our inclusion criteria with a mean age of 48.0 years (range 30-62 years). There were 15 males and 6 females and all except 2 were on the maximal dose of tolvaptan (90 mg/30 mg) (Table 1). 14 patients had evidence of optimal blood pressure control below target of 130/80 and 7 had blood pressures above this target range. Two patients had intercurrent urinary tract infections (Table 1). The mean rate of eGFR decline prior to tolvaptan initiation was -6.02 ml/min/1.73m2 per year. This is consistent with this cohort having rapid progression of their disease (Figure 1A). Post tolvaptan initiation the eGFR decline was -2.47 ml/min/1.73m2 per year. Tolvaptan led to a mean change in the slope of decline by 3.56 ml/min/1.73m2 per year (Figure 1A). Individual patients and their eGFR plots demonstrate a wide range of responses (Figure 2). The majority of patients responded to treatment, with 1 who had a worsening of eGFR slope (ID 20) and 3 with an apparent non-response to the therapy with no change in eGFR slope (IDs 5, 12 and 15) (Figure 1B). The reasons for this lack of effect of response to tolvaptan are not fully apparent but it is noteworthy that 2 of these 4 cases had an ADPKD risk classification of 1E, the most severe category and that only 1 was less than 40 years of age.
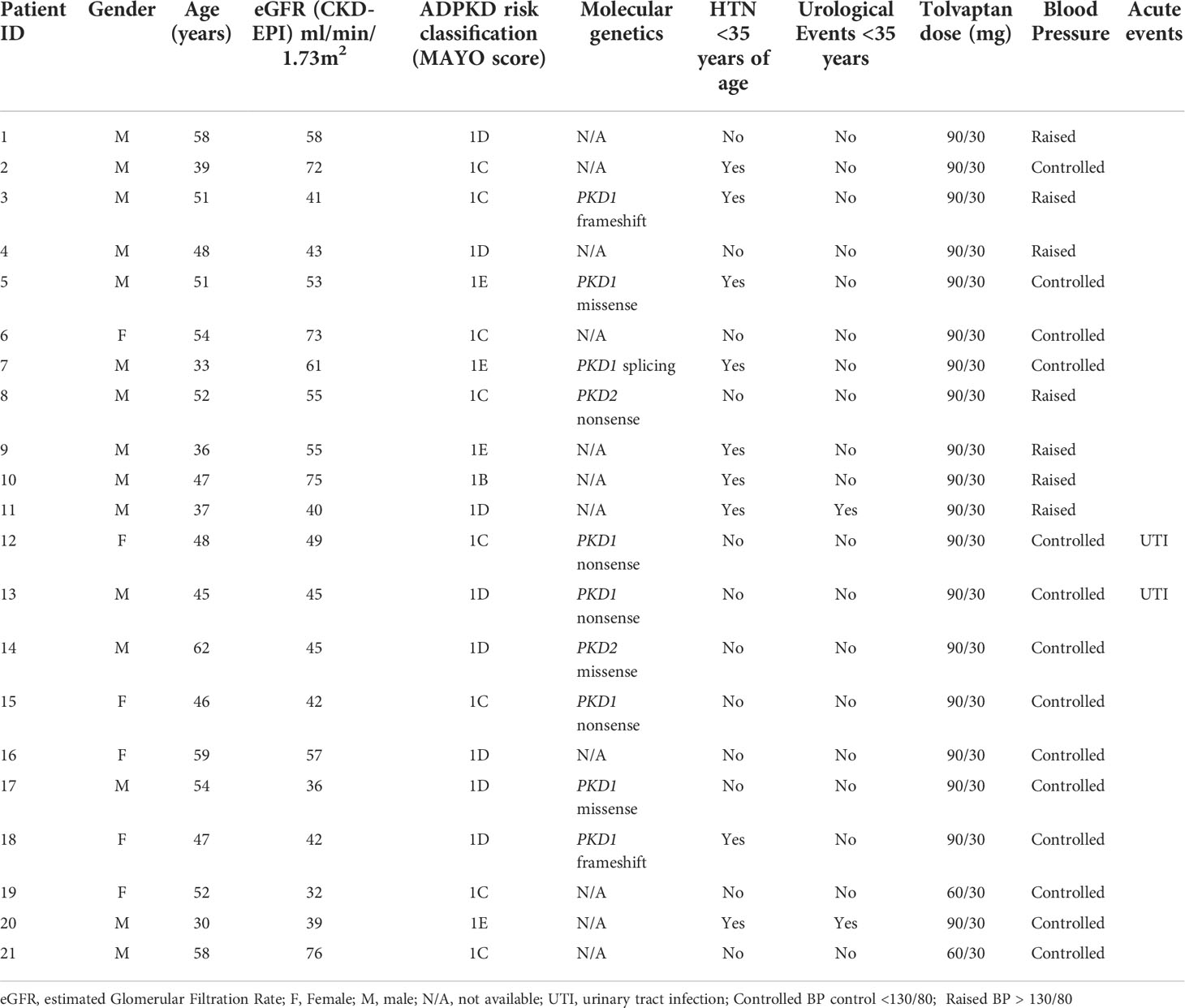
Table 1 ADPKD patient baseline characteristics and tolvaptan dose.
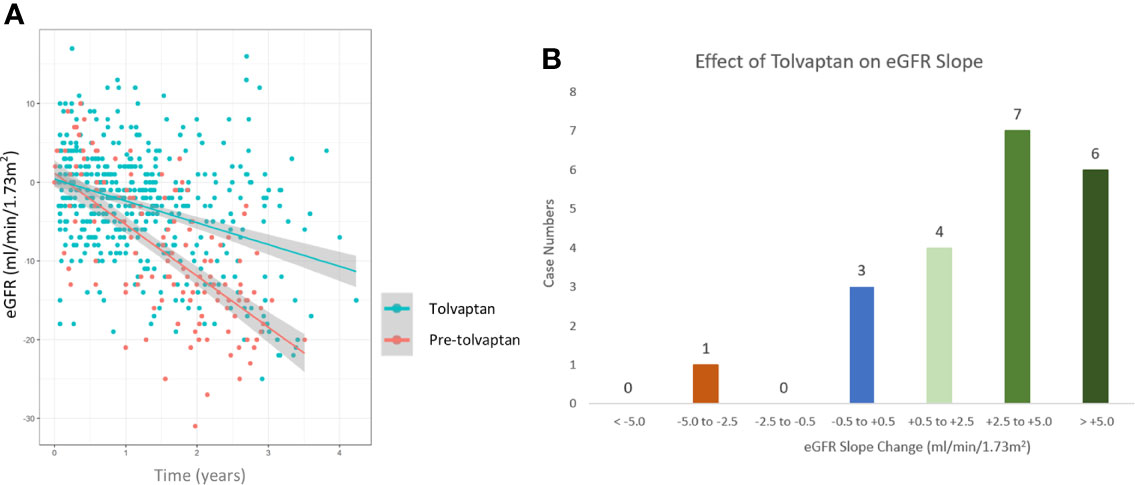
Figure 1 Comparison of estimated glomerular filtration (eGFR) slopes pre and post tolvaptan treatment in ADPKD patients (A) A scatter plot of eGFR (ml/min/1.73m2/year) over time in pre-tolvaptan and post-tolvaptan patients annotated with a line of best fit with 95% confidence intervals (CI) shown in grey shade. The rate of decline pre-tolvaptan was -6.51 ml/min/1.73m2 (slopes for 2.5% -7.53 & 97.5% -5.49) and the rate of decline on tolvaptan was -2.77 ml/min/1.73m2 (2.5% -3.39 & 97.5% -2.15) (B) The range of eGFR (ml/min/1.73m2/year) slope change and the number of patients in whom this was achieved in each range is shown.
