 Iván Cortés1†
Iván Cortés1† Cristina Cuadrado
Cristina Cuadrado Antonio Hernández Daranas
Antonio Hernández Daranas Ariel M. Sarotti
Ariel M. Sarotti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nat. Prod., 27 January 2023
Sec. Structural and Stereochemical Analysis
Volume 2 - 2023 | https://doi.org/10.3389/fntpr.2023.1122426
This article is part of the Research TopicInsights in Structural and Stereochemical Analysis: 2022View all 5 articles
Structure elucidation is a stage of paramount importance in the discovery of novel compounds because molecular structure determines their physical, chemical and biological properties. Computational prediction of spectroscopic data, mainly NMR, has become a widely used tool to help in such tasks due to its increasing easiness and reliability. However, despite the continuous increment in CPU calculation power, classical quantum mechanics simulations still require a lot of effort. Accordingly, simulations of large or conformationally complex molecules are impractical. In this context, a growing number of research groups have explored the capabilities of machine learning (ML) algorithms in computational NMR prediction. In parallel, important advances have been made in the development of machine learning-inspired methods to correlate the experimental and calculated NMR data to facilitate the structural elucidation process. Here, we have selected some essential papers to review this research area and propose conclusions and future perspectives for the field.
Determination of molecular structure is one of the most complex and important stages in the discovery of natural products. Traditionally, this has been done using various spectroscopic techniques, NMR being the most important and decisive one (Gil, 2011). However, even with the advent of increasingly powerful equipment and new multidimensional experiments (Liu et al., 2019), structural misassignment is far from being eradicated, and unfortunately persists in current literature (Nicolaou and Snyder, 2005; Chhetri et al., 2018). In this context, computational chemistry has been synergistically coupled with experimental NMR, giving rise to a huge variety of hybrid methods that greatly facilitate elucidation (Napolitano et al., 2011; Gutiérrez-Cepeda et al., 2014). From earlier contributions of Bifulco, Bagno, and Saielli (Barone et al., 2002a; Barone et al., 2002b; Bagno et al., 2006), to the explosion in the post-DP4 era (Smith and Goodman, 2010; Grimblat et al., 2015; Ermanis et al., 2017; Grimblat et al., 2019), the process has undergone continuous improvements (Lodewyk et al., 2012a; Grimblat and Sarotti, 2016; Lauro and Bifulco, 2020; Marcarino et al., 2020; Costa et al., 2021; Marcarino et al., 2022). Among them, perhaps one of the most disruptive has been the implementation of machine learning (ML), which has undoubtedly revolutionized molecular simulation (Noé et al., 2020). Application of machine learning to computational NMR in the context of structural elucidation can be roughly divided into 2 main categories, namely, prediction and correlation (Figure 1). In the first, ML is used to obtain quantum-quality NMR chemical shifts at a remarkably lower computational cost. In the second category, ML facilitates the correlation between experimental and calculated data in order to determine the most likely structures. In this minireview, the latest developments in both approaches are discussed, focusing on methods that combine ML with quantum NMR calculations. For other applications of ML to NMR, including molecular phenotyping and clustering (Peng et al., 2020a; Peng, 2021; dos Santos et al., 2022; Peng et al., 2020b), among others, we refer the interested reader to other recent reviews on the subject (Cobas, 2019; Jonas et al., 2022). We begin here with a description of the most important ML procedures for NMR prediction (Section 2) and then discuss exciting examples of ML in data correlation (Section 3). A final conclusion and future perspectives are also provided (Section 4).
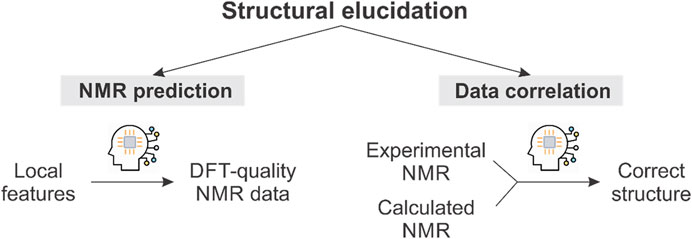
FIGURE 1. Schematic representation of the use of ML in computational NMR-aided structural elucidation.
There are many reasons to calculate NMR chemical shifts with high accuracy. For example, NMR simulations can be helpful during spectroscopic assignment; that is, to determine which NMR signal belongs to which nucleus in the molecule. They can also provide insightful information in mechanistic and biogenetic studies (Cen-Pacheco et al., 2021; Li et al., 2021; Simonetti et al., 2021), conformational analysis (Domínguez et al., 2014a; Nguyen et al., 2018; Li et al., 2020; Sosa-Rueda et al., 2021), structural revisions (Lodewyk et al., 2012b; Cen-Pacheco et al., 2012; Sarotti, 2020), and structural elucidation (perhaps one of the most widely used applications today) (Napolitano et al., 2009; Cen-Pacheco et al., 2013; Domínguez et al., 2014b; Cen-Pacheco et al., 2014; Marcarino et al., 2020; Wang et al., 2020; Domínguez et al., 2021; Zanardi and Sarotti, 2021; Marcarino et al., 2022). Empirical approaches present the fastest alternatives, such as additive methods based on the cumulative effect of substituents (Fürst and Pretsch, 1990). However, the quality of those predictions can be insufficient for some applications. More refined methods have been developed to enhance predictive performance. For instance, hierarchically ordered spherical description of environments (HOSE) encodes the neighborhood of each atom from a 2D representation of the molecule (Bremser, 1978; Jonas et al., 2022), achieving good predictive accuracies (mean absolute errors, MAE, 1.7 ppm for 13C and 0.2 ppm for 1H) (Smurnyy et al., 2008). Using graph neural networks (GNN), Jonas and Kuhn developed ML based on 2D molecular connectivity with good results (1.43 ppm 13C and 0.28 ppm 1H) (Jonas and Kuhn, 2019). In spite of these excellent results, stereochemical and conformational effects are often ignored by most empirical approaches, being limited to accounting for the impact of geometrical factors on chemical shifts. At this point, it is important to highlight that highly accurate predictions are required to differentiate between similar structures, such as that provided by NMR at density functional theory (DFT) level. The main drawback of such approaches is their high computational cost, in terms of resources and time. The latter rapidly becomes longer with a greater number of atoms, so description of large systems often becomes prohibitive. One of the best ways to reduce computational cost while maintaining predictive capacity is to employ ML-based schemes. This will be discussed with the following methods as typical examples.
Paruzzo et al. (2018) Solid-state NMR is a powerful tool for analyzing powdered and amorphous solids at the atomic level, with great utility in pharmaceutical sciences. The discipline has been revolutionized by the advent of quantum methods to calculate chemical shifts with high accuracy. Chemical shift-based NMR crystallography has therefore become a popular strategy to identify polymorphs, and also for de novo determination of crystal structures from powders (Facelli and Grant, 1993). This has been enabled by plane wave DFT methods developed for periodic systems based on gauge, including projected augmented wave (GIPAW), which provides good accuracy to emulate the local atomic environments (Blöchl, 1994). However, computational cost is again prohibitive for large systems and/or for higher levels of theory. In this seminal work, Paruzzo and co-workers developed a local environment-ML based method to predict chemical shifts of molecular solids with an accuracy comparable to DFT (Paruzzo et al., 2018). Due to limitations in experimental databases for NMR of solids, the authors decided to train and validate their ML using GIPAW DFT-calculated chemical shifts of a wide variety of structures taken from the Cambridge Structural Database (CSD) (Groom et al., 2016). Bypassing the experimental information has several advantages, which include avoiding biased or incorrectly reported data, as well as offering an unlimited number of virtual structures. In fact, this interesting practical idea has been replicated by other subsequent studies, as detailed below. From an initial database of 61,000 structures, the authors selected 2,000 diverse molecules for training, and 500 for validation. The first subset was selected using the farthest point sampling algorithm (FPS), and the second one randomly. The NMR properties of the resulting structures were calculated at the GIPAW DFT level, the local environments being based on the smooth overlap of atomic positions (SOAP) kernel (Bartók et al., 2013; De et al., 2016). This approach is based on encoding the atomic environment by a 3D neighborhood density defined by a superimposition of Gaussians, one centered at each atom located within a spherical boundary from the core atom. The ML was trained using the Gaussian process regression (GPR) framework, which previously performed well when coupled with SOAP (Figure 2). (Bartók et al., 2022) Once trained, ShiftML was highly accurate, particularly for 1H (MAE 0.49 ppm). The other nuclei showed larger errors relative to DFT (4.3 ppm for 13C, 13.3 ppm for 15N, and 17.7 ppm for 17O). According to the authors, this was due to the significantly fewer training environments for heteronuclei than for 1H. However, the reduction in computational cost is remarkable (less than 1 min vs. 62–150 CPU hours), demonstrating its amazing predictive ability in short periods of time.
