 Marcos Marçal Ferreira Queiroz1,2†
Marcos Marçal Ferreira Queiroz1,2† Robin Huber1,2†
Robin Huber1,2† Laurence Marcourt1,2
Laurence Marcourt1,2 Laure Guénée3
Laure Guénée3 Pierre-Marie Allard1,2,4
Pierre-Marie Allard1,2,4 Adriano Rutz1,2Louis-Félix Nothias1,2
Adriano Rutz1,2Louis-Félix Nothias1,2 Concetta Carlotta De Ruvo1,2
Concetta Carlotta De Ruvo1,2 Jonathan Kissling5
Jonathan Kissling5 Mahabir Prashad Gupta6§
Mahabir Prashad Gupta6§ Emerson Ferreira Queiroz1,2*‡
Emerson Ferreira Queiroz1,2*‡ Jean-Luc Wolfender1,2*‡
Jean-Luc Wolfender1,2*‡- 1School of Pharmaceutical Sciences, University of Geneva, CMU, Geneva, Switzerland
- 2Institute of Pharmaceutical Sciences of Western Switzerland, University of Geneva, CMU, Geneva, Switzerland
- 3Laboratory of Crystallography, University of Geneva, Geneva, Switzerland
- 4Department of Biology, University of Fribourg, Fribourg, Switzerland
- 5Institute of Biology, University of Neuchâtel, Neuchâtel, Switzerland
- 6Center of Pharmacognostic Research on Panamanian Flora (CIFLORPAN), College of Pharmacy, University of Panama, Panama City, Panama
Alzatea verticillata Ruiz & Pav. (Alzateaceae) is a tropical tree from Central and South America. It is the only living species of Alzatea genus and the Alzateaceae family, all others being extinct. With the aim to investigate the possibility to find unusual natural products, the chemical content of the dichloromethane and methanolic extracts (stems and leaves) of A. verticillata have been investigated. Apolar and polar extracts were purified by semi-preparative HPLC using appropriate stationary phase columns allowing the isolation of 12 compounds: walterolactone B (2) walterolactone A/B β-D-pyranoglucoside (3), gallic acid (4), caffeic acid 4-O-β-D-glucopyranoside (6), walterolactone A/B 6-O-gallate-β-D-glucopyranoside (8), caffeic acid (9), 8-desmethylsideroxylin (11), sideroxylin (12) and 7,7′-bis(3,4-dihydroxyphenyl)-8,8′-cyclobutanedicarboxylic acid (7). Three isolated compounds are natural products described here for the first time: dimethyl-anemonin (1) and two β-truxinic acid derivatives (rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane dicarboxylic acid (5) and rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane-9-methyl dicarboxylic acid (10). The structures of the isolated compounds were elucidated by NMR and HRMS. The structure of compound 1 was confirmed by X-ray crystallography. A MS-based metabolite analysis of the A. verticillata extracts revealed additional truxinic acid derivatives that were putatively annotated with the help of feature-based molecular network. The presence of phenolic compounds such as truxinic acid derivatives could explain the traditional use of this plant as these compounds are known to possess anti-inflammatory and anti-nociceptive properties.
Introduction
Alzateaceae (Myrtales) [Q13634079] is a peculiar botanical family of Central and South America, represented by the single species Alzatea verticillata Ruiz & Pavón (Ruiz and Pavón, 1798) [Q131843]. It is a small tree typically found on steep slopes of the low mountain rain forest at an elevation between 1,000 and 2,200 m. Its closest relatives are the South-East Asian Crypteroniaceae and the African Penaeaceae (Graham, 1984; Conti et al., 2002; Schonenberger and Conti, 2003). This led both Rutschmann et al. (Rutschmann et al., 2004; Rutschmann et al., 2007) and Berger et al. (Berger et al., 2016) to suggest that the ancestor of the clade formed by those three families lived on the super-continent Gondwana millions of years ago and that following the Gondwana breakup, the ancestors of the Alzateaceae was isolated on the South American continent (c. 100 mya). Thus, Alzatea has evolved along an independent course over a long period of time acquiring many specialized attributes (e.g., a different chromosome number, n = 14 for A. verticillata subsp. amplifolia versus a base chromosome number of x = 12 for the other families of the order (Almeda, 1997)), ultimately occupying an isolated position in the order Myrtales. This genus is of phytochemical interest because its distant genetical background (and thus its biochemical phenotypes) might have chosen unique evolutionary pathways. Interestingly, the leaves of this plant are used as antipruritic for itching, scaly scalp, shampoo, and for venereal disease, by the Guyana Patamona Indians (Tiwari, 2002). Inspired from the unique diterpenes (ginkgolide derivatives) observed in Ginkgo biloba, which is also the sole species of its family (Forman et al., 2022), we undertook a phytochemical investigation of A. verticillata. To this aim, the chemical profile of the dichloromethane (DCM) and methanolic (MeOH) extracts of A. verticillata stems or leaves were analyzed by untargeted high-resolution mass spectrometry (UHPLC-HRMS/MS) and the results interpreted by molecular networking. The main constituents were then isolated by high-resolution semi-preparative HPLC.
Results and discussion
To establish a preliminary chemical profile of the specialized metabolites produced by A. verticillata, the DCM and MeOH extracts obtained from the leaves and stems were analysed by HPLC-PDA (Figure 1) and UHPLC-HRMS/MS (Supplementary Figure S1). Given that the species and genus weren’t previously investigated, we have carried out a preliminary isolation and structure elucidation of the main compounds of the extract observed prior to a more advanced annotation procedure of most metabolites detected.
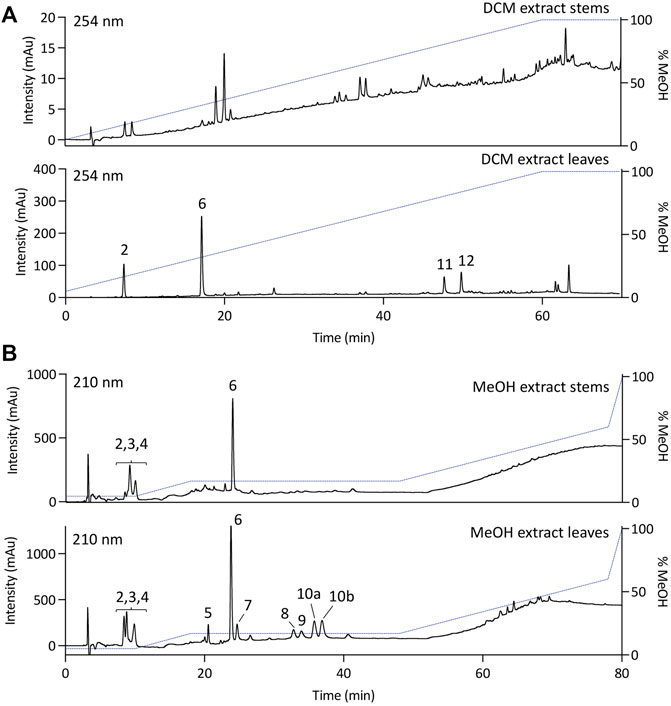
FIGURE 1. HPLC-PDA analysis of the (A) dichloromethane (DCM) or (B) methanolic (MeOH) extracts of stems or leaves of Alzatea verticillata.
The DCM and MeOH extracts of the leaves and stems of A. verticillata were purified in one step by semi-preparative HPLC-UV (see experimental section). The conditions were first established by analytical HPLC and then transferred to the semi-preparative HPLC using a gradient transfer method (Guillarme et al., 2008). To obtain a sufficient resolution for the one step-isolation procedure of the different compounds, the samples were injected using a dry load method (Queiroz et al., 2019). This approach resulted in the successful isolation of twelve compounds (1–12). The structure elucidation of the isolated compounds was performed based on the NMR and HRMS analyses. The isolated metabolites were identified as walterolactone B (2) (Nguyen et al., 2018), walterolactone A/B β-D-pyranoglucoside (3) (Nguyen et al., 2018), gallic acid (4) (Gottlieb et al., 1991), caffeic acid 4-O-β-D-glucopyranoside (6) (Lai et al., 2008), walterolactone A/B 6-O-gallate-β-D-glucopyranoside (8) (Nguyen et al., 2018), caffeic acid (9) (Exarchou et al., 2001), 8-desmethylsideroxylin (11) (Junio et al., 2011), sideroxylin (12) (Junio et al., 2011) and 7,7′-bis(3,4-dihydroxyphenyl)-8,8′-cyclobutanedicarboxylic acid (7) (Figure 2). As the NMR data for the latter compound has not been described, they are presented in the experimental part. Beside the known isolated compounds, the purification of the stems DCM extract also yielded a new dimeric lactone, dimethyl anemonin (1), while two unusual dimeric diphenyl cyclobutane dicarboxylic acids (5 and 10) were isolated from the MeOH extract.
