 Elton L. Cao
Elton L. Cao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nat. Prod., 15 January 2024
Sec. Informatics and Computational Methods
Volume 2 - 2023 | https://doi.org/10.3389/fntpr.2023.1309994
Drug combination therapies have shown effective performance in treating cancer through increased efficacy and circumvention of drug resistance through synergy. Two avenues can be used to discover drug combinations: a novel approach that utilizes natural products compared with the textbook approach of utilizing existing chemotherapy drug combinations. Many natural products achieve efficacy due to synergistic interactions between the active ingredients. Therefore, the pharmacophore relationships in herbal compounds which synergize can potentially be applied to chemotherapy drugs to drive combination discovery. Machine learning approaches have been developed to identify drug combinations, especially deep neural networks (DNN), which have achieved state-of-the-art performance in many drug discovery tasks. Here, a drug protein interaction (DPI) prediction DNN, DeepDPI, was developed to employ DPI drug representations and achieved state-of-the-art performance using the DrugBank database. Two DNNs were also developed to predict novel drug combinations: DeepNPD, which predicts combinations in natural products, and DeepCombo, which predicts synergy in chemotherapy drugs, using the HERB and DrugCombDB databases respectively. An ensemble architecture enhanced with a novel similarity based weight adjustment (SBWA) approach was used and both models accurately predicted drug combinations for both known and unknown drugs. Lastly, a screening was conducted using each model where DeepNPD predicted combinations where drugs had similar targets, while DeepCombo predicted combinations where one agent potentiated the other, with both models’ predicted combinations investigated through a network-based analysis and identifying as a synergistic combinations in literature. DeepNPD notably identified Thioguanine and Hydroxyurea and DeepCombo discovered Vinblastine and Dasatinib as hits for potential new anticancer drug combinations. DeepNPD illustrates how natural products are a novel path where new drug combinations can be discovered.
In 2020, there were an estimated 19.3 million new cancer cases and 10.0 million deaths (Ferlay et al., 2021). It is also estimated that there is a 20% chance of getting cancer and a 10% chance of dying from cancer before age 75 (Ferlay et al., 2021). The “one drug, one target, one disease” strategy for drugs has been the dominating principle in pharmaceutical approaches and drug treatments (Zhou et al., 2016). However, there has been a shift in the past decade to combination therapies involving the use of several drugs to achieve greater therapeutic effects in treating complex diseases such as cancer (Chen et al., 2015; Zhou et al., 2016). The advantages of combinations are largely attributed to synergy, in that a combination of drugs results in a significantly stronger than additive effect (Chen et al., 2015). Synergy is typically a result of complex pharmacokinetic interactions between drugs affecting different pathways in the body (Bulusu et al., 2016). Various studies have shown that multicomponent therapies have greater efficacy in treating cancer. Thus, discovery of these multicomponent drug combinations is crucial to continue fighting cancer (Bulusu et al., 2016; Zhang et al., 2019).
Natural products have been shown to effectively treat many diseases and thus may provide an avenue for discovering new, effective drug combinations (Caesar and Cech, 2019; Sharma et al., 2023). Recently, natural products such as Opuntia ficus-indica and Ganoderma lucidum have been investigated and found to be effective against various cancers (Liu et al., 2021; Wang et al., 2023). Medicine systems such as traditional Chinese medicine (TCM) similarly provide extensive sets of natural products, many of which have been used as pharmaceutical drugs including berberine, capsaicin, and icariin (Zhu et al., 2022). Natural products are able to achieve good efficacy while reducing side effects through its active ingredients (Li, 2009; Atanasov et al., 2021). With natural products often containing many active ingredients, their efficacy is partly attributed to the effect of the natural compounds’ complex synergistic interactions (Zhou et al., 2016; Caesar and Cech, 2019). 5-Demethylnobiletin, an active ingredient found in orange peel, was found to synergize with Paclitaxel and greatly reduced the dosage to achieve effective anticancer properties (Tan et al., 2019). Furthermore, curcumin and hinokitiol, two natural products, were found to display synergistic anticancer properties at proportional low dosages, with limited cytotoxicity against normal fibroblast (Lee et al., 2018).
In fact, efforts to isolate a single active ingredient in natural products are often unsuccessful due to the removal of synergy (Caesar and Cech, 2019) and the complex organic matrices involved with the development of the natural product (Bucar et al., 2013). Compounds isolated from Rauwolfia serpentina, Hypericum perforatum, and Passiflora incarnata were unable to replicate activity of the plants’ crude extracts (Gilbert and Alves, 2003). Specifically, in Hypericum perforatum, hypericin’s oral bioavailability improved significantly when combined with several other H. perforatum active compounds such as procyanidin B2 and hyperocide (Caesar and Cech, 2019). Furthermore, the Artemisia annua herb’s antimalarial properties were significantly enhanced when using an herb extract compared to only its primary active ingredient, artemisinin (Rasoanaivo et al., 2011).
There is also increasing evidence that natural products display synergistic effects based on experiments conducted between active ingredients of herbs (Lin et al., 2007; Xu et al., 2014). In a study conducted by Lin et al. (2007), the compounds in Wedelia chinensis including indole-3-carboxaldehyde, wedelolactone, luteolin and apigenin were found to synergize with one another towards suppressing androgen activity. Certain plant metabolites of the Dorstenia family of herbs initially lacked trypanocidal activity when administered separately, yet became active when administered in a combination (Sandjo et al., 2016). Specifically, 4-hydroxylonchocarpine and 6,8-deprenyl eriodictyol displayed significant trypanocidal synergy, especially when administered in a 1:3 ratio (Sandjo et al., 2016).
There is also evidence that each component in a natural product contributes to the overall synergistic effect (Xu et al., 2014). Therefore, learning from this “combination rule” in which active ingredients combine in natural products can lead to discovery of new synergistic combinations. Over 60% of currently existing chemotherapy drugs are either natural products or derivatives of natural products, with natural products continuing to be useful for modern chemotherapy drug development (Demain and Vaishnav, 2011; Zhang et al., 2020). Therefore, natural products may have large applicability to modern pharmaceutical medicine.
In vitro investigations of drug synergism are typically conducted through high throughput screening (HTS), a method that can test a large number of combinations in a reasonable amount of time (He et al., 2018). However, HTS still cannot completely explore the large synergistic space of all existing drugs and potential combinations (Preuer et al., 2018). Therefore, computational methods of predicting synergism between drugs have appeal in completely exploring the synergistic space and identifying hits for new drug combinations (Preuer et al., 2018). Machine learning models built to predict synergy have already been employed effectively in several applications, including support vector machines (SVM) and random forest (RF) classifiers (Li et al., 2015). With the rise in computational models, many large datasets of HTS screenings of synergistic drug combinations have also been developed for use, making the development of such models much easier (Holbeck et al., 2017). Furthermore, deep neural networks (DNN), which are machine learning models designed after the human neuron network, can be utilized to achieve greater performance. Deep learning models are neural networks with multiple hidden layers, which serve to allow the model to create more complex relationships. Deep learning has affected many areas of science with its superior learning capacity (LeCun et al., 2015). Deep learning has also been applied to synergy predictions with better success than traditional machine learning models (Preuer et al., 2018). With synergy relationships being a complex subject, deep learning is able to learn better due to its superior ability to understand complex relationships than traditional machine learning methods. With the massive amount of natural products and compounds within those natural products, there is a large amount of data that can be used in the DNN, which benefits from large datasets (LeCun et al., 2015).
Beyond Preuer et al. (2018)’s DNN implementation using chemical structure, Liu and Xie (2021) also incorporated protein target information. Yang et al. (2021) developed a synergy prediction model using graph neural networks (GNN) to represent molecules, and Li et al. (2023) incorporated a novel siamese network and random matrix projection model to predict synergy. Liu et al. (2022) also developed a network based approach by representing the interaction network as a hypergraph, with drug-drug-cell edges. However, these models struggle with predicting for “unseen” drugs, or drugs that do not appear in the training data (Preuer et al., 2018; Yang et al., 2021; Li et al., 2023). This issue is problematic when employing models for newly developed drugs, or drugs that do not have adequate literature covering them. To mediate this issue, I implemented a novel ensemble and similarity based framework to allow for the proposed model to be broadcasted to completely novel drugs.
