 Nicole A. d’Almeida
Nicole A. d’Almeida Marla Tipping*
Marla Tipping*- Providence College, Providence, RI, United States
Advancements in understanding the pathogenesis of C9orf72-associated frontotemporal dementia (C9orf72-FTD) have highlighted the role of repeat-associated non-ATG (RAN) translation and dipeptide repeat proteins (DPRs), with Drosophila melanogaster models providing valuable insights. While studies have primarily focused on RAN translation and DPR toxicity, emerging areas of investigation in fly models have expanded to neuronal dysfunction, autophagy impairment, and synaptic dysfunction, providing potential directions for new therapeutic targets and mechanisms of neurodegeneration. Despite this progress, there are still significant gaps in Drosophila models of C9orf72-FTD, namely in the areas of metabolism and circadian rhythm. Metabolic dysregulation, particularly lipid metabolism, autophagy, and insulin signaling, has been implicated in disease progression with findings from animal models and human patients with C9orf72 repeat expansions. Moreover, circadian disruptions have been observed in C9of72-FTD, with alterations in rest-activity patterns and cellular circadian machinery, suggesting a potential role in disease pathophysiology. Drosophila models offer unique opportunities to explore these aspects of C9orf72-FTD and identify novel therapeutic targets aimed at mitigating neurodegeneration.
Introduction
Frontotemporal dementia (FTD) comprises a spectrum of rare neurodegenerative disorders resulting from neuronal damage in frontal and temporal lobes of the brain (Surguchov, 2021). It is marked by progressive deterioration in executive functioning, behavior, language, and personality. Specific patterns of cognitive and behavioral deficits characterize the three FTD subtypes: behavioral variant FTD (bvFTD), which is most common, and nonfluent and semantic variant primary progressive aphasia. Symptom onset typically occurs between ages 45 and 60 (DeJesus-Hernandez et al., 2011; Moore et al., 2020), making FTD one of the leading causes of early-onset dementia.
Despite advancements in understanding FTD, challenges remain in early and accurate diagnosis, as well as the development of effective disease-modifying treatments. Exploring the underlying molecular and genetic mechanisms is critical for understanding FTD pathophysiology and advancing therapeutic strategies.
Drosophila as a model of FTD
Drosophila model and C9orf72-associated FTD
Drosophila melanogaster models offer unique advantages for studying disease pathology and therapeutic intervention due to their short lifespan, fast reproduction, well-characterized genetics, and genetic similarity to humans. Flies have been particularly instrumental in neurodegeneration research, with their conserved gene pathology elements such as chaperone modification, protein aggregation, tissue deterioration, and behavioral characteristics (Bonini, 2022). These conserved properties combined with Drosophila’s robust genetic toolkit allow for manipulation of genes linked to FTD heritability.
The main heritable mutations are autosomal dominant and associated with three genes: chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), and microtubule-associated protein tau (MAPT). Deciding which genetic variant model to employ depends on the topic of interest, as the prominent aspects of the FTD phenotype and the geographical variability differ across variants (Greaves and Rohrer, 2019).
Across Europe and North America, the predominant genetic factor underlying FTD is a GGGGCC hexanucleotide repeat expansion in intron 1 of C9orf72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The fundamental pathology involves both loss- and gain-of-function. Hypotheses on its mechanism include haploinsufficiency, creation of repeat RNA foci, and dipeptide repeat proteins (DPRs) formed via repeat-associated non-ATG (RAN) translation (DeJesus-Hernandez et al., 2011; Balendra and Isaacs, 2018). Notably, RAN translation leads to five distinct DPRs: poly-GA, poly-GP, poly-GR, poly-PA, and poly-PR (Ash et al., 2013; Zu et al., 2013).
Recent data reveals these three major hypotheses are not mutually exclusive. One study found that reduced C9orf72 activity increases susceptibility to degenerative stimuli, specifically glutamate-induced excitotoxicity and compromised DPR clearance (Shi et al., 2018). In essence, haploinsufficiency exacerbates toxic gain-of-function effects. This has been corroborated by similar studies establishing that C9orf72 reduction suppresses autophagy, resulting in DPR accumulation and neuronal death (Boivin et al., 2020; Zhu et al., 2020). Together, the literature offers that loss- and gain-of-function mechanisms synergize in C9orf72-associated pathology.
Introducing the C9orf72 mutation into the fly genome replicates key elements of the human FTD phenotype, facilitating exploration of cellular and molecular pathways implicated in C9orf72-associated neurotoxicity. Since Drosophila lack a C9orf72 homolog, studying loss-of-function is impeded but avoids risk of redundancy. By introducing this gene into flies, great strides have been made in exploring C9orf72-FTD.
Current focus
Major mechanisms underlying C9orf72-FTD and insights from Drosophila have been extensively reviewed elsewhere (Balendra and Isaacs, 2018). Recent progress with fly models continues to focus primarily on RAN translation and DPRs; due to their role in C9orf72 hexanucleotide repeat expansions, factors regulating RAN translation are under investigation. Inhibited translation via interaction between ribosomal proteins and DPRs, specifically poly-GR and poly-PR, is proposed as a mechanism for DPR-induced toxicity in C9orf72-associated neurodegeneration. Therapeutic targeting of this translational repression shows promise, with studies indicating that translation initiation factor eIF1A expression can rescue toxicity (Moens et al., 2019). Additionally, the identification of Protein RPS25A, a component of the small (40S) ribosomal subunit required for RAN translation, suggests inhibiting this subunit as a promising therapeutic approach (Yamada et al., 2019).
There has also been interest in the post-translational modification of DPR’s and its contribution to FTD progression. One study identified three types of poly-GR arginine methylation: monomethylation, symmetric dimethylation, and asymmetric dimethylation. Analysis reveals both types of dimethylation reduce phase separation, with symmetric dimethylation occurring most frequently and correlating with longer disease duration. Therefore, poly-GR inclusion toxicity is influenced by methylation and correlates with clinical manifestations (Gittings et al., 2020).