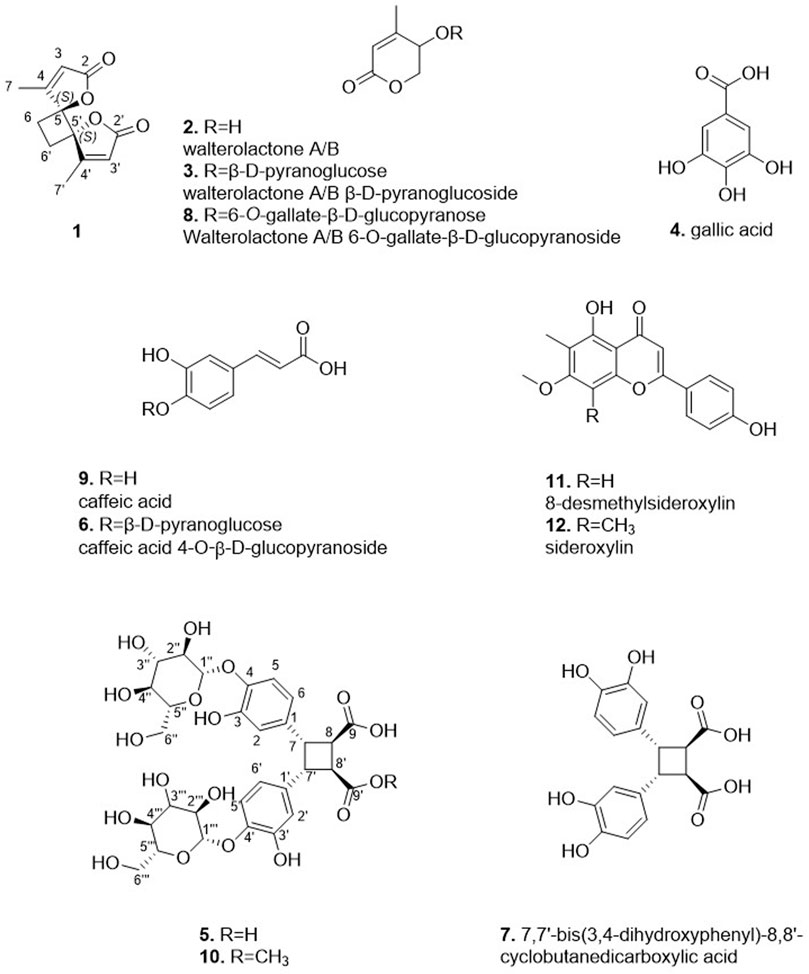
FIGURE 2. Isolated compounds from the different extracts of Alzatea verticillata.
Compound 1 was isolated as an amorphous solid. The HRMS spectrum with an atmospheric pressure chemical ionization source (APCI) showed a protonated molecule at m/z 221.0800 [M + H]+. The 1H NMR and edited-HSQC spectra of 1 showed a vinylic signal at δH 5.90 (each, 1H, q, J = 1.5 Hz, H-3, H-3′), a methylene at δH 2.52 (each, 2H, s, H2-6, H2-6′) and a methyl at δH 2.41 (each, 3H, d, J = 1.5 Hz, H3-7, H3-7′). The HMBC correlations from the methyl to the vinylic C-3 (δC 117.5), the sp2 carbon C-4 (δC 169.6) and the oxygenated quaternary carbon C-5 (δC 92.4), from H-3 to C-5 and the ester carbonyl C-2 (δC 172.6) and from H-6 to C-6 (δC 25.0) and C-4 indicated the presence of a 4-methylfuran-2(5H)-one with extra methylene in C-5, accounting for six carbons. The molecular formula and the fact that the methylene protons correlated in HMBC with their own carbon indicated that 1 should be a symmetric dimer. To confirm its structure and establish its configuration, compound 1 was successfully crystalized in ethyl acetate and the crystals were subjected to X-ray crystallography (Figure 3). Compound 1 has crystallized in the centrosymmetric space group P-1 and therefore is a S,S and R,R racemic mixture. Dimethyl-anemonin (1) is a dimethylated derivative of the known compound anemonin which was first isolated by Heyer in 1792 (Heyer, 1792). It is well known that, in plant, anemonin is formed by the spontaneous dimerization of protoanemonin, which comes from the enzymatic deglycosylation of raninculin (Pirvu et al., 2022). Suga and Hirata showed that protoanemonin in Ranunculus glaber comes from 2-oxo-glutaric acid (Suga and Hirata, 1982). In our case, we could imagine a methylation of 2-oxo-glutaric acid before cyclisation to obtain methyl-protoanemonin and then the dimethyl-anemonin (1) (Supplementary Figure S2). Moreover, one of the fractions obtained from the purification of the dichloromethane extract of the leaves contained in mixture with 8-desmethylsideroxylin (11), the 5-hydroxy-4,5-dimethylfuran-2(5H)-one and 4,5-dimethylfuran-2(5H)-one which are in line with this biosynthesis proposal (data in Supplementary Figure S3). This type of dimerisation explains that 1 was, as anemonin, isolated as a racemic mixture. One note that walterolactone B (2) could also be obtain by rearrangement of methyl-protoanemonine. Anemonin has been shown to have anti-inflammatory antibacterial, antiviral, antitoxic, and cytopathogenic properties (Duan et al., 2006).
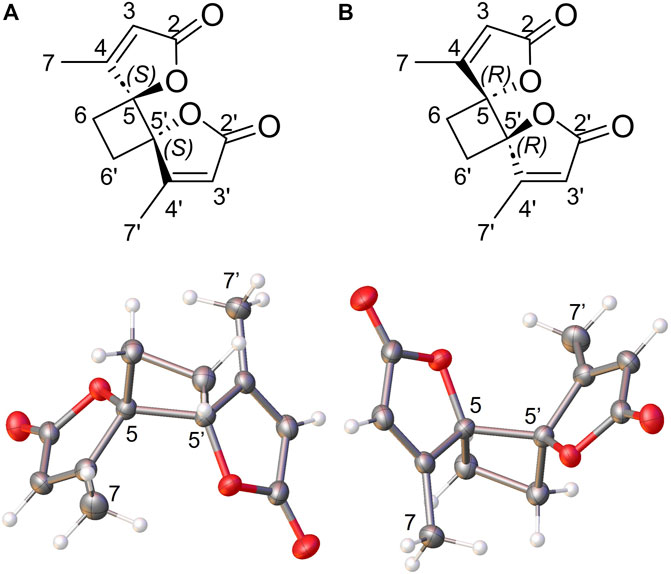
FIGURE 3. Chemical structure and Ortep view of (A) S,S enantiomer and (B) R,R enantiomer of compound 1 (Thermal ellispoids are drawn at 50% probability level).