If the patterns of combinations in natural products can be discovered, they can potentially be applied to chemotherapy drugs to identify new and powerful synergistic drug combinations in treating cancer. Currently, the main issue with natural compounds is that they are often large, complex molecules which are difficult to isolate and produce for mass pharmaceutical uses (Chan, 1995; Meng et al., 2009). Due to the Nagoya Protocol and potential conflicts with countries where the natural compound originates from, researching natural products is difficult and their mass production is unrealistic (Atanasov et al., 2015). However, if the synergistic mechanisms of these natural compounds can be applied to chemotherapy drugs, which are easy to mass produce for pharmaceutical uses, there is potential to achieve both lower drug toxicity and effective synergistic interactions achieved by natural products on a mass pharmaceutical scale (Kong et al., 2008). Natural products and modern pharmaceutical medicines already have structural similarities and overlaps, with many natural compounds appearing in and used in pharmaceutical medicine (Kong et al., 2008). For example, galangal rhizome, a natural analgesic, appears in medicine as the eugenol drug which has analgesic properties (Kong et al., 2008).
Machine learning approaches can also be used to predict compound combination patterns in natural products. Preuer et al. (2018) have conducted a similar approach to predict synergy in chemotherapy drugs using chemical structure information, achieving good results. Compound chemical information along with protein targets were incorporated into the model. Protein targets play a key role in synergistic mechanisms in drugs, and models involving target proteins have also displayed positive results (Liu and Xie, 2021). Due to lack of reliable information for natural compound DPIs, a feature based machine learning approach was developed to predict DPIs in natural compounds. In addition, the downstream protein-protein interactions (PPI) of compounds were also simulated through a random walk with restart (RWR) algorithm to account for complex downstream protein pathways that affect synergistic interactions.
DeepNPD, a model to predict drug combinations within natural products, and DeepCombo, a model to predict synergy in chemotherapy drugs, were developed to explore a novel avenue of combination discovery and a more textbook avenue of combination discovery using the HERB and DrugCombDB databases respectively (Fang et al., 2018; Liu et al., 2020). A screening was run on a dataset of chemotherapy drugs to identify synergistic hits based on natural product combination principles. These hits were then investigated through literature review and a network based analysis to determine the validity of the new combinations.
To represent compounds’ chemical structure, each compound’s digital structural representation (SMILES) was obtained from their respective databases. Data of natural compounds was obtained from the HERB database, which contains 7,263 TCM natural product herbs and 49,258 of the compounds making up the herbs (Fang et al., 2018). Conversely, data of chemotherapy drugs was obtained from the DrugCombDB databases, which contains 2,887 unique chemotherapy drugs (Liu et al., 2020).
Using the SMILES key, each drug was encoded using the Mordred molecular descriptor calculator, a diverse and efficient descriptor calculator (Moriwaki et al., 2018). Molecular descriptors are mathematical representations of various properties of a compound, ranging from simple representation such as the number of acids and bases to more complex calculations such as lipophilicity and autocorrelation (Moriwaki et al., 2018). In total, 1613 2D descriptors were calculated using Mordred for representation of molecules.
Molecular fingerprints were also used for representation of molecules. Molecular fingerprints are a list of specific substructures and a fingerprint determines if each substructure is present in the molecule (Morgan, 1965). The Morgan fingerprint was implemented using the RDKit module in Python (Morgan, 1965; Landrum, 2010; Capecchi et al., 2020). In total, the Morgan fingerprint resulted in 2048 bits, which combined with the Mordred descriptors, resulted in 3661 features for each molecule representation.
Each DNN was engineered using a conic structure of neurons, with the first layer having n number of neurons, a second layer with n neurons, the third layer having n/2 number of neurons, and a final softmax output layer. Different optimizers were tested, including stochastic gradient descent (SGD), momentum boosted SGD, and the Adam optimizer, along with ReLU and Tanh initializers and dropout layers.
Additionally, a DPI based compound representation for combination prediction was developed. Since many natural compounds lack reliable DPI information, I developed DeepDPI, a DPI prediction model using the DrugBank database, a gold standard database with over 30,000 DPIs (Knox et al., 2023). DeepDPI followed a feature based approach, with the drug representation being concatenated with the protein representation. Deep learning has shown great success in many DPI prediction applications, including a compound’s binding affinity to a protein, therefore it was used to build the model (Yu et al., 2012; Mayr et al., 2018).
The drug representation consisted of the chemical representation described in the earlier section. The protein representation was calculated using PyDPI, a descriptor calculator capable of predicting descriptors for target proteins such as amino acid composition, autocorrelation, and quasi-sequence order (Cao et al., 2013). The protein sequences of each target gene were obtained from the STRING database to then input into PyDPI (Cao et al., 2013). With PyDPI calculating a total of 2049 protein descriptors concatenated with the 3661 features of the compound representation, there were a total of 5710 features.
After calculation of target proteins, the DPI representation also included downstream effects of target proteins and PPIs. Data of PPIs was obtained from the STRING database, a state-of-the-art database consisting of over one million human PPIs (Szklarczyk et al., 2021). To predict downstream effects, a random walk with restart algorithm was implemented to determine the relationship between the target protein and its PPIs based on a graph network with proteins as the nodes, PPIs as the edges, and the STRING confidence scores as the edge weight (Tong et al., 2008). Liu and Xie (2021) have implemented a similar RWR algorithm to good success.
I constructed a DNN, DeepNPD, to predict combinations within natural products. Data of natural products and their ingredients was obtained from the HERB database, with each drug combination being two compounds that appear in the same natural product (Fang et al., 2021). By following this method of obtaining drug combinations, millions of potential drug combinations are available to train DNNs, rather than just a couple thousand unique combinations from chemotherapy drug datasets.
Additionally, a screening was conducted to remove non-druglike molecules. For a drug to be druglike, it must fulfill aspects such as bioavailability. A method to assess if a drug is druglike is quantitative estimate of drug-likeness (QED), which has shown major advantages over typical assessments of drug-likeness (Bickerton et al., 2012).
Out of the products from the HERB database, there were over three million possible herb ingredient pairs. Since it is unrealistic to fit that much data in a single model, it was elected to choose subsets of ingredients and include all the possible pairs involving the subset of ingredients. By limiting the number of unique compounds, the model can focus on predicting patterns for a smaller set. The set of positive drug combinations consisted of all possible pairs in a set of 1000 herb compounds. The set of negative drug combinations consisted of two randomly selected drugs that do not appear in the same herb together in a 1:1 ratio with the number of positive combinations. In the dataset, a representation of a drug combination consisted of the features of Drug 1 and the features of Drug 2 concatenated together (Drug 1 + Drug 2). The inverted combination was also integrated (Drug 2 + Drug 1) to ensure that drug order does not play a role in the resulting prediction.
Two representations of drugs were tested: one solely using the chemical structure representation and another using the combined representation with chemical structure and DPI information to determine if DPIs can provide a boost to performance. The DPI representation consisted of the DPIs predicted by DeepDPI enhanced with the RWR algorithm.
Additionally, ensemble approaches were used to optimize the model to unseen drugs, or drugs that do not appear in the model’s training data. Ensemble approaches increase the number of unique models to consider a novel drug, which can therefore reduce the overfitting that merely a single model may bring. After a set of 2,688 natural products linked to treat cancer resulting in 15,149 ingredients were isolated and reduced to 6,148 druglike structures, those druglike structures were divided into six sets of approximately 1,000 structures in a bootstrapping approach. Each dataset then included all possible positive combinations of appearing in the same herb and an equal number of negative combinations of not appearing in the same herb, resulting in approximately 80,000 samples per model. Isolating one dataset for validation on unseen drugs, five models were trained and ensembled together by averaging the DNN confidence scores with one another.
I developed a novel similarity based weight adjustment (SBWA), which involves increasing the weights of the models which are trained on an ingredient similar to the drugs that are being predicted. In addition to the ensemble network, SBWA develops a weighted average to emphasize the models which could potentially predict better for a given novel drug, thereby resulting in better unseen drug predictions. Tanimoto similarity is calculated between the ingredients in each model and the drugs being predicted, which judges similarity by the number of shared substructures. A Tanimoto score of 0.7 or above is considered to be similar. If similar, the model weights of the model containing the similar drug were adjusted according to the following equation I developed:
where i indicated the model whose weight is being determined, W is the weight, s is the set of Tanimoto similarity scores of similar ingredients in the model’s training data that are above 0.70, t is a constant determined by how similar the predicted drug set is to the training data in general (higher t = more similar, lower t = less similar), n is the number of appearances of the similar drugs in the dataset, d is the dataset size, and m is the number of ensemble models in total. The equation not only takes into account whether similar drugs exist, but also how well the model has learned from those similar drugs. The more instances of the similar drugs in the model’s dataset, the better the model has learned from those drugs, therefore, the more weight that is assigned.