Latest advancements in the field
Newer focuses on Drosophila C9orf72-FTD highlight neuronal dysfunction, autophagy, and synaptic dysfunction. In terms of neuronal dysfunction, C9orf72 hexanucleotide repeat RNAs localize to dynamic mRNA transport granules in neurites, disrupting machinery responsible for transporting and translating mRNA. This results in transport granule dysfunction and branching defects, with two components modulating repeat toxicity: FMRP and Orb2 (Burguete et al., 2015). These findings successfully identify the involvement of transport granules in the disease pathology and their components as a potential therapeutic target.
Research on autophagy in Drosophila C9orf72 found that in motor neurons, 30-repeat DPRs disrupt the morphology and dynamics of the endoplasmic reticulum (ER), impairing autophagosome formation. Despite ER disruption in both axons and synapses, autophagosomes remained intact in axons, yet their biogenesis was hindered in synaptic termini (Sung and Lloyd, 2024).
Motor neurons can also be studied for synaptic dysfunction. A novel cell-autonomous excitotoxicity mechanism selectively associated with arginine-rich DPRs in glutaminergic neurons has been discovered using Drosophila C9orf72. These DPRs—poly-GR and poly-PR—were moderately toxic at 36 repeats, increasing synaptic boutons, activity zones, extracellular glutamate, intracellular calcium, and presynaptic NMDA receptor activation (Xu and Xu, 2018). This suggests neurodegeneration via glutamate excitotoxicity and synaptic overgrowth, which was presynaptic NMDA receptor-dependent and therefore cell-autonomous. More toxic 100-repeat DPRs result in loss of active zones, suggesting severe neurodegeneration via synaptic degeneration (Xu and Xu, 2018). These findings support glutamate inhibition therapies and warrant further investigation into synaptic dysfunction in C9orf72-FTD pathology.
Research needs
Metabolism
Metabolic model of neurodegeneration
Historically, medical research primarily emphasized understanding diseases’ molecular and genetic basis, targeting genes, proteins, and signaling pathways to uncover pathology and potential therapies. Cellular metabolism, though crucial, was not as extensively studied for its role in pathophysiology nor recognized as a primary driver of disease. However, since the late 20th and early 21st century, there has been a striking shift toward investigating metabolism in disease, spurred by technological advancements. Metabolic changes are now recognized as significant in disease development and progression, notably in cancer research, where metabolic reprogramming is a recognized hallmark of cancer cells (Hanahan and Weinberg, 2011). This trend has since expanded to other areas, such as autoimmune and endocrine disorders. Despite this, the emphasis on metabolism in neurodegenerative disorders has been strikingly less pronounced, especially in Drosophila models.
The lack of research is concerning, given insights from other animal models linking neurodegeneration and metabolism. C9orf72 involvement in lysosomal-autophagy pathway dysregulation has been robustly established via interaction with various proteins, including CARM1 involved in lipid metabolism (Liu et al., 2018), WDR41, SMCR8, Rab proteins, and the ULK1 complex involved in autophagy initiation (Sellier et al., 2016; Sullivan et al., 2016; Webster et al., 2016; Yang et al., 2016), and a master regulator of metabolism, mTOR (Amick et al., 2016; Ugolino et al., 2016; Ji et al., 2017). Further studies have connected C9orf72 neurodegeneration with altered lipid catabolism (Marian et al., 2023).
These findings are reflected in human patients with C9orf72 repeat expansion, where glucose metabolism is altered during clinical and preclinical disease stages (Cistaro et al., 2014; de Vocht et al., 2020; Popuri et al., 2021; Xia et al., 2023). Along with the distinct metabolic profile of the C9orf72 mutation, there is also a toxic decrease in metabolic flexibility regarding substrate transport and glycogen, adenosine, and fructose metabolism (Allen et al., 2019). This translates to impaired resilience to altered nutrient availability, energy demand, or environmental conditions, where cells struggle to effectively alternate fuel sources and adjust metabolic pathways to support cellular function and homeostasis. Together, these findings strongly support metabolic dysregulation as a pathogenic factor in C9orf72-related neurodegeneration.
Drosophila metabolism and disease
Compared to clinical research and other animal models, Drosophila research on this is sparse, however promising. Considerable potential can be seen in a recent study using C9orf72 to investigate autophagy and proteasome pathways. Results showed spironolactone, an aldosterone antagonist, effectively lowers DPR levels through the autophagy pathway. A similar reduction in DPR levels was achieved via the proteasome pathway using geldanamycin, an HSP90 inhibitor. The study also implicated cAMP and inhibition of protein kinase A (PKA) activity in reducing DPR, hinting at PKA inhibition’s therapeutic potential (Licata et al., 2022). Other research explores insulin/IGF signaling, revealing consistent impairment in C9orf72 repeat expansion flies. They observed a decrease in GGGGCC repeat and poly-GR toxicity with heightened insulin signaling, suggesting a neuroprotective role for insulin signaling (Atilano et al., 2021). Together, these fly models identify significant metabolic components in neurodegenerative pathogenesis and potential therapeutic avenues for C9orf72-FTD.
Like any model, flies present limitations, such as potential energy consumption differences due to physiological temperature disparities (Warr et al., 2018). This distinction arises because flies are ectotherms, while humans are endotherms. Nevertheless, the similarities are extensive. For instance, Drosophila fat body, oenocytes, malpighian tubes, proventriculus, and midgut parallel in function to human adipose hepatocyte, kidney, stomach, and intestinal tissue, respectively (Bharucha, 2009; Rajan and Perrimon, 2013; Warr et al., 2018). Flies also exhibit similar metabolic pathways and associated phenotypes, including insulin-signaling and lipolysis in diabetes and obesity, as well as conserved hormones, enzymes, and homeostatic mechanisms (Bharucha, 2009; Chatterjee and Perrimon, 2021). Just as fly metabolism has contributed to studies of other neurodegenerative diseases, such as Parkinson’s (Solana-Manrique et al., 2022) and Huntington’s disease (Aditi et al., 2016), these similarities support clinical relevance for fly metabolic studies.