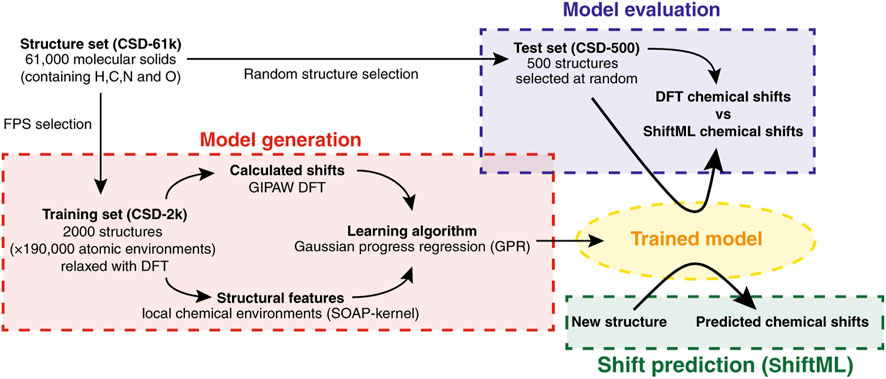
FIGURE 2. Scheme of the ML model used in ShiftML. Reproduced from Ref (Paruzzo et al., 2018).
Gerrard et al. (2020) A few years after ShiftML, Craig Butts and co-workers developed their first generation solution-state NMR prediction machines entitled IMPRESSION (Intelligent Machine PREdiction of Shift and Scalar Information Of Nuclei). Inspired by ShiftML, the training and validation was done using DFT-predicted values rather than scarce and potentially misassigned experimental data. The key step of selecting the example molecules followed an interesting adaptive sampling procedure starting from a superset of 75,382 structures taken from CSD, with the boundary condition of only comprising C, H, N, O, and F atoms. The active learning sampling took 100 randomly selected structures, and then the trained ML predicted the NMR shifts of the remaining structures in the superset. The 100 with the highest variance (after a 5-fold cross validation) were added to the training set, and the procedure was iterated, leading to 882 final structures. Each structure was submitted to geometry optimizations at the mPW1PW91/6-311G** level, for further NMR calculations at the wB97XD/6-311G** level. The training procedure applied KRR (Kernel Ridge Regression) (Vu et al., 2015), with three different methods to encode similarity between atomic environments: CM (Coulomb Matrixes) (Rupp et al., 2015), aSLATM, and FCHL (Faber et al., 2018). As expected, better results were obtained with aSLATM and FCHL (which involve 3-body interactions) than CM (2-body interactions). The FCHL method was selected for its minimal computational cost. After optimization, IMPRESSION achieved MAE of 0.23 ppm/2.45 ppm/0.87 Hz for 1H/13C/1JCH predictions and a root mean squared error (RMSE) of 0.35 ppm/3.88 ppm/1.39 Hz against the validation set. These results were considerably better than with ShiftML (Paruzzo et al., 2018), confirming that selection of environments and training models are fundamental elements in the ML process. Nevertheless, the authors detected some chemical environments in the test set (around 2.5% of the total) that were not successfully emulated by IMPRESSION, with errors up to 11 ppm (1H), 63 ppm (13C) and 25 Hz (J). To overcome this problem, an “prediction variance filter” was applied to improve the quality of the results by removing poorly described environments that show high variance across a 5-fold cross validation. With this modification, IMPRESSION achieved an improvement in accuracy relative to DFT comparable with that of DFT relative to experiment. However, note that this version of IMPRESSION only accelerated NMR prediction, it still required DFT-optimized structures that demand from hours to days, depending on the system. The authors recognized that this could be improved by using 3D structures derived directly from the molecular mechanics, with a resultant time saving. When IMPRESSION was re-trained under this modification, the average errors increased ∼30%–50% for 1H and J data, whereas 13C data remained almost insensitive. The method was successfully tested in the prediction of experimental NMR data, and in structural discrimination.
Guan et al. (2021) Paton’s group developed a ML model to tackle the usual difficulties in predicting 1H and 13C chemical shifts, namely: computation time demand and reaching the required accuracy to select the correct structure from among several candidates. For this purpose, a huge amount of experimental data is necessary, but are not always easily accessible, complete, well assigned or parseable. Therefore, to surpass these problems the authors decided to use their own dataset obtained from DFT calculations to train a neural network (NN). However, such an approach is restricted by the DFT methodology used (basis set and solvation model, among others). Therefore, to solve these limitations, they used a Transfer Learning (TL) approach (Taylor and Stone, 2009). In that direction, they developed three GNN models (St. John et al., 2019), namely: DFTNN, ExpNN-dft and ExpNN-ff. Software architectures and hyper-parameters are identical for all of these, but they were developed using different input structures (Figures 3A,B). DFTNN used a vast array of structurally diverse organic molecules from the DFT8K dataset. Their NMR chemical shifts were calculated at the mPW1PW91/6-311+G(d,p) level of theory, implementing optimized geometries at the M06-2X/def2-TZVP level, which were subsequently used to train the GNN (1H and 13C separately). However, a weak point of this first approach was that the neural network was only trained against DFT-calculated data. To face this problem, the authors used TL to retrain the DFTNN model against experimental NMR data from the Exp5K dataset, creating a new model named ExpNN-dft. However, this model also needed structure optimization that caused a performance bottleneck. The authors’ final answer was the ExpNN-ff model, where ExpNN-dft was retrained replacing the starting geometries with those directly obtained from molecular mechanics conformational searches using the MMFF94 force field. This replacement drastically reduced CPU time. The ExpNN-ff model was tested with good results in three different applications: i) structure elucidation by comparison between predicted and experimental NMR data, ii) NMR data reassignment and iii) forecast of regioselectivity of electrophilic aromatic substitution sites using simulated NMR data as descriptors. Moreover, the model differentiated between stereoisomers and even showed distinct predictions for different conformations of the same molecule. Differences between GNN predicted NMR chemical shifts and those obtained from DFT calculations resulted in mean average errors (MAE) of 1.26 ppm for 13C and 0.16 ppm for 1H. Importantly, ExpNN-ff showed a comparable accuracy to normal DFT calculations but with a 6000-fold reduction in CPU time. Therefore, this model can perform NMR data predictions for large flexible structures that are unfeasible for DFT calculations (Daranas and Sarotti, 2021). According to the authors, there is still room for improvement in its results. Their main concern is regarding dependency of the outcome on the quality of the input candidate 3D structures obtained in the molecular mechanics conformational search step. Recent work on this issue confirms the importance of this stage (Cuadrado et al., 2022). Thus, the authors suggest the use of semi-empirical structures as an alternative. It is worth noting that they make this analysis tool easily available in a webpage (http://nova.chem.colostate.edu/cascade/) to perform chemical shift predictions.
