 Payal Chauhan
Payal Chauhan Karan Wadhwa
Karan Wadhwa Govind Singh
Govind Singh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nanotechnol. , 10 October 2022
Sec. Biomedical Nanotechnology
Volume 4 - 2022 | https://doi.org/10.3389/fnano.2022.1018754
The impact of neurodegenerative illnesses on society is significant, but the mechanisms leading to neuronal malfunction and death in these conditions remain largely unknown despite identifying essential disease genes. To pinpoint the mechanisms behind the pathophysiology of neurodegenerative diseases, several researchers have turned to nematode C. elegans instead of using mammals. Since C. elegans is transparent, free-living, and amenable to culture, it has several benefits. As a result, all the neurons in C. elegans can be easily identified, and their connections are understood. Human proteins linked to Neurodegeneration can be made to express in them. It is also possible to analyze how C. elegans orthologs of the genes responsible for human neurodegenerative diseases function. In this article, we focused at some of the most important C. elegans neurodegeneration models that accurately represent many elements of human neurodegenerative illness. It has been observed that studies using the adaptable C. elegans have helped us in better understanding of human diseases. These studies have used it to replicate several aspects of human neurodegeneration. A nanotech approach involves engineering materials or equipments interacting with biological systems at the molecular level to trigger physiological responses by increasing stimulation, responding, and interacting with target sites while minimizing side effects, thus revolutionizing the treatment and diagnosis of neurodegenerative diseases. Nanotechnologies are being used to treat neurological disorders and deliver nanoscale drugs. This review explores the current and future uses of these nanotechnologies as innovative therapeutic modalities in treatment of neurodegenerative diseases using C elegans as an experimental model.
Various model systems are being used to understand the genesis and progression of human diseases. Better understanding is needful in drug screening/development. Humans would undoubtedly be the finest study subjects, but this is typically not the case due to practical and moral/ethical considerations. Other mammals are the next apparent option and are good in many aspects, mainly when emulating behavioral characteristics. Various mammalian models, including rodents, are critically established to understand the pathophysiology and to explore new therapeutic interventions for neurodegenerative disorders (NDs) because of their ability to mimic various clinical features like neuronal loss, alteration in neuronal signaling and transmission, motor and non-motor dysfunctions (Torres and Dunnett, 2011; Thiele et al., 2012; Ribeiro et al., 2013; Schirinzi et al., 2016). But, several toxin-induced ND models in mammals still fail to mimic many pathological hallmarks like gross morphological transformation and steady neurodegenerative progression (Ribeiro et al., 2013; Schirinzi et al., 2016; Visanji et al., 2016). However, they also have few other significant drawbacks, including how well they imitate particular disease, the sluggish pathological development, and the length of time needed to conclude. To better understand these pathological hallmarks in ND and overcome the limitations of well-established rodent models, numerous invertebrate models like those of the Drosophila melanogaster or Caenorhabditis elegans has gained popularity recently and are being developed as first-round preclinical investigation followed by mammalian assay (Brenner, 1974; Sulston et al., 1975).
C. elegans is an effective tool for development of different experimental models because of its various characteristics and simple culturing/maintenance. This free-living, tiny, non-lethal, 1 mm long self-fertile hermaphrodite nematode has a 3.5-day reproduction cycle with survival at 20°C for roughly 3 weeks. It typically grows in the soil but can easily be raised in a lab environment on Escherichia coli diet (Garigan et al., 2002; Olsen et al., 2006; Chew et al., 2017). C. elegans share similar cellular mechanisms and pathways to humans and other mammals, which can be genetically analyzed and studied using RNA interference technologies (Fire et al., 1998), Furthermore, ease of its laboratory preservation, transparent anatomical observation body, high genetic homology (60–80%) to humans, complete genetic sequence detection, stored biological cell responses, high fertility rates (240 eggs/worm in a few days), and access to cellular biological tools (such as transgenic, gene knockouts) makes C. elegans a considerable invertebrate model for NDs and its associated genetic research over fundamental ND neurobehavioral studies (Matsunami, 2018; Youssef et al., 2019). C. elegans is also considered as an advanced model of biology in the study of aging. Apart from a few anatomical variations from mammals, the nervous system of the C. elegans embraces a circumpharyngeal nerve ring, along with neurotransmitter systems for acetylcholine (ACh), glutamate, dopamine (DA), and gamma-aminobutyric acid (GABA), including their receptor and synaptic features, like that of primary cellular and molecular features in mammalian neurons (Markaki and Tavernarakis, 2010; Maulik et al., 2017). The nervous system of nematodes contains 302 neurons in adult female hermaphrodites while 383 in males. More than 7,600 synapses are formed by 118 morphologically different classes and 56 glia cells (White et al., 1986; Bargmann, 1998; Barclay et al., 2012). Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD) are the few major NDs that have been modeled using C. elegans (Sattelle and Buckingham, 2006; Kim et al., 2019).
Furthermore, the use of a nanoparticulated drug delivery system is a cutting-edge technology that is widely employed to transport medications right to the brain, and has been proven to be incredibly effective in treating various CNS illnesses. Nano-sized particles have attracted great interest because of their capacity to stop chemical and enzymatic drug degradation. These nano drugs also improve drug solubility and ease drug transport across biological membranes via direct drug delivery to the site of action, which consequently minimize drug side effects and raise its therapeutic index. Various nanoparticulate systems including polymeric nanoparticles, nanoemulsions, liposomes, etc. have been developed so far that opened up new possibilities beyond simply enhancing traditional treatments’ pharmacokinetics (Wadhwa et al., 2022). Nanomaterial toxicity is a growing concern in nanotechnology as more and more nanomaterials are produced and used in a variety of applications. Due to their strong antibacterial action, Silver nanoparticles (AgNPs) are one of the most popular commercial nanomaterials at the moment (Durán et al., 2016). AgNPs’ toxicity has been proven in numerous models over the years, including bacteria (Xiu et al., 2012), cell culture systems (Wang et al., 2013), zebrafish (Rahman et al., 2009), and mice (Maurer et al., 2016). Due to its unique characteristics, C. elegans is quickly becoming one of the most valuable model systems for determining the potential toxicity of nanoparticles. Due to the absence of established protocols, there have been a number of difficulties up until this point. Consideration must be taken seriously to improve the experimental settings for nanotoxicity to get reliable and accurate results (Roh et al., 2009; Maurer et al., 2016). Thus, the present manuscript highlights the current research that endeavored C. elegans as a promising screening model to underline the pathogenecity of NDs and associated molecular mechanism of therapeutic inventions that can be used for the treatments of various NDs, with a special attention to the implementation of these C. elegans models in nanomedicine and nanotechnology.
Researches affirmed that several NDs have a significant genetic influence, and is particularly true of familial forms of these diseases, where genetics plays a more critical role in disease pathology. The central part of C. elegans research on NDs has been devoted to understand the effects of genes involved in AD, PD, ALS, and HD. Most mutations in these genes associated with the diseases are autosomal dominant mutations, since one copy of each mutation causes disease (White et al., 1986; Chalfie et al., 1994). C. elegans may be treated as advanced model of biology in the study of neurodegeneration associated with different diseases. Apart from above discussed advantages, the short lifespan (about 3 weeks) and small size of this organism reduce experimental cost, and can be used for high-throughput screening studies, which is advantageous to screen neuroprotective nanoformulations/drugs. Also, no ethical consideration and requirements with experimentations will led to many breakthrough discoveries in the field of neuroprotective and aging research using C. elegans (David et al., 2010).
Alzheimer’s disease (AD) is the most common form of dementia, characterized by amyloid plaque formation through β-secretase and γ-secretes enzymes and hyperphosphorylation of tau protein. Frontotemporal dementia is a distinct neurodegenerative condition caused by mutations in the tau gene, while presenilin mutation causes familiar forms of the disease. Amyloid precursor protein (APP) gene is a crucial factor for AD that helps in the cleavage of the amyloid peptide. C. elegans has an APP orthologous APL-1, but this protein lacks the amyloid peptide. Additionally, nematodes’ genome does not contain these β-Secretase cleaving enzymes. Human peptide expression in the worms has always been used to create elegans models of amyloid Aβ toxicity. By producing the Aβ-42 peptide in body-wall muscle, the first C. elegans model of a ND was created (Link, 1995; Alexander et al., 2014). As a result, transgenic worms suffer from paralysis, and amyloid peptide accumulation was observed in their muscles. Many models in worms were developed to investigate the neuroprotective effects of herbal drugs and many synthetic compounds. To test the neuron activity of compounds worm models with either muscle or neuronal Aβ-42 expression have since been developed (Li and Le, 2013; Alexander et al., 2014; Ma et al., 2018).
Notably in 1993, the first gene, presenilin, was identified in C. elegans, which later on found linked with early-onset (Levitan et al., 1996; De Strooper et al., 1999; Wittenburg et al., 2000). In C. elegans three presenilin genes i.e. sel-12, hop-1, and spe-4 have been found, among which Hop-1 and sel-12 are broadly expressed, whereas, spe-4 is only displayed in the male germ line (Calahorro and Ruiz-Rubio, 2011; Alexander et al., 2014; Sarasija and Norman, 2018). Furthermore, higher sequence similarity to the human presenilins gene, which controls APP processing, can be seen in sel-12. APL-1 overexpression results in 70% mortality, which is reversed in sel-12 mutants, suggesting that Sel-12 regulates APL-1 cleavage. Due to the dysregulation of mitochondrial calcium (Ca2+) homeostasis caused by sel-12 mutations, the worms develop AD (Sarasija et al., 2018; Sarasija and Norman, 2018). Only ptl-1, an orthologue of tau with significant sequence similarity to mammalian tau, is present in C. elegans (shown in Figure 1). There is proof that human tau and ptl-1 both play crucial roles in preserving the integrity of the nervous system. Tau mutations cause frontotemporal dementia, and increasing uncoordinated phenotypes and Neurodegeneration are produced in worm models by transgenic production of human tau variations (Alexander et al., 2014; Pir et al., 2017).
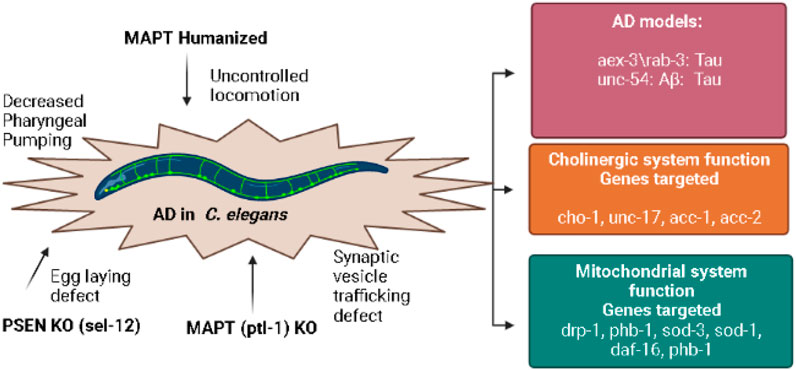
FIGURE 1. C. elegans models for Alzheimer’s disease by targeting different genes.