In addition to developing an herb predicting model, a model was also developed that predicts synergism in chemotherapy drugs. Using the same methods as DeepNPD, DeepCombo was developed using the DrugCombDB database, a publicly available database which includes a large number of cancer drug combinations tested on various cancer cell lines (Liu et al., 2020). A drug combination was considered to be any two drugs which synergized against a cancer cell line. The entire set of 2,887 chemotherapy drugs provided by DrugCombDB was used.
I also developed an ensemble approach for the chemotherapy drugs. In this case, three models were developed using different datasets: one model for the whole dataset, an ensemble of two models for the halved dataset, and an ensemble of four models for the quartered dataset. As the positive drug space is not as large for chemotherapy drugs, a SBWA approach was unable to be utilized.
It is useful to understand how confident the model is in its predictions. In DNNs, a softmax output is used, which outputs two decimals which indicate the model’s confidence in being a positive and negative sample. However, these confidence scores outputted by the softmax are often overconfident and not accurately representing.
Such an issue can be solved by calibrating the neural network through a method called temperature scaling. Temperature scaling involves dividing the logits of a classification neural network by a variable T (Guo et al., 2017). T is calibrated using cross entropy loss on a model’s validation dataset by comparing the predicted confidence scores with the actual results.
The equation is based off of the softmax equation in Gao et al. (2017)‘s softmax function, where the i subscript indicates the class being calibrated, q is the calibrated probability, λ is the logits of a class, n is the number of classes (2), and T is the temperature. In temperature scaling, a portion of the validation set is used to calibrate the value of T, a number typically greater than 1 which scales the softmax scores down to interpretable values. An example of an unscaled softmax score is [0.95, 0.05], and when scaled, the score is calibrated to a more useful value, for example,: [0.75, 0.25].
To evaluate the performance of the models, a variety of metrics should be used in order to develop an accurate idea of the model’s predictive capacity. For problems where class imbalance is not an issue (number of positive = number of negative), accuracy can be used which tells the proportion of predicted samples correct. However, for problems where class imbalance is an issue, F1 score, defined as the harmonic mean of precision and recall, is preferred, where precision is the proportion of positive samples predicted by the model which are correct and recall is the proportion of total positive samples identified by the model. F1 score is defined as the product of precision and recall divided by the sum.
Additionally, AUROC (area under receiving operator curve) and AUPRC (area under precision-recall curve) are both metrics which are built by measuring the area of respective curves (ROC plots true positive rate and true negative rate, PRC plots precision and recall). Beyond that, precision in itself is useful to report, as well as sensitivity/recall and specificity (proportion of negative samples identified).
It is possible to trace the measure of overlap between two drugs in a combination by mapping each drug’s DPIs onto a graph network of PPIs used in the RWR algorithm. Cheng et al. (2019) implemented the separation measure which was used in this study to quantify the amount of overlap between two drugs using the following equation:
which compares the mean shortest difference within the protein targets of each drug
In order to use a target protein based representation of drugs for combination prediction, a DPI prediction model must first be developed to predict DPI’s in natural compounds. DrugBank, a verified, gold standard drug database was used (Knox et al., 2023). In total, when considering each drug in which descriptors could be calculated, I used a set of 5,651 drugs in the dataset. Additionally, a list of 2,895 protein targets which appeared in the DrugBank data and whose descriptors could be calculated were used. In total, 24,930 DPIs were collected from DrugBank to use for the model creation. Those DPIs from DrugBank represented the positive samples in the dataset. The negative samples were chosen by randomly selecting a drug and a protein that it does not bind to, resulting in a dataset size of 49,860.
A DeepDPI, a DNN, was engineered to accomplish this task. To measure the performance of each model, five-fold cross validation was conducted to get the best measurement of the model’s performance. DeepDPI achieved an average accuracy of 0.84, with an average recall/sensitivity (positive binding accuracy) of 0.84 and an average specificity (negative binding accuracy) of 0.87. DeepDPI also achieved an average AUROC of 0.92 and AUPRC of 0.93 (Figure 1) These results indicate that the model was able to successfully learn from binding and also non-binding patterns.
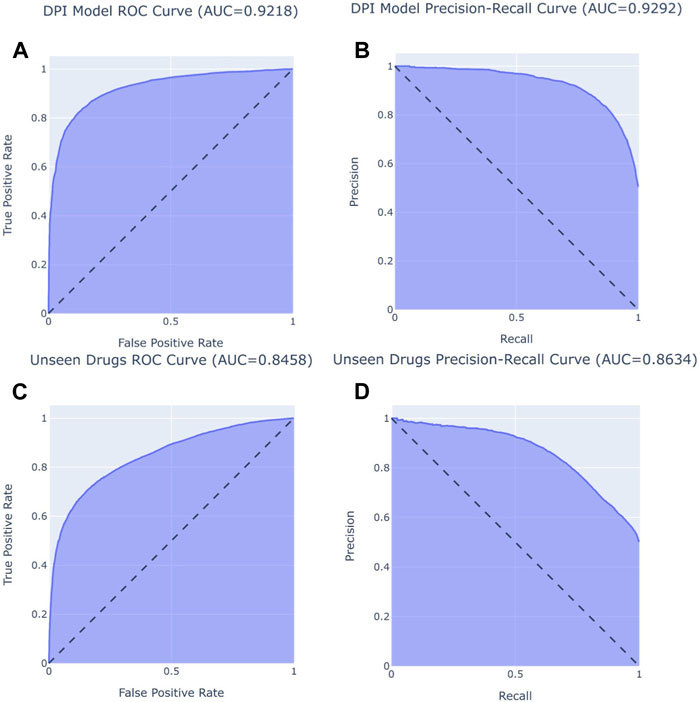
FIGURE 1. Left graphs (A, C) display AUROC curves, and right graphs (B, D) display AUPRC curves. Top graphs (A, B) display DeepDPI model performance (AUROC of 0.92, AUPRC of 0.91) and bottom graphs (C, D) show DeepDPI performance on unseen drugs (AUROC of 0.85, AUPRC of 0.86).
Since the model’s intended use is for unknown herb ingredients, it is important that the model is able to determine if unknown drugs would bind to known proteins. Therefore, a modified dataset which set aside a random sample of 1,000 drugs was created, including those 1,000 drugs’ binding proteins and randomly selected non-binding proteins. The rest of the DPI’s not included in this validation dataset were used to train the DNN model which would evaluate the modified dataset.
On the unseen drugs, DeepDPI achieved an accuracy of 0.77, an AUROC of 0.85, and an AUPRC of 0.86 (Figure 1). Additionally, the model achieved a high precision of 0.84 and also a high specificity of 0.87. Although there is a dropoff in performance, the model still performs well for unseen drugs and seen proteins.
The deep learning approach to the DPI prediction outperformed a similar feature based approach by Yu et al. (2012). Yu et al. utilized a RF model to predict DPIs with the DrugBank dataset, in which DeepDPI outperformed in most performance metrics (Table 1). Not only does accuracy show that DeepDPI performed better, the AUROC scores also show that DeepDPI’s confidence scores yielded better results than Yu et al.‘s RF approach.

TABLE 1. DeepDPI model performance vs. other models. Bolded values indicate the best performing model in each metric.
Additionally, a couple other models similarly trained on the Drugbank dataset are compared to DeepDPI, including Ezzat et al. (2016)’s ensemble learning approach and Sharma and Rani (2018)’s active learning ensemble framework (BE-DTI’). DeepDPI outperforms two of these state-of-the-art models while achieving similar performance to the most recently developed model, BE-DTI’.
To predict drug combinations within natural products, DeepNPD was developed to predict if two ingredients appear in the same natural product. DeepNPD represents the natural product avenue to combination discovery. As the total ingredient list had a size of 6,148, six models were developed and evaluated using 5-fold cross validation. The results of these six splits for both representations were averaged and can be found in Table 2. Overall, DeepNPD achieved excellent performance, with the combined representation only providing a small, nearly negligible boost in the overall performance of the model.