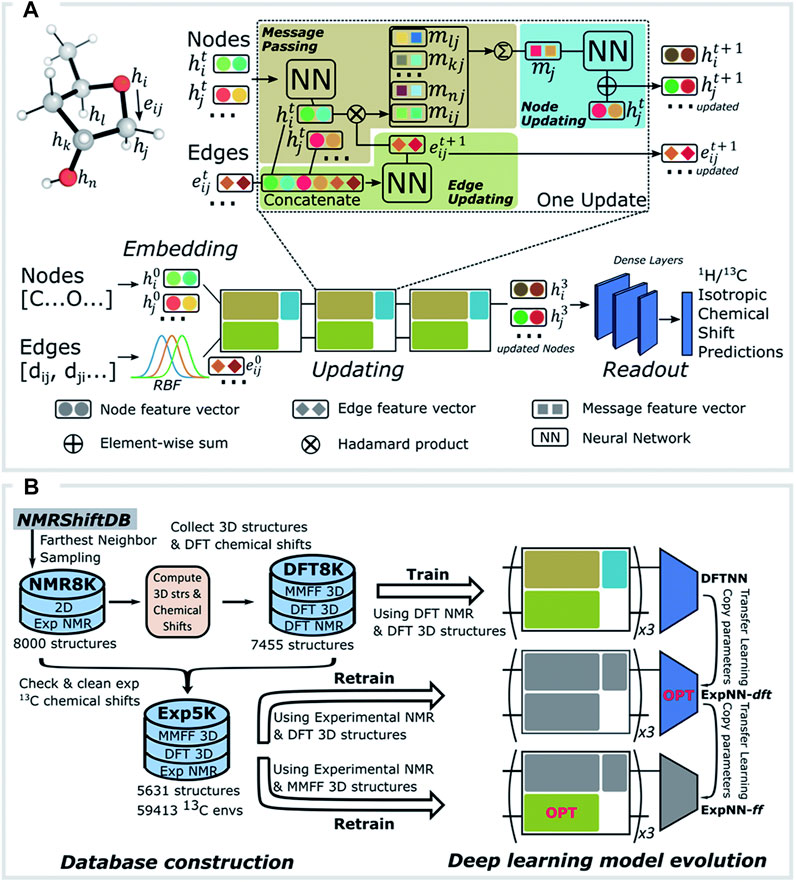
FIGURE 3. Scheme of the ML model used in CASCADE. Reproduced from Ref (Guan et al., 2021). With permission from the Royal Society of Chemistry.
(Tsai et al., 2022) In 2019, Hernández Daranas, Sarotti, and co-workers reported J-DP4 (Grimblat et al., 2019), an updated version of DP4 (10) that incorporates J values into the method´s architecture in 2 different ways. First, the J values are used to restrict conformational sampling, keeping only those structures with dihedral angles in agreement with the experimental data. This not only reduces computational cost considerably, but also improves the conformational landscape description by neglecting spurious conformations that otherwise might make high Boltzmann contributions. Next, the remaining shapes are submitted to chemical shifts and J calculations at DFT level. The J calculations include the Fermi contact term (FC), being correlated with the experimental values by using an additional Bayesian component to account for the probability term, given by 3JHH. Despite the excellent results obtained, J-DP4 was computationally costly. We accelerated it with a new workflow in 2022, involving fast Karplus-type J calculations (Navarro-Vázquez et al., 2018). These were coupled with NMR chemical shift predictions at the cheapest HF/STO-3G level, enhanced by machine learning (ML). The decision to use a hybrid representation of the molecular environments was inspired in the work of Beran and co-workers (Unzueta et al., 2021). This representation included the isotropic shielding constants computed at the very fast HF/STO-3G//MMFF level coupled with local descriptors. The research demonstrated that a Δ-ML approach can be highly accurate. In Δ-ML, the chemical shifts calculated at a lower level (PBE0/6-31G//ωB97XD) can be improved to PBE0/6-311+G (2d,p)//ωB97XD (high level) through artificial neural networks using the AEV (atomic environment vector) to encode the geometric data of the atoms. Based on this background, we hypothesized that the negligible additional cost involved in running NMR calculations at a fast quantum level would be justified by the quality of the NMR predictions, suitable for stereochemical discrimination. The workflow (Figure 4) involved selecting 27,000 diverse structures by computing the Morgan fingerprint (Rogers and Hahn, 2010) of the 170,000 original structures taken from COCONUT (Sorokina et al., 2021) and then using the MinMax algorithm to pick the most diverse of them. The data were randomly divided into 17,000 molecules for training (232,560 13C and 280,446 1H values, T17k set) and 10,000 molecules for validation (150,760 13C and 183,612 1H values, V10k set). In this hybrid approach, the GIAO NMR shielding constants computed at the HF/STO-3G//MMFF level were complemented with different geometric and electronic descriptors that capture the local environments, including charges, hybridizations, distances, angles, long-range interactions (Coulomb and tensorial matrices, etc). As in IMPRESSION (Gerrard et al., 2020), KRR was the data correlation strategy that afforded the best results. However, the main difference was that the adaptive training used the individual environments (rather than individual molecules) that maximized the predictive capacity of the ML. This was supported by the fact that NMR properties are local in nature, so it is not considered mandatory to use all environments from a test molecule, but rather the most important ones. After selecting the most influential descriptors, adaptive learning was conducted to select the best set of environments based on a 25-step iterative incorporation of the 1,000 worst-predicted environments provided by the previous training set. The hyperparameters of the resulting machines (composed of 25 K selected 13C and 1H environments) were further optimized using a 5-fold approach, and the optimal machines were tested against the independent test set (V10k, 150,760 13C and 183,612 1H values). The predictions were highly satisfactory, with MAE of 1.21/0.14 ppm, RMS of 1.63/0.19 ppm, and MaxErr of 20.74/1.89 ppm for 13C and 1H data, respectively. These results were excellent compared to those obtained with other recent ML approaches discussed above. It is true that ML requires quantum computation of isotropic shielding values, but we consider that the quality of the results justifies that extremely low additional cost. The entire process was automated in the form of a Python script and released under an open-source MIT license available at https://github.com/Sarotti-Lab/ML_J_DP4.
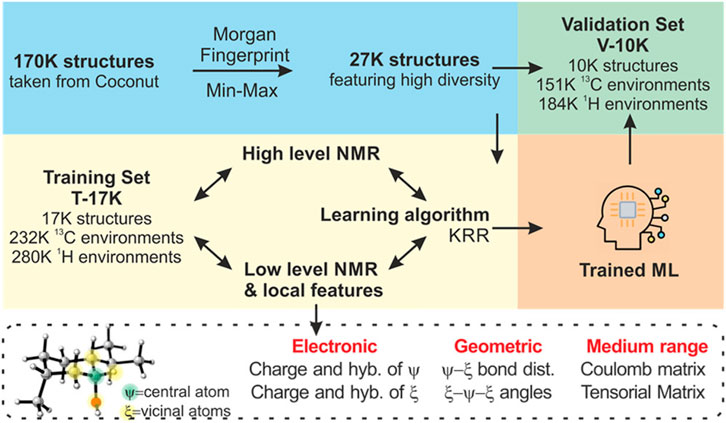
FIGURE 4. Scheme of the ML model used in ML-J-DP4. Reprinted with permission from (Tsai et al., 2022). Copyright (2022) American Chemical Society.
