 Johannes Lederbauer
Johannes Lederbauer Sarada Das2
Sarada Das2 Amelie Piton
Amelie Piton Davor Lessel
Davor Lessel Hans-Jürgen Kreienkamp
Hans-Jürgen Kreienkamp
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 01 August 2024
Sec. Neuroplasticity and Development
Volume 17 - 2024 | https://doi.org/10.3389/fnmol.2024.1414949
This article is part of the Research Topic Come as You R(NA): Post-transcriptional Regulation Will Do the Rest View all 12 articles
Neurodevelopmental disorders (NDDs) represent a large group of disorders with an onset in the neonatal or early childhood period; NDDs include intellectual disability (ID), autism spectrum disorders (ASD), attention deficit hyperactivity disorders (ADHD), seizures, various motor disabilities and abnormal muscle tone. Among the many underlying Mendelian genetic causes for these conditions, genes coding for proteins involved in all aspects of the gene expression pathway, ranging from transcription, splicing, translation to the eventual RNA decay, feature rather prominently. Here we focus on two large families of RNA helicases (DEAD- and DExH-box helicases). Genetic variants in the coding genes for several helicases have recently been shown to be associated with NDD. We address genetic constraints for helicases, types of pathological variants which have been discovered and discuss the biological pathways in which the affected helicase proteins are involved.
RNA helicases use energy from the hydrolysis of ATP to unwind double stranded sections of RNA/RNA or RNA/DNA hybrids, and may also assist in the restructuring of RNA/protein complexes (ribonucleoproteins, RNPs). They carry out essential cellular functions, many of which are conserved from yeast to humans (Linder and Jankowsky, 2011). Accordingly, the distinct families of these helicases are also highly conserved throughout eukaryotic evolution (Pyle, 2008). Six major helicase superfamilies have been identified (SF1-SF6) which have distinct functions in several aspects of DNA and RNA metabolism (Fairman-Williams et al., 2010). While members of SF3-6 superfamilies are active in a “toroidal,” hexameric form, SF1 and SF2 are active as monomers. SF1 and SF2 helicases are rather similar to each other, but individual differences in conserved helicase core motifs allow for a clear differentiation between the two families (Gorbalenya and Koonin, 1993; Tanner and Linder, 2001; Fairman-Williams et al., 2010). We will focus here on the DEAD-and DExH-box helicase families which are part of the SF2 superfamily, and which in humans consist of 54 different members. While it appears surprising that our transcriptome (and genome) requires such a diversity of different helicase activities, it should be noted that every single aspect, e.g., of the life cycle of an RNA requires in most cases not only one, but several different helicases. Quite often a complete knockout of one of the RNA helicase coding genes is lethal early in mouse development (Li et al., 2014; Zheng et al., 2015; Kim et al., 2022). On the other hand, heterozygous, loss-of-function or missense variants in helicase genes are often associated with a neuronal, or neurodevelopmental phenotype in humans (Snijders Blok et al., 2015; Lessel et al., 2017; Balak et al., 2019). This may reflect the specific requirements of the developing nervous system for precise regulation of gene expression and RNA metabolism.
NDDs represent a large, clinically and genetically, heterogeneous group of human disorders with an onset in the neonatal or early childhood period. NDDs include intellectual disability (ID), autism spectrum disorders (ASD), attention deficit hyperactivity disorders (ADHD), seizures, various motor disabilities and abnormal muscle tone (Morris-Rosendahl and Crocq, 2020). NDDs are usually characterized by impairments in cognition, communication, adaptive behavior and psychomotor skills.
NDDs are often associated with Mendelian, single genetic events such as chromosomal rearrangements, copy number variations, small insertions/deletions, nonsense or missense variants. NDDs have been estimated to affect 3% of the general population (Gilissen et al., 2014), with 0.5% of all newborn affected by severe ID (Parenti et al., 2020). Although each of the underlying genetic causes of NDD is rare, their accumulated number is high enough worldwide to cause a serious socio-economic problem for health care systems. The genetic testing now routinely relies on next generation sequencing (NGS) techniques, i.e., whole exome/whole genome sequencing, or a panel based approach focused on known NDD genes. Identification of novel Mendelian, genetic causes requires a complex process, which includes evaluation of databases of human genetic variations such as the gnomAD database (Chen S. et al., 2024), identification of similarly affected individuals harboring similar genetic variants and in most instances, confirmatory functional analyses. Due to the increased use of NGS, the last decade has seen a tremendous increase in identification of novel genetic causes for NDDs. However, for the majority of NDDs two main challenges remain: reliable assessment of the pathogenicity of identified variants and meaningful clinical interventions. Thus, there is currently an urgent need for improved understanding of NDD pathology (Gilissen et al., 2014; Niemi et al., 2018; Parenti et al., 2020).
One might have thought that genes involved in neuron-specific functions would contribute to the prevalence of NDDs. Indeed, pathogenic variants in genes coding for synaptic proteins have been implicated in autism spectrum disorders and in ID (Bourgeron, 2009). However, a more quantitative analysis points to genes coding for proteins involved in control of the different steps of the gene expression pathway, such as transcriptional regulators, splicing factors, translational regulators or aspects of miRNA pathways (Greene et al., 2023). In fact, the most prevalent cause for ID now appears to be a pathogenic variant affecting the non-coding RNA RNU4-2, which is a component of the spliceosome (Chen Y. et al., 2024; Greene et al., 2024). It should be noted that in contrast to other tissues, the nervous system consists of an amazingly large number of different cell types. These can be differentiated based on single cell transcriptomics. Thus, one recent study identified 461 clusters of different cell types in the human brain, with 3,313 individual subclusters of cell types defined by a specific pattern of transcripts (Siletti et al., 2023). Due to this extreme transcriptomic diversity, the developing brain may require the precise regulation of gene expression pathways much more than other tissues. Furthermore, neurons engage in localized protein synthesis both in dendrites and in axons (Steward and Schuman, 2001; Cajigas et al., 2012). Local protein synthesis in dendrites, close to postsynaptic sites, is believed to contribute to synaptic plasticity, i.e., to activity dependent changes in synaptic strength which should be specific to those synapses which have been activated (Kindler and Kreienkamp, 2012; Sun et al., 2021). Local protein synthesis in axons is necessary due to the long distance of axon terminals from the cell body, making somatic protein synthesis followed by protein transport to synaptic terminals too slow for replenishment of and adaptation of protein levels. Many mRNAs have been shown to be not only present, but also translated locally near pre-or postsynaptic sites (Hafner et al., 2019; Glock et al., 2021). This concept entails transport of ribosomes, tRNAs and mRNAs to dendrites or axons, localized control of translation as well as the eventual degradation of the localized mRNAs. Indeed, specific structures involved in the regulation of mRNA translation such as stress granule components and P-bodies have been detected in neuronal axons and dendrites (Shiina et al., 2005; Cougot et al., 2008; Zeitelhofer et al., 2008; Sahoo et al., 2018; Lessel et al., 2020). Again, the requirements for proteins involved in RNA metabolism appear to be more complex in neurons when compared to non-neuronal cells. RNA helicases of the DExH/DEAD-box families constitute a large group of proteins which may be present in dendrites (Kanai et al., 2004), which contribute to these processes, and which may cause neurological problems upon the occurrence of damaging alterations in their coding genes.
The core functions of DexH/DEAD-box RNA helicases (ATP binding and hydrolysis, nucleic acid binding and unwinding) are carried out by two adjacent core domains which show structural similarity to the recombination protein RecA. Within these helicase core domains, up to 14 conserved helicase core motifs (HCMs) can be identified (Fairman-Williams et al., 2010). Out of these, Ia – Ib, IV, IVa, V and Vb bind nucleic acid substrates. HCMs Q, I, II, IIIa and VI are involved in ATP binding and hydrolysis. Interestingly, not all HCMs are present in all members of these helicase families (Fairman-Williams et al., 2010). Thus, the Q-HCM, a 9 amino acid (aa) sequence containing an invariant glutamine residue along with a conserved phenylalanine residue 17 aa further upstream, is found only in DEAD-box RNA helicases. The namesake DEAD or DExH sequence motifs constitute motif II. In motif II, specifically the Asp-Glu part is involved in coordinating the ATP associated Mg2+ ion, and in positioning the water molecule which performs the ATP hydrolysis. In 3D structures of both DEAD-box as well as DExH box proteins, the side chains of the C-terminal Asp/His residues of motif III are in direct contact to the Ser and Thr side chains of motif III (sequence SAT), which couples NTP binding and hydrolysis to nucleic acid binding and unwinding (Tanner et al., 2003; Cordin et al., 2006; Figure 1; see Supplementary Figure S1 for a complete view on human DEAD box proteins).