The effects of chondroitin sulfate E on neuronal adhesion, neurite outgrowth, and neuroprotection are regulated. Chondroitin sulfate E prevents amyloid peptide from aggregating and causing toxicity in transgenic C. elegans, according to a research (Wang et al., 2022). In a different study, transgenic C. elegans producing amyloid genic proteins are treated with silver nanoparticles. The C. elegans strains such as CL2120 and CPV10, which express the human b2-microglobulin (b2-m) and the Ab3-42 peptide, respectively, simulate illnesses related to the deposition of amyloid, one of the most significant categories of chronic disorders linked to population aging. As a result, silver nanoparticles’ toxicity on several physiological parameters in both C. elegans in the wild type and in transgenic form was established. The sensitivity of the tests conducted on the worms was compared to that produced from the well-established human brain D384 and lung A549 cell lines (Diomede et al., 2012; Wang et al., 2022).
It is hypothesized that the accumulation of β-amyloid causes AD, therefore, by expressing human Aβ peptide constructs in worm muscle cells, numerous transgenic strains of C. elegans can be developed for underlining the pathogenesis of AD (Link, 1995). Treusch et al. (2011) have discovered many neurodegenerative modifiers that are functionally linked to cytoskeleton genes (YAP 1802, INP52, SLA1, CRM1, GRR1, KEM1, and RTS1). In 1995, the first C. elegans transgenic model CL2006 was developed by expressing human Aβ1-42 in body-wall muscle using the unc-54 promoter, which is widely employed nowadays to illustrate the neuroprotective properties of various natural and synthetic compounds (Link, 1995). Two 8-OHQs, PBT2, and clioquinol, which were earlier, found to have neuroprotective effects in mice models of AD, also demonstrated effective in treating Aβ-42 toxicity in neurons and body wall muscle cells of C. elegans (Mccoll et al., 2012; Matlack et al., 2014). Currently, three types of C. elegans models for AD such as unc-54, myo-3, and snb-1 are widely employed for screening neuroprotective effect of drug against Aβ toxicity.
Due to age-dependent paralysis in CL2006 C. elegans, it is difficult to explain whether protective compounds are altering Aβ peptide toxicity or aging (Cohen et al., 2006) Several myo-3 promoters have been added to C. elegans to overcome this constraint (Link et al., 2003). Dostal and Link (2010) reported that CL4176 transgenic C. elegans paralyzes immediately, repeatedly, and completely at 25°C. Apart from paralysis, this model exhibits oxidative stress, Aβ deposits, and increased autophagy (Link et al., 2003; Florez-McClure et al., 2007). Muscle cells express Aβ in both types, but neuronal Aβ expression may better represent AD pathophysiology. Later on CL2355 strain was used as an AD model to examine the neuroprotective properties of seven new 2-aryl ethenyl quinoline derivatives. In addition to improving learning memory and suppressing Aβ monomer production, two of the seven chemicals decreased the stress response induced by Aβ in the elegans AD model (He et al., 2017).
AD is linked to the accumulation of NFT, which aggregates into insoluble hyperphosphorylated Tau protein (Mandelkow and Mandelkow, 1998). Several transgenic worms expressing Tau components have been developed to study the consequences of neuronal Tau expression (Kraemer et al., 2003; Miyasaka et al., 2005; Brandt et al., 2009). Ptl-1 is the gene in C. elegans that is homologous to the tau protein. However, the ptl-1 function deficiency cannot entirely imitate the clinical characteristic of tauopathy (Krieg et al., 2017). The neurons of C. elegans exhibit wild-type and frontal, temporal cognitive impairment with parkinson chromosome 17 type mutant human tau protein (Kraemer et al., 2003). It's interesting to note that frontotemporal dementia with parkinsonism chromosome 17 type (FTDP-17), a different neurodegenerative illness, appears to be caused by tau gene mutations rather than Alzheimer’s disease (Rademakers et al., 2012). With age, insoluble phosphorylated tau protein deposition increases along with neuron loss and inconsistent movement (Liachko et al., 2016). When compared to non-transgenic C. elegans, the lifespan of axe-3/tau and FTLD-17 mutant axe-3/tau was significantly reduced. The C. elegans axe-3/tau V337M strain was utilized for RNAi screening to find the abnormal phenotype brought on by tau protein (Kraemer et al., 2006). The mec-7/tau C. elegans mutants R406W and P301L express tau protein in tactual neurons, that consequently induces progressive loss of tactual sensibility with aging, however, neurons do not degenerate, instead, they acquire neuritic flaws, and gradually lose their functionality (Miyasaka et al., 2016). Furthermore, over expressing GSK-3 can make tau protein more toxic, over expressing HSP70 can only slightly lower tau protein toxicity (Miyasaka et al., 2005). Brandt et al. (2009) generated human tau and a pseudo hyperphosphorylated (PHP) tau pan-neuronal transgenic C. elegans model to investigate the role of phosphorylation in tau protein toxicity. As the animals grow older, both wild and PHP types suffer from identical mobility disorders. Tau protein aggregation only developed in PHP type, not wild type, but caused faulty motor neuron development .
According to Fatouros et al. (2012) these tau models also inhibit tau aggregation and aid in discovering substances that have neuroprotective action in the C. elegans model. Tetracycline, coffee extract (without caffeine), copper, and traditional Chinese medicine are substances that have demonstrated anti-aggregation protective properties (Li and Le, 2013). On the human Aβ transgenic worms, Link et al. (2003) used microarrays to analyze gene expression. They have discovered 240 down-regulated genes and 67 up-regulated genes. Transgenic worms exhibit strong induction of the transcript levels of B-crystallin (CRYAB) and tumor necrosis factor-induced protein 1. Postmortem AD brain tissue also indicates upregulation of the human homologs of the genes. Noteworthy, a DA receptor antagonist antipsychotic, azaperon, used to treat schizophrenia, improve transgenic worm movement, and reduce the insoluble tau level in C. elegans AD model, indicating dopamine D2 receptor antagonism may be a promising method for preventing tau-induced neurotoxicity (McCormick et al., 2013).
Parkinson’s disease is considered the second most rapidly growing neurological disorder affecting millions of lives and is now being intrigued as a burden to healthy life owing to its progression (Saewanee et al., 2021). Pathophysiologically, PD is pigeonholed by continuous loss of DA neurons with pervasive intracellular accumulation of the α-synuclein protein in the substantia nigra pars compacta causing irregularities in motor behavior (Thome et al., 2016; Ullah and Khan, 2018), and can be easily diagnosed with four significant clinical cardinal signs, i.e., rigidity, resting tremor, bradykinesia, and loss of postural reflexes (Maulik et al., 2017). Since DA neuronal loss affects both the central and peripheral nervous system, certain non-motor clinical features, including memory loss, depression, anxiety, dysphagia, sleep disorder, and adipsia, are also common in PD (Maulik et al., 2017; Saewanee et al., 2021). The etiology of PD is unclear, yet familial (genetic) and sporadic (environmental) are considered two principal factors triggering the development of PD, and involves predisposition of various genes like α-synuclein, Parkin (PARK2), PTEN-induced putative kinase 1 (PINK1), ubiquitin C-terminal hydrolase L1(UCHL1), leucine-rich repeat kinase 2 (LRRK2), glucocerebrosidase (GBA), DJ-1, etc. (Trinh and Farrer, 2013) (Figure 2). Noteworthy, these genes are at least present in one C. elegans homolog (Harrington et al., 2010; Chege and McColl, 2014; Lee and Cannon, 2015). Interestingly, hermaphroditic C. elegans consists of around 302 neurons, out of which eight are DA in nature, associated with four DA receptors and are homologs to mammalian DA system (Sulston et al., 1975; Chase and Koelle, 2007). Although, α-synuclein, which is responsible for DA neuronal degradation, is not endogenous to C. elegans (Lakso et al., 2003; Karpinar et al., 2009).
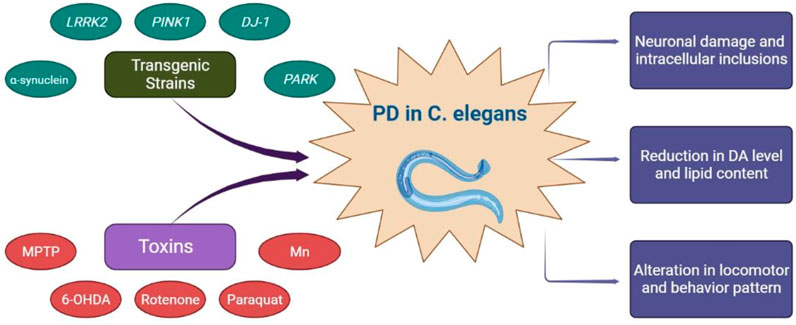
FIGURE 2. C. elegans models for Parkinson’s disease and their pathological outcomes.
C. elegans transgenic models are used to study the genetic basis of PD and its allied neuronal deficits. The α-Synuclein gene is one of the genes responsible for regulating several enzymes associated with DA transportation and release (Maulik et al., 2017). It is one of the significant causative genes for the PD, as any duplicate or triplication α-synuclein locus induces familial type PD (Van Ham et al., 2008). Several transgenic C elegans models with mammalian α-synuclein homologues have been generated to investigate the role of α-synuclein overexpression and aggregation in Parkinson’s disease. (Maulik et al., 2017). C. elegans transgenic α-synuclein strains NL5901, having phenotype unc-54p:: α-synuclein::YFP + unc-119 is widely employed to selectively underline the mechanism of new and potent anti- PD drugs (Jadiya et al., 2011; Bodhicharla et al., 2013; García-Moreno et al., 2019; Anjaneyulu et al., 2020; Chalorak et al., 2021; Schmidt et al., 2021). Anjaneyulu et al. (2020) explored the six ayurvedic nootropics extracts using NL5901 transgenic strain to understand their anti-PD and neuroprotective mechanisms. In addition, Wild isolate Bristol Type N2, OW13, and DDP1 are a few other transgenic strains, having α-synuclein overexpression as their pathological hallmark, which are also being used for understanding the pathogenesis of PD and for screening anti-PD drugs (Bodhicharla et al., 2013; Fu et al., 2014a; Fu et al., 2014b; Chen et al., 2015). Despite overexpressed wild-type α-synuclein strains, mutant α-synuclein (A53T, E46K, and A30P) and α-synuclein pre-formed fibril transgenic strains are also extensively employed as PD transgenic models for C. elegans (Lakso et al., 2003; Maulik et al., 2017; Polinski et al., 2018; Zhang et al., 2018; Gaeta et al., 2019).