Drosophila tools for studying metabolism
Various tools for studying fly metabolism are widely available, such as genetic manipulation, metabolic assays, locomotor activity assays, metabolomics, and imaging techniques. These methods are well-established and benefit from Drosophila’s unique advantages as a model organism, namely their short lifespan, fast reproduction, and well-characterized genetics. Widely used tools include tissue-specific RNAi, colorimetric triglyceride quantification, capillary feeder assays, lipid staining with Oil Red O, Sudan Black, and Nile Red, cholesterol extraction, glucose assays, luciferase-based ATP assays, and gas chromatography–mass spectrometry for metabolomic profiling (Figures 1B–D; Tennessen et al., 2014). More recently, barcode feeding assays have been developed (Park et al., 2018). This quantifies feeding history via qPCR of the fly body, measuring specific oligonucleotides that are incorporated into the food (Figure 1F).
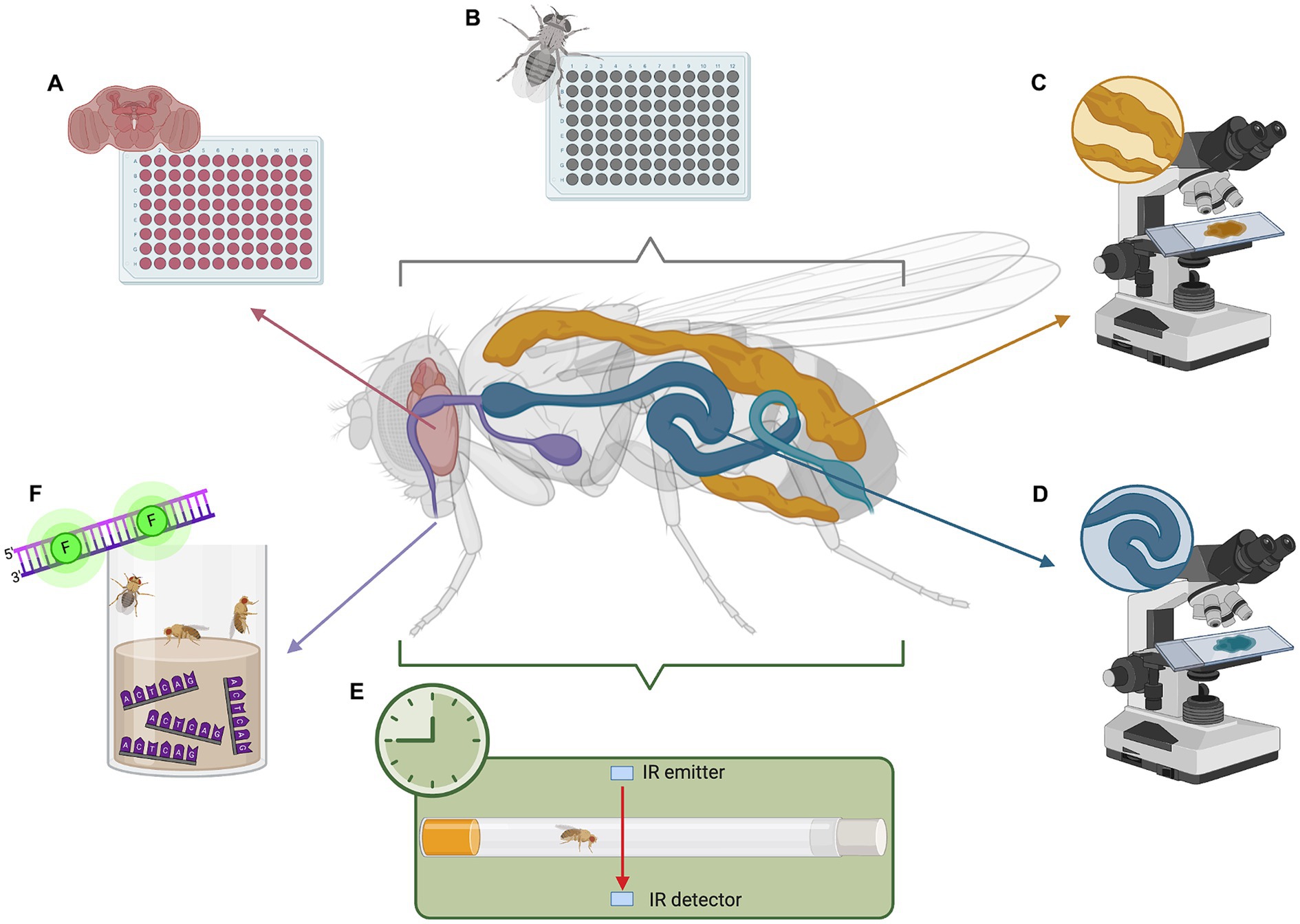
Figure 1. Analyzing metabolism and circadian rhythm in Drosophila model of C9orf72-FTD. (A) Whole-brain metabolic assays to measure oxygen consumption and extracellular acidification. (B) Whole-body metabolic assays to determine non-cell autonomous effects due to neurodegeneration. (C) Fat body staining to visualize lipid storage. (D) Gut staining to visualize microbiome and lipid digestion. (E) Drosophila activity monitor (DAM) to analyze activity and disruptions in circadian rhythm. (F) Barcode feeding assay to precisely measure food intake in a quantitative manner using qPCR. This image was created with BioRender (https://biorender.com/).