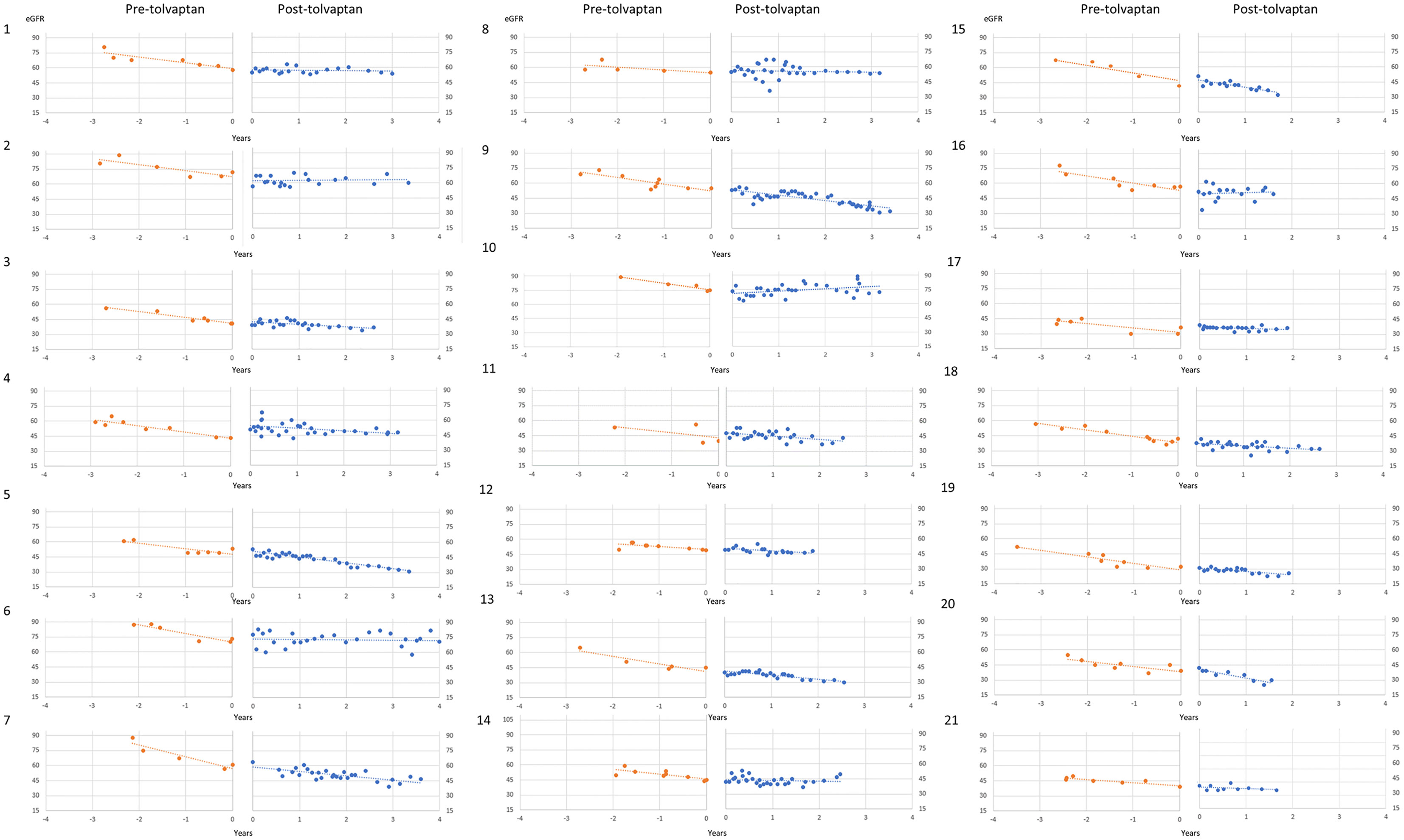
Figure 2 Individual patient eGFR plots pre- and post-tolvaptan eGFR (ml/min/1.73m2/year) is plotted against time in years for each of the patients.