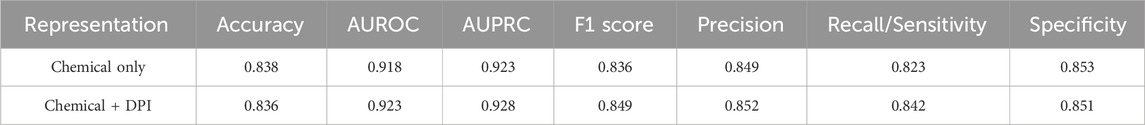
TABLE 2. DeepNPD average performance of bootstrapped models based on different representations.
However, when evaluating performance for such models which perform very well for drugs it has seen before, it is also crucial to evaluate performance for sets of drugs in which the model has not seen before. The above performance is based on validation sets which include drugs the model has seen before.
Although this model performs with high accuracy for seen data, it performs very poorly on unseen data. This is an issue seen in many state-of-the-art drug combination prediction models (Preuer et al., 2018). A novel way to remedy this issue lies in ensemble approaches. The bootstrapped models used for evaluation can be combined to form an ensemble model.
In the DNNs, ensemble approaches are conducted by averaging the confidence scores of each model. Furthermore, the weights of the ensemble model can be selectively changed to improve the model in the novel SBWA approach.
Ensemble approaches are highly successful in increasing a model’s precision, resulting in a more selective model (Table 3). Precision is crucial in drug combination prediction models since when the model predicts a positive sample, it is important that the model is correct, as it may be evaluated in vitro. Additionally, the SBWA applied models overall performed better than the model with equal weights. Although the precision and specificity dipped slightly, the recall saw large increases, indicating that the model is able to identify more positive samples when models that are familiar with the drugs at hand are given more weight. Although overall, the recall is still rather low, a high precision serves the purpose of screening new drug combinations for combination “hits” to test.
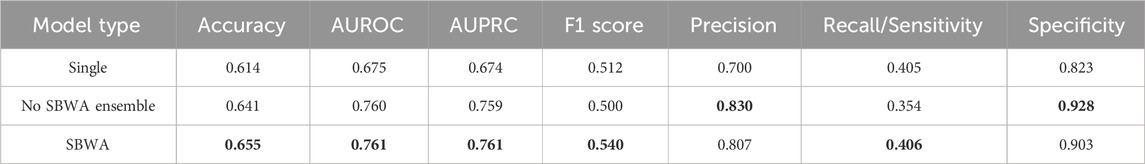
TABLE 3. DeepNPD performance on unseen drugs for single model, no SBWA ensemble model, and SBWA applied model. Bolded values indicate the best performing model in each metric.
The DrugCombDB drug combination database was used to find synergistic chemotherapy drug combinations in vitro (Liu et al., 2020). DeepCombo, a DNN, was developed to predict synergistic combinations in chemotherapy drugs, in order to investigate the avenue of drug combination discovery through existing chemotherapy drug combinations. A drug combination in DeepCombo was classified as any drug combination that displays a synergistic effect for any cell line. The positive drug combinations resulted in a set of 1925 drugs and 23,224 combinations. Since data of official cancer drug combinations is limited, this approach of cancer cell line data was opted for in order to gather more data.
Negative combinations were selected by randomly selecting pairs of drugs from the 1925 drug set which do not appear in the positive set in an equal ratio to the number of positive samples. A 5-fold cross-validation was conducted to evaluate the performance of each representation of the model. The average performances of the folds can be found in Table 4. Similar to DeepNPD, DeepCombo achieved excellent performance, but the DPIs failed to significantly increase performance.
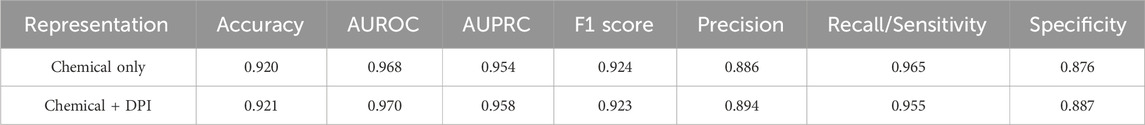
TABLE 4. DeepCombo performance.
Additionally, performance for unseen drugs must be tested in order to determine how accurate the model can be during deployment. In DeepCombo, SBWA approaches are not feasible due to smaller datasets; however, an equal weighted ensemble approach can still be conducted. Three different types of models were tested against sets of unseen drugs: the model trained on the whole dataset, an ensemble of two models each trained on half of the dataset, and an ensemble of four models trained on a quarter of the dataset. The results of a cross validation of four different sets of 500 drugs as unseen validation drugs are displayed in Table 5. Generally, the four model ensembles did the best in balancing precision and recall, therefore, it was used for eventual screening.

TABLE 5. DeepCombo performance on unseen drugs for single model, halved data ensemble model, and quartered data ensemble model. Bolded values indicate the best performing model in each metric.
A straightforward way to understand the overlap between these two medicine types is to run each model on the other model’s dataset(s). It could potentially show how well natural product combination properties fit to synergistic chemotherapy cancer drug combinations.
When running DeepNPD on the chemotherapy drug dataset, DeepNPD recorded a precision of 0.87 but a very low recall of just 0.05. This indicates that the drug pairs that fit the qualifications of a natural product pair are synergistic 87% of the time; however, only 5% of drugs manage to fulfill this criteria. This is likely due to the inherent structural differences between many of the natural compounds vs. chemotherapy compounds; natural compounds are much more complex; therefore, descriptor values tend to follow different patterns. However, when those patterns were picked out within the chemotherapy drug pairs, the rate of being synergistic was very high.
Additionally, when running DeepCombo on the DeepNPD datasets, DeepCombo recorded a precision of 0.65. DeepCombo was much more accepting of the natural product data, predicting 22% of samples as synergistic and achieving a recall of 0.27. Although DeepCombo is hardly a good overall predictor of natural product combinations, such results show the overlap of the chemotherapy combinations and natural product combinations.
Lastly, a screening for new drug combinations was conducted to discover new potential drug combinations using each model. A list of 488 anti-neoplastic agents was collected from DrugBank, and a screening which tested all possible combinations of the drug set was conducted using the chemical representation SBWA DeepNPD and four model combined representation of DeepCombo. The resulting screening tested the ability to use natural product combination patterns to identify new drug combinations in chemotherapy drugs that are easily mass produced. Resulting confidence scores were calculated by averaging the scores of a combination scanned twice with different orders of drug pairs (Drug 1 + Drug 2, Drug 2 + Drug 1). First, a screening using DeepNPD was conducted and a list of curated combinations appears in Table 6. Top DeepNPD predictions do not have particularly high confidence scores; this again results from the inherent structural differences resulting in somewhat imperfect matches. Therefore, unsurprisingly, the model largely favored natural compounds, or derivatives of natural compounds. In an effort to discover fully chemotherapy drugs, only a couple sets of these natural compounds were shown. Additionally, DeepNPD also did manage to identify natural product pairs, such as vinblastine and vincristine, which were isolated from the periwinkle plant (Kumar, 2016).
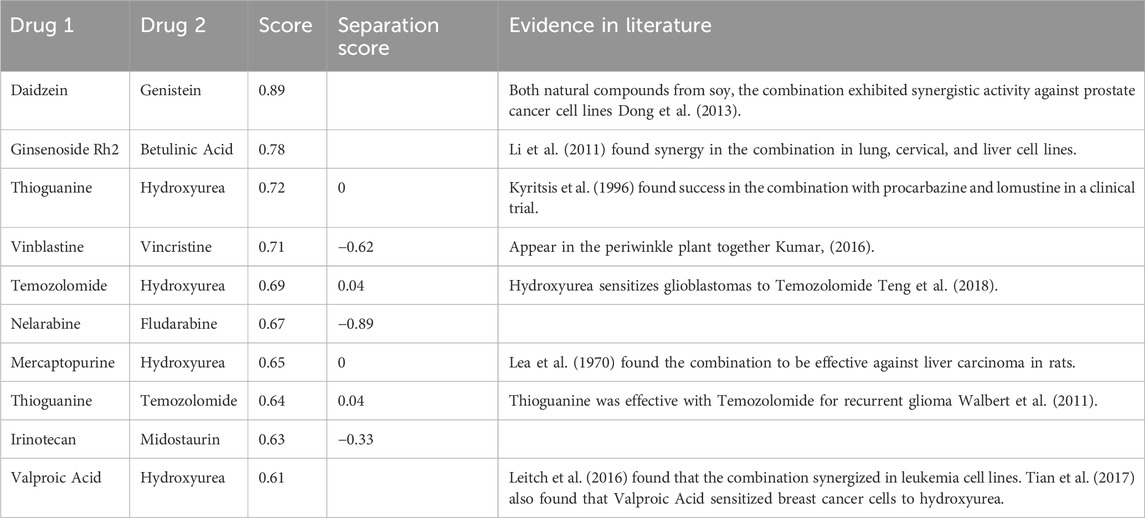
TABLE 6. Table of selected DeepNPD drug screening predictions.