Novitskiy and Kutateladze (2022a) In this paper, Novitskiy and Kutateladze started from their previously developed DU8+ hybrid DFT-parametric method (Kutateladze and Reddy, 2017). DU8+ incorporates binomial correction functions to improve the calculation of NMR parameters of carbons attached to heavy atoms. According to the authors, their approach was the seed of what was later called ML-augmented DFT (Gao et al., 2020). Thus, adding ML methods they developed a DU8+ augmented method called DU8ML, which calculates NMR chemical shifts and spin-spin coupling constants (SSCC) of large natural products with high accuracy, in short computational times. The RMSD deviations calculated from correct structures were 0.95 ppm for 13C (11.000 values were used as training set) and 0.28 Hz for SSCC (from 4,000 experimental values). To enhance accuracy, molecular fragments from these datasets showing the highest deviations were selected as the ML training set (using experimental chemical shifts and SSCC) to identify and correct any inconsistencies. Specifically, a first step of optimization and calculations was nuclear magnetic shieldings at ωB97XD/6-31G (d) PCM and Fermi contact at B3LYP/DU8 under Gaussian computations. This step was followed by the necessary ML-derived corrections for both NMR parameters. The authors present several examples to demonstrate the applicability of DU8ML. In most cases, 13C chemical shift RMSD values were chosen to detect misassignments. In this field, the selected examples illustrate problems related to bad atom connectivities, the type of substituents selected or those associated with flipped fused rings. However, not only were incorrect 2D assignments confronted, but also stereochemical ones. The later are much more challenging and continue to be the most usual source of errors in structural elucidation. Thus, inversion of stereoconfigurations (including an N-oxide), fused rings, and the tricky epoxide rings were tackled. Moreover, the authors also show the usefulness of the method in detecting wrong assignments due to molecule protonation by NMR solvents, SSCC and disagreements between NMR and X-ray or mass spectroscopy data. They also introduce a novel application of DU8ML that amends a previously proposed reaction mechanism, by correcting the assignment of structures involved in the process (Novitskiy and Kutateladze, 2022b). Some aspects that could improve the workflow—discussed in the paper—were the design of a fully automated program for every kind of molecule and the addition of a probability calculation for each candidate structure.
The accuracy of quantum NMR calculations using affordable levels of theory can be more than enough to differentiate very different structures, as in the case of constitutional isomers. However, for stereoisomers the situation becomes more challenging because of their spectroscopic resemblance. For that reason, in addition to the advances made in improving and accelerating NMR predictions, the development of robust data correlation methods is essential (Grimblat and Sarotti, 2016). To date, a wide variety of methods have been reported (Grimblat and Sarotti, 2016; Lauro and Bifulco, 2020; Marcarino et al., 2020; Costa et al., 2021; Marcarino et al., 2022). In this section, state-of-the-art ML-based methods will be discussed.
Sarotti (2013), Zanardi and Sarotti, 2015) These methods were developed by Sarotti´s group to tackle the structural validation problem. That is, to decide the correctness of structural proposals based on the correlation of the experimental NMR data collected for a given molecule and the theoretical chemical shifts calculated for it. By that time, the leading strategy in DFT-based structural elucidation was based on a direct comparison between potential candidates (for example, CP3 or DP4) (Smith and Goodman, 2009; Smith and Goodman, 2010). Regardless of the performance of each method, the underlying hypothesis assumes that the correct structure is included within the candidate set. The approach followed to determine a potential structural misassignment using one set of experimental and calculated data was based on the use of pattern recognition analysis (PRA), with the aid of artificial neural networks (ANNs). The latter are mathematical models in which interconnected artificial neurons emulate the function of a biological brain able to learn from the data. The proof-of-concept was based on monodimensional 13C NMR data correlation with the aid of two-layer feed-forward ANNs, using a test set of 200 structures. Different descriptors were assessed as reference standards, including R2, MAE, maximum error (MaxErr), each computed using TMS and MSTD (multi-standard approach) (Sarotti and Pellegrinet, 2009; Sarotti and Pellegrinet, 2012). A large number of ANNs featuring different numbers of input and hidden layers were built and trained, then those with optimal classification ability were kept for validation using a set of 26 natural products originally misassigned, with their respective revised structures. This first generation of ANNs performed excellently in identifying connectivity mistakes (such as constitutional isomerism), though they were not conceived to tackle subtler differences like stereoisomerism (Sarotti, 2013). This motivated development of a new generation of ANNs by merging mono-dimensional 1H and 13C NMR data with 2D HSQC correlations (Figures 5A,B). Hundreds of different ANNs were trained using the standard correlation parameters described above, as well as 18 new descriptors accounting for the global correlation between experimental and simulated HSQC data. The training set was composed of 200 structures (100 correct and 100 artificially-made incorrect ones). The most efficient ANNs were validated using a set of 32 originally misassigned natural products, along with their revised structures. The performance achieved was noteworthy, identifying subtle structural errors in an efficient and simple manner (Zanardi and Sarotti, 2015).
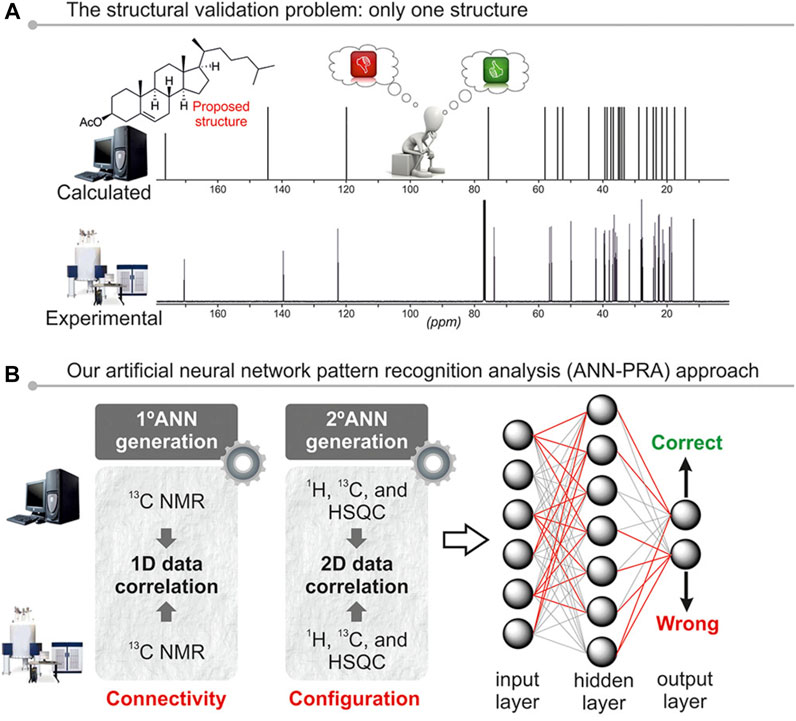
FIGURE 5. Schematic representation of the ANN-PRA method. Adapted with permission from Marcarino et al. (2020). Copyright (2020) American Chemical Society.
Howarth et al. (2020) A common feature of structure elucidation of small molecules assisted by computational methods is that they all need candidate structures, as well as user-assigned 1H and/or 13C NMR experimental data as input (Smith and Goodman, 2010; Grimblat et al., 2015; Lauro and Bifulco, 2020; Marcarino et al., 2020; Zanardi and Sarotti, 2021). These data are then used in different ways to find the best match between measured and computed values. Currently, the most human-time consuming stage within this workflow is NMR data assignment. DP4-AI is an attempt to solve this by means of an automatic interpretation of NMR spectra, coupled to a standard DP4 calculation (Smith and Goodman, 2010). This is a complex task that involves several stages, where the result of each affects subsequent steps (Cobas, 2019). Therefore, after Fourier transformation of NMR data, a hybrid method to phase the resulting spectra was selected. The resulting baseline distortions are corrected, and peaks are picked using first and second derivative methods. Next, the detected peaks are grouped into multiples and integrated (Chen et al., 2002; Wang et al., 2013; Zorin et al., 2017), following similar procedures for both 1H and 13C spectra. However, the core of DP4-AI is the assignment algorithm (AA) that is responsible for assigning the atoms in each candidate structure, according to the previously detected experimental peaks. The system also predicts chemical shift values by means of DFT GIAO methods. Using these values, the AA calculates the assignment probability matrix M to find the most likely identity of each experimental peak (Kuhn, 1955). The M derives from a statistical model that considers error distribution of DFT-predicted values at the selected computational settings. DP4-AI was evaluated using 47 molecules with an average of 3.49 asymmetric centers each, and a diversity of carbon backbones. Their NMR spectra were recorded in different solvents, adding other analytical difficulties such as low signal to noise ratio spectra or even using mixtures of compounds. Four different statistical models were tested. The best results were found for a single region three Gaussian model, fitted to an empirical prediction error distribution obtained from the same test set. Importantly, the efficacy of this tool depends greatly on the level of theory, since accuracy of the chemical shift calculation underpins both the assignments and the subsequent DP4 calculation. As expected, the best overall results were obtained with the most accurate chemical shift predictions tested, after geometry optimization by the B3LYP functional followed by chemical shift prediction using the previous structures utilizing PCM/mPW1PW91/6-311G(d) and single point energies calculated at the M06-2X/def2-TZVP levels of theory (Ermanis et al., 2019). At this level, the probability of obtaining the results by chance was about 3 × 10–8, indicating high performance. The authors provide DP4-AI as an open-source software with a GUI. The capability of this system to greatly increase processing speed with minimal human intervention enables high-throughput data analysis. It was estimated that one molecule per minute can be processed. Therefore, DP4-AI facilitates exploration of large data sets and the discovery of new structural information via machine learning techniques. This software tool also could be potentially used to support CASE software.