The HRMS spectrum with an electrospray ionization source (ESI) of 5 showed a deprotonated molecule at m/z 683.1837 [M-H]- corresponding to the molecular formula C30H35O18. The 1H NMR spectrum displayed aromatic signals at δH 6.48 (each, 1H, dd, J = 8.3, 2.6 Hz, H-6, H-6′), 6.52 (each, 1H, d, J = 2.6 Hz, H-2, H-2′) and 6.96 (each, 1H, d, J = 8.3 Hz, H-5, H-5′) corresponding to 1,3,4-trisubstituted benzene but with some slightly splitting multiplicities, suggesting the presence of two close groups. The HSQC NMR spectrum showed a glucose moieties as indicated with the signals at δH/δC 4.64/104.3;104.4 (H/C-1′′, H/C-1‴), 3.42/74.9 (H/C-2′′, H/C-2‴), 3.42/77.6 (H/C-3′′, H/C-3‴), 3.37/71.3 (H/C-4′′, H/C-4‴), 3.37/78.2 (H/C-5′′, H/C-5‴), 3.87;3.69/62.4 (H/C-6′′, H/C-6‴). These signals were also slightly splitting as, for example, as at δH 4.64 where two doublets with a coupling constant of 7.4 Hz were observed for the anomeric protons of each glucose. The ROESY correlation from H-1′′/H-1‴ to H-5/H-5′ allowed positioning the glucose groups in C-4 and C-4′. In addition to these signals, four methines were observed at δH 3.72 (2H, d, J = 6.0 Hz, H-8, H-8′), 4.16 (2H, d, J = 6.0 Hz, H-7, H-7′) and δC 45.4 (C-8, C-8′), 46.3 (C-7, C-7′) suggesting the presence of a cyclobutane ring. The HMBC correlation from H-2/H-2′ and H-6/H-6′ to C-7/C-7′ positioned the phenolic groups in C-7 and C-7′. Despite the lack of acid carbonyl signal in both HMBC and the 13C spectrum, the HRMS data confirmed the presence of two acid functions. At this stage, two types of compounds were possible, a truxillic acid derivative (head-to-tail dimer) or a truxinic acid one (head-to-head dimer). The examination of MS/MS fragmentation pattern in positive mode indicated the presence of the fragment ions m/z 245.0804 and 117.0185 (Figure 4), supporting a head-to-head dimerization for compound 5 which was thus assigned to a truxinic acid derivative (Stewart et al., 1992; Fujiwara et al., 2016). The values of the chemical shifts of H-7 and H-8 and the coupling constant between H-7 and H-8 of 6.0 Hz were in good agreement with the data of β-truxinic acid (Ghosn and Wolf, 2010). These compounds are known to be probably obtained by a [2 + 2] cycloaddition reaction between the two olefinic carbon bonds of the caffeic acids through an enzymatic catalysis or a photocatalysis in nature (Yang et al., 2022). It is in our case difficult to know if these compounds are obtained as a single enantiomer, as a racemic or as a scalemic mixture. While compound 7, due to the presence of a plane of symmetry, is an achiral meso compound, 5, due to the presence of the β-glucoses, loses its symmetry and could thus exist as one or two enantiomers. The specific rotation was measured with a negative value of −70, indicates that 5 has probably been isolated as a single enantiomer. Compound 5 was thus identified as (−)-rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane dicarboxylic acid, a new β-truxinic acid derivative.
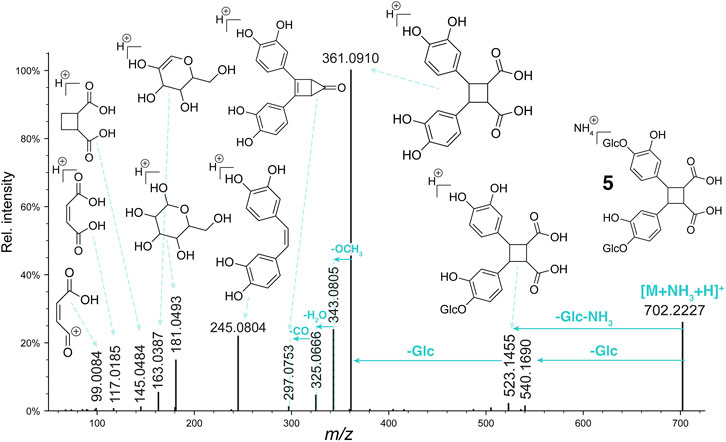
FIGURE 4. Fragmentation pattern of compound 5.
Compound 10 presented a deprotonated molecule at m/z 697.1991 [M-H]- in the ESI-HRMS spectra, suggesting the molecular formula C31H37O18. The 1H NMR spectra of 10 showed close similarities to that of 5, except for the presence of a methoxy group at δH 3.72 for 10 and a splitting of the cyclobutane signals for H-7 and H-7′ as well as H-8 and H-8′ protons. In 10, the methylation of only one of the two acid functions results in a loss of symmetry and thus the cyclobutane protons became non-equivalent. The COSY correlations from H-8 at δH 3.78 to H-7 at δH 4.19, from H-7 to H-7′ at δH 4.13 and from H-7′ to H-8′ at δH 3.75, the HMBC correlations from H-7 and H-7′ to the aromatic carbons C-1/1′(δC 136.1/136.3), C-2/2′(δC 116.8/116.9) and C-6/6′(δC 120.5/120.6), and from H-7, H-8 and the methoxy H-10 (δH 3.72) to C-9 at δC 175.2 allowed to identify a truxinic acid derivative (Figure 5). This was confirmed by the observation of the fragment ions at m/z 113.0224, 131.0313 and 245.0753 in ESI-HRMS in positive ionization mode (precursor ion [M+NH4]+, m/z 716.2321) (Figure 6). The ROE correlations from H-8 to H-2/H-6 and from H-8′ to H-2′/H-6′ indicated a trans configuration of each monomer. The configuration was finally assigned to that of β-truxinic acid based on the values of H-8/H-8′ chemical shifts (between δH 3.75 and 4.23) and coupling constants (3JH-7-H-7' = 3JH-8-H-8' = 10.2 Hz and 3JH-7-H-8 = 3JH-7'-H-8' = 6.2–6.6 Hz) (Fujiwara et al., 2016). Two different diastereoisomers of this compound could exist depending on the methoxy position in C-9 or C-9′. Indeed, the almost same spectra were obtained from two very close HPLC peaks (10a, 10b) (Figure 1; Supplementary Figure S4) so that it wasn’t possible to distinguish these two diastereoisomers. Based on the data described above, compound 10 was thus identified as rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane-9-methyl dicarboxylic acid, a new β-truxinic acid derivative.
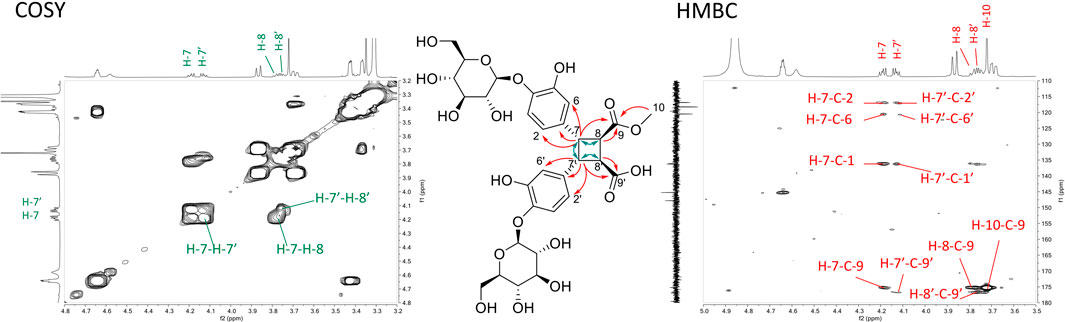
FIGURE 5. Key COSY (green arrows, left spectrum) and HMBC (red arrows, right spectrum) correlations of compound 10.
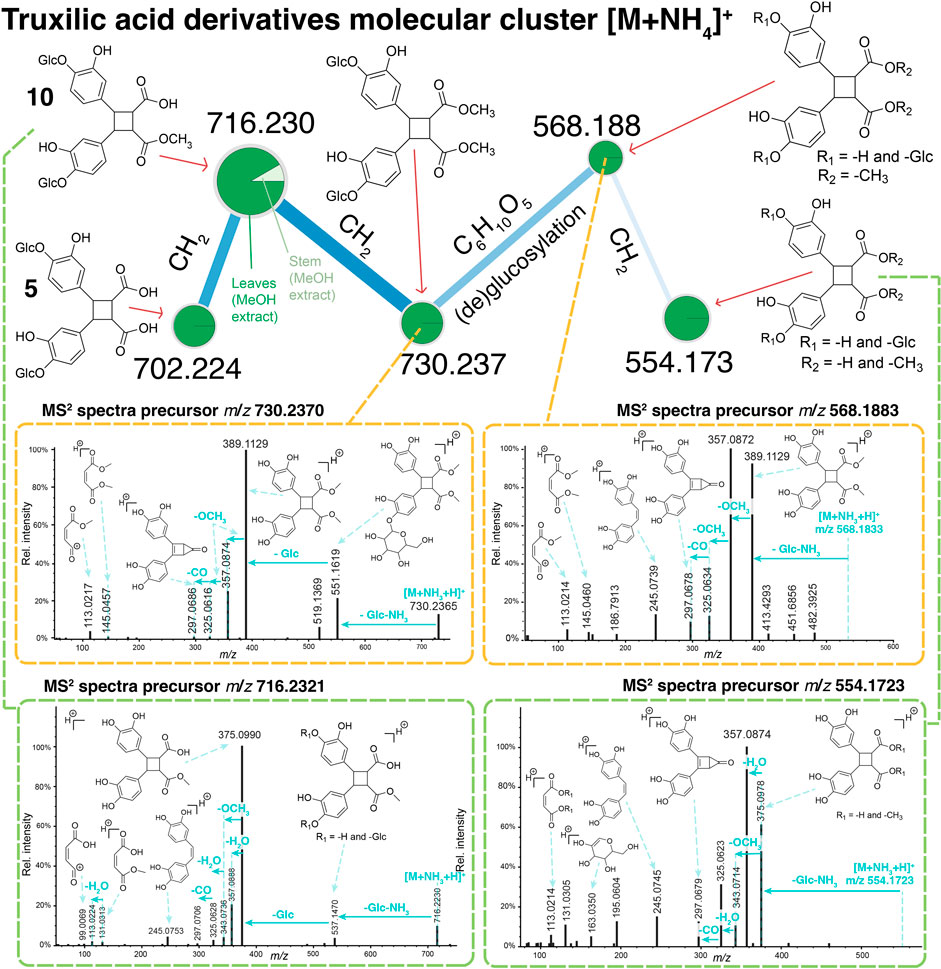
FIGURE 6. Molecular network cluster containing 5 and 10, as well as other putative truxinic acid derivatives. These data were generated from LC-ESI-HRMS/MS data of the DCM or MeOH extracts of stems or leaves of Alzatea verticillata. The classical molecular networking workflow was used where the node size represents the relative intensity of the precursor ions for each extract.