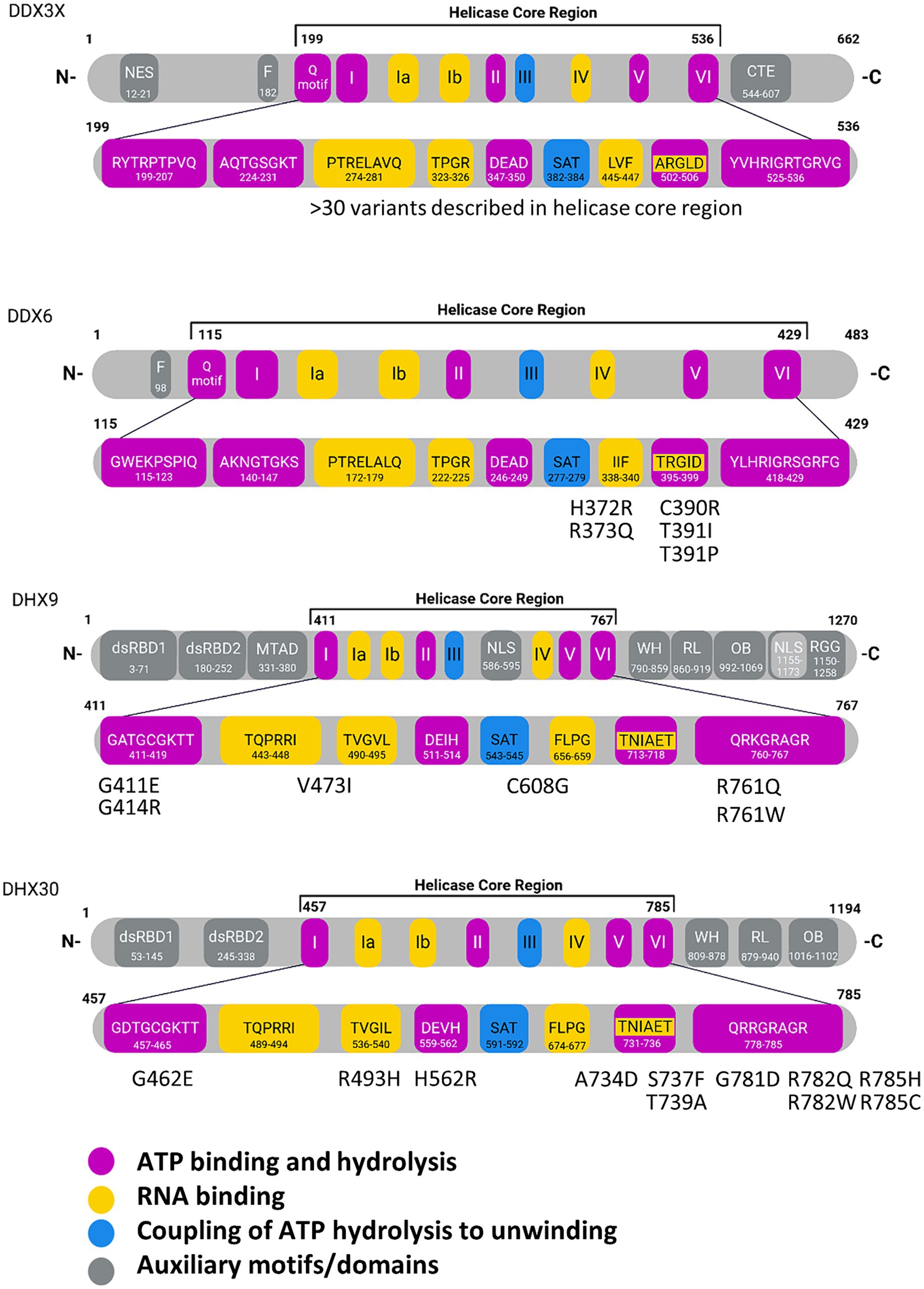
Figure 1. Domain structures of individual RNA helicases involved in NDDs. In the enlargement of the helicase core regions, individual core motifs are indicated with amino acid sequences and positions, based on Uniprot entries for the human proteins. Missense variants identified in NDD patients are indicated for DDX6, DHX9 and DHX30. For DDX3X, more than 30 variants have been described in the helicase core region. Motif V has been implicated in RNA and ATP binding. WH, Winged-helix; RL, Rachet-like; OB, Oligosaccharide binding; Znf, Zinc finger; dsRBD, double stranded RNA binding domain; mtad, minimal transactivation domain; NLS, nuclear localization signal; NES, nuclear export signal; NTD, N-terminal domain; RGG, arginine-glycine–glycine domain. Created with Biorender.com.
DExH helicases are processive helicases which move along a dsRNA substrate, capable of performing several unwinding steps along the way. In contrast, DEAD-box proteins dissociate from their RNA substrates after a single unwinding step (Bohnsack et al., 2023). The unique C-terminal domains of DExH helicases, i.e., the winged-helix (WH) and ratchet-like domains (often described together as helicase associated 2 or HA2 domain), as well as the oligonucleotide/oligosaccharide-binding (OB)-fold domain are relevant here (Murakami et al., 2017; Jagtap et al., 2023; Figure 1; see Supplementary Figure S2 for a complete view on human DExH box proteins). For these helicases, HA2 and OB motifs are integral parts of the helicase function as they contribute to a tunnel for single stranded RNA. In the case of the Drosophila maleless (MLE) helicase, a member of the DExH family which has been studied in much detail, the core RecA domains bind the RNA substrate via non sequence specific interaction with the sugar/phosphate backbone. In contrast, the OB domain is involved in base-specific contacts at the 5′ end of the tunnel. Here, also the second of the two N-terminal dsRBD domains (dsRBD2) is essential for activity as it is involved in regulating helicase activity, and also provides for proper positioning of dsRNA substrates at the entrance of the RNA tunnel (Prabu et al., 2015; Jagtap et al., 2023).
Other accessory domains, including additional dsRNA binding domains or RNA recognition motifs (RRMs), are typically not conserved within a given family of these RNA helicases. Both, N-and C-terminal domains frequently determine the integration of helicase proteins into larger functional complexes and are therefore highly relevant for the physiological function.
Different stages of the RNA life cycle are shown in Figure 2. Transcription produces the crude RNA that will go through several interactions with various proteins or different kinds of RNA in its path that shapes its journey. Transcription is coordinated by several RNA helicases which act as coactivators or corepressors by binding to key transcriptional machinery (Rajendran et al., 2003; Rossow and Janknecht, 2003; Yan et al., 2003). This is relevant for both Pol1 and Pol2-transcribed genes, as has been shown for DDX21 which associates with genes coding for ribosomal RNA, as well as ribosomal proteins and positively regulates their transcription (Calo et al., 2015). In addition, several RNA helicases (e.g., DDX1, DDX17 and DHX9) have been suggested to contribute to the formation and resolving of so-called R-loops. These three-stranded nucleic acids, consisting of an RNA–DNA hybrid and a displaced single-stranded DNA, occur during transcription and DNA replication, and must be resolved to avoid epigenetic misregulation (Al-Hadid and Yang, 2016) and genome instability (Yang et al., 2023).
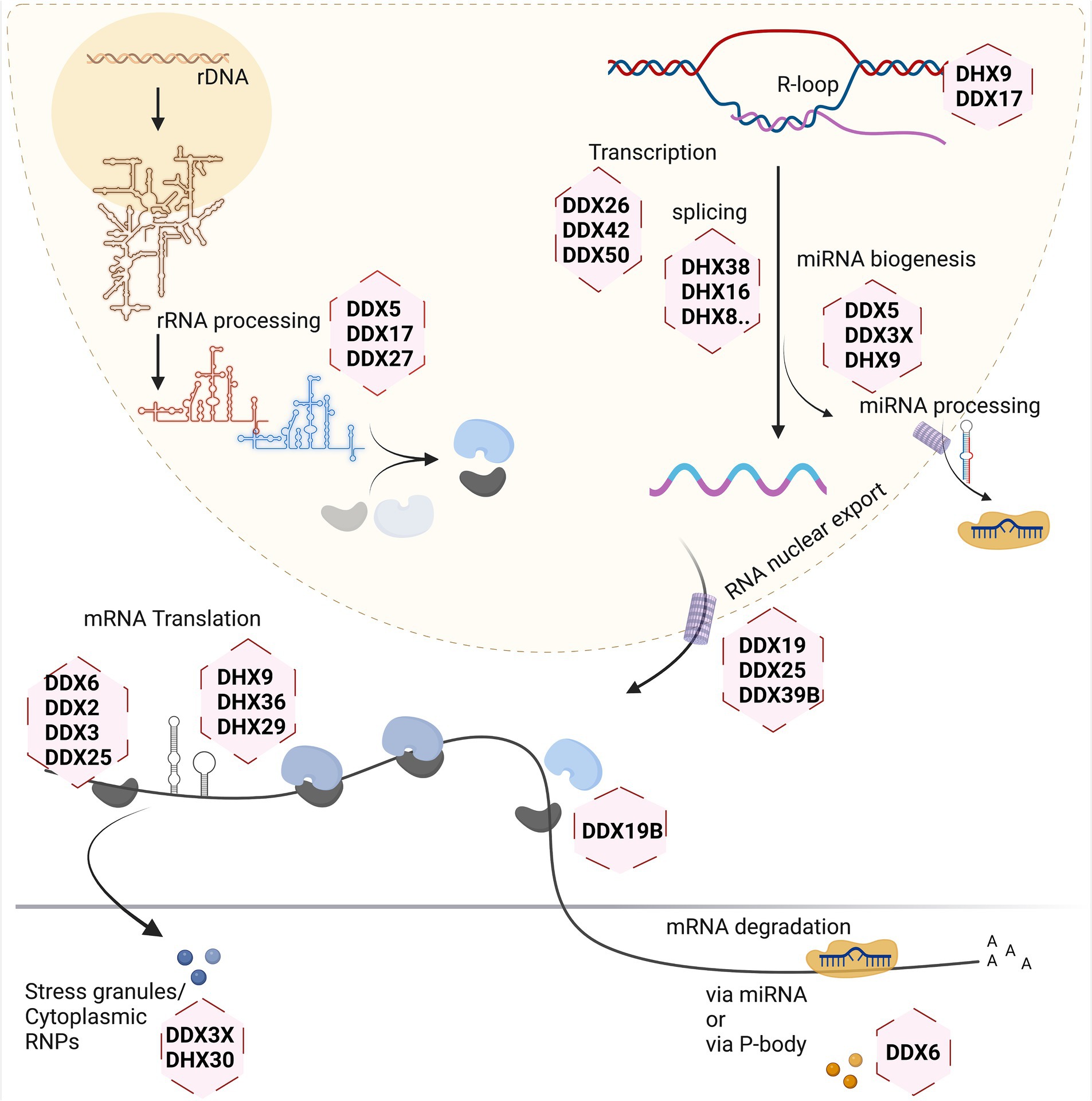
Figure 2. Involvement of RNA helicases in different stages of eukaryotic RNA metabolism. Created with Biorender.com.