Only two combinations of natural products were included (the top two highest scores), both of which synergized. Among the predicted results, hydroxyurea appeared very often due it being an analog to urea, a natural compound. In the curated list, I attempted to include a diverse set of drugs (more unique drugs) and refrained from including too many natural products. Overall, eight out of these ten predictions were found in literature, with five of the combinations having synergized; two of these being natural compounds, and three of these chemotherapy drug combinations.
Many of DeepNPD’s predictions seem to involve two drugs which act on the same target through different pathways, termed “specific” synergy by Cokol et al. (2011).
In the natural compound combination of Ginsenoside Rh2 and Betulinic Acid, both agents’ anticancer effects are from direct inducement of apoptosis, where Ginsenoside Rh2 targets the Bcl-2 gene and Betulinic Acid targets a mitochondrial pathway (Li et al., 2011). Within the chemotherapy drug combination of Temozolomide and Hydroxyurea, both agents similarly target different sections of DNA synthesis, resulting in their synergy.
The Nelarabine and Fludarabine combination has shown promise in clinical trials, showing less toxicity and increased efficacy despite lacking evidence of direct synergy (Gandhi et al., 2008). Nelarabine and Fludarabine, when synthesized into ara-GTP and F-ara-ATP respectively, act through different mechanisms to inhibit DNA synthesis.
The combination of Hydroxyurea and Thioguanine similarly work to inhibit DNA synthesis. Hydroxyurea disables the ribonucleotide reductase enzyme, inhibiting DNA synthesis, whereas Thioguanine competes to bind to the hypoxanthine-guanine phosphoribosyltransferase enzyme. Thioguanine nucleotides additionally link with DNA and RNA, contributing to cytotoxic effects. Although the two drugs have not been tested for synergy, they have displayed synergy with similar psoriasis therapies (Lebwohl et al., 2004). Similarly, Mercaptopurine, which was also predicted in a combination with Hydroxyurea, also competes to bind to hypoxanthine-guanine phosphoribosyltransferase.
Additionally, a screening using DeepCombo using the same set of drugs was conducted (Table 7).
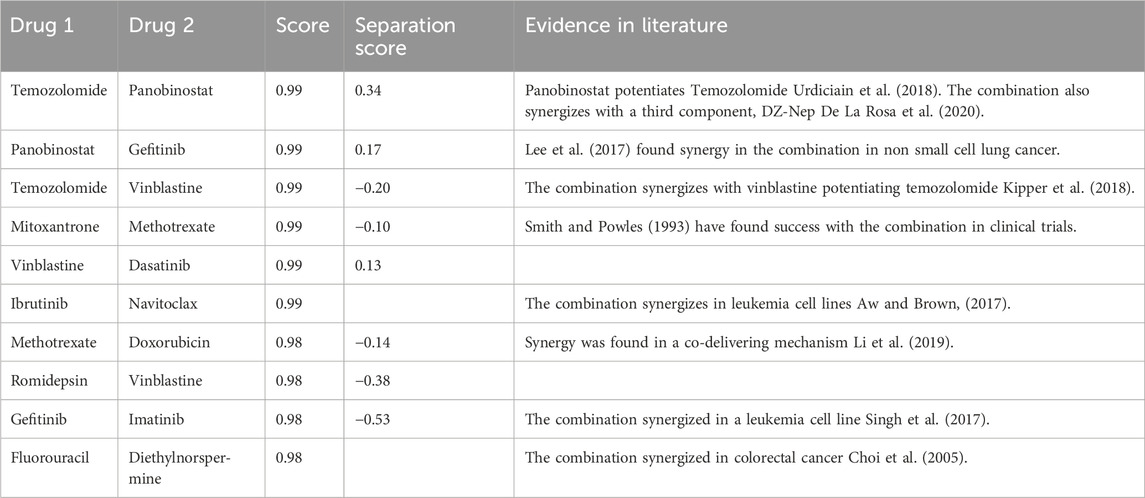
TABLE 7. Table of selected DeepCombo drug screening predictions.
Among the top DeepCombo predictions, eight of the ten combinations were found in literature and seven of those were shown to synergize against cancer cells in vitro. Interestingly, the model heavily favored certain drugs, especially panobinostat and temozolomide. Due to that occurrence, the number of appearances of those drugs in the list was limited to focus on displaying other drug combinations. In the curated list, I similarly attempted to include a diverse drug set in the combination space.
Contrary to DeepNPD predictions, DeepCombo screening results seem to indicate sets of drugs which potentiate the other, rather than act on the same target. Foremost of the predictions involve Temozolomide and Panobinostat, which are two drugs which act on different pathways. Where Temozolomide is a DNA inhibitor, Panobinostat is a histone deacetylase (HDAC) inhibitor (Urdiciain et al., 2018). A study between the two drugs found that Panobinostat had a limited effect on glioblastoma cells, but greatly boosted the cytotoxic effects of Temozolomide against the same cells. Similarly, Panobinostat also potentiates the effects of Gefitinib by inhibiting a pathway which downregulates the epidermal growth factor receptor, the primary target of Gefitinib (Lee et al., 2017). Panobinostat is a common potentiating agent, which was favored heavily by DeepCombo.
Regarding the Mitoxantrone and Methotrexate combination, the two agents also act on different targets. Where Mitoxantrone directly interferes with DNA, Methotrexate interferes with nucleotide synthesis. The nature of the combination again differs with the patterns seen in DeepNPD. The same differing mechanisms are seen in Vinblastine and Dasatinib, with Vinblastine directly interfering with mitosis and Dasatinib being a kinase inhibitor. Such patterns suggest that if the combination does indeed synergize, it would be due to potentiation.
Using the screening predictions from DeepNPD and DeepCombo, the successful combinations were run through the PPI network and a separation score calculated. DeepNPD’s positive predictions for drugs with available protein target information resulted in an average separation score of −0.03, while DeepCombo’s predictions resulted in an average separation score of 0.15. A t-test of the two scores resulted in a p-value <0.001. This result further confirms that DeepNPD predicts drugs with more similar/overlapping targets while DeepCombo tends to predict potentiation interactions where drugs target separate target neighborhoods.
In this study, DNNs were built to predict combinations within natural products and chemotherapy drugs, exploring two avenues of drug combination discovery. First, a DPI prediction DNN, termed DeepDPI, was built to predict DPIs using the DrugBank database. DeepDPI achieved state-of-the-art performance in DPI prediction tasks. Then, DeepNPD, a model which predicts if a pair of ingredients would appear in a natural product together, was successfully developed using the HERB database (Fang et al., 2018). Using an ensemble approach and SBWA techniques, DeepNPD was optimized to predict combinations in unseen drug combinations, helping to solve a problem seen in many past drug combination prediction models, including DeepSynergy, GraphSynergy, and TranSynergy (Preuer et al., 2018; Liu and Xie, 2021; Yang et al., 2021). Similarly, DeepCombo, a model which predicts synergistic chemotherapy drug combinations in cancer cell lines, was also built successfully and optimized to unseen drugs, using the DrugCombDB database (Liu et al., 2020).
In the model integration stage where DeepNPD was tested on DeepCombo data and vice versa, both models achieved decent precision scores. Most notably, DeepNPD had a very high precision in DeepCombo data, which has implications in using DeepNPD as a novel method to develop new drug combinations. Although DeepNPD prefers natural products, the patterns that DeepNPD identifies may lead to synergistic drug combinations in chemotherapy drugs. In DeepNPD’s chemotherapy drug combination screening, eight of the ten chemotherapy drug combinations were found in literature, with five of them showing evidence of synergy in cancer cell lines.