Mutations in the LRRK2 gene in human is also a prevalent cause of autosomal dominant and idiopathic PD, and its homologous gene, lrk-1, is present in C. elegans (Sämann et al., 2009; Liu et al., 2011; Cooper et al., 2017). Yao et al., 2010, developed transgenic LRRK2 C. elegans models by mutating R1441C/G and G2019S within GTPase and kinase domains, which later showed depletion in DA level, behavioral and locomotor dysfunction in worms due to over-expression of LRRK2 proteins (Yao et al., 2010). Similarly, mutation of the PINK1 gene, encoded as pink-1 in C. elegans, portrayed a significant loss in dopamine-dependent behaviors in C. elegans along with age-dependent- mitochondrial dysfunction (Cooper et al., 2017). Deletion of djr-1.1, an ortholog of DJ-1 gene in C. elegans, can also impact dopamine-dependent behaviors with elevated sensitivity to oxidative stress (Cooper et al., 2017). Noteworthy, several neuronal transgenic strains such as BZ555, pRF4, BY200, and TG2435, tagged explicitly with a green fluorescent protein (GFP) and a DAT-1 promoter, have been used with environmental toxins to trigger PD in C. elegans (Masoudi et al., 2014).
The neurotoxin 1-methyl-1, 2, 3, 6-tetrahydropyidine (MPTP) induced-PD model is one of the well-established and widely used screening models for the detection of potential anti-PD drugs in rodents which triggers PD-like symptoms by selectively demolishing DA neurons in the substantia nigra. Biologically, its metabolite 1-methyl-4-phenylpyridinium ion (MPP+), produced via oxidation with monoamine oxidase (MAO)-B, persuades mitochondrial damage by impeding complex I during the mitochondrial electron transport chain, which as a result, impairs autophagic degradation and reduces mitochondrial DNA level (Zhu et al., 2012; Miyara et al., 2016). Studies affirmed that incubation of C. elegans with neurotoxin MPTP augments lethality and dwindles mobility, and the developments of these sturdy symptomatic flaws can be correlated to peculiar deterioration of the DA neurons (Braungart et al., 2004). Based on the following outcomes, several investigational studies utilized the MPTP-triggered C. elegans PD model to understand and screen new anti-PD drugs (Lu et al., 2010; Johnson et al., 2018; Lee et al., 2021). Lee et al. (2021) explored the neuroprotective effect of damaurone D in a C. elegans model of PD, and the results revealed significant alleviation in MPTP- induced neuronal damage and α-synuclein expression.
Likewise, MPTP, catecholaminergic neurotoxin 6-hydroxydopamine (6-OHDA) critically triggers PD-like pathology. Despite similar chemical structure to DA, the presence of hydroxyl group in 6-OHDA induces mitochondrial dysfunction, resulting in elevation of oxidative stress and ATP depletion, which consequently degrade DA neuronal activity (Glinka et al., 1997; Glinka et al., 1998; Blesa et al., 2012; Offenburger et al., 2018). Administration of 6-OHDA to C. elegans hamper DA cell bodies and their associated processes (Masoudi et al., 2014). Marvanova and Nichols (2007), screened the neuroprotective potential of several DA, NMDA, and GABA receptor agonists for 6-OHDA-induced DA neurotoxicity in C. elegans, and results showed that two D2 receptor agonists, quinpirole and bromocriptine, dose-dependently diminish 6-OHDA toxicity through receptor-independent mechanisms. TSP-17, tetraspanin family membrane proteins also restrain the DA transporter, DAT-1, in C. elegans and preserve DA neurons from 6-OHDA-induced neurotoxicity (Masoudi et al., 2014). Isoflorentin, an isoflavone from the roots of Belamcnda chinensis (L.) DC, significantly prevents DA neuronal deterioration and elevated life span in the 6-OHDA triggered PD model in C. elegans. Results also affirmed that isoflorentin also ameliorates α-synuclein accumulation in the C. elegans model, indicating it as a possible target agent for PD (Chen et al., 2015). Similarly, Betulin prevents neuronal damage and reverses lifespan dwindleness in the 6-OHDA-induced PD model in C. elegans with significant improvement in proteasomes activity with down-regulation of apoptotic pathways genes (Tsai et al., 2017). Furthermore, several other phytochemicals have been evaluated for their potent anti-PD activity using the 6-OHDA-induced PD model in C. elegans (Chalorak et al., 2018; Ma et al., 2020; Tsai et al., 2020; Chalorak et al., 2021; Li et al., 2021; Saewanee et al., 2021; Long et al., 2022; Muhammad et al., 2022).
Apart from neurotoxins, several insecticides and herbicides have also been employed to induce PD-like symptoms in C. elegans (Settivari et al., 2009; VanDuyn et al., 2010; Jadiya et al., 2013; Jadiya and Nazir, 2013; Jafri Ali and Sharda Rajini, 2013; Settivari et al., 2013). Studies confirmed that rotenone, a broad-spectrum insecticide, develops PD-like pathogenesis in NL5901 C. elegans by significantly elevating oxidative stress and α-synuclein aggregation. Moreover, the reduction in lipid content and mitochondrial activity in the worms indicate it as an environmental neurotoxin that triggers PD in C. elegans (Jadiya et al., 2013; Jadiya and Nazir, 2013; Zhou et al., 2013; González-Hunt et al., 2014). Similarly, monocrotophos, an Organophosphorus insecticide, declines DA neuronal integrity in both N2 and BZ555 type C. elegans, along with a significant reduction in locomotor rate and life span, when compared with MPTP (Jafri Ali and Sharda Rajini, 2013). Paraquat herbicides also elicit DA neurodegeneration in C. elegans by depleting mitochondrial DNA (González-Hunt et al., 2014). Marsova et al., 2020 demonstrated that lactobacillus fermentum U-21 protects C elegans from paraquat-induced oxidative stress and neurodegeneration (Marsova et al., 2020). Manganism, a neurological condition caused by overexposure to manganese, provokes oxidative stress, irregular DA signaling, and cellular death and significantly induces PD-like symptoms in C. elegans. Administration of a high level of manganese to C. elegans can also be used as a toxin-induced C. elegans model of PD (Settivari et al., 2009; Settivari et al., 2013).
In Parkinson disease it has been difficult to deliver drugs to the brain in effective and well controlled manner. Nanotechnology’s development has opened up new possibilities for the treatment of PD. A study by de Guzman et al. (2022), evaluated the impact of functional food nanocomplexes on PD prevention using a C elegans model system. The developed curcumin-loaded human serum albumin nanoparticles improve the body movement, basal slowing response, and dopaminergic neuron degeneration in the C. elegans model of PD, thus affirming the importance of nanomedicine in suppressing the emergence of symptoms of PD (de Guzman et al., 2022).
ALS is a genetically diverse ND Identify by the loss of motor neurons (brain and spinal cord), ultimately imparting ongoing paralysis of the body (Turner et al., 2013; Vérièpe et al., 2015). Neuroinflammation, mitochondrial dysfunction, and impairment in axonal transports are featured as cellular and molecular pathogenesis of ALS (Ferraiuolo et al., 2011; Rojas et al., 2020). Likewise PD, the etiology of ALS is familial and sporadic in nature, which involves numerous genes and proteins, including fused in sarcoma (FUS,) transactive response DNA Binding Protein 43 (TDP-43), and superoxide dismutase-1 (SOD-1) (Alexander et al., 2014; Vérièpe et al., 2015). ALS rodent models exhibit significant ALS pathology, but difficulty in manipulating several genes of ALS at once turned of C. elegans genes researchers to use simple organisms like C. elegans to model ALS toxicity (Therrien and Parker, 2014). Since 80% have human homologs, both Cholinergic and GABAergic neurons play a significant part in the locomotor activity of C. elegans (Jorgensen, 2005), thus making it a suitable model for the exploration of ALS and its pathogenesis.
Mutation in the SOD1 gene alters the enzyme’s folding and stability, causing aggregation in motor neurons following paralysis (Wang et al., 2009; Caldwell et al., 2020). There are more than 160 mutations in the SOD1 gene since 1993 (Al-Chalabi et al., 2012), making it the most common mutation in familial ALS, and the human SOD1 gene has a similar function to that of the C elegans gene. (Wroe et al., 2008; Turner et al., 2013). To date, numerous transgenic lines C. elegans models having mutant human SOD1 gene have been effectively produced with motor neuron degeneration and paralysis characteristics like that of ALS patients (Oeda et al., 2001; Gidalevitz et al., 2009; Wang et al., 2009; Li et al., 2013; Thompson et al., 2014; Baskoylu et al., 2018; Osborne et al., 2021). Both G85R and G93R mutations in the SOD1 transgenic worm exhibited severe Endoplasmic Reticulum (ER)-stress (Wang et al., 2009; Li et al., 2013). Interestingly, Baskoylu et al. (2018) generated a single-copy SOD1 knock-in C. elegans strain with two mutations, G85R and G93R, using a novel CRISPR/Cas9-mediated genome editing technique. Li et al. (2013) developed a novel transgenic G93A mutant SOD1 C. elegans model to underline the role of autophagy in ALS. A4V mutation has also been discovered to induce ER stress, yet the mechanism remains unclear (Perri et al., 2020). Recently, Xu et al., (2022) affirmed the therapeutic potential of metformin in treating ALS by improving autophagy and lengthening lifespan via the daf-16 pathway, using SOD-1 mutant transgenic C. elegans.
Mutation in TDP-43 stimulates protein aggregation along with cytoplasmic mislocalization, which induces impaired motility (Liu et al., 2017). TDP-43 orthologue tdp-1 presents in C. elegans postulate a promising relationship between genetic mutation and cellular pathology (Wegorzewska and Baloh, 2011). Firstly, Ash et al. (2010) developed the first TDP-43 mutated transgenic C. elegans model for ALS, and the over-expression of TDP-1 in worms induces GABAergic motor neuronal degradation with uncoordinated movement. Afterward, various TDP-43 mutated strains have been developed, including TDP-43A315T, being widely used in understanding the pathogenesis of ALS (Vaccaro et al., 2012a; Zhang et al., 2012; Liachko et al., 2013). Noteworthy, Tauffenberger et al. (2013) explored the protective effects of several compounds, i.e., resveratrol, reserpine, trolox, propyl gallate, rolipram, and ethosuximide (earlier affirmed to boost longevity in C. elegans) against mutant TDP-43 toxicity in motor neurons using transgenic TDP-43 models.