Prior to treatment, the mean predicted time to reaching ESKD in this cohort of ADPKD patients was 6.94 years. Following treatment with tolvaptan, the mean time to reach ESKD was 15.30 years. Tolvaptan therefore lead to a mean estimated delay of 8.36 (8 years and 4 months) years to reach ESKD in this cohort of rapidly progressing ADPKD patients.
Discussion
ADPKD is the most common genetic condition leading to ESKD (13) and the most common life-threatening inherited disease worldwide. In recent years there have been significant gains in our understanding of the disease pathogenesis, its natural history and therapeutic interventions. The HALT-PKD trial (14, 15) demonstrated the benefits of strict blood pressure control and the effectiveness of blockade of the renin-angiotensin-aldosterone system in the reduction of progression of early ADPKD. The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) was a prospective, multicenter, observational study of ADPKD patients which determined the relationship between total kidney volume (TKV) and the decline of kidney function using cross sectional imaging. Now, baseline height-adjusted TKV can be used to predict decline of kidney function and progression to ESKD. Then randomized controlled studies TEMPO 3:4 (7) and REPRISE (10) introduced the use of a disease modifying drug, tolvaptan into clinical practice.
These data supporting the use of tolvaptan as a therapeutic intervention for ADPKD has been acquired from carefully designed multinational trials. The overall effect size on eGFR was relatively modest in both studies, suggesting that firstly patients were required to be on the drug for long periods of time to gain the most benefit and secondly that there may be significant variance in response rates to tolvaptan. The long-term gains of tolvaptan have been established in small cohorts of patients (11). A recent post hoc analysis of TEMPO 3:4 in Japanese ADPKD patients documented a change in eGFR of -8.5 ml/min/1.73m2 following tolvaptan treatment (compared to -12.6 in the placebo group) using a Japanese eGFR equation, suggesting that there may be population specific effects of this drug. A helpful update on the use of tolvaptan for ADPKD has recently been published which includes a treatment algorithm to guide patient selection (16).
We set out to assess its use in a real-world outpatient clinic setting by retrospectively reviewing our clinical service in the Northeast of England. Tolvaptan was found to be well tolerated in our patient cohort with the majority of patients receiving maximal dose of tolvaptan (120 mg per day). We excluded from this study patients who were unable to tolerate tolvaptan long term or had treatment breaks/holidays. We chose to monitor renal function using the CKD-EPI equation as this was the reported eGFR for our patients within our hospitals and represents real world data. We are aware of the inaccuracies in using eGFR equations in ADPKD patients. The CKD-EPI has an accuracy of 90%, and differences in accuracy were seen especially when the GFR was greater than 60 ml/min/1.73m2 (17).
We also chose to consider the slope of eGFR as a linear progression, however deviations from linearity (unrelated to acute events) can occur in a proportion of patients (18, 19). In an 10 year retrospective analysis of individual eGFR slopes, the majority of patients had a linear decline in eGFR, but some had more complex patterns including prolonged stable phases and negative exponential declines were seen, which are informative for clinical management, mechanistic studies and future clinical trial designs (19).
An alternative specific measure for ADPKD progression is serial renal volume measurement using MRI scanning (20, 21) but this was not routinely performed in our clinical practice and serial TKV measurement has not been recommended for the monitoring of response to tolvaptan (22). There is some evidence that greatest renal benefit is linked to the greatest suppression of urine osmolality (23), however we did not assess this in our cohort. We had no reports of deranged liver enzymes or other severe reactions. In our analysis, we demonstrate good overall efficacy of tolvaptan in our patient cohort whereby it led to a mean gain of > 10 ml/min/1.73m2 over a 3 year treatment period. The main strengths of our study are its “in practice”, clinic-based data which lends itself to real world application. In this regard we were able to define individual response rates to tolvaptan on eGFR slopes. There were 4 patients who failed to respond to tolvaptan or in whom it had no discernible effect on the decline in eGFR. Two of these patients had massively enlarged kidneys leading to an ADPKD prediction score of 1E. Of note, the measurement of eGFR whilst taking tolvaptan is reduced, due to the reversible inhibition if the tubulo-glomerular feedback mechanisms (24) and may have masked a small response in some of these cases. As this was a retrospective study, we did not include measurement of eGFR after a washout period. It may be important for future use of tolvaptan and other pharmacological agents to determine biomarkers of responsiveness. Measurement of urine osmolality can be used as a tool to assess compliance but may also provide a window into the effect of tolvaptan on the kidney. In the TEMPO 3:4 study baseline urinary osmolarity in ADPKD reflected age, kidney function and TKV and the greatest kidney benefit occurred in patients achieving greater suppression of urine osmolarity (23). Routine measurement of urinary osmolality before and after treatment might be a useful tool to guide the efficacy of tolvaptan. The main weaknesses of this study are the small sample size and retrospective data collection with inclusion of only patients who were able to tolerate the drug.