Ribosome biogenesis starts in the nucleolus, with a few final steps occurring in the cytoplasm. As it involves the dynamic rearrangement of ribonucleoprotein complexes, it requires a multitude of RNA helicases to help avoid undesirable configuration of RNA and its interactions. In the yeast model system, at least 19 RNA helicases are required for the maturation of ribosomes, and even more helicases are involved in this process in human cells (Martin et al., 2013). A single large precursor rRNA molecule undergoes systematic cleavage by endo-and exonucleases along with several other hundreds of transacting factors to produce the mature rRNAs. The main roles of the DExH/DEAD-box helicases during this process are to mechanistically fold the precursor rRNA for formation of several RNP complexes, careful removal of specific small nuclear RNAs guiding rRNA folding, and mediating structural alteration in ribosomal subunits during the ribosome assembly (Jalal et al., 2007; Martin et al., 2014; Kellner et al., 2015; Xing et al., 2019).
Splicing of newly transcribed pre-mRNAs to form the mature mRNA involves structural rearrangements and folding which requires assistance by several RNA helicases. Studies of yeast as well as human spliceosomes have shown that at least eight RNA helicases act in a sequential manner, with seven of them belonging to DExH/DEAD-box families (Fleckner et al., 1997; Gencheva et al., 2010; English et al., 2012; Zanini et al., 2017; Hug et al., 2022; Obuća et al., 2022). Often these helicases share more than one role in the cellular system. For example, RNA helicase DDX39B is part of the spliceosome affecting splicing and also regulates nuclear export of mRNAs by being part of the TREX mRNA export complex (Li et al., 2005; Kota et al., 2008; Hautbergue et al., 2009; Shen, 2009). On the other hand, DDX39B also contributes to translation by regulating pre-ribosomal RNA levels (Awasthi et al., 2018).
The translation machinery associates with most of the major kinds of RNA in the cell. This is one process that involves the three major types of RNA in the cell: the mRNA, rRNA, and tRNA. So naturally, the helicases involved here inter-share their roles to maintain a coherent system. The eIF4A RNA helicase (also known as DDX2) constitutes one of the smallest DEAD-box helicases. Its evolutionary conservation is due to its indispensable role for translation initiation, to unwind the 5’UTR of mRNA, thus facilitating scanning of the 5’UTR by small ribosomal subunits to identify the start codon (Pause and Sonenberg, 1992). Other RNA helicases such as DDX3, DHX9 and DHX36 are needed to overcome highly structured 5’UTRs on large transcripts during the scanning process (Sheng et al., 2006; Lee et al., 2008; Calviello et al., 2021). Another RNA helicase recently found crucial for protein biosynthesis is DHX19, which assists in formation of the termination complex and release of the newly formed protein from the ribosomal complex (Mikhailova et al., 2017).
Several regulatory mechanisms act on mRNAs to control translation. Translational shutdown upon various cellular stresses leads to sequestration of mRNAs in large protein/RNA complexes within the cytoplasm called stress granules (SGs). Several RNA helicases such as DDX3X, DHX30, DHX36 act in the assembly, and possibly also in the disassembly or clearance of SGs (Chalupnikova et al., 2008; Valentin-Vega et al., 2016; Sauer et al., 2019; Mannucci et al., 2021). In addition, mRNAs may be degraded in processing (P-) bodies, which also exist under basal (non-stressed) conditions and are associated with miRNA dependent silencing of mRNAs. The helicase DDX6 plays major role in P-bodies and is involved in several aspects of mRNA degradation (Nissan et al., 2010; Ostareck et al., 2014; Rouya et al., 2014; Weber et al., 2024).
Both SGs and P-bodies, together with nucleoli and nuclear speckles, constitute membrane-less organelles which perform a large portion of cellular RNA processing. These cellular bodies are held together by a multitude of interactions of RNAs with RNA binding proteins, and frequently involve a biomolecular condensation process termed liquid-/liquid phase separation (LLPS) (Dorner and Hondele, 2024). Proteins can contribute to condensate formation through their various interaction domains, but also through larger segments of intrinsically disordered regions (Rosa et al., 2024). Most if not all DEAD-and DExH helicases are present in such condensates at some point of the RNA life cycle, and they are probably needed for both the assembly as well as the disassembly of such organelles (Hondele et al., 2019). This is exemplified by the two sexually dimorphic DDX3 variants, DDX3x and DDX3y. Here, it was recently shown that the disordered region in DDX3y promoted LLPS more strongly, while its weaker ATPase activity was less active in SG dsassembly. This leads to increased LLPS, reduced translation and increased mRNA aggregation, e.g., under stress in a sex-specific manner (Shen et al., 2022). Aberrant formation of stress granules is an important feature of NDD associated pathogenic variants in DHX30 and DDX3x (Lessel et al., 2017; Lennox et al., 2020; Mannucci et al., 2021).
In order to provide a detailed overview of the DExH/DEAD-box RNA helicases involved in NDDs we first queried the Online Mendelian Inheritance in Man (OMIM)1 database. OMIM is a comprehensive compendium that continuously updates human genes and their association to human genetic disorders according to the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC).2 This search revealed nine RNA helicase genes, that can be regarded as definitely associated with a human disorders. Out of these six genes were associated with a primary neurodevelopmental disorder. In more detail, missense and loss-of-function variants in EIF4A2 (DDX2B) are associated with the Neurodevelopmental disorder with hypotonia and speech delay, with or without seizures (NEDHSS, #620455) (Paul et al., 2023). Similarly, a broad spectrum of variants in DDX3X is associated with Syndromic X-linked intellectual developmental disorder of the Snijders Blok type [MRXSSB, #300958 (Snijders Blok et al., 2015)]. Missense variants in DDX6 and DHX16 are associated with Intellectual developmental disorder with impaired language and dysmorphic facies [IDDILF, #618653 (Balak et al., 2019)] and Neuromuscular oculoauditory syndrome [NMOAS, #618733 (Paine et al., 2019)], respectively. Missense variants in DHX30 are associated with Neurodevelopmental disorder with variable motor and language impairment (NEDMIAL, #617804), with loss-of-function variants causing a milder phenotype (Lessel et al., 2017; Mannucci et al., 2021). Missense variants in DHX37 are associated with Neurodevelopmental disorder with brain anomalies and with or without vertebral or cardiac anomalies [NEDBAVC, #618731 (Paine et al., 2019)]. In addition, DDX11, EIF4A3 and DDX59 have been associated with complex human disorders that involve a variable degree of neurodevelopmental delay. These genes have bene associated with Warsaw breakage syndrome [#601150 (van der Lelij et al., 2010)], Robin sequence with cleft mandible and limb anomalies [#268305 (Favaro et al., 2014)] and Orofaciodigital syndrome V [OFD5, #174300 (Shamseldin et al., 2013)], respectively (Table 1).
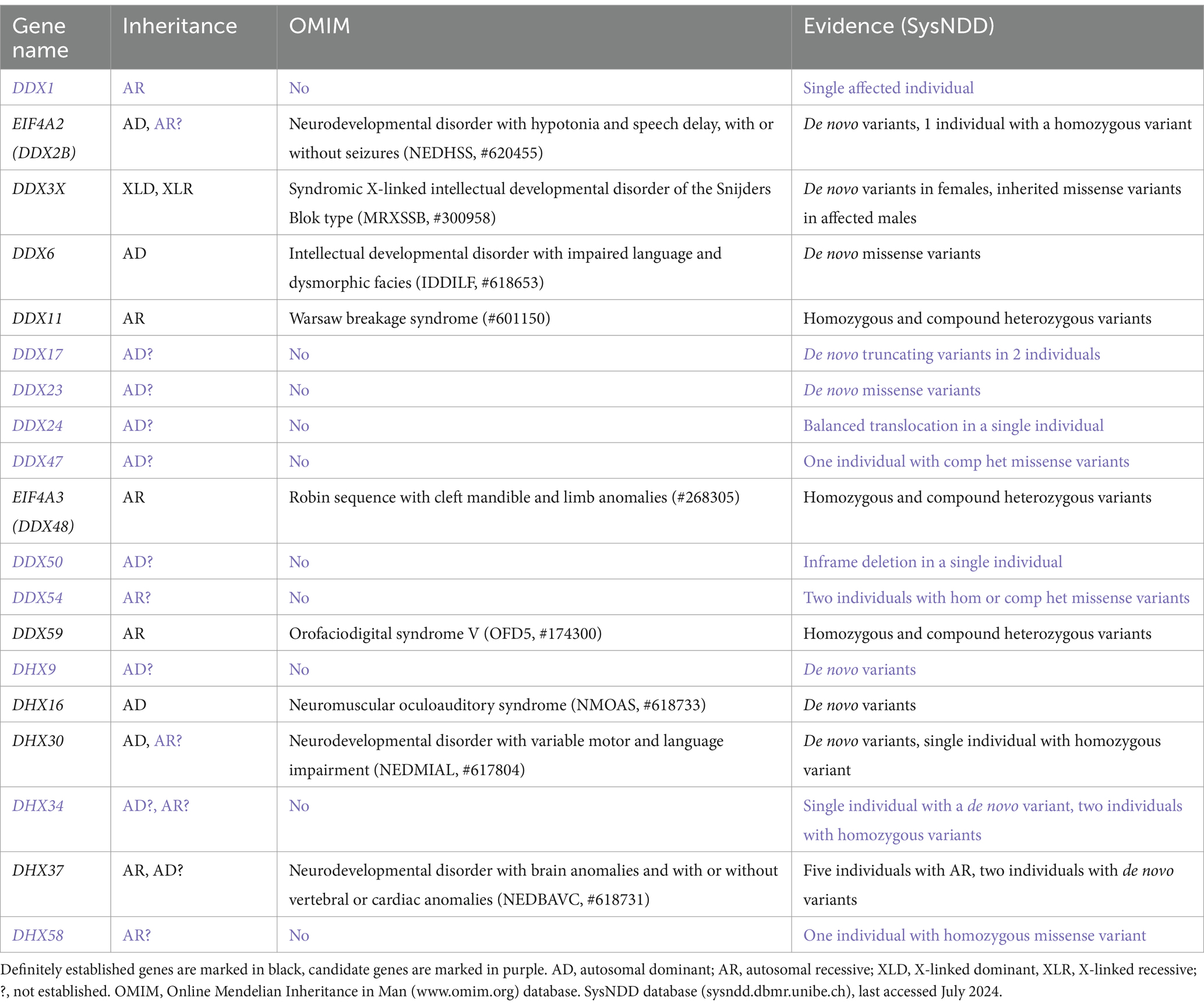
Table 1. DExH/DEAD-box RNA helicases associated with NDDs.
