 Huajie Zhu
Huajie Zhu Yufang Wang
Yufang Wang Laurence A. Nafie3*
Laurence A. Nafie3*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nat. Prod., 12 January 2023
Sec. Structural and Stereochemical Analysis
Volume 1 - 2022 | https://doi.org/10.3389/fntpr.2022.1086897
This article is part of the Research TopicInsights in Structural and Stereochemical Analysis: 2022View all 5 articles
With the rapid development of high performance computers and computational methods, including software, an increasing number of experimental chemists have tried to use computational methods such as optical rotation (OR, including the matrix model), optical rotatory dispersion (ORD), electronic circular dichroism (ECD or CD), vibrational circular dichroism (VCD), and magnetic shielding constants—nuclear magnetic resonance (NMR)—to explain and/or assign absolute configuration (AC) for various compounds. Such reports in the field of natural products have increased dramatically. However, every method has its range of application. This leads, in some cases, to incorrect conclusions by researchers who are not familiar with these methods. In this review, we provide experimental chemists and researchers with more computational details and suitable suggestions, and especially hope that this experience may help readers avoid computational pitfalls. Finally, we discuss the use of simplified models to replace original complex structures with a long side chain. The fundamental basis for using models to represent complex chiral compounds, such as in OR calculations, is the existence of conformation pairs with near canceling conformer contributions that justify the use of models rather than the original compounds. Using examples, we here introduce the transition state (TS) calculation, which may benefit readers in this area for use and mastery for their AC study. This review will summarize the general concepts involved in the study of AC determinations.
With the rapid development of high performance computers and computational methods, including software, an increasing number of experimental chemists have tried to use computational methods such as optical rotation (OR, including optical rotatory dispersion (ORD)), electronic circular dichroism (ECD)—also known as CD before vibrational circular dichroism (VCD) was widely used—and magnetic shielding constants—nuclear magnetic resonance (NMR)—to explain and/or assign absolute or relative configurations for various compounds and polymers (fibers) (Kurouski et al., 2014; Marty et al., 2014; Li et al., 2016a; Fernández et al., 2019), including metallic complex (Merten et al., 2014; Li et al., 2019a). Reports of such work in bioactive compounds and natural products chemistry has dramatically increased (Sherer et al., 2014a; Sherer et al., 2014b; Yang et al., 2018), while traditional methods, such as Mosher ester (Hoye et al., 2007; Zheng et al., 2019) and X-rays (Wang et al., 2022)—in which Cu-Kα (1.5418 Å) radiation is preferred to Mo-Kα (0.7107 Å) radiation, due to its stronger anomalous scattering effect (Bijvoet et al., 1951)—are also widely used in absolute configuration (AC) studies. Natural products have also been called the “engines of its development and … links to the domain of biology” in organic chemistry (E. J. Corey, cited in Barton et al., 1999). However, every method has its range of use (María-Magdalena and Jorge, 2015). This can lead to incorrect conclusions by some researchers who are not familiar with such methods. On the other hand, TS calculations are often used in organic reaction mechanism studies. They have been applied in AC studies in order to understand the AC of final reaction product results. This review also discusses the use of simplified models to replace original complex structures in calculations. The fundamental basis for using models to represent complex chiral compounds, such as in OR calculations, is the existence of conformation pairs that can be investigated rather than the original compounds. It is important and necessary to provide experimental chemists and researchers with more computational details and suitable suggestions, especially to avoid pitfalls in their computations.
The first, most important, step is to find the lowest energy conformation for a specific chiral molecule. For example, a 2-butanol should have four stable conformations at different energy that can be found manually. The most stable conformation has a zigzag carbon chain. However, the maximum conformation number is proportional to the bond numbers (n) that can rotate freely (3 n−2, n = 3, 4...). Thus, with the increase in free rotation bonds, the possible conformations increase quickly. Therefore, it is necessary to have software which uses various force fields to investigate all possible conformations and list these geometries according to their relative energy.
Much commercially available software is frequently used, such as ComputeVOA (ComputeVOA, 2022), GaussView16 (GaussView6, 2022; Schrodinger LLC, 2022; RDKIT, 2022), and Merck molecular force field (MMFF94) and its developed MMFF94S (Halgren, 1996; Halgren, 1999). Other force fields or methods can also be used.
Each piece of software has its advantages and limitations. Thus, it is suggested that researchers use at least two software packages in conformational searches. This can ensure that the lowest energy conformations can be found. If one or two important conformations are missed in a procedure, the 13C NMR, OR, ORD, ECD, or VCD simulations may be affected. If the missed conformation is too different from the other conformation’s physical data, this missed conformation may lead to significant error in the simulated 13C NMR, OR, ORD, ECD, or VCD. Comparing this simulated data with the experimental results may lead to an incorrect conclusion.
Although everyone knows the importance of computations of an OR, ECD, and VCD in AC study, the errors can also appear in different situations. To avoid such mistakes, it is important to list reasons for the errors and the methods that can prevent them. As a very typical example, 5-(tert-butyl)benzo[c]quinolino[1,2,3-fg]acridine-3,7-dione (Figure 1A) was used as one example of conformational analysis.
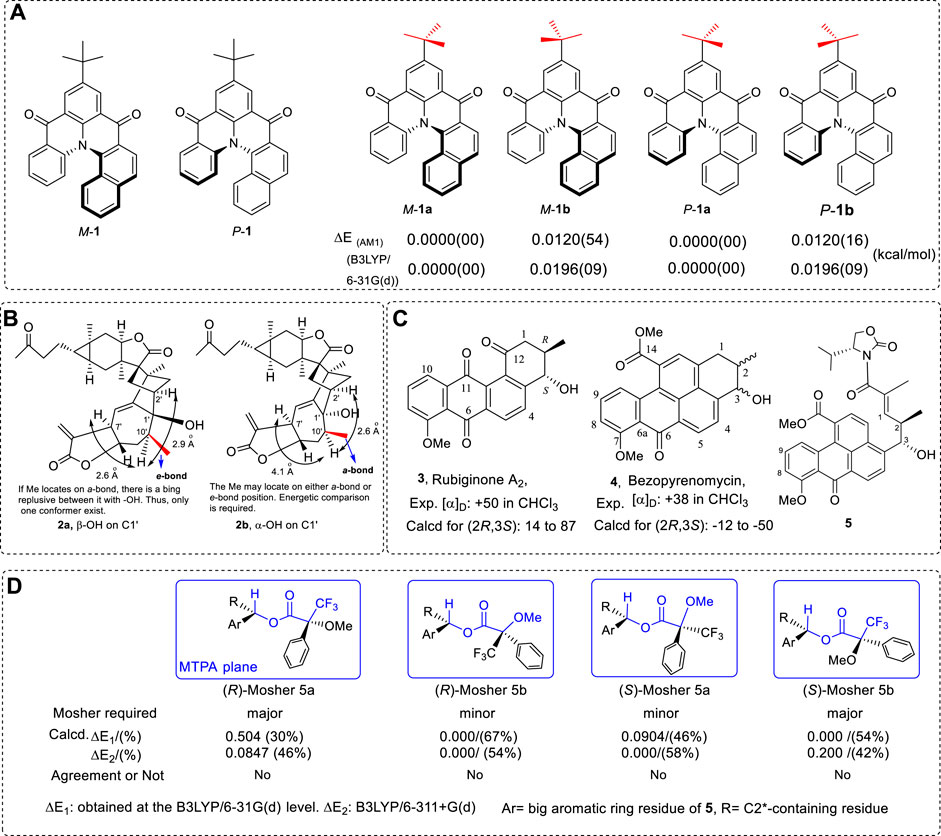
FIGURE 1. Structures of 1–5. (A) The M and P configurations of 5-(tert-butyl)benzo[c]quinolino[1,2,3-fg]acridine-3,7-dione of one and four conformers and their relative energy data using AM1 and B3LYP/6-31G (d) methods. (B) Structure 2 and its conformers reported. (C) Compounds 3 and 4 and the intermediate 5 used for AC of C3 via Mosher ester. (D) The expected major conformer structures required by the Mosher method, the relative energy of the two conformers’ relative energy, and the agreement between the expected conformers and the computed results. The blue frame corresponds to the MTPA plane. The bonds and atoms in blue are located in the MTPA plane.
It has been reported that the relative energy of M-1 is more stable than that of P-1 by about 0.02 kcal/mol (Pandith et al., 2011). This is an incredible result since the structures are enantiomers. Theoretically, they should have the same energy. The reason for reaching this result must be a mistake in the conformational analysis.
In this example, the key substituent is the t-butyl group in calculations. The tertiary butyl group should have two stable conformations in the M and P structures, as illustrated below (Figure 1A). For M-1, the geometry M-1a had lower energy of 0.0196 kcal/mol than that of M-1b at the B3LYP/6-31G(d) level in the gas phase (Zhu, 2015). Once the total mirror-image geometries M-1a and P-1a were used in calculations, they both exhibited almost the same energy, the difference between the two geometries being less than 10–6 Kcal/mol in calculation. To conclude that M-1 has lower energy of 0.012 Kcal/mol than P-1 must have been due to incorrect use of the geometries of M-1a and P-1b (different conformers) from the energy calculations.
In OR, ECD, or VCD calculations, such a small energy difference conformer may be ignored in simulations. However, if it is said that this pair of enantiomers has different energies, then this is an absolutely wrong conclusion. It must strongly indicate that the origin of the chirality does not require any other force, such as chiral catalyst, or other power to form a small excessive enantiomer (%ee) in the prebiotic environment (pool). This is because the 0.012 kcal/mol energy difference is a definite large effect which may bring the %ee value up to of 0.49%ee for the enantiomers. Indeed, the two enantiomers must have the same energy in nature. Professional software is helpful for complex molecular conformational study; however, it is not sufficient to completely find all conformations with relatively low energy.
Another similar example is helicene structures, which consist of non-planar polycyclic aromatic hydrocarbons that form an ortho-condensed aromatic ring. Many helicenes exhibit interesting characteristics, such as their use as organic light-emitting diodes (Riobé et al., 2015; Joly et al., 2016). They also exhibit circularly polarized luminescence (CPL) activities (Goto et al., 2012; Hellou et al., 2017). Thus, this is an important kind of intermediate or chiral material.
The following Example 2 contains a seven-membered ring which may have different geometries (Figure 1B) (Xu et al., 2016a). To assign the configuration of C1′, NOE signals between the H7′ and H10′ via H10′ with H2′ were used. As illustrated in Figure 1B, two conformers were reported. Since the distance of H7′ to H10′ in conformer 2a (β-OH) is shorter than that of H10′ to H2′ in structure 2b (α-OH), the AC of C1′ was assigned as β-OH. This is a good example of using geometries to support an experimental conclusion. This compound has a seven-membered ring. The Me on C10’ may orientate on an e-bond or a-bond. If the energetics of these conformers were investigated and reported, the conclusion should be clearer.
Historically, both rubiginone A2 (3) and benzopyrenomycin (4) have very close OR values, and it was suggested that both have the same AC on C2 and C3 (Figure 1C) (Pezzuto et al., 1976). Benzopyrenomycin (4) was recently synthesized (Oka et al., 1990; Huang et al., 2008). By determining the AC of C3 in intermediate 5 using Mosher methods (Ancheeva et al., 2017), researchers thought that 4 should have (2R,3S) configurations. It seems that the AC of (2R,3S)-4 should be correct. However, it is incorrect, based on the computed OR value and sign. Why does the Mosher method used here give the wrong conclusion?
The reason involves which conformer had the major population in solution. As an empirical method, the Mosher tool is normally valid, and most compounds have the correct conformation distribution in solution. For this specific case 5 (Figure 1C), its conformational searches were performed using three pieces of software and two more MM methods. Thousands of geometries were found for the Mosher ester of 5 using MMFF94 and AMBER force fields and followed by DFT methods (Figure 1D) in order to find the lowest and the second-lowest conformers. The expected major conformers were (R)-Mosher 5a and (S)-Mosher 5b according to the Mosher methods required. However, the major conformers were (R)-Mosher 5b and (S)-Mosher 5a by referring to the MTPA plane. The specific energy data and distributions are listed in Figure 1D. The major conformers that Mosher methods require are not those predicted by computational results. This caused the errors in the use of Mosher methods.
In the example, it is an additionally surprising result that the Mosher method requires that the substituent near the stereogenic center not be a large group, such as the substituent of IMI. Indeed, if the substituent is extremely rigid and close to a linear structure, such as two or two more -C≡C- connected directly, one would need to be careful to use the Mosher method to assign their ACs since this group may change the conformer distribution of the expected major or minor conformers that Mosher methods require in solution.
The three examples are given at the beginning of this review to show the importance of the conformational search not only in AC study but also in other research areas. Indeed, it is very important for chemists in structural study (Perez-Mellor et al., 2019; Wang and Zhu, 2021; Zhu, 2022a), and it is also hard to find some conformers of rigid chiral compounds using software. The reason why some software cannot find the corresponding conformers is very complex. In some cases, some compounds have slightly different conformations exhibiting computed spectra that are almost mirror image (Bringmann et al., 2009; Zajac et al., 2015). Thus, obtaining the conformers with low energy and the correct geometries are very important. We recommend using multiple software to perform the same conformational search for a molecule in order to reduce the possibility of a wrong conclusion. Developing powerful conformational search software is still a big challenge for computational chemists.
Finally, the concept of “most stable conformation” must be mentioned here. There are many stable conformers in a solution. However, only one most-stable conformer exists in solution instead of multiple most-stable conformers—followed by a second most-stable and third most-stable conformation. However, some authors mention “the most stable conformations” during OR, ECD, or VCD calculations. This is in no way acceptable. If it is a racemic chiral compound in solution, then there are only two most-stable enantiomers. As an example, a general statement is listed here: 38 conformations were found with low energy from 0–3.5 kcal/mol using a MMFF94 force field. These geometries were then used in further optimizations at the B3LYP/6–31+G(d) level (in the gas phase), obtaining one most-stable conformer. To posit “the most stable conformations” is wrong; however, this statement is frequently found in reports.
NMR calculations involve the position of all atoms in a space and the chemical environment. Many atoms’ chemical shifts can be computed, such as 1H (Wang et al., 2003), 3He (Ramalho and Buehl, 2005), and 15N (ComputeVOA, 2022). The study of 13C NMR is more attractive for organic chemists—the number methods developed for 13C NMR computations is more than the number of NMR methods for other atoms. The gauge including the atomic orbitals (GIAO) model has been used widely.
Firstly, it must be clear that 13C NMR can be used to assign the relative configuration of a chiral molecule having two or two more stereogenic centers. If one chiral compound has a known stereogenic center, it is possible to use 13NMR to assign its AC by comparing the other stereogenic center(s) with the known center. These computed 13C NMR can be compared with the experimental data to assign the configurations (Lauro and Bifulco, 2020). The use of 13C NMR to study the configuration of chiral organic compounds is effective in stereochemistry study (Bifulco et al., 2007; Lodewyk et al., 2012; Gussem et al., 2014; Lauro and Bifulco, 2020).
The maximum principle must be applied to the examination of structures. If the coefficients between two structures are small enough, they cannot be used as evidence for judging structural evaluation. In some cases, more evidence is needed for further confirmation of structures.
The originally computed 13C NMR data may require treatment for further uses (Forsyth and Sebag, 1997; Barone et al., 2002; Liu et al., 2006; Yang et al., 2011; Cao et al., 2020a; Li et al., 2022). For example, after linear corrections (Forsyth and Sebag, 1997), there are two methods for judging whether the structure is correct. The first is to use to maximum of Δδ values. If the maximum of Δδ is over 8.0 ppm, the structure is not reliable; if it is less than 8.0 ppm, then the structure is located in the reliable structure range. However, more evidence may be required for further confirmation of its configuration, such as comparing coefficients of two structures: the bigger the coefficient, the more reliable the structure will be (Yang et al., 2011).
For example, chiral compound 6 was originally obtained and assigned as either 6a or 6b since no interaction signal was obtained between the protons H5 and H8 in ROESY spectra (Figure 2A) (Liu et al., 2006). The 13C NMR was investigated. After conformational searches were performed using the MMFF94 force field, all six conformations were used for the Boltzmann sum.
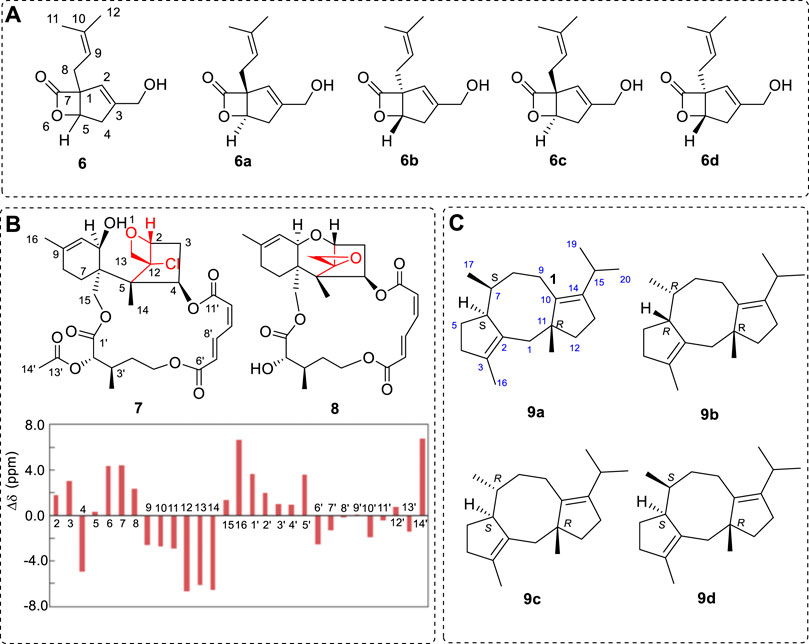
FIGURE 2. (A) Reported compound 6 and its four possible structures (6a to 6d). Compounds 6a and 6d are one pair of enantiomers, and 6b and 6c are the other pair enantiomers. (B) Structures 7, 8, and the 13C NMR differences between the predicted 7 with the experimental chemical shifts. (C) The four diastereomers of 9a to 9d.
After conformational searches were performed using a MMFF94 force field and optimizations for the geometries using DFT methods, six conformations were used in 13C NMR calculations. After linear corrections (Forsyth and Sebag, 1997), the coefficients were 0.9947 and 0.9940 (Liu et al., 2006). Other methods could be used to assign the AC (Cao et al., 2020a; Li et al., 2022). It appears that 6a should be the preferred structure. However, the maximum of the chemical shift errors between C2, C8, and the experiments were −9.1 −10.2, and −11.2 −11.4 ppm in 6a and 6b, respectively. Thus, both 6a and 6b are not the correct structures. The other two cis-structures (6c and 6d) have bigger coefficients (0.99932 and 0.99935). All the shift errors were smaller than 8.0 ppm. In this case, if it is regarded as 6d (coefficient was 0.99935), it is still not correct. The OR was then computed. The experimental OR was −135, and the predicted OR for 6c was −127o at the B3LYP/6–311++G(2d,p) level; compound 6d was +127o. Therefore, the correct structure is 6c after its enantiomer 6b was excluded at the beginning. The structure 6c was synthesized and the synthetic 6c had the OR value of −129 o (Zhou and Snider, 2008a; Zhou and Snider, 2008b).
Other methods are used in the assignment of the chiral compounds. For example, the mean absolute error (MAE), corrected mean absolute error (CMAE), and the root-mean square deviation (RMSD) (Smith and Goodman, 2010; Grimblat et al., 2015) were applied for the study, including DP4 and DP4+. The concepts used here, for the ith nulcei, has a chemical shift error of Δδi = δ i cald-δ i exp. MAE = ∑(Δδi)/n. CMAE = ∑(Δδiscaled)/n. Here, n is the total number of nuclei, and δiscaled can be computed using a linear fit of the calculated (δi calcd) versus experimental (δi exp) chemical shifts for calculating the related Δδiscal values. For more details, see Baassou et al. (1983), Li et al. (2022), and Cao et al. (2020a), who also compared the experimental shifts with the predicted values in the study. Consequently, comparing the computed 13C NMR is quite valid for assigning chiral compounds’ relative configurations, including some AC assignments (Lauro and Bifulco, 2020).
If one compound’s configuration is known, it can be used to determine the AC of another moiety’s structures for a compound. This is more conveniently used for organic synthetic compound study. For example, compound 7 is derived from natural product 8 by reaction with 2-chloroacetyl chloride (Jia et al., 2018). This three-membered oxirane structure disappeared and a new four-membered oxetane moiety formed while the six-membered ring in 8 decomposed in the reaction. Thus, it had a big difference in 13C NMR from those of 8. This example is a strong hint that, during the modification of a natural product, even the reaction condition is mild and the reagent may be frequently used, such as where example 8 was reacted with 2-Cl-acetyl chloride, leading the three-membered-ring opened (red moiety) to 7. Use of the 13C NMR can easily identify the structure of 7. In this case, the whole aliphatic carbon chemical shift difference is less than 8.0 ppm, within a reasonable error range (Figure 2B).
After their development (Goodman Group, 2017), of DP4 methods have been applied to the AC determination of many chiral compounds by computing the possibility of assigning their structures, such as fusicoca-2,10(14)diene (9) (Merten et al., 2017). It has three other diastereomers besides 9a (6S,7S,11R): 9b (6R,7R,11R), 9c (6S,7R,11R), and 9d (6R,7S,11R) (Figure 2C).
After comprehensive analysis, diasteromer 9a is preferred. As the authors indicated, “The DP4 analysis should not be used as a black-box method by simply evaluating the calculated and experimental 13C NMR shifts in the order of appearance (e.g., lowest calculated with lowest experimental shift, and so on). In case of FCdiene, neglect of a correct connectivity-based assignment leads to different results for both methods, as shown in Merten et al. (2017): Using the unassigned 13C chemical shifts in a DP4 analysis, diastereomer 9b becomes favored over diastereomer 9a, with a probability of 94.9%. It is therefore important to point out the importance to include the connectivity information obtained from 2D NMR spectroscopy in the assignment of the experimental to the calculated 13C NMR shifts in order to ensure a reliable, unambiguous assignment of a relative configuration”.
In addition, comparing the calculated 13C NMR with the experimental results determines that the organic compounds are not only used in AC but also applied for planar structure determinations since 13C NMR calculations involve the position of all atoms in space and the chemical environment. In some cases, it can be used to determine the cis or trans structure for a compound with C=C bonds. For example, cyclopeptide anthranilic acid (10) was assigned as a cis structure by comparing its 13C NMR with the calculated data (Figure 3A) (Zheng et al., 2009). In the first case (Z), all of the maximum relative errors of chemical shift were below 8.0 ppm. However, in the second case (E-), the maximum errors at C-21 were 8.9 ppm. Thus, it was assigned as (Z)-10a. As another method, ECD, which will be referred to later, it is also a good way to assign the E/Z structures (Rode and Frelek, 2017).
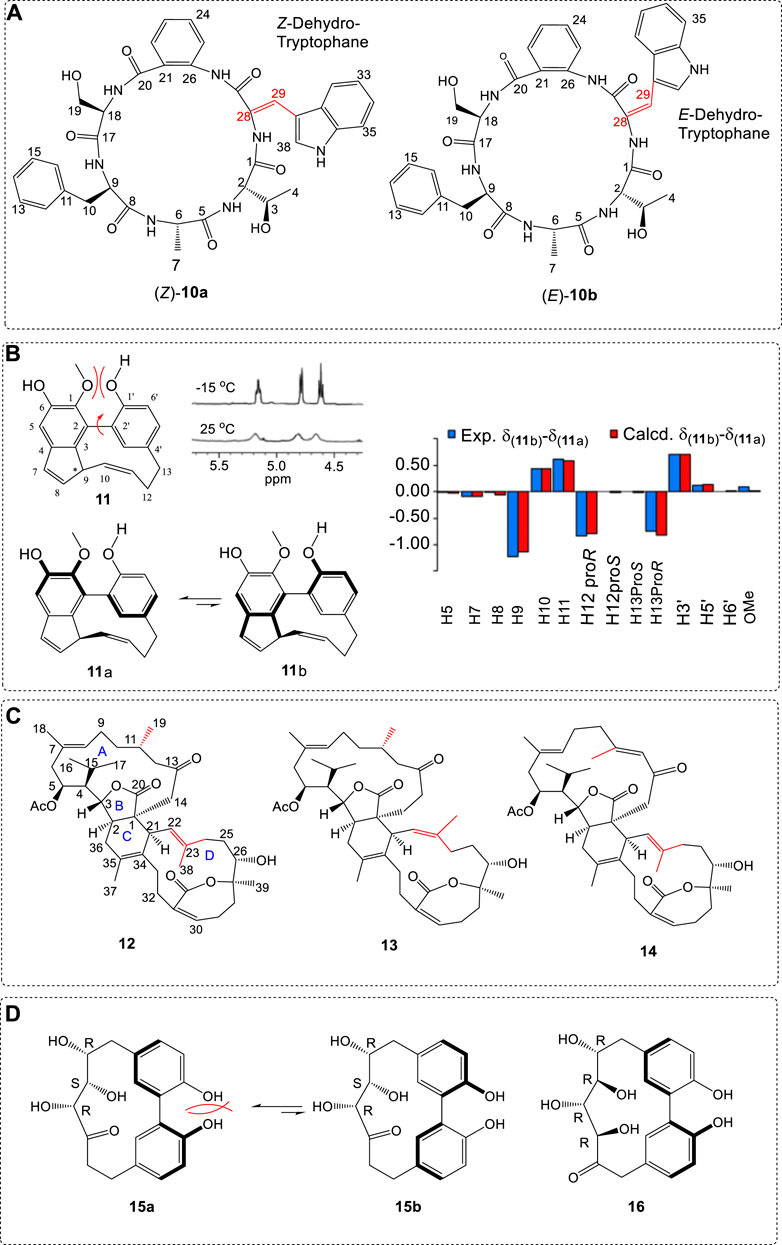
FIGURE 3. (A) (Z) and (E)-structures of cyclopeptide anthranilic acid (10). (B) Structure of zosteraphenol A (11) and its two rotamers (11a and 11b) in equilibrium, together with the experimental and DFT-calculated differences of 1H NMR between chemical shifts of the minor (11b) and the major (11a) rotamers. All shielding and de-shielding effects shown are reproduced within 0.08 ppm. (C) The structures of 12 to 14 used in NMR calculations. (D) Structures 15 and 16 and the two conformers 15a and 15b of atropisomer 15.
By comparing the relative chemical shifts of 1H and/or 13C NMR, valid information in the chiral compound’s configuration assignments can be provided. An example is zosteraphenol A (11) from the seagrass Zostera marina (Grauso et al., 2020). One minor conformer (11b) and major (11a) rotamer were at equilibrium in solution due to the repulsion between the -MeO in ring A and the -OH in ring B, the rotation of the single bond C2-C2' being restricted. This barrier is big enough at room temperature to block the single-bond rotation and leads the exchange signals at 4.65. 4.80 and 5.20 ppm to appear as a broad peak. Once the temperature decreased to 258 K (-15 °C), the signal became very sharp (Yu et al., 2015). After computing its rotamers, 13C NMR at the chemical shifts were subsequently calculated at the PBE0/6–311+G(2d,p) level using B3LYP/6–31+G(d,p)-optimized geometries via the PCM model in chloroform (Figure 3B). The computational results supported the hypothesized conformational equilibrium for 11.
Because the shift of nuclei is sensitive to their position in the molecule, we can compare the differences of the same nuclei in different diastereoisomers. This is very useful when some epimers were obtained. In this case, structural assignments could be made by comparing the experimental shift difference between two epimers with the experimental shift differences between the two diastereoisomers. For example, glaucumolides B (12) and bistrochelides A (13) were a pair of structurally similar epimers (Figure 3C) (Sun et al., 2019). MAEs between the investigated diastereoisomers were used to assign the relative configuration of C11 by comparing the error pattern and referring the X-ray crystalline of key compound 14. The AC of glaucumolide B (12) was assigned as (1S,2R,3R,4R,5S,21S,26S,27R) in the assistance of ECD spectra comparison of experimental and theoretical results.
Similarly, the relative configuration of streroisomers 15 and 16 can also be assigned by comparing the experimental and theoretical 13C/1H NMR spectra (Figure 3D) (Cerulli et al., 2017). Eight isomers for 15 and 16 were used in the computations. After the 13C NMR shifts were obtained by a Boltzmann statistics sum, the DP4+ method was applied for comparison. Their relative configurations were assigned by a combined quantum mechanical/NMR approach, comparing the experimental 13C/1 H NMR chemical shift data and the related predicted values. It was found that they are atropisomers due to the restricted single-bond rotation where the resistance came from the repulsion of the two hydroxyl groups on the phenyl rings. The predicted transition state (TS) barrier between 15a and 15b was ΔG⧧ = 20.7 kcal/mol. This barrier may be high enough to block its rotation r = freely at room temperature.
There is another method for assigning the relative configurations for diastereoisomers by using the chemical shift error patterns of the experimental and computed 13C NMR data. For example, there are two stereoisomers 17 and 18 from Parepigynum funingense (Hua et al., 2004); their configurations were assigned by comparing the theoretical 13C NMR with the experimental data using the current configurations in Figure 4A (Hua et al., 2007). Only C20 had a different configuration. In this case, if we listed the shift differences from top to bottom, it could be seen that the sign and shift difference values change pattern almost similarly (Supplementary Table S2). Therefore, the real relative configurations—for example, 17A via 18A—should have the same relative configurations with those that were used in 13C NMR computations of 17A and 18A. If the sign is reversed, the real relative configurations of 17A and 18A should be the configurations of 18A and 17A that used in the 13C NMR computations. This can be re-drawn as shift difference pattern plot as illustrated in Figure 4A. If the error pattern was used, it would be found almost the same between the linear fit corrections. Thus, in this case, using the error pattern is relatively convenient. Importantly, if the HF/6-31G(d)//HF/6-31G (d) was used in the computations, the shift error differences would almost be the same between the experimental and computational results. This is a cheap method that can be applied to this study.