Moreover, recent and ongoing research is lending to new techniques that facilitate further exploration. Of particular note is a novel method for simple, quick, and precise ex vivo whole-brain metabolic analysis (Figure 1A), enabling investigation of metabolism in small organisms in a tissue-dependent context (Neville et al., 2018). Adapted from the Agilent XFe96 metabolic analyzer, this method proves a useful tool for exploring fly metabolic dysfunction. Other methods have been optimized to dependably measure lactate, pyruvate, and 2-oxoglutarate in vivo using fluorescence resonance energy transfer (FRET) signals from metabolic sensors (Gándara et al., 2019). This toolkit presents an avenue for studying intact fly tissues and whole organs.
Circadian rhythms
Circadian model of neurodegeneration
Circadian rhythms are observable in a plethora of organisms, as they are a defining feature across virtually all life forms. Initially overlooked, their significance in pathogenesis emerged with advancements in molecular biology and genetics. These 24-h cycles, governed by internal timekeeping mechanisms influenced by environmental, behavioral, and endogenous factors, impact metabolism, hormone, immune, and sleep–wake regulation. Disturbances in rhythmicity correlate with disease risk and severity (Fishbein et al., 2021) in a bidirectional manner. This has been validated for numerous pathologies, including immune (Liu et al., 2017; Shivshankar et al., 2020; Awuah et al., 2023), cardiovascular (Gottlieb et al., 2019; Hayter et al., 2021), psychiatric (Wehr et al., 1987; Meyer et al., 2024; Scheer and Chellappa, 2024), and neurodegenerative disorders (Fernández-Arcos et al., 2019; Sharma et al., 2020; Ibrahim et al., 2024). Given this wide reach, investigating the circadian dimension of disease is imperative.
This dimension of FTD has been robustly established in humans using sleep studies. Through actigraphy and sleep reports, FTD patients have been found to have reduced morning activity, total sleep, and sleep efficiency, and increased nighttime activity and sleep fragmentation (Anderson et al., 2009; Walker and Vaughn, 2021; Larsen et al., 2023). Unlike Alzheimer’s Disease, sleep disturbances were not reserved for those with severe cognitive and behavioral impairment, suggesting suprachiasmatic nucleus (SCN)-related neuropathology in FTD (Anderson et al., 2009).
In bvFTD, rest-activity rhythm alterations were time-locked to mornings and early afternoons (Filardi et al., 2024). Additionally, bilateral cortical thickness reductions in frontal brain regions were correlated with bvFTD patients’ prolonged sleep duration (Filardi et al., 2024). Given the role of cortical thickness in neurodegeneration and cognitive decline (Vidal-Pineiro et al., 2020; Subotic et al., 2021; Pletcher et al., 2023), these results imply a circadian influence in FTD pathogenesis.
C9orf72-FTD in particular is implicated in sleep disturbances, both at behavioral and cellular levels. Behaviorally, disruptive sleep events like restlessness and shouting have uniform associations with C9orf72-FTD but not GRN-FTD or MAPT-FTD (Sani et al., 2019). At the cellular level, circadian analysis of sleep/wake-associated cells in post-mortem human brain tissue with C9orf72-pathology reveals abundant DPR inclusions in pinealocytes and, to a lesser degree, SCN neurons (Dedeene et al., 2019). Via pineal gland dysfunction and the disturbed SCN-pineal gland axis, these findings suggest circadian implications in C9orf72-pathology.
Further, a potential connection between C9orf72 repeat expansions and Rapid Eye Movement Sleep Behavior Disorder (RBD) has been established. RBD is a sleep disorder considered to be an early indicator of neurodegeneration in Parkinson’s Disease, and, to a lesser extent, Amyotrophic Lateral Sclerosis (ALS) and FTD (Zhang et al., 2020). Among 344 RBD patients, researchers identified two carriers of C9orf72 repeat expansion with a risk haplotype associated with FTD (Daoud et al., 2014). As indicated by these findings, there may be a rare but potential link between these two disorders, suggesting C9orf72-FTD patients may be susceptible to sleep abnormalities.
Drosophila circadian rhythms and disease
Despite these outcomes, circadian studies are rare in C9orf72 animal models. Using Drosophila, researchers found sleep disruptions caused by poly-PR but not poly-GA DPRs. In poly-GR flies, altered sleep patterns included increased daytime sleep and decreased nighttime sleep, with nocturnal episodes elevated in number but reduced in duration, suggesting flies woke up more and slept less (Uy et al., 2024). These changes parallel the sleep disruptions in clinical patients (McCarter et al., 2016), suggesting a potential role of poly-GR in regulating sleep. This research highlights the pioneering potential of circadian research using C9orf72 Drosophila models.
As a highly conserved process, sleep regulation in flies resembles that of humans. Specifically, Drosophila exhibit all the fundamental characteristics of mammalian sleep (Hendricks et al., 2000; Shaw et al., 2000). This includes protein channel function, catecholamine wake-promoting effects, hypothalamic-like regulation, differential brain activity patterns, and the major clock genes period and timeless (Cirelli and Bushey, 2008; Dubowy and Sehgal, 2017). In mammals, the SCN regulates physiological and behavioral cycles and integrates external information, serving as the primary circadian pacemaker. Though Drosophila lack an SCN, they possess a functionally analogous set of approximately 150 neurons that regulate clock genes and behavioral activity (Dubowy and Sehgal, 2017). Overall, these similarities suggest clinical relevance of Drosophila circadian studies for human diseases.
Drosophila tools for studying rhythmicity
Years of research on Drosophila identified daytime activity and nighttime immobility, initially speculated as quiet wakefulness. In 2000, studies confirmed sustained nighttime immobility is a sleep-like state rather than quiet wakefulness (Hendricks et al., 2000; Shaw et al., 2000). This state exhibits an elevated, reversible arousal threshold and decreased responsiveness to external stimuli (Huber et al., 2004). It can last for hours, though a long duration is not a qualifying factor. Rather, sleep in Drosophila is operationally defined as at least 5 min of behavioral quiescence (Cirelli and Bushey, 2008).