To search for further candidate genes, we next enquired the SysNDD database (sysndd.dbmr.unibe.ch) that curates gene disease relationships in NDDs. This search revealed 10 potential candidate NDD genes, variants in which have been identified in at least one affected individual (Table 1). Out of these, an association of DHX9 (Calame et al., 2023; Yamada et al., 2023) and DDX23 (Burns et al., 2021) with a NDD has been published only recently.
The gross majority of genes involved in human NDDs are highly intolerant to genetic variation, both to missense and loss-of-function variants (Samocha et al., 2014). This is nicely exemplified by the DHX30 gene, being one of the most variation-intolerant genes in the human genome (Lessel et al., 2017). We have therefore utilized the large sequencing data from the Genome Aggregation Database (gnomAD V2.1)3 (Karczewski et al., 2020) to document constraint metrics of the 54 human DExH/DEAD-box RNA helicases (Table 2). We were interested in the tolerance to missense variants (missense Z-scores), where a score of >3 is regarded as intolerant, and tolerance to loss-of-function variants [probability of being loss-of-function intolerant (pLI)], where a score of >0.90 is regarded as intolerant (Samocha et al., 2014; Lek et al., 2016). Genes with either high Z score or pLI values, or both, are regarded as strong candidates for disorders caused by heterozygous, de novo variants (Samocha et al., 2014). Indeed, out of the already established NDD-related DExH/DEAD-box RNA helicase genes, primarily caused by de novo variants, all bear high scores (DDX3X, DDX6, DHX16 and DHX30). Out of the candidate genes listed in Table 1, DDX17, DDX23 and DHX9 show high missense Z scores and pLI’s.
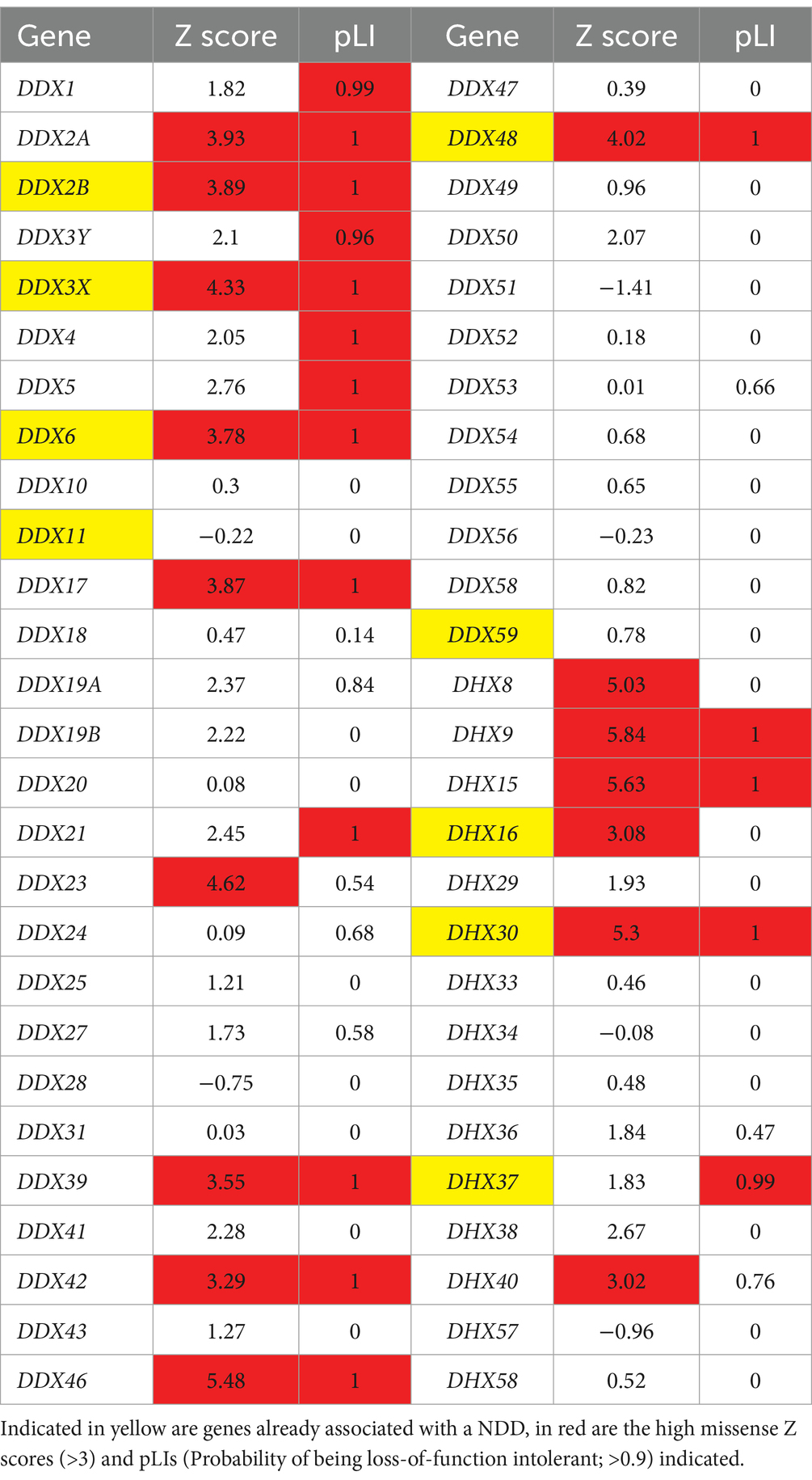
Table 2. Constraint metrics of DExH/DEAD-box RNA helicases according to gnomAD V2.1.
Neurodevelopmental disorders arise due to various perturbations during brain development (Khodosevich and Sellgren, 2023). Thus, we were also interested in the expression profiles of the DExH/DEAD-box RNA helicases in various brain regions during brain development. For this, we utilized the Human Brain Transcriptome (HBT) database,4 which provides transcriptome data for the developing and adult human brain (Kang et al., 2011) in six brain regions (neocortex, hippocampus, amygdala, striatum, mediodorsal nucleus of the thalamus and cerebellar cortex). We documented the mean expression levels at four different time points during development, namely during the embryonic period (TP1; 4 PCW ≤ Age < 8 PCW), late fetal period (TP7, 24 PCW ≤ Age < 38 PCW), neonatal and early infancy period (TP8, 0 M (birth) ≤ Age < 6 M) and early childhood (TP10, 1 Y ≤ Age < 6 Y) (Table 3). Out of the already established NDD-associated DExH/DEAD-box RNA helicase genes, EIF4A2 (DDX2B), DDX3X, DDX6 and DHX30, showed a strong expression during all developmental periods. Genes associated with complex human disorders, DDX11 and EIF4A3, displayed strong expression only at certain periods. DDX59, DHX16 and DHX37 are not highly expressed during brain development. Out of the candidate genes, DDX1, DDX17, DDX24, DDX47 and DHX9 display a strong expression during all developmental periods, whereas DDX23 displays a somewhat lower expression only at TP7.