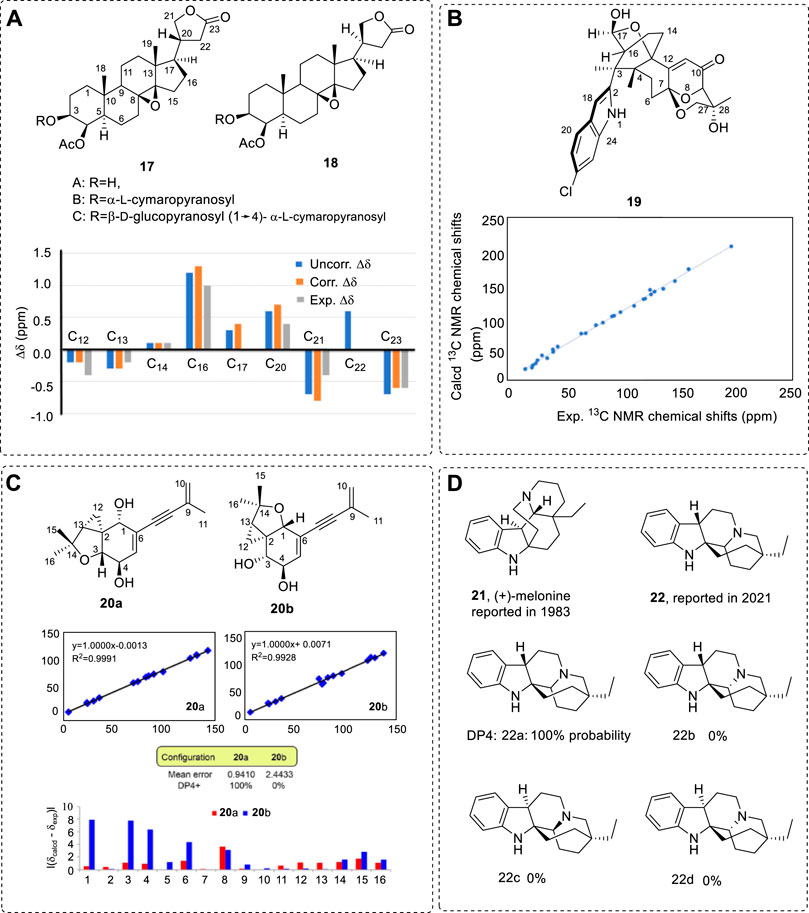
FIGURE 4. (A) Six diastereoisomers found from a plant and the shift difference pattern plot re-drawn for this review (based on the data in Supplementary Table S2). Only data from Method D and the experimental shift difference between 17A and 18A were used (in Column A of Supplementary Table S2). (B) Structure of 19 and its linear fit of the experimental chemical shifts vs. the computed shifts of 13C NMR. (C) Structures of 20 and three methods used in 13C NMR data treatments and the corresponding structure’s reliability. (D) Revised structure 22 from 21 using the DP4 method, and DP4 probability scores obtained from the 13C NMR data of the different candidate diastereoisomers (22a to 22d) of melonine.
Note that a deeper truth that can be found in the compounds. The different stereogenic center was C20; however, the shift difference of C20 was only 0.4 ppm between the 17 and 18 in their corresponding A, B, and C compounds. The largest shift difference was 1.0 ppm at C16. This is a typical example, and one must be very careful in handling experimental data. Generally, the different chiral center may have small to medium chemical shifts among the neighboring carbon atoms in the diastereoisomers.
A novel indole-diterpenoid 19 was obtained and its relative configuration was assigned based on NOESY spectra, with assistance provided by comparing the experimental 13C NMR with that computed (Figure 4B) (Zhou et al., 2021). Its coefficient was 0.9968 after the linear correlations. In this case, its AC can be determined using ECD.
One more example shows that several methods can reach the same conclusion if the structure is reliable. For example, the following two possible structures were identified using the method mentioned above. Diisoprenyl-cyclohexene/ane-type meroterpenoids are obtained from Biscogniauxia sp. 71-10-1-1 w. Compound 20 may have either a 20a or 20b structure (Figure 4C). As a typical example, it affords us a general viewpoint: cross-testing using several methods can provide more confident conclusions, and, if the structure is correct, these methods must give the same conclusion.
Finally, a structure revision example is illustrated here using DP4 and other assistance. The original structure melonine (+)-21 was reported early (Baassou et al., 1983). After nearly 40 years, it was corrected as (2R,3S,7S,20R)-22 using NMR data (Figure 4D) (Kouamé et al., 2021). This is easily understood since the structure elucidation mostly depended on 1D NMR data from 1983. This structure correction also led to its different biogenetic pathway.
Specific optical rotation, which has the longest history in AC study, has played an important role in AC research. Even in today, it is still a useful method. Along with progress in electronic circular dichroism (ECD), vibrational circular dichroism (VCD), and Ramaon optical activity (ROA)—including some derivative technologies like magnetic circular dichroism (MCD)—these modern technologies have become increasingly important tools in stereochemistry. As a valid method, OR has been widely used as a necessary datum in many reports of chiral compounds.
Specific optical rotation ([α]D) is simply called “optical rotation” (OR) in this text. Different OR values measured at different wavelengths are termed “optical rotatory dispersion” (ORD). In the absence of an ORD spectrometer, OR at several wavelengths is used in ORD study—633, 589, 546, 436, and 365 nm. Quantum method theories are widely applied for AC study (Grauso et al., 2019). Some typical chiral molecules were used in the study (Egidi et al., 2019). As one of the simplest and cheapest tools in AC determination, calculating chiral molecular OR and then comparing it with the experimental value is a valid and effective method for OR values that are generally not less than 20 degrees (Raghavan and Polavarapu, 2017). OR and ORD have been widely used in the study of AC (Reinscheid and Reinscheid, 2016; Saito and Schreiner, 2020). For example, the chiral compounds 23–25 were assigned using OR methods; they are listed in Figure 5A (Li et al., 2011a; Li et al., 2011b; Ding et al., 2016).
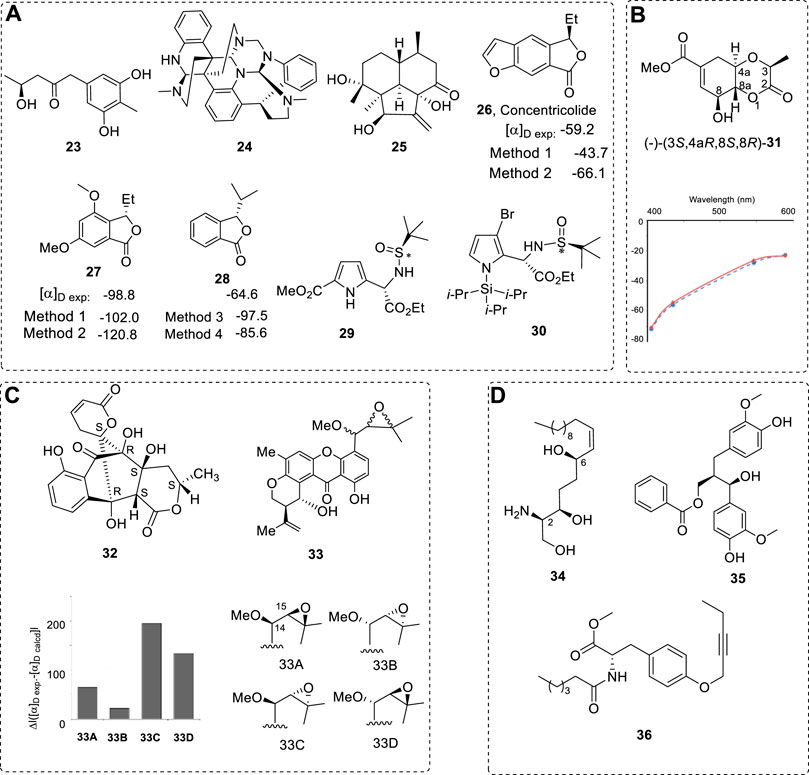
FIGURE 5. (A) Compounds 23–30 and partial experimental and computed OR values for 26–28 using different methods. (B) Structure of compound 31 and the calculated ORD of 31 with (3S,4aR,8S,8R) configuration (●, dashed blue line) and experimental ORD for (−)-31 (⧫, solid red line). (C) Structures of 32 and 33, and the OR differences of four isomers of 33A–33D between their experimental and calculated OR values. (D) Compounds 34–36.
Compound 27 was obtained because natural concentricolide (26) has an anti-HIV property; it was assigned as (R) configuration with a −59.2 of OR (Qin et al., 2006). Quantum methods were used in the AC study (Ren et al., 2009). Method 1: B3LYP/aug-cc-pVDZ//B3LYP/6-31+G(d), and Method 2: B3LYP/aug-cc-pVDZ//MP2/6-311+G(d) were used for OR computations of (S)-26. Both methods predicted −43.7 and −66.1. Thus, the configuration of 26 must be (S). The other compound 28 with OR value of −64.6 (Li and Si, 2012) provided other evidence to support this conclusion (Method 3: (B3LYP/6-31+G(d,p)/PCM//B3LYP/6-31+G(d,p) and Method 4 PCM/B3LYP/6-31++G(d,p)//B3LYP/6-1+G(d,p)) (Figure 5A). Other kinds of chiral compounds, such as sulfone (29) or Si-, Br-atom-containing compound (30) could be used in OR computations using current theoretical methods (Figure 5A) (Haghdani et al., 2016). The interesting chirality of >S=O in 29 and 30 have been well investigated. Like CAM-B3LYP functional and the second-order approximate coupled cluster singles and doubles (CC2) method. Currently, OR values for any chiral compounds that contain most elements of the periodic table could be computed using quantum methods.
Firstly, it should be clear that the assigned AC concluded from OR must be the same as the other quantum theories or experimental results. The bioactive metabolite phyllostin (31) was isolated from Phyllosticta cirsii fungus (Evidente et al., 2008); its relative configuration was established as (3R,4aS,8R,8aS) by NMR and X-ray analyses (Tuzi et al., 2010) while its AC remained unknown. Total synthesis (Muralidharam et al., 1990) and the ECD exciton chirality method (Isogai et al., 1985; Mu et al., 2021) permitted the determination of the AC of its C8 epimer. The ORD study gave the same conclusion: (-)-(3S,4aR,8S,8R)-31 (Figure 5B) (Mazzeo et al., 2013).
It is simple to assign one compound’s AC if one chiral compound is relatively rigid and its relative configuration is known. It is only necessary to compute its OR directly and then compare its OR value with the experimental results to assign its AC. Compound 32 was obtained with a known relative configuration via X-ray (Mo radiation) (Ding et al., 2010); its OR was +57.4 in MeOH. The computed OR for (R)-32 was +74.3 in the gas phase and +66.2 in methanol using the PCM model (Figure 5C). This means the (+)-32 must have the configuration as illustrated in Figure 5C. Indeed, ideal situations are rare.
In some cases, there are several diastereoisomers—for example, 33 (Figure 5C) (Figueroa et al., 2009). Its side-chain configurations with ethylene oxide moiety were not assigned. By comparing the Δ[α]D values among the four diastereoisomers, isomer 33B with the smallest Δ[α]D value was assigned as the most likely structure.
Many chiral compounds may have flexible structures and their AC assignment is not easy due to many conformations in solution. For example, with chiral compound halisphingosine A (34) (Molinski et al., 2013), a very flexible linear chiral compound with three stereogenic centers, it is hard to compute its OR to assign its AC. A simpler model is needed—to be discussed latter—or making its derivatives for ECD exciton chirality spectra. Compounds (7R,8R)-35 (Zhang et al., 2021a) and 36 (Zaman et al., 2021) are not as flexible as those of 34 (Figure 5D); they can be used directly in computations of OR or ECD.
It has been reported that the structurally similar compounds rubiginone A2 (37) and benzopyrenomycin (38) have close OR values; 37 had an OR of +50 and 38 had +38 in CHCl3 (Figure 6). However, the computed OR values were 14 to 87 for 37 and −12 to −50 for 38, using different quantum methods (Li et al., 2013). Therefore, 37 had the (2R,3S) configuration and 38 must have (2S,3R) configurations. Why is there such a big difference from the viewpoint of structural features? The key point is that these two molecules formed different helical chirality, as illustrated below (Figure 6). When both had (2R,3S) configuration, their conformation analyses were performed. The minor conformers S-37a had an OR value of −240.8 and major conformer M-37b had 211.6. After the Boltzmann statistics, the OR value depended on the major conformer M-37b′s OR sign. In contrast, the major conformer S-38a had −605 and the minor M-38b had 622.8. Finally, the simulated OR value and sign were decided by the major conformer S-38a. Thus, if both had (2R,3S) configuration, they should have a reversed OR sign. Consequently, when both had the same OR sign, they must have different ACs.
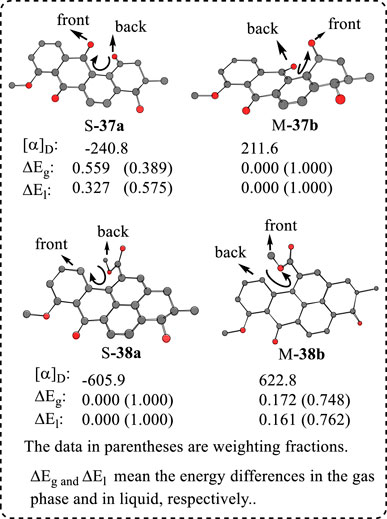
FIGURE 6. Helical structures [(S) and (M)] for rubiginone A2 (37) and benzopyrenomycin (38) when 38 had (2R,3S) configuration (H atoms were hidden for clarity).
This example provided an important hint: when two structures are very similar, the real conformer structures must be carefully examined before identifying their AC using their OR values. Helical structures exist widely and, importantly, generally have large OR values. This example again indicates that conformational study is very important, as we have emphasized previously.
ORD provided more information in other wavelengths for various structures. Since different structures have different sensitivity at different wavelengths, different structures may have obvious and different ORD curves. The AC of C8 of jonquailine (39) was assigned as (R) (Vergura et al., 2018) based on the computations of ORD at the level of B3LYP/aug-cc-pVDZ//B3LYP/TZVP level in the gas phase. The (8S)-39 had a large difference in ORD from the (8R)-39 and the experimental ORD (Figure 7). The ORD values of 39 in methanol were larger than those in chloroform.
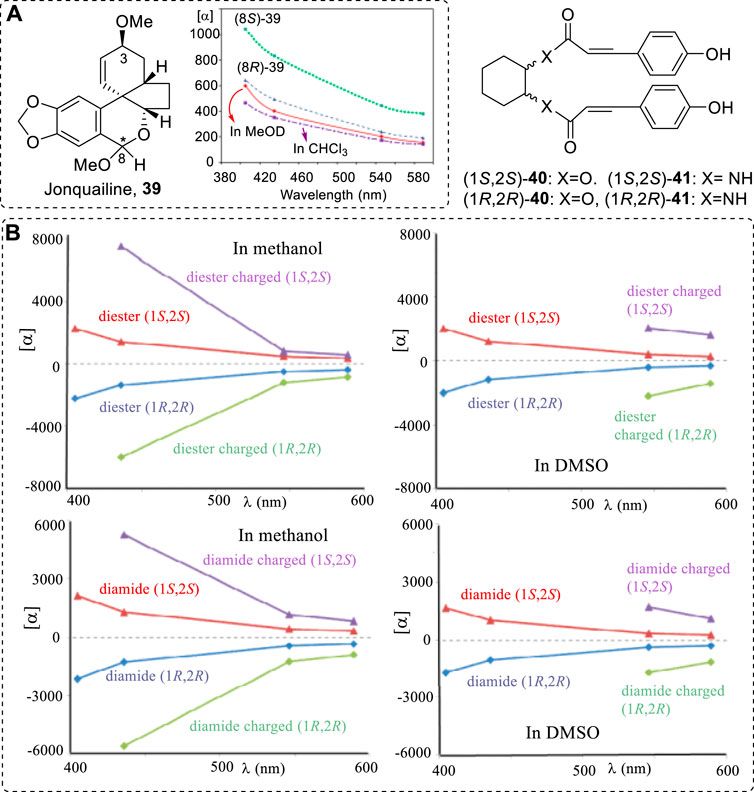
FIGURE 7. (A) Experimental ORD of 39 (methanol, solid red curve, chloroform, dash-dotted purple curve) and computed spectra (gas phase) for (8S)-39 (dotted green curve) and (8R)-39 (dashed blue curve). (B) Experimental ORD curves of neutral and charged diesters of (1S,2S)- and (1R,2R)-40 (up panels) and (1R,2R) and (1S,2S)-41 (down panels) in MeOH and DMSO, respectively.
Because, for some chiral phenol compounds, the -OH is acidic, its ORD or other properties are dependent on the pH values of the solution. In this case, different solvents may have varying effects on the OR. For example, different ORD curves were recorded for the diesters of (1R,2R)- and (1S,2S)-cyclohexanediols and diamides of (1R,2R)- and (1S,2S)-diaminocyclohexane (40 and 41) with p-hydroxycinnamic acid (Figure 7) (Mazzeo et al., 2016). This is due to the equilibrium under different pH values (Eq. 1). The compounds also exhibited different chiroptical spectroscopies in solution.
In addition, the ORD of any chiral compounds, such as the reaction atropisomer 45 from 42, could be also used in the AC assignment (Figure 8A) (Mirco et al., 2019). The enantiomers (+)-45 and (-)-45 exhibited a very good mirror image as expected. The (S)-45 was used in the ORD study.
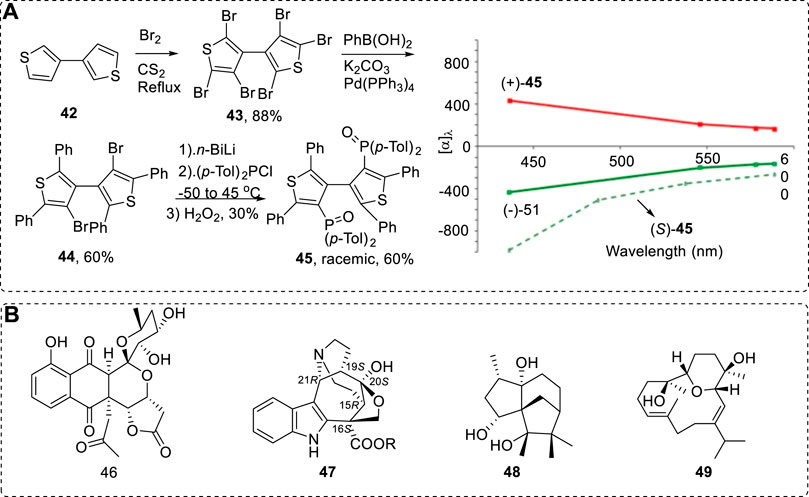
FIGURE 8. (A) Left is the reaction scheme for the synthesis of racemic; right is the experimental and calculated (dashed lines) ORD spectra of enantiomers (45) in chloroform. (B) Structures of compounds 46–49.
Finally, an important phenomenon should be noted. Many linear chiral compounds have only very small OR values (Lu et al., 2020). In these cases, it is common to use quantum methods to compute OR values to assign AC unless there is a so-called known analogue’s OR (theoretical and experimental OR values used as reference). For more about this study, please see the simplified model section. The OR analysis of four additional chiral compounds (46–49) are summarized below (Figure 8B) (Taniguchi et al., 2009; Ding et al., 2012; Yang et al., 2013; Sun et al., 2021a).
A notable case is the computed OR value being generally larger than the experimental datum. In some cases, the computed OR is two or more times larger than the experimental OR values using current DFT methods. Therefore, it is better to refer to other methods in AC determinations for complex chiral compounds, particularly if any of the OR values are small, such as less than 20°.
OR has been widely used for a long time and it will continue to be, as has the ORD method. However, its application for AC study is not as frequent as OR or ECD.
Electronic circular dichroism (ECD) spectra have been used for AC determination since the 1960s. ECD was originally known more simply as CD, and, more recently, many reports use the term ECD for AC assignment (Guo et al., 2020; Zhang et al., 2021b; Kim et al., 2021; Nhoek et al., 2021; Quan et al., 2021). As mentioned above, a detailed conformational search should first be performed (Mazzeo et al., 2014). Many known chiral compounds have been used this way in chiroptical spectroscopic study (Molteni et al., 2015; Stepanek and Bour, 2015; Pescitelli and Bruhn, 2016; Rossi et al., 2016; Johnson et al., 2018) while new synthetic chiral compounds have also been studied (Paolino et al., 2021; Yajima et al., 2021). Chiral compounds require one or two UV-vis chromophores in order to use ECD in their configuration assignments.
The DFT theory has been widely used in ECD computations for various chiral organic compounds (Polavarapu and Covington, 2014; Jiang et al., 2021a; Ji et al., 2021; Niu et al., 2021), including metallic chiral compounds (Enamullah et al., 2016; Saha et al., 2017; Ravutsov et al., 2021). This method provides not only a good balance in treating different molecular systems but also gives relatively accurate computational results when applied to real molecules. For example, using the functional B3LYP at the 6-311+G(d) set level basis can obtain relatively good agreement between theoretical prediction and experimental results. An improved theory, such as cam-DFT (Yanai et al., 2004), is also used in ECD computation. Other methods, like simplified TD-DFT, may give fast and accurate assignment for some chiral compounds (Cerra et al., 2019; Martynov et al., 2019; Ianni et al., 2022). For some chiral molecules, this method can provide more accurate results, although this varies depending on different molecules. A general mixed quantum/classical method for the calculation of the vibronic ECD has been reported as valid tests (Cerezo et al., 2018). Compared to DFT calculations, its computational cost is relatively larger, but it is useful in cases where the ECD shape arises from a subtle balance between vibronic effects and conformational varieties. Indeed, as an active research area, many developed methods have been reported (Liu et al., 2016a; Hodecker et al., 2016; Hong et al., 2016; Nørby et al., 2017; Jimenez et al., 2019; Scott et al., 2021; Wibowo et al., 2021), including the efficient simplified time-dependent density functional theory (sTD-DFT) that has been used to compute large molecular systems with more than 100 atoms (María-Magdalena and Jorge, 2015) such as helicene (Bannwarth et al., 2016), which consists of 102 atom and 16 annulated benzene rings. Simulations of ECD for nucleic acids have also effectively used quantum theory (Zhu et al., 2014; Padula et al., 2016a; Zou et al., 2021).
DFT methods should first be used and, if the predicted ECD curves are greatly different from the experimental results after UV correction, cam-DFT, such as cam-B3LYP theory, can be used in computations to compare both measured and predicted ECD spectra.
DFT is an efficient tool used in AC determinations. Some typical compounds and their ECD spectra are summarized below, such as an unprecedented C9-spiro-fused 7,8-seco-ent-abietane, decandrinin (50) (Figure 9A) (Wang et al., 2014a). The ECD signals had almost no red or blue shifts. The ECD of organic salts, such as clavulanate potassium, can also be calculated using DFT method shift (Li et al., 2018). The structure of some chiral compounds, such as anacolosins A (51), contain some isolated C=C or C=O groups, and ECD signals mostly appear from 200–250 nm (Cai et al., 2019). The best evidence is its match of the predicted VCD with experimental results.
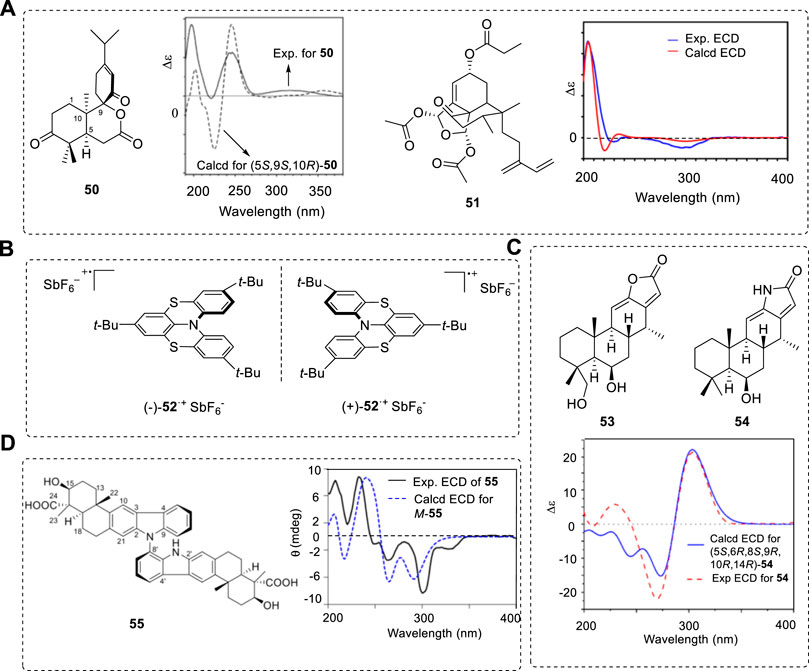
FIGURE 9. (A) Structures of 50 and 51 and their ECD spectra. (B) Chemical structures of investigated dithia-bridged hetero[4]helicene radical cations (−)-52+SbF6− and (+)-52+SbF6−. (C) Structures 53 and 54 and experimental and theoretical ECD spectra. (D) Dimeric form of carbolines (55) and its ECD obtained at the CAM-B3LYP/6-31G(d)//B97D/TZVP.
In ECD study generally, chiral molecules have one or more vis-UV chromophores in the UV wavelength range of 200–400 nm for organic compounds. The short wavelength range, such as 180–210 nm, can also provide valid information for aliphatic compounds, such as terpenoids, peptides, or proteins (Farkas et al., 2016; Banerjee and Sheet, 2017). However, signals of 180–210 nm are more difficult to measure without distortions. Most ECD study involves the range of 210–400 nm for organic compounds (Li et al., 2014; Nicu et al., 2014; Podlech et al., 2014; Yu, 2014; Han et al., 2021; Omar et al., 2021; Saetang et al., 2021). The ECD wavelength range for chiral metallic ion-containing organic compounds or poly-conjugated compounds may cover 500 nm or more (Wu et al., 2014; Saleh et al., 2015; Enamullah et al., 2016; Vesga and Hernandez, 2016).
One configurationally stable chiral dithia-bridged hetero (Marty et al., 2014) helicene radical cation (52) was studied (Figure 9B) (Gliemann et al., 2017) involving nitrogen N-chirality. This molecule had a bulky negative ion SbF6, which is located close to the N atom to fix its conformation. This provides stability to the N chirality formation—natural compounds containing N-cation in natural products are not rare.
Some novel types of chiral compounds were recently obtained from natural sources. Anti-inflammatory cassane-type diterpenoids from the seed kernels of Caesalpinia sinensis have a novel framework formed with the new ring D. This rigid framework (+)-53 was confirmed by X-ray, and its analogues (+)-54 must have the same AC; this has been confirmed by comparing the computed ECD for (5S,6R,8R,9S,10R,14S)-54 with the experimental ECD of (+)-54 as (5S,6R,8R,9S,10R,14S) (Figure 9C) (Wang et al., 2021a) The ACs confirmed by X-ray structure and ECD spectra are the same.
The dimeric form of axial carboline compound 55 has been isolated and identified using ECD (CAM-B3LYP/6-31G(d)//B97D/TZVP) (Jin et al., 2021), which was obtained from the culture broth of a cold-seep derived actinomycete, Streptomyces olivaceus OUCLQ19-3 (Figure 9D). Its other axial diastereisomer was also obtained and the predicted ECD spectrum agreed well with the experimental result. The N-C single bond rotation was seriously restricted; if the rotation barrier was provided there, we could more clearly understand the atropisomer’s properties of NMR in solution.
Finally, we consider the standard deviation (σ) factor of the data. Among the various factors that affect the AC determinations, one problem that has been ignored in the teaching process is the data of σ. The wrong assignment of AC was caused by a wrong use of the sigma. Thus, this factor must be used carefully by beginners to this method.
The data of σ could be 0.2 to 0.5 ev, but 0.3 and/or 0.4 ev has mostly been used in ECD simulations. The general principle of using σ depends on the ECD signals recorded in experiments. Unless the experimental ECD resolution is very high, 0.3 or 0.4 ev can be used in most cases. Once the signal resolution is very high, where the signal’s half-height is small, 0.2 or 0.25 ev can be used to simulate the ECD. In contrast, 0.5 ev may be used in simulation of ECD spectra. Generally, the smaller the σ value, the sharper the signals of ECD will be and the resolution of the ECD will be high.
However, when the ECD signals are complex, it is not easy to select their σ values. For example, one wrong assignment of chiral compound 56 was reported (Ji et al., 2013). The conformational search for all geometries was performed correctly, and the excitation state numbers were covered from 200 nm to 400 nm in the calculations. Its ECD simulation was performed at the B3LYP/6-31G(d) level. The wrong σ value of 0.2 ev was used. After its analogues A–C were obtained, the ACs were assigned again. The original AC of 56 was corrected to 57 by using 0.35 ev (Figure 10A) (Li et al., 2016b). Obviously, the small σ value of 0.2 ev in the report led to the wrong assignment of the compound 56.
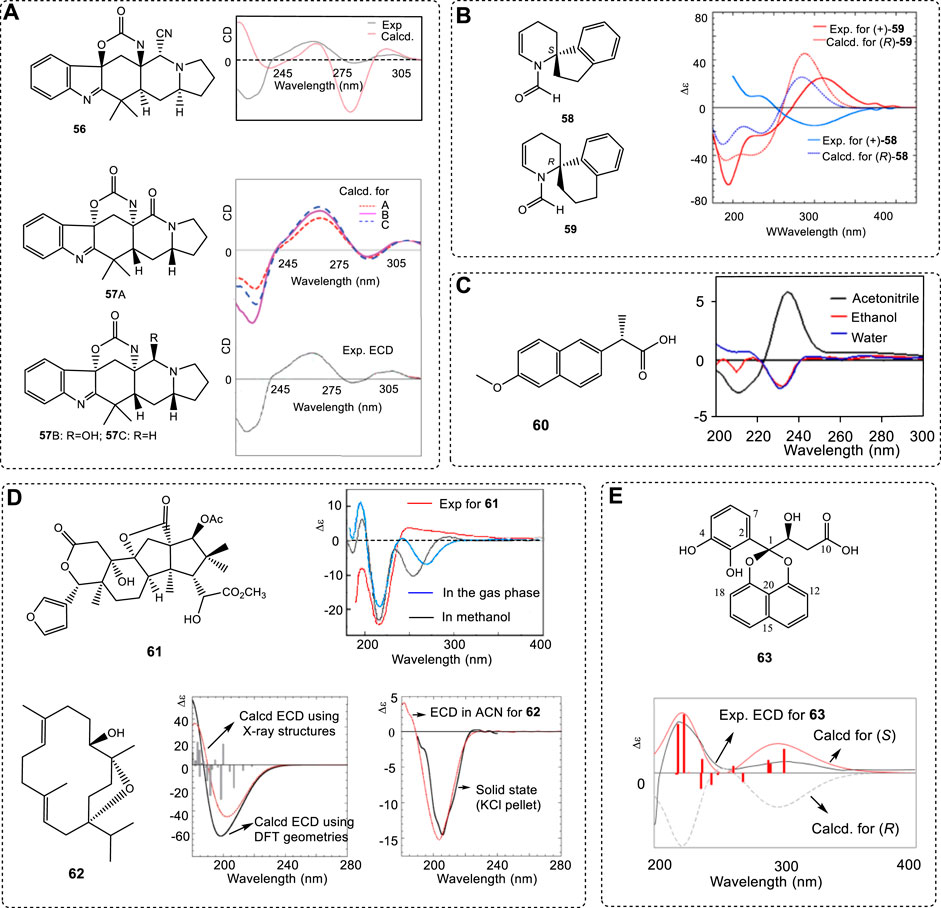
FIGURE 10. (A) Structures of 56 and 57 and their ECD experimental and computed ECD spectra using different σ values. (B) Two similar structures 58 and 59 and the experimental ECD spectra in ethanol and dotted traces giving ECD spectra calculated for compounds (S)-58 and (R)-59 at the CAM-B3LYP/def2-TZVP level in vacuo optimized at the ωB97X-D/6-311+G(d,p) level. (C) (S)-naproxen (60) and its ECD spectra in acetonitrile, ethanol and water, respectively. (D) Isolated products 61 and 62 and comparison of their experimental ECD spectra with the computed ECD spectra. (E) Structure of sparticols A (63) and its experimental ECD spectrum (black line) compared with B3LYP/6-31G(d)-calculated ECD spectra (red solid line) for the B3LYP/6-31G(d) (PCM/MeCN)-optimized conformers of (S)-63 (overlaid 3D structures) and B3LYP/6-31G(d)-calculated ECD spectra of (R)-63 isomer (gray broken line).