Monitoring human circadian rhythms involves various methods, including actigraphy, body temperature, melatonin levels, hormone biomarkers, and rest-activity behavior (Yousefzadehfard et al., 2022). Sleep studies typically rely on recording eye movements and brain waves using electroencephalography (EEG) and polysomnography. These tools are adaptable to various animal models, such as pigs, dogs, rabbits, rats, and mice (Zong et al., 2023) but not to Drosophila. An alternative method uses local field potentials in brain regions involved in locomotion (Nitz et al., 2002; Qiu et al., 2021).
A common method used in flies is behavior-based paradigms. This includes actograms, temperature preference, courtship behavior, feeding behavior, or locomotor activity (Tataroglu and Emery, 2014). Locomotor activity is often measured using the Drosophila Activity Monitor (DAM) by Trikinetics. This houses a single fly in a locomotor tube where infrared light detects each time the fly breaks the beam, indicating its activity (Figure 1E). The DAM has been effectively utilized in studies (Chouhan et al., 2021; Karam et al., 2022), and various analysis software and protocols have been established (Chiu et al., 2010; Pfeiffenberger et al., 2010; Cichewicz and Hirsh, 2018; Silva et al., 2022). Thus, the DAM toolkit provides an effective means for studying circadian behavior in flies.
Discussion
While progress has been made in unraveling FTD pathology, challenges remain in diagnosis and treatment, underscoring the importance of continued research. Drosophila are a powerful model for studying FTD pathogenesis, particularly in the C9orf72 mutation, a common genetic factor. By introducing this mutation into flies, key aspects of the human FTD phenotype are replicated, enabling exploration of cellular and molecular pathways implicated in C9orf72-neurotoxicity.
Historically, Drosophila studies focused on RAN translation and DPR toxicity in C9orf72-FTD pathology. Research then expanded to neuronal dysfunction, autophagy, and synaptic dysfunction, suggesting transport granule components and glutamate inhibition as possible therapeutic targets. Despite these advances, gaps in FTD pathogenesis, especially in metabolism and circadian rhythms, remain. Limited studies in Drosophila models show metabolic dysregulation, including altered lipid metabolism and autophagy, and circadian disturbances, urging further exploration. With evidence that metabolism is altered prior to symptomatic disease (Popuri et al., 2021), there is promise that metabolic biomarkers identified in Drosophila studies could be used for early diagnosis, and potentially preventative treatment.
Addressing these gaps is crucial for a comprehensive understanding of FTD. Considering that the fly community has accessible tools to study metabolism and circadian rhythms, investigating their ramifications on neurodegeneration is filled with potential. Exploring non-cell autonomous effects is also vital, as neuronal pathologies have systemic consequences. Leveraging the unique advantages of Drosophila models is crucial to advancing our understanding of FTD and uncovering novel therapeutic targets, ultimately benefiting FTD patients.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
NAD: Writing – original draft, Writing – review & editing. MT: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aditi, K., Shakarad, M. N., and Agrawal, N. (2016). Altered lipid metabolism in Drosophila model of Huntington’s disease. Sci. Rep. 6:31411. doi: 10.1038/srep31411
Allen, S. P., Hall, B., Woof, R., Francis, L., Gatto, N., Shaw, A. C., et al. (2019). C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142, 3771–3790. doi: 10.1093/brain/awz302
Amick, J., Roczniak-Ferguson, A., and Ferguson, S. M. (2016). C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell 27, 3040–3051. doi: 10.1091/mbc.E16-01-0003
Anderson, K. N., Hatfield, C., Kipps, C., Hastings, M., and Hodges, J. R. (2009). Disrupted sleep and circadian patterns in frontotemporal dementia. Eur. J. Neurol. 16, 317–323. doi: 10.1111/j.1468-1331.2008.02414.x
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atilano, M. L., Grönke, S., Niccoli, T., Kempthorne, L., Hahn, O., Morón-Oset, J., et al. (2021). Enhanced insulin signalling ameliorates C9orf72 hexanucleotide repeat expansion toxicity in Drosophila. eLife 10:e58565. doi: 10.7554/eLife.58565
Awuah, W. A., Huang, H., Kalmanovich, J., Mehta, A., Mikhailova, T., Ng, J. C., et al. (2023). Circadian rhythm in systemic autoimmune conditions: potential of chrono-immunology in clinical practice: a narrative review. Medicine 102:e34614. doi: 10.1097/MD.0000000000034614
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Bharucha, K. N. (2009). The epicurean Fly: using Drosophila melanogaster to study metabolism. Pediatr. Res. 65, 132–137. doi: 10.1203/PDR.0b013e318191fc68
Boivin, M., Pfister, V., Gaucherot, A., Ruffenach, F., Negroni, L., Sellier, C., et al. (2020). Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 39:e100574. doi: 10.15252/embj.2018100574
Bonini, N. M. (2022). A perspective on Drosophila genetics and its insight into human neurodegenerative disease. Front. Mol. Biosci. 9:1060796. doi: 10.3389/fmolb.2022.1060796