Howarth and Goodman (2022) The DP5 probability is a new methodology complemented by a software package that draws on DP4-AI sources (Howarth et al., 2020). DP5 goes conceptually further than other methods such as CP3 or DP4, since it faces the important challenge of assessing the probability of a single structure being correct (Figure 6). This is a very important step forward because previous methods must assume that the correct structure has been included within the panel of candidate structures. In other words, in earlier approaches, if all the proposed structures are erroneous they cannot be applied because they are designed to necessary select one of them. Whereas ANN-PRA (Sarotti, 2013; Zanardi and Sarotti, 2015) categorizes candidate structures in a binary fashion either as correct or incorrect, DP5 estimates normalized stand-alone probabilities without assuming that one of the possibilities must necessarily be correct. To do this, DP5 considers the spatial geometry for each atom, to calculate the probability of a DFT prediction error individually. This advance solves the problem that the associated errors vary in complex ways depending on their atomic environments. DFT calculations were undertaken using the same levels of theory employed in DP4-AI. It must be noted that the CASCADE training data were the source of the optimized geometries and NMR data predictions (Guan et al., 2021). Interestingly, a single conformation was selected for each molecule. At the core of this method there is a prediction error distribution for each atom that was found empirically by a Kernel Density Estimation, using a test-set of 5,140 molecules obtained from NMRShiftDB. Importantly, DP5 was developed using only 13C NMR data. DP5 global efficacy was evaluated using all molecules in a leave-one-out cross validation experimental design. The system works well even when tested using incorrect proposals with errors comparable to those obtained for DFT predictions derived from the correct structures. This is because the statistical model applied considers the proposed structure, something not possible in previous error analysis. A very interesting feature of this study was the maximum possible DP5 probability. Thus, a maximum confidence of 72% was found that a proposed structure is correct. On the other hand, the user can sometimes be 100% sure that a certain proposal is erroneous if further data is available. The DP5 workflow was further tested with 13 examples of reassigned molecular structures obtained from the literature. Notably in all of them, this methodology showed an average 41% more confidence in the correct structures than in the rejected ones. Moreover, 42 examples of stereochemical problems were faced and the results were almost equal to those using DP4.
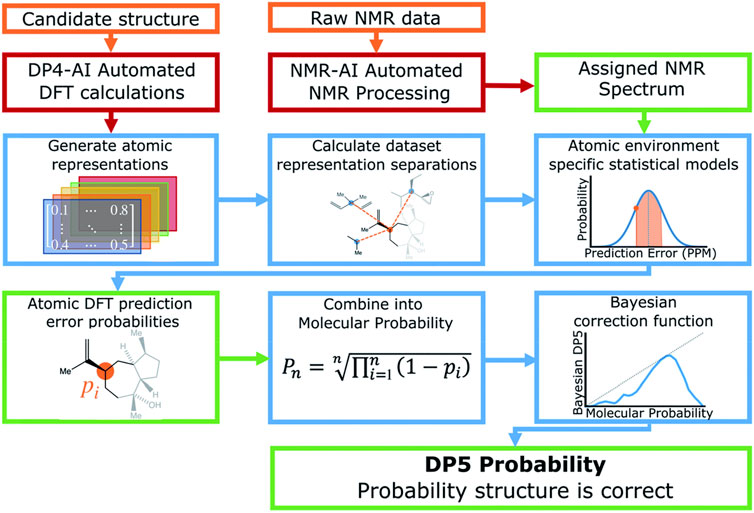
FIGURE 6. Schematic representation of DP5. Reproduced from Ref (Howarth and Goodman, 2022). with permission from the Royal Society of Chemistry.
On assessing the evolution of ML applied in the field of NMR, one can be totally optimistic towards the results that will certainly appear in the coming years. The development of new ML procedures, augmented with more powerful computers, will surely improve the capabilities of current methods. However, as stated by Cobas (Cobas, 2019), one of the most important challenges to overcome is the enormously immense diversity of molecular environments, coupled with the lack of massive and reliable experimental NMR data sets. This is the main reason why most ML-NMR methods use DFT NMR chemical shifts as the output layer, which might be good for some applications but will not provide the ultimate solution to the problem. After all, it has been well documented that in many cases DFT predictions can be poor for certain systems (Zanardi et al., 2020). Based on the above, perhaps a next stage in this discipline would be merging the two so far disconnected approaches discussed in this article (prediction and correlation). That is, a fully based and automated ML method that predicts, at real time, the NMR data with high accuracy (relative to the experimental NMR data) and simultaneously correlates it with the experimental information to facilitate the assignment. To achieve that goal, it is critically important to improve the predictive levels of current ML approaches, as well as to solve the calculation of the right Boltzmann amplitudes of flexible molecules. If we ever achieve that, many problems in structural elucidation will be solved. Perhaps this may sound utopian, but as the Uruguayan writer Eduardo Galeano said “Utopia is on the horizon. I move two steps closer; she moves two steps further away. I walk ten steps and the horizon runs ten steps further away. No matter how much I walk, I´ll never reach her. So, what´s the point of utopia? The point is this: to keep walking”.
IC, CC, AD, and AS contributed to the study concept and design. IC, CC, AD, and AS wrote the sections of the manuscript. All authors contributed to manuscript revision and review, and approved the submitted version.
Our research was funded by the UNR (BIO 500 and 567), ANPCyT (PICT-2016-0116, PICT-2017-1524, and PICT-2019-4052), CONICET (PIP 11220200102205CO), MICINN (PID 2019-109476RB-C21), and ACIISI-FEDER (ProID2021010118).
IC thanks CONICET for a postdoctoral fellowship, and CC thanks ACIISI and FSE (Programa Operativo Integrado de Canarias 2014–2020, Eje 3, Tema Prioritario 74%–85%) for a predoctoral fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bagno, A., Rastrelli, F., and Saielli, G. (2006). Toward the complete prediction of the 1H and 13C NMR spectra of complex organic molecules by DFT methods: Application to natural substances. Chem. – A Eur. J. [Internet] 12 (21), 5514–5525. Available from. doi:10.1002/chem.200501583
Barone, G., Duca, D., Silvestri, A., Gomez-Paloma, L., Riccio, R., and Bifulco, G. (2002). Determination of the relative stereochemistry of flexible organic compounds by ab initio methods: Conformational analysis and Boltzmann-averaged GIAO 13C NMR chemical shifts. Chem. – A Eur. J. [Internet] 8 (14), 3240–3245. Available from:14%3C3240:AID-CHEM3240%3E3.0.CO[. doi:10.1002/1521-3765(20020715)8:14<3240:AID-CHEM3240>3.0.CO;2-G
Barone, G., Gomez-Paloma, L., Duca, D., Silvestri, A., Riccio, R., and Bifulco, G. (2002). Structure validation of natural products by quantum-mechanical GIAO calculations of 13C NMR chemical shifts. Chemistry 8 (14), 3233–3239. doi:10.1002/1521-3765(20020715)8:14<3233::AID-CHEM3233>3.0.CO;2-0
Bartók, A. P., De, S., Poelking, C., Bernstein, N., Kermode, J. R., Csányi, G., et al. (2022). Machine learning unifies the modeling of materials and molecules. Sci. Adv. [Internet] 3 (12), e1701816. Available from. doi:10.1126/sciadv.1701816
Bartók, A. P., Kondor, R., and Csányi, G. (2013). On representing chemical environments. Phys. Rev. B [Internet] 87 (18), 184115. Available from: https://link.aps.org/doi/10.1103/PhysRevB.87.184115.