Likewise TDP-43, the mutation in the DNA/RNA-binding proteins, FUS, also aggregates protein to induce mortality impairment, and its overexpression alters synaptic functions (Ling et al., 2019). Several transgenic C. elegans models with mutated and overexpressed FUS gene has been developed (Vaccaro et al., 2012b; Murakami et al., 2012; Vérièpe et al., 2015; Ma et al., 2018; Markert et al., 2020; Labarre et al., 2021; Baskoylu et al., 2022). R524S and P525L mutated FUS transgenic ALS models for C. elegans have been created with impaired neuromuscular function and locomotion (Baskoylu et al., 2022). With the advancement in genetic manipulation, Labarre et al. (2021) recently developed a single copy FUS mutant transgenic strain of C. elegans, exhibiting similar ALS phenotypes, including GABAergic neurodegeneration with progressive paralysis. Knockdown dnc-1/dynactin 1 ameliorates autophagosome transportation and stimulates motor neuron degeneration. Based on this, Ikenaka and their team developed a novel dnc-1 knockdown transgenic model of C. elegans and furthermore employed this behavior-based model to identify and evaluate drugs having a potential neuroprotective effect against motor neuron disease (Ikenaka et al., 2013; Ikenaka et al., 2019).
An extension of the hexanucleotide GGGGCC replication in the first intron of the C9ORF72 gene was recently linked to ALS; however, its mechanism is still unclear. The use of C. elegans can be a suitable approach to underline their association with ALS because of the presence of C9ORF72 homolog as alfa-1 (ALS/FTD associated gene homolog) (Therrien et al., 2013; Therrien and Parker, 2014; Rudich et al., 2017). Therrien et al., 2013 firstly developed a transgenic model that induces motor deficits in C. elegans by a mutation in alfa-1. homologe. Furthermore, it was also observed that the model demonstrated a synergistic toxic effect with TDP-43 mutation (Therrien et al., 2013).
Overexposure to metalloid selenium in the body has been concerned as an etiological factor of ALS (Vinceti et al., 2009; Kamel et al., 2012; Malek et al., 2012). Based on this, Estevez et al. (2012) developed a toxin-induced ALS model in C. elegans using sodium selenite as a neurotoxin. Exposure to a high dose of sodium selenite triggers neurodegeneration of cholinergic neurons, followed by paralysis (Estevez et al., 2014). Furthermore, it was also observed that a decrease in insulin/insulin-like (IIS) signaling by elevating PTEN and PINK1 gene expression overcomes selenium-induced motor defects (Estevez et al., 2014).
HD is an incurable, autosomal-dominant adult-onset ND characterized by a reduction in motor and memory functions (Orr and Zoghbi, 2007; Dayalu and AlbinHuntington, 2015). Polyglutamine (polyQ) stretch and expansion in the N terminus of Huntington protein (HTT), by an atypical CAG triplet, replicated extended mutant in Huntington gene, is the prime cause for its induction, with oxidative stress, irregular neuronal metabolism, and cytoplasmic inclusions as major pathological manifestations (MacDonald et al., 1993; DiFiglia et al., 1997). The developed polyQ expansions promote aggregation and misfolding of HTT, which subsequently alters the neurotransmitter uptake and release (Poirier et al., 2005). Despite several mammalian models available, C. elegans may endow better polyQ-induced toxic outcomes and help explore new therapeutic targets for HD.
Although HTT ortholog is absent in C. elegans, numerous polyQ tract and human HTT fragments expressing transgenic C. elegans models have been designed in different neuronal subtypes to investigate the pathogenesis of HD (Faber et al., 1999; Satyal et al., 2000; Parker et al., 2001; Morley et al., 2002; Nollen et al., 2004; Parker et al., 2004; Poirier et al., 2005; Lee et al., 2017). Faber et al. (1999), developed the first transgenic HD model in C. elegans, having 150 repeat polyQ replicates (HTT-Q150) and inducing sensory neuron degradation. Later on, it was also observed that loss of polyQ enhancer-1 gene (pqe-1) further worsens the neurodegeneration; however, its overexpression attenuates Htt-Q150- induced neurotoxicity (Faber et al., 2002). Using these transgenic models, several phytochemical and chemical moieties have been evaluated to be developed as a potential therapy for the treatment of HD (Voisine et al., 2007; Tauffenberger et al., 2012; Boasquívis et al., 2018; Landon et al., 2020; Cordeiro et al., 2021). Glucose exhibited DAF-16 dependent neuroprotective effect in the 128-repeat polyQ HD model (Tauffenberger et al., 2012). Recently, Corerio et al. (2021), demonstrated the neuroprotective potential of rutin against polyQ-induced neurodegeneration using various transgenic C. elegans strains, i.e., Bristol N2 (wild-type), AM141, AM101 (Q40 over-expressed), HA759 (H150 over-expressed), CL 2070, and CF1553, and results showed a significant reduction in polyQ-induced neuronal death with DAF-16 up-regulation (Cordeiro et al., 2021). A study using primary cell culture neurons and C. elegans polyQ poisoning model also showed that acetylation of mutant Htt at K444 enhanced mutant Htt clearance and neuroprotection (Jeong et al., 2009).
The size of the CAG repeats also affects the aggregation phenotype in C. elegans, just as it does in humans. At least three models have been developed to produce polyglutamine-associated neurotoxicity in neurons by enlarging polyglutamine sequences in sensory neurons, touch receptor neurons, or the whole nervous system of C worms (Faber et al., 1999; Parker et al., 2001; Brignull et al., 2006). The body wall muscle cells of the worm C. elegans have been used like in A.D. to imitate polyglutamine aggregation (Morley et al., 2002).
Implication of nanotechnology and nanomedicine can be a possible intervention to treat HD by preventing aggregation of Huntington protein. Selenium nanoparticles developed by Cong et al. (2019) significantly diminishes neuronal death, restores behavioral dysfunction, and protected C. elegans neuronal damage by impeding oxidative stress, huntington protein aggregation, and down–regulation of histone deacetylase enzyme. The results affirmed that nanomedicine can be used as a effective approach to improve HD therapy.
The widespread prevalence of NDs highlights the urgent need of ingenious approaches to identify novel therapeutic targets and disease-modifying elements. AD, PD, ALS, and HD have become more common in recent years. As models of human nervous system, worms share structural and functional similarities. Also, visualizations of neurons and synapses in worms, correlation between neuronal activity and behavior in worms attract interest for development of experimental models with C. elegans. Genetic manipulations make it possible to recognize genes involved in neuronal formation, migration or other functions. Past half century, numerous significant discoveries have completely changed our understanding of biology using the nematode C. elegans as the experimental subject. The C. elegans is an excellent model for studying neurodegeneration and aging, and discovering numerous signaling pathways involved in longevity. There have also been innumerable models of neurodegenerative illnesses created using either worm genome mutations or the expression of human proteins linked to neurodegeneration (such as β-amyloid, α-synuclein, polyglutamine) in specific worm tissues.
The development of C. elegans models to investigate the genetic causes of NDs continues to open new doors for medical research and helped in identifying possible therapeutic targets for specific conditions. The C. elegans models discussed in this review demonstrate that malfunctioning and misfolded proteins cause harmful aggregation and impair critical cellular functions. Although C. elegans exhibits several advantageous characteristics for aging and neurodegenerative studies, yet there are several drawbacks with C. elegans as a model that should not be overlooked while considering its usefulness. Many distinct organs, including the liver, lungs, skin, and blood circulation system, are absent in this primitive organism. Nevertheless, they lack epinephrine, norepinephrine, histamine signaling, and several other notable differences in sodium-dependent channels. Researchers continue to use the benefits of utilizing C. elegans to study various aspects of neurodegeneration, from the genetic routes leading to neuronal death to how various disease-associated molecular pathways might cause neuronal damage. However, further investigation is required to determine which molecular processes cause and contribute in aging.
Increasing access to high-throughput chemical-genetic screenings and the possibility of assessing toxic mixture effects have led to the increased use of C. elegans in toxicological investigations. Because mammalian models have long life cycles, it is challenging to measure chronic and delayed effects of environmental toxins. Short lifespan of C. elegans is making it easy to measure these effects, yet still require more consideration in toxicological investigations.
Furthermore, due to the restricted ability of therapeutic molecules to penetrate the blood-brain barrier, the treatment of neurodegenerative illness continues to pose a formidable challenge. Because of their unique physico-chemical characteristics and capacity to traverse the blood-brain barrier, nanoparticles offer multifunctional accommodations for resolving these biomedical and pharmacological problems. Numerous CNS-related illnesses like AD, PD, ALS and HD may benefit from integrating nanomedicine and neuroscience. The variety of nanoparticles now in the market needs to go through rigorous stability and toxicity testing. They must also be tailored for gene or medication delivery to the CNS. To better under the nanoformulation efficacy, C. elegans models are the novel and suitable approach. However, more studies should be done to describe improved tracking of the origin of NDs and nanoparticle mobility. In the near future, we earnestly anticipate an increase in the use of nanomedicine to treat neurodegenerative diseases.