The default sigma value of 0.4 ev is used in ECD data treatment in GaussView software; there is much no-cost software can be used for ECD simulations. For more details, please refer to the corresponding websites and download the appropriate software. The corresponding website for ECD spectra is at https://github.com/jbloino/estampes. Readers who need to use it may contact the corresponding person there. Other software may exist on different websites.
As mentioned above, correct conformer structures are extremely important for ACs. This procedure must use quantum theory. Due to the computational limits from before 2000 or earlier, the use of a high basis set, such as 6–311+G(d) for a large chiral molecule, was then difficult. In 1997, two spiro compounds 58 and 59 with very similar skeletons exhibited almost identical ECD spectra (Figure 10B) (Ripa et al., 1997). Compound 59 just had one more CH2 than 58; both had the same two chromophoric moieties. It was expected that both (S)-58 and (S)-59 would have the same ECD spectra since their spiro-center structure was identical. However, an early report argued that both would have mirror-image ECD spectra using Schellman matrix methods (Sandström, 2000a)—a popular approach in 1990s based on independent system approximation (ISA). For more details about ISA, refer the corresponding materials (Raabe et al., 2012; Sandström, 2000b).
The two structures were then carefully examined using DFT theory (Padula et al., 2016b). The new study “…demonstrates that the main source of the error in the original investigation lies in the fact that the aromatic and N-formyl enamine chromophores are not independent in the sense implied by the ISA; therefore, the ECD spectra of both compounds 58 and 59 cannot be correctly treated by the matrix method approach (Padula et al., 2016b).” Obviously, the early conclusion was not correct. After using all the new conformers in the ECD simulations, the correct results were obtained (Figure 10B). The key reason was that a TDDFT-based fragmentation approach (ISA) led to very different results from full TDDFT calculations. All theories of B3LYP, CAM-B3LYP, and M06-2X with the def2-TZVP basis sets predicted the same conclusion. With the development of computers, full chiral molecules are now used in computations unless the chiral molecules are extremely large.
In general, solvents may have some effect on ECD spectra. In a very unusual case, naproxen (60) had completely different ECD in different solvents (Figure 10C) (Ximenes et al., 2018). It had a (+)-Cotton signal at near 235 nm in acetonitrile and one (−)-Cotton signal at 210 nm. In contrast, it had a (−)-Cotton signal at 230 and 231 nm in ethanol and water and an unremarkable (+)-Cotton effect from 200 to 220 nm. However, the simulated ECD spectra in the three solvents gave very similar curves. All predicted ECD had a (+)-Cotton effect at a 240–245 nm range and a (−)-Cotton effect of 202–205 nm. This is an extremely unusual case, in that the predicted ECD did not match the experimental results in different solvents. Its importance may be that
This finding deserves great attention because polar protic and aprotic solvents are widely used for HPLC analysis in quality control of pharmaceutical drugs. If we consider the importance of discrimination between the active drug [(S)-naproxen, anti- inflammatory] and the toxic one [(R)-naproxen, hepatotoxic] using ECD detector coupled to HPLC, the findings reported here could represent an important source of error in these analyses (authors’ statement).
The reason was not analyzed until now. It is possible that there was a very strong solvation effect on the formation of different geometries with different energies.
The use of ECD to assign a chiral molecule’s AC requires the chiral center(s) to be close to the chromophore. If the stereogenic center is too far from the chromophores, it can be only used when this molecular relative configuration is reliable. An example is compound 62 (Yuan et al., 2010), which has very limited chromophores. If one had one to two different configurations from the current one (61) illustrated below, it is impossible to record the obvious ECD spectra difference from the 62s ECD. Only when its relative configuration is reliable, such as the X-ray information, then can its AC using ECD be significant and correct (62, Figure 10D) (Avula et al., 2016).
Once again, it must be emphasized that, although compound 61 has one more sugar moiety with five chiral centers, it has no chromophores and its stereogenic centers have very little effect on its ECD spectra. This raises another question: if a chiral molecule has polychiral centers but its relative configuration is not identified, then, if anyone wants to use ECD to identify its AC, it is impossible, since its ECD curves would hardly change, no matter which stereogenic center changes its configuration,.
When the chiral center is located on the side chain, then it is the favored relative to the neighboring ring structures. For example, sparticols A (63) has a chiral center at C8 connected to a hydroxyl group, and its computed ECD and experimental ECD spectrum matched well (Figure 10E) (Garcia et al., 2021). Due to the through space interaction of the -OH with the phenyl ring and the intramolecular H-bond, it has limited flexibility. In this case, its major contribution to the conformer generally played a key role in ECD spectra. Other examples, like asperglaucin A, have been reported (Lin et al., 2021).
Diols with a diol moiety often have biological activities, such as antibiotics (Fischbach and Walsh, 2009) and steroidal hormones (Norman et al., 2004; Niwa and Niwa, 2014). In organic chemistry, a diol fragment can be used as a chiral building block (Hartung and Grubbs, 2014) and promotor used in asymmetric synthesis (Huo et al., 2014). On the other hand, many diol-containing compounds are transparent in the UV-vis spectral range. Thus, they need to be converted into a derivative with a chromophoric group. Chiroptical methods can be used to identify the ACs of some diols (Jawiczuk et al., 2015). It is convenient to convert diols, which may be located on one ring moiety or on a linear carbon chain, into a new cyclic moiety to reduce the conformers in various sugar-derivatives or poly-alcohol-containing compounds in AC study (Pardo-Novoa et al., 2016).
The method of determining the AC of diols is to use dimolybdenum methodology with dimolybdenum tetracarboxylates which act as auxiliaries (Jawiczuk et al., 2014; Masuda et al., 2021). This consists of the in situ formation of chiral complexes (Mo2-core). The measured ECD spectra of the chiral complexes minus the free-ligand spectrum afford a different spectrum. The determination is strictly associated with the dependence of the signs of the Cotton effect (Górecki et al., 2007). Identical signs occurred respectively at near 400 nm and 310 nm, which is consistent with the sign of the O-C-C-O torsion angle. Examples are introduced here using 64 and 65 (Popik et al., 2014). Hexose 65 negative signs of decisive ECD band at 310 nm and 380 nm conform to the negative sign of the O-C-C-O torsion angle. The second requirement—the antiperiplanar orientation of both hydroxyl groups versus 1,2-O-isopropylidene ring and acetyl group—is also met (Figure 11). On this basis, the assignment of 3S,4S AC in compound 65 was directly possible from the level of the ECD spectrum. However, it is difficult to assign the AC of 64 because it has two possible Newman projections with two states (+) and (-); when two hydroxy groups and two connected Mo atoms were located at the bottom, the large groups had two different positions in space. Thus, it is important to give the proper Newman projection for a given chiral molecule using this method.
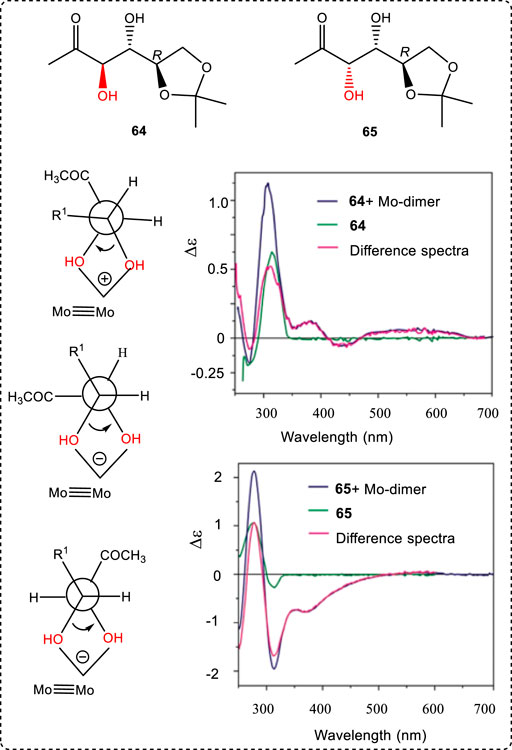
FIGURE 11. Structures of 64 and 65. ECD spectrum of hexose 64: chiral Mo2-complex, free ligand; difference spectrum (complex spectrum minuses free ligand spectrum) and two possible arrangements of the erythro 1,2-diol unit in the chiral adduct formed after ligation with the Mo-dime; ECD spectrum of hexose 65: chiral Mo2-complex, free ligand; difference spectrum (complex spectrum minuses free ligand spectrum); arrangement of the threo 1,2-diol unit in the chiral adduct formed after ligation with the Mo-dimer.
A popular method of analysis is to calculate ECD and compare it with the experimental spectrum. Many examples have been studied and the predicted ECD curves agreed well with the experimental results (Junior et al., 2014; Wang et al., 2014b; Fleming et al., 2015; Pescitelli and Bruhn, 2016; Masnyk et al., 2016; Lee et al., 2018; Zhu et al., 2018; Liu et al., 2019a; Liu et al., 2019b; Guo et al., 2021a; Arreaga-González et al., 2021; Guo et al., 2021b; Wang et al., 2021b; Rao et al., 2021; Tan et al., 2021; Xie et al., 2021); this includes the compounds in which the ECD were compared with the compound’s configuration that was assigned using quantum theory methods (Socolsky et al., 2016). Some examples (66–84) are summarized in Figure 12 (Zhang et al., 2008; Pazderkova et al., 2014; Zhang et al., 2014; Bultinck et al., 2015; Sherer et al., 2015; Liu et al., 2016b; Xu et al., 2016b; Evidente et al., 2016; Nakashima et al., 2016; Yu et al., 2016; Endo et al., 2017; Maslovskaya et al., 2017; Xu et al., 2019; Zhou et al., 2019; He et al., 2021a; Sun et al., 2021b; Lu et al., 2021; Xue et al., 2021; Yang et al., 2021).
FIGURE 12. Structures 66–84.
Another CD method is CD exciton chirality (Pescitelli, 2018). This method generally requires two chromophores to be present in the molecule (could be obtained by synthesis). It is different from the ECD calculations using Gaussian or another package via quantum equations, since the CD exciton method does not give a specific ECD curve or a series of excitation state energy for any chiral compounds. Instead, it uses the relative position of di-chromophores, such as the dibenzoates of acyclic 1,2-glycols in space to determine the ACs. This may entail uncertainties for some chiral compounds where it is not easy to correctly orientate the chromophores’ position. It is useful to determine the AC of the acyclic 1,2-glycols, acetylene alcohols (Naito et al., 2005), or similar chiral compounds. As emphasized above, a key step is to locate the relative positions of two chromophores for a specific chiral compound by conformational considerations.
The Mosher ester method is valid for the AC assignment of secondary alcohols or others (Dale and Mosher, 1973; Seco et al., 2004). This method provides very valid assistance in the study of ACs. In most cases, this method requires the substituents adjacent to the stereogenic center to be small. A previous example clearly stated this point. If there is a very bulky group, it may change the most and second-most stable conformers’ distribution and will finally lead to a wrong conclusion. This point must be clearly remembered.
Both methods are out of the scope of the present review; therefore, the two useful methods are just mentioned here rather than described in detail. Readers interested in the two methods can look for the corresponding reports (Gao et al., 2021), including other experimental methods (Fujii et al., 1998).
Generally, vibrational circular dichroism (VCD) spectroscopy can be used for the AC determinations of any chiral compounds. It differs from ECD in that ECD spectrometers were quickly commercialized in the 1970s and were widely applied to the study of AC. VCD was first applied to AC determinations in 1990s by using magnetic field perturbation equations (Nafie, 1983; Stephens, 1985; Nafie, 1992; Stephens et al., 1994), especially when quantum theory was developed and computational methods became available by use of the Gaussian package (commercially available from 1998, and then more widely for Gaussian 03 in 2003). Modern VCD instruments provide important assistance for AC study (Zhu et al., 2015; Keiderling and Lakhani, 2018; Ren et al., 2019) while computational methods (Polavarapu and Covington, 2014), including anharmonic VCD methods or other theoretical methods (Barone et al., 2012; Domingos et al., 2015; Fuse et al., 2019), make it possible for scientists to perform calculations and comparisons with a wide range of experimental results (Rossi et al., 2017; Taniguchi, 2017; Kohout et al., 2016; Góbi et al., 2015; Abbate et al., 2014; Frelek et al., 2014; Batista et al., 2015; Giovannini et al., 2016; Nieto et al., 2010; Böselt et al., 2019; Demarque et al., 2020; Ren and Zhu, 2009; Monde et al., 2003; Cichewicz et al., 2005; Batista et al., 2011a).
VCD spectroscopy is a valid method used in ACs determination for various chiral compounds (Polavarapu and Santoro, 2020). Since it uses IR light as the source for covering vibrational transitions in all parts of a molecule, it does not require any chromophores in the vis-UV region. It has been widely used in the past decade in the AC assignment of various natural products (Nafie, 2008; Batista et al., 2011b; Debie et al., 2011; Felippe et al., 2012; Lourenço et al., 2012; Cheng et al., 2020; Nafie, 2020).
Raman optical activity (ROA) spectroscopy is hampered by lower sensitivity than VCD for small molecules; however, there are some possibilities for enhancing its signal. Here, we note a recently developed mechanism wherein chirality is enhanced using the resonance that results from supramolecular aggregation. This mechanism is aggregation-induced resonance Raman optical activity (AIRROA). As an example, J-aggregates of astaxanthin (AXT) strongly absorb circularly polarized light in the range of ROA excitation at 532 nm. The implications of aggregation-induced signal enhancement for chiroptical spectroscopy are discussed by Nafie (1996), Vargek et al. (1998), and Zajac et al. (2016). Some ROA studies have been reported (Daugey et al., 2014), including the AC determination of ribifolin (Pinto et al., 2015), which is a kind of cyclopeptide with moderate effectiveness (IC50 = 42 μM) against the Plasmodium falciparum strain 3D7. However, this method has not been widely used in AC study until now, and will not be further discussed in this review.
It has been theoretically shown using nuclear velocity perturbation theory that crystal packing induces the enhancement of VCD or other methods (He et al., 2001; Nafie, 2004; Domingos et al., 2014a; Jahnigen et al., 2018). A so-called “switchable amplification of VCD” for chiral structures was reported to amplify VCD signals (Domingos et al., 2014b). Another way is to use lanthanide tris(β-diketonates) as useful probes in the stereogenic center determination of biological amino alcohols near 1500 cm−1 (β-diketonate IR absorption region) due to the formation of ternary complexation with racemic lanthanide tris(β-diketonates) (Miyake et al., 2014).
In some cases, flexible chiral compounds, such as β-peptides and foldamers, could be analyzed by careful conformational study (Farkas et al., 2019)—for example, compounds 85 (Santoro et al., 2019) to 98 (Mazzeo et al., 2017a; Taniguchi et al., 2017; Gimenesa et al., 2019). Typical structures are 86 (Gimenesa et al., 2019) and 87 (Taniguchi et al., 2017), boron-containing chiral compound 88 (Mazzeo et al., 2017a), chiral thiophene sulfonamide 89 (Rode et al., 2017), chiral Si-containing compound 90 (Xia et al., 2018), pseudoenantiomeric atropisomers anti-(S)-streptorubin B (91A) and syn-(S)-streptorubin (91B) (Andrade et al., 2015), axial chiral ligand 92 (Zhu et al., 2015), 93 (Qiu et al., 2013), bioactive compound 94 (Junior et al., 2015), axial N-BF2 compound 95 (Abbate et al., 2017), Möbius-shaped cycloparaphenylenes 96 (Nishigaki et al., 2019), and axial 3,3′-bithiophene atropisomeric scaffold 97 (Gabrieli et al., 2016). The chirality of isotope of 12C and 13C-containing compound (98) can be assigned using VCD (Miura et al., 2019). These compounds nearly cover most important organic compound types (Figure 13). Many other natural products have been well investigated using VCD methods (Nafie, 2011; Ji et al., 2014; Kessler et al., 2014; Mazzeo et al., 2015; Taniguchi et al., 2015; Cheng et al., 2016; Covington et al., 2016; Zhang et al., 2016).
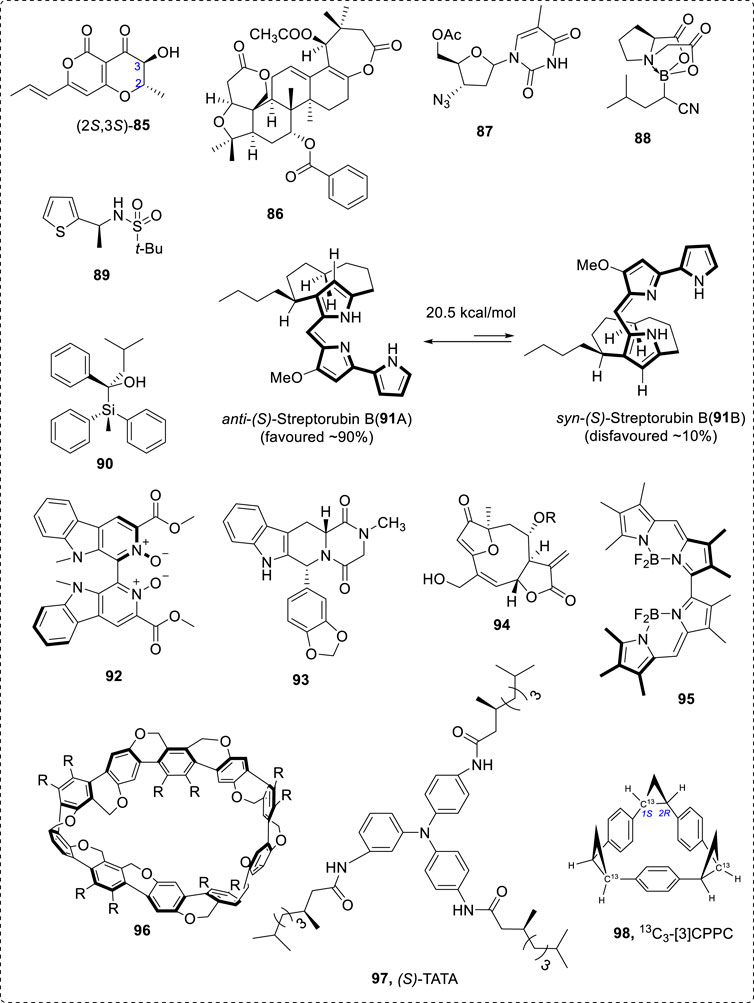
FIGURE 13. AC assigned for some typical structures 85–98.
As a useful tool, VCD has been widely examined experimentally and theoretically by theoretical chemists (Ortega et al., 2015a; Nafie et al., 2018; Nafie and Laane, 2018). An interesting example is the N-chirality molecule, the analogue (99) of Tröger’s base. It can be obtained using HPLC with mixtures of CH2Cl2 and n-heptane as eluents (Runarsson et al., 2015). A frequency-scaling factor was not applied to the predicted spectra for correcting the calculated spectra appearing at a higher wave number (Figure 14A). In this case, the calculated VCD/IR spectra were calculated at the B3LYP/6–311++G(d,p) level of theory, and the calculation results matched the experimental results well.
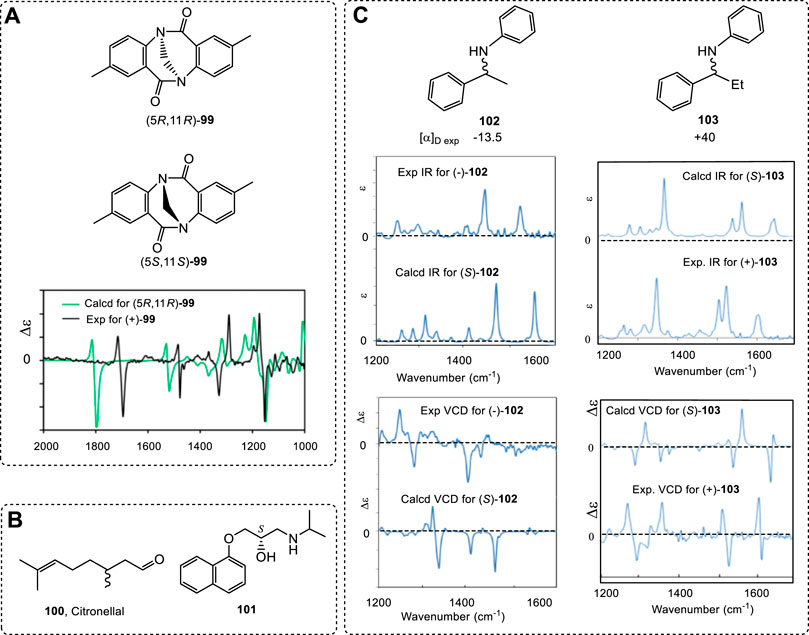
FIGURE 14. (A) Experimental VCD and IR spectra of 99 in CD2Cl2 (black) and the DFT calculations with implicit CH2Cl2 solvent (green). (B) Structures of citronella (100) and compound 101. (C) Comparison of experimental and computed IR and VCD spectra of 102 and 103.
As mentioned previously, it is hard to find all conformers for linear chiral compounds. It is also difficult to compute all found geometries in simulations of VCD. The conformer numbers increased quickly with the free rotated single bonds in linear chiral compounds. In one example, the conformers of citronellal (100, Figure 14B), a kind of flavor, were 162 in liquid (Koenis et al., 2019). The total geometries were used in VCD simulations with good agreement between the experimental and computed spectra. Similar examples included compound 101 (Górecki et al., 2017) and others (Zinna and Pescitelli, 2016).
In some cases, chiral molecules are quite large. As with ECD study, significant progress in the study of VCD using quantum mechanical theory for large molecules has been made in the past decade using fragment-based methods (Gordon et al., 2012). The molecules-in-molecules (MIM) fragment-based method is now suggested for handling large molecular VCD treatment (Mayhall and Raghavachari, 2011; Jose et al., 2015). It employs a multilayer method with multiple levels of theory using a generalized hybrid energy expression, or when considering many solvent molecules mixed with chiral molecules using QM/MM methods (Ghidinelli et al., 2018). This is similar to the ONIOM methods used in optimization for large molecules. For more details, readers can refer to the corresponding reports.
For 1,2-diol compounds, VCD can use the di-Mo-intermediates as used in ECD (Jawiczuk et al., 2015). On the other hand, converting the molecule into a five-ring structure is also helpful when the two hydroxyl groups are located on a linear carbon chain, such as pseurotin A3 with an unusual hetero-spirocyclic system (Xu et al., 2016c).
To compute a VCD spectrum requires calculating both atomic polar and atomic axial tensors based on the force constant matrix, which is generally time-consuming. Density functional-based tight binding (DFTB) theory was recently developed by Visscher (Teodoro et al., 2018), making it possible to obtain accurate VCD spectra with a much lower computational demand than standard DFT methods.
As mentioned above, the ECD spectrum is sensitive to chromophores. In a series of chiral analogues, these molecules have very similar ECD spectra. However, the corresponding VCD spectra are quite different. Even though two chiral compounds have extremely similar structures, they may have totally different VCD spectra. For example, compounds 102 and 103 are two extremely similar analogues (Zhu and Zhao, 2015; Cao et al., 2017), but, while their ECD spectra are similar, their VCD spectra are extremely different (Figure 14C).
Therefore, it is impossible to compare the VCD spectra among a similar series of chiral compounds to assign their AC by comparing the unknown compound’s VCD with the known structure’s VCD. One must compute the VCD using the quantum methods and then compare this with the experimental result.
An advantage of VCD is that it can be used to treat the interaction of two molecules in space. One early report studied the structural arrangement of four molecules of 6,6′-dimethyl-[1,1′-biphenyl]-2,2′-dicarboxylic acid in space (Urbanová et al., 2005). It is now possible to study the self-assembly of supramolecular polymers of N-centered triarylamine trisamides (Koenis et al., 2020). In a catalytic reaction, solvent-induced conformational changes of the thiourea and the evidence for an unexpected binding topology were discussed between the thiourea and an acetate anion interaction in space (Kreienborg et al., 2016). In real systems, the interaction of water with chiral molecules such as dipeptides is quite strong. Careful investigation of the water-molecule interaction has been performed. For example, there are six water molecules around the alanine dipeptide using the VCD probe (Mirtic et al., 2014). Many similar cases, like conformational equilibrium in (-)-S-nicotine, have been reported (Ortega et al., 2015b).
When determining the AC of various chiral compounds, other methods, such as transition state barrier calculation, may prove useful for understanding the assignments of some special compounds. For example, the alkaloid brevianamide M (104) was assigned as a (2S,13S) configuration using X-ray (Mo radiation) and hydrolysis experiments to afford L-phenyalanine (Figure 15A) (Li et al., 2009). This assignment is very solid. The recorded experimental OR was –146; however, the correct AC was (2R,13R) instead of (2S,13S). This re-assignment was performed using the OR, ECD (using Gibbs free energy data in Boltzmann statistics), and VCD methods (Ren et al., 2013). Computations were performed at the B3LYP/6–311++G(2d,p)//B3LYP/6–311++G(2d,p) level. The correct structure was finally confirmed by X-ray using Cu-radiation. It is a very interesting question as to why using the AC of L-phenyalanine from the hydrolysis of (-)-104 led to a wrong conclusion.
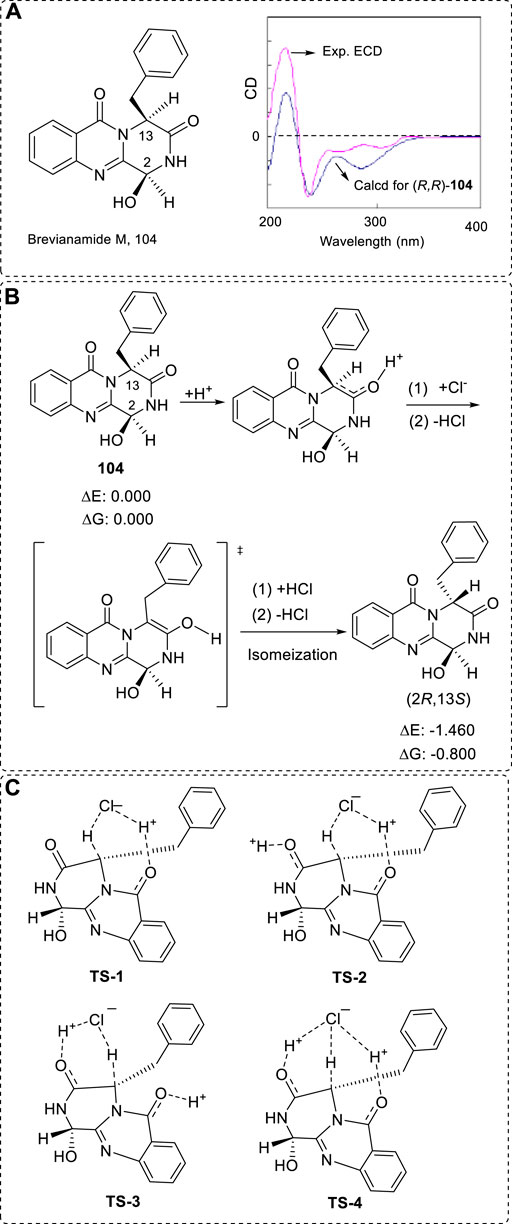
FIGURE 15. (A) Structure of reported brevianamide M (104) and the revised structure’s ECD spectra. (B) Plausible transition procedure for the conversion. (C) All possible transition state structures. TS-1: non-catalyzed by H+. TS-2 to TS-4, catalyzed by H+.
This may possibly be because of the configuration conversion of C13 from R to S during the hydrolysis of (-)-104 (Figure 15B) via four possible TS structures (TS-1 to TS-4), since the two groups of -OH and benzyl ring are cis orientated (Figure 15C). In this procedure, if the final product’s energy is higher than the starting material, this conversion would not happen. Thus, firstly, the relative energy of (2R,13R)-104 and (2R,13S)- 104 must be compared before the TS analyzation. Fortunately, the (2R,13S)- 104 was lower by 0.8 kcal/mol than the (2R,13R)- 104 using Gibbs free energy at the B3LYP/6–31++G(2d,p) level in the gas phase after conformational searches. Hence, if the TS barrier is not high enough, 92% of the (2R,13R)- 104 using total electronic energy or 80% using Gibbs free energy would convert into (2R,13S)- 104.
The calculated barrier was about 25.1 kcal/mol via TS-1 in water without any acidic promotion. The acidic catalysis procedures via TS-2 and TS-4 were 16.5 and 17.9 kcal/mol, respectively. This barrier of 25.1 kcal/mol may mean that the reaction may possibly happen when the water temperature increases to about 65℃. After the configuration conversion, the next step is the hydrolysis of (2R,13S)- brevianamide M in water—this barrier was only 21.3 kcal/mol in water. Thus, after the configuration conversion, it immediately decomposed into the corresponding L-phenyalanine.
It is very popular to hydrolyze one complex compound into one or two simple known compounds in order to assign the complex compound’s structure. Although there are not many examples with configuration conversion, the possibility does exist in some examples. To raise vigilance in daily study may help reduce the possibility of wrong conclusions.
Interestingly, another similar example (105) was reported (Eq. 2). This compound racemized in room temperature under a strong base KOH condition (Figure 16A) (Xu et al., 2021). However, it is stable under acetic acid at room temperature. This shows that acidic promotion needs about 25 kcal/mol (HCl). It would be better if there were a TS barrier calculation for this conversion; in this case, the barrier could be compared with the barrier under acidic catalysis.