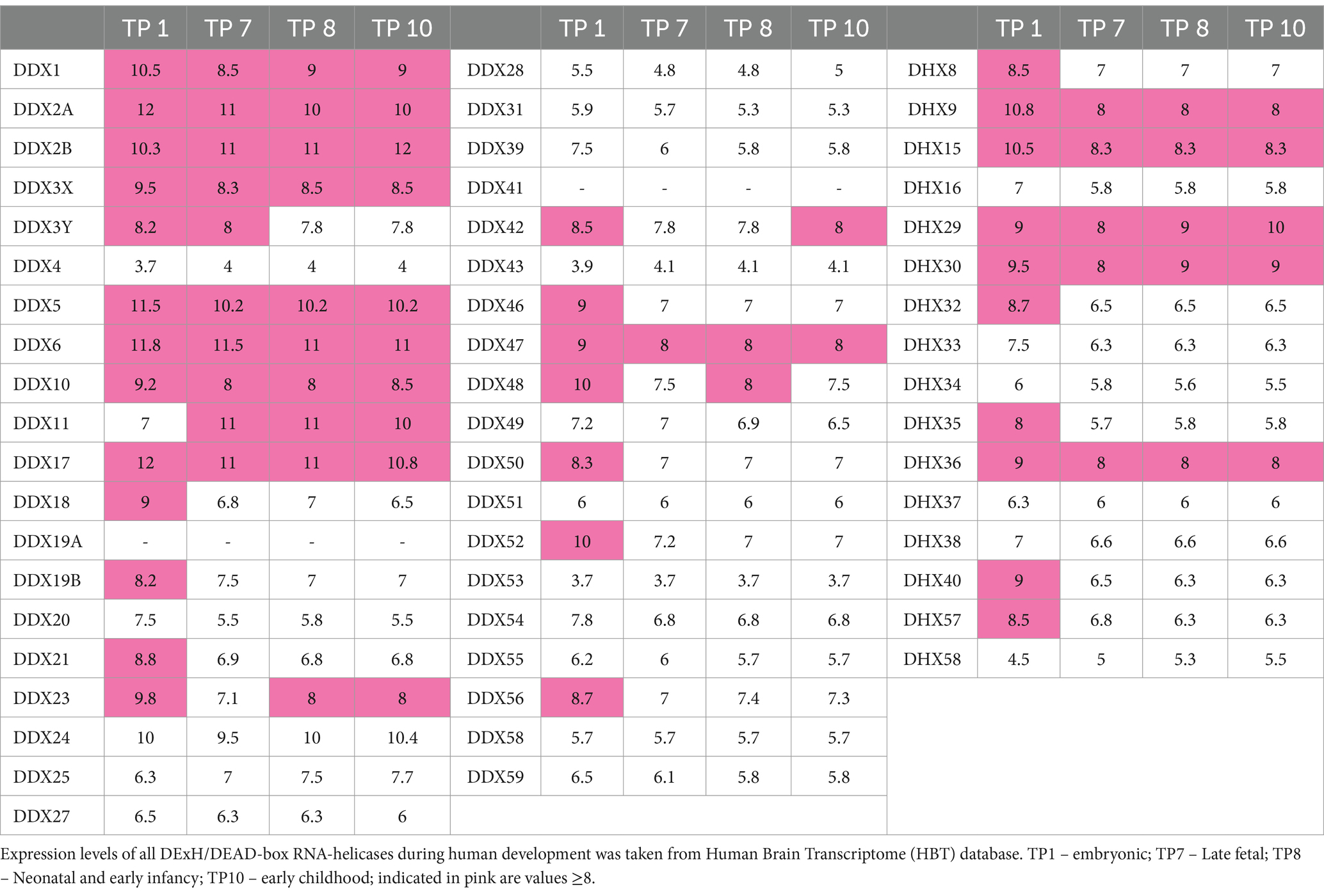
Table 3. Gene expression data of DExH/DEAD-box RNA helicases in 6 brain areas during human development.
Taken together the constraint metrics and brain expression data provide evidence for heterozygous, de novo variants in DDX17, DDX23 and DHX9 as being associated with NDDs. Based on both datasets we suggest that heterozygous, de novo variants in DDX2A, DDX42, DDX46, DHX8, DHX15 and DHX40 might represent additional candidates for this group of disorders. Finally, based on brain expression levels, DDX1, DDX5, DDX24, DDX47 and DHX36 might constitute NDD associated genes following an autosomal recessive mode of inheritance. Clearly, further high-throughput sequencing studies are needed to confirm these hypotheses.
Below we provide a brief overview of the four well-studied NDDs associated with pathogenic variants in DExH/DEAD-box RNA helicases.
This DExH helicase has received little attention until recently; early studies in mice revealed that the Dhx30 gene is essential for survival, as complete Dhx30 ko mice die early in embryonic development (Zheng et al., 2015). Several transcript variants arise due to the use of alternative promoters, and possibly also alternative splicing. Variations in the N-termini allow for either import into mitochondria or targeting to the cytosol of the expressed protein (Lessel et al., 2017; Bosco et al., 2021). As a consequence, a substantial portion of the protein resides in mitochondrial RNA granules which play a role in RNA processing and biogenesis of mitochondrial ribosomes (Antonicka and Shoubridge, 2015). Like several other DExH-type helicases, DHX30 carries additional domains besides the two RecA domains which constitute the helicase core. There are two dsRBDs in the N-terminal part of the protein, and the winged helix, ratchet like and OB fold domains in the C-terminus which are typical for DExH helicases.
We have previously established de novo, heterozygous, DHX30 missense variants, affecting highly conserved residues within its HCMs, as a cause of a severe neurodevelopmental disorder, Neurodevelopmental disorder with variable motor and language impairment (NEDMIAL; #OMIM 617804) (Lessel et al., 2017). This condition is primarily characterized by severe global developmental delay (GDD), intellectual disability (ID), absent speech or speech limited to single words along with severe gait abnormalities (if walking is acquired at all). In contrast, individuals harboring a loss-of-function (frameshift or nonsense) variant develop a milder clinical course (Mannucci et al., 2021). The latter individuals have a mild GDD and ID, learn to speak full sentences and learn to walk in the second year of life. Two other missense variants outside HCMs have been described which are associated with a different clinical course. However, their causality still remains to be fully confirmed.
By performing in-depth functional analyses we were able to provide a molecular understanding for this genotype–phenotype correlation. Missense variants within the helicase core motifs (HCMs) of DHX30 impair either its ATPase activity or RNA binding capacity, and thereby its RNA helicase activity (Lessel et al., 2017; Mannucci et al., 2021). However, in addition to this clear loss-of-function, these missense variants additionally lead to a detrimental gain-of-function by inducing stress granule (SG) formation with concomitant global translation impairment. Utilizing CRISPR/Cas9 based technology, analyses of two DHX30 knockdown/knockout models, HEK293T cells and zebrafish model, revealed an impairment of SG formation (Mannucci et al., 2021). These data strongly suggest that the severe DHX30-associated phenotype (NEDMIAL) is due to the selective gain-of-function by triggering SG formation.
The gene coding for the DEAD-box helicase DDX3X is localized on the X-chromosome and is one of the few genes known to escape X inactivation in females; a second gene on the Y-chromosome codes for the almost identical DDX3Y protein, apparently ensuring equal gene dosage for this type of helicase in males (Lahn and Page, 1997). Germline variants in DDX3X are one of the most common causes for intellectual disability in females; indeed more than a hundred affected individuals have been reported (Snijders Blok et al., 2015; Lennox et al., 2020; Greene et al., 2023). Variants in DDX3X are associated with a wide spectrum of neuronal phenotypes, ranging from ID and loss of speech to severe failures of cortical development including microcephaly and polymicrogyria (a condition characterized by too many, but too small folds in the surface of the cortex). In most cases, females carry de novo, heterozygous loss-of-function or missense variants. In addition, hemizygous variants inherited from unaffected mothers have also been identified in rare male patients. Missense variants lead to a significantly more severe outcome, suggesting a dominant effect of these variants (Lennox et al., 2020). Similar to the situation in DHX30, several missense variants alter residues in one of the conserved HCMs; thus, there are four cases with a severe phenotype carrying the T532M variant in motif VI. In addition, there is a number of cases with variants outside of the HCMs but within the two RecA helicase core domains. Functional analysis shows that lack of ATPase and RNA helicase activity in missense variants correlates with severity of disease (Lennox et al., 2020).
Studies of the mammalian DDX3X protein, as well as the yeast homolog Ded1p, suggest that this helicase is involved in translation and may play a role in unwinding complex 5′ untranslated regions (5’UTRs) during scanning of the preinitiation complex (Chuang et al., 1997; Hilliker et al., 2011). Work by Soto-Rifo et al. (2012) suggested that DDX3X needs to unwind secondary structures close to the 5′ end which occlude the 5’cap structure, thereby preventing access of the cap binding protein eIF4F. An additional role in the formation of stress granules was also discussed which might be directly related to the role of DDX3X in translation (Hilliker et al., 2011). Interestingly, somatic missense variants found in medulloblastoma, as well as germline variants in NDD patients, lead to excessive formation of stress granules even in the absence of cellular stressors such as heat or oxidative stress (Valentin-Vega et al., 2016; Lennox et al., 2020). In this respect, pathogenic variants in DDX3X to some extent mimic missense variants in DHX30 which also lead to enhanced stress granule formation [see above (Lessel et al., 2017; Mannucci et al., 2021)]. However, a recent analysis in neuronal progenitor cells suggested that some of the NDD-associated missense variants trigger formation of ribonucleoprotein (RNP) granules that may not be stress granules (Lennox et al., 2020).
In the mammalian brain, lack of DDX3X leads to reduced neurogenesis during embryonic development, likely explaining the polymicrogyria (Lennox et al., 2020). A conditional knockout line in mice with deletion of Ddx3x expression in neural progenitor cells in early embryonic development showed that the encoded protein is necessary for cell cycle control and for the generation of a sufficient number of neuronal cells. DDX3x does so by promoting translation of a small set of mRNAs relevant for neurogenesis (Calviello et al., 2021; Hoye et al., 2022). Similar defects in cortical neurogenesis were observed in mice lacking another helicase gene, EIF43/DDX48, suggesting a common pathological mechanism (Lupan et al., 2023). In zebrafish, mutant Ddx3x causes a deficit in the Wnt signaling pathway (Snijders Blok et al., 2015). A previous study had shown that the DDX3X protein performs some “moonlighting” in this pathway as an essential positive regulator of casein kinase 1 (CK1ε), which is required for phosphorylation of Disheveled and activation of the transcriptional role of β-catenin (Cruciat et al., 2013). This raises the question, whether the helicase activity or the CK1ε-dependent activity of DDX3 is relevant for human pathologies. Activation of CK1ε by DDX3 does not require RNA binding or helicase activity of the DDX3 protein, as many of the conserved motifs involved in RNA or ATP binding can be deleted without affecting CK1ε binding or activation of the signaling pathway. Instead, the DDX3X function in this pathway depends on its ability to interact with CK1ε though a sequence element in its C-terminus (Cruciat et al., 2013). Thus, current evidence indicates that a disrupted RNA helicase function of DDX3X, primarily caused by pathogenic variants in the helicase core region, is the main cause of NDD in patients.