Blöchl, P. E. (1994). Projector augmented-wave method. Phys. Rev. B [Internet] 50 (24), 17953–17979. Available at: https://link.aps.org/doi/10.1103/PhysRevB.50.17953.
Bremser, W. (1978). Hose — A novel substructure code. Anal. Chim. Acta [Internet] 103 (4), 355–365. Available at: https://www.sciencedirect.com/science/article/pii/S0003267001831007doi:10.1016/s0003-2670(01)83100-7
Cen-Pacheco, F., Mollinedo, F., Villa-Pulgarín, J. A., Norte, M., Fernández, J. J., and Hernández DaranasSaiyacenols, A. A. B. (2012). Saiyacenols A and B: The key to solve the controversy about the configuration of aplysiols. Tetrahedron 68 (36), 7275–7279. doi:10.1016/j.tet.2012.07.005
Cen-Pacheco, F., Norte, M., Fernández, J. J., and Daranas, A. H. (2014). Zoaramine, a zoanthamine-like alkaloid with a new skeleton. Org. Lett. 16 (11), 2880–2883. doi:10.1021/ol500860v
Cen-Pacheco, F., Rodríguez, J., Norte, M., Fernández, J. J., and Hernández Daranas, A. (2013). Connecting discrete stereoclusters by using DFT and NMR spectroscopy: The case of nivariol. Chem. - A Eur. J. 19 (26), 8525–8532. doi:10.1002/chem.201204272
Cen-Pacheco, F., Santiago-Benítez, A. J., Tsui, K. Y., Tantillo, D. J., Fernández, J. J., and Daranas, A. H. (2021). Structure and computational basis for backbone rearrangement in marine oxasqualenoids. J. Org. Chem. 86 (3), 2437–2446. doi:10.1021/acs.joc.0c02600
Chen, L., Weng, Z., Goh, L., and Garland, M. (2002). An efficient algorithm for automatic phase correction of NMR spectra based on entropy minimization. J. Magn. Reson 158 (1–2), 164–168. doi:10.1016/s1090-7807(02)00069-1
Chhetri, B. K., Lavoie, S., Sweeney-Jones, A. M., and Kubanek, J. (2018). Recent trends in the structural revision of natural products. Nat. Prod. Rep. [Internet] 35 (6), 514–531. Available from. doi:10.1039/c8np00011e
Cobas, C. (2019). NMR signal processing, prediction, and structure verification with machine learning techniques. Magn. Reson Chem. 2020, 1–8.
Costa, F. L. P., de Albuquerque, A. C. F., Fiorot, R. G., Lião, L. M., Martorano, L. H., Mota, G. V. S., et al. (2021). Structural characterisation of natural products by means of quantum chemical calculations of NMR parameters: New insights. Org. Chem. Front. [Internet] 8 (9), 2019–2058. Available from. doi:10.1039/d1qo00034a
Cuadrado, C., Daranas, A. H., and Sarotti, A. M. (2022). May the force (field) Be with you: On the importance of conformational searches in the prediction of NMR chemical shifts. Mar. Drugs 20, 699. doi:10.3390/md20110699
Daranas, A. H., and Sarotti, A. M. (2021). Are computational methods useful for structure elucidation of large and flexible molecules? Belizentrin as a case study. Org. Lett. 23, 503–507. doi:10.1021/acs.orglett.0c04016
De, S., Bartók, A. P., Csányi, G., and Ceriotti, M. (2016). Comparing molecules and solids across structural and alchemical space. Phys. Chem. Chem. Phys. [Internet] 18 (20), 13754–13769. Available from. doi:10.1039/c6cp00415f
Domínguez, H. J., Cabrera-García, D., Cuadrado, C., Novelli, A., Fernández-Sánchez, M. T., Fernández, J. J., et al. (2021). Prorocentroic acid, a neuroactive super-carbon-chain compound from the dinoflagellate prorocentrum hoffmannianum. Org. Lett. 23 (1), 13–18. doi:10.1021/acs.orglett.0c03437
Domínguez, H. J., Crespín, G. D., Santiago-Beńitez, A. J., Ga&vacute;in, J. A., Norte, M., Fernández, J. J., et al. (2014). Stereochemistry of complex marine natural products by quantum mechanical calculations of NMR chemical shifts: Solvent and conformational effects on okadaic acid. Mar. Drugs 12 (1), 176–192. doi:10.3390/md12010176
Domínguez, H. J., Napolitano, J. G., Fernández-Sánchez, M. T., Cabrera-García, D., Novelli, A., Norte, M., et al. (2014). Belizentrin, a highly bioactive macrocycle from the dinoflagellate Prorocentrum belizeanum. Org. Lett. 16 (17), 4546–4549. doi:10.1021/ol502102f
dos Santos, V. R., Goncalves, V., Deng, P., Ribeiro, A. C., Teigao, M. M., Dias, B., et al. (2022). Novel time-domain NMR-based traits for rapid, label-free Olive oils profiling. npj Sci. Food [Internet] 6 (1), 59. Available from. doi:10.1038/s41538-022-00173-z
Ermanis, K., Parkes, K. E. B., Agback, T., and Goodman, J. M. (2017). Doubling the power of DP4 for computational structure elucidation. Org. Biomol. Chem. 15 (42), 8998–9007. doi:10.1039/c7ob01379e
Ermanis, K., Parkes, K. E. B., Agback, T., and Goodman, J. M. (2019). The optimal DFT approach in DP4 NMR structure analysis-pushing the limits of relative configuration elucidation. Org. Biomol. Chem. 17 (24), 5886–5890. doi:10.1039/c9ob00840c
Faber, F. A., Christensen, A. S., Huang, B., and von Lilienfeld, O. A. (2018). Alchemical and structural distribution based representation for universal quantum machine learning. J. Chem. Phys. [Internet] 148 (24), 241717. Available from. doi:10.1063/1.5020710
Facelli, J. C., and Grant, D. M. (1993). Determination of molecular symmetry in crystalline naphthalene using solid-state NMR. Nat. [Internet] 365 (6444), 325–327. Available from. doi:10.1038/365325a0
Fürst, A., and Pretsch, E. (1990). A computer program for the prediction of 13-C-NMR chemical shifts of organic compounds. Anal. Chim. Acta [Internet] 229, 17–25. Available at: https://www.sciencedirect.com/science/article/pii/S0003267000851053 doi:10.1016/s0003-2670(00)85105-3
Gao, P., Zhang, J., Peng, Q., Zhang, J., and Glezakou, V. A. (2020). General protocol for the accurate prediction of molecular 13C/1H NMR chemical shifts via machine learning augmented DFT. J. Chem. Inf. Model 60 (8), 3746–3754. doi:10.1021/acs.jcim.0c00388
Gerrard, W., Bratholm, L. A., Packer, M. J., Mulholland, A. J., Glowacki, D. R., and Butts, C. P. (2020). IMPRESSION-prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. Chem. Sci. 11 (2), 508–515. doi:10.1039/c9sc03854j
Gil, R. R. (2011). Constitutional, configurational, and conformational analysis of small organic molecules on the basis of NMR residual dipolar couplings. Angew. Chem. Int. Ed. 50 (32), 7222–7224. [Internet]Available from. doi:10.1002/anie.201101561
Grimblat, N., Gavín, J. A., and Hernandez Daranas, A. (2019). Combining the power of J coupling and DP4 analysis on stereochemical assignments: The J-DP4 methods. Org. Lett. 21 (11), 4003–4007. doi:10.1021/acs.orglett.9b01193
Grimblat, N., and Sarotti, A. M. (2016). Computational chemistry to the rescue: Modern toolboxes for the assignment of complex molecules by GIAO NMR calculations. Chem. - A Eur. J. 22 (35).