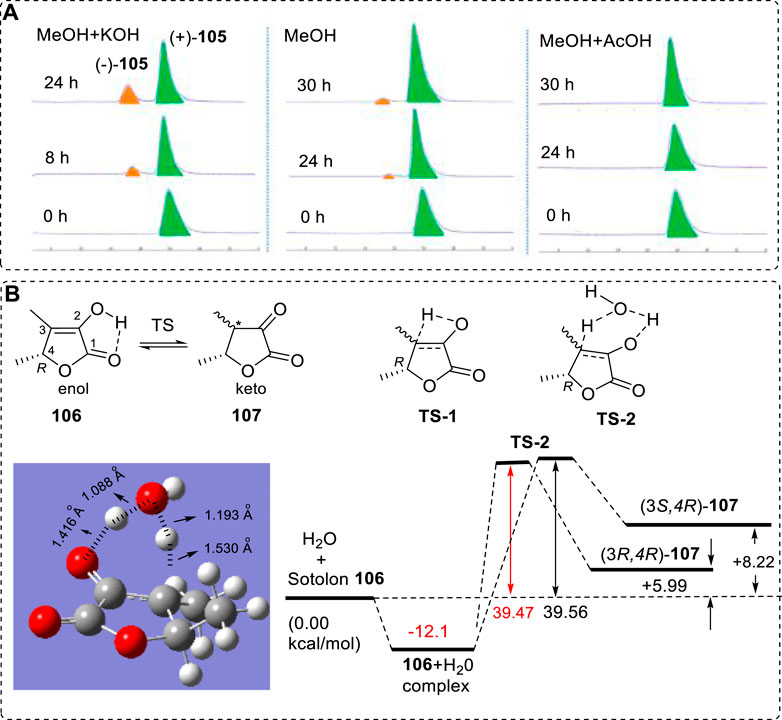
FIGURE 16. (A) Transformation of (+)-(S)-105 and (−)-(R)-105 in MeOH with different pH levels. (B) Transition state structure (TS-2) from 106 to 107 and the reaction coordinates and relative energetics of 106 to (3S,4R)-107 and (3R,4R)-107 predicted from DFT/B3LYP/6–311++G(2d,p) calculations.
In contrast to the above case, another example can exclude the possibility of a natural product to form the other structure. For example, Sotolon (106) is a naturally occurring chiral furanone with a well measured VCD. However, the predicted VCD was little different from the experimental results at 1080 and 1060 cm−1 (Nakahashi et al., 2011). Should it then form the corresponding enol structures and should the enol structure bring the corresponding contribution to the VCD spectra? Before investigating the OR, ECD, and VCD spectra of 106, the possible conversion of enol 106 to its keto tautomer 107 was theoretically investigated. (R)-106 could transform into either (3R,4R)-107 or (3S,4R)-107 (Figure 2), depending on the values of the different transition state (TS) barriers in the pathway. Direct conversion via TS-1 would face a large barrier since it involves a four-membered ring in TS. Therefore, only TS-2 was considered. The barrier to generate (3R,4R)-107 was 39.48 Kcal/mol while the formation of (3S,4R)-107 required overcoming a 39.56 Kcal/mol barrier (Yang et al., 2017). Both barriers are high, suggesting that this conversion is impossible. Furthermore, even if this barrier could be overcome, the keto forms (3R,4R)-107 or (3S,4R)-107 were predicted to be 5.99 or 8.22 Kcal/mol less stable, respectively, than the sum of the starting material 106 and a water molecule (Figure 16B). Hence, 106 is predicted to greatly dominate over either (3R,4R)-107 or (3S,4R)-107 at equilibrium and it or its other structural form may be considered in its OR or CD calculation.
Obviously, using TS barrier data can provide many benefits for understanding the AC assignment procedure. It is also obvious that computing TS barriers is not only the job of synthetic chemists. Correct use of TS barriers can accelerate the understanding of AC study.
In some cases, the biogenetic pathway is an important way of performing the organic synthesis of some bioactive compounds. Theoretical methods can provide, to a certain degree, direct knowledge of whether the pathway is possible. For example, after obtaining the diastereoisomers 108 and 109, compound 108 may be converted from 109 via an ammonia molecule present in the reaction (Figure 17A). The computed TS coordinates for the conversion were fully investigated and the largest TS barrier was only 19.2 kcal/mol, using the B3LYP-D3BJ/def2-SVP level of theory with the PCM for H2O.
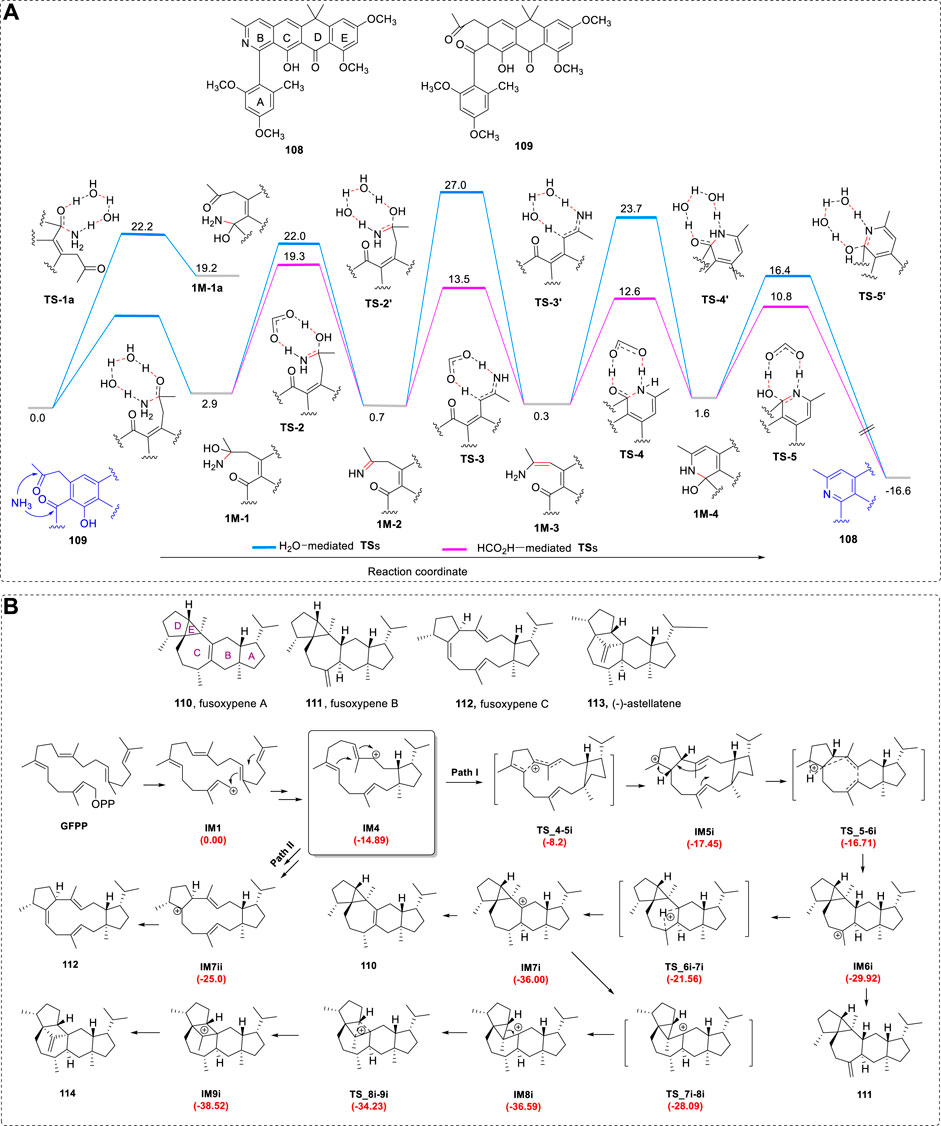
FIGURE 17. (A) Reaction coordinate of 1,5-dicarbonyl condensation with ammonia for conversion of 109 to 108 (data represent ΔG values in kcal/mol). (B) Simplified computed reaction pathways of 110–114. IM4 is the bifurcation point to generate Path I and Path II. IM = InterMediate. The potential energy profiles relative to IM1 are shown in red.
The genome-based discovery of two previously unreported fungal bifunctional terpene synthases (BFTSs) from phytopathogenic fungi has been reported. The formation of fusoxypenes A−C (110–112) and (−)-astellatene (113) has been investigated using quantum theory (Figure 17B) (Jiang et al., 2021b). The Gibbs free energies were calculated from the electron energies at the mPW1PW91/6–31+G(d,p) level plus the thermal correction at the M06-2X/6-31G(d,p) level.
Generally, a single method may settle some chiral molecular ACs. However, for some chiral compounds, one method cannot provide enough evidence to support the AC assignments. It is thus better if two, three, or all of the OR, ECD, VCD and 13CNMR methods are used. This is more important for determining the AC of chiral medicines, where many drugs have been investigated using various methods (Polavarapu, 2016). Example 6 used the 13C NMR and OR methods. There are many examples used with various combinations of the tools—for example, rare sesquiterpene (+)-3-ishwarone (115) was investigated using NMR, ORD, ECD, and VCD (Junior et al., 2014; Hu et al., 2021; Lee et al., 2021), and other examples include 116 and 117 (Cao et al., 2019; Cao et al., 2020b) (Figure 18A).
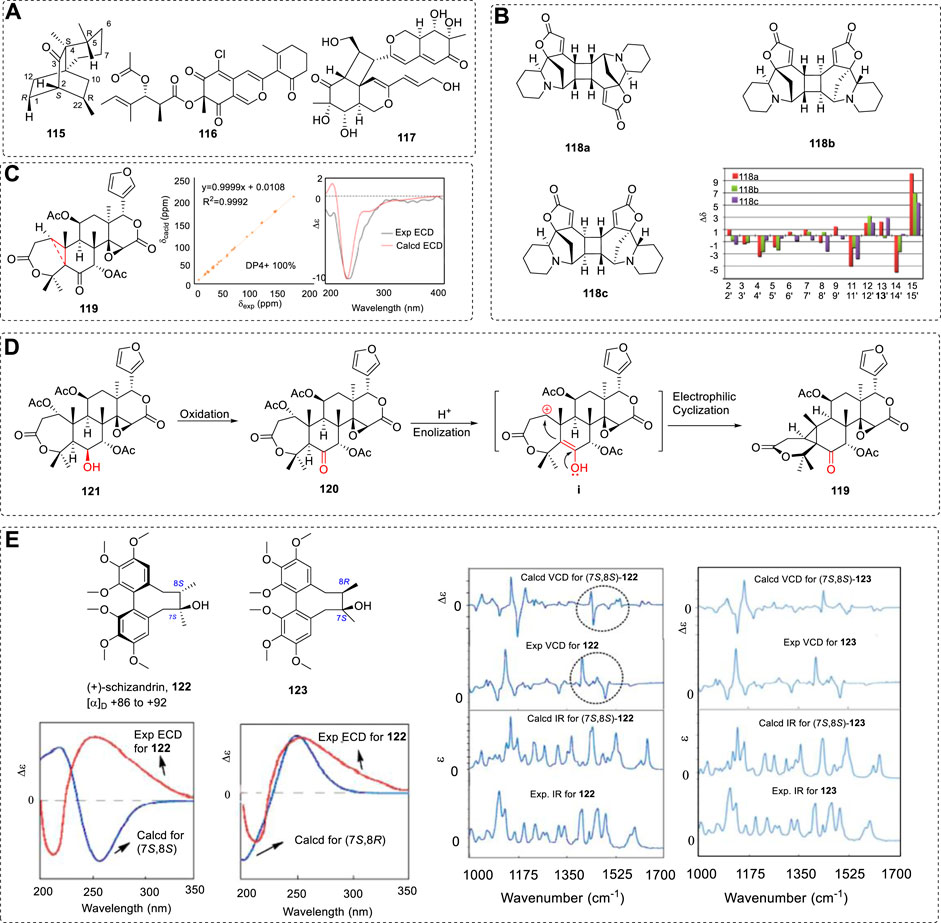
FIGURE 18. (A) Structures of natural products 115–117. (B) Relative chemical shift errors between the calculated 13C NMR of the three possible structures (118a to 118c) and the experimental 13C NMR data. (C) Structure of 119, results of calculations of 13C NMR with DP4+ analyses and ECD spectra for 119. (D) Plausible conversion routes from 121 to 119. (E) Structures of 122 and 123; (Right) comparison of both experimental and computed IR and VCD spectra; (Left) comparison of both experimental and computed ECD spectra.
Flueggedine, isolated from Flueggea virosa (Zhao et al., 2013), has one set of NMR but two units of molecular weight—a very rigid compound. During the conformational searches, one conformer was manually obtained. The theoretical 13C NMR data of 118a, 118b, and 118c were calculated at the B3LYP/6–311++G(2d,p)//B3LYP/ 6–31+G(d) level in the gas phase. Structure 118a must be excluded due to the relatively high chemical shift at up to 10.2 ppm at C15 (Figure 18B). Structure 118c had the smallest relative errors at C15; its linear coefficient should be the largest. The calculated OR value of 118c (-38.8) was close to the recorded one (−33.5). In contrast, the calculated OR magnitude for 118b was +62.5. Thus, the stereochemistry of 118 was determined to be 118c—2(2′)S, 7(7′)R, 9(9′)R, 14(14′)S, and 15(15′)S.
For an unusual aglatestine A 1 (119) with a cyclopropenyl moiety, its relative and AC was assigned via 13C NMR and ECD methods (Figures 18B, C) (Sun et al., 2021c). In the series of analogues, it was found that the three compounds obtained may have biosynthetic routes (Figure 18D). The relative configuration of the three compounds matched well and this confirmed the relationship among them (119–121).
However, the use of ECD for some specific chiral compounds for their AC assignment may not be enough. For example, schizandrin 122 was assigned as (7S,8S) in a previous report but was corrected as (7S,8R). Its ECD spectra is illustrated below (Figure 18E), predicting that its original configuration assignment was wrong. Based on the calculated ECD spectrum curve shape, it is tempting to predict its real configuration as (7R,8R), the opposite of the original assignment. However, VCD study shows that its real configuration is (7S,8R) (Figure 18E). In this case, the predicted ECD for 123 matched well with the experimental results in both ECD and VCD studies (He et al., 2014). The DFT calculations were performed at the B3LYP/6–311+G(d)//B3LYP/6–311 + G(d) level in the gas phase.
In some cases, the chiral molecules are not large and rigid but are inflexible, such as diplobifuranylones A−C (124–126) (Figure 19A) (Mazzeo et al., 2017b). In the assignment procedures, the Mosher method was used while the ECD, VCD, and ORD were applied for the AC study.
FIGURE 19. (A) Structures of 124 to 126. (B) Original structures 127 and 128, and revised structures 129 and 130.
It is now very popular to use two or more chiroptical spectroscopies in AC determinations (Reinhardt et al., 2019). Some chiral compounds have their stereogenic centers on a linear carbon chain. In these cases, Mosher methods may provide good assistance during AC assignment—for example, the compounds 1 and 4 (El-Kashef et al., 2019). It can be seen that the groups near the stereogenic center (on chain) are not as large as in compound 5.
Two bioactive spirooxindole alkaloids, (−)-citrinadin A (127) and (+)-citrinadin B (128), were isolated and reported (Tsuda et al., 2004; Mugishima et al., 2005). Their AC assignment used the ROSEY, ECD, and VCD methods. However, after a total synthesis of both compounds, it was found that the correct structures should be 129 and 130 (Figure 19B) (Bian et al., 2014). It is unresolved why the study of combinational uses of NMR, ECD, and VCD do not afford the correct ACs.
Organic synthesis is still a good way of testing the stereochemistry of some compounds accompanied by quantum calculation. The revision of the absolute stereochemistry of pycnidione was achieved via the NMR and ECD methods (Bemis et al., 2021).
With the development of computations and synthetic methods, the AC assignments of many different chiral compounds have used two more chiroptical spectroscopies (Zhang et al., 2021c). Some typical examples are listed in Table 1.
TABLE 1. AC assignment for structures of 131 to 142 using various methods.
Use of a simplified model is popular in the interpretation of chiroptical spectroscopies or in TS barrier calculations. For example, researchers frequently use a vinyl group or allyl to replace a benzyl group in a TS calculation. A methyl, an ethyl, or a propyl is used to represent a long carbon chain which has no stereogenic center. There are two possible reasons for researchers to collect in this way. One is that current computational methods, even DFT methods, are not the most accurate: they are all approximate calculations which introduce computational errors in energy and chiroptical spectroscopy calculations. Another reason is that there are fundamental reasons to shorten the long carbon chain to a short chain, or a methyl. Although the concept has been suggested in OR computations (Zhao et al., 2019; Zhu, 2022b), this principle could be extended to ECD or VCD calculations using simplified models.
The aim of using simplified models in chiroptical calculations is to reduce computational time without greatly reducing the accuracy of the calculation. This involves a new concept: a conformation pair (or conformer pair) (Zhao et al., 2019; Zhu, 2022b). For example, butane (S) has four conformers: a, b, c, and d (Figure 20A). Conformation c has no symmetric element, so its OR will not be 0, as with d. Since c and d are mirror-image symmetric, their relative energy and OR magnitudes will be same, but the OR signs will be reversed. Thus, the net OR value from c and d is 0. Therefore, the total OR for 143 will be 0.
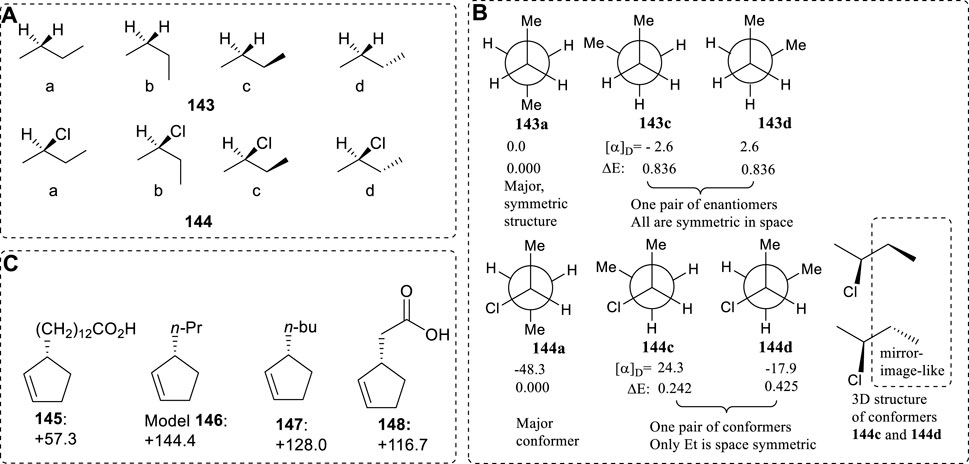
FIGURE 20. (A) Conformation analysis of achiral compound butane and chiral 2-Cl-butane. (B) Conformations of n-butane and (R)-2-Cl-butane. The OR values were computed at the level of B3LYP/6-31G(d)//B3LYP/6-31G(d) to exhibit the contributions to molecular ORs played by all the enantiomer conformations, including the very similar existing conformers. The relative energy (ΔE) unit is in kcal/mol. The long chain substituent Et structures (in the dashed line) in conformers 144c and 144d of (R)-2-chlorobutane are mirror-image-like. (C) Structure of 145 and its simplified models 146–148.
In the next example, (R)-2-chlorobutane 144 (Figure 20A) has one pair of conformers (144c and 144d) that have symmetric Et substituent structures (Figure 20B); this pair has opposite OR signs. This is very similar to the case in 143c and 143d, which are complete mirror-images. The symmetric structures that appear in 144c and 144d exist within their Et substituents. Therefore, the sum of the OR contributions of conformations 144c and 144d have a very small magnitude of only 7.4 degrees, calculated from their individual OR values at the B3LYP/6-31G(d)//B3LYP/6-31G(d) level and based on their relative conformer populations derived from their relative energies. Thus, the calculated OR ([α]D) contribution (7.4o) of this 144c and 144d pair of conformers was only 15% of the OR value of –48.3 from conformation 144a.
Conformer pairs like 143c and 143d, or 144c and 144d, are expected in any chiral linear molecules existing in solution. The final contribution from a summation of all the enantiomeric conformer pairs and the closely symmetric pairs will only represent a small fraction of the total measured molecular [α]D value. The major [α]D contribution is from the most stable conformer, as illustrated by conformer 144a in (R)-2-chlorobutane.
As the number of a chain's carbons increase, the number of stable conformations increases by 3(n−2) (here n is the number of single bond rotations and must be larger than 3). This formula is applicable to many chiral linear compounds like the above molecules. No matter how long the chain is, only a unique conformer with the lowest energy exists; it has the largest OR values. The conformer pairs must exist in double numbers, and their Boltzmann statistic fraction will not be larger than the most stable conformation’s OR values. Theoretically, the major OR contribution is from this OR: the most stable conformer’s OR sign closely represents the chiral molecular AC. Thus, the fundamental basis for using simplified models is the existence of conformer pairs for chiral compounds with a long chain.
When the carbon chain numbers are more than six, it is difficult to find all conformations. This may lead to computational errors since some conformer pairs, where there is not another conformer to form a pair to the conformer pair, are not found in the conformational search. At the same time, every theoretical method must have its accuracy level in energy and OR computations. This point again entails computation errors. Therefore, both sides entail such errors. Thus, when using a short carbon chain to representative a long chain, it is easier to simulate a large OR value than use a long carbon chain. To use an ethyl to representative a long chain with an even carbon number or a propyl to represent an odd carbon chain number may be a good choice. For example, the chiral compound 145 was assigned based on the following the OR values of three model molecules (models 146–148) (Figure 20C) (Wang et al., 2010). The computed OR of 147 with four carbons was almost 16 degrees smaller than the compound 146 with a three-carbon chain. If the side chain was simplified as an acetyl (148), its OR became 8 degrees less than 147. All of the models have over-estimated OR values compared to the OR of the actual molecule of +57.3o. Some suggested simplified substituents to replace the corresponding large and complex substituents are summarized in Table 2.
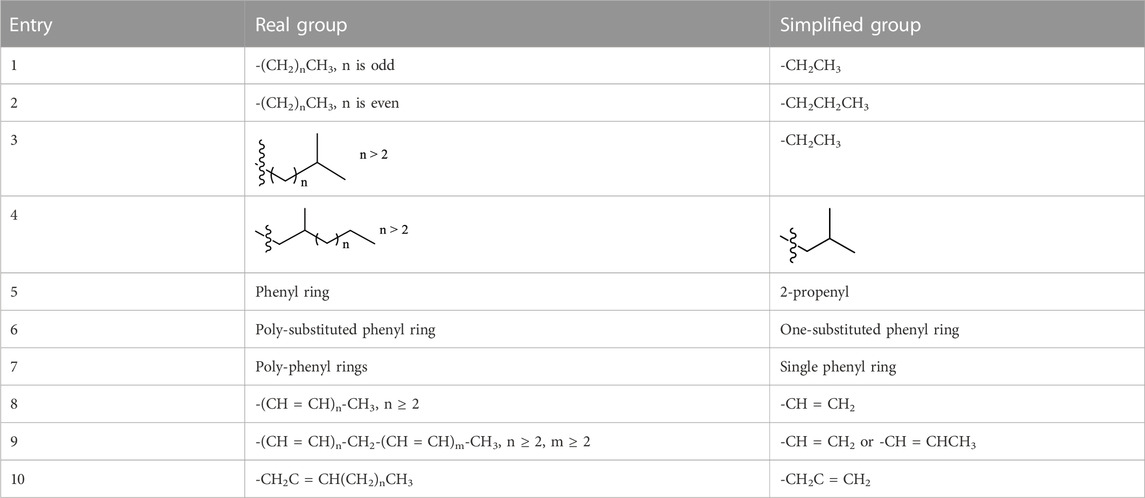
TABLE 2. Suggested simplified groups used to replace the big groups in the calculations.
Although the use of simplified models is determined above from the OR, it can also be used in related calculations for ECD and VCD. Many natural products are too complex or flexible to use their large number of low energy conformations in calculations of ECD, OR, or VCD spectra. Thus, model molecules must be selected that are representative of the original structure. The choice of satisfactory model structures is the focus of this section. For example, the compounds 149 and 150 can be simplified as the models 151 and 152 in the ECD study (Figure 21A) (Tanaka et al., 2017), including 13C NMR simulations.
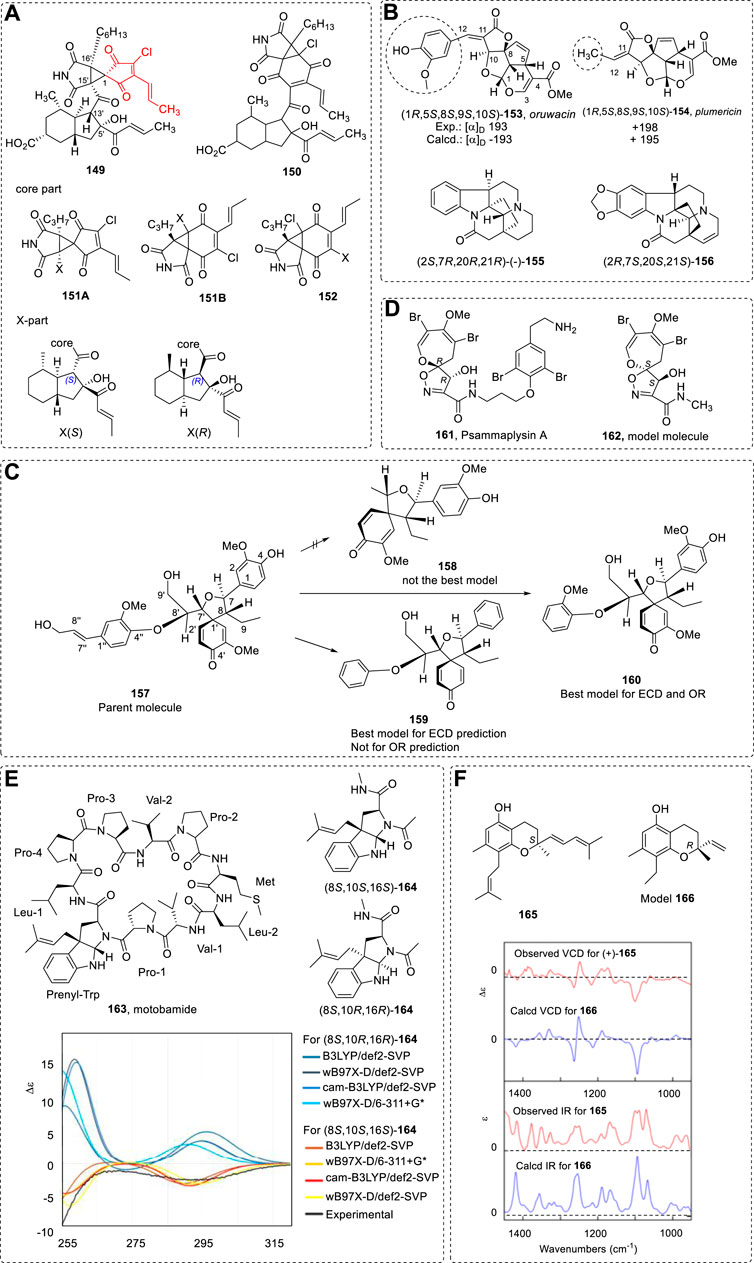
FIGURE 21. (A) Structures of cyclohelminthol X (149) and AD0157 (150) and their simplified model parts. (B) Two pairs of similar analogues of 153 vs. 154 and 155 vs. 156. (C) Natural product 157 and its three simplified models used in OR and ECD study. (D) Psammaplysin A (161) and its simplified model compound (162) used for ECD predication. The model is smaller and, more importantly, much less conformationally mobile than the natural product. (E) Experimental ECD spectrum of motobamide (163) and theoretical ECD spectra of the model compounds (8S, 10S, 16S)-164 and (8S, 10R, 16R)-164. (F) Structure 165 and its simplified model 166 and their experimental and computed VCD spectra.
The highlighted red is the core framework of cyclohelminthols in 149. It was thus separated as two parts: 151A and 151B and X (Figure 21A) for 149. The 152 part and X part were used for 150. Conformational searches were performed using semiempirical AM1 MMFF provided for only a limited number of candidate conformers for some models. The DFT theory of BHLYP/def2-TZVP was used for ECD calculations, and ωB97X-D17 was used at the 6-31G* basis set in the 13C NMR calculation.
The two parts were independently computed. Thus, different combinations were used for the calculations (Supplementary Table S3); good agreement between the theoretical results and the experimental data was achieved.
However, use of a wrong simplified group may result in the wrong assignment. For example, ourwacin and plumerichi are very similar natural products (Stephens et al., 2007a). Ourwacin’s (153) experimental OR is 193. (1R,5S,8S,9S,10S)-plumerichi (154) had a +198. It is easy to assign (+)-ourwacin as (1R,5S,8S,9S,10S). If the (1R,5S,8S,9S,10S)-ourwacin was used in OR calculations, –193 was obtained. However, if (1R,5S,8S,9S,10S)-plumerichi was used in the calculations, the +195 of OR value was recorded. This clearly shows that a phenyl ring cannot be simplified as a methyl group in calculations. As an opposite example, the compound 155 had –25.4 of OR values (Hajıcek and Trojanek, 1981; Laguna et al., 1984), and its structural similar compound 156 had [α]D of +15.5 in CHCl3) (Renner and Kernweisz, 1963). Its AC was assigned as the (2R,7S,20S,21S) using ECD, VCD, and ORD (Stephens et al., 2007b). It has no phenyl group. The same configuration had the same OR sign.
In this case, it is easy to see that simplifying a phenyl group as a non-aromatic substitute like methyl leads to a wrong conclusion. This can be observed again in another example: 157 (Figure 21C) (Zhao et al., 2016). The model molecule 158 was not the best in the simulations of ECD or OR.
In this model study, consider for computing OR that the -OMe on the phenyl ring may restrict some stable conformation formations. Thus, it is necessary to keep this OMe at its ortho-position instead of replacing it as a H atom since OR is more sensitive to geometries and spatial locations while ECD is more sensitive to the positions of double bond or phenyl ring chromophores.
The structure psammaplysin A (161) has a large side chain (Mandi et al., 2015). It was simplified as the model molecule 162 in ECD computations (Figure 21D). In this way, the chromophore is a long distance from the stereogenic centers. Its effect on ECD curves is not large enough to affect the assignment of AC.
A cyclic peptide (motobamide 163) was simplified as 164 below and used in ECD study (Figure 21E) (Takahashi et al., 2021). This simplification is quite bold, since the >C=O is also a chromophore, although it has a weak contribution to entire molecular ECD spectra. It should be noted that the model is a linear structure and the peptide is cyclic. Although conformer structures of the linear model are expected to be different from cyclic ones, it remains enigmatic whether this difference should yield a ECD difference or not.
As mentioned above, the effect of the carbon chain length on the OR has its limitation, which means that, even if the chain is very long, its OR value will not increase significantly. This clearly leads to the question of why the chain increase does not have an effect on its OR value.
Although this conclusion is obtained from OR calculations, it can be used in other calculations as mentioned above, such as VCD. For example, chiral compound 165 can be simplified as 166 in its VCD calculations (Figure 21F) (Batista et al., 2009; Mota et al., 2009; Batista et al., 2010).