PC: reviewing, writing. KW: reviewing, writing. GS: conceptualization, framing, reviewing, proof reading.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alexander, A. G., Marfil, V., and Li, C. (2014). Use of Caenorhabditis elegans as a model to study Alzheimer's disease and other neurodegenerative diseases. Front. Genet. 5, 279. doi:10.3389/fgene.2014.00279
Anjaneyulu, J., R, V., and Godbole, A. (2020). Differential effect of ayurvedic nootropics on C. Elegans models of Parkinson’s disease. J. Ayurveda Integr. Med. 11, 440–447. doi:10.1016/j.jaim.2020.07.006
Ash, P. E. A., Zhang, Y. J., Roberts, C. M., Saldi, T., Hutter, H., Buratti, E., et al. (2010). Neurotoxic effects of TDP-43 overexpression in C. Elegans. Hum. Mol. Genet. 19, 3206–3218. doi:10.1093/HMG/DDQ230
Barclay, J. W., Morgan, A., and Burgoyne, R. D. (2012). Neurotransmitter release mechanisms studied in Caenorhabditis elegans. Cell Calcium 52, 289–295. doi:10.1016/j.ceca.2012.03.005
Bargmann, C. I. (1998). Neurobiology of the Caenorhabditis elegans genome. Sci. (80) 282, 2028–2033. doi:10.1126/science.282.5396.2028
Baskoylu, S. N., Chapkis, N., Unsal, B., Lins, J., Schuch, K., Simon, J., et al. (2022). Disrupted autophagy and neuronal dysfunction in C. Elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 38, 110195. doi:10.1016/j.celrep.2021.110195
Baskoylu, S. N., Yersak, J., O’Hern, P., Grosser, S., Simon, J., Kim, S., et al. (2018). Single copy/knock-in models of ALS SOD1 in C. Elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 14, e1007682. doi:10.1371/JOURNAL.PGEN.1007682
Blesa, J., Phani, S., Jackson-Lewis, V., and Przedborski, S. (2012). Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 1–10. doi:10.1155/2012/845618
Boasquívis, P. F., Silva, G. M. M., Paiva, F. A., Cavalcanti, R. M., Nunez, C. V., and De Paula Oliveira, R. (2018). Guarana (paullinia cupana) extract protects Caenorhabditis elegans models for alzheimer disease and huntington disease through activation of antioxidant and protein degradation pathways. Oxid. Med. Cell. Longev. 2018, 1–16. doi:10.1155/2018/9241308
Bodhicharla, R., Nagarajan, A., Winter, J., Adenle, A., Nazir, A., Brady, D., et al. (2013). Effects of α-synuclein overexpression in transgenic Caenorhabditis elegans strains. CNS Neurol. Disord. - Drug Targets 11, 965–975. doi:10.2174/1871527311211080005
Brandt, R., Gergou, A., Wacker, I., Fath, T., and Hutter, H. (2009). A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 30, 22–33. doi:10.1016/j.neurobiolaging.2007.05.011
Braungart, E., Gerlach, M., Riederer, P., Baumeister, R., and Hoener, M. C. (2004). Caenorhabditis elegans MPP+ model of Parkinson’s disease for high-throughput drug screenings. Neurodegener. Dis. 1, 175–183. doi:10.1159/000080983
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. doi:10.1093/genetics/77.1.71
Brignull, H. R., Moore, F. E., Tang, S. J., and Morimoto, R. I. (2006). Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J. Neurosci. 26, 7597–7606. doi:10.1523/JNEUROSCI.0990-06.2006
Calahorro, F., and Ruiz-Rubio, M. (2011). Caenorhabditis elegans as an experimental tool for the study of complex neurological diseases: Parkinson’s disease, Alzheimer’s disease and autism spectrum disorder. Invert. Neurosci. 11, 73–83. doi:10.1007/s10158-011-0126-1
Caldwell, K. A., Willicott, C. W., and Caldwell, G. A. (2020). Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 13, dmm046110. doi:10.1242/dmm.046110
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Sci. (80) 263, 802–805. doi:10.1126/science.8303295
Chalorak, P., Jattujan, P., Nobsathian, S., Poomtong, T., Sobhon, P., and Meemon, K. (2018). Holothuria scabra extracts exhibit anti-Parkinson potential in C. Elegans: A model for anti-Parkinson testing. Nutr. Neurosci. 21, 427–438. doi:10.1080/1028415X.2017.1299437
Chalorak, P., Sanguanphun, T., Limboonreung, T., and Meemon, K. (2021). Neurorescue effects of frondoside a and ginsenoside Rg3 in c. Elegans model of Parkinson’s disease. Molecules 26, 4843. doi:10.3390/molecules26164843
Chase, D. L., and Koelle, M. R. (2007). Biogenic amine neurotransmitters in C. Elegans. WormBook 2007, 1–15. doi:10.1895/wormbook.1.132.1
Chege, P. M., and McColl, G. (2014). Caenorhabditis elegans: A model to investigate oxidative stress and metal dyshomeostasis in Parkinson’s disease. Front. Aging Neurosci. 6, 89. doi:10.3389/fnagi.2014.00089
Chen, Y. M., Liu, S. P., Lin, H. L., Chan, M. C., Chen, Y. C., Huang, Y. L., et al. (2015). Irisflorentin improves α-synuclein accumulation and attenuates 6-OHDA-induced dopaminergic neuron degeneration, implication for Parkinson’s disease therapy. BioMed. 5, 24–32. doi:10.7603/s40681-015-0004-y
Chew, Y. L., Walker, D. S., Towlson, E. K., Vértes, P. E., Yan, G., Barabási, A. L., et al. (2017). Recordings of Caenorhabditis elegans locomotor behaviour following targeted ablation of single motorneurons. Sci. Data 4, 170156. doi:10.1038/sdata.2017.156
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Sci. (80) 313, 1604–1610. doi:10.1126/science.1124646
Cong, W., Bai, R., Li, Y. F., Wang, L., and Chen, C. (2019). Selenium nanoparticles as an efficient nanomedicine for the therapy of huntington’s disease. ACS Appl. Mat. Interfaces 11, 34725–34735. doi:10.1021/acsami.9b12319
Cooper, J. F., Machiela, E., Dues, D. J., Spielbauer, K. K., Senchuk, M. M., and Van Raamsdonk, J. M. (2017). Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 7, 16441–16516. doi:10.1038/s41598-017-16637-2
Cordeiro, L. M., Soares, M. V., da Silva, A. F., Machado, M. L., Bicca Obetine Baptista, F., da Silveira, T. L., et al. (2021). Neuroprotective effects of rutin on ASH neurons in Caenorhabditis elegans model of huntington’s disease. Nutr. Neurosci. 2021, 1–14. doi:10.1080/1028415X.2021.1956254
David, D. C., Ollikainen, N., Trinidad, J. C., Cary, M. P., Burlingame, A. L., and Kenyon, C. (2010). Widespread protein aggregation as an inherent part of aging in C. Elegans. PLoS Biol. 8, 10004500–e1000548. doi:10.1371/journal.pbio.1000450
Dayalu, P., and AlbinHuntington, R. L. (2015). Disease: Pathogenesis and treatment. Neurol. Clin. 33, 101–114. doi:10.1016/j.ncl.2014.09.003
de Guzman, A. C. V., Razzak, M. A., Cho, J. H., Kim, J. Y., and Choi, S. S. (2022). Curcumin-loaded human serum albumin nanoparticles prevent Parkinson’s disease-like symptoms in C. Elegans. Nanomaterials 12, 758. doi:10.3390/nano12050758
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., et al. (1999). A presenilin-1-dependent γ-Secretase-like protease mediates release of notch intracellular domain. Nature 398, 518–522. doi:10.1038/19083
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Sci. (80-. ) 277, 1990–1993. doi:10.1126/science.277.5334.1990
Diomede, L., Soria, C., Romeo, M., Giorgetti, S., Marchese, L., Mangione, P. P., et al. (2012). C. elegans expressing human β2-microglobulin: A novel model for studying the relationship between the molecular assembly and the toxic phenotype. PLoS One 7, e52314. doi:10.1371/journal.pone.0052314
Dostal, V., and Link, C. D. (2010). Assaying β-amyloid toxicity using a transgenic C. elegans model. J. Vis. Exp. 2010, e2252. doi:10.3791/2252
Durán, N., Durán, M., de Jesus, M. B., Seabra, A. B., Fávaro, W. J., and Nakazato, G. (2016). Silver nanoparticles: A new view on mechanistic aspects on antimicrobial activity. Nanomedicine Nanotechnol. Biol. Med. 12, 789–799. doi:10.1016/j.nano.2015.11.016
Estevez, A. O., Morgan, K. L., Szewczyk, N. J., Gems, D., and Estevez, M. (2014). The neurodegenerative effects of selenium are inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like growth factor signaling in Caenorhabditis elegans. Neurotoxicology 41, 28–43. doi:10.1016/j.neuro.2013.12.012
Estevez, A. O., Mueller, C. L., Morgan, K. L., Szewczyk, N. J., Teece, L., Miranda-Vizuete, A., et al. (2012). Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans. Neurotoxicology 33, 1021–1032. doi:10.1016/j.neuro.2012.04.019
Faber, P. W., Alter, J. R., Macdonald, M. E., and Hart, A. C. (1999). Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. U. S. A. 96, 179–184. doi:10.1073/PNAS.96.1.179
Faber, P. W., Voisine, C., King, D. C., Bates, E. A., and Hart, A. C. (2002). Glutamine/proline-rich PQE-1 proteins protect Caenorhabditis elegans neurons from huntingtin polyglutamine neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 99, 17131–17136. doi:10.1073/pnas.262544899
Fatouros, C., Pir, G. J., Biernat, J., Koushika, S. P., Mandelkow, E., Mandelkow, E. M., et al. (2012). Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 21, 3587–3603. doi:10.1093/hmg/dds190
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi:10.1038/nrneurol.2011.152
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. doi:10.1038/35888
Florez-McClure, M. L., Hohsfield, L. A., Fonte, G., Bealor, M. T., and Link, C. D. (2007). Decreased insulin-receptor signaling promotes the autophagic degradation of β-amyloid peptide in C. Elegans. Autophagy 3, 569–580. doi:10.4161/auto.4776
Fu, R. H., Harn, H. J., Liu, S. P., Chen, C. S., Chang, W. L., Chen, Y. M., et al. (2014). N-butylidenephthalide protects against dopaminergic neuron degeneration and α-synuclein accumulation in Caenorhabditis elegans models of Parkinson’s disease. PLoS One 9, e85305. doi:10.1371/JOURNAL.PONE.0085305
Fu, R. H., Wang, Y. C., Chen, C. S., Tsai, R. T., Liu, S. P., Chang, W. L., et al. (2014). Acetylcorynoline attenuates dopaminergic neuron degeneration and α-synuclein aggregation in animal models of Parkinson’s disease. Neuropharmacology 82, 108–120. doi:10.1016/J.NEUROPHARM.2013.08.007
Gaeta, A. L., Caldwell, K. A., and Caldwell, G. A. (2019). Found in translation: The utility of C. Elegans alpha-synuclein models of Parkinson’s disease. Brain Sci. 9, 73. doi:10.3390/brainsci9040073
García-Moreno, J. C., Porta de la Riva, M., Martínez-Lara, E., Siles, E., and Cañuelo, A. (2019). Tyrosol, a simple phenol from EVOO, targets multiple pathogenic mechanisms of neurodegeneration in a C. Elegans model of Parkinson’s disease. Neurobiol. Aging 82, 60–68. doi:10.1016/j.neurobiolaging.2019.07.003
Garigan, D., Hsu, A. L., Fraser, A. G., Kamath, R. S., Abringet, J., and Kenyon, C. (2002). Genetic analysis of tissue aging in Caenorhabditis elegans: A role for heat-shock factor and bacterial proliferation. Genetics 161, 1101–1112. doi:10.1093/GENETICS/161.3.1101
Gidalevitz, T., Krupinski, T., Garcia, S., and Morimoto, R. I. (2009). Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 5, e1000399. doi:10.1371/JOURNAL.PGEN.1000399
Glinka, Y., Gassen, M., and Youdim, M. B. H. (1997). “Mechanism of 6-hydroxydopamine neurotoxicity,” in Advances in research on neurodegeneration. Editors P. Riederer, D. B. Calne, R. Horowski, Y. Mizuno, W. Poewe, and M. B. H. Youdim (Springer Vienna), 55–66.