The DDX6 gene (MIM: 600326) encodes the DEAD-box helicase 6, involved in the regulation of mRNA decay and translation. DDX6 is an essential component of P-bodies, cytoplasmic granules containing enzymes necessary for the post-transcriptional regulation of mRNA. In fact, DDX6 is one of the very few proteins which are actually essential for P-body formation (Weston and Sommerville, 2006; Ayache et al., 2015). An initial publication from 2019 reported five de novo missense variants in DDX6 in individuals with neurodevelopmental disorders (Balak et al., 2019). All the variants were located in the same exon of the gene and affected four amino acids from two conserved motifs (amino acids 372–373 and 390–391) of the second RecA domain of the protein, namely the QxxR domain and the motif V. These variants affect the ability of DDX6 to form P-bodies and to interact with several of its partners involved in translation control. All individuals present with global developmental delay, intellectual disability, hypotonia, gait instability with a delay in walk acquisition, and similar dysmorphic features including a high-bossing forehead, bulbous nasal tip, hypertelorism, epicanthus, arched eyebrows and low-set ears, associated with a small head circumference. They also present with additional non-neurological symptoms, such as cardiac, hand/foot, and urogenital anomalies. The identification of additional nonsynonymous variants in individuals with NDD will be necessary to refine the syndrome associated with DDX6 variants and to establish if missense changes located outside the QxxR and the motif V domains and truncating variants might also be pathogenic. So far, little is known about the role of DDX6 in the brain and how its dysfunction could alter normal processes of brain development, but few studies have reported its involvement in neuronal differentiation and synaptic plasticity. In mouse neural stem cells, DDX6 is needed for neuronal differentiation by regulating let7a activity through cooperation with TRIM32 (Nicklas et al., 2015). DDX6 also regulates the retinoic acid-induced neuronal differentiation of human neuroblastoma cell lines SH-SY5Y and SK-N-SH (Shih et al., 2023). Finally, a role of DDX6 in mediating NMDAR-dependent spine shrinkage via the Ago2 dependent silencing of Limk1 has been recently described in rat neurons (Perooli et al., 2024).
DHX9 is involved in transcription, in the regulation of R-loops and in the repair of DNA double strand breaks by BRCA1 (Chakraborty et al., 2018; Cristini et al., 2018). In addition DHX9 represses the effects of Alu elements in the human genome on RNA processing (Aktas et al., 2017). For these purposes, DHX9 needs to be targeted to the nucleus by a nuclear localization sequence (NLS) in the C-terminal part of the protein. However, recent work in hippocampal neurons has shown that a substantial part of the protein may be present in the cytosol, specifically associated with the dendritically localized mRNA coding for Dendrin (Yang et al., 2024). Dhx9 deficient mice are viable but display distinct behavioral and neurological abnormalities (Calame et al., 2023). Although, OMIM still does not list DHX9 as associated with human disease, two very recent studies identified pathogenic variants in DHX9 in human patients. Thus, actually already formally establishing the link to NDD. Again, some variants associated with a moderate NDD phenotype alter conserved residues in motif I or motif VI and interfere with ATPase activity (Calame et al., 2023; Yamada et al., 2023). More severe NDD is observed with variants which alter the NLS and interfere with nuclear localization of DHX9; these lead to a higher number of R-loops and double strand breaks, indicating that the function of DHX9 in these processes is indeed required for neuronal homeostasis and function. Adding to phenotypic complexity, Calame et al. (2023) also identified missense variants in the winged helix and the C-terminal RGG domains of DHX9 which associate with hereditary motor and sensory neuropathy (also known as Charcot–Marie Tooth type 2 disease).
The recent identification of pathogenic variants in several genes coding for DExH/DEAD-box RNA helicases has raised a strong interest in the function of this group of enzymes. Due to extensive studies in model organisms such as yeast, the functional relevance of the conserved helicase core domains has been elucidated in much detail (Linder and Jankowsky, 2011). These data from basic science strongly aided the interpretation of individual missense variants found in patients. Thus, it became clear that quite often the genetic variants affected key residues in HCMs, thereby interfering with the RNA binding, ATPase and eventually helicase activity of the encoded proteins. Nevertheless, in many cases it is still unclear whether a specific variant identified in a patient is pathogenic, or a harmless polymorphism. Inexpensive, non-complicated assays are needed to assess molecular relevance. Furthermore, there is still a large gap between understanding the molecular relevance of individual genetic variants, and understanding the relevance of these variants on a neuronal or systems level. Quite often, the particular cellular process which is relevant for disease is unknown. Thus, while DHX30 is partially present in mitochondria (Antonicka and Shoubridge, 2015; Bosco et al., 2021), the phenotypes of patients carrying variants in helicase core motifs of DHX30 are not typical for a mitochondrial disorder (Lessel et al., 2017). As the function of non-mitochondrial DHX30 is currently unknown, it is difficult to determine why variants in this gene lead to such a severe phenotype. In the case of DDX3X, most data now point to the relevance of translational control, as this helicase is needed for efficient translation of mRNAs with longer, structured 5’UTRs (Hoye et al., 2022). Nevertheless, it is unclear how the role of DDX3X in Wnt signaling may contribute to the development of intellectual disability in carriers of pathogenic variants. Further work both in mouse models as well as in induced pluripotent stem cell models derived from patient cells will be necessary to determine which particular aspects of neuronal RNA metabolism are affected by particular variants in DEAD-and DExH-box helicases.
Finally, it is mostly unclear how treatment can be achieved for these very rare disorders. Small molecule inhibitors have been identified for some helicases (reviewed by Naineni et al., 2023), which may be helpful in cases where the pathogenic variants cause a clear gain-of-function. Alternatively, specific antisense oligonucleotides may be considered in cases where a missense variant causes a more severe phenotype than the loss-of-function variants (as observed in DHX30; Mannucci et al., 2021).
JL: Writing – original draft, Data curation, Formal analysis, Visualization. SD: Formal analysis, Visualization, Writing – original draft. AP: Writing – original draft, Conceptualization, Writing – review & editing. DL: Conceptualization, Writing – original draft, Writing – review & editing, Data curation, Funding acquisition, Supervision. H-JK: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Work in the author’s laboratories is funded by grants from Deutsche Forschungsgemeinschaft (Kr1321/11-1 to H-JK; Le 4223/3-1 to DL) and The Austrian Science Fund (FWF; I 6657-B to DL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2024.1414949/full#supplementary-material
Supplementary Figure S1 | Helicase core motifs of human DEAD box helicases.
Supplementary Figure S2 | Helicase core motifs of human DExH helicases.