Fluralaner (167), a racemic animal health product used to prevent fleas and ticks, was used in a VCD study that used the entire molecule and its simplified model (168) (Kong et al., 2017). Six conformers were found using the entire molecule and only two for the simplified model molecule. There is good agreement using the simplified model in simulations of its VCD spectra (Figure 22A).
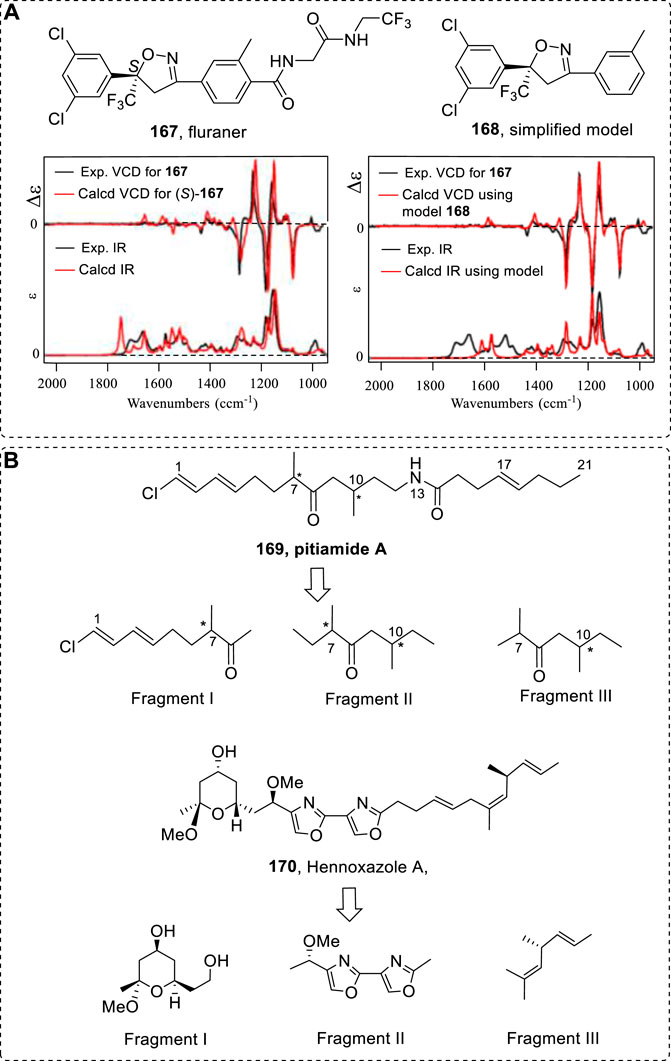
FIGURE 22. (A) Structures of 167 and 168; the experimental VCD for (+)-fluralaner (black line) and the calculated VCD (red line) for (S)-fluralaner (left). Comparison of the experimental VCD and the computed VCD using the simplified (S)- model (right). (B) Original structure 169 and 170 and the corresponding separated fragments.
If a chiral molecule has several stereogenic centers on its carbon chain, such as pitiamide A (169) (Nagle et al., 1997), one possible method is to cut the structure into several small molecules that have one or two stereogenic centers in OR calculations (Ribe et al., 2000).Another example hennoxazole A (170) had been treated similarly (Figure 22B) (Kondru et al., 1997; Kondru et al., 1998). These are only model molecules and it is not easy to cut similar molecules into small molecules in practice if one has had no experience in this study.
Consider a chiral molecule with a single stereogenic center: molecule 171 (Lu et al., 2010). If the entire molecule was used in ECD calculation, the number of conformers would be large. To reduce the computation time, it was simplified as the model 172 for ECD computations. In this case, the ortho-OH cannot be cut off since it restricts the rotation of the phenyl ring and thereby affects the geometry’s overall structure and energy distribution. Simplification is possible by using an ethyl to replace the isopentyl group. Considering that the chiral molecule has only three isolated phenyl rings, its ECD maximum of CD signal must appear at the 220–240 nm range. Here, the long chain may have a relatively large effect on the observed CD. Indeed, to use model B in the ECD simulation agrees well with the experimental results obtained (Figure 23A). This conclusion also agrees with the matrix method results.
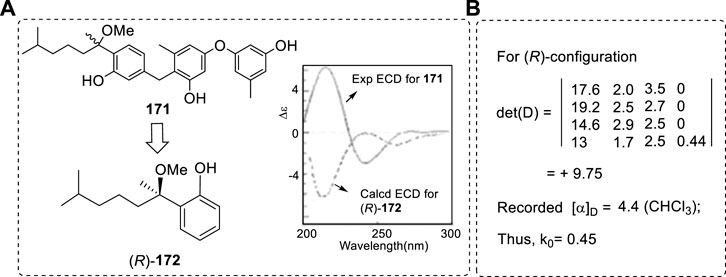
FIGURE 23. (A) Structure 171 and its simplified model 172; comparison of experimental and computed ECD spectra; (B) Determinant [det(D)] for compound 171 using (R)-configuration.
The matrix may thus be another approach to this study. For example, molecule 171 yielded a very small OR value of 4.4o. If the entire molecule is used in OR calculations, the large number of conformers needs to be investigated. On the other hand, use of the matrix model is relatively quick. For the current group order (S) configuration, its determinant was +9.75. Based on the positive k0 value (0.45) for a tertiary-alcohol derivative, it is concluded that it has S configuration. This conclusion agrees with that from the ECD calculations using simplified model molecule 172.
Many other chiral compounds have been similarly reported. For example, the compound 173 is bioactive; its AC was assigned using Mosher and ECD analysis (Liu et al., 2015). If a simplified model can be used, it may be easily assigned since it can form a six-membered ring structure via an intramolecular H-bond if using the model 174 as illustrated below (Figure 24A).
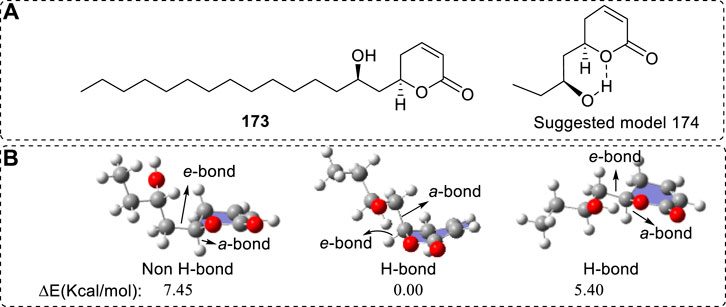
FIGURE 24. (A) Structure of 173 and its simplified model 174. (B) Energy difference of the conformers with or without intramolecular H-bond (blue section is the ring structure moiety).
A total of 46 conformers were found for 174 using the Gaussian package. The lowest energy conformer which had an intramolecular H-bond was about 7.4 kcal/mol lower than the lowest energy conformer without this bond (Figure 24B). It is surprising that the energy of the conformer with a side chain located on an axial-bond is lower by about 5.4 kcal/mol than the conformer that has its H-bond as an equatorial-bond. If the large group is on equatorial-bond, the intramolecular conformer is only about 2.0 kcal/mol lower than the one without H-bond and also has the large group located on equatorial-bond. To clearly state the conformer question, the conformational search was performed using a MMFF94S force field. All the conformers were then computed at HF/3-21G*. Both kinds of conformer with or without H-bonds with relative energy from 0–2.0 kcal/mol were then re-optimized at B3LYP/6-31G* level in the gas phase. These B3LYP/6-31G*-optimized conformers with relative energy from 0.0 to 2.0 kcal/mol were finally optimized at the B3LYP/6-311G* level. All optimizations were performed in the gas phase. Since the conformers with low energy are very limited, they could be used for ECD or other spectroscopic computations.
Finally, a possible case to consider is that of chiral flavor molecules that may form dimers (Slaughter, 1999; Collin et al., 2012; Yang et al., 2017). Generally, the four bases A, G, T, C are found in DNA structures where the largest substituent is methyl group-base thymine (T). This contains a Me group instead of a larger substituent such as Et among the four base pairs C, G, T, and A in DNA. Very stable H-bonds can form among the four base pairs. Could the derivative of T, with an ethyl group instead of T in DNA, form a strong H-bond in living systems? Why is only methyl involved instead of Et or a larger group in DNA evolution? In reference to this interesting question, compound 106, which has two methyl groups, can form dimers in solution, but compound 175, which has one ethyl group, cannot. This result might provide some hints to understanding such questions. Since OR values are very sensitive to their conformers in solution, monomer versus a dimer structure may have a large calculated difference in OR values (Jiang et al., 2021b). Five different methods (B3LYP/6–31+G(d)//B3LYP/6–31+G(d) (Method 1, M1), B3LYP/6–311++G (2d,p)//B3LYP/6–311++G(2d,p) (Method 2, M2), B3LYP/6–311++G (2d,p)(PCM-CHCl3)//B3LYP/6–311++G(2d,p) (PCM-CHCl3) (Method 3, M3), MPW1PW91/6–311+G(d)//MPW1PW91/6–311++G(2d,p) (Method 4, M4), and MPW1PW91/6–311+G(d) (PCM-CHCl3)//MPW1PW91/6–311+G(d) (PCM-CHCl3) (Method 5, M5)) were used in OR calculations. Only the OR values of 106a approached the experimental OR value range (reported OR is –21.5). Method 3 gave a much more accurate estimation of the OR value (–24.6) (Table 3).
TABLE 3. OR simulations of sotolon (106) and furanone (175) and their dimer structures (106a and 175a).
The ECD of 106 and 106a, and 175 and 175a were little different. However, the predicted VCD had a very interesting difference in strength at the vibration numbers of 1020 and 1040 cm−1 (Figure 25B). In experiments, mode 1 is stronger than mode 2. In the monomer’s VCD signals, mode 1 is weaker than mode 2. However, signal 1 is stronger than signal 2 in dimeric structure 106a.
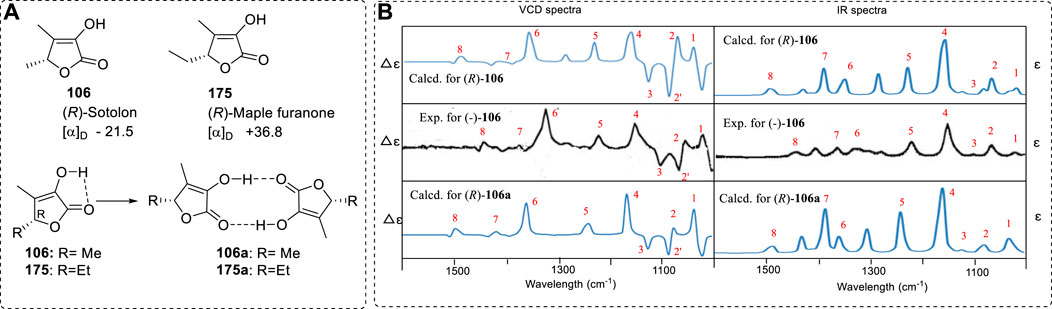
FIGURE 25. (A) Structures of 106 and 175 and their possible dimeric form. (B) Comparison of the experimental VCD and IR spectra of sotolon (106) to the calculated VCD spectra of both 106 and its dimer 106a using Method 3.
The VCD of 175 and 175a had a very large difference between the vibration numbers from 1300–1420 cm−1. Their OR, ECD, VCD, or ROA calculations should involve the dimeric structure in the calculations. However, if there is Et or a larger group on the structure, a dimeric structure may not be possible for consideration in computations.
The VCD study of isoindolinones (such as 176) gave more information about the 3D structures organized by dimeric structures, forming poly-unit connected structures (polymeric form) (Rode et al., 2018). The most stable conformers were obtained by conformational searches for each isoindolinone, and each of the most stable conformations were used to organize the corresponding poly-unit structure layer by layer ((S)-I- stack and (S)-I-stack2 via (S)-II-stack and (S)-II-stack2), as illustrated below (Figure 26A). The IR and VCD ranges were near 1700 cm−1 for focusing on the C=O vibration modes.
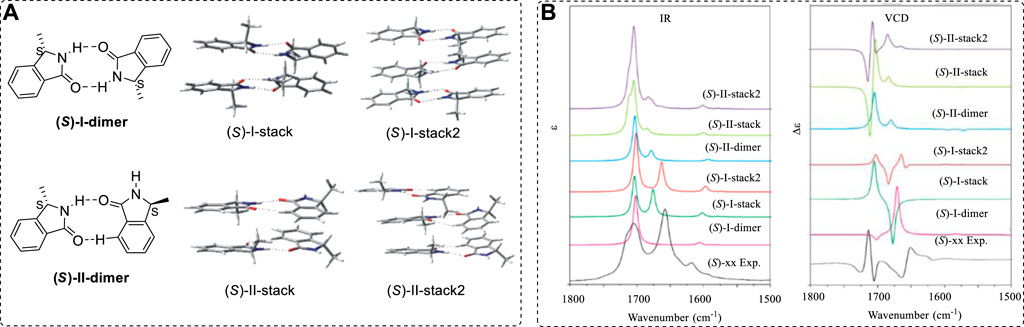
FIGURE 26. (A) Schemes of possible isoindolinone 176 dimers of types I and II. (B) Comparison of experimental IR and VCD spectra of (S)–isoindolinone with those calculated obtained for dimers (B3LYP/TZVP) and stacking structures (B3LYP-D3/TZVP). The VCD intensities of the (S)-I-dimer and (S)-II-dimer structures were multiplied by factors of 20 and 40, respectively.
The IR results clearly show that the (S)-I-stack and/or (S)-I-stack2 were the possible structures for (S)-176. Furthermore, the VCD spectra showed that the (S)-I-stack2 was preferred in spatial arrangement (Figure 26B). Clearly, for the rigid structures, it is easy to form the dimeric structures in solution.
When the methyl of isoindolinone was replaced by other large substituents, they may assume polymeric form in solution by H-bonds. Their 3D structures were quite complex and are not easily computed in VCD study. Amides can easily form dimeric or polymeric structure, like diastereomer diketopiperazine peptides derived from phenylalanine in VCD study (Pérez-Mellor and Zehnacker, 2017).
This review summarizes the OR (ORD), matrix, ECD, VCD, TS, and simplified models used in AC study for natural products. The monomer and dimeric forms of the chiral compounds were also introduced to readers. Some guidance is given to prevent wrong conclusions in the study of organic stereochemistry.
HZ is responding for almost writing the entire review. YW is responding for partial compound structure preparation and handling references. LN is responding for the VCD outline and English proofreading of the whole review.
HZ thanks the financial support of the NSFC (NSFC#21877025) and Key Project of Joint Medicines of Hebei Province from the Committee of Science and Technology of Hebei Province (#H2020201029 and #B2022208057), and from the Hebei University of Science and Technology for the “Muxing Scholar Fund.”
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fntpr.2022.1086897/full#supplementary-material
Abbate, S., Bruhn, T., Pescitelli, G., and Longhi, G. (2017). Vibrational optical activity of BODIPY dimers: The role of magnetic-electric coupling in vibrational excitons. J. Phys. Chem. A 121, 394–400. doi:10.1021/acs.jpca.6b11327
Abbate, S., Longhi, G., Lebon, F., Castiglioni, E., Superchi, S., Pisani, L., et al. (2014). Helical sense-responsive and substituent-sensitive features in vibrational and electronic circular dichroism, in circularly polarized luminescence, and in Raman spectra of some simple optically active hexahelicenes. J. Phys. Chem. C 118, 1682–1695. doi:10.1021/jp4105695
Ancheeva, E., Küppers, L., Akone, S. H., Ebrahim, W., Liu, Z., Mandi, A., et al. (2017). Expanding the metabolic profile of the fungus Chaetomium sp. through co-culture with autoclaved Pseudomonas aeruginosa. Eur. J. Org. Chem. 2017, 3256–3264. doi:10.1002/ejoc.201700288
Andrade, M. S., Silva, V. S., Lourenço, A. M., Lobo, A. M., and Rzepa, H. S. (2015). Chiroptical properties of streptorubin B: The synergy between theory and experiment. Chirality 27, 745–751. doi:10.1002/chir.22486
Arreaga-González, H. M., Oliveros-Ortiz, A. J., Río, R. E. D., Rodríguez-García, G., Torres-Valencia, J. M., Cerda-García-Rojas, C. M., et al. (2021). Methodology for the absolute configuration determination of epoxythymols using the constituents of Piptothrix areolare. J. Nat. Prod. 84, 707–712. doi:10.1021/acs.jnatprod.0c01113
Avula, S. K., Hussain, H., Csuk, R., Sommerwerk, S., Liebing, P., Górecki, M., et al. (2016). 5-epi-Incensole: Synthesis, X-ray crystal structure and absolute configuration by means of ECD and VCD studies in solution and solid state. Tetrahedron Asymmetry 27, 829–833. doi:10.1016/j.tetasy.2016.07.007
Baassou, S., Mehri, H. M., Rabaron, A., and Plat, M. (1983). (+) Melonine and NB-oxy melonine, a new indoline skeleton. Tetrahedron Lett. 24, 761–762. doi:10.1016/S0040-4039(00)81519-1
Banerjee, R., and Sheet, T. (2017). Ratio of ellipticities between 192 and 208 nm (R1): An effective electronic circular dichroism parameter for characterization of the helical components of proteins and peptides. Proteins Struct. Funct. Bioinforma. 85, 1975–1982. doi:10.1002/prot.25351
Bannwarth, C., Seibert, J., and Grimme, S. (2016). Electronic circular dichroism of [16]helicene with simplified TD-DFT: Beyond the single structure approach. Chirality 28, 365–369. doi:10.1002/chir.22594
Barone, G., Gomez-Paloma, L., Duca, D., Silvestri, A., Riccio, R., and Bifulco, G. (2002). Structure validation of natural products by quantum-mechanical GIAO calculations of 13C NMR chemical shifts. Chem. A Eur. J. 8, 3233–3239. doi:10.1002/1521-3765(20020715)8:14<3233:AID-CHEM3233>3.0.CO;2-0
Barone, V., Biczysko, M., Bloino, J., Panek, M. B., Carnimeo, I., and Panek, P. (2012). Toward anharmonic computations of vibrational spectra for large molecular systems. Int. J. Quantum Chem. 112, 2185–2200. doi:10.1002/qua.23224
Barton, D., Nakanishi, K., and Meth-Cohn, O. (1999). Comprehensive natural products chemistry. New York: Elsevier.
Batista, J. J. M., Wang, B., Castelli, M. V., Blanch, E. W., and López, S. N. (2015). Absolute configuration assignment of an unusual homoisoflavanone from Polygonum ferrugineum using a combination of chiroptical methods. Tetrahedron Lett. 56, 6142–6144. doi:10.1016/j.tetlet.2015.09.102
Batista, J. M., Batista, A. N., Rinaldo, D., Vilegas, W., Cass, Q. B., Bolzani, V. S., et al. (2010). Absolute configuration reassignment of two chromanes from Peperomia obtusifolia (piperaceae) using VCD and DFT calculations. Tetrahedron Asymmetry 21, 2402–2407. doi:10.1016/j.tetasy.2010.09.004
Batista, J. M., López, S. N., Mota, J. S., Vanzolini, K. L., Cass, Q. B., Rinaldo, D., et al. (2009). Resolution and absolute configuration assignment of a natural racemic chromane from Peperomia obtusifolia (piperaceae). Chirality 21, 799–801. doi:10.1002/chir.20676
Batista, J. M. J., Batista, A. N. L., Mota, J. S., Cass, Q. B., Kato, M. J., Bolzani, V. S., et al. (2011). Structure elucidation and absolute stereochemistry of isomeric monoterpene chromane esters. J. Org. Chem. 76, 2603–2612. doi:10.1021/jo1025089
Batista, J. M. J., Batista, A. N. L., Rinaldo, D., Vilegas, W., Ambrósio, D. L., Cicarelli, R. M. B., et al. (2011). Absolute configuration and selective trypanocidal activity of gaudichaudianic acid enantiomers. J. Nat. Prod. 74, 1154–1160. doi:10.1021/np200085h
Bemis, C. Y., Ungarean, C. N., Shved, A. S., Jamieson, C. S., Hwang, T., Lee, K. S., et al. (2021). Total synthesis and computational investigations of sesquiterpene tropolones ameliorate stereochemical inconsistencies and resolve an ambiguous biosynthetic relationship. J. Am. Chem. Soc. 143, 6006–6017. doi:10.1021/jacs.1c02150
Bian, Z. G., Marvin, C. C., Pettersson, M., and Martin, S. F. (2014). Enantioselective total syntheses of citrinadins A and B. stereochemical revision of their assigned structures. J. Am. Chem. Soc. 136, 14184–14192. doi:10.1021/ja5074646
Bifulco, G., Dambruoso, P., Gomez-Paloma, L., and Riccio, R. (2007). Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 107, 3744–3779. doi:10.1021/cr030733c
Bijvoet, J. M., Peerdeman, A. F., and van Bommel, A. J. (1951). Determination of the absolute configuration of optically active compounds by means of X-rays. Nature 168, 271–272. doi:10.1038/168271a0
Böselt, L., Sidler, D., Kittelmann, T., Stohner, J., Zindel, D., Wagner, T., et al. (2019). Determination of absolute stereochemistry of flexible molecules using a vibrational circular dichroism spectra alignment algorithm. J. Chem. Inf. Model. 59, 1826–1838. doi:10.1021/acs.jcim.8b00789
Bringmann, G., Maksimenka, K., Bruhn, T., Reichert, M., Harada, T., and Kuroda, R. (2009). Quantum chemical CD calculations of dioncophylline A in the solid state. Tetrahedron 65, 5720–5728. doi:10.1016/j.tet.2009.05.024
Brotin, T., Daugey, N., Vanthuyne, N., Jeanneau, E., Ducasse, L., and Buffeteau, T. (2015). Chiroptical properties of cryptophane-223 and -233 investigated by ECD, VCD, and ROA spectroscopy. J. Phys. Chem. B 119, 8631–8639. doi:10.1021/acs.jpcb.5b04539
Cai, S. X., Risinger, A. L., Petersen, C. L., Grkovic, T., O’Keefe, B. R., Mooberry, S. L., et al. (2019). Anacolosins A–F and corymbulosins X and Y, clerodane diterpenes from Anacolosa clarkii exhibiting cytotoxicity toward pediatric cancer cell lines. J. Nat. Prod. 82, 928–936. doi:10.1021/acs.jnatprod.8b01015
Cam-dft theory Yanai, T., Tew, D. P., and Handy, N. C. (2004). A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57. doi:10.1016/j.cplett.2004.06.011
Cao, F., Meng, Z., Wang, P., Luo, D., and Zhu, H. J. (2020). Dipleosporalones A and B, dimeric azaphilones from a marine-derived Pleosporales sp. fungus. J. Nat. Prod. 83, 1283–1287. doi:10.1021/acs.jnatprod.0c00132
Cerezo, J., Aranda, D., Ferrer, F. J. A., Prampolini, G., Mazzeo, G., Longhi, G., et al. (2018). Toward a general mixed quantum/classical method for the calculation of the vibronic ECD of a flexible dye molecule with different stable conformers: Revisiting the case of 2, 2, 2-trifluoro-anthrylethanol. Chirality 30, 730–743. doi:10.1002/chir.22853
Cerra, B., Carotti, A., Passeri, D., Passeri, D., Sardella, R., Moroni, G., et al. (2019). Exploiting chemical toolboxes for the expedited generation of tetracyclic quinolines as a novel class of PXR agonists. ACS Med. Chem. Lett. 10, 677–681. doi:10.1021/acsmedchemlett.8b00459
Cerulli, A., Lauro, G., Masullo, M., Cantone, V., Olas, B., Kontek, B., et al. (2017). Cyclic diarylheptanoids from Corylus avellana green leafy covers: Determination of their absolute configurations and evaluation of their antioxidant and antimicrobial activities. J. Nat. Prod. 80, 1703–1713. doi:10.1021/acs.jnatprod.6b00703
Cheng, G. G., Li, D., Hou, B., Li, X. N., Liu, L., Chen, Y. Y., et al. (2016). Melokhanines A–J, bioactive monoterpenoid indole alkaloids with diverse skeletons from Melodinus khasianus. Melodinus khasianus J. Nat. Prod. 79, 2158–2166. doi:10.1021/acs.jnatprod.6b00011
Cheng, M. M., Li, P. L., Jiang, Y., Tang, X. L., Zhang, W. J., Wang, Q., et al. (2021). Penitol A and penicitols E-I: Citrinin derivatives from Penicillium citrinum and the structure revision of previously proposed analogues. J. Nat. Prod. 84, 1345–1352. doi:10.1021/acs.jnatprod.1c00082
Cheng, Y., Ding, W., Yan, Y., Meng, X., Nafie, L. A., Xu, T., et al. (2020). Isolation, total synthesis, and absolute configuration determination of renoprotective dimeric N-Acetyldopamine–Adenine hybrids from the insect Aspongopus chinensis. Org. Lett. 22, 5726–5730. doi:10.1021/acs.orglett.0c01593
Cichewicz, R. H., Clifford, L. J., Lassen, P. R., Cao, X., Freedman, T. B., Nafie, L. A., et al. (2005). Stereochemical determination and bioactivity assessment of (S)-(+)-curcuphenol dimmers isolated from the marine sponge didiscus aceratus and synthesized through laccase biocatalysis. Bioorg. Med. Chem. 13, 5600–5612. doi:10.1016/j.bmc.2005.06.020
Collin, S., Nizet, S., Bouuaert, T. C., and Despatures, P. M. (2012). Main odorants in jura flor-sherry wines. Relative contributions of sotolon, abhexon, and theaspirane-derived compounds. J. Agri. Food Chem. 60, 380–387. doi:10.1021/jf203832c
Conformation pairs Zhao, D., Ren, J., Xiong, Y. F., Dong, M. X., Zhu, H. J., and Pittman, C. U. (2019). Conformer pairs contributions to the optical rotations in a series of chiral linear aliphatic alcohols. Chem. Res. Chin. U. 35, 109–119. doi:10.1007/s40242-018-8182-2
Compounds 66-84 references: for 66, see Evidente, M., Santoro, E., Petrovic, A. G., Cimmino, A., Koshoubu, J., Evidente, A., et al. (2016). Absolute configurations of phytotoxic inuloxins B and C based on experimental and computational analysis of chiroptical properties. Phytochem 130, 328–334. doi:10.1016/j.phytochem.2016.07.012
ComputeVOA (2022). ComputeVOA. Available at: https://legacy.biotools.us/product/computevoa-comprehensive-software-gaussian-software-not-included.