To investigate the presence of other truxinic acid analogues, a molecular network (Wang et al., 2016) was generated from the DCM and MeOH extracts of leaves and stems of A. verticillata using an UHPLC-HRMS/MS ESI, positive ion mode). A molecular network cluster containing the two isolated truxinic derivatives was observed as [M+NH4]+ adducts (Figure 6). Interestingly, other non-isolated derivatives were also part of this cluster. An ion at m/z 730.237 suggested a molecular formula differing by an additional CH2 compared to 10. The examination of the fragmentation spectra allowed to determine that the compound at m/z 730.237 corresponded to a methoxylated derivative of 5 on the free acid function. Further examination of the fragmentation pattern for the remaining ions of this cluster enabled to putatively annotate them. The ion at m/z 568.188 corresponded to the deglucosylated derivative of m/z 730.237. Similarly, the interpretation of the fragmentation spectra of the ion at m/z 554.173 agreed with a deglucosylated derivative of 10. In the case of the latter, the precise location of the deglucosylation on one specific trihydroxybenzen unit could not be established. It is important to note that the methoxylated derivatives such as compound 10 or other annotated derivatives, may be artefacts due to methylation during the extraction process.
Although many compounds belonging to the phenylpropanoid structural class have been isolated, phenylpropanoid dimers that contain a cyclobutane moiety are relatively rare (Yang et al., 2022). These compounds possess strong radical scavenging properties (Shahidi and Chandrasekara, 2010), and they are also involved in the plant tolerance to both biotic and abiotic stresses (Shahidi and Chandrasekara, 2010).
Further insight into the chemical profile of A. verticillata was enabled by annotating the mass spectrometry data by spectral library matching with public libraries. These annotated ions were not isolated and thus most likely corresponded to minor metabolites. The spectral matches obtained were classified with NPClassifier (Supplementary Figure S5) and indicated the presence of multiple derivatives belonging to shikimates and phenylpropanoids (32.3%, n = 19), fatty acids (29.2%, n = 19), terpenoids (21.5%, n = 14), alkaloids (9.23%, n = 6), and polyketides (6.15%, n = 4).
Conclusion
The chemical study of polar and apolar extracts of A. verticillata revealed specialized metabolites obtained from the shikimate acid pathway: gallic acid (4), rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane dicarboxylic acid (5), caffeic acid 4-O-β-D-glucopyranoside (6), 7,7′-bis(3,4-dihydroxyphenyl)-8,8′-cyclobutanedicarboxylic acid (7), caffeic acid (9), rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane-9-methyl dicarboxylic acid 8-desmethylsideroxylin (11), and sideroxylin (12). This study also showed us the presence of a series of walterolactone compounds (2, 3, and 8).The presence of diphenyl cyclobutane dicarboxylic acids (truxinic acid derivatives) obtained from the dimerization of two phenylpropane units remains interesting since these compounds play a vital role in plant physiology by providing cell wall stiffness, which is an important factor in plant interactions with the environment (Caffall and Mohnen, 2009). Truxilic acids have been also reported from Melastomataceae species, a family from the same order (Myrtales) than Alzateaceae (Serna and Martinez, 2015). Truxinic acid derivatives are known to possess anti-inflammatory and anti-nociceptive properties and their presence in the polar extracts of A. verticillata might explain the use of this plant as antipruritic for itching in traditional medicine (Chi et al., 2006; Priebe et al., 2018).
Experimental section
General experimental procedures
NMR spectroscopic data were recorded on a 500 MHz Varian (Palo Alto, CA, United States) INOVA NMR spectrometer and on a Bruker Avance III HD 600 MHz NMR spectrometer equipped with a QCI 5 mm Cryoprobe and a SampleJet automated sample changer (Bruker BioSpin, Rheinstetten, Germany). Chemical shifts are reported in parts per million (δ) using the residual CD3OD signal (δH 3.31; δC 49.0) or DMSO-d6 signal (δH 2.50; δC 39.5) as internal standards for 1H and 13C NMR, respectively, and coupling constants (J) are reported in Hz. Complete assignments were obtained based on 2D-NMR experiments (COrrelation SpectroscopY (COSY), Nuclear Overhauser Effect SpectroscopY (NOESY), Heteronuclear Single Quantum Correlation (HSQC) and Heteronuclear Multiple Bond Correlation (HMBC)). ESI-HRMS data were obtained on a Micromass LCT Premier time-of-flight mass spectrometer from Waters with an electrospray ionization (ESI) interface (Waters, Milford, MA, United States). HPLC-PDA data were obtained with an Agilent HP 1,260 series system consisting of an autosampler, high-pressure mixing pump and DAD detector (Agilent Technologies, Santa Clara, CA, United States). HPLC-fractionation was performed with an Armen modular spot prep II (Saint-Avé, France) equipped with an UV detector and fraction collector and on a Shimadzu system equipped with a LC-20A module pumps, an SPD-20A UV/VIS, a 7725I Rheodyne® valve, and an FRC-10A fraction collector (Shimadzu, Kyoto, Japan). The system was controlled by the LabSolutions software, also from Shimadzu.
Plant material
Leaves and stems of A. verticillata were collected at Parque Nacional General de División Omar Torrijos Herrera, in the provincial of Coclé, Panama in July 2013. The species was identified by De Gracia J. and voucher was deposited at the National Herbarium of Panama (FLORPAN, Voucher at PBNB: n° 8747).
Extraction
The air-dried plant material (500 g) was pulverized with a Wiley Mill and extracted at room temperature successively with dichloromethane and methanol to give 32 g, and 48.5 g, respectively. The extracts were concentrated under pressure and later lyophilized.
HPLC-PDA analyses
HPLC-PDA analyses were conducted on an Agilent 1,260 system equipped with a photodiode array detector. The separations of the dichloromethane and methanolic extracts of stems and leaves of A. verticillata were performed on a Waters XBridge C18 column (250 × 4.6 mm i.d., 5 µm) (Waters, Milford, MA, United States) equipped with a Waters C18 pre-column cartridge holder (20 × 4.6 mm i.d., 5 µm) with MeOH (B) and H2O (A), both containing 0.1% formic acid as solvents. The flow rate was set at 1 mL/min, the injection volumes were 10 µL with samples at about 10 mg/mL. The separation for the dichloromethane extracts was performed in gradient mode using the following conditions: 5%–100% B in 60 min, followed by 10 min of washing at 100% B. The separation for the methanolic extracts was also performed in gradient mode using the following conditions: 5% B for 10 min, 5%–17% B in 18 min, 17% B for 30 min, 17%–60% B in 30 min and 60%–100% in 2 min, followed by 10 min washing at 100% B.