Glinka, Y., Tipton, K. F., and Youdim, M. B. H. (1998). Mechanism of inhibition of mitochondrial respiratory complex I by 6- hydroxydopamine and its prevention by desferrioxamine. Eur. J. Pharmacol. 351, 121–129. doi:10.1016/S0014-2999(98)00279-9
González-Hunt, C. P., Leung, M. C. K., Bodhicharla, R. K., McKeever, M. G., Arrant, A. E., Margillo, K. M., et al. (2014). Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. PLoS One 9, e114459. doi:10.1371/journal.pone.0114459
Harrington, A. J., Hamamichi, S., Caldwell, G. A., and Caldwell, K. A. C. (2010). C. elegansas a model organism to investigate molecular pathways involved with Parkinson's disease. Dev. Dyn. 239, 1282–1295. doi:10.1002/dvdy.22231
He, Q., Huang, G., Chen, Y., Wang, X., Huang, Z., and Chen, Z. (2017). The protection of novel 2-arylethenylquinoline derivatives against impairment of associative learning memory induced by neural Aβ in C. Elegans Alzheimer’s disease model. Neurochem. Res. 42, 3061–3072. doi:10.1007/s11064-017-2339-0
Ikenaka, K., Kawai, K., Katsuno, M., Huang, Z., Jiang, Y. M., Iguchi, Y., et al. (2013). Dnc-1/Dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS One 8, e54511. doi:10.1371/journal.pone.0054511
Ikenaka, K., Tsukada, Y., Giles, A. C., Arai, T., Nakadera, Y., Nakano, S., et al. (2019). A behavior-based drug screening system using a Caenorhabditis elegans model of motor neuron disease. Sci. Rep. 9, 10104. doi:10.1038/s41598-019-46642-6
Jadiya, P., Khan, A., Sammi, S. R., Kaur, S., Mir, S. S., and Nazir, A. (2011). Anti-parkinsonian effects of bacopa monnieri: Insights from transgenic and pharmacological Caenorhabditis elegans models of Parkinson’s disease. Biochem. Biophys. Res. Commun. 413, 605–610. doi:10.1016/j.bbrc.2011.09.010
Jadiya, P., Mir, S., and Nazir, A. (2013). Effect of various classes of pesticides on expression of stress genes in transgenic C. Elegans model of Parkinson’s disease. CNS Neurol. Disord. - Drug Targets 11, 1001–1005. doi:10.2174/1871527311211080009
Jadiya, P., and Nazir, A. (2013). Environmental toxicants as extrinsic epigenetic factors for parkinsonism: Studies employing transgenic C. Elegans model. CNS Neurol. Disord. - Drug Targets 11, 976–983. doi:10.2174/1871527311211080006
Jafri Ali, S., and Sharda Rajini, P. (2013). Elicitation of dopaminergic features of Parkinson’s disease in C. Elegans by monocrotophos, an organophosphorous insecticide. CNS Neurol. Disord. - Drug Targets 11, 993–1000. doi:10.2174/1871527311211080008
Jeong, H., Then, F., Melia, T. J., Mazzulli, J. R., Cui, L., Savas, J. N., et al. (2009). Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137, 60–72. doi:10.1016/j.cell.2009.03.018
Johnson, S., Park, H., DaSilva, N., Vattem, D., Ma, H., and Seeram, N. (2018). Levodopa-reduced mucuna pruriens seed extract shows neuroprotective effects against Parkinson’s disease in murine microglia and human neuroblastoma cells, Caenorhabditis elegans, and Drosophila melanogaster. Nutrients 10, 1139. doi:10.3390/nu10091139
Kamel, F., Umbach, D. M., Bedlack, R. S., Richards, M., Watson, M., Alavanja, M. C. R., et al. (2012). Pesticide exposure and amyotrophic lateral sclerosis. Neurotoxicology 33, 457–462. doi:10.1016/j.neuro.2012.04.001
Karpinar, D. P., Balija, M. B. G., Kügler, S., Opazo, F., Rezaei-Ghaleh, N., Wender, N., et al. (2009). Pre-fibrillar α-synuclein variants with impaired Β-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 28, 3256–3268. doi:10.1038/emboj.2009.257
Kim, C., Kim, J., Kim, S., Cook, D. E., Evans, K. S., Andersen, E. C., et al. (2019). Long-read sequencing reveals intra-species tolerance of substantial structural variations and new subtelomere formation in C. Elegans. Genome Res. 29, 1023–1035. doi:10.1101/gr.246082.118
Kraemer, B. C., Burgess, J. K., Chen, J. H., Thomas, J. H., and Schellenberg, G. D. (2006). Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 15, 1483–1496. doi:10.1093/hmg/ddl067
Kraemer, B. C., Zhang, B., Leverenz, J. B., Thomas, J. H., Trojanowski, J. Q., and Schellenberg, G. D. (2003). Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. U. S. A. 100, 9980–9985. doi:10.1073/pnas.1533448100
Krieg, M., Stühmer, J., Cueva, J. G., Fetter, R., Spilker, K., Cremers, D., et al. (2017). Genetic defects in β-spectrin and tau sensitize C. Elegans axons to movement-induced damage via torque-tension coupling. Elife 6, e20172. doi:10.7554/eLife.20172
Labarre, A., Tossing, G., Maios, C., Doyle, J. J., and Parker, J. A. A single copy transgenic mutant FUS strain reproduces age-dependent ALS phenotypes in C. Elegans. Micropubl. Biol. 2021, 2021, 000473. doi:10.17912/MICROPUB.BIOLOGY.000473
Lakso, M., Vartiainen, S., Moilanen, A. M., Sirviö, J., Thomas, J. H., Nass, R., et al. (2003). Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J. Neurochem. 86, 165–172. doi:10.1046/j.1471-4159.2003.01809.x
Landon, G., Whitney, W., Priya, R., and Mindy, F. (2020). Glucose effects on polyglutamine-induced proteotoxic stress in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 522, 709–715. doi:10.1016/j.bbrc.2019.11.159
Lee, A. L., Ung, H. M., Sands, L. P., and Kikis, E. A. (2017). A new Caenorhabditis elegans model of human huntingtin 513 aggregation and toxicity in body wall muscles. PLoS One 12, e0173644. doi:10.1371/journal.pone.0173644
Lee, J. W., and Cannon, J. R. (2015). LRRK2 mutations and neurotoxicant susceptibility. Exp. Biol. Med. 240, 752–759. doi:10.1177/1535370215579162
Lee, S. H., Han, Y. T., and Cha, D. S. (2021). Neuroprotective effect of damaurone D in a C. Elegans model of Parkinson’s disease. Neurosci. Lett. 747, 135623. doi:10.1016/j.neulet.2021.135623
Levitan, D., Doyle, T. G., Brousseau, D., Lee, M. K., Thinakaran, G., Slunt, H. H., et al. (1996). Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 93, 14940–14944. doi:10.1073/pnas.93.25.14940
Li, H., Feng, Y., Chen, Z., Jiang, X., Zhou, Z., Yuan, J., et al. (2021). Pepper component 7-ethoxy-4-methylcoumarin, a novel dopamine D2 receptor agonist, ameliorates experimental Parkinson’s disease in mice and Caenorhabditis elegans. Pharmacol. Res. 163, 105220. doi:10.1016/j.phrs.2020.105220
Li, J., Huang, K. X., and Le, W. D. (2013). Establishing a novel C. Elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta Pharmacol. Sin. 34, 644–650. doi:10.1038/aps.2012.190
Li, J., and Le, W. (2013). Modeling neurodegenerative diseases in Caenorhabditis elegans. Exp. Neurol. 250, 94–103. doi:10.1016/J.EXPNEUROL.2013.09.024
Liachko, N. F., McMillan, P. J., Guthrie, C. R., Bird, T. D., Leverenz, J. B., and Kraemer, B. C. (2013). CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol. 74, 39–52. doi:10.1002/ANA.23870
Liachko, N. F., Saxton, A. D., McMillan, P. J., Strovas, T. J., Currey, H. N., Taylor, L. M., et al. (2016). The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathol. 132, 545–561. doi:10.1007/s00401-016-1600-y
Ling, S.-C., Dastidar, S. G., Tokunaga, S., Ho, W. Y., Lim, K., Ilieva, H., et al. (2019). Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome Axis. Elife 8, e40811. doi:10.7554/eLife.40811
Link, C. D. (1995). Expression of human β-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 92, 9368–9372. doi:10.1073/pnas.92.20.9368
Link, C. D., Taft, A., Kapulkin, V., Duke, K., Kim, S., Fei, Q., et al. (2003). Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol. Aging 24, 397–413. doi:10.1016/S0197-4580(02)00224-5
Liu, E. Y., Cali, C. P., and Lee, E. B. (2017). RNA metabolism in neurodegenerative disease. Dis. Model. Mech. 10, 509–518. doi:10.1242/DMM.028613
Liu, Z., Hamamichi, S., Lee, B. D., Yang, D., Ray, A., Caldwell, G. A., et al. (2011). Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 20, 3933–3942. doi:10.1093/HMG/DDR312
Long, T., Wu, Q., Wei, J., Tang, Y., He, Y.-N., He, C.-L., et al. (2022). Ferulic acid exerts neuroprotective effects via autophagy induction in C. Elegans and cellular models of Parkinson’s disease. Oxid. Med. Cell. Longev. 2022, 1–19. doi:10.1155/2022/3723567
Lu, X. lin, Yao, X. li, Liu, Z., Zhang, H., Li, W., Li, Z., et al. (2010). Protective effects of xyloketal B against MPP+-Induced neurotoxicity in Caenorhabditis elegans and PC12 cells. Brain Res. 1332, 110–119. doi:10.1016/J.BRAINRES.2010.03.071
Ma, L., Zhao, Y., Chen, Y., Cheng, B., Peng, A., and Huang, K. (2018). Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur. J. Pharmacol. 819, 169–180. doi:10.1016/j.ejphar.2017.11.051
Ma, X., Li, J., Cui, X., Li, C., and Wang, Z. (2020). Dietary supplementation with peptides from sesame cake alleviates Parkinson’s associated pathologies in Caenorhabditis elegans. J. Funct. Foods 65, 103737. doi:10.1016/J.JFF.2019.103737
MacDonald, M. E., Barnes, G., Srinidhi, J., Duyao, M. P., Ambrose, C. M., Myers, R. H., et al. (1993). Gametic but not somatic instability of CAG repeat length in huntington’s disease. J. Med. Genet. 30, 982–986. doi:10.1136/jmg.30.12.982
Malek, A. M., Barchowsky, A., Bowser, R., Youk, A., and Talbott, E. O. (2012). Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: A meta-analysis of epidemiological studies. Environ. Res. 117, 112–119. doi:10.1016/j.envres.2012.06.007
Mandelkow, E. M., and Mandelkow, E. (1998). Tau in Alzheimer’s disease. Trends Cell Biol. 8, 425–427. doi:10.1016/S0962-8924(98)01368-3