Grimblat, N., Zanardi, M. M., and Sarotti, A. M. (2015). Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. [Internet] 80 (24), 12526–12534. Available from. doi:10.1021/acs.joc.5b02396
Groom, C. R., Bruno, I. J., Lightfoot, M. P., and Ward, S. C. (2016). The Cambridge structural database. Acta Crystallogr. Sect. B [Internet] 72 (2), 171–179. Available from. doi:10.1107/s2052520616003954
Guan, Y., Shree Sowndarya, S. V., Gallegos, L. C., St. John, P. C., and Paton, R. S. (2021). Real-time prediction of 1H and 13C chemical shifts with DFT accuracy using a 3D graph neural network. Chem. Sci. [Internet] 12 (36), 12012–12026. Available from. doi:10.1039/d1sc03343c
Gutiérrez-Cepeda, A., Hernández Daranas, A., Fernández, J. J., Norte, M., and Souto, M. L. (2014). Stereochemical determination of five-membered cyclic ether acetogenins using a spin-spin coupling constant approach and DFT calculations. Mar. Drugs 12 (7), 4031–4044. doi:10.3390/md12074031
Howarth, A., Ermanis, K., and Goodman, J. M. (2020). DP4-AI automated NMR data analysis: Straight from spectrometer to structure. Chem. Sci. 11, 4351–4359. doi:10.1039/d0sc00442a
Howarth, A., and Goodman, J. M. (2022). The DP5 probability, quantification and visualisation of structural uncertainty in single molecules. Chem. Sci. [Internet] 13 (12), 3507–3518. Available from. doi:10.1039/d1sc04406k
Jonas, E., and Kuhn, S. (2019). Rapid prediction of NMR spectral properties with quantified uncertainty. J. Cheminform [Internet] 11 (1), 50. Available from. doi:10.1186/s13321-019-0374-3
Jonas, E., Kuhn, S., and Schlörer, N. (2022). Prediction of chemical shift in NMR: A review. Magn. Reson Chem. [Internet] 60 (11), 1021–1031. Available from. doi:10.1002/mrc.5234
Kuhn, H. W. (1955). The Hungarian method for the assignment problem. Nav. Res. Logist. Q. 2 (1-2), 83–97. doi:10.1002/nav.3800020109
Kutateladze, A. G., and Reddy, D. S. (2017). High-throughput in silico structure validation and revision of halogenated natural products is enabled by parametric corrections to DFT-computed 13C NMR chemical shifts and spin-spin coupling constants. J. Org. Chem. 82 (7), 3368–3381. doi:10.1021/acs.joc.7b00188
Lauro, G., and Bifulco, G. (2020). Elucidating the relative and absolute configuration of organic compounds by quantum mechanical approaches. Eur. J. Org. Chem. 1–14.
Li, S. W., Cuadrado, C., Yao, L. G., Daranas, A. H., and Guo, Y. W. (2020). Quantum mechanical-NMR-aided configuration and conformation of two unreported macrocycles isolated from the soft coral lobophytum sp.: Energy calculations versus coupling constants. Org. Lett. 22 (11), 4093–4096. doi:10.1021/acs.orglett.0c01155
Li, S. W., Mudianta, I. W., Cuadrado, C., Li, G., Yudasmara, G. A., Setiabudi, G. I., et al. (2021). Litosetoenins A-E, diterpenoids from the soft coral litophyton setoensis, backbone-rearranged through divergent cyclization achieved by epoxide reactivity inversion. J. Org. Chem. 86 (17), 11771–11781. doi:10.1021/acs.joc.1c01218
Liu, Y., Navarro-Vázquez, A., Gil, R. R., Griesinger, C., Martin, G. E., and Williamson, R. T. (2019). Application of anisotropic NMR parameters to the confirmation of molecular structure. Nat. Protoc. 14 (1), 217–247. doi:10.1038/s41596-018-0091-9
Lodewyk, M. W., Siebert, M. R., and Tantillo, D. J. (2012). Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. [Internet] 112 (3), 1839–1862. Available from. doi:10.1021/cr200106v
Lodewyk, M. W., Soldi, C., Jones, P. B., Olmstead, M. M., Rita, J., Shaw, J. T., et al. (2012). The correct structure of aquatolide—experimental validation of a theoretically-predicted structural revision. J. Am. Chem. Soc. [Internet] 134 (45), 18550–18553. Available from. doi:10.1021/ja3089394
Marcarino, M. O., Cicetti, S., Zanardi, M. M., and Sarotti, A. M. (2022). A critical review on the use of DP4+ in the structural elucidation of natural products: The good, the bad and the ugly. A Pract. guide Nat. Prod. Rep. [Internet]. Available from. doi:10.1039/D1NP00030F
Marcarino, M. O., Zanardi, M. M., Cicetti, S., and Sarotti, A. M. (2020). NMR calculations with quantum methods: Development of new tools for structural elucidation and beyond. Acc. Chem. Res. [Internet] 53 (9), 1922–1932. Available from. doi:10.1021/acs.accounts.0c00365
Napolitano, J. G., Gavín, J. A., García, C., Norte, M., Fernández, J. J., and Daranas, A. H. (2011). On the configuration of five-membered rings: A spin-spin coupling constant approach. Chem. - A Eur. J. 17 (23), 6338–6347. doi:10.1002/chem.201100412
Napolitano, J. G., Norte, M., Padrón, J. M., and Fernández, J. J. (2009). Hernández Daranas A. Belizeanolide, a cytotoxic macrolide from the dinoflagellate Prorocentrum belizeanum. Angew. Chem. - Int. Ed. 48 (4), 796–799.
Navarro-Vázquez, A., Santamaría-Fernández, R., and Sardina, F. J. (2018). MSpin-JCoupling. A modular program for prediction of scalar couplings and fast implementation of Karplus relationships. Magn. Reson Chem. [Internet] 56 (6), 505–512. Available from. doi:10.1002/mrc.4667
Nguyen, Q. N. N., Schwochert, J., Tantillo, D. J., and Lokey, R. S. (2018). Using 1H and 13C NMR chemical shifts to determine cyclic peptide conformations: A combined molecular dynamics and quantum mechanics approach. Phys. Chem. Chem. Phys. [Internet] 20 (20), 14003–14012. Available from. doi:10.1039/c8cp01616j
Nicolaou, K. C., and Snyder, S. A. (2005). Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem. - Int. Ed. 44 (7), 1012–1044.