UHPLC-ESI-HRMS/MS analyses
The UHPLC-ESI-HRMS/MS analysis was carried out on a Waters Acquity UPLC IClass system interfaced to a Q Exactive Focus mass spectrometer (Thermo Scientific, Bremen, Germany), using a heated electrospray ionization (HESI-II) source. Chromatographic separation was performed on a column of Waters BEH C18 50 × 2.1 mm i.d., 1.7 µm, mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B), flow rate was 600 μL/min, injection volume was 1 μL, and linear gradient elution from 5% to 100% B in 7 min, followed by isocratic at 100% B for 1 min, and decreased to 5% B at the final step for 2 min. Positive and negative ionization mode were applied in this study. The diisooctyl phthalate C24H38O4 [M-H]− ion (m/z 389.2697) was used as an internal lock mass. The optimized HESI-II parameters were set as follows: source voltage, 3.5 kV (pos) or 2.5 kV (neg); sheath gas flow rate (N2), 48 units; auxiliary gas flow rate, 11 units; spare gas flow rate, 2.0; capillary temperature, 300°C (pos), S‒Lens RF Level, 55. The mass analyzer was calibrated using a mixture of caffeine, methionine‒arginine‒phenylalanine‒alanine‒acetate (MRFA), sodium dodecyl sulfate, sodium taurocholate, and Ultramark 1,621 in an acetonitrile/methanol/water solution containing 1% formic acid by direct injection. The data‒dependent MS/MS events were performed on the three most intense ions detected in full scan MS (Top3 experiment). The MS/MS isolation window width was 2 Da, and the normalized collision energy (NCE) was set to 35 units. In data-dependent MS/MS experiments, full scans were acquired at a resolution of 35,000 fwhm (at m/z 200) and MS/MS scans at 17,500 fwhm both with a maximum injection time of 50 ms. After being acquired in a MS/MS scan, parent ions were placed in a dynamic exclusion list for 2.0 s.
Mass spectrometry and fragmentation spectra annotation
The UPHLC−HRMS/MS raw data was converted to.mzXML format using the MSConvert software, part of the ProteoWizard package. Molecular networks were generated on the GNPS web-platform using the classical molecular networking workflow (Wang et al., 2016) from the mass spectrometry of the extracts of A. verticillata (DCM or MeOH extracts of stems or leaves). The fragmentation spectra were annotated by spectral library matching using public libraries and reference spectra of the isolated compounds. The interactive view of the entirety of molecular networks generated and the parameters used are available via the job link at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=7e5e5b3e23de4e2395a247a73ff18a85. The reference MS/MS fragmentation spectra (ESI positive ionization mode) of compounds 5–8, 10–12 were added to the GNPS public spectral library (Wang et al., 2016). The NPClassifier taxonomy (Kim et al., 2021) was used to classify the annotated compounds.
Purification of the dichloromethane extract of stems by semi-preparative HPLC-UV
The dichloromethane extract of stems was purified using the semi-preparative HPLC-UV equipment and was performed on an Armen modular spot prep II system equipped with an UV detector and fraction collector. The separation was performed with an Interchim Silica HP column (250 × 21 mm i.d., 10 μm; Interchim, Montluçon, France). The flow rate was set at 21 mL/min. The solvent system used was a mixture of hexane (A) and ethyl acetate (B) in gradient mode: 5%–100% of A in 61.4 min followed by 100% of A for 10 min. The UV absorbance was measured at 210 nm. The sample (90 mg) was injected by loop injection (3 injections of 30 mg in 500 µL of ethyl acetate). After collection, each fraction was evaporated to dryness using a SpeedVac (HT-4X Genevac®, Stone Ridge, NY, United States). The separation yielded 7 mg of compound 1.
Purification of the dichloromethane extract of the leaves by semi-preparative HPLC-UV
For the separation of the dichloromethane extract of the leaves of A. verticillata, the semi-preparative HPLC was performed on a Shimadzu system equipped with a LC-20A module pumps, an SPD-20A UV/VIS, a 7725I Rheodyne® valve, and an FRC-10A fraction collector. The system was controlled by the LabSolutions software from Shimadzu. The separation was performed as follows: Waters XBridge C18 column (250 × 19 mm i.d., 5 µm); solvent system MeOH (B) and H2O (A), both containing 0.1% formic acid. The separation was performed in gradient mode as follows: 5%–100% B in 60 min. After this the column was washed with 100% of B during 10 min. Flow rate was 17 mL/min. The sample (50 mg of the extract) was injected by dry load using the protocol recently published (Queiroz et al., 2019). The UV absorbance was measured at 210 and 254 nm. The separation yielded compound 6 (1.8 mg), 9 (0.7 mg), 11 (0.6 mg), and 12 (0.4 mg).
Purification of the methanolic extract of the leaves by semi-preparative HPLC-UV
For the separation of the methanolic leave and stem extracts of A. verticillata, the semi-preparative HPLC conditions was performed on a Shimadzu system equipped with a LC-20A module pumps, an SPD-20A UV/VIS, a 7725I Rheodyne® valve, and a FRC-10A fraction collector. The system was controlled by the LabSolutions software from Shimadzu. The separation was performed as follows: Waters XBridge C18 column (250 × 19 mm i.d., 5 µm); solvent system MeOH (B) and H2O (A), both containing 0.1% formic acid. The separation was performed in gradient mode as follows: 5% B for 10 min, 5%–17% B in 18 min, 17% B for 30 min, 17%–60% B in 30 min and 60%–100% in 2 min, followed by 10 min washing at 100% B. Flow rate was 17 mL/min. The sample (50 mg for each injection) was injected by dry load using the protocol recently published (Queiroz et al., 2019). The UV absorbance was measured at 210 and 254 nm. The separation of the methanolic leaves extract (3 × 50 mg) yielded compounds 2 (0.5 mg), 3 (1.4 mg), 4 (2.1 mg), 5 (0.7 mg), 6 (1.1 mg), 7 (0.3 mg), 8 (0.3 mg), 9 (0.6 mg), 10 (1.3 mg). The separation of the methanolic stems extract yielded compounds 2 (1.2 mg), 3 (2.2 mg), and 4 (1.7 mg).
Description of the isolated compounds
Dimethyl-anemonin (1)
1H NMR (DMSO-d6, 500 MHz) δ 2.34 (each, 3H, d, J = 1.3 Hz, H3-7, H3-7′), 2.45 (each, 2H, m, H2-6, H2-6′), 6.04 (each, 1H, q, J = 1.3 Hz, H-3, H-3′); 13C NMR (DMSO-d6, 126 MHz) δ 13.8 (CH3-7, CH3-7′), 23.9 (CH2-6, CH2-6′), 90.6 (C-5, C-5′), 116.4 (CH-3, CH-3′), 168.3 (C-4, C-4′), 170.5 (C-2, C-2′). 1H NMR (CD3OD, 500 MHz) δ 2.41 (each, 3H, d, J = 1.5 Hz, H3-7, H3-7′), 2.52 (each, 2H, s, H2-6, H2-6′), 5.90 (each, 1H, q, J = 1.5 Hz, H-3, H-3′); 13C NMR (CD3OD, 126 MHz) δ 14.1 (CH3-7, CH3-7′), 25.0 (CH2-6, CH2-6′), 92.4 (C-5, C-5′), 117.5 (CH-3, CH-3′), 169.6 (C-4, C-4′), 172.6 (C-2, C-2′) (Supplementary Figures S5–S12); APCI-HRMS m/z 221.0800 [M+ H]+ (calcd for C12H12O4, 221.0808, Δ = −3.6 ppm).
Walterolactone B (2)
[α]25D +837 ± 39 (c 0.003, H2O/MeOH); 1H NMR (CD3OD, 600 MHz) δ 2.07 (3H, d, J = 1.5 Hz, H3-7), 4.16 (1H, dd, J = 4.5, 3.9 Hz, H-5), 4.28 (1H, dd, J = 12.0, 4.5 Hz, H-6′′), 4.41 (1H, dd, J = 12.0, 3.9 Hz, H-6′), 5.81 (1H, q, J = 1.5 Hz, H-3); 13C NMR (CD3OD, 151 MHz) δ 19.9 (CH3-7), 65.0 (CH-5), 72.9 (CH2-6), 117.5 (CH-3), 161.4 (C-4), 166.4 (C-2) (Supplementary Figures S13–S18); ESI-HRMS m/z 129.0550 [M+H]+ (calcd for C6H9O3, 129.0546, Δ = 2.9 ppm).