Markaki, M., and Tavernarakis, N. (2010). Modeling human diseases in Caenorhabditis elegans. Biotechnol. J. 5, 1261–1276. doi:10.1002/biot.201000183
Markert, S. M., Skoruppa, M., Yu, B., Mulcahy, B., Zhen, M., Gao, S., et al. (2020). Overexpression of an ALS-associated FUS mutation in C. Elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol. Open 9, bio055129. doi:10.1242/bio.055129
Marsova, M., Poluektova, E., Odorskaya, M., Ambaryan, A., Revishchin, A., Pavlova, G., et al. (2020). Protective effects of lactobacillus fermentum U-21 against paraquat-induced oxidative stress in Caenorhabditis elegans and mouse models. World J. Microbiol. Biotechnol. 36, 104. doi:10.1007/s11274-020-02879-2
Marvanova, M., and Nichols, C. D. (2007). Identification of neuroprotective compounds of Caenorhabditis elegans dopaminergic neurons against 6-OHDA. J. Mol. Neurosci. 31, 127–137. doi:10.1385/JMN/31:02:127
Masoudi, N., Ibanez-Cruceyra, P., Offenburger, S. L., Holmes, A., and Gartner, A. (2014). Tetraspanin (TSP-17) protects dopaminergic neurons against 6-OHDA-induced neurodegeneration in C. Elegans. PLoS Genet. 10, e1004767. doi:10.1371/journal.pgen.1004767
Matlack, K. E. S., Tardiff, D. F., Narayan, P., Hamamichi, S., Caldwell, K. A., Caldwell, G. A., et al. (2014). Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. U. S. A. 111, 4013–4018. doi:10.1073/pnas.1402228111
Matsunami, K. (2018). Frailty and Caenorhabditis elegans as a benchtop animal model for screening drugs including natural herbs. Front. Nutr. 5, 111. doi:10.3389/fnut.2018.00111
Maulik, M., Mitra, S., Bult-Ito, A., Taylor, B. E., and Vayndorf, E. M. (2017). Behavioral phenotyping and pathological indicators of Parkinson’s disease in C. Elegans models. Front. Genet. 8, 77. doi:10.3389/fgene.2017.00077
Maurer, L. L., Yang, X., Schindler, A. J., Taggart, R. K., Jiang, C., Hsu-Kim, H., et al. (2016). Intracellular trafficking pathways in silver nanoparticle uptake and toxicity in Caenorhabditis elegans. Nanotoxicology 10, 831–835. doi:10.3109/17435390.2015.1110759
Mccoll, G., Roberts, B. R., Pukala, T. L., Kenche, V. B., Roberts, C. M., Link, C. D., et al. (2012). Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 7, 57–59. doi:10.1186/1750-1326-7-57
McCormick, A. V., Wheeler, J. M., Guthrie, C. R., Liachko, N. F., and Kraemer, B. C. (2013). Dopamine D2 receptor antagonism suppresses tau aggregation and neurotoxicity. Biol. Psychiatry 73, 464–471. doi:10.1016/j.biopsych.2012.08.027
Miyara, M., Kotake, Y., Tokunaga, W., Sanoh, S., and Ohta, S. (2016). Mild MPP+ exposure impairs autophagic degradation through a novel lysosomal acidity-independent mechanism. J. Neurochem. 139, 294–308. doi:10.1111/jnc.13700
Miyasaka, T., Ding, Z., Gengyo-Ando, K., Oue, M., Yamaguchi, H., Mitani, S., et al. (2005). Progressive neurodegeneration in C. Elegans model of tauopathy. Neurobiol. Dis. 20, 372–383. doi:10.1016/j.nbd.2005.03.017
Miyasaka, T., Xie, C., Yoshimura, S., Shinzaki, Y., Yoshina, S., Kage-Nakadai, E., et al. (2016). Curcumin improves tau-induced neuronal dysfunction of nematodes. Neurobiol. Aging 39, 69–81. doi:10.1016/j.neurobiolaging.2015.11.004
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 99, 10417–10422. doi:10.1073/pnas.152161099
Muhammad, F., Liu, Y., Wang, N., Zhao, L., Zhou, Y., Yang, H., et al. (2022). Anti-α-Synuclein toxicity and anti-neurodegenerative role of chrysin in transgenic Caenorhabditis elegans models of Parkinson’s disease. ACS Chem. Neurosci. 13, 442–453. doi:10.1021/acschemneuro.1c00548
Murakami, T., Yang, S. P., Xie, L., Kawano, T., Fu, D., Mukai, A., et al. (2012). Als mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 21, 1–9. doi:10.1093/hmg/ddr417
Nollen, E. A. A., Garcia, S. M., Van Haaften, G., Kim, S., Chavez, A., Morimoto, R. I., et al. (2004). Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U. S. A. 101, 6403–6408. doi:10.1073/pnas.0307697101
Oeda, T., Shimohama, S., Kitagawa, N., Kohno, R., Imura, T., Shibasaki, H., et al. (2001). Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 10, 2013–2023. doi:10.1093/HMG/10.19.2013
Offenburger, S.-L., Ho, X. Y., Tachie-Menson, T., Coakley, S., Hilliard, M. A., and Gartner, A. (2018). 6-OHDA-Induced dopaminergic neurodegeneration in Caenorhabditis elegans is promoted by the engulfment pathway and inhibited by the transthyretin-related protein TTR-33. PLoS Genet. 14, e1007125. doi:10.1371/journal.pgen.1007125
Olsen, A., Vantipalli, M. C., and Lithgow, G. J. (2006). Using Caenorhabditis elegans as a model for aging and age-related diseases. Ann. N. Y. Acad. Sci. 1067, 120–128. doi:10.1196/annals.1354.015
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621. doi:10.1146/annurev.neuro.29.051605.113042
Osborne, J. F., Yanagi, K. S., and Hart, A. C. (2021). Genetic interactions in a C. Elegans sod-1 ALS model: Glutamatergic neuron degeneration. Micropubl. Biol. 2021, 000338. doi:10.17912/MICROPUB.BIOLOGY.000338
Parker, J. A., Connolly, J. B., Wellington, C., Hayden, M., Dausset, J., and Neri, C. (2001). Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. U. S. A. 98, 13318–13323. doi:10.1073/pnas.231476398
Parker, J. A., Holbert, S., Lambert, E., Abderrahmane, S., and Néri, C. (2004). Genetic and pharmacological suppression of polyglutamine-dependent neuronal dysfunction in Caenorhabditis elegans. J. Mol. Neurosci. 23, 061–068. doi:10.1385/jmn:23:1-2:061
Perri, E. R., Parakh, S., Vidal, M., Mehta, P., Ma, Y., Walker, A. K., et al. (2020). The cysteine (cys) residues cys-6 and cys-111 in mutant superoxide dismutase 1 (SOD1) A4V are required for induction of endoplasmic Reticulum stress in amyotrophic lateral sclerosis. J. Mol. Neurosci. 70, 1357–1368. doi:10.1007/s12031-020-01551-6
Pir, G. J., Choudhary, B., and Mandelkow, E. (2017). Caenorhabditis elegans models of tauopathy. FASEB J. 31, 5137–5148. doi:10.1096/fj.201701007
Poirier, M. A., Jiang, H., and Ross, C. A. (2005). A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: Evidence for a compact beta-sheet structure. Hum. Mol. Genet. 14, 765–774. doi:10.1093/hmg/ddi071
Polinski, N. K., Volpicelli-Daley, L. A., Sortwell, C. E., Luk, K. C., Cremades, N., Gottler, L. M., et al. (2018). Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J. Park. Dis. 8, 303–322. doi:10.3233/JPD-171248
Rademakers, R., Neumann, M., and MacKenzie, I. R. (2012). Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434. doi:10.1038/nrneurol.2012.117
Rahman, M. F., Wang, J., Patterson, T. A., Saini, U. T., Robinson, B. L., Newport, G. D., et al. (2009). Expression of genes related to oxidative stress in the mouse brain after exposure to silver-25 nanoparticles. Toxicol. Lett. 187, 15–21. doi:10.1016/j.toxlet.2009.01.020
Ribeiro, F. M., Camargos, E. R., da, S., De Souza, L. C., and Teixeira, A. L. (2013). Animal models of neurodegenerative diseases. Rev. Bras. Psiquiatr. 35, S82–S91. doi:10.1590/1516-4446-2013-1157
Roh, J. Y., Sang, J. S., Yi, J., Park, K., Kyu, H. C., Ryu, D. Y., et al. (2009). Ecotoxicity of silver nanoparticles on the soil nematode Caenorhabditis elegans using functional ecotoxicogenomics. Environ. Sci. Technol. 43, 3933–3940. doi:10.1021/es803477u
Rojas, P., Ramírez, A. I., Fernández-Albarral, J. A., López-Cuenca, I., Salobrar-García, E., Cadena, M., et al. (2020). Amyotrophic lateral sclerosis: A neurodegenerative motor neuron disease with ocular involvement. Front. Neurosci. 14, 566858. doi:10.3389/FNINS.2020.566858
Rudich, P., Snoznik, C., Watkins, S. C., Monaghan, J., Pandey, U. B., and Lamitina, S. T. (2017). Nuclear localized C9orf72-associated arginine containing dipeptides exhibit age-dependent toxicity in C. Elegans. Hum. Mol. Genet. 26, 4916–4928. doi:10.1093/hmg/ddx372
Saewanee, N., Praputpittaya, T., Malaiwong, N., Chalorak, P., and Meemon, K. (2021). Neuroprotective effect of metformin on dopaminergic neurodegeneration and α-synuclein aggregation in C. Elegans model of Parkinson’s disease. Neurosci. Res. 162, 13–21. doi:10.1016/j.neures.2019.12.017
Sämann, J., Hegermann, J., von Gromoff, E., Eimer, S., Baumeister, R., and Schmidt, E. (2009). Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 284, 16482–16491. doi:10.1074/jbc.M808255200
Sarasija, S., Laboy, J. T., Ashkavand, Z., Bonner, J., Tang, Y., and Norman, K. R. (2018). Presenilin mutations deregulate mitochondrial Ca2+ homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife 7, e33052. doi:10.7554/elife.33052
Sarasija, S., and Norman, K. R. (2018). Role of presenilin in mitochondrial oxidative stress and neurodegeneration in Caenorhabditis elegans. Antioxidants 7, 111. doi:10.3390/antiox7090111
Sattelle, D. B., and Buckingham, S. D. (2006). Invertebrate studies and their ongoing contributions to neuroscience. Invert. Neurosci. 6, 1–3. doi:10.1007/s10158-005-0014-7
Satyal, S. H., Schmidt, E., Kitagawa, K., Sondheimer, N., Lindquist, S., Kramer, J. M., et al. (2000). Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 97, 5750–5755. doi:10.1073/pnas.100107297
Schirinzi, T., Madeo, G., Martella, G., Maltese, M., Picconi, B., Calabresi, P., et al. (2016). Early synaptic dysfunction in Parkinson’s disease: Insights from animal models. Mov. Disord. 31, 802–813. doi:10.1002/mds.26620