Covington, C. L., Junior, F. M. S., Silva, J. H. S., Kuster, R. M., Amorim, M. B. D., and Polavarapu, P. L. (2016). Atropoisomerism in biflavones: The absolute configuration of (-)-agathisflavone via chiroptical spectroscopy. J. Nat. Prod. 79, 2530–2537. doi:10.1021/acs.jnatprod.6b00395
Dale, J. A., and Mosher, H. S. (1973). Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and .alpha.-methoxy-.alpha.-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 95, 512–519. doi:10.1021/ja00783a034
Daugey, N., Brotin, T., Vanthuyne, N., Cavagnat, D., and Buffeteau, T. (2014). Raman optical activity of enantiopure cryptophanes. J. Phys. Chem. B 118, 5211–5217. doi:10.1021/jp502652p
Debie, E., Gussem, E. D., Dukor, R. K., Herrebout, W., Nafie, L. A., and Bultinck, P. (2011). A confidence level algorithm for the determination of absolute configuration using vibrational circular dichroism or Raman optical activity. ChemPhysChem 12, 1542–1549. doi:10.1002/cphc.201100050
Demarque, D. P., Heinrich, S., Schulz, F., and Merten, C. (2020). Sensitivity of VCD spectroscopy for small structural and stereochemical changes of macrolide antibiotics. Chem. Commun. 56, 10926–10929. doi:10.1039/D0CC03838E
For compound 23: Ding, S. S., Zhang, C. C., Shi, W. S., Liang, M. M., Yang, Q., Zhu, H. J., et al. (2016). Absolute configuration of two novel compounds from the Talaromyces aculeatus using optical rotation, electronic circular dichroism and vibrational circular dichroism. Tetrahedron Lett. 57, 75–79. doi:10.1016/j.tetlet.2015.11.066
Ding, Z. G., Ren, J., Li, M. G., Zhao, J. Y., Huang, R., Cui, X. L., et al. (2010). Naphthospironone A: An unprecedented and highly functionalized polycyclic metabolite from an alkaline mine waste extremophile. Chem. J. Eur. 16, 3902–3905. doi:10.1002/chem.200903198
Ding, Z. G., Zhao, J. Y., Li, M. G., Huang, R., Li, Q. M., Cui, X. L., et al. (2012). Griseusins F and G, spiro-naphthoquinones from a tin mine tailings-derived alkalophilic Nocardiopsis species. J. Nat. Prod. 75, 1994–1998. doi:10.1021/np3004936
Domingos, S. R., Hartl, F., Buma, W. J., and Woutersen, S. (2015). Elucidating the structure of chiral molecules by using amplified vibrational circular dichroism: From theory to experimental realization. ChemPhysChem 16, 3363–3373. doi:10.1002/cphc.201500551
Domingos, S. R., Huerta-Viga, A., Baij, L., Amirjalayer, S., Dunnebier, D. A. E., Walters, A. J. C., et al. (2014). Amplified vibrational circular dichroism as a probe of local biomolecular structure. J. Am. Chem. Soc. 136, 3530–3535. doi:10.1021/ja411405s
Domingos, S. R., Sanders, H. J., Hartl, F., Buma, W. J., and Woutersen, S. (2014). Cover picture: Switchable amplification of vibrational circular dichroism as a probe of local chiral structure (Angew. Chem. Int. Ed. 51/2014). Angew. Chem. Int. Ed. 126, 14266–14269. doi:10.1002/ange.201407376
Du, L., King, J. B., Morrow, B. H., Shen, J. K., Miller, A. N., and Cichewicz, R. H. (2012). Diarylcyclopentendione metabolite obtained from a Preussia typharum isolate procured using an unconventional cultivation approach. J. Nat. Prod. 75, 1819–1823. doi:10.1021/np300473h
ECD application example Zhang, Y., Yu, Y. Y., Peng, F., Duan, W. T., Wu, C. H., Li, H. T., et al. (2021). Neolignans and diarylheptanoids with anti-inflammatory activity from the rhizomes of Alpinia zerumbet. J. Agric. Food Chem. 69, 9229–9237. doi:10.1021/acs.jafc.1c02271
Egidi, F., Giovannini, T., Frate, G. D., Lemler, P. M., Vaccaro, P. H., and Cappelli, C. (2019). A combined experimental and theoretical study of optical rotatory dispersion for (R)-glycidyl methyl ether in aqueous solution. Phys. Chem. Chem. Phys. 21, 3644–3655. doi:10.1039/C8CP04445G
El-Kashef, D. H., Daletos, G., Plenker, M., Hartmann, R., Mandi, A., Kurtan, T., et al. (2019). Polyketides and a dihydroquinolone alkaloid from a marine-derived strain of the fungus Metarhizium marquandii. J. Nat. Prod. 82, 2460–2469. doi:10.1021/acs.jnatprod.9b00125
Enamullah, M., Makhloufi, G., Ahmed, R., Joy, B. A., Islam, M. A., Padula, D., et al. (2016). Synthesis, X-ray, and spectroscopic study of dissymmetric tetrahedral Zinc(II) complexes from chiral schiff base naphthaldiminate ligands with apparent exception to the ECD exciton chirality. Inorg. Chem. 55, 6449–6464. doi:10.1021/acs.inorgchem.6b00403
Evidente, A., Cimmino, A., Andolfi, A., Vurro, M., Zonno, M. C., and Motta, A. (2008). Phyllostoxin and phyllostin, bioactive metabolites produced by Phyllosticta cirsii, a potential mycoherbicide for Cirsium arvense Biocontrol. J. Agric. Food Chem. 56, 884–888. doi:10.1021/jf0731301
Fang, S. T., Liu, X. H., Yan, B. F., Miao, F. P., Yin, X. L., Li, W. Z., et al. (2021). Terpenoids from the marine-derived fungus Aspergillus sp. RR-YLW-12, associated with the red alga Rhodomela confervoides. J. Nat. Prod. 84, 1763–1771. doi:10.1021/acs.jnatprod.1c00021
Farkas, V., Jakli, I., Toth, G. K., and Perczel, A. (2016). Aromatic cluster sensor of protein folding: Near-UV electronic circular dichroism bands assigned to fold compactness. Chem. Eur. J. 22, 13871–13883. doi:10.1002/chem.201602455
Farkas, V., Nagy, A., Menyhard, D. K., and Perczel, A. (2019). Assignment of vibrational circular dichroism cross-referenced electronic circular dichroism spectra of flexible foldamer building blocks: Towards assigning pure chiroptical properties of foldamers. Chem. Eur. J. 25, 14890–14900. doi:10.1002/chem.201903023
Felippe, L. G., Batista, J. M. J., Baldoqui, D. C., Nascimento, I. R., Kato, M. J., He, Y., et al. (2012). VCD to determine absolute configuration of natural product molecules: Secolignans from P. eperomia Bl. Org. Biomolec. Chem. 10, 4208–4214. doi:10.1039/c2ob25109d
Fernández, B., Rodríguez, R., Quiñoá, E., Riguera, R., and Freire, F. (2019). Decoding the ECD spectra of poly (phenylacetylene)s: Structural significance. ACS Omega 4, 5233–5240. doi:10.1021/acsomega.9b00122
Figueroa, M., González, M. C., Rodríguez-Sotres, R., Sosa-Peinado, A., González-Andrade, M., Cerda-García-Rojas, C. M., et al. (2009). Calmodulin inhibitors from the fungus Emericella sp. Bioorg. Med. Chem. 17, 2167–2174. doi:10.1016/j.bmc.2008.10.079
Fischbach, M. A., and Walsh, C. T. (2009). Antibiotics for emerging pathogens. Science 325, 1089–1093. doi:10.1126/science.1176667
Fleming, A. M., Alshykhly, O., Orendt, A. M., and Burrows, C. J. (2015). Computational studies of electronic circular dichroism spectra predict absolute configuration assignments for the guanine oxidation product 5-carboxamido-5-formamido-2-iminohydantoin. Tetrahedron Lett. 56, 3191–3196. doi:10.1016/j.tetlet.2014.12.052
For compound 24: Li, X. N., Zhang, Y., Cai, X. H., Feng, T., Liu, Y. P., Li, Y., et al. (2011). Psychotripine: A new trimeric pyrroloindoline derivative from Psychotria pilifera. Org. Lett. 13, 5896–5899. doi:10.1021/ol202536b
For 67, See: Liu, Z. M., Chen, Y., Chen, S. H., Liu, Y. Y., Lu, Y. J., Chen, D. N., et al. (2016). Aspterpenacids A and B, two sesterterpenoids from a mangrove endophytic fungus Aspergillus terreus H010. Org. Lett. 18, 1406–1409. doi:10.1021/acs.orglett.6b00336
For 68, see: Maslovskaya, L. A., Savchenko, A. I., Gordon, V. A., Reddell, P. W., Pierce, C. J., Parsons, P. G., et al. (2017). The first casbane hydroperoxides EBC-304 and EBC-320 from the Australian rainforest. Chem. Eur. J. 23, 537–540. doi:10.1002/chem.201604674
For 69, see: Nakashima, T., Miyano, R., Matsuo, R. H., Iwatsuki, M., Shirahata, T., Kobayashi, Y., et al. (2016). Absolute configuration of iminimycin B, a new indolizidine alkaloid, from Streptomyces griseus OS-3601. Tetrahedron Lett. 57, 3284–3286. doi:10.1016/j.tetlet.2016.06.040
For 70, see: Xu, L. L., Zhao, Q. Q., Yu, H., Wang, J. C., Wanf, H. J., Yang, Q., et al. (2016). Absolute configuration determination of one new compound trichoderol A from Trichoderma sp fungus. Chem. J. Chin. Univ. 37, 1972–1976. doi:10.7503/cjcu20160499
For 71, see: Bultinck, P., Cherblanc, F. L., Fuchter, M. J., Herrebout, W. A., Lo, Y. P., Rzepa, H. S., et al. (2015). Chiroptical studies on brevianamide B: Vibrational and electronic circular dichroism confronted. J. Org. Chem. 80, 3359–3367. doi:10.1021/jo5022647
For 72, see: Sherer, E. C., Cheeseman, J. R., and Williamson, R. T. (2015). Absolute configuration of remisporines A & B. Org. Biomol. Chem. 13, 4169–4173. doi:10.1039/C5OB00082C
For 73, see: Yu, S., Li, F., Jeon, H., Lee, S., Shin, J., and Kim, S. (2016). Total syntheses of isowondonins based on a biosynthetic pathway. Org. Lett. 18, 2986–2989. doi:10.1021/acs.orglett.6b01336
For 74, see: Endo, Y., Kasahara, T., Harada, K., Kubo, M., Etoh, T., Ishibashi, M., et al. (2017). Furanocassane diterpenoids from the seeds of Bowdichia virgilioides. J. Nat. Prod. 80, 3120–3127. doi:10.1021/acs.jnatprod.7b00249
For 75, see: Xu, W. J., Tang, P. F., Lu, W. J., Zhang, Y. Q., Wang, X. B., Zhang, H., et al. (2019). Hyperberins A and B, type B polycyclic polyprenylated acylphloroglucinols with bicyclo[5.3.1]hendecane core from Hypericum beanie. Org. Lett. 21, 8558–8562. doi:10.1021/acs.orglett.9b03098
For 76, see: Pazderkova, M., Profant, V., Seidlerova, B., Dlouha, H., Hodacova, J., Javorfi, T., et al. (2014). Electronic circular dichroism of the chiral rigid tricyclic dilactam with nonplanar tertiary amide groups. J. Phys. Chem. B 118, 11100–11108. doi:10.1021/jp5063463
For 77, see: Zhang, Y. L., Ge, H. M., Zhao, W., Dong, H., Xu, Q., Li, S. H., et al. (2008). Unprecedented immunosuppressive polyketides from Daldinia eschscholzii, a mantis-associated fungus. Angew. Chem. Int. Ed. 47, 5823–5826. doi:10.1002/anie.200801284
For 78, see: Zhang, H., Zhang, D. D., Lao, Y. Z., Fu, W. W., Liang, S., Yuan, Q. H., et al. (2014). Cytotoxic and anti-inflammatory prenylated benzoylphloroglucinols and xanthones from the twigs of Garcinia esculenta. J. Nat. Prod. 77, 1700–1707. doi:10.1021/np5003498
For 79: Zhou, D., Chen, G., Ma, Y. P., Wang, C. G., Lin, B., Yang, Y. Q., et al. (2019). Isolation, structural elucidation, optical resolution, and antineuroinflammatory activity of phenanthrene and 9,10-dihydrophenanthrene derivatives from Bletilla striata. J. Nat. Prod. 82, 2238–2245. doi:10.1021/acs.jnatprod.9b00291
For 80, see: Lu, W. J., Xu, W. J., Zhang, M. H., Zhang, Y. Q., Li, Y. R., Zhang, H., et al. (2021). Diverse polycyclic polyprenylated acylphloroglucinol congeners with anti-nonalcoholic steatohepatitis activity from Hypericum forrestii. J. Nat. Prod. 84, 1135–1148. doi:10.1021/acs.jnatprod.0c01202
For 81, see: He, Q., Fan, Y., Liu, Y., Rao, L., You, Y. X., Su, Y., et al. (2021). Cryptoyunnanones A–H, complex flavanones from Cryptocarya yunnanensis. J. Nat. Prod. 84, 2209–2216. doi:10.1021/acs.jnatprod.1c00287
For 82, see: Sun, Y. P., Li, Q. R., Sun, Y. J., Cui, L. T., Wang, Y. Y., Li, Y. Y., et al. (2021). Limonoids with diverse oxidation patterns of C-12 indicating a complete ring C-seco biogenetic pathway from Munronia unifoliolata. J. Nat. Prod. 84, 2352–2365. doi:10.1021/acs.jnatprod.1c00519
For 83, see: Xue, P. H., Zhang, N., Liu, D., Zhang, Q. R., Duan, J. S., Yu, Y. Q., et al. (2021). Cytotoxic and anti-inflammatory sesquiterpenes from the whole plants of Centipeda minima. J. Nat. Prod. 84, 247–258. doi:10.1021/acs.jnatprod.0c00884
For 84, see: Yang, F., Su, B. J., Hu, Y. J., Liu, J. L., Li, H., Wang, Y. Q., et al. (2021). Piperhancins A and B, two pairs of antineuroinflammatory cycloneolignane enantiomers from Piper hancei. J. Org. Chem. 86, 5284–5291. doi:10.1021/acs.joc.1c00240
For compound 117, see: Cao, F., Meng, Z., Wang, P., Luo, D., and Zhu, H. J. (2020). Dipleosporalones A and B, dimeric azaphilones from a marine-derived Pleosporales sp. Fungus. J. Nat. Prod. 83, 1283–1287. doi:10.1021/acs.jnatprod.0c00132
For compound 116, see: Cao, F., Meng, Z. H., Mu, X., Yue, Y. F., and Zhu, H. J. (2019). Absolute configuration of bioactive azaphilones from the marine-derived fungus Pleosporales sp. CF091. J. Nat. Prod. 82, 386–392. doi:10.1021/acs.jnatprod.8b01030
Forsyth, D. A., and Sebag, A. B. (1997). Computed 13C NMR chemical shifts via empirically scaled GIAO shieldings and molecular mechanics geometries. Conformation and configuration from 13C shifts. J. Am. Chem. Soc. 119, 9483–9494. doi:10.1021/ja970112z
Frelek, J., Karchier, M., Madej, D., Michalak, K., Różański, P., and Wicha, J. (2014). Chemoenzymatic approach to optically active 4-hydroxy-5-alkylcyclopent-2-en-1-one derivatives: An application of a combined circular dichroism spectroscopy and DFT calculations to assignment of absolute configuration. Chirality 26, 300–306. doi:10.1002/chir.22322
Fujii, K., Shimoya, T., Ikai, Y., Oka, H., and Harada, K. (1998). Further application of advanced Marfey's method for determination of absolute configuration of primary amino compound. Tetrahedron Lett. 39, 2579–2582. doi:10.1016/S0040-4039(98)00273-1
Fuse, M., Mazzeo, G., Longhi, G., Abbate, S., Masi, M., Evidente, A., et al. (2019). Unbiased determination of absolute configurations by vis-à-vis comparison of experimental and simulated spectra: The challenging case of diplopyrone. J. Phys. Chem. B 123, 9230–9237. doi:10.1021/acs.jpcb.9b08375
Gabrieli, S., Mazzeo, G., Longhi, G., Abbate, S., and Benincori, T. (2016). Discrimination of axial and central stereogenic elements in chiral bis(oxazolines) based on atropisomeric 3, 3’-bithiophene scaffolds through chiroptical spectroscopies. Chirality 28, 686–695. doi:10.1002/chir.22633
Gao, Y., Duan, F. F., Liu, L., Peng, X. G., Meng, X. G., and Ruan, H. L. (2021). Hypothemycin-type resorcylic acid lactones with Immunosuppressive activities from a Podospora sp. J. Nat. Prod. 84, 483–494. doi:10.1021/acs.jnatprod.0c01344
Garcia, K. Y. M., Phukhamsakda, C., Quimque, M. T. J., Hyde, K. D., Stadler, M., and Macabeo, A. P. G. (2021). Catechol-bearing polyketide derivatives from Sparticola junci. J. Nat. Prod. 84, 2053–2058. doi:10.1021/acs.jnatprod.1c00415
GaussView6 (2022). GaussView6. Available at: https://gaussian.com.
Ghidinelli, S., Abbate, S., Boiadjiev, S. E., Lightner, D. A., and Longhi, G. (2018). l-stercobilin-HCl and d-urobilin-HCl. analysis of their chiroptical and conformational properties by VCD, ECD, and CPL experiments and MD and DFT calculations. J. Phys. Chem. B 122, 12351–12362. doi:10.1021/acs.jpcb.8b07954
Gimenesa, L., Juniora, J. M. B., Junior, F. M. D. S., Souza, M. D. S., Dulcey, L. L., Ellena, J. E., et al. (2019). Picraviane A and B: Nortriterpenes with limonoid-like skeletons containing a heptanolide e-ring system from picramnia glazioviana. Phytochem 163, 38–45. doi:10.1016/j.phytochem.2019.03.024
Giovannini, T., Olszowka, M., and Cappelli, C. (2016). Effective fully polarizable QM/MM approach to model vibrational circular dichroism spectra of systems in aqueous solution. J. Chem. Theory Comput. 12, 5483–5492. doi:10.1021/acs.jctc.6b00768
Gliemann, B. D., Petrovic, A. G., Zolnhofer, E. M., Dral, P. O., Hampel, F., Breitenbruch, G., et al. (2017). Configurationally stable chiral dithia-bridged hetero[4]helicene radical cation: Electronic structure and absolute configuration. Chem. Asian J. 12, 31–35. doi:10.1002/asia.201601452
Góbi, S., Magyarfalvi, G., and Tarczay, G. (2015). VCD robustness of the amide-I and amide-II vibrational modes of small peptide models. Chirality 27, 625–634. doi:10.1002/chir.22475
Goodman Group (2017). DP4 website provided by Goodman and coworkers. Avilable at: http://www-jmg.ch.cam.ac.uk/tools/nmr/DP4/.
Gordon, M. S., Fedorov, D. G., Pruitt, S. R., and Slipchenko, L. V. (2012). Fragmentation methods: A route to accurate calculations on large systems. Chem. Rev. 112, 632–672. doi:10.1021/cr200093j
Górecki, M., Groszek, G., and Frelek, J. (2017). Chirality sensing of bioactive compounds with amino alcohol unit via circular dichroism. Chirality 29, 589–598. doi:10.1002/chir.22736
Górecki, M., Jabłonska, E., Kruszewska, A., Suszczynska, A., Urbanczyk-Lipkowska, Z., Gerards, M., et al. (2007). Practical method for the absolute configuration assignment of tert/tert 1, 2-Diols using their complexes with Mo2(OAc)4. J. Org. Chem. 72, 2906–2916. doi:10.1021/jo062445x
Goto, K., Yamaguchi, R., Hiroto, S., Ueno, H., Kawai, T., and Shinokubo, H. (2012). Intermolecular oxidative annulation of 2-aminoanthracenes to diazaacenes and aza[7]helicenes. Angew. Chem. Int. Ed. 51, 10333–10336. doi:10.1002/anie.201204863
Grauso, L., Li, Y., Scarpato, S., Shulha, O., Rarova, L., Strnad, M., et al. (2020). Structure and conformation of zosteraphenols, tetracyclic diarylheptanoids from the seagrass zostera marina: An NMR and DFT study. Org. Lett. 22, 78–82. doi:10.1021/acs.orglett.9b03964
Grauso, L., Teta, R., Esposito, G., Menna, M., and Mangoni, A. (2019). Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 36, 1005–1030. doi:10.1039/C9NP00018F
Grimblat, N., Zanardi, M. M., and Sarotti, A. M. (2015). Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 80, 12526–12534. doi:10.1021/acs.joc.5b02396
Guo, X., Meng, Q. Y., Niu, S. W., Liu, J., Guo, X. C., Sun, Z. L., et al. (2021). Epigenetic manipulation to trigger production of guaiane-type sesquiterpenes from a marine-derived Spiromastix sp. fungus with antineuroinflammatory effects. J. Nat. Prod. 84, 1993–2003. doi:10.1021/acs.jnatprod.1c00293
Guo, X. C., Zhang, Y. H., Gao, W. B., Pan, L., Zhu, H. J., and Cao, F. (2020). Absolute configurations and chitinase inhibitions of quinazoline-containing diketopiperazines from the marine-derived fungus Penicillium polonicum. Mar. Drugs 18, 479. doi:10.3390/md18090479
Guo, Y. J., Ding, L., Ghidinelli, S., Gotfredsen, C. H., Cruz, M., Mackenzie, T. A., et al. (2021). Taxonomy driven discovery of polyketides from Aspergillus californicus. J. Nat. Prod. 84, 979–985. doi:10.1021/acs.jnatprod.0c00866
Gussem, E. D., Herrebout, W., Specklin, S., Meyer, C., Cossy, J., and Bultinck, P. (2014). Strength by joining methods: Combining synthesis with NMR, IR, and vibrational circular dichroism spectroscopy for the determination of the relative configuration in hemicalide. Chem. Eur. J. 20, 17385–17394. doi:10.1002/chem.201404822
Haghdani, S., Gautun, O. R., Koch, H., and Åstrand, P. O. (2016). Optical rotation calculations for a set of pyrrole compounds. J. Phys. Chem. A 120, 7351–7360. doi:10.1021/acs.jpca.6b07004
Hajıcek, J., and Trojanek, J. (1981). Stereospecific total synthesis of (±)-strempeliopine. Tetrahedron Lett. 22, 2927–2928. doi:10.1016/S0040-4039(01)81789-5
Halgren, T. A. (1996). Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519. doi:10.1002/(SICI)1096-987X(199604)17:5/6<490:AID-JCC1>3.0.CO;2-P
Halgren, T. A. (1999). MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 20, 720–729. doi:10.1002/(sici)1096-987x(199905)20:7<720:aid-jcc7>3.0.co;2-x
Han, J. J., Li, Y., Zhou, J. C., Zhang, J. Z., Qiao, Y. N., Fang, K. L., et al. (2021). Terpenoids from Chinese liverworts Scapania spp. J. Nat. Prod. 84, 1210–1215. doi:10.1021/acs.jnatprod.0c01284
Hartung, J., and Grubbs, R. H. (2014). Catalytic, enantioselective synthesis of 1, 2-anti-diols by asymmetric ring-opening/cross-metathesis. Angew. Chem. Int. Ed. Engl. 126, 3885–3888. doi:10.1002/anie.201310767
He, J., Xu, J. K., Guo, L. B., Xia, C. Y., Lian, W. W., Tian, H. Y., et al. (2021). Fischdiabietane A, an antitumoral diterpenoid dimer featuring an unprecedented carbon skeleton from Euphorbia fischeriana. J. Org. Chem. 86, 5894–5900. doi:10.1021/acs.joc.1c00305
He, P., Wang, X. F., Guo, X. J., Ji, Y. N., Zhou, C. Q., Shen, S. G., et al. (2014). Vibrational circular dichroism study for natural bioactive schizandrin and reassignment of its absolute configuration. Tetrahedron Lett. 55, 2965–2968. doi:10.1016/j.tetlet.2014.03.108
He, Y., Cao, X., Nafie, L. A., and Freedman, T. B. (2001). Ab initio VCD calculation of a transition-metal containing molecule and a new intensity enhancement mechanism for VCD. J. Am. Chem. Soc. 123, 11320–11321. doi:10.1021/ja016218i
Hellou, N., Srebro-Hooper, M., Favereau, L., Zinna, F., Caytan, E., Toupet, L., et al. (2017). Enantiopure cycloiridiated complexes bearing a pentahelicenic N-heterocyclic carbene and displaying long-lived circularly polarized phosphorescence. Angew. Chem. Int. Ed. 56, 8236–8239. doi:10.1002/anie.201704263
Hodecker, M., Biczysko, M., Dreuw, A., and Barone, V. (2016). Simulation of vacuum UV absorption and electronic circular dichroism spectra of methyl oxirane: The role of vibrational effects. J. Chem. Theory Comput. 12, 2820–2833. doi:10.1021/acs.jctc.6b00121
Hong, A., Jang, H., Jeong, C., Choi, M. C., Heo, J., and Kim, N. J. (2016). Electronic circular dichroism spectroscopy of jet-cooled phenylalanine and its hydrated clusters. J. Phys. Chem. Lett. 7, 4385–4390. doi:10.1021/acs.jpclett.6b01894
Hoye, T. R., Jeffrey, C. S., and Shao, F. (2007). Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2, 2451–2458. doi:10.1038/nprot.2007.354
Hu, B. Y., Zhao, Y. L., Xiong, D. S., He, Y. J., Zhou, Z. S., Zhu, P. F., et al. (2021). Potent antihyperuricemic triterpenoids based on two unprecedented scaffolds from the leaves of Alstonia scholaris. Org. Lett. 23, 4158–4162. doi:10.1021/acs.orglett.1c01102
Hua, Y., Han, L. D., and Chen, C. X. (2004). Six novel 5α-adynerin-type cardenolides from Parepigynum funingense. Helvetica Chim. Acta 87, 516–523. doi:10.1002/hlca.200490049
Hua, Y., Ren, J., Chen, C. X., and Zhu, H. J. (2007). Determinations of the configuration of C-20 in derivatives of adynerin using DFT/HF methods. Chem. Res. Chin. U. 23, 592–597. doi:10.1016/S1005-9040(07)60129-9
Huang, X. S., He, J., Niu, X. M., Menzel, K. D., Dahse, H. M., Grabley, S., et al. (2008). Benzopyrenomycin, a cytotoxic bacterial polyketide metabolite with a benzo[a]pyrene-type carbocyclic ring system. Angew. Chem. Int. Ed. 47, 3995–3998. doi:10.1002/anie.200800083
Huo, H. X., Duvall, J. R., Huang, M. Y., and Hong, R. (2014). Catalytic asymmetric allylation of carbonyl compounds and imines with allylic boronates. Org. Chem. Front. 1, 303–320. doi:10.1039/C3QO00081H
Ianni, F., Cerra, B., Moroni, G., Varfaj, I., Michele, A. D., Gioiello, A., et al. (2022). Combining molecular modeling approaches to establish the chromatographic enantiomer elution order in the absence of pure enantiomeric standards: A study case with two tetracyclic quinolines. SSCP 5, 662–670. doi:10.1002/sscp.202200073
Isogai, A., Washizu, M., Murakoshi, S., and Suzuki, A. (1985). A new shikimate derivative, methyl 5-lactyl shikimate lactone, from Penicillium sp. Agric. Biol. Chem. 49, 167–169. doi:10.1080/00021369.1985.10866673
Jahnigen, S., Scherrer, A., Vuilleumier, R., and Sebastian, D. (2018). Chiral crystal packing induces enhancement of vibrational circular dichroism. Angew. Chem. Int. Ed. 57, 13344–13348. doi:10.1002/anie.201805671
Jawiczuk, M., Górecki, M., Masnyk, M., and Frelek, J. (2015). Complementarity of electronic and vibrational circular dichroism based on stereochemical studies of vic-diols. Trends Anal. Chem. 73, 119–128. doi:10.1016/j.trac.2015.04.028
Jawiczuk, M., Rode, J. E., Suszczynska, A., Szugajew, A., and Frelek, J. (2014). The utility of dimolybdenum tetrakis(μ-isovalerate) and tetrakis(μ-pivalate) in the stereochemical studies of various transparent compounds. RSC Adv. 4, 43691–43707. doi:10.1039/C4RA07408D
Ji, N. Y., Liu, X. H., Miao, F. P., and Qiao, M. F. (2013). Aspeverin, a new alkaloid from an algicolous strain of Aspergillus versicolor. Org. Lett. 15, 2327–2329. doi:10.1021/ol4009624
Ji, Y. N., He, P., Li, J. L., Guo, X. J., Wang, X. F., Yu, H., et al. (2014). Stereochemistry of hydroxyscytalone isolated from Penicillium Oxalicum using VCD, ECD and OR methods. Chem. J. Chin. Univ. 35, 746–749. doi:10.7503/cjcu20131023
Ji, Y. N., Zhou, Q. Q., Liu, G. S., Zhu, T. H., Wang, Y. F., Fu, Y., et al. (2021). New protein tyrosine phosphatase inhibitors from fungus Aspergillus gorakhpurensis F07ZB1707. RSC Adv. 11, 10144–10153. doi:10.1039/d1ra00788b
Jia, Y. J., Shi, W. S., Hu, F. L., Zhu, H. J., Liu, L., and Ma, Z. Y. (2018). Cytotoxic activity of trichothecene compounds and derivatives from Myrothecium sp. Chem. J. Chin. Univ. 39, 1668–1675. doi:10.7503/cjcu20180007
Jiang, C. X., Yu, B., Miao, Y. M., Ren, H., Xu, Q. H., Zhao, C., et al. (2021). Indole alkaloids from a soil-derived Clonostachys rosea. J. Nat. Prod. 84, 2468–2474. doi:10.1021/acs.jnatprod.1c00457
Jiang, L., Zhang, X., Sato, Y. Y., Zhu, G. L., Minami, A., Zhang, W. Y., et al. (2021). Genome-based discovery of enantiomeric pentacyclic sesterterpenes catalyzed by fungal bifunctional terpene synthases. Org. Lett. 23, 4645–4650. doi:10.1021/acs.orglett.1c01361
Jimenez, D. E. Q., Barreiro, J. C., dos Santos, F. M., de Vasconcellos, S. P., Porto, A. L. M., and Batista, J. M. (2019). Enantioselective ene-reduction of E-2-cyano-3-(furan-2-yl) acrylamide by marine and terrestrial fungi and absolute configuration of (R)-2-cyano-3-(furan-2-yl) propanamide determined by calculations of electronic circular dichroism (ECD) spectra. Chirality 31, 534–542. doi:10.1002/chir.23078
Jin, E. J., Li, H. Y., Liu, Z. Z., Xiao, F., and Li, W. L. (2021). Antibiotic dixiamycins from a cold-seep-derived Streptomyces olivaceus. J. Nat. Prod. 84, 2606–2611. doi:10.1021/acs.jnatprod.1c00411
Johnson, J. L., Nair, D. S., Pillai, S. M., Johnson, D., Kallingathodi, Z., Ibnusaud, I., et al. (2019). Dissymmetry factor spectral analysis can provide useful diastereomer discrimination: Chiral molecular structure of an analogue of (-)-crispine A. ACS Omega 4, 6154–6164. doi:10.1021/acsomega.8b03678
Johnson, J. L., Raghavan, V., Cimmino, A., Moeini, A., Petrovic, A. G., Santoro, E., et al. (2018). Absolute configurations of chiral molecules with multiple stereogenic centers without prior knowledge of the relative configurations: A case study of inuloxin C. Chirality 30, 1206–1214. doi:10.1002/chir.23013
Joly, D., Bouit, P. A., and Hissler, M. (2016). Organophosphorus derivatives for electronic devices. J. Mat. Chem. C 4, 3686–3698. doi:10.1039/C6TC00590J
Jose, K. V. J., Beckett, D., and Raghavachari, K. (2015). Vibrational circular dichroism spectra for large molecules through molecules-in-molecules fragment-based approach. J. Chem. Theory Comput. 11, 4238–4247. doi:10.1021/acs.jctc.5b00647
Junior, F. M. S., Covington, C. L., Albuquerque, A. C. F. D., Lobo, J. F. R., Borges, R. M., Amorim, M. B. D., et al. (2015). Absolute configuration of (-)-centratherin, a sesquiterpenoid lactone, defined by means of chiroptical spectroscopy. J. Nat. Prod. 78, 2617–2623. doi:10.1021/acs.jnatprod.5b00546
Junior, F. M. S., Covington, C. L., Amorim, M. B. D., Velozo, L. S. M., Kaplan, M. A. C., and Polavarapu, P. L. (2014). Absolute configuration of a rare sesquiterpene: (+)-3-Ishwarone. J. Nat. Prod. 77, 1881–1886. doi:10.1021/np500363e
Kasamatsu, K., Yoshimura, T., Mandi, A., Taniguchi, T., Monde, K., Furuta, T., et al. (2017). α-Arylation of α-amino acid derivatives with arynes via memory of chirality: Asymmetric synthesis of benzocyclobutenones with tetrasubstituted carbon. Org. Lett. 19, 352–355. doi:10.1021/acs.orglett.6b03533
Keiderling, T. A., and Lakhani, A. (2018). Mini review: Instrumentation for vibrational circular dichroism spectroscopy, still a role for dispersive instruments. Chirality 30, 238–253. doi:10.1002/chir.22799
Kessler, J., Keiderling, T. A., and Bour, P. (2014). Arrangement of fibril side chains studied by molecular dynamics and simulated infrared and vibrational circular dichroism spectra. J. Phys. Chem. B 118, 6937–6945. doi:10.1021/jp502178d