Walterolactone A/B β-D-pyranoglucoside (3)
[α]25D +158 ± 18 (c 0.02, H2O/MeOH);1H NMR (CD3OD, 600 MHz) δ 2.12 (3H, d, J = 1.5 Hz, H3-7), 3.18 (1H, dd, J = 9.1, 7.7 Hz, H-2′), 3.30 (2H, m, H-4′, H-5′), 3.36 (1H, t, J = 8.8 Hz, H-3′), 3.68 (1H, dd, J = 12.0, 4.9 Hz, H-6′b), 3.89 (1H, dd, J = 12.0, 1.6 Hz, H-6'a), 4.43 (3H, m, H-5, H-6b, H-1′), 4.58 (1H, dd, J = 11.8, 2.8 Hz, H-6a), 5.87 (1H, q, J = 1.5 Hz, H-3); 13C NMR (CD3OD, 151 MHz) δ 20.4 (CH3-7), 62.8 (CH2-6′), 70.3 (CH2-6), 71.1 (CH-5), 71.5 (CH-4′), 74.8 (CH-2′), 78.1 (CH-3′), 78.2 (CH-5′), 103.0 (CH-1′), 118.9 (CH-3), 158.6 (C-4), 166.1 (C-2) (Supplementary Figures S19–S24); ESI-HRMS m/z 335.0987 [M+FA-H]- (calcd for C13H19O10, 335.0973, Δ = 4.2 ppm).
Gallic acid (4)
1H NMR (CD3OD, 600 MHz) δ 7.05 (2H, s, H-2, H-6); 13C NMR (CD3OD, 151 MHz) δ 110.3 (CH-2, CH-6), 122.1 (C-1), 140.3 (C-4), 146.4 (C-3, C-5), 170.4 (C-7) (Supplementary Figures S25–S28); ESI-HRMS m/z 169.0132 [M-H]- (calcd for C7H5O5, 169.0131, Δ = 0.5 ppm).
rel-(7S, 8R, 7′R, 8′S)-7,7′-Bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane dicarboxylic acid (5)
[α]25D −70 ± 28 (c 0.013, H2O/MeOH); 1H NMR (CD3OD, 600 MHz) δ 3.37 (4H, m, H-4′′, H-5'', H-4‴, H-5‴), 3.42 (4H, m, H-2′′, H-3′′, H-2‴, H-3‴), 3.69 (2H, m, H-6′′b, H-6‴b), 3.72 (each, 1H, d, J = 6.0 Hz, H-8, H-8′), 3.87 (2H, m, H-6′′a, H-6‴a), 4.16 (each, 1H, d, J = 6.0 Hz, H-7, H-7′), 4.64 (each, 1H, d, J = 7.6 Hz, H-1′′, H-1‴), 6.48 (each, 1H, dd, J = 8.3, 2.6 Hz, H-6, H-6′), 6.52 (each, 1H, d, J = 2.6 Hz, H-2, H-2′), 6.96 (each, 1H, d, J = 8.3 Hz, H-5, H-5′); 13C NMR (CD3OD, 151 MHz) δ 45.4 (CH-8, CH-8′), 46.3 (CH-7, CH-7′), 62.4 (CH2-6′′, CH2-6‴), 71.3 (CH-4′′, CH-4‴), 74.9 (CH-2′′, CH-2‴), 77.6 (CH-3′′, CH-3‴), 78.2 (CH-5′′, CH-5‴), 104.3/104.4 (CH-1′′, CH-1‴), 116.8/116.9 (CH-2, CH-2′), 118.2/118.3 (CH-5, CH-5′), 120.5/120.6 (CH-6, CH-6′), 136.5 (C-1, C-1′), 145.2 (C-4, C-4′), 147.8 (C-3, C-3′) (Supplementary Figures S30–S34); ESI-HRMS m/z 683.1837 [M-H]- (calcd for C30H35O18, 683.1818, Δ = 2.7 ppm); MS/MS spectrum: CCMSLIB00010128653.
Caffeic acid 4-O-β-D-glucopyranoside (6)
[α]25D −64 ± 5 (c 0.1, H2O/MeOH); 1H NMR (CD3OD, 600 MHz) δ 3.41 (1H, dd, J = 9.7, 8.2 Hz, H-4′), 3.45 (1H, ddd, J = 8.2, 5.4, 2.2 Hz, H-5′), 3.48 (1H, dd, J = 9.7, 7.5 Hz, H-3′), 3.52 (1H, dd, J = 9.3, 7.5 Hz, H-2′), 3.72 (1H, dd, J = 12.1, 5.4 Hz, H-6'b), 3.91 (1H, dd, J = 1,212, 2.2 Hz, H-6'a), 4.85 (1H, overlapped. H-1′), 6.32 (1H, d, J = 15.9 Hz, H-8), 7.04 (1H, dd, J = 8.4, 2.1 Hz, H-6), 7.10 (1H, d, J = 2.1 Hz, H-2), 7.20 (1H, d, J = 8.4 Hz, H-5), 7.55 (1H, d, J = 15.9 Hz, H-7); 13C NMR (CD3OD, 151 MHz) δ 62.4 (CH2-6′), 71.3 (CH-4′), 74.8 (CH-2′), 77.6 (CH-3′), 78.4 (CH-5′), 103.6 (CH-1′), 115.9 (CH-2), 118.0 (CH-8), 118.2 (CH-5), 122.1 (CH-6), 131.3 (C-1), 145.9 (CH-7), 148.5 (C-3), 148.8 (C-4), 170.7 (C-9) (Supplementary Figures S35–S40); ESI-HRMS m/z 341.0879 [M-H]- (calcd for C15H17O9, 341.0867, Δ = 3.6 ppm); MS/MS spectrum: CCMSLIB00010129502.
3,4-Bis(3,4-dihydroxyphenyl)-1,2-cyclobutanedicarboxylic acid (7) in mixture with compound 6
1H NMR (CD3OD, 600 MHz) 3.75 (each, 1H, dd, J = 10.5, 7.2 Hz, H-8/H-8′), 4.18 (each, 1H, dd, J = 10.5, 7.2 Hz, H-7/H-7′), 6.66 (each, 1H, dd, J = 8.1, 2.1 Hz, H-6, H-6′), 6.70 (each, 1H, d, J = 8.1 Hz, H-5, H-5′), 6.78 (each, 1H, d, J = 2.1 Hz, H-2, H-2′); 13C NMR (CD3OD, 151 MHz) δ 42.5 (CH-7, CH-7′), 48.7 (CH-8, CH-8′), 115.9 (CH-2, CH-2′), 116.2 (CH-5, CH-5′), 120.1 (CH-6, CH-6′), 132.4 (C-1, C-1′), 145.3 (C-4, C-4′), 146.1 (C-3, C-3′), 176.2 (C-9, C-9′) (Supplementary Figures S41–S46); ESI-HRMS m/z 359.0775 [M-H]- (calcd for C18H15O8, 359.0761, Δ = 3.8 ppm); MS/MS spectrum: CCMSLIB00010128655.
Walterolactone A/B 6-O-gallate-β-D-glucopyranoside (8)
[α]25D +44 ± 7 (c 0.02, H2O/MeOH); 1H NMR (CD3OD, 600 MHz) δ 2.00 (3H, d, J = 1.6 Hz, H3-7), 3.21 (1H, t, J = 7.8 Hz, H-2′), 3.42 (2H, m, H-3′, H-4′), 3.60 (1H, m, H-5′), 4.24 (1H, t, J = 3.4, 2.9 Hz, H-5), 4.40 (1H, dd, J = 12.6, 3.4 Hz, H-6b), 4.43 (1H, dd, J = 12.0, 5.6 Hz, H-6'b), 4.45 (1H, d, J = 7.8 Hz, H-1′), 4.56 (1H, dd, J = 12.6, 2.9 Hz, H-6a), 4.59 (1H, dd, J = 12.0, 2.1 Hz, H-6′a), 5.82 (1H, q, J = 1.6 Hz, H-3), 7.07 (2H, s, H-2′′, H-6′′); 13C NMR (CD3OD, 151 MHz) δ 20.4 (CH3-7), 64.4 (CH2-6′), 70.5 (CH2-6), 71.5 (CH-4′, CH-5), 74.7 (CH-2′), 75.5 (CH-5′), 77.7 (CH-3′), 103.2 (CH-1′), 110.1 (CH-2′′, CH-6′′), 118.9 (CH-3), 139.7 (C-4′′), 157.9 (C-4), 165.9 (C-2), 168.0 (C-7′′) (Supplementary Figures S47–S51); ESI-HRMS m/z 441.1040 [M-H]- (calcd for C19H21O12, 441.1028, Δ = 2.8 ppm); MS/MS spectrum: CCMSLIB00010129501.