Aktas, T., Avsar Ilik, I., Maticzka, D., Bhardwaj, V., Pessoa Rodrigues, C., Mittler, G., et al. (2017). DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 544, 115–119. doi: 10.1038/nature21715
Al-Hadid, Q., and Yang, Y. (2016). R-loop: an emerging regulator of chromatin dynamics. Acta Biochim. Biophys. Sin. Shanghai 48, 623–631. doi: 10.1093/abbs/gmw052
Antonicka, H., and Shoubridge, E. A. (2015). Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 10, 920–932. doi: 10.1016/j.celrep.2015.01.030
Awasthi, S., Chakrapani, B., Mahesh, A., Chavali, P. L., Chavali, S., and Dhayalan, A. (2018). DDX39B promotes translation through regulation of pre-ribosomal RNA levels. RNA Biol. 15, 1157–1166. doi: 10.1080/15476286.2018.1517011
Ayache, J., Benard, M., Ernoult-Lange, M., Minshall, N., Standart, N., Kress, M., et al. (2015). P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Mol. Biol. Cell 26, 2579–2595. doi: 10.1091/mbc.E15-03-0136
Balak, C., Benard, M., Schaefer, E., Iqbal, S., Ramsey, K., Ernoult-Lange, M., et al. (2019). Rare de novo missense variants in RNA helicase DDX6 cause intellectual disability and dysmorphic features and Lead to P-body defects and RNA dysregulation. Am. J. Hum. Genet. 105, 509–525. doi: 10.1016/j.ajhg.2019.07.010
Bohnsack, K. E., Yi, S., Venus, S., Jankowsky, E., and Bohnsack, M. T. (2023). Cellular functions of eukaryotic RNA helicases and their links to human diseases. Nat. Rev. Mol. Cell Biol. 24, 749–769. doi: 10.1038/s41580-023-00628-5
Bosco, B., Rossi, A., Rizzotto, D., Hamadou, M. H., Bisio, A., Giorgetta, S., et al. (2021). DHX30 coordinates cytoplasmic translation and mitochondrial function contributing to cancer cell survival. Cancers 13:4412. doi: 10.3390/cancers13174412
Bourgeron, T. (2009). A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. doi: 10.1016/j.conb.2009.06.003
Burns, W., Bird, L. M., Heron, D., Keren, B., Ramachandra, D., Thiffault, I., et al. (2021). Syndromic neurodevelopmental disorder associated with de novo variants in DDX23. Am. J. Med. Genet. A 185, 2863–2872. doi: 10.1002/ajmg.a.62359
Cajigas, I. J., Tushev, G., Will, T. J., tom Dieck, S., Fuerst, N., and Schuman, E. M. (2012). The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74, 453–466. doi: 10.1016/j.neuron.2012.02.036
Calame, D. G., Guo, T., Wang, C., Garrett, L., Jolly, A., Dawood, M., et al. (2023). Monoallelic variation in DHX9, the gene encoding the DExH-box helicase DHX9, underlies neurodevelopment disorders and Charcot-Marie-tooth disease. Am. J. Hum. Genet. 110, 1394–1413. doi: 10.1016/j.ajhg.2023.06.013
Calo, E., Flynn, R. A., Martin, L., Spitale, R. C., Chang, H. Y., and Wysocka, J. (2015). RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature 518, 249–253. doi: 10.1038/nature13923
Calviello, L., Venkataramanan, S., Rogowski, K. J., Wyler, E., Wilkins, K., Tejura, M., et al. (2021). DDX3 depletion represses translation of mRNAs with complex 5' UTRs. Nucleic Acids Res. 49, 5336–5350. doi: 10.1093/nar/gkab287
Chakraborty, P., Huang, J. T. J., and Hiom, K. (2018). DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 9:4346. doi: 10.1038/s41467-018-06677-1
Chalupnikova, K., Lattmann, S., Selak, N., Iwamoto, F., Fujiki, Y., and Nagamine, Y. (2008). Recruitment of the RNA helicase RHAU to stress granules via a unique RNA-binding domain. J. Biol. Chem. 283, 35186–35198. doi: 10.1074/jbc.M804857200
Chen, Y., Dawes, R., Kim, H. C., Stenton, S. L., Walker, S., Ljungdahl, A., et al. (2024). De novo variants in the non-coding spliceosomal snRNA gene RNU4-2 are a frequent cause of syndromic neurodevelopmental disorders. medRxiv. doi: 10.1101/2024.04.07.24305438
Chen, S., Francioli, L. C., Goodrich, J. K., Collins, R. L., Kanai, M., Wang, Q., et al. (2024). A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100. doi: 10.1038/s41586-023-06045-0
Chuang, R. Y., Weaver, P. L., Liu, Z., and Chang, T. H. (1997). Requirement of the DEAD-box protein ded1p for messenger RNA translation. Science 275, 1468–1471. doi: 10.1126/science.275.5305.1468
Cordin, O., Banroques, J., Tanner, N. K., and Linder, P. (2006). The DEAD-box protein family of RNA helicases. Gene 367, 17–37. doi: 10.1016/j.gene.2005.10.019
Cougot, N., Bhattacharyya, S. N., Tapia-Arancibia, L., Bordonne, R., Filipowicz, W., Bertrand, E., et al. (2008). Dendrites of mammalian neurons contain specialized P-body-like structures that respond to neuronal activation. J. Neurosci. 28, 13793–13804. doi: 10.1523/JNEUROSCI.4155-08.2008
Cristini, A., Groh, M., Kristiansen, M. S., and Gromak, N. (2018). RNA/DNA hybrid Interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 23, 1891–1905. doi: 10.1016/j.celrep.2018.04.025
Cruciat, C. M., Dolde, C., de Groot, R. E., Ohkawara, B., Reinhard, C., Korswagen, H. C., et al. (2013). RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-beta-catenin signaling. Science 339, 1436–1441. doi: 10.1126/science.1231499
Dorner, K., and Hondele, M. (2024). The story of RNA unfolded: the molecular function of DEAD-and DExH-box ATPases and their complex relationship with membraneless organelles. Annu. Rev. Biochem. 93:11.1-11.30. doi: 10.1146/annurev-biochem-052521-121259
English, M. A., Lei, L., Blake, T., Wincovitch, S. M. Sr., Sood, R., Azuma, M., et al. (2012). Incomplete splicing, cell division defects, and hematopoietic blockage in dhx8 mutant zebrafish. Dev. Dyn. 241, 879–889. doi: 10.1002/dvdy.23774
Fairman-Williams, M. E., Guenther, U. P., and Jankowsky, E. (2010). SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 20, 313–324. doi: 10.1016/j.sbi.2010.03.011
Favaro, F. P., Alvizi, L., Zechi-Ceide, R. M., Bertola, D., Felix, T. M., de Souza, J., et al. (2014). A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am. J. Hum. Genet. 94, 120–128. doi: 10.1016/j.ajhg.2013.11.020
Fleckner, J., Zhang, M., Valcarcel, J., and Green, M. R. (1997). U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 11, 1864–1872. doi: 10.1101/gad.11.14.1864
Gencheva, M., Kato, M., Newo, A. N., and Lin, R.-J. (2010). Contribution of DEAH-box protein DHX16 in human pre-mRNA splicing. Biochem. J. 429, 25–32. doi: 10.1042/BJ20100266
Gilissen, C., Hehir-Kwa, J. Y., Thung, D. T., van de Vorst, M., van Bon, B. W., Willemsen, M. H., et al. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347. doi: 10.1038/nature13394
Glock, C., Biever, A., Tushev, G., Nassim-Assir, B., Kao, A., Bartnik, I., et al. (2021). The translatome of neuronal cell bodies, dendrites, and axons. Proc. Natl. Acad. Sci. U. S. A. 118:e2113929118. doi: 10.1073/pnas.2113929118
Gorbalenya, A. E., and Koonin, E. V. (1993). Helicases: amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 3, 419–429. doi: 10.1016/S0959-440X(05)80116-2
Greene, D., Genomics England Research, C., Pirri, D., Frudd, K., Sackey, E., Al-Owain, M., et al. (2023). Genetic association analysis of 77,539 genomes reveals rare disease etiologies. Nat. Med. 29, 679–688. doi: 10.1038/s41591-023-02211-z
Greene, D., Thys, C., Berry, I. R., Jarvis, J., Ortibus, E., Mumford, A. D., et al. (2024). Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders. Nat. Med. doi: 10.1038/s41591-024-03085-5
Hafner, A. S., Donlin-Asp, P. G., Leitch, B., Herzog, E., and Schuman, E. M. (2019). Local protein synthesis is a ubiquitous feature of neuronal pre-and postsynaptic compartments. Science 364:eaau3644. doi: 10.1126/science.aau3644
Hautbergue, G. M., Hung, M.-L., Walsh, M. J., Snijders, A. P., Chang, C.-T., Jones, R., et al. (2009). UIF, a new mRNA export adaptor that works together with REF/ALY, requires FACT for recruitment to mRNA. Curr. Biol. 19, 1918–1924. doi: 10.1016/j.cub.2009.09.041
Hilliker, A., Gao, Z., Jankowsky, E., and Parker, R. (2011). The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol. Cell 43, 962–972. doi: 10.1016/j.molcel.2011.08.008
Hondele, M., Sachdev, R., Heinrich, S., Wang, J., Vallotton, P., Fontoura, B. M. A., et al. (2019). DEAD-box ATPases are global regulators of phase-separated organelles. Nature 573, 144–148. doi: 10.1038/s41586-019-1502-y
Hoye, M. L., Calviello, L., Poff, A. J., Ejimogu, N. E., Newman, C. R., Montgomery, M. D., et al. (2022). Aberrant cortical development is driven by impaired cell cycle and translational control in a DDX3X syndrome model. eLife 11:e78203. doi: 10.7554/eLife.78203
Hug, N., Aitken, S., Longman, D., Raab, M., Armes, H., Mann, A. R., et al. (2022). A dual role for the RNA helicase DHX34 in NMD and pre-mRNA splicing and its function in hematopoietic differentiation. RNA 28, 1224–1238. doi: 10.1261/rna.079277.122
Jagtap, P. K. A., Muller, M., Kiss, A. E., Thomae, A. W., Lapouge, K., Beck, M., et al. (2023). Structural basis of RNA-induced autoregulation of the DExH-type RNA helicase maleless. Mol. Cell 83, 4318–4333.e10. doi: 10.1016/j.molcel.2023.10.026
Jalal, C., Uhlmann-Schiffler, H., and Stahl, H. (2007). Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Res. 35, 3590–3601. doi: 10.1093/nar/gkm058
Kanai, Y., Dohmae, N., and Hirokawa, N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. doi: 10.1016/j.neuron.2004.07.022