Kim, J. G., Lee, J. W., Le, T. P. L., Han, J. S., Kwon, H., Lee, D., et al. (2021). Diterpenoids and diacetylenes from the roots of Aralia cordata with inhibitory effects on nitric oxide production. J. Nat. Prod. 84, 230–238. doi:10.1021/acs.jnatprod.0c00842
Koenis, M. A. J., Osypenko, A., Fuks, G., Giuseppone, N., Nicu, V. P., Visscher, L., et al. (2020). Self-assembly of supramolecular polymers of N-centered triarylamine trisamides in the light of circular dichroism: Reaching consensus between electrons and nuclei. J. Am. Chem. Soc. 142, 1020–1028. doi:10.1021/jacs.9b11306
Koenis, M. A. J., Xia, Y. Y., Domingos, S. R., Visscher, L., Buma, W. J., and Nicu, V. P. (2019). Taming conformational heterogeneity in and with vibrational circular dichroism spectroscopy. Chem. Sci. 10, 7680–7689. doi:10.1039/c9sc02866h
Kohout, M., Vandenbussche, J., Roller, A., Tůma, J., Bogaerts, J., Bultinck, P., et al. (2016). Absolute configuration of the antimalarial erythro-mefloquine - vibrational circular dichroism and X-ray diffraction studies of mefloquine and its thiourea derivative. RSC Adv. 6, 81461–81465. doi:10.1039/C6RA19367F
Kondru, R. K., Lim, S., Wipf, P., and Beratan, D. N. (1997). Synthetic and model computational studies of molar rotation additivity for interacting chiral centers: A reinvestigation of van't hoff's principle. Chirality 9, 469–477. doi:10.1002/(SICI)1520-636X(1997)9:5/6<469:AID-CHIR13>3.0.CO;2-M
Kondru, R. K., Wipf, P., and Beratan, D. N. (1998). Theory-assisted determination of absolute stereochemistry for complex natural products via computation of molar rotation angles. J. Am. Chem. Soc. 120, 2204–2205. doi:10.1021/ja973690o
Kong, J., Joyce, L. A., Liu, J. C., Jarrell, T. M., Culberson, J. C., and Sherer, E. C. (2017). Absolute configuration assignment of (+)-fluralaner using vibrational circular dichroism. Chirality 29, 854–864. doi:10.1002/chir.22770
Kouamé, T., Bernadat, G., Turpin, V., Litaudon, M., Okpekon, A. T., Gallard, J. F., et al. (2021). Structure reassignment of melonine and quantum-chemical calculations-based assessment of biosynthetic scenarios leading to its revised and original structures. Org. Lett. 23, 5964–5968. doi:10.1021/acs.orglett.1c02055
Kreienborg, N. M., Pollok, C. H., and Merten, C. (2016). Towards an observation of active conformations in asymmetric catalysis: Interaction-induced conformational preferences of a chiral thiourea model compound. Chem. Eur. J. 22, 12455–12463. doi:10.1002/chem.201602097
Kurouski, D., Lu, X. F., Popova, L., Wan, W., Shanmugasundaram, M., Stubbs, G., et al. (2014). Is supramolecular filament chirality the underlying cause of major morphology differences in amyloid fibrils? J. Am. Chem. Soc. 136, 2302–2312. doi:10.1021/ja407583r
Laguna, A., Novotný, L., Dolejš, L., and Buděšínský, M. (1984). Alkaloids from roots of Strempeliopsis strempelioides - structures of strempeliopine and strempeliopidine. Planta Med. 50, 285–288. doi:10.1055/s-2007-969710
Lauro, G., and Bifulco, G. (2020). Elucidating the relative and absolute configuration of organic compounds by quantum mechanical approaches. Eur. J. Org. Chem. 2020, 3929–3941. doi:10.1002/ejoc.201901878
Lee, D. H., Shin, J. S., Kang, S. Y., Lee, S. B., Lee, J. S., Ryu, S. M., et al. (2018). Iridoids from the roots of Patrinia scabra and their inhibitory potential on LPS-induced nitric oxide production. J. Nat. Prod. 81, 1468–1473. doi:10.1021/acs.jnatprod.8b00229
Lee, S., Kim, C. S., Yu, J. S., Kang, H., Yoo, M. J., Youn, U. J., et al. (2021). Ergopyrone, a styrylpyrone-fused steroid with a hexacyclic 6/5/6/6/6/5 skeleton from a mushroom Gymnopilus orientispectabilis. Org. Lett. 23, 3315–3319. doi:10.1021/acs.orglett.1c00790
Li, C. W., Wu, C. J., Cui, C. B., Xu, L. L., Cao, F., and Zhu, H. J. (2016). Penicimutamides A–C: Rare carbamate-containing alkaloids from a mutant of the marine-derived Penicillium purpurogenum G59. RSC Adv. 6, 73383–73387. doi:10.1039/C6RA14904A
Li, G., Li, H., Zhang, Q., Yang, M., Gu, Y. C., Liang, L. F., et al. (2019). Rare cembranoids from Chinese soft coral sarcophyton ehrenbergi: Structural and stereochemical studies. J. Org. Chem. 84, 5091–5098. doi:10.1021/acs.joc.9b00030
For compound 25: Li, G. H., Liu, F. F., Shen, L., Zhu, H. J., and Zhang, K. Q. (2011). Stereumins H-J, stereumane-type sesquiterpenes from the fungus Stereum sp. J. Nat. Prod. 74, 296–299. doi:10.1021/np100813f
Li, G. J., Kessler, J., Cheramy, J., Wu, T., Poopari, M. R., Bour, P., et al. (2019). Transfer and amplification of chirality within the “ring of fire” observed in resonance Raman optical activity experiments. Angew. Chem. Int. Ed. 58, 16647–16650. doi:10.1002/ange.201909603
Li, G. Y., Yang, T., Luo, Y. G., Chen, X. Z., Fang, D. M., and Zhang, G. L. (2009). Brevianamide J, A new indole alkaloid dimer from fungusAspergillus versicolor. Org. Lett. 11, 3714–3717. doi:10.1021/ol901304y
Li, J. J., Du, M., Zhao, Z. Q., and Liu, H. W. (2016). Cyclopolymerization of disiloxane-tethered divinyl monomers to synthesize chirality-responsive helical polymers. Macromolecules 49, 445–454. doi:10.1021/acs.macromol.5b02142
Li, L., Chang, Q. H., Zhang, S. S., Yang, K., Chen, F. L., Zhu, H. J., et al. (2022). (±)-Brevianamides Z and Z1, new diketopiperazine alkaloids from the marine-derived fungus Aspergillus versicolor. J. Mol. Struct. 1261, 132904. doi:10.1016/j.molstruc.2022.132904
Li, L., and Si, Y. K. (2012). Study on the absolute configurations of 3-alkylphthalides using TDDFT calculations of chiroptical properties. Chirality 24, 987–993. doi:10.1002/chir.22086
Li, Q. M., Ren, J., Shen, L., Bai, B., Li, Q. M., Liu, X. C., et al. (2013). Determining the absolute configuration of benzopyrenomycin by optical rotation, electronic circular dichroism, and population analysis of different conformations via DFT methods and experiments. Tetrahedron 69, 3067–3074. doi:10.1016/j.tet.2013.01.082
Li, W., Zhou, S. P., Jin, Y. P., Huang, X. F., Zhou, W., Han, M., et al. (2014). Salvianolic acids T and U: A pair of atropisomeric trimeric caffeic acids derivatives from root of Salvia miltiorrhiza. Fitoterapia 98, 248–253. doi:10.1016/j.fitote.2014.08.018
Li, Z., Li, L. W., Zheng, Y., Chen, C., and Sun, T. (2018). Diagnostic absolute configuration determination of clavulanate potassium: A comprehensive investigation of chiroptical spectroscopies and theoretical calculations. J. Pharm. Biomed. Anal. 160, 351–359. doi:10.1016/j.jpba.2018.08.010
Lin, L. B., Gao, Y. Q., Han, R., Xiao, J., Wang, Y. M., Zhang, Q., et al. (2021). Alkylated salicylaldehydes and prenylated indole alkaloids from the endolichenic fungus Aspergillus chevalieri and their bioactivities. J. Agric. Food Chem. 69, 6524–6534. doi:10.1021/acs.jafc.1c01148
Liu, D. Z., Wang, F., Liao, T. G., Tang, J. G., Steglich, W., Zhu, H. J., et al. (2006). Vibralactone: A lipase inhibitor with an unusual fused β-lactone produced by cultures of the basidiomycete Boreostereum vibrans. Org. Lett. 8, 5749–5752. doi:10.1021/ol062307u
Liu, M. X., Chen, L. X., Tian, T. T., Zhang, Z. G., and Li, X. J. (2019). Identification and quantitation of enantiomers by capillary electrophoresis and circular dichroism independent of single enantiomer standard. Anal. Chem. 91, 13803–13809. doi:10.1021/acs.analchem.9b03276
Liu, Y. F., Yue, Y. F., Feng, L. X., Zhu, H. J., and Cao, F. (2019). Asperienes A–D, bioactive sesquiterpenes from the marine-derived fungus Aspergillus flavus. Mar. Drugs 17, 550. doi:10.3390/md17100550
Liu, Y. L., Cerezo, J., Mazzeo, G., Lin, N., Zhao, X., Longhi, G., et al. (2016). Vibronic coupling explains the different shape of electronic circular dichroism and of circularly polarized luminescence spectra of hexahelicenes. J. Chem. Theory Comput. 12, 2799–2819. doi:10.1021/acs.jctc.6b00109
Liu, Y. X., Rakotondraibe, L. H., Brodie, P. J., Wiley, J. D., Cassera, M. B., Miller, J. S., et al. (2015). Antimalarial 5, 6-dihydro-α-pyrones from Cryptocarya rigidifolia: Related bicyclic tetrahydro-α-pyrones are artifacts. J. Nat. Prod. 78, 1330–1338. doi:10.1021/acs.jnatprod.5b00187
Lodewyk, M. W., Siebert, M. R., and Tantillo, D. J. (2012). Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 112, 1839–1862. doi:10.1021/cr200106v
Lourenço, T. C., Batista, J. M. J., Furlan, M., He, Y., Nafie, L. A., Santana, C. C., et al. (2012). Albendazole sulfoxide enantiomers: Preparative chiral separation and absolute stereochemistry. J. Chrom. A 1230, 61–65. doi:10.1016/j.chroma.2012.01.070
Lu, J. M., Yang, B. B., and Li, L. (2020). Specific optical rotation and absolute configuration of flexible molecules containing a 2-methylbutyl residue. Eur. J. Org. Chem. 2020, 4768–4774. doi:10.1002/ejoc.202000736
Lu, Z. Y., Zhu, H. J., Fu, P., Wang, Y., Zhang, Z. H., Lin, H. P., et al. (2010). Cytotoxic polyphenols from the marine-derived fungus Penicillium expansum. J. Nat. Prod. 73, 911–914. doi:10.1021/np100059m
Mandi, A., Mudianta, I. W., Kurtan, T., and Garson, M. J. (2015). Absolute configuration and conformational study of psammaplysins A and B from the Balinese marine sponge Aplysinella strongylata. Aplysinella strongylata J. Nat. Prod. 78, 2051–2056. doi:10.1021/acs.jnatprod.5b00369
María-Magdalena, C., and Jorge, B. (2015). Structure elucidation in organic chemistry: The search for the right tools. Weinheim, Germany: Wiley-VCH, Verlag GmbH & Co. KGaA, 65–104.
Marty, R., Frauenrath, H., and Helbing, J. (2014). Aggregates from perylene bisimide oligopeptides as a test case for giant vibrational circular dichroism. J. Phys. Chem. B 118, 11152–11160. doi:10.1021/jp506837c
Martynov, A. G., Mack, J., May, A. K., Nyokong, T., Gorbunova, Y. G., and Tsivadze, A. Y. (2019). Methodological survey of simplified td-dft methods for fast and accurate interpretation of UV–Vis–NIR spectra of phthalocyanines. ACS Omega 4, 7265–7284. doi:10.1021/acsomega.8b03500
Masnyk, M., Butkiewicz, A., Gorecki, M., Luboradzki, R., Bannwarth, C., Grimme, S., et al. (2016). Synthesis and comprehensive structural and chiroptical characterization of enones derived from (−)-α-Santonin by experiment and theory. J. Org. Chem. 81, 4588–4600. doi:10.1021/acs.joc.6b00416
Masnyk, M., Butkiewicz, A., Gorecki, M., Luboradzki, R., Paluch, P., Potrzebowski, M. J., et al. (2018). In depth analysis of chiroptical properties of enones derived from abietic acid. J. Org. Chem. 83, 3547–3561. doi:10.1021/acs.joc.7b02911
Masuda, Y., Fujihara, K., Hayashi, S., Sasaki, H., Kino, Y., Kamauchi, H., et al. (2021). Inhibition of BACE1 and amyloid-β aggregation by meroterpenoids from the mushroom Albatrellus yasudae. J. Nat. Prod. 84, 1748–1754. doi:10.1021/acs.jnatprod.0c01329
Mayhall, N. J., and Raghavachari, K. (2011). Molecules-in-molecules: An extrapolated fragment-based approach for accurate calculations on large molecules and materials. J. Chem. Theory Comput. 7, 1336–1343. doi:10.1021/ct200033b
Mazzeo, G., Abbate, S., Longhi, G., Castiglioni, E., Boiadjiev, S. E., and Lightner, D. A. (2016). pH dependent chiroptical properties of (1R, 2R)- and (1S, 2S)-trans-cyclohexane diesters and diamides from VCD, ECD, and CPL spectroscopy. J. Phys. Chem. B 120, 2380–2387. doi:10.1021/acs.jpcb.5b11223
Mazzeo, G., Abbate, S., Longhi, G., Castiglioni, E., and Villani, C. (2015). Vibrational circular dichroism (VCD) reveals subtle conformational aspects and intermolecular interactions in the carnitine family. Chirality 27, 907–913. doi:10.1002/chir.22532
Mazzeo, G., Cimmino, A., Andolfi, A., Evidente, A., and Superchi, S. (2014). Computational ECD spectrum simulation of the phytotoxin scytalone: Importance of solvent effects on conformer populations. Chirality 26, 502–508. doi:10.1002/chir.22311
Mazzeo, G., Cimmino, A., Masi, M., Longhi, G., Maddau, L., Memo, M., et al. (2017). Importance and difficulties in the use of chiroptical methods to assign the absolute configuration of natural products: The case of phytotoxic pyrones and furanones produced by Diplodia corticola. J. Nat. Prod. 80, 2406–2415. doi:10.1021/acs.jnatprod.7b00119
Mazzeo, G., Longhi, G., Corless, V. B., Zajdlik, A., Yudin, A. K., and Abbate, S. (2017). Vibrational circular dichroism unveils chiroptical, electrical, and magnetic properties of borylated isocyanides and aldehydes. Eur. J. Org. Chem. 2017, 5262–5268. doi:10.1002/ejoc.201701044
Mazzeo, G., Santoro, E., Andolfi, A., Cimmino, A., Troselj, P., Petrovic, A. G., et al. (2013). Absolute configurations of fungal and plant metabolites by chiroptical methods. ORD, ECD, and VCD studies on phyllostin, scytolide, and oxysporone. J. Nat. Prod. 76, 588–599. doi:10.1021/np300770s
Merten, C., Dirkmann, M., and Schulz, F. (2017). Stereochemical assignment of fusiccocadiene from NMR shielding constants and vibrational circular dichroism spectroscopy. Chirality 29, 409–414. doi:10.1002/chir.22708
Merten, C., McDonald, R., and Xu, Y. J. (2014). Strong solvent-dependent preference of Δ and Λ stereoisomers of a tris(diamine)nickel(II) complex revealed by vibrational circular dichroism spectroscopy. Inorg. Chem. 53, 3177–3182. doi:10.1021/ic4031766
Mirco, A. V., Maurizio, B., Sergio, R., Tiziana, B., Roberto, C., and Marco, P. (2019). TetraPh-tol-BITIOPO: A new atropisomeric 3, 3’-bithiophene based phosphine oxide as an organocatalyst in lewis base-catalyzed lewis acid mediated reactions. Org. Biomol. Chem. 17, 7474–7481. doi:10.1039/C9OB01297D
Mirtic, A., Merzel, F., and Grdadolnik, J. (2014). The amide III vibrational circular dichroism band as a probe to detect conformational preferences of alanine dipeptide in water. Biopolymers 101, 814–818. doi:10.1002/bip.22460
Miura, T., Nakamuro, T., Nagata, Y., Moriyama, D., Stewart, S. G., and Murakami, M. (2019). Asymmetric synthesis and stereochemical assignment of 12C/13C isotopomers. J. Am. Chem. Soc. 141, 13341–13345. doi:10.1021/jacs.9b07181
Miyake, H., Terada, K., and Tsukube, H. (2014). Lanthanide tris (β-diketonates) as useful probes for chirality determination of biological amino alcohols in vibrational circular dichroism: Ligand to ligand chirality transfer in lanthanide coordination sphere. Chirality 26, 293–299. doi:10.1002/chir.22319
Molinski, T. F., Biegelmeyer, R., Stout, E. P., Wang, X., Frota, M. L. C., and Henriques, A. T. (2013). Halisphingosines A and B, modified sphingoid bases from Haliclona tubifera. Assignment of configuration by circular dichroism and van’t Hoff’s principle of optical superposition. J. Nat. Prod. 76, 374–381. doi:10.1021/np300744y
Molteni, E., Onida, G., and Tiana, G. (2015). Conformational dependence of the circular dichroism spectra of single amino acids from plane-waves-based density functional theory calculations. J. Phys. Chem. B 119, 4803–4811. doi:10.1021/jp5118568
Monde, K., Taniguchi, T., Miura, N., Nishimura, S. I., Harada, N., Dukor, R. K., et al. (2003). Preparation of cruciferous phytoalexin related metabolites, (-)-dioxibrassinin and (-)-3-cyanomethyl-3-hydroxyoxindole, and determination of their absolute configurations by vibrational circular dichroism (VCD). Tetrahedron Lett. 44, 6017–6020. doi:10.1016/S0040-4039(03)01513-2
Mota, J. D. S., Leite, A. C., Batista, J. M., López, S. N., Ambrósio, D. L., Passerini, G. D., et al. (2009). In vitro trypanocidal activity of phenolic derivatives from Peperomia obtusifolia. Planta Med. 75, 620–623. doi:10.1055/s-0029-1185364
Mu, H. Y., Tang, S., Zuo, Q., Huang, M., and Zhao, W. M. (2021). Dihydro-β-agarofuran-type sesquiterpenoids from the seeds of Celastrus virens and their multidrug resistance reversal activity against the KB/VCR cell line. J. Nat. Prod. 84, 588–600. doi:10.1021/acs.jnatprod.0c01182
Mugishima, T., Tsuda, M., Kasai, Y., Ishiyama, H., Fukushi, E., Kawabata, J., et al. (2005). Absolute stereochemistry of citrinadins A and B from marine-derived fungus. J. Org. Chem. 70, 9430–9435. doi:10.1021/jo051499o
Muralidharam, V. B., Wood, H. B., and Ganem, B. (1990). Enantioselective synthesis of (−)-methyl 5-lactylshikimate lactone. Tetrahedron Lett. 31, 183–184. doi:10.1016/S0040-4039(00)94365-X
Nafie, L. A. (1983). Adiabatic molecular properties beyond the Born–Oppenheimer approximation. Complete adiabatic wave functions and vibrationally induced electronic current density. J. Chem. Phys. 79, 4950–4957. doi:10.1063/1.445588
Nafie, L. A., and Dukor, R. K. (2018). Vibrational optical activity in chiral analysis. Editor P. L Polavarapu Second Edition (Amsterdam: Elsevier).
Nafie, L. A. (1996). Theory of resonance Raman optical activity: The single electronic state limit. Chem. Phys. 205, 309–322. doi:10.1016/0301-0104(95)00400-9
Nafie, L. A. (2004). Theory of vibrational circular dichroism and infrared absorption: Extension to molecules with low-lying excited electronic states. J. Phys. Chem. A 108, 7222–7231. doi:10.1021/jp0499124
Nafie, L. A. (2008). Vibrational circular dichroism: A new tool for the solution-state determination of the structure and absolute configuration of chiral natural product molecules. Nat. Prod. Commun. 3, 1934578X0800300–466. doi:10.1177/1934578X0800300322
Nafie, L. A. (2020). Vibrational optical activity: From discovery and development to future challenges. Chirality 32, 667–692. doi:10.1002/chir.23191
Nafie, L. A. (2018). “Vibrational optical activity: From small chiral molecules to protein pharmaceuticals and beyond,” in Frontiers and advances in molecular spectroscopy. Editor J. Laane (New York: Elsevier), 421–469.
Nafie, L. A. (2011). Vibrational optical activity: Principles and applications. Chichester: John Wiley & Sons.
Nafie, L. A. (1992). Velocity-Gauge Formalism in the theory of vibrational circular dichroism and infrared absorption. J. Chem. Phys. 96, 5687–5702. doi:10.1063/1.462668
Nagle, D. G., Park, P. U., and Paul, V. J. (1997). Pitiamide A, a new chlorinated lipid from a mixed marine cyanobacterial assemblage. Tetrahedron Lett. 38, 6969–6972. doi:10.1016/S0040-4039(97)01666-3
Naito, J., Yamamoto, Y., Akagi, M., Sekiguchi, S., Watanabe, M., and Harada, N. (2005). Unambiguous Determination of the absolute configurations of acetylene alcohols by combination of the Sonogashira reaction and the CD exciton chirality method - exciton coupling between phenylacetylene and benzoate chromophores. Chem. Mon. 136, 411–445. doi:10.1007/s00706-005-0281-3
Nakahashi, A., Yaguchi, Y., Miura, N., Emura, M., and Monde, K. (2011). A vibrational circular dichroism approach to the determination of the absolute configurations of flavorous 5-substituted-2(5H)-furanones. J. Nat. Prod. 74, 707–711. doi:10.1021/np1007763
Nhoek, P., Chae, H. S., Kim, Y. M., Pel, P., Huh, J., Kim, H. W., et al. (2021). Sesquiterpenoids from the aerial parts of Salvia plebeia with inhibitory activities on proprotein convertase subtilisin/kexin type 9 expression. J. Nat. Prod. 84, 220–229. doi:10.1021/acs.jnatprod.0c00829
Nicu, V. P., Mándi, A., Kurtán, T., and Polavarapu, P. L. (2014). On the complementarity of ECD and VCD techniques. Chirality 26, 525–531. doi:10.1002/chir.22330
Nieto, B., Ramırez, F. J., Hennrich, G., Gomez-Lor, B., Casado, J., and Navarrete, J. T. L. (2010). Aggregation behavior of a conjugated C3-symmetric molecule: A description based on chiro-optical experimental and theoretical spectroscopies. J. Phys. Chem. B 114, 5710–5717. doi:10.1021/jp100628s
Nishigaki, S., Shibata, Y., Nakajima, A., Okajima, H., Masumoto, Y., Osawa, T., et al. (2019). Synthesis of belt- and möbius-shaped cycloparaphenylenes by rhodium-catalyzed alkyne cyclotrimerization. J. Am. Chem. Soc. 141, 14955–14960. doi:10.1021/jacs.9b06197
Niu, C. S., Leavitt, L. S., Lin, Z. J., Paguigan, N. D., Sun, L. L., Zhang, J., et al. (2021). Neuroactive type-A γ-aminobutyric acid receptor allosteric modulator steroids from the hypobranchial gland of marine mollusk, Conus geographus. J. Med. Chem. 64, 7033–7043. doi:10.1021/acs.jmedchem.1c00562
Niwa, R., and Niwa, Y. S. (2014). Enzymes for ecdysteroid biosynthesis: Their biological functions in insects and beyond. Biosci. Biotechnol. Biochem. 78, 1283–1292. doi:10.1080/09168451.2014.942250
Norman, A. W., Mizwicki, M. T., and Norman, D. P. G. (2004). Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat. Rev. Drug Discov. 3, 27–41. doi:10.1038/nrd1283
Oka, M., Kamei, H., Hamagishi, Y., Tomita, K., Miyaki, T., Konishi, M., et al. (1990). Chemical and biological properties of rubiginone, a complex of new antibiotics with vincristine-cytotoxicity potentiating activity. J. Antibiot. 43, 967–976. doi:10.7164/antibiotics.43.967
Omar, A. M., Sun, S. J., Kim, M. J., Phan, N. D., Tawila, A. M., and Awale, S. (2021). Benzophenones from Betula alnoides with antiausterity activities against the PANC-1 human pancreatic cancer cell line. J. Nat. Prod. 84, 1607–1616. doi:10.1021/acs.jnatprod.1c00150
Ortega, P. G. R., Montejo, M., and Gonzalez, J. J. L. (2015). Vibrational circular dichroism and theoretical study of the conformational equilibrium in (-)-s-nicotine. Chem. Phys. Chem. 16, 342–352. doi:10.1002/cphc.201402652
Ortega, P. G. R., Montejo, M., Mrquez, F., and Gonzlez, J. J. L. (2015). DFT-aided vibrational circular dichroism spectroscopy study of (-)-s-cotinine. ChemPhysChem 16, 1416–1427. doi:10.1002/cphc.201500018
Other corresponding packages Schrodinger LLC (2022). Schrodinger LLC. Available at: http://www.schrodinger.com.
Padula, D., Bari, L. D., and Pescitelli, G. (2016). The “Case of two compounds with similar configuration but nearly mirror image CD spectra” refuted. Reassignment of the absolute configuration of N-formyl-3’, 4’-dihydrospiro[indan-1, 2’(1’H)-pyridine]. J. Org. Chem. 81, 7725–7732. doi:10.1021/acs.joc.6b01416
Padula, D., Jurinovich, S., Bari, L. D., and Mennucci, B. (2016). Simulation of electronic circular dichroism of nucleic acids: From the structure to the spectrum. Chem. Eur. J. 22, 17011–17019. doi:10.1002/chem.201602777
Pandith, A. H., Islam, N., Syed, Z. F., Suhail-ul, R., Bandaru, S., and Anoop, A. (2011). Density functional theory prediction of geometry and vibrational circular dichroism of bridged triarylamine helicenes. Chem. Phy. Lett. 516, 199–203. doi:10.1016/j.cplett.2011.09.074
Paolino, M., Giovannini, T., Manathunga, M., Latterini, L., Zampini, G., Pierron, R., et al. (2021). On the transition from a biomimetic molecular switch to a rotary molecular motor. J. Phys. Chem. Lett. 12, 3875–3884. doi:10.1021/acs.jpclett.1c00526
Pardo-Novoa, J. C., Arreaga-Gonzalez, H. M., Gomez-Hurtado, M. A., Rodríguez-García, G., Cerda-García-Rojas, C. M., Joseph-Nathan, P., et al. (2016). Absolute configuration of menthene derivatives by vibrational circular dichroism. J. Nat. Prod. 79, 2570–2579. doi:10.1021/acs.jnatprod.6b00491
Passarello, M., Abbate, S., Longhi, G., Lepri, S., Ruzziconi, R., and Nicu, V. P. (2014). Importance of C*–H based modes and large amplitude motion effects in vibrational circular dichroism spectra: The case of the chiral adduct of dimethyl fumarate and anthracene. J. Phys. Chem. A 118, 4339–4350. doi:10.1021/jp502544v
Perez-Mellor, A., Alata, I., Lepere, V., and Zehnacker, A. (2019). Conformational study of the jet-cooled diketopiperazine peptide cyclo tyrosyl-prolyl. J. Phys. Chem. B 123, 6023–6033. doi:10.1021/acs.jpcb.9b04529
Pérez-Mellor, A., and Zehnacker, A. (2017). Vibrational circular dichroism of a 2, 5-diketopiperazine (DKP) peptide: Evidence for dimer formation in cyclo LL or LD diphenylalanine in the solid state. Chirality 29, 89–96. doi:10.1002/chir.22674
Pescitelli, G., and Bruhn, T. (2016). Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 28, 466–474. doi:10.1002/chir.22600
Pescitelli, G. (2018). For a correct application of the CD exciton chirality method: The case of laucysteinamide A. Mar. Drugs 16, 388–399. doi:10.3390/md16100388
Pezzuto, J. M., Lea, M. A., and Yang, C. S. (1976). Binding of metabolically activated benzo(a)pyrene to nuclear macromolecules. Cancer Res. 36, 3647–3653.
Pinto, M. E. F., Batista, J. M., Koehbach, J., Gaur, P., Sharma, A., Nakabashi, M., et al. (2015). Ribifolin, an orbitide from Jatropha ribifolia, and its potential antimalarial activity. J. Nat. Prod. 78, 374–380. doi:10.1021/np5007668
Podlech, J., Fleck, S. C., Metzler, M., Burck, J., and Ulrich, A. S. (2014). Determination of the absolute configuration of perylene quinone-derived mycotoxins by measurement and calculation of electronic circular dichroism spectra and specific rotations. Chem. Eur. J. 20, 11463–11470. doi:10.1002/chem.201402567
Polavarapu, P. L., and Covington, C. L. (2014). Comparison of experimental and calculated chiroptical spectra for chiral molecular structure determination. Chirality 26, 539–552. doi:10.1002/chir.22316
Polavarapu, P. L. (2016). Determination of the absolute configurations of chiral drugs using chiroptical spectroscopy. Molecules 21, 1056–1069. doi:10.3390/molecules21081056
Polavarapu, P. L., and Santoro, E. (2020). Vibrational optical activity for structural characterization of natural products. Nat. Prod. Rep. 37, 1661–1699. doi:10.1039/D0NP00025F
Popik, O., Pasternak-Suder, M., Lesniak, K., Jawiczuk, M., Górecki, M., Frelek, J., et al. (2014). Amine-catalyzed direct aldol reactions of hydroxy- and dihydroxyacetone: Biomimetic synthesis of carbohydrates. J. Org. Chem. 79, 5728–5739. doi:10.1021/jo500860g
Progresses in ECD study Nørby, M. S., Olsen, J. M. H., Steinmann, C., and Kongsted, J. (2017). Modeling electronic circular dichroism within the polarizable embedding approach. J. Chem. Theory Comput. 13, 4442–4451. doi:10.1021/acs.jctc.7b00712
Qin, X. D., Dong, Z. J., Liu, J. K., Yang, L. M., Wang, R. R., Zheng, Y. T., et al. (2006). Concentricolide, an anti-HIV agent from the Ascomycete Daldinia concentrica. Helv. Chim. Acta. 89, 127–133. doi:10.1002/hlca.200690004
Qiu, S., Gussem, E. D., Tehrani, K. A., Sergeyev, S., Bultinck, P., and Herrebout, W. (2013). Stereochemistry of the tadalafil diastereoisomers: A critical assessment of vibrational circular dichroism, electronic circular dichroism, and optical rotatory dispersion. J. Med. Chem. 56, 8903–8914. doi:10.1021/jm401407w
Quan, K. T., Park, H. B., Yuk, H., Lee, S. J., and Na, M. (2021). Paratrimerins J–Y, dimeric coumarins isolated from the stems of Paramignya trimera. J. Nat. Prod. 84, 310–326. doi:10.1021/acs.jnatprod.0c00978
Raabe, G., Fleischhauer, J., and Woody, R. W. (2012). Comprehensive chiroptical spectroscopy. New York: Wiley, 543–591.
Raghavan, V., and Polavarapu, P. L. (2017). Specific optical rotation is a versatile tool for the identification of critical micelle concentration and micellar growth of tartaric acid–based diastereomeric amphiphiles. Chirality 29, 836–846. doi:10.1002/chir.22767
Ramalho, T. C., and Buehl, M. (2005). Probing NMR parameters, structure and dynamics of 5-nitroimidazole derivatives. Density functional study of prototypical radiosensitizers. Magn. Reson Chem. 43, 139–146. doi:10.1002/mrc.1514
Rao, L., Li, Y., He, Q., Liu, Y., Su, Y., You, Y. X., et al. (2021). Iridoid constituents of viburnum brachybotryum. J. Nat. Prod. 84, 1915–1923. doi:10.1021/acs.jnatprod.1c00042
Ravutsov, M., Dobrikov, G. M., Dangalov, M., Nikolova, R., Dimitrov, V., Mazzeo, G., et al. (2021). 1, 2-Disubstituted planar chiral ferrocene derivatives from sulfonamide-directed ortho-lithiation: Synthesis, absolute configuration, and chiroptical properties. Organometallics 40, 578–590. doi:10.1021/acs.organomet.0c00712
RDKIT (2022). RDKIT: Open-source cheminformatics. Available at: http://www.rdkit.org.