Caffeic acid (9)
1H NMR (CD3OD, 600 MHz) δ 6.22 (1H, d, J = 15.9 Hz, H-8), 6.77 (1H, d, J = 8.1 Hz, H-5), 6.93 (1H, dd, J = 8.1, 2.1 Hz, H-6), 7.03 (1H, d, J = 2.1 Hz, H-2), 7.51 (1H, d, J = 15.9 Hz, H-7); 13C NMR (CD3OD, 151 MHz) δ 115.0 (CH-2), 116.1 (CH-8), 116.5 (CH-5), 122.8 (CH-6), 127.9 (C-1), 146.6 (CH-7), 146.8 (C-3), 149.4 (C-4), 171.4 (C-9) (Supplementary Figures S52–S57); ESI-HRMS m/z 179.0340 [M-H]- (calcd for C9H7O4, 179.0339, Δ = 0.8 ppm).
rel-(7S, 8R, 7′R, 8′S)-7,7′-bis(4-glucosyloxy-3-hydroxyphenyl)-8,8′-cyclobutane-9-methyl dicarboxylic acid (10)
1H NMR (CD3OD, 600 MHz) δ 3.37 (4H, m, H-4'', H-4‴, H-5'', H-5‴), 3.43 (4H, m, H-2'', H-2‴, H-3'', H-3‴), 3.69 (2H, dd, J = 12.0, 4.4 Hz, H-6‴a, H-6''b), 3.72 (3H, s, H3-10), 3.75 (1H, m, H-8′), 3.78 (1H, m, H-8), 3.87 (2H, dd, J = 12.0, 1.5 Hz, H-6''a, H-6‴a), 4.13 (1H, dd, J = 10.2, 6.2 Hz, H-7′), 4.19 (1H, dd, J = 10.2, 6.6 Hz, H-7), 4.64 (2H, m, H-1'', H-1‴), 6.47 (1H, m, H-6, H- 6′), 6.50 (1H, d, J = 2.4 Hz, H-2), 6.52 (1H, dd, J = 2.2 Hz, H-2′), 6.96 (1H, d, J = 8.4 Hz, H-5′), 6.96 (1H, d, J = 8.4 Hz, H-5); 13C NMR (CD3OD, 151 MHz) δ 44.5 (CH-8), 45.3 (CH-8′), 45.5 (CH-7), 46.2 (CH-7′), 52.4 (CH3-10), 62.4 (CH2-6'', CH2-6‴), 71.3 (CH-4′′, CH-4‴), 74.9 (CH-2′′, CH-2‴), 77.5 (CH-3′′, CH-3‴), 78.2 (CH-5′′, CH-5‴), 104.3/104.4 (CH-1′′, CH-1‴), 116.8/116.9 (CH-2, CH-2′), 118.2/118.3 (CH-5, CH-5′), 120.5/120.6 (CH-6, CH-6′), 136.1/136.3 (C-1, C-1′), 145.3 (C-4, C-4′), 147.8 (C-3, C-3′), 175.2 (C-9), 176.7 (C-9′) (Supplementary Figures S58–S63); ESI-HRMS m/z 697.1991 [M-H]- (calcd for C31H37O18, 697.1974, Δ = 2.4 ppm); MS/MS spectrum: CCMSLIB00010128654.
8-Desmethylsideroxylin (11)
1H NMR (DMSO-d6, 600 MHz) δ 1.97 (3H, s, H3-11), 3.89 (3H, s, H3-12), 6.78 (1H, s, H-3), 6.80 (1H, s, H-8), 6.92 (2H, d, J = 8.7 Hz, H-3′, H-5′), 7.92 (2H, d, J = 8.7 Hz, H-2′, H-6′), 13.05 (1H, s, OH5); 13C NMR (DMSO-d6, 151 MHz) δ 7.5 (CH3-11), 56.5 (CH3-12), 90.5 (CH-8), 103.2 (CH-3), 104.5 (C-10), 107.8 (C-6), 116.2 (CH-3′, CH-5′), 121.3 (C-1′), 128.7 (CH-2′, CH-6′), 155.6 (C-9), 157.7 (C-5), 161.4 (C-4′), 163.2 (C-7), 164.0 (C-2), 182.1 (C-4) (Supplementary Figures S64–S69); ESI-HRMS m/z 299.0904 [M + H]+ (calcd for C17H15O5, 299.0914, Δ = −3.3 ppm); MS/MS spectrum: CCMSLIB00010129499.
Sideroxylin (12)
1H NMR (DMSO-d6, 600 MHz) δ 2.08 (3H, s, H3-11), 2.32 (3H, s, H3-13), 3.74 (3H, s, H3-12), 6.85 (1H, s, H-3), 6.94 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.94 (2H, d, J = 8.8 Hz, H-2′, H-6′), 10.39 (1H, s, 4′-OH), 13.06 (1H, s, 5-OH); 13C NMR (DMSO-d6, 151 MHz) δ 8.1 (CH3-11), 8.3 (CH3-13), 60.3 (CH3-12), 102.8 (CH-3), 106.5 (C-10), 108.7 (C-8), 113.1 (C-6), 116.1 (CH-3′, CH-5′), 121.3 (C-1′), 128.5 (CH-2′, CH-6′), 152.3 (C-9), 156.4 (C-5), 161.4 (C-4′), 162.1 (C-7), 164.1 (C-2), 182.6 (C-4) (Supplementary Figures S70–S75); ESI-HRMS m/z 313.1070 [M+H]+ (calcd for C18H17O5, 313.1071, Δ = −0.1 ppm); MS/MS spectrum: CCMSLIB00010129500.
X ray analysis of compound 1
The compound (5 mg) was solubilized in 1 mL of a hexane and ethyl acetate mixture (1:1) in a small glass vial (5 mL). The vial was wrapped with aluminium foil and small holes were made in the cover for slow evaporation of the solvent. The solution was left at room temperature overnight. The next day the crystals appeared in the bottom of the bottle. Summary of crystal data, intensity measurements and structure refinements for 1 were collected in Supplementary Tables S1, S2 (Supplementary information). Supplementary Figures S74 shows the Ortep of compound 1 (Thermal ellipsoids are drawn at 50% probability level). The crystals were mounted on MiTeGen Kapton cryoloops with protection oil. X-ray data collection were performed with a Rigaku Super Nova Dual diffractometer equipped with a CCD Atlas detector (Cu[Kα] radiation). The structures were solved by using direct methods and full-matrix least-square refinements on F2 were performed with SHELXL-2014 (Sheldrick, 2015). CCDC 1873730 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
Conceived and designed the experiments: MF, EFQ, LM, and J-LW. Performed the experiments: MF, AR, RH, LM, P-MA, LG, CC, L-FN, and JK. Analyzed the data: MF, AR, EFQ, RH, LM, P-MA, LG, CC, L-FN, and JK. Contributed reagents/materials/analysis tools: J-LW and EFQ. Contributed to the writing of the manuscript: MF, AR, RH, LM, P-MA, LG, CC, JK, L-FN, MPG, EFQ, and J-LW.
Funding
Open access funding by University Of Geneva.