Burguete, A. S., Almeida, S., Gao, F. B., Kalb, R., Akins, M. R., and Bonini, N. M. (2015). GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife 4:e08881. doi: 10.7554/eLife.08881
Chatterjee, N., and Perrimon, N. (2021). What fuels the fly: energy metabolism in Drosophila and its application to the study of obesity and diabetes. Sci. Adv. 7:eabg4336. doi: 10.1126/sciadv.abg4336
Chiu, J. C., Low, K. H., Pike, D. H., Yildirim, E., and Edery, I. (2010). Assaying locomotor activity to study circadian rhythms and sleep parameters in Drosophila. J. Vis. Exp. 43:2157. doi: 10.3791/2157
Chouhan, N. S., Griffith, L. C., Haynes, P., and Sehgal, A. (2021). Availability of food determines the need for sleep in memory consolidation. Nature 589, 582–585. doi: 10.1038/s41586-020-2997-y
Cichewicz, K., and Hirsh, J. (2018). ShinyR-DAM: a program analyzing Drosophila activity, sleep and circadian rhythms. Commun. Biol. 1:25. doi: 10.1038/s42003-018-0031-9
Cirelli, C., and Bushey, D. (2008). Sleep and wakefulness in Drosophila melanogaster. Ann. N. Y. Acad. Sci. 1129, 323–329. doi: 10.1196/annals.1417.017
Cistaro, A., Pagani, M., Montuschi, A., Calvo, A., Moglia, C., Canosa, A., et al. (2014). The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur. J. Nucl. Med. Mol. Imaging 41, 844–852. doi: 10.1007/s00259-013-2667-5
Daoud, H., Postuma, R. B., Bourassa, C. V., Rochefort, D., Gauthier, M. T., Montplaisir, J., et al. (2014). C9orf72 repeat expansions in rapid eye movement sleep behaviour disorder. Can. J. Neurol. Sci. 41, 759–762. doi: 10.1017/cjn.2014.39
de Vocht, J., Blommaert, J., Devrome, M., Radwan, A., van Weehaeghe, D., de Schaepdryver, M., et al. (2020). Use of multimodal imaging and clinical biomarkers in presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol. 77, 1008–1017. doi: 10.1001/jamaneurol.2020.1087
Dedeene, L., van Schoor, E., Vandenberghe, R., van Damme, P., Poesen, K., and Thal, D. R. (2019). Circadian sleep/wake-associated cells show dipeptide repeat protein aggregates in C9orf72-related ALS and FTLD cases. Acta Neuropathol. Commun. 7:189. doi: 10.1186/s40478-019-0845-9
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide repeat in noncoding region of C9ORF72Causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dubowy, C., and Sehgal, A. (2017). Circadian rhythms and sleep in Drosophila melanogaster. Genetics 205, 1373–1397. doi: 10.1534/genetics.115.185157
Fernández-Arcos, A., Morenas-Rodríguez, E., Santamaria, J., Sánchez-Valle, R., Lladó, A., Gaig, C., et al. (2019). Clinical and video-polysomnographic analysis of rapid eye movement sleep behavior disorder and other sleep disturbances in dementia with Lewy bodies. Sleep 42:zsz086. doi: 10.1093/sleep/zsz086
Filardi, M., Gnoni, V., Tamburrino, L., Nigro, S., Urso, D., Vilella, D., et al. (2024). Sleep and circadian rhythm disruptions in behavioral variant frontotemporal dementia. Alzheimers Dement. 20, 1966–1977. doi: 10.1002/alz.13570
Fishbein, A. B., Knutson, K. L., and Zee, P. C. (2021). Circadian disruption and human health. J. Clin. Invest. 131:e148286. doi: 10.1172/JCI148286
Gándara, L., Durrieu, L., Behrensen, C., and Wappner, P. (2019). A genetic toolkit for the analysis of metabolic changes in Drosophila provides new insights into metabolic responses to stress and malignant transformation. Sci. Rep. 9:19945. doi: 10.1038/s41598-019-56446-3
Gittings, L. M., Boeynaems, S., Lightwood, D., Clargo, A., Topia, S., Nakayama, L., et al. (2020). Symmetric dimethylation of poly-GR correlates with disease duration in C9orf72 FTLD and ALS and reduces poly-GR phase separation and toxicity. Acta Neuropathol. 139, 407–410. doi: 10.1007/s00401-019-02104-x
Gottlieb, E., Landau, E., Baxter, H., Werden, E., Howard, M. E., and Brodtmann, A. (2019). The bidirectional impact of sleep and circadian rhythm dysfunction in human ischaemic stroke: a systematic review. Sleep Med. Rev. 45, 54–69. doi: 10.1016/j.smrv.2019.03.003
Greaves, C. V., and Rohrer, J. D. (2019). An update on genetic frontotemporal dementia. J. Neurol. 266, 2075–2086. doi: 10.1007/s00415-019-09363-4
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hayter, E. A., Wehrens, S. M. T., van Dongen, H. P. A., Stangherlin, A., Gaddameedhi, S., Crooks, E., et al. (2021). Distinct circadian mechanisms govern cardiac rhythms and susceptibility to arrhythmia. Nat. Commun. 12:2472. doi: 10.1038/s41467-021-22788-8
Hendricks, J. C., Finn, S. M., Panckeri, K. A., Chavkin, J., Williams, J. A., Sehgal, A., et al. (2000). Rest in Drosophila is a sleep-like state. Neuron 25, 129–138. doi: 10.1016/s0896-6273(00)80877-6
Huber, R., Hill, S. L., Holladay, C., Biesiadecki, M., Tononi, G., and Cirelli, C. (2004). Sleep homeostasis in Drosophila melanogaster. Sleep 27, 628–639. doi: 10.1093/sleep/27.4.628
Ibrahim, A., Högl, B., and Stefani, A. (2024). The bidirectional relationship between sleep and neurodegeneration: actionability to improve brain health. Clin. Transl. Neurosci. 8:11. doi: 10.3390/ctn8010011
Ji, Y. J., Ugolino, J., Brady, N. R., Hamacher-Brady, A., and Wang, J. (2017). Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy 13, 1254–1255. doi: 10.1080/15548627.2017.1299312