In summary, we report significant improvement in eGFR slope and time to CKD 5 in patients on Tolvaptan in our cohort but highlight that there are sub-groups of tolvaptan non-responders who may benefit from alternative approaches. The maximum dose of tolvaptan was well tolerated. The precise reasons for both non-response and exaggerated response need to be evaluated carefully to determine how individualization of tolvaptan therapy can be utilized for patient benefit.
Data availability statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
Author contributions
JS and SS contributed to conception and design of the study. EG and TYTT organized the database and performed the analysis. SS wrote the first draft of the manuscript. EG, TYTT, AG, HD and RF wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
SS is funded by PKD Charity, ref. ADPKD-19-01. JAS is funded by PKD Charity, Kidney Research UK, ref Paed_RP_001_20180925 and the Northern Counties Kidney Research Fund, ref. 2020-01. We thank Dr Ian Wilson, Newcastle University for statistical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Abad JM, et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival–an analysis of data from the ERA-EDTA registry. Nephrol Dial Transplant (2014) 29 Suppl 4(Suppl 4):iv15–25. doi: 10.1093/ndt/gfu017
2. Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: Core curriculum 2016. Am J Kidney Dis (2016) 67(5):792–810. doi: 10.1053/j.ajkd.2015.07.037
3. Grantham JJ. Clinical practice. autosomal dominant polycystic kidney disease. N Engl J Med (2008) 359(14):1477–85. doi: 10.1056/NEJMcp0804458
4. Senum SR, Li YSM, Benson KA, Joli G, Olinger E, Lavu S, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet (2022) 109(1):136–56. doi: 10.1016/j.ajhg.2021.11.016
5. Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol (2006) 17(11):3013–9. doi: 10.1681/ASN.2006080835
6. Chapman AB, Torres VE, Perrone RD, Steinman TI, Bae KT, Miller JP, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol (2010) 5(1):102–9. doi: 10.2215/CJN.04310709
7. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med (2012) 367(25):2407–18. doi: 10.1056/NEJMoa1205511
8. Müller RU, Haas CS, Sayer JA. Practical approaches to the management of autosomal dominant polycystic kidney disease patients in the era of tolvaptan. Clin Kidney J (2018) 11(1):62–9. doi: 10.1093/ckj/sfx071
9. Gansevoort RT, Arici M, Benzing T, Birn H, Capasso G, Covic A, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA working groups on inherited kidney disorders and European renal best practice. Nephrol Dial Transplant (2016) 31(3):337–48. doi: 10.1093/ndt/gfv456
10. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med (2017) 377(20):1930–42. doi: 10.1056/NEJMoa1710030
11. Edwards ME, Chebib FT, Irazabal MV, Ofstie TG, Bungum LA, Metzger AJ, et al. Long-term administration of tolvaptan in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol (2018) 13(8):1153–61. doi: 10.2215/CJN.01520218
12. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med (2009) 150(9):604–12. doi: 10.7326/0003-4819-150-9-200905050-00006
13. Radhakrishnan Y, Duriseti P, Chebib FT. Management of autosomal dominant polycystic kidney disease in the era of disease-modifying treatment options. Kidney Res Clin Pract (2022). 41(4):422–431 doi: 10.23876/j.krcp.21.309
14. Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med (2014) 371(24):2255–66. doi: 10.1056/NEJMoa1402685
15. Brosnahan GM, Abebe KZ, Moore CG, Bae KT, Braun WE, Chapman AB, et al. Determinants of progression in early autosomal dominant polycystic kidney disease: Is it blood pressure or renin-Angiotensin-Aldosterone-System blockade? Curr Hypertens Rev (2018) 14(1):39–47. doi: 10.2174/1573402114666180322110209
16. Müller RU, Messchendorp AL, Birn H, Capasso G, Cornec-Le Gall E, Devuyst O, et al. An update on the use of tolvaptan for autosomal dominant polycystic kidney disease: consensus statement on behalf of the ERA working group on inherited kidney disorders, the European rare kidney disease reference network and polycystic kidney disease international. Nephrol Dial Transplant (2022) 37(5):825–39. doi: 10.1093/ndt/gfab312
17. Orskov B, Borresen ML, Feldt-Rasmussen B, Østergaard O, Laursen I, Strandgaard S. Estimating glomerular filtration rate using the new CKD-EPI equation and other equations in patients with autosomal dominant polycystic kidney disease. Am J Nephrol (2010) 31(1):53–7. doi: 10.1159/000256657
18. Higashihara E, Horie S, Muto S, Mochizuki T, Nishio S, Nutahara K. Renal disease progression in autosomal dominant polycystic kidney disease. Clin Exp Nephrol (2012) 16(4):622–8. doi: 10.1007/s10157-012-0611-9
19. Neagu M, Coca D, Ong ACM. Linear and nonlinear estimated GFR slopes in ADPKD patients reaching ESRD. Am J Kidney Dis (2018) 71(6):912–3. doi: 10.1053/j.ajkd.2018.01.052
20. Bae KT, Tao C, Zhu F, Bost JE, Chapman AB, Grantham JJ, et al. MRI-Based kidney volume measurements in ADPKD: reliability and effect of gadolinium enhancement. Clin J Am Soc Nephrol (2009) 4(4):719–25. doi: 10.2215/CJN.03750708
21. Higashihara E, Nutahara K, Okegawa T, Shishido T, Tanbo M, Kobayasi K, et al. Kidney volume and function in autosomal dominant polycystic kidney disease. Clin Exp Nephrol (2014) 18(1):157–65. doi: 10.1007/s10157-013-0834-4
22. Mei CL, Xue C, Yu SQ, Dai B, Chen JH, Li Y, et al. Executive summary: Clinical practice guideline for autosomal dominant polycystic kidney disease in China. Kidney Dis (Basel) (2020) 6(3):144–9. doi: 10.1159/000506288
23. Devuyst O, Chapman AB, Gansevoort RT, Higashihara E, Perrone RD, Torres VE, et al. Urine osmolality, response to tolvaptan, and outcome in autosomal dominant polycystic kidney disease: Results from the TEMPO 3:4 trial. J Am Soc Nephrol (2017) 28(5):1592–602. doi: 10.1681/ASN.2016040448
Keywords: ADPKD (autosomal dominant polycystic kidney disease), tolvaptan treatment, end stage kidney disease (ESKD), disease progression, real world
Citation: Gkekas E, Tang TYT, Green A, Davidson H, Fraser R, Sayer JA and Srivastava S (2022) Outcomes from the Northeast England cohort of autosomal dominant polycystic kidney disease (ADPKD) patients on tolvaptan. Front. Nephrol. 2:984165. doi: 10.3389/fneph.2022.984165
Received: 01 July 2022; Accepted: 07 September 2022;
Published: 23 September 2022.
Edited by:
Michal Nowicki, Medical University of Lodz, PolandReviewed by:
Silvia Lai, Sapienza University of Rome, ItalyMagdalena Jankowska, Medical University of Gdansk, Poland
Copyright © 2022 Gkekas, Tang, Green, Davidson, Fraser, Sayer and Srivastava. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Sayer, John.sayer@ncl.ac.uk
†These authors have contributed equally to this work