Acknowledgments
The School of Pharmaceutical Sciences of the University of Geneva (J-LW) is thankful to the Swiss National Science Foundation for the support in the acquisition of the NMR 600 MHz (SNF R’Equip grant 316030_164095). The authors would like to dedicate this article to our colleague MPG from the University of Panama with whom they collaborated for over 25 years, who passed away in December 2020. The authors express their gratitude to Dr. Georges Massiot (University of Reims Champagne-Ardenne, France) for the discussions concerning the biosynthesis of the compounds.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fntpr.2023.1147195/full#supplementary-material
References
Almeda, F. (1997). Chromosomal observations on the Alzateaceae (myrtales). Ann. Mo. Bot. Gard. 84, 305. doi:10.2307/2400006
Berger, B. A., Kriebel, R., Spalink, D., and Sytsma, K. J. (2016). Divergence times, historical biogeography, and shifts in speciation rates of Myrtales. Mol. Phylogenet. Evol. 95, 116–136. doi:10.1016/j.ympev.2015.10.001
Caffall, K. H., and Mohnen, D. (2009). The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr. Res. 344, 1879–1900. doi:10.1016/j.carres.2009.05.021
Chi, Y. M., Nakamura, M., Zhao, X. Y., Yoshizawa, T., Yan, W. M., Hashimoto, F., et al. (2006). Antinociceptive activities of.ALPHA. Truxillic acid and.BETA. Truxinic acid derivatives. Biol. Pharm. Bull. 29, 580–584. doi:10.1248/bpb.29.580
Conti, E., Eriksson, T., Schonenberger, J., Sytsma, K. J., and Baum, D. A. (2002). Early tertiary out-of-India dispersal of Crypteroniaceae: Evidence from phylogeny and molecular dating. Evolution 56, 1931–1942. doi:10.1111/j.0014-3820.2002.tb00119.x
Duan, H. Q., Zhang, Y. D., Xu, J. Q., Qiao, J., Suo, Z. W., Hu, G., et al. (2006). Effect of anemonin on NO, ET-1 and ICAM-1 production in rat intestinal microvascular endothelial cells. J. Ethnopharmacol. 104, 362–366. doi:10.1016/j.jep.2005.09.034
Exarchou, V., Troganis, A., Gerothanassis, I. P., Tsimidou, M., and Boskou, D. (2001). Identification and quantification of caffeic and rosmarinic acid in complex plant extracts by the use of variable-temperature two-dimensional nuclear magnetic resonance spectroscopy. J. Agric. Food Chem. 49, 2–8. doi:10.1021/jf990928e
Forman, V., Luo, D., Geu-Flores, F., Lemcke, R., Nelson, D. R., Kampranis, S. C., et al. (2022). A gene cluster in Ginkgo biloba encodes unique multifunctional cytochrome P450s that initiate ginkgolide biosynthesis. Nat. Commun. 13, 5143. doi:10.1038/s41467-022-32879-9
Fujiwara, A., Nishi, M., Yoshida, S., Hasegawa, M., Yasuma, C., Ryo, A., et al. (2016). Eucommicin A, a beta-truxinate lignan from Eucommia ulmoides, is a selective inhibitor of cancer stem cells. Phytochemistry 122, 139–145. doi:10.1016/j.phytochem.2015.11.017
Ghosn, M. W., and Wolf, C. (2010). Stereocontrolled photodimerization with congested 1,8-Bis(4 '-anilino)naphthalene templates. J. Org. Chem. 75, 6653–6659. doi:10.1021/jo101547w
Gottlieb, H. E., Kumar, S., Sahai, M., and Ray, A. B. (1991). Ethyl brevifolin carboxylate from Flueggea microcarpa. Phytochemistry 30, 2435–2438. doi:10.1016/0031-9422(91)83676-C
Graham, S. A. (1984). Alzateaceae, a new family of Myrtales from the American tropics. Ann. Mo. Bot. Gard. 71, 757–779.
Guillarme, D., Nguyen, D. T. T., Rudaz, S., and Veuthey, J. L. (2008). Method transfer for fast liquid chromatography in pharmaceutical analysis: Application to short columns packed with small particle. Part II: Gradient experiments. Eur. J. Pharm. Biopharm. 68, 430–440. doi:10.1016/j.ejpb.2007.06.018
Junio, H. A., Sy-Cordero, A. A., Ettefagh, K. A., Burns, J. T., Micko, K. T., Graf, T. N., et al. (2011). Synergy-directed fractionation of botanical medicines: A case study with goldenseal (Hydrastis canadensis). J. Nat. Prod. 74, 1621–1629. doi:10.1021/np200336g
Kim, H. W., Wang, M. X., Leber, C. A., Nothias, L. F., Reher, R., Kang, K. B., et al. (2021). NPClassifier: A deep neural network-based structural classification tool for natural products. J. Nat. Prod. 84, 2795–2807. doi:10.1021/acs.jnatprod.1c00399
Lai, G., Zhao, P., Ni, Z., Xu, Y., Wang, M., Luo, S., et al. (2008). A new fructofuranoside from Helwingia chinensis (Cornaceae). Yunnan Zhiwu Yanjiu 30, 115–120.
Nguyen, V. B., Wang, S. L., Nguyen, A. D., Lin, Z. H., Doan, C. T., Tran, T. N., et al. (2018). Bioactivity-guided purification of novel herbal antioxidant and anti-NO compounds from Euonymus laxiflorus Champ. Molecules 24, 120. doi:10.3390/molecules24010120
Pirvu, L., Stefaniu, A., Neagu, G., and Pintilie, L. (2022). Studies on Anemone nemorosa L. extracts; polyphenols profile, antioxidant activity, and effects on Caco-2 cells by in vitro and in silico studies. Open Chem. 20, 299–312. doi:10.1515/chem-2022-0137
Priebe, A., Hunke, M., Tonello, R., Sonawane, Y., Berta, T., Natarajan, A., et al. (2018). Ferulic acid dimer as a non-opioid therapeutic for acute pain. J. Pain Res. 11, 1075–1085. doi:10.2147/Jpr.S161161
Queiroz, E. F., Alfattani, A., Afzan, A., Marcourt, L., Guillarme, D., and Wolfender, J. L. (2019). Utility of dry load injection for an efficient natural products isolation at the semi-preparative chromatographic scale. J. Chromatogr. A 1598, 85–91. doi:10.1016/j.chroma.2019.03.042
Ruiz, H., and Pavón, J. (1798). Systema vegetabilium florae peruvianae et chilensis. Madrid, Spain: Typis Gabrielis de Sancha.
Rutschmann, F., Eriksson, T., Salim, K. A., and Conti, E. (2007). Assessing calibration uncertainty in molecular dating: The assignment of fossils to alternative calibration points. Syst. Biol. 56, 591–608. doi:10.1080/10635150701491156
Rutschmann, F., Eriksson, T., Schonenberger, J., and Conti, E. (2004). Did crypteroniaceae really disperse out of India? Molecular dating evidence from rbcL, ndhF, and rpl16 intron sequences. Int. J. Plant Sci. 165, S69–S83. doi:10.1086/383335
Schonenberger, J., and Conti, E. (2003). Molecular phylogeny and floral evolution of Penaeaceae, oliniaceae, rhynchocalycaceae, and Alzateaceae (myrtales). Am. J. Bot. 90, 293–309. doi:10.3732/ajb.90.2.293
Serna, D. M. O., and Martinez, J. H. I. (2015). Phenolics and polyphenolics from Melastomataceae species. Molecules 20, 17818–17847. doi:10.3390/molecules201017818
Shahidi, F., and Chandrasekara, A. (2010). Hydroxycinnamates and their in vitro and in vivo antioxidant activities. Phytochem. Rev. 9, 147–170. doi:10.1007/s11101-009-9142-8
Sheldrick, G. M. (2015). Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C. Struct. Chem. 71, 3–8. doi:10.1107/S2053229614024218
Stewart, D., Robertson, G. W., and Morrison, I. M. (1992). Identification of cyclobutane-type dimers of substituted cinnamic-acids by gas-chromatography mass-spectrometry. Rapid Commun. Mass Spectrom. 6, 46–53. doi:10.1002/rcm.1290060110
Suga, T., and Hirata, T. (1982). The biosynthesis of protoanemonin in Ranunculus glaber. The pivotal biosynthetic intermediate and the stereospecific hydrogen elimination from the intermediate. Bull. Chem. Soc. Jpn. 55, 1584–1587. doi:10.1246/bcsj.55.1584
Tiwari, S. (2002). Ethnomedicine of the Patamona Indians of Guyana. UMI: The City University of New-York.
Wang, M. X., Carver, J. J., Phelan, V. V., Sanchez, L. M., Garg, N., Peng, Y., et al. (2016). Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 34, 828–837. doi:10.1038/nbt.3597
Keywords: Alzatea verticillata, Alzateaceae, semi-preparative HPLC-UV, metabolic profile and LC-MS/MS, molecular network analysis, truxinic acid derivative
Citation: Ferreira Queiroz MM, Huber R, Marcourt L, Guénée L, Allard P-M, Rutz A, Nothias L-F, Carlotta De Ruvo C, Kissling J, Gupta MP, Ferreira Queiroz E and Wolfender J-L (2023) Phytochemical study of Alzatea verticillata, the sole species belonging to the Alzateaceae family. Front. Nat. Prod. 2:1147195. doi: 10.3389/fntpr.2023.1147195
Received: 18 January 2023; Accepted: 24 February 2023;
Published: 09 March 2023.
Edited by:
Robert Alan Burrow, Universidade Federal De Santa Maria, BrazilReviewed by:
Smith B. Babiaka, University of Buea, CameroonDaniel Pecoraro Demarque, Universidade de São Paulo, Brazil
Gabin Bitchagno, Mohammed VI Polytechnic University, Morocco
Copyright © 2023 Ferreira Queiroz, Huber, Marcourt, Guénée, Allard, Rutz, Nothias, Carlotta De Ruvo, Kissling, Gupta, Ferreira Queiroz and Wolfender. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emerson Ferreira Queiroz, ZW1lcnNvbi5mZXJyZWlyYUB1bmlnZS5jaA==; Jean-Luc Wolfender, SmVhbi1MdWMuV29sZmVuZGVyQHVuaWdlLmNo
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
§Deceased author