Karam, C. S., Williams, B. L., Jones, S. K., and Javitch, J. A. (2022). The role of the dopamine transporter in the effects of amphetamine on sleep and sleep architecture in Drosophila. Neurochem. Res. 47, 177–189. doi: 10.1007/s11064-021-03275-4
Larsen, E., Jiang, J., Monica, C. D., Hassanin, H., Atzori, G., Dijk, D. J., et al. (2023). Sleep disturbance associated with temporal lobe degeneration in frontotemporal dementia. Alzheimers Dement. 19:e073890. doi: 10.1002/alz.073890
Licata, N. V., Cristofani, R., Salomonsson, S., Wilson, K. M., Kempthorne, L., Vaizoglu, D., et al. (2022). C9orf72 ALS/FTD dipeptide repeat protein levels are reduced by small molecules that inhibit PKA or enhance protein degradation. EMBO J. 41:e105026. doi: 10.15252/embj.2020105026
Liu, Y., Wang, T., Ji, Y. J., Johnson, K., Liu, H., Johnson, K., et al. (2018). A C9orf72-CARM1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes Dev. 32, 1380–1397. doi: 10.1101/gad.315564.118
Liu, X., Yu, R., Zhu, L., Hou, X., and Zou, K. (2017). Bidirectional regulation of circadian disturbance and inflammation in inflammatory bowel disease. Inflamm. Bowel Dis. 23, 1741–1751. doi: 10.1097/MIB.0000000000001265
Marian, O. C., Teo, J. D., Lee, J. Y., Song, H., Kwok, J. B., Landin-Romero, R., et al. (2023). Disrupted myelin lipid metabolism differentiates frontotemporal dementia caused by GRN and C9orf72 gene mutations. Acta Neuropathol. Commun. 11:52. doi: 10.1186/s40478-023-01544-7
McCarter, S. J., St Louis, E. K., and Boeve, B. F. (2016). Sleep disturbances in frontotemporal dementia. Curr. Neurol. Neurosci. Rep. 16:85. doi: 10.1007/s11910-016-0680-3
Meyer, N., Lok, R., Schmidt, C., Kyle, S. D., McClung, C. A., Cajochen, C., et al. (2024). The sleep–circadian interface: a window into mental disorders. Proc. Natl. Acad. Sci. U. S. A. 121:e2214756121. doi: 10.1073/pnas.2214756121
Moens, T. G., Niccoli, T., Wilson, K. M., Atilano, M. L., Birsa, N., Gittings, L. M., et al. (2019). C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 137, 487–500. doi: 10.1007/s00401-018-1946-4
Moore, K. M., Nicholas, J., Grossman, M., McMillan, C. T., Irwin, D. J., Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 19, 145–156. doi: 10.1016/S1474-4422(19)30394-1
Neville, K. E., Bosse, T. L., Klekos, M., Mills, J. F., Weicksel, S. E., Waters, J. S., et al. (2018). A novel ex vivo method for measuring whole brain metabolism in model systems. J. Neurosci. Methods 296, 32–43. doi: 10.1016/j.jneumeth.2017.12.020
Nitz, D. A., van Swinderen, B., Tononi, G., and Greenspan, R. J. (2002). Electrophysiological correlates of rest and activity in Drosophila melanogaster. Curr. Biol. 12, 1934–1940. doi: 10.1016/S0960-9822(02)01300-3
Park, A., Tran, T., and Atkinson, N. S. (2018). Monitoring food preference in Drosophila by oligonucleotide tagging. Proc. Natl. Acad. Sci. U. S. A. 115, 9020–9025. doi: 10.1073/pnas.1716880115
Pfeiffenberger, C., Lear, B. C., Keegan, K. P., and Allada, R. (2010). Locomotor activity level monitoring using the Drosophila activity monitoring (DAM) system. Cold Spring Harb. Protoc. 2010:pdb.prot5518. doi: 10.1101/pdb.prot5518
Pletcher, C., Dabbs, K., Barzgari, A., Pozorski, V., Haebig, M., Wey, S., et al. (2023). Cerebral cortical thickness and cognitive decline in Parkinson’s disease. Cereb. Cortex Commun. 4:tgac044. doi: 10.1093/texcom/tgac044
Popuri, K., Beg, M. F., Lee, H., Balachandar, R., Wang, L., Sossi, V., et al. (2021). FDG-PET in presymptomatic C9orf72 mutation carriers. Neuroimage 31:102687. doi: 10.1016/j.nicl.2021.102687
Qiu, S., Sun, K., and Di, Z. (2021). Collective dynamics of neural networks with sleep-related biological drives in Drosophila. Front. Comput. Neurosci. 15:616193. doi: 10.3389/fncom.2021.616193
Rajan, A., and Perrimon, N. (2013). Of flies and men: insights on organismal metabolism from fruit flies. BMC Biol. 11:38. doi: 10.1186/1741-7007-11-38
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A Hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Sani, T. P., Bond, R. L., Marshall, C. R., Hardy, C. J. D., Russell, L. L., Moore, K. M., et al. (2019). Sleep symptoms in syndromes of frontotemporal dementia and Alzheimer’s disease: a proof-of-principle behavioural study. eNeurologicalSci 17:100212. doi: 10.1016/j.ensci.2019.100212
Scheer, F. A. J. L., and Chellappa, S. L. (2024). Endogenous circadian rhythms in mood and well-being. Sleep Health 10, S149–S153. doi: 10.1016/j.sleh.2023.07.012
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Sharma, A., Lee, S., Kim, H., Yoon, H., Ha, S., and Kang, S. U. (2020). Molecular crosstalk between circadian rhythmicity and the development of neurodegenerative disorders. Front. Neurosci. 14:844. doi: 10.3389/fnins.2020.00844
Shaw, P. J., Cirelli, C., Greenspan, R. J., and Tononi, G. (2000). Correlates of sleep and waking in Drosophila melanogaster. Science 287, 1834–1837. doi: 10.1126/science.287.5459.1834
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shivshankar, P., Fekry, B., Eckel-Mahan, K., and Wetsel, R. A. (2020). Circadian clock and complement immune system—complementary control of physiology and pathology? Front. Cell. Infect. Microbiol. 10:418. doi: 10.3389/fcimb.2020.00418