Noé, F., Tkatchenko, A., Müller, K-R., and Clementi, C. (2020). Machine learning for molecular simulation. Annu. Rev. Phys. Chem. [Internet] 71 (1), 361–390. Available from. doi:10.1146/annurev-physchem-042018-052331
Novitskiy, I. M., and Kutateladze, A. G. (2022). DU8ML: Machine learning-augmented density functional theory nuclear magnetic resonance computations for high-throughput in silico solution structure validation and revision of complex alkaloids. J. Org. Chem. [Internet] 87 (7), 4818–4828. Available from. doi:10.1021/acs.joc.2c00169
Novitskiy, I. M., and Kutateladze, A. G. (2022). Peculiar reaction products and mechanisms revisited with machine learning-augmented computational NMR. J. Org. Chem. 87 (13), 8589–8598. doi:10.1021/acs.joc.2c00749
Paruzzo, F. M., Hofstetter, A., Musil, F., De, S., Ceriotti, M., and Emsley, L. (2018). Chemical shifts in molecular solids by machine learning. Nat. Commun. [Internet] 9 (1), 4501. Available from. doi:10.1038/s41467-018-06972-x
Peng, W. K., Chen, L., Boehm, B. O., Han, J., and Loh, T. P. (2020). Molecular phenotyping of oxidative stress in diabetes mellitus with point-of-care NMR system. npj Aging Mech. Dis. [Internet] 6 (1), 11. Available from. doi:10.1038/s41514-020-00049-0
Peng, W. K. (2021). Clustering Nuclear Magnetic Resonance: Machine learning assistive rapid two-dimensional relaxometry mapping. Eng. Rep. [Internet] 3 (10), e12383. Available from. doi:10.1002/eng2.12383
Peng, W. K., Ng, T-T., and Loh, T. P. (2020). Machine learning assistive rapid, label-free molecular phenotyping of blood with two-dimensional NMR correlational spectroscopy. Commun. Biol. [Internet] 3 (1), 535. Available from. doi:10.1038/s42003-020-01262-z
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inf. Model [Internet] 50 (5), 742–754. Available from. doi:10.1021/ci100050t
Rupp, M., Ramakrishnan, R., and von Lilienfeld, O. A. (2015). Machine learning for quantum mechanical properties of atoms in molecules. J. Phys. Chem. Lett. [Internet] 6 (16), 3309–3313. Available from. doi:10.1021/acs.jpclett.5b01456
Sarotti, A. M., and Pellegrinet, S. C. (2009). A multi-standard approach for GIAO 13C NMR calculations. J. Org. Chem. 74 (19), 7254–7260. doi:10.1021/jo901234h
Sarotti, A. M., and Pellegrinet, S. C. (2012). Application of the multi-standard methodology for calculating 1H NMR chemical shifts. J. Org. Chem. 77 (14), 6059–6065. doi:10.1021/jo3008447
Sarotti, A. M. (2020). Silico reassignment of (+)-Diplopyrone by NMR calculations: Use of a DP4/J-DP4/dp4+/DIP tandem to revise both relative and absolute configuration. J. Org. Chem. 85 (17). In.
Sarotti, A. M. (2013). Successful combination of computationally inexpensive GIAO 13C NMR calculations and artificial neural network pattern recognition: A new strategy for simple and rapid detection of structural misassignments. Org. Biomol. Chem. 11 (29), 4847–4859. doi:10.1039/c3ob40843d
Simonetti, S. O., Kaufman, T. S., Rasia, R. M., Sarotti, A. M., and Grimblat, N. (2021). Thermal decomposition of hexamethylenetetramine: Mechanistic study and identification of reaction intermediates via a computational and NMR approach. Org. Biomol. Chem. [Internet] 19 (34), 7374–7378. Available from. doi:10.1039/d1ob01522b
Smith, S. G., and Goodman, J. M. (2010). Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. [Internet] 132 (37), 12946–12959. Available from. doi:10.1021/ja105035r
Smith, S. G., and Goodman, J. M. (2009). Assigning the stereochemistry of pairs of diastereoisomers using GIAO NMR shift calculation. J. Org. Chem. [Internet] 74 (12), 4597–4607. Available from. doi:10.1021/jo900408d
Smurnyy, Y. D., Blinov, K. A., Churanova, T. S., Elyashberg, M. E., and Williams, A. J. (2008). Toward more reliable 13C and 1H chemical shift Prediction: A systematic comparison of neural-network and least-squares regression based approaches. J. Chem. Inf. Model [Internet] 48 (1), 128–134. Available from. doi:10.1021/ci700256n
Sorokina, M., Merseburger, P., Rajan, K., Yirik, M. A., and Steinbeck, C. (2021). COCONUT online: Collection of open natural products database. J. Cheminform [Internet] 13 (1), 2. Available from. doi:10.1186/s13321-020-00478-9
Sosa-Rueda, J., Domínguez-Meléndez, V., Ortiz-Celiseo, A., López-Fentanes, F. C., Cuadrado, C., Fernández, J. J., et al. (2021). Squamins C–F, four cyclopeptides from the seeds of Annona globiflora. Phytochemistry 2022 (194), 4–10.
St. John, P. C., Phillips, C., Kemper, T. W., Wilson, A. N., Guan, Y., Crowley, M. F., et al. (2019). Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. [Internet] 150 (23), 234111. Available from. doi:10.1063/1.5099132
Taylor, M. E., and Stone, P. (2009). Transfer learning for reinforcement learning domains: A survey. J. Mach. Learn Res. 10 (7).
Tsai, Y-H., Amichetti, M., Zanardi, M. M., Grimson, R., Daranas, A. H., and Sarotti, A. M. (2022). ML-J-DP4: An integrated quantum mechanics-machine learning approach for ultrafast NMR structural elucidation. Org. Lett. [Internet] 24, 7487–7491. Available from. doi:10.1021/acs.orglett.2c01251
Unzueta, P. A., Greenwell, C. S., and Beran, G. J. O. (2021). Predicting density functional theory-quality nuclear magnetic resonance chemical shifts via Δ-machine learning. J. Chem. Theory Comput. 17 (2), 826–840. doi:10.1021/acs.jctc.0c00979
Vu, K., Snyder, J. C., Li, L., Rupp, M., Chen, B. F., Khelif, T., et al. (2015). Understanding kernel ridge regression: Common behaviors from simple functions to density functionals. Int. J. Quantum Chem. [Internet] 115 (16), 1115–1128. Available from. doi:10.1002/qua.24939
Wang, F., Sarotti, A. M., Jiang, G., Huguet-Tapia, J. C., Zheng, S-L., Wu, X., et al. (2020). Waikikiamides A–C: Complex diketopiperazine dimer and diketopiperazine–polyketide hybrids from a Hawaiian marine fungal strain Aspergillus sp. FM242. Org. Lett. [Internet] 22 (11), 4408–4412. Available from. doi:10.1021/acs.orglett.0c01411
Wang, K-C., Wang, S-Y., Kuo, C., and Tseng, Y. J. (2013). Distribution-based classification method for baseline correction of metabolomic 1D proton nuclear magnetic resonance spectra. Anal. Chem. 85 (2), 1231–1239. doi:10.1021/ac303233c
Zanardi, M. M., Marcarino, M. O., and Sarotti, A. M. (2020). Redefining the impact of Boltzmann analysis in the stereochemical assignment of polar and flexible molecules by NMR calculations. Org. Lett. 22, 52–56. doi:10.1021/acs.orglett.9b03866
Zanardi, M. M., and Sarotti, A. M. (2015). GIAO C-H COSY simulations merged with artificial neural networks pattern recognition analysis. Pushing the structural validation a step forward. J. Org. Chem. 80 (19), 9371–9378. doi:10.1021/acs.joc.5b01663
Zanardi, M. M., and Sarotti, A. M. (2021). Sensitivity analysis of DP4+ with the probability distribution terms: Development of a universal and customizable method. J. Org. Chem. [Internet] 86 (12), 8544–8548. Available from. doi:10.1021/acs.joc.1c00987
Keywords: NMR, GIAO, machine learning, structural elucidation, artificial intelligence
Citation: Cortés I, Cuadrado C, Hernández Daranas A and Sarotti AM (2023) Machine learning in computational NMR-aided structural elucidation. Front. Nat. Prod. 2:1122426. doi: 10.3389/fntpr.2023.1122426
Received: 12 December 2022; Accepted: 12 January 2023;
Published: 27 January 2023.
Edited by:
Robert Alan Burrow, Universidade Federal De Santa Maria, BrazilReviewed by:
Stefan Kuhn, University of Tartu, EstoniaCopyright © 2023 Cortés, Cuadrado, Hernández Daranas and Sarotti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Hernández Daranas, YWRhcmFuYXNAaXBuYS5jc2ljLmVz; Ariel M. Sarotti, c2Fyb3R0aUBpcXVpci1jb25pY2V0Lmdvdi5hcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.