Schmidt, M. Y., Chamoli, M., Lithgow, G. J., and Andersen, J. K. (2021). Swimming exercise reduces native-synuclein protein species in a transgenic C. Elegans model of Parkinson’s disease. Micropubl. Biol. 2021, 000413. doi:10.17912/MICROPUB.BIOLOGY.000413
Settivari, R., LeVora, J., and Nass, R. (2009). The divalent metal transporter homologues SMF-1/2 mediate dopamine neuron sensitivity in Caenorhabditis elegans models of manganism and Parkinson disease. J. Biol. Chem. 284, 35758–35768. doi:10.1074/jbc.M109.051409
Settivari, R., VanDuyn, N., LeVora, J., and Nass, R. (2013). The Nrf2/SKN-1-Dependent glutathione S-transferase π homologue GST-1 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of manganism. Neurotoxicology 38, 51–60. doi:10.1016/j.neuro.2013.05.014
Sulston, J., Dew, M., and Brenner, S. (1975). Dopaminergic neurons in the nematode Caenorhabditis elegans. J. Comp. Neurol. 163, 215–226. doi:10.1002/cne.901630207
Tauffenberger, A., Julien, C., and Parker, J. A. (2013). Evaluation of longevity enhancing compounds against transactive response DNA-binding protein-43 neuronal toxicity. Neurobiol. Aging 34, 2175–2182. doi:10.1016/j.neurobiolaging.2013.03.014
Tauffenberger, A., Vaccaro, A., Aulas, A., Velde, C. V., and Parker, J. A. (2012). Glucose delays age-dependent proteotoxicity. Aging Cell 11, 856–866. doi:10.1111/j.1474-9726.2012.00855.x
Therrien, M., and Parker, J. A. (2014). Worming forward: Amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 5, 85. doi:10.3389/fgene.2014.00085
Therrien, M., Rouleau, G. A., Dion, P. A., and Parker, J. A. (2013). Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. Elegans. PLoS One 8, e83450. doi:10.1371/journal.pone.0083450
Thiele, S. L., Warre, R., and Nash, J. E. (2012). Development of a unilaterally-lesioned 6-OHDA mouse model of Parkinson’s disease. J. Vis. Exp. e3234, 3234. doi:10.3791/3234
Thome, A. D., Harms, A. S., Volpicelli-Daley, L. A., and Standaert, D. G. (2016). MicroRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J. Neurosci. 36, 2383–2390. doi:10.1523/JNEUROSCI.3900-15.2016
Thompson, M. L., Chen, P., Yan, X., Kim, H., Borom, A. R., Roberts, N. B., et al. (2014). TorsinA rescues ER-associated stress and locomotive defects in C. Elegans models of ALS. DMM Dis. Model. Mech. 7, 233–243. doi:10.1242/dmm.013615
Torres, E. M., and Dunnett, S. B. (2011). 6-OHDA lesion models of Parkinson’s disease in the rat. Neuromethods 61, 267–279. doi:10.1007/978-1-61779-298-4_13
Treusch, S., Hamamichi, S., Goodman, J. L., Matlack, K. E. S., Chung, C. Y., Baru, V., et al. (2011). Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Sci. (80-. ) 334, 1241–1245. doi:10.1126/science.1213210
Trinh, J., and Farrer, M. (2013). Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 9, 445–454. doi:10.1038/nrneurol.2013.132
Tsai, C. W., Tsai, R. T., Liu, S. P., Chen, C. S., Tsai, M. C., Chien, S. H., et al. (2017). Neuroprotective effects of Betulin in pharmacological and transgenic Caenorhabditis elegans models of Parkinson’s disease. Cell Transpl. 26, 1903–1918. doi:10.1177/0963689717738785
Tsai, R. T., Tsai, C. W., Liu, S. P., Gao, J. X., Kuo, Y. H., Chao, P. M., et al. (2020). Maackiain ameliorates 6-hydroxydopamine and snca pathologies by modulating the pink1/parkin pathway in models of Parkinson’s disease in Caenorhabditis elegans and the sh-sy5y cell line. Int. J. Mol. Sci. 21, 4455–4534. doi:10.3390/ijms21124455
Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chio, A., De Carvalho, M., et al. (2013). Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322. doi:10.1016/s1474-4422(13)70036-x
Ullah, H., and Khan, H. (2018). Anti-Parkinson potential of silymarin: Mechanistic insight and therapeutic standing. Front. Pharmacol. 9, 422–516. doi:10.3389/fphar.2018.00422
Vaccaro, A., Tauffenberger, A., Aggad, D., Rouleau, G., Drapeau, P., and Parker, J. A. (2012). Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. Elegans. PLoS One 7, e31321. doi:10.1371/journal.pone.0031321
Vaccaro, A., Tauffenberger, A., Ash, P. E. A., Carlomagno, Y., Petrucelli, L., and Parker, J. A. (2012). TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 8, e1002806. doi:10.1371/JOURNAL.PGEN.1002806
Van Ham, T. J., Thijssen, K. L., Breitling, R., Hofstra, R. M. W., Plasterk, R. H. A., and Nollen, E. A. A. C. (2008). C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genet. 4, e1000027. doi:10.1371/journal.pgen.1000027
VanDuyn, N., Settivari, R., Wong, G., and Nass, R. (2010). SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol. Sci. 118, 613–624. doi:10.1093/toxsci/kfq285
Vérièpe, J., Fossouo, L., and Parker, J. A. (2015). Neurodegeneration in C. Elegans models of ALS requires TIR-1/sarm1 immune pathway activation in neurons. Nat. Commun. 6, 7319–9. doi:10.1038/ncomms8319
Vinceti, M., Maraldi, T., Bergomi, M., and Malagoli, C. (2009). Risk of chronic low-dose selenium overexposure in humans: Insights from epidemiology and biochemistry. Rev. Environ. Health 24, 231–248. doi:10.1515/REVEH.2009.24.3.231
Visanji, N. P., Brotchie, J. M., Kalia, L. V., Koprich, J. B., Tandon, A., Watts, J. C., et al. (2016). α-Synuclein-Based animal models of Parkinson’s disease: Challenges and opportunities in a new era. Trends Neurosci. 39, 750–762. doi:10.1016/j.tins.2016.09.003
Voisine, C., Varma, H., Walker, N., Bates, E. A., Stockwell, B. R., and Hart, A. C. (2007). Identification of potential therapeutic drugs for huntington’s disease using Caenorhabditis elegans. PLoS One 2, e504. doi:10.1371/journal.pone.0000504
Wadhwa, K., Kadian, V., Puri, V., Bhardwaj, B. Y., Sharma, A., Pahwa, R., et al. (2022). New insights into quercetin nanoformulations for topical delivery. Phytomedicine Plus 2, 100257. doi:10.1016/j.phyplu.2022.100257
Wang, J., Farr, G. W., Hall, D. H., Li, F., Furtak, K., Dreier, L., et al. (2009). An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 5, e1000350. doi:10.1371/journal.pgen.1000350
Wang, X., Yang, Y., Zou, J., Li, Y., and Zhang, X. G. (2022). Chondroitin sulfate E alleviates β-amyloid toxicity in transgenic Caenorhabditis elegans by inhibiting its aggregation. Int. J. Biol. Macromol. 209, 1280–1287. doi:10.1016/j.ijbiomac.2022.04.124
Wang, Z., Liu, S., Ma, J., Qu, G., Wang, X., Yu, S., et al. (2013). Silver nanoparticles induced RNA polymerase-silver binding and RNA transcription inhibition in erythroid progenitor cells. ACS Nano 7, 4171–4186. doi:10.1021/nn400594s
Wegorzewska, I., and Baloh, R. H. (2011). TDP-43-Based animal models of neurodegeneration: New insights into ALS pathology and pathophysiology. Neurodegener. Dis. 8, 262–274. doi:10.1159/000321547
White, J., Southgate, E., Thomson, J., and Brenner, S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340. doi:10.1098/rstb.1986.0056
Wittenburg, N., Elmer, S., Lakowski, B., Röhrig, S., Rudolph, C., and Baumelster, R. (2000). Presenilin is required for proper morphology and function of neurons in C. Elegans. Nature 406, 306–309. doi:10.1038/35018575
Wroe, R., Wai-Ling Butler, A., Andersen, P. M., Powell, J. F., and Al-Chalabi, A. (2008). Alsod: The amyotrophic lateral sclerosis online database. Amyotroph. Lateral Scler. 9, 249–250. doi:10.1080/17482960802146106
Xiu, Z. M., Zhang, Q. B., Puppala, H. L., Colvin, V. L., and Alvarez, P. J. J. (2012). Negligible particle-specific antibacterial activity of silver nanoparticles. Nano Lett. 12, 4271–4275. doi:10.1021/nl301934w
Xu, H., Jia, C., Cheng, C., Wu, H., Cai, H., and Le, W. (2022). Activation of autophagy attenuates motor deficits and extends lifespan in a C. Elegans model of ALS. Free Radic. Biol. Med. 181, 52–61. doi:10.1016/J.FREERADBIOMED.2022.01.030
Yao, C., El Khoury, R., Wang, W., Byrd, T. A., Pehek, E. A., Thacker, C., et al. (2010). LRRK2-Mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 40, 73–81. doi:10.1016/j.nbd.2010.04.002
Youssef, K., Tandon, A., and Rezai, P. (2019). Studying Parkinson’s disease using Caenorhabditis elegans models in microfluidic devices. Integr. Biol. 11, 186–207. doi:10.1093/INTBIO/ZYZ017
Zhang, T., Hwang, H. Y., Hao, H., Talbot, C., and Wang, J. (2012). Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem. 287, 8371–8382. doi:10.1074/JBC.M111.311977
Zhang, Z., Shen, Y., Luo, H., Zhang, F., Jing, L., Wu, Y., et al. (2018). MANF protects dopamine neurons and locomotion defects from a human α-synuclein induced Parkinson’s disease model in C. Elegans by regulating ER stress and autophagy pathways. Exp. Neurol. 308, 59–71. doi:10.1016/j.expneurol.2018.06.016
Zhou, S., Wang, Z., and Klaunig, J. E. (2013). Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. Int. J. Biochem. Mol. Biol. 4, 191–200.
Keywords: Alzheimer’s disease, amyotrophic lateral scelerosis, C. elegans, Huntington’s disease, Parkinson’s disease, nanotechnology, neurodegeneration
Citation: Chauhan P, Wadhwa K and Singh G (2022) Caenorhabditis elegans as a model system to evaluate neuroprotective potential of nano formulations. Front. Nanotechnol. 4:1018754. doi: 10.3389/fnano.2022.1018754
Received: 13 August 2022; Accepted: 23 September 2022;
Published: 10 October 2022.
Edited by:
Abhijit Biswas, USA Prime Biotech, United StatesReviewed by:
Jp Singh, Indian Institute of Technology Delhi, IndiaCopyright © 2022 Chauhan, Wadhwa and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Govind Singh, ZHJnb3ZpbmQucGhhcm1hQG1kdXJvaHRhay5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.