Silva, R. F. O., Pinho, B. R., Monteiro, N. M., Santos, M. M., and Oliveira, J. M. A. (2022). Automated analysis of activity, sleep, and rhythmic behaviour in various animal species with the Rtivity software. Sci. Rep. 12:4179. doi: 10.1038/s41598-022-08195-z
Solana-Manrique, C., Sanz, F. J., Torregrosa, I., Palomino-Schätzlein, M., Hernández-Oliver, C., Pineda-Lucena, A., et al. (2022). Metabolic alterations in a Drosophila model of Parkinson’s disease based on DJ-1 deficiency. Cells 11:331. doi: 10.3390/cells11030331
Subotic, A., McCreary, C. R., Saad, F., Nguyen, A., Alvarez-Veronesi, A., Zwiers, A. M., et al. (2021). Cortical thickness and its association with clinical cognitive and neuroimaging markers in cerebral amyloid angiopathy. J. Alzheimer. Dis. 81, 1663–1671. doi: 10.3233/JAD-210138
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Sung, H., and Lloyd, T. E. (2024). Disrupted endoplasmic reticulum-mediated autophagosomal biogenesis in a Drosophila model of C9-ALS-FTD. Autophagy 20, 94–113. doi: 10.1080/15548627.2023.2249750
Surguchov, A. (2021). Invertebrate models untangle the mechanism of neurodegeneration in Parkinson's disease. Cells 10:407. doi: 10.3390/cells10020407
Tataroglu, O., and Emery, P. (2014). Studying circadian rhythms in Drosophila melanogaster. Methods 68, 140–150. doi: 10.1016/j.ymeth.2014.01.001
Tennessen, J. M., Barry, W. E., Cox, J., and Thummel, C. S. (2014). Methods for studying metabolism in Drosophila. Methods 68, 105–115. doi: 10.1016/j.ymeth.2014.02.034
Ugolino, J., Ji, Y. J., Conchina, K., Chu, J., Nirujogi, R. S., Pandey, A., et al. (2016). Loss of C9orf72 enhances Autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 12:e1006443. doi: 10.1371/journal.pgen.1006443
Uy, G., Farrell, L. N., Faheem, S. F., Kinne, L. E., Adore, M. G., Im, S. H., et al. (2024). The effects of poly-GA and poly-PR C9orf72 dipeptide repeats on sleep patterns in Drosophila melanogaster. MicroPubl. Biol. 2024:000973. doi: 10.17912/micropub.biology.000973 no date
Vidal-Pineiro, D., Parker, N., Shin, J., French, L., Grydeland, H., Jackowski, A. P., et al. (2020). Cellular correlates of cortical thinning throughout the lifespan. Sci. Rep. 10:21803. doi: 10.1038/s41598-020-78471-3
Walker, N., and Vaughn, B. (2021). 803 sleep disturbances in patients with frontotemporal dementia. Sleep 44, A312–A314. doi: 10.1093/sleep/zsab072.800
Warr, C. G., Shaw, K., Azim, A., Piper, M., and Parsons, L. (2018). Using mouse and Drosophila models to investigate the mechanistic links between diet, obesity, type II diabetes, and Cancer. Int. J. Mol. Sci. 19:4110. doi: 10.3390/ijms19124110
Webster, C. P., Smith, E. F., Bauer, C. S., Moller, A., Hautbergue, G. M., Ferraiuolo, L., et al. (2016). The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. doi: 10.15252/embj.201694401
Wehr, T. A., Sack, D. A., and Rosenthal, N. E. (1987). Sleep reduction as a final common pathway in the genesis of mania. Am. J. Psychiatry 144, 201–204. doi: 10.1176/ajp.144.2.201
Xia, K., Witzel, S., Witzel, C., Klose, V., Fan, D., Ludolph, A. C., et al. (2023). Mutation-specific metabolic profiles in presymptomatic amyotrophic lateral sclerosis. Eur. J. Neurol. 30, 87–95. doi: 10.1111/ene.15584
Xu, W., and Xu, J. (2018). C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in Drosophila glutamatergic neurons. J. Neurosci. 38, 7741–7752. doi: 10.1523/JNEUROSCI.0908-18.2018
Yamada, S. B., Gendron, T. F., Niccoli, T., Genuth, N. R., Grosely, R., Shi, Y., et al. (2019). RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat. Neurosci. 22, 1383–1388. doi: 10.1038/s41593-019-0455-7
Yang, M., Liang, C., Swaminathan, K., Herrlinger, S., Lai, F., Shiekhattar, R., et al. (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2:e1601167. doi: 10.1126/sciadv.1601167
Yousefzadehfard, Y., Wechsler, B., and DeLorenzo, C. (2022). Human circadian rhythm studies: practical guidelines for inclusion/exclusion criteria and protocol. Neurobiol. Sleep Circad. Rhyth. 13:100080. doi: 10.1016/j.nbscr.2022.100080
Zhang, F., Niu, L., Liu, X., Liu, Y., Li, S., Yu, H., et al. (2020). Rapid eye movement sleep behavior disorder and neurodegenerative diseases: an update. Aging Dis. 11, 315–326. doi: 10.14336/AD.2019.0324
Zhu, Q., Jiang, J., Gendron, T. F., McAlonis-Downes, M., Jiang, L., Taylor, A., et al. (2020). Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624. doi: 10.1038/s41593-020-0619-5
Zong, S., du, P., Li, H., Wang, M., and Xiao, H. (2023). Advances in animal models of obstructive sleep apnea. Front. Med. 10:988752. doi: 10.3389/fmed.2023.988752
Keywords: frontotemporal dementia (FTD), Drosophila melanogaster, neurodegeneration, C9orf72, metabolism, circadian rhythm
Citation: d’Almeida NA and Tipping M (2024) Flight to insight: maximizing the potential of Drosophila models of C9orf72-FTD. Front. Mol. Neurosci. 17:1434443. doi: 10.3389/fnmol.2024.1434443
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
Irina G. Sourgoutcheva, University of Kansas Medical Center, United StatesCopyright © 2024 d’Almeida and Tipping. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marla Tipping, bXRpcHBpbmdAcHJvdmlkZW5jZS5lZHU=