In DeepCombo’s chemotherapy drug combination screening, seven of the ten drug combinations were found to display synergy in literature. This potentially indicates the strength of DeepCombo to discover novel drug combinations. Compared to other cancer drug combination prediction models such as DeepSynergy and TranSynergy, which predicts the strength of synergy in a regression format for a specific cell line, DeepCombo follows a more general approach by indicating if a drug synergizes in any cell line (Preuer et al., 2018; Liu et al., 2021). Additionally, the classification format that DeepCombo follows may also be beneficial, especially for screening new drugs. DeepSynergy and TranSynergy, which are designed as regression tasks, have poor performance for screening new drugs. However, DeepCombo, designed as a classification task, shows a high precision for screening new drugs. Therefore, by using a classification framework, synergy prediction models can be more useful when screening unseen drugs. Furthermore, when applying the ensemble structure and SBWA, better optimization towards unseen drugs can potentially be achieved in future drug combination predicting models.
Additionally, DeepNPD tends to predict drugs which have similar targets but differing mechanisms of action. Synergy which occurs through that mechanism was termed “specific” by Cokol et al. (2011), and specific synergy is less common than “promiscuous” synergy, where each drug acts on a different target, typically synergizing through potentiation. It is important to note that DeepNPD does not simply predict a drug pair which are similar with one another, but rather differing drugs that act on a similar target through different mechanisms. There may also be implications in the mechanisms of natural products, which may tend to have a group of ingredients which act on a single target through differing mechanisms.
On the other hand, DeepCombo predictions tend to follow more typical promiscuous synergy where one agent potentiates the other. This is displayed in many of the screening predictions. Interestingly, Panobinostat appeared very frequently with high confidence. HDAC inhibitors like Panobinostat have been shown to potentiate various DNA inhibitors while having a low anticancer effect itself (Maiso et al., 2009). Such an occurrence may implicate the model’s preference towards potentiation interactions. Additionally, many of the other predicted combinations showed drugs with differing targets, suggesting that if the combination indeed synergizes, then it would be due to promiscuous synergy rather than specific synergy.
This pattern is further confirmed by the network-based analysis, which ran separation scores on the resulting predictions. DeepNPD’s predictions resulting in drugs with heavier target protein overlap and lower separation scores than DeepCombo’s predictions is further evidence of the differing patterns.
Due to this difference in synergy predictions, DeepNPD could potentially predict combinations with less intense polypharmacological properties due to specific synergy targeting a less widespread set of targets. Interestingly, this contrasts with common ideas of natural products promoting polypharmacological approaches (Fang et al., 2018). Regardless, polypharmacology presents both benefits and risks of increased efficacy and adverse side effects respectively (Peters, 2013). However, natural products can often reduce toxicity due to low active compound concentrations. In the future, it may be more beneficial to characterize a natural product by a group of active ingredients rather than a pair, to fully allow the network interactions to be taken advantage of.
In vitro testing is still needed to fully determine the viability of combination predictions, especially the DeepNPD predictions. Although a general pattern was observed in DeepNPD, it is still unclear whether such mechanisms would truly be effective.
The weaknesses of DeepCombo lie in that it does not indicate which cell lines a combination may synergize for. Many models such as DeepSynergy and TranSynergy account for genomic expression profiles of cell lines which relate to the drug representations (Preuer et al., 2018; Liu et al., 2021). Although there are strengths to DeepCombo’s approach, a selected combination must be further tested in vitro against cell lines to determine where it may synergize in DeepCombo, where in DeepSynergy, a combination could be immediately tested against the cell line it was predicted to synergize against.
The combined representation failed to increase accuracy in both models. The boost was very small and almost negligible, possible even due to random chance. Since the DPI representation is based on the chemical representation, the model may have already learned from much of the information provided by the DPI representation. Overall, the added computational power to predict DPIs using DeepDPI is not worth such a small boost in performance.
DeepNPD and DeepCombo both successfully identified synergistic drug combinations, indicating the strength of both avenues of natural products and existing synergistic drug combinations. Using novel model architectures to optimize models towards unseen drugs, DeepCombo displays an edge against typical chemotherapy drug combination prediction models, implying potential future usage in predicting for newer drugs and discovering synergistic mechanisms faster and more efficiently. Regarding the “combination rule” in natural products there is potential to further investigate mechanisms of drug synergy within natural products, and in utilizing the synergy patterns in DeepNPD to drive modern drug discovery in chemotherapy drugs. Despite the general difficulty of using natural products, there may be deeper and more useful pharmacokinetic patterns inherent in them, which can be utilized to advance modern drug discovery as a whole.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
EC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
I would like to thank Dr. Daniel LaBarbera (University of Colorado Denver) for his advice regarding natural product based drug discovery, as well as Dr. Paul K. Strode (Fairview High School) for his advice towards manuscript writing. Lastly, I would also like to thank ChemRxiv for publishing the manuscript as a preprint.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Atanasov, A. G., Waltenberger, B., Pferschy-Wenzig, E. M., Linder, T., Wawrosch, C., Uhrin, P., et al. (2015). Discovery and resupply of pharmacologically active plant-derived natural products: a review. Biotechnol. Adv. 33 (8), 1582–1614. doi:10.1016/j.biotechadv.2015.08.001
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., and Supuran, C. T. (2021). Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov. 20 (3), 200–216. doi:10.1038/s41573-020-00114-z
Aw, A., and Brown, J. R. (2017). The potential combination of BCL-2 inhibitors and ibrutinib as frontline therapy in chronic lymphocytic leukemia. Leukemia Lymphoma 58 (10), 2287–2297. doi:10.1080/10428194.2017.1312387
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S., and Hopkins, A. L. (2012). Quantifying the chemical beauty of drugs. Nat. Chem. 4 (2), 90–98. doi:10.1038/nchem.1243
Bucar, F., Wube, A., and Schmid, M. (2013). Natural product isolation–how to get from biological material to pure compounds. Nat. Product. Rep. 30 (4), 525–545. doi:10.1039/c3np20106f
Bulusu, K. C., Guha, R., Mason, D. J., Lewis, R. P., Muratov, E., Motamedi, Y. K., et al. (2016). Modelling of compound combination effects and applications to efficacy and toxicity: state-of-the-art, challenges and perspectives. Drug Discov. Today 21 (2), 225–238. doi:10.1016/j.drudis.2015.09.003
Caesar, L. K., and Cech, N. B. (2019). Synergy and antagonism in natural product extracts: when 1+ 1 does not equal 2. Nat. Product. Rep. 36 (6), 869–888. doi:10.1039/c9np00011a
Cao, D. S., Liang, Y. Z., Yan, J., Tan, G. S., Xu, Q. S., and Liu, S. (2013). PyDPI: freely available python package for chemoinformatics, bioinformatics, and chemogenomics studies.
Capecchi, A., Probst, D., and Reymond, J. L. (2020). One molecular fingerprint to rule them all: drugs, biomolecules, and the metabolome. J. Cheminformatics 12 (1), 43–15. doi:10.1186/s13321-020-00445-4
Chan, K. (1995). Progress in traditional Chinese medicine. Trends Pharmacol. Sci. 16 (6), 182–187. doi:10.1016/s0165-6147(00)89019-7
Chen, D., Liu, X., Yang, Y., Yang, H., and Lu, P. (2015). Systematic synergy modeling: understanding drug synergy from a systems biology perspective. BMC Syst. Biol. 9 (1), 56–10. doi:10.1186/s12918-015-0202-y
Cheng, F., Kovács, I. A., and Barabási, A. L. (2019). Network-based prediction of drug combinations. Nat. Commun. 10 (1), 1197. doi:10.1038/s41467-019-09186-x
Choi, W., Gerner, E. W., Ramdas, L., Dupart, J., Carew, J., Proctor, L., et al. (2005). Combination of 5-fluorouracil and N1, N11-diethylnorspermine markedly activates spermidine/spermine N1-acetyltransferase expression, depletes polyamines, and synergistically induces apoptosis in colon carcinoma cells. J. Biol. Chem. 280 (5), 3295–3304. doi:10.1074/jbc.m409930200
Cokol, M., Chua, H. N., Tasan, M., Mutlu, B., Weinstein, Z. B., Suzuki, Y., et al. (2011). Systematic exploration of synergistic drug pairs. Mol. Syst. Biol. 7 (1), 544. doi:10.1038/msb.2011.71
De La Rosa, J., Urdiciain, A., Zazpe, I., Zelaya, M. V., Meléndez, B., Rey, J. A., et al. (2020). The synergistic effect of DZ-NEP, panobinostat and temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 56 (1), 283–300. doi:10.3892/ijo.2019.4905
Demain, A. L., and Vaishnav, P. (2011). Natural products for cancer chemotherapy. Microb. Biotechnol. 4 (6), 687–699. doi:10.1111/j.1751-7915.2010.00221.x
Dong, X., Xu, W., Sikes, R. A., and Wu, C. (2013). Combination of low dose of genistein and daidzein has synergistic preventive effects on isogenic human prostate cancer cells when compared with individual soy isoflavone. Food Chem. 141 (3), 1923–1933. doi:10.1016/j.foodchem.2013.04.109
Ezzat, A., Wu, M., Li, X. L., and Kwoh, C. K. (2016). Drug-target interaction prediction via class imbalance-aware ensemble learning. BMC Bioinforma. 17 (19), 509–276. doi:10.1186/s12859-016-1377-y
Fang, J., Liu, C., Wang, Q., Lin, P., and Cheng, F. (2018). In silico polypharmacology of natural products. Briefings Bioinforma. 19 (6), 1153–1171. doi:10.1093/bib/bbx045
Fang, S., Dong, L., Liu, L., Guo, J., Zhao, L., Zhang, J., et al. (2021). HERB: a high-throughput experiment-and reference-guided database of traditional Chinese medicine. Nucleic Acids Res. 49 (D1), D1197–D1206. doi:10.1093/nar/gkaa1063
Ferlay, J., Colombet, M., Soerjomataram, I., Parkin, D. M., Piñeros, M., Znaor, A., et al. (2021). Cancer statistics for the year 2020: an overview. Int. J. Cancer 149 (4), 778–789. doi:10.1002/ijc.33588
Gandhi, V., Tam, C., O'Brien, S., Jewell, R. C., Rodriguez, C. O., Lerner, S., et al. (2008). Phase I trial of nelarabine in indolent leukemias. J. Clin. Oncol. 26 (7), 1098–1105. doi:10.1200/jco.2007.14.1986
Gao, B., and Pavel, L. (2017). On the properties of the softmax function with application in game theory and reinforcement learning. arXiv preprint arXiv:1704.00805.