Reinhardt, J. K., Klemd, A. M., Danton, O., Mieri, M. D., Smiesko, M., Huber, R., et al. (2019). Sesquiterpene lactones from artemisia argyi: Absolute configuration and immunosuppressant activity. J. Nat. Prod. 82, 1424–1433. doi:10.1021/acs.jnatprod.8b00791
Reinscheid, F., and Reinscheid, U. M. (2016). Stereochemical analysis of (+)-limonene using theoretical and experimental NMR and chiroptical data. J. Mol. Struct. 1106, 141–153. doi:10.1016/j.molstruc.2015.10.061
Ren, J., Jiang, J. X., Li, L. B., Liao, T. G., Tian, R. R., Chen, X. L., et al. (2009). Assignment of the absolute configuration of concentricolide–absolute configuration determination of its bioactive analogs using DFT methods. Eur. J. Org. Chem. 23, 3987–3991. doi:10.1002/ejoc.200900422
Ren, J., Li, G. L., Shen, L., Zhang, G. L., Nafie, L., and Zhu, H. J. (2013). Challenges in the assignment of relative and absolute configurations of complex molecules: Computation can resolve conflicts between theory and experiment. Tetrahedron 69, 10351–10356. doi:10.1016/j.tet.2013.10.004
Ren, J., Zhao, D., Wu, S. J., Wang, J., Jia, Y., Li, W., et al. (2019). Reassigning the stereochemistry of bioactive cepharanthine using calculated versus experimental chiroptical spectroscopies. Tetrahedron 75, 1194–1202. doi:10.1016/j.tet.2019.01.028
Ren, J., and Zhu, H. J. (2009). Application of the computational chemistry in identification of chiral compounds. Chem. J. Chin. Univ. 30, 1907–1918. Available at: http://www.cjcu.jlu.edu.cn/CN/.
Renner, U., and Kernweisz, P. (1963). Alkaloide ausSchizozygia caffaeoides (boj.) baill. Experientia 19, 244–246. doi:10.1007/BF02151358
Ribe, S., Kondru, R. K., Beratan, D. N., and Wipf, P. (2000). Optical rotation computation, total synthesis, and stereochemistry assignment of the marine natural product pitiamide A. J. Am. Chem. Soc. 122, 4608–4617. doi:10.1021/ja9945313
Riobé, F., Szucs, R., Bouit, P. A., Tondelier, D., Geffroy, B., Aparicio, F., et al. (2015). Synthesis, electronic properties and WOLED devices of planar phosphorus-containing polycyclic aromatic hydrocarbons. Chem. Eur. J. 21, 6547–6556. doi:10.1002/chem.201500203
Ripa, L., Hallberg, A., and Sandström, J. (1997). Determination of absolute configurations of N-formyl-3, 3‘, 4, 4‘-tetrahydrospiro[naphthalene-1(2H), 2‘(1‘H)-pyridine] (2) and N-Formyl-3‘, 4‘-dihydrospiro[indan-1, 2‘(1‘H)-pyridine] (3) by analysis of circular dichroism spectra. A case of two compounds with similar configuration but nearly mirror image CD spectra. J. Am. Chem. Soc. 119, 5701–5705. doi:10.1021/ja9635995
Rode, J. E., Dobrowolski, J. C., Lyczko, K., Wasiewicz, A., Kaczorek, D., Kawecki, R., et al. (2017). Chiral thiophene sulfonamide—A challenge for VOA calculations. J. Phys. Chem. A 121, 6713–6726. doi:10.1021/acs.jpca.6b11015
Rode, J. E., and Frelek, J. (2017). Circular dichroism spectroscopy and DFT calculations in determining absolute configuration and E/Z isomers of conjugated oximes. Chirality 29, 653–662. doi:10.1002/chir.22750
Rode, J. E., Lyczko, K., Jawiczuk, M., Kawęcki, R., Stańczyk, W., Jaglińska, A., et al. (2018). The vibrational circular dichroism pattern of the ν(C=O) bands in isoindolinones. ChemPhysChem 19, 2411–2422. doi:10.1002/cphc.201800244
Rossi, D., Nasti, R., Collin, S., Mazzeo, G., Ghidinelli, S., Longhi, G., et al. (2017). The role of chirality in a set of key intermediates of pharmaceutical interest, 3-aryl-substituted-γ-butyrolactones, evidenced by chiral HPLC separation and by chiroptical spectroscopies. J. Pharm Biomed. Analy. 144, 41–51. doi:10.1016/j.jpba.2017.01.007
Rossi, D., Nasti, R., Marra, A., Meneghini, S., Mazzeo, G., Longhi, G., et al. (2016). Enantiomeric 4-acylamino-6-alkyloxy-2 alkylthiopyrimidines as potential A3 adenosine receptor antagonists: HPLC chiral resolution and absolute configuration assignment by a full set of chiroptical spectroscopy. Chirality 28, 434–440. doi:10.1002/chir.22599
Runarsson, ÖV., Benkhauser, C., Christensen, N. J., Ruiz, J. A., Ascic, E., Harmata, M., et al. (2015). Resolution and determination of the absolute configuration of a twisted bis-lactam analogue of tröger’s base: A comparative spectroscopic and computational study. J. Org. Chem. 80, 8142–8149. doi:10.1021/acs.joc.5b01236
Saetang, P., Rukachaisirikul, V., Phongpaichit, S., Preedanon, S., Sakayaroj, J., Hadsadee, S., et al. (2021). Antibacterial and antifungal polyketides from the fungus Aspergillus unguis PSU-MF16. J. Nat. Prod. 84, 1498–1506. doi:10.1021/acs.jnatprod.0c01308
Saha, B., Ikbal, S. A., Petrovic, A. G., Berova, N., and Rath, S. P. (2017). Complexation of chiral zinc-porphyrin tweezer with achiral diamines: Induction and two-step inversion of interporphyrin helicity monitored by ECD. Inorg. Chem. 56, 3849–3860. doi:10.1021/acs.inorgchem.6b02686
Saito, F., and Schreiner, P. R. (2020). Determination of the absolute configurations of chiral alkanes–an analysis of the available tools. Eur. J. Org. Chem. 2020, 6328–6339. doi:10.1002/ejoc.202000711
Saleh, N., Moore, B., Srebro, M., Vanthuyne, N., Toupet, L., Williams, J. A. G., et al. (2015). Acid/base-triggered switching of circularly polarized luminescence and electronic circular dichroism in organic and organometallic helicenes. Chem. Eur. J. 21, 1673–1681. doi:10.1002/chem.201405176
Sandström, J. (2000). Determination of absolute configurations and conformations of organic compounds by theoretical calculations of CD spectra. Chirality 12, 162–171. doi:10.1002/(SICI)1520-636X(2000)12:4<162:AID-CHIR2>3.0.CO;2-C
Sandström, J. (2000). In circular dichroism-principles and applications. New York: Wiley VCH, 459–490.
Santoro, E., Mazzeo, G., Marsico, G., Masi, M., Longhi, G., Superchi, S., et al. (2019). Assignment through chiroptical methods of the absolute configuration of fungal dihydropyranpyran-4-5-diones phytotoxins, potential herbicides for buffelgrass (Cenchrus ciliaris) biocontrol. Molecules 24, 3022–3033. doi:10.3390/molecules24173022
Scott, M., Rehn, D. R., Norman, P., and Dreuw, A. (2021). Ab initio excited-state electronic circular dichroism spectra exploiting the third-order algebraic-diagrammatic construction scheme for the polarization propagator. J. Phys. Chem. Lett. 12, 5132–5137. doi:10.1021/acs.jpclett.1c00839
Seco, J. M., Quiñoá, E., and Riguera, R. (2004). The assignment of absolute configuration by NMR. Chem. Rev. 104, 17–118. doi:10.1021/cr000665j
Sherer, E. C., Lee, C. H., Shpungin, J., Cuff, J. F., Da, C., Ball, R., et al. (2014). Systematic approach to conformational sampling for assigning absolute configuration using vibrational circular dichroism. J. Med. Chem. 57, 477–494. doi:10.1021/jm401600u
Sherer, E. C., Lee, C. H., Shpungin, J., Cuff, J. F., Da, C. X., Ball, R., et al. (2014). Systematic approach to conformational sampling for assigning absolute configuration using vibrational circular dichroism. J. Med. Chem. 57, 477–494. doi:10.1021/jm401600u
Slaughter, J. C. (1999). The naturally occurring furanones: Formation and function from pheromone to food. Biol. Rev. 74, 259–276. doi:10.1111/j.1469-185x.1999.tb00187.x
Smith, S. G., and Goodman, J. M. (2010). Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 132, 12946–12959. doi:10.1021/ja105035r
Socolsky, C., Salamanca, E., Gimenez, A., Borkosky, S. A., and Bardoń, A. (2016). Prenylated acylphloroglucinols with leishmanicidal activity from the fern Elaphoglossum lindbergii. J. Nat. Prod. 79, 98–105. doi:10.1021/acs.jnatprod.5b00767
Stepanek, P., and Bour, P. (2015). Origin-independent sum over states simulations of magnetic and electronic circular dichroism spectra via the localized orbital/local origin method. J. Comput. Chem. 36, 723–730. doi:10.1002/jcc.23845
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627. doi:10.1021/j100096a001
Stephens, P. J., Pan, J. J., Devlin, F. J., Krohn, K., and Kurtán, T. (2007). Determination of the absolute configurations of natural products via density functional theory calculations of vibrational circular dichroism, electronic circular dichroism, and optical rotation: The iridoids plumericin and isoplumericin. J. Org. Chem. 72, 3521–3536. doi:10.1021/jo070155q
Stephens, P. J., Pan, J. J., Devlin, F. J., Urbanová, M., and Hájíček, J. (2007). Determination of the absolute configurations of natural products via density functional theory calculations of vibrational circular dichroism, electronic circular dichroism and optical rotation: The schizozygane alkaloid schizozygine. J. Org. Chem. 72, 2508–2524. doi:10.1021/jo062567p
Stephens, P. J. (1985). Theory of vibrational circular dichroism. J. Phys. Chem. 89, 748–752. doi:10.1021/j100251a006
Sun, L. L., Li, W. S., Li, J., Zhang, H. Y., Yao, L. G., Luo, H., et al. (2021). Uncommon diterpenoids from the South China Sea soft coral Sinularia humilis and their stereochemistry. J. Org. Chem. 86, 3367–3376. doi:10.1021/acs.joc.0c02742
Sun, P., Cai, F. Y., Lauro, G., Tang, H., Su, L., Wang, H. L., et al. (2019). Immunomodulatory biscembranoids and assignment of their relative and absolute configurations: Data set modulation in the density functional theory/nuclear magnetic resonance approach. J. Nat. Prod. 82, 1264–1273. doi:10.1021/acs.jnatprod.8b01037
Sun, Y. J., Yin, Y., Sun, Y. P., Li, Q. R., Cui, L. T., Xu, W. J., et al. (2021). Aglatestine A, a rearranged limonoid with a 3/6/6 tricarbocyclic framework from the fruits of Aglaia edulis. J. Org. Chem. 86, 11263–11268. doi:10.1021/acs.joc.1c00968
Takahashi, H., Iwasaki, A., Kurisawa, N., Suzuki, R., Jeelani, G., Matsubara, T., et al. (2021). Motobamide, an antitrypanosomal cyclic peptide from a Leptolyngbya sp. marine cyanobacterium. J. Nat. Prod. 84, 1649–1655. doi:10.1021/acs.jnatprod.1c00234
Tan, X., Han, X. Y., Teng, H., Li, Q. Q., Chen, Y., Lei, X. X., et al. (2021). Structural elucidation of garcipaucinones A and B from garcinia paucinervis using quantum chemical calculations. J. Nat. Prod. 84, 972–978. doi:10.1021/acs.jnatprod.0c00883
Tanaka, S., Honmura, Y., Uesugi, S., Fukushi, E., Tanaka, K., Maeda, H., et al. (2017). Cyclohelminthol X, a hexa-substituted spirocyclopropane from helminthosporium velutinum yone96: Structural elucidation, electronic circular dichroism analysis, and biological properties. J. Org. Chem. 82, 5574–5582. doi:10.1021/acs.joc.7b00393
Taniguchi, T. (2017). Analysis of molecular configuration and conformation by (electronic and) vibrational circular dichroism: Theoretical calculation and exciton chirality method. Bull. Chem. Soc. Jpn. 90, 1005–1016. doi:10.1246/bcsj.20170113
Taniguchi, T., Manai, D., Shibata, M., Itabashi, Y., and Monde, K. (2015). Stereochemical analysis of glycerophospholipids by vibrational circular dichroism. J. Am. Chem. Soc. 137, 12191–12194. doi:10.1021/jacs.5b05832
Taniguchi, T., Martin, C. L., Monde, K., Nakanishi, K., Berova, N., and Overman, L. E. (2009). Absolute configuration of actinophyllic acid as determined through chiroptical data. J. Nat. Prod. 72, 430–432. doi:10.1021/np800665s
Taniguchi, T., Nakano, K., Baba, R., and Monde, K. (2017). Analysis of configuration and conformation of furanose ring in carbohydrate and nucleoside by vibrational circular dichroism. Org. Lett. 19, 404–407. doi:10.1021/acs.orglett.6b03626
Teodoro, T. Q., Koenis, M. A. J., Rüger, R., Galembeck, S. E., Buma, W. J., Nicu, V. P., et al. (2018). Use of density functional based tight binding methods in vibrational circular dichroism. J. Phys. Chem. A 122, 9435–9445. doi:10.1021/acs.jpca.8b08218
The structures are cited in: Cao, F., Gao, T., Xu, L. L., and Zhu, H. J. (2017). Progress on vibration circular dichroism in organic chemistry. Sci. Sin. Chim. 47, 801–815. doi:10.1360/N032016-00231
Tsuda, M., Kasai, Y., Komatsu, K., Sone, T., Tanaka, M., Mikami, Y., et al. (2004). Citrinadin A, a novel pentacyclic alkaloid from marine-derived fungus Penicillium citrinum. J. Org. Lett. 6, 3087–3089. doi:10.1021/ol048900y
Tuzi, A., Andolfi, A., Cimmino, A., and Evidente, A. (2010). X-Ray crystal structure of phyllostin, a metabolite produced by phyllosticta cirsii, a potential mycoherbicide of Cirsium arvense. J. Chem. Crystallogr. 40, 15–18. doi:10.1007/s10870-009-9597-x
Urbanová, M., Setnička, V., Devlin, F. J., and Stephens, P. J. (2005). Determination of molecular structure in solution using vibrational circular dichroism spectroscopy: The supramolecular tetramer of s-2, 2’-dimethyl-biphenyl-6, 6’-dicarboxylic acid. J. Am. Chem. Soc. 127, 6700–6711. doi:10.1021/ja050483c
Vargek, M., Freedman, T. B., Lee, E., and Nafie, L. A. (1998). Experimental observation of resonance Raman optical activity. Chem. Phys. Lett. 287, 359–364. doi:10.1016/S0009-2614(98)00017-7
Vergura, S., Santoro, E., Masi, M., Evidente, A., Scafato, P., Superchi, S., et al. (2018). Absolute configuration assignment to anticancer Amaryllidaceae alkaloid jonquailine. Fitoterapia 129, 78–84. doi:10.1016/j.fitote.2018.06.013
Vesga, Y., and Hernandez, F. E. (2016). Study of the effect of the pulse width of the excitation source on the two-photon absorption and two-photon circular dichroism spectra of biaryl derivatives. J. Phys. Chem. A 120, 6774–6779. doi:10.1021/acs.jpca.6b06925
Wang, B., Bruhn, J. F., Weldeab, A., Wilson, T. S., McGilvray, P. T., Mashore, M., et al. (2022). Absolute configuration determination of pharmaceutical crystalline powders by MicroED via chiral salt formation. Chem. Commun. 58, 4711–4714. doi:10.1039/D2CC00221C
Wang, G. W., Zhan, X. H., Zhan, H., Guo, Q. X., and Wu, Y. D. (2003). Accurate calculation, prediction, and assignment of 3He NMR chemical shifts of helium-3-encapsulated fullerenes and fullerene derivatives. J. Org. Chem. 68, 6732–6738. doi:10.1021/jo0341259
Wang, H., Li, M. Y., Katele, F. Z., Satyanandamurty, T., Wu, J., and Bringmann, G. (2014). Decandrinin, an unprecedented C9-spiro-fused 7, 8-seco-ent-abietane from the Godavari mangrove Ceriops decandra. Beilstein J. Org. Chem. 10, 276–281. doi:10.3762/bjoc.10.23
Wang, J. F., Dai, H. Q., Wei, Y. L., Zhu, H. J., Yana, Y. M., Wang, Y. H., et al. (2010). Antituberculosis agents and an inhibitor of the para-aminobenzoic acid biosynthetic pathway from Hydnocarpus anthelminthica seeds. Chem. Biodiv. 7, 2046–2053. doi:10.1002/cbdv.201000072
Wang, L., Yu, Z. Y., Guo, X. W., Huang, J. P., Yan, Y. J., Huang, S. X., et al. (2021). Bisaspochalasins D and E: Two heterocycle-gused cytochalasan homodimers from an endophytic Aspergillus flavipes. J. Org. Chem. 86, 11198–11205. doi:10.1021/acs.joc.1c00425
Wang, M., Yu, S. Y., Qi, S. Z., Zhang, B. H., Song, K. R., Liu, T., et al. (2021). Anti-inflammatory cassane-type diterpenoids from the seed kernels of Caesalpinia sinensis. J. Nat. Prod. 84, 2175–2188. doi:10.1021/acs.jnatprod.1c00233
Wang, X., Peng, X., Tang, C. P., Zhou, S. Z., Ke, C. Q., Liu, Y. L., et al. (2021). Anti-inflammatory eudesmane sesquiterpenoids from Artemisia hedinii. J. Nat. Prod. 84, 1626–1637. doi:10.1021/acs.jnatprod.1c00177
Wang, X. X., and Zhu, H. J. (2021). Discussion of the key and common academic questions in absolute configuration determination of chiral compounds. Chem. J. Chin. Univ. 42, 1685–1693. doi:10.7503/cjcu20200726
Wang, Z. Q., Zhao, L. X., Chen, Y., Xu, W., and Sun, T. M. (2014). Determination of absolute configurations of bedaquiline analogs by quantum chemical electronic circular dichroism calculations and an X-ray diffraction study. Eur. J. Org. Chem. 2014, 3814–3821. doi:10.1002/ejoc.201400135
Wibowo, M., Irons, T. J. P., and Teale, A. M. (2021). Modeling ultrafast electron dynamics in strong magnetic fields using real-time time-dependent electronic structure methods. J. Chem. Theory Comput. 17, 2137–2165. doi:10.1021/acs.jctc.0c01269
Wu, T., Zhang, X. P., You, X. Z., Li, Y. Z., and Bour, P. (2014). Chirality transfer in magnetic coordination complexes monitored by vibrational and electronic circular dichroism. Chem. Plus Chem. 79, 698–707. doi:10.1002/cplu.201300429
Xia, Y. Y., Koenis, M. A. J., Colladosb, J. F., Ortiz, P., Harutyunyan, S. R., Visscherc, L., et al. (2018). Regional susceptibility in VCD spectra to dynamic molecular motions: The case of a benzyl α-hydroxysilane. Chem. Phys. Chem. 19, 561–565. doi:10.1002/cphc.201701335
Xie, Y. H., Guo, L., Huang, J., Huang, X. L., Cong, Z. W., Liu, Q. Q., et al. (2021). Cyclopentenone-containing tetrahydroquinoline and geldanamycin alkaloids from Streptomyces malaysiensis as potential anti-androgens against prostate cancer Cells. J. Nat. Prod. 84, 2004–2011. doi:10.1021/acs.jnatprod.1c00297
Ximenes, V. F., Morgon, N. H., and Souza, A. R. D. (2018). Solvent-dependent inversion of circular dichroism signal in naproxen: An unusual effect. Chirality 30, 1049–1053. doi:10.1002/chir.22988
Xu, L. L., Cao, F., Yang, Q., Li, W., Tian, S. S., Zhu, H. J., et al. (2016). Experimental and theoretical study of stereochemistry for new pseurotin A3 with an unusual hetero-spirocyclic system. Tetrahedron 72, 7194–7199. doi:10.1016/j.tet.2016.09.053
Xu, W. F., Chao, R., Hai, Y., Guo, Y. Y., Wei, M. Y., Wang, C. Y., et al. (2021). 17-hydroxybrevianamide N and its N1-methyl derivative, quinazolinones from a soft-coral-derived Aspergillus sp. fungus: 13S enantiomers as the true natural products. J. Nat. Prod. 84, 1353–1358. doi:10.1021/acs.jnatprod.1c00098
Xu, X. K., Ye, J., Chen, L. P., Zhang, W. D., Yang, Y. X., and Li, H. L. (2016). Three new sesquiterpene lactone dimers from Carpesium faberi. Phytochem. Lett. 16, 277–282. doi:10.1016/j.phytol.2016.05.006
Yajima, A., Shimura, M., Saito, T., Katsuta, R., Ishigami, K., Huffaker, A., et al. (2021). Synthesis and determination of absolute configuration of zealexin A1, a sesquiterpenoid phytoalexin from Zea mays. Eur. J. Org. Chem. 2021, 1174–1178. doi:10.1002/ejoc.202001596
Yang, C. S., Wang, X. B., Wang, J. S., Luo, J. G., Luo, J., and Kong, L. Y. (2011). A [2 + 2] cycloaddition dimer and a Diels–Alder adduct from Alpinia katsumadai. Org. Lett. 13, 3380–3383. doi:10.1021/ol201137v
Yang, Q., Liang, M. M., Wang, H. J., Zhao, Q. Q., Zhu, H. J., Liu, L., et al. (2017). Investigating cyclic sotolon, maple furanone and their dimers in solution using optical rotation, electronic circular dichroism and vibrational circular dichroism. Tetrahedron 73, 2432–2438. doi:10.1016/j.tet.2017.03.025
Yang, X. W., Grossman, R. B., and Xu, G. (2018). Research progress of polycyclic polyprenylated acylphloroglucinols. Chem. Rev. 118, 3508–3558. doi:10.1021/acs.chemrev.7b00551
Yang, X. W., Peng, K., Liu, Z., Zhang, G. Y., Li, J., Wang, N., et al. (2013). Strepsesquitriol, a rearranged zizaane-type sesquiterpenoid from the deep-sea-derived Actinomycete Streptomyces sp. SCSIO 10355. J. Nat. Prod. 76, 2360–2363. doi:10.1021/np400923c
Yu, H., Li, W. X., Wang, J. C., Yang, Q., Wang, H. J., Zhang, C. C., et al. (2015). Pestalotiopsin C, stereochemistry of a new caryophyllene from a fungus of Trichoderma sp. and its tautomerization characteristics in solution. Tetrahedron 71, 3491–3494. doi:10.1016/j.tet.2015.03.063
Yu, H. G. (2014). Origin of anomalous electronic circular dichroism spectrum of RuPt2(tppz)2Cl2(PF6)4 in acetonitrile. J. Phys. Chem. A 118, 5400–5406. doi:10.1021/jp502957z
Yuan, T., Zhu, R. X., Zhang, H., Odeku, O. A., Yang, S. P., Liao, S. G., et al. (2010). Structure determination of grandifotane A from Khaya grandifoliola by NMR, X-ray Diffraction, and ECD calculation. Org. Lett. 12, 252–255. doi:10.1021/ol902565s
Yun, Y. S., Fukaya, H., Nakane, T., Takano, A., Takahashi, S., Takahashi, Y., et al. (2014). A new bis-seco-abietane diterpenoid from Hyptis crenata pohl ex benth. Org. Lett. 16, 6188–6191. doi:10.1021/ol503086n
Zajac, G., Kaczor, A., Buda, S., Mlynarski, J., Frelek, J., Dobrowolski, J. C., et al. (2015). Prediction of ROA and ECD related to conformational changes of astaxanthin enantiomers. J. Phys. Chem. B 119, 12193–12201. doi:10.1021/acs.jpcb.5b07193
Zajac, G., Kaczor, A., Zazo, A. P., Mlynarski, J., Dudek, M., and Baranska, M. (2016). Aggregation-induced resonance Raman optical activity (AIRROA): A new mechanism for chirality enhancement. J. Phys. Chem. B 120, 4028–4033. doi:10.1021/acs.jpcb.6b02273
Zaman, K. H. A. U., Park, J. H., DeVine, L., Hu, Z. Q., Wu, X. H., Kim, H. S., et al. (2021). Secondary metabolites from the leather coral-derived fungal strain Xylaria sp. FM1005 and their glycoprotein IIb/IIIa inhibitory activity. J. Nat. Prod. 84, 466–473. doi:10.1021/acs.jnatprod.0c01330
Zhang, W. G., Lu, X. X., Huo, L. Q., Zhang, S., Chen, Y., Zou, Z. X., et al. (2021). Sesquiterpenes and steroids from an endophytic Eutypella scoparia. J. Nat. Prod. 84, 1715–1724. doi:10.1021/acs.jnatprod.0c01167
Zhang, Y., Yu, Y. Y., Peng, F., Duan, W. T., Wu, C. H., Li, H. T., et al. (2021). Neolignans and diarylheptanoids with anti-Inflammatory activity from the rhizomes of Alpinia zerumbet. J. Agric. Food Chem. 69, 9229–9237. doi:10.1021/acs.jafc.1c02271
Zhang, Y. F., Poopari, M. R., Cai, X., Savin, A., Dezhahang, Z., Cheramy, J., et al. (2016). IR and vibrational circular dichroism spectroscopy of matrine- and artemisinin-type herbal products: Stereochemical characterization and solvent effects. J. Nat. Prod. 79, 1012–1023. doi:10.1021/acs.jnatprod.5b01082
Zhao, B. X., Wang, Y., Li, C., Wang, G. C., Huang, X. J., Fan, C. L., et al. (2013). Flueggedine, a novel axisymmetric indolizidine alkaloid dimer from. Flueggea virosa Tetrahedron Lett. 54, 4708–4711. doi:10.1016/j.tetlet.2013.06.097
Zhao, D., Li, Z. Q., Cao, F., Liang, M. M., Pittman, C. U., Zhu, H. J., et al. (2016). Revised absolute configuration of sibiricumin A: Substituent effects in simplified model structures used for quantum mechanical predictions of chiroptical properties. Chirality 28, 612–617. doi:10.1002/chir.22621
Zheng, J. K., Zhu, H. J., Hong, K., Wang, Y., Liu, P. P., Wang, X., et al. (2009). Novel cyclic hexapeptides from marine-derived fungus, Aspergillus sclerotiorum PT06-1. Org. Lett. 11, 5262–5265. doi:10.1021/ol902197z
Zheng, Z. Q., Wei, W. J., Zhang, J., Li, H. Y., Xu, K., Xu, J., et al. (2019). Heliaquanoids A–E, five sesquiterpenoid dimers from Inula helianthus-aquatica. J. Org. Chem. 84, 4473–4477. doi:10.1021/acs.joc.8b03284
Zhou, G. L., Sun, C. X., Hou, X. W., Che, Q., Zhang, G. J., Gu, Q. Q., et al. (2021). Ascandinines A–D, indole diterpenoids, from the sponge-derived fungus Aspergillus candidus HDN15-152. J. Org. Chem. 86, 2431–2436. doi:10.1021/acs.joc.0c02575
Zhou, Q., and Snider, B. B. (2008). Synthesis of (±)- and (−)-vibralactone and vibralactone C. J. Org. Chem. 73, 8049–8056. doi:10.1021/jo8015743
Zhou, Q., and Snider, B. B. (2008). Synthesis of (±)-vibralactone. Org. Lett. 10, 1401–1404. doi:10.1021/ol800118c
Zhu, A., Zhang, X. W., Zhang, M., Li, W., Ma, Z. Y., Zhu, H. J., et al. (2018). Aspergixanthones I–K, new anti-Vibrio prenylxanthones from the marine-derived fungus Aspergillus sp. ZA-01. Mar. Drugs 16, 312. doi:10.3390/md16090312
Zhu, H. J., Li, W., Hu, D., and Wen, M. (2014). Discussion of absolute configuration for bioactive griseusins by comparing computed optical rotations and electronic circular dichroism with the experimental results. Tetrahedron 70, 8236–8243. doi:10.1016/j.tet.2014.09.032
Zhu, H. J., Liu, L., and Yang, Q. (2015). Vibrational circular dichroism in study of stereochemistry of chiral β-biscarboline with N-O functional group. Chem. J. Chin. Univ. 36, 1559–1562. doi:10.7503/cjcu20150151
Zhu, H. J., and Zhao, D. (2015). Theoretical methods in configuration determinations for natural chiral compounds: Research advances. J. Int. Pharm. Res. 42, 669–685. doi:10.13220/j.cnki.jipr.2015.06.001
Zhu, H. J. (2015). Organic stereochemistry---experimental and computational methods. Weinheim, Germany: Wiley-VCH Verlag GmbHCo. KGaA.
Zhu, H. J. (2022). Progress of the theoretical research on absolute configuration assignment for chiral medicines. J. Hebei Uni. Sci. Tech. 43, 401–414. doi:10.7535/hbkd.2022yx04007
Zhu, H. J. (2022). The role of the conformer pairs in optical rotation. Available at: https://researchfeatures.com/role-conformer-pairs-optical-rotation/.
Zinna, F., and Pescitelli, G. (2016). Towards the limits of vibrational circular dichroism spectroscopy: VCD spectra of some alkyl vinylethers. Chirality 28, 143–146. doi:10.1002/chir.22555
Keywords: optical rotation, matrix model, electronic circular dichroism, vibrational circular dichroism, carbon NMR, conformer pair, simplified model, transition state
Citation: Zhu H, Wang Y and Nafie LA (2023) Computational methods and points for attention in absolute configuration determination. Front. Nat. Prod. 1:1086897. doi: 10.3389/fntpr.2022.1086897
Received: 01 November 2022; Accepted: 24 November 2022;
Published: 12 January 2023.
Edited by:
Quezia B. Cass, Federal University of São Carlos, BrazilReviewed by:
Marcin Górecki, Institute of Organic Chemistry, Polish Academy of Sciences, PolandCopyright © 2023 Zhu, Wang and Nafie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huajie Zhu, emh1aHVhamllQGhvdG1haWwuY29t; Laurence A. Nafie, bGFuYWZpZUBhb2wuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.