Gilbert, B., and Alves, L. (2003). Synergy in plant medicines. Curr. Med. Chem. 10 (1), 13–20. doi:10.2174/0929867033368583
Guo, C., Pleiss, G., Sun, Y., and Weinberger, K. Q. (2017). “On calibration of modern neural networks,” in International conference on machine learning (PMLR), 1321–1330.
He, L., Kulesskiy, E., Saarela, J., Turunen, L., Wennerberg, K., Aittokallio, T., et al. (2018). “Methods for high-throughput drug combination screening and synergy scoring,” in Cancer systems biology (New York, NY: Humana Press), 351–398.
Holbeck, S. L., Camalier, R., Crowell, J. A., Govindharajulu, J. P., Hollingshead, M., Anderson, L. W., et al. (2017). The national cancer Institute almanac: a comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res. 77 (13), 3564–3576. doi:10.1158/0008-5472.can-17-0489
Kipper, F. C., Silva, A. O., Marc, A. L., Confortin, G., Junqueira, A. V., Neto, E. P., et al. (2018). Vinblastine and antihelmintic mebendazole potentiate temozolomide in resistant gliomas. Investig. New Drugs 36, 323–331. doi:10.1007/s10637-017-0503-7
Knox, C., Wilson, M., Klinger, C. M., Franklin, M., Oler, E., Wilson, A., et al. (2023). DrugBank 6.0: the DrugBank knowledgebase for 2024. Nucleic Acids Res., gkad976. doi:10.1093/nar/gkad976
Kong, D. X., Li, X. J., Tang, G. Y., and Zhang, H. Y. (2008). How many traditional Chinese medicine components have been recognized by modern Western medicine? A chemoinformatic analysis and implications for finding multicomponent drugs. ChemMedChem Chem. Enabling Drug Discov. 3 (2), 233–236. doi:10.1002/cmdc.200700291
Kyritsis, A. P., Yung, W. K., Jaeckle, K. A., Bruner, J., Gleason, M. J., Ictech, S. E., et al. (1996). Combination of 6-thioguanine, procarbazine, lomustine, and hydroxyurea for patients with recurrent malignant gliomas. Neurosurgery 39 (5), 921–926. doi:10.1227/00006123-199611000-00006
Landrum, G. (2010). “RDKit.” Q2. Available at: https://wwwrdkit.org/.
Lea, M. A., Sasovetz, D., Musella, A., and Morris, H. P. (1970). Effects of hydroxyurea and 6-mercaptopurine on growth and some aspects of carbohydrate metabolism in regenerating and neoplastic liver. Cancer Res. 30 (7), 1994–1999.
Lebwohl, M., Menter, A., Koo, J., and Feldman, S. R. (2004). Combination therapy to treat moderate to severe psoriasis. J. Am. Acad. Dermatology 50 (3), 416–430. doi:10.1016/j.jaad.2002.12.002
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep learning. Nature 521 (7553), 436–444. doi:10.1038/nature14539
Lee, T. B., Seo, E. J., Lee, J. Y., and Jun, J. H. (2018). Synergistic anticancer effects of curcumin and hinokitiol on gefitinib resistant non-small cell lung cancer cells. Nat. Product. Commun. 13 (12), 1934578X1801301. doi:10.1177/1934578x1801301223
Lee, W. Y., Chen, P. C., Wu, W. S., Wu, H. C., Lan, C. H., Huang, Y. H., et al. (2017). Panobinostat sensitizes KRAS-mutant non-small-cell lung cancer to gefitinib by targeting TAZ. Int. J. Cancer 141 (9), 1921–1931. doi:10.1002/ijc.30888
Leitch, C., Osdal, T., Andresen, V., Molland, M., Kristiansen, S., Nguyen, X. N., et al. (2016). Hydroxyurea synergizes with valproic acid in wild-type p53 acute myeloid leukaemia. Oncotarget 7 (7), 8105–8118. doi:10.18632/oncotarget.6991
Li, P., Huang, C., Fu, Y., Wang, J., Wu, Z., Ru, J., et al. (2015). Large-scale exploration and analysis of drug combinations. Bioinformatics 31 (12), 2007–2016. doi:10.1093/bioinformatics/btv080
Li, Q., Li, Y., Wang, X., Fang, X., He, K., Guo, X., et al. (2011). Co-treatment with ginsenoside Rh2 and betulinic acid synergistically induces apoptosis in human cancer cells in association with enhanced capsase-8 activation, bax translocation, and cytochrome c release. Mol. Carcinog. 50 (10), 760–769. doi:10.1002/mc.20673
Li, S. (2009). Network systems underlying traditional Chinese medicine syndrome and herb formula. Curr. Bioinforma. 4 (3), 188–196. doi:10.2174/157489309789071129
Li, T. H., Wang, C. C., Zhang, L., and Chen, X. (2023). SNRMPACDC: computational model focused on Siamese network and random matrix projection for anticancer synergistic drug combination prediction. Briefings Bioinforma. 24 (1), bbac503. doi:10.1093/bib/bbac503
Li, Y., Chen, S., Chang, X., He, F., and Zhuo, R. (2019). Efficient co-delivery of doxorubicin and methotrexate by pH-sensitive dual-functional nanomicelles for enhanced synergistic antitumor efficacy. ACS Appl. Bio Mater. 2 (5), 2271–2279. doi:10.1021/acsabm.9b00230
Lin, F. M., Chen, L. R., Lin, E. H., Ke, F. C., Chen, H. Y., Tsai, M. J., et al. (2007). Compounds from Wedelia chinensis synergistically suppress androgen activity and growth in prostate cancer cells. Carcinogenesis 28 (12), 2521–2529. doi:10.1093/carcin/bgm137
Liu, H., Zhang, W., Zou, B., Wang, J., Deng, Y., and Deng, L. (2020). DrugCombDB: a comprehensive database of drug combinations toward the discovery of combinatorial therapy. Nucleic Acids Res. 48 (D1), D871–D881. doi:10.1093/nar/gkz1007
Liu, M. M., Liu, T., Yeung, S., Wang, Z., Andresen, B., Parsa, C., et al. (2021). Inhibitory activity of medicinal mushroom Ganoderma lucidum on colorectal cancer by attenuating inflammation. Precis. Clin. Med. 4 (4), 231–245. doi:10.1093/pcmedi/pbab023
Liu, Q., and Xie, L. (2021). TranSynergy: mechanism-driven interpretable deep neural network for the synergistic prediction and pathway deconvolution of drug combinations. PLoS Comput. Biol. 17 (2), e1008653. doi:10.1371/journal.pcbi.1008653
Liu, X., Song, C., Liu, S., Li, M., Zhou, X., and Zhang, W. (2022). Multi-way relation-enhanced hypergraph representation learning for anti-cancer drug synergy prediction. Bioinformatics 38 (20), 4782–4789. doi:10.1093/bioinformatics/btac579
Maiso, P., Colado, E., Ocio, E. M., Garayoa, M., Martin, J., Atadja, P., et al. (2009). The synergy of panobinostat plus doxorubicin in acute myeloid leukemia suggests a role for HDAC inhibitors in the control of DNA repair. Leukemia 23 (12), 2265–2274. doi:10.1038/leu.2009.182
Mayr, A., Klambauer, G., Unterthiner, T., Steijaert, M., Wegner, J. K., Ceulemans, H., et al. (2018). Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 9 (24), 5441–5451. doi:10.1039/c8sc00148k
Meng, W., Xiaoliang, R., Xiumei, G., Vincieri, F. F., and Bilia, A. R. (2009). Stability of active ingredients of traditional Chinese medicine (TCM). Nat. Product. Commun. 4 (12), 1934578X0900401. doi:10.1177/1934578x0900401229
Morgan, H. L. (1965). The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Documentation 5 (2), 107–113. doi:10.1021/c160017a018
Moriwaki, H., Tian, Y. S., Kawashita, N., and Takagi, T. (2018). Mordred: a molecular descriptor calculator. J. Cheminformatics 10 (1), 4–14. doi:10.1186/s13321-018-0258-y
Peters, J. U. (2013). Polypharmacology–foe or friend? J. Med. Chem. 56 (22), 8955–8971. doi:10.1021/jm400856t
Preuer, K., Lewis, R. P., Hochreiter, S., Bender, A., Bulusu, K. C., and Klambauer, G. (2018). DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 34 (9), 1538–1546. doi:10.1093/bioinformatics/btx806
Rasoanaivo, P., Wright, C. W., Willcox, M. L., and Gilbert, B. (2011). Whole plant extracts versus single compounds for the treatment of malaria: synergy and positive interactions. Malar. J. 10 (1), S4–S12. doi:10.1186/1475-2875-10-s1-s4
Sandjo, L. P., de Moraes, M. H., Kuete, V., Kamdoum, B. C., Ngadjui, B. T., and Steindel, M. (2016). Individual and combined antiparasitic effect of six plant metabolites against Leishmania amazonensis and Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 26 (7), 1772–1775. doi:10.1016/j.bmcl.2016.02.044
Sharma, A., and Rani, R. (2018). BE-DTI’: ensemble framework for drug target interaction prediction using dimensionality reduction and active learning. Comput. Methods Programs Biomed. 165, 151–162. doi:10.1016/j.cmpb.2018.08.011
Sharma, R., Singla, R. K., Banerjee, S., and Sharma, R. (2023). Revisiting Licorice as a functional food in the management of neurological disorders: bench to trend. Neurosci. Biobehav. Rev. 155, 105452. doi:10.1016/j.neubiorev.2023.105452
Singh, V. K., Chang, H. H., Kuo, C. C., Shiao, H. Y., Hsieh, H. P., and Coumar, M. S. (2017). Drug repurposing for chronic myeloid leukemia: in silico and in vitro investigation of DrugBank database for allosteric Bcr-Abl inhibitors. J. Biomol. Struct. Dyn. 35 (8), 1833–1848. doi:10.1080/07391102.2016.1196462
Smith, I. E., and Powles, T. J. (1993). MMM (mitomycin/mitoxantrone/methotrexate): an effective new regimen in the treatment of metastatic breast cancer. Oncology 50 (1), 9–15. doi:10.1159/000227241
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49 (D1), D605–D612. doi:10.1093/nar/gkaa1074
Tan, K. T., Li, S., Li, Y. R., Cheng, S. L., Lin, S. H., and Tung, Y. T. (2019). Synergistic anticancer effect of a combination of paclitaxel and 5-demethylnobiletin against lung cancer cell line in vitro and in vivo. Appl. Biochem. Biotechnol. 187, 1328–1343. doi:10.1007/s12010-018-2869-1
Teng, J., Hejazi, S., Hiddingh, L., Carvalho, L., de Gooijer, M. C., Wakimoto, H., et al. (2018). Recycling drug screen repurposes hydroxyurea as a sensitizer of glioblastomas to temozolomide targeting de novo DNA synthesis, irrespective of molecular subtype. Neuro-Oncology 20 (5), 642–654. doi:10.1093/neuonc/nox198
Tian, Y., Liu, G., Wang, H., Tian, Z., Cai, Z., Zhang, F., et al. (2017). Valproic acid sensitizes breast cancer cells to hydroxyurea through inhibiting RPA2 hyperphosphorylation-mediated DNA repair pathway. DNA Repair 58, 1–12. doi:10.1016/j.dnarep.2017.08.002
Tong, H., Faloutsos, C., and Pan, J. Y. (2008). Random walk with restart: fast solutions and applications. Knowl. Inf. Syst. 14 (3), 327–346. doi:10.1007/s10115-007-0094-2
Urdiciain, A., Meléndez, B., Rey, J. A., Idoate, M. A., and Castresana, J. S. (2018). Panobinostat potentiates temozolomide effects and reverses epithelial–mesenchymal transition in glioblastoma cells. Epigenomes 2 (1), 5. doi:10.3390/epigenomes2010005
Walbert, T., Gilbert, M. R., Groves, M. D., Puduvalli, V. K., Alfred Yung, W. K., Conrad, C. A., et al. (2011). Combination of 6-thioguanine, capecitabine, and celecoxib with temozolomide or lomustine for recurrent high-grade glioma. J. Neuro-Oncology 102, 273–280. doi:10.1007/s11060-010-0313-7
Wang, J., Rani, N., Jakhar, S., Redhu, R., Kumar, S., Kumar, S., et al. (2023). Opuntia ficus-indica (L.) Mill.-anticancer properties and phytochemicals: current trends and future perspectives. Front. Plant Sci. 14, 1236123. doi:10.3389/fpls.2023.1236123
Xu, X., Li, F., Zhang, X., Li, P., Zhang, X., Wu, Z., et al. (2014). In vitro synergistic antioxidant activity and identification of antioxidant components from Astragalus membranaceus and Paeonia lactiflora. PloS One 9 (5), e96780. doi:10.1371/journal.pone.0096780
Yang, J., Xu, Z., Wu, W. K. K., Chu, Q., and Zhang, Q. (2021). GraphSynergy: a network-inspired deep learning model for anticancer drug combination prediction. J. Am. Med. Inf. Assoc. 28 (11), 2336–2345. doi:10.1093/jamia/ocab162
Yu, H., Chen, J., Xu, X., Li, Y., Zhao, H., Fang, Y., et al. (2012). A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data. PloS One 7 (5), e37608. doi:10.1371/journal.pone.0037608
Zhang, W., Huai, Y., Miao, Z., Qian, A., and Wang, Y. (2019). Systems pharmacology for investigation of the mechanisms of action of traditional Chinese medicine in drug discovery. Front. Pharmacol. 10, 743. doi:10.3389/fphar.2019.00743
Zhang, Y., Li, H., Zhang, J., Zhao, C., Lu, S., Qiao, J., et al. (2020). The combinatory effects of natural products and chemotherapy drugs and their mechanisms in breast cancer treatment. Phytochem. Rev. 19, 1179–1197. doi:10.1007/s11101-019-09628-w
Zhou, X., Seto, S. W., Chang, D., Kiat, H., Razmovski-Naumovski, V., Chan, K., et al. (2016). Synergistic effects of Chinese herbal medicine: a comprehensive review of methodology and current research. Front. Pharmacol. 7, 201. doi:10.3389/fphar.2016.00201
Keywords: deep learning, natural products, drug combinations, network analysis, drug discovery, machine learning, synergism
Citation: Cao EL (2024) Natural product based anticancer drug combination discovery assisted by deep learning and network analysis. Front. Nat. Produc. 2:1309994. doi: 10.3389/fntpr.2023.1309994
Received: 09 October 2023; Accepted: 29 December 2023;
Published: 15 January 2024.
Edited by:
Marcus Scotti, Federal University of Paraíba, BrazilReviewed by:
Smith B. Babiaka, University of Tuebingen, GermanyCopyright © 2024 Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elton L. Cao, ZWx0b25jYW8wMUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.