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. doi: 10.1038/nature10523
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kellner, M., Rohrmoser, M., Forné, I., Voss, K., Burger, K., Mühl, B., et al. (2015). DEAD-box helicase DDX27 regulates 3′ end formation of ribosomal 47S RNA and stably associates with the PeBoW-complex. Exp. Cell Res. 334, 146–159. doi: 10.1016/j.yexcr.2015.03.017
Khodosevich, K., and Sellgren, C. M. (2023). Neurodevelopmental disorders-high-resolution rethinking of disease modeling. Mol. Psychiatry 28, 34–43. doi: 10.1038/s41380-022-01876-1
Kim, J., Muraoka, M., Okada, H., Toyoda, A., Ajima, R., and Saga, Y. (2022). The RNA helicase DDX6 controls early mouse embryogenesis by repressing aberrant inhibition of BMP signaling through miRNA-mediated gene silencing. PLoS Genet. 18:e1009967. doi: 10.1371/journal.pgen.1009967
Kindler, S., and Kreienkamp, H. J. (2012). Dendritic mRNA targeting and translation. Adv. Exp. Med. Biol. 970, 285–305. doi: 10.1007/978-3-7091-0932-8_13
Kota, K. P., Wagner, S. R., Huerta, E., Underwood, J. M., and Nickerson, J. A. (2008). Binding of ATP to UAP56 is necessary for mRNA export. J. Cell Sci. 121, 1526–1537. doi: 10.1242/jcs.021055
Lahn, B. T., and Page, D. C. (1997). Functional coherence of the human Y chromosome. Science 278, 675–680. doi: 10.1126/science.278.5338.675
Lee, C. S., Dias, A. P., Jedrychowski, M., Patel, A. H., Hsu, J. L., and Reed, R. (2008). Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 36, 4708–4718. doi: 10.1093/nar/gkn454
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lennox, A. L., Hoye, M. L., Jiang, R., Johnson-Kerner, B. L., Suit, L. A., Venkataramanan, S., et al. (2020). Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development. Neuron 106, 404–420.e8. doi: 10.1016/j.neuron.2020.01.042
Lessel, D., Schob, C., Kury, S., Reijnders, M. R. F., Harel, T., Eldomery, M. K., et al. (2017). De novo missense mutations in DHX30 impair global translation and cause a neurodevelopmental disorder. Am. J. Hum. Genet. 101, 716–724. doi: 10.1016/j.ajhg.2017.09.014
Lessel, D., Zeitler, D. M., Reijnders, M. R. F., Kazantsev, A., Hassani Nia, F., Bartholomaus, A., et al. (2020). Germline AGO2 mutations impair RNA interference and human neurological development. Nat. Commun. 11:5797. doi: 10.1038/s41467-020-19572-5
Li, Y., Wang, X., Zhang, X., and Goodrich, D. W. (2005). Human hHpr1/p84/Thoc1 regulates transcriptional elongation and physically links RNA polymerase II and RNA processing factors. Mol. Cell. Biol. 25, 4023–4033. doi: 10.1128/MCB.25.10.4023-4033.2005
Li, Q., Zhang, P., Zhang, C., Wang, Y., Wan, R., Yang, Y., et al. (2014). DDX3X regulates cell survival and cell cycle during mouse early embryonic development. J. Biomed. Res. 28, 282–291. doi: 10.7555/JBR.27.20130047
Linder, P., and Jankowsky, E. (2011). From unwinding to clamping - the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 12, 505–516. doi: 10.1038/nrm3154
Lupan, B. M., Solecki, R. A., Musso, C. M., Alsina, F. C., and Silver, D. L. (2023). The exon junction complex component EIF4A3 is essential for mouse and human cortical progenitor mitosis and neurogenesis. Development 150:dev201619. doi: 10.1242/dev.201619
Mannucci, I., Dang, N. D. P., Huber, H., Murry, J. B., Abramson, J., Althoff, T., et al. (2021). Genotype-phenotype correlations and novel molecular insights into the DHX30-associated neurodevelopmental disorders. Genome Med. 13:90. doi: 10.1186/s13073-021-00900-3
Martin, R., Hackert, P., Ruprecht, M., Simm, S., Brüning, L., Mirus, O., et al. (2014). A pre-ribosomal RNA interaction network involving snoRNAs and the Rok1 helicase. RNA 20, 1173–1182. doi: 10.1261/rna.044669.114
Martin, R., Straub, A. U., Doebele, C., and Bohnsack, M. T. (2013). DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 10, 4–18. doi: 10.4161/rna.21879
Mikhailova, T., Shuvalova, E., Ivanov, A., Susorov, D., Shuvalov, A., Kolosov, P. M., et al. (2017). RNA helicase DDX19 stabilizes ribosomal elongation and termination complexes. Nucleic Acids Res. 45, 1307–1318. doi: 10.1093/nar/gkw1239
Morris-Rosendahl, D. J., and Crocq, M. A. (2020). Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin. Neurosci. 22, 65–72. doi: 10.31887/DCNS.2020.22.1/macrocq
Murakami, K., Nakano, K., Shimizu, T., and Ohto, U. (2017). The crystal structure of human DEAH-box RNA helicase 15 reveals a domain organization of the mammalian DEAH/RHA family. Acta Crystallogr. F Struct. Biol. Commun. 73, 347–355. doi: 10.1107/S2053230X17007336
Naineni, S. K., Robert, F., Nagar, B., and Pelletier, J. (2023). Targeting DEAD-box RNA helicases: the emergence of molecular staples. Wiley Interdiscip. Rev. RNA 14:e1738. doi: 10.1002/wrna.1738
Nicklas, S., Okawa, S., Hillje, A. L., Gonzalez-Cano, L., Del Sol, A., and Schwamborn, J. C. (2015). The RNA helicase DDX6 regulates cell-fate specification in neural stem cells via miRNAs. Nucleic Acids Res. 43, 2638–2654. doi: 10.1093/nar/gkv138
Niemi, M. E. K., Martin, H. C., Rice, D. L., Gallone, G., Gordon, S., Kelemen, M., et al. (2018). Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271. doi: 10.1038/s41586-018-0566-4
Nissan, T., Rajyaguru, P., She, M., Song, H., and Parker, R. (2010). Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol. Cell 39, 773–783. doi: 10.1016/j.molcel.2010.08.025
Obuća, M., Cvačková, Z., Kubovčiak, J., Kolář, M., and Staněk, D. (2022). Retinitis pigmentosa-linked mutation in DHX38 modulates its splicing activity. PLoS One 17:e0265742. doi: 10.1371/journal.pone.0265742
Ostareck, D. H., Naarmann-de Vries, I. S., and Ostareck-Lederer, A. (2014). DDX6 and its orthologs as modulators of cellular and viral RNA expression. Wiley Interdiscip. Rev. RNA 5, 659–678. doi: 10.1002/wrna.1237
Paine, I., Posey, J. E., Grochowski, C. M., Jhangiani, S. N., Rosenheck, S., Kleyner, R., et al. (2019). Paralog studies augment gene discovery: DDX and DHX genes. Am. J. Hum. Genet. 105, 302–316. doi: 10.1016/j.ajhg.2019.06.001
Parenti, I., Rabaneda, L. G., Schoen, H., and Novarino, G. (2020). Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci. 43, 608–621. doi: 10.1016/j.tins.2020.05.004
Paul, M. S., Duncan, A. R., Genetti, C. A., Pan, H., Jackson, A., Grant, P. E., et al. (2023). Rare EIF4A2 variants are associated with a neurodevelopmental disorder characterized by intellectual disability, hypotonia, and epilepsy. Am. J. Hum. Genet. 110, 120–145. doi: 10.1016/j.ajhg.2022.11.011
Pause, A., and Sonenberg, N. (1992). Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 11, 2643–2654. doi: 10.1002/j.1460-2075.1992.tb05330.x
Perooli, F. M., Wilkinson, K. A., Pring, K., and Hanley, J. G. (2024). An essential role for the RNA helicase DDX6 in NMDA receptor-dependent gene silencing and dendritic spine shrinkage. Sci. Rep. 14:3066. doi: 10.1038/s41598-024-53484-4
Prabu, J. R., Muller, M., Thomae, A. W., Schussler, S., Bonneau, F., Becker, P. B., et al. (2015). Structure of the RNA helicase MLE reveals the molecular mechanisms for uridine specificity and RNA-ATP coupling. Mol. Cell 60, 487–499. doi: 10.1016/j.molcel.2015.10.011
Pyle, A. M. (2008). Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 37, 317–336. doi: 10.1146/annurev.biophys.37.032807.125908
Rajendran, R. R., Nye, A. C., Frasor, J., Balsara, R. D., Martini, P. G., and Katzenellenbogen, B. S. (2003). Regulation of nuclear receptor transcriptional activity by a novel DEAD box RNA helicase (DP97). J. Biol. Chem. 278, 4628–4638. doi: 10.1074/jbc.M210066200
Rosa, E. S. I., Smetana, J. H. C., and de Oliveira, J. F. (2024). A comprehensive review on DDX3X liquid phase condensation in health and neurodevelopmental disorders. Int. J. Biol. Macromol. 259:129330. doi: 10.1016/j.ijbiomac.2024.129330
Rossow, K. L., and Janknecht, R. (2003). Synergism between p68 RNA helicase and the transcriptional coactivators CBP and p300. Oncogene 22, 151–156. doi: 10.1038/sj.onc.1206067
Rouya, C., Siddiqui, N., Morita, M., Duchaine, T. F., Fabian, M. R., and Sonenberg, N. (2014). Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA 20, 1398–1409. doi: 10.1261/rna.045302.114
Sahoo, P. K., Lee, S. J., Jaiswal, P. B., Alber, S., Kar, A. N., Miller-Randolph, S., et al. (2018). Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Commun. 9:3358. doi: 10.1038/s41467-018-05647-x
Samocha, K. E., Robinson, E. B., Sanders, S. J., Stevens, C., Sabo, A., McGrath, L. M., et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950. doi: 10.1038/ng.3050
Sauer, M., Juranek, S. A., Marks, J., De Magis, A., Kazemier, H. G., Hilbig, D., et al. (2019). DHX36 prevents the accumulation of translationally inactive mRNAs with G4-structures in untranslated regions. Nat. Commun. 10:2421. doi: 10.1038/s41467-019-10432-5
Shamseldin, H. E., Rajab, A., Alhashem, A., Shaheen, R., Al-Shidi, T., Alamro, R., et al. (2013). Mutations in DDX59 implicate RNA helicase in the pathogenesis of orofaciodigital syndrome. Am. J. Hum. Genet. 93, 555–560. doi: 10.1016/j.ajhg.2013.07.012
Shen, H.-H. (2009). UAP56-a key player with surprisingly diverse roles in pre-mRNA splicing and nuclear export. BMB Rep. 42, 185–188. doi: 10.5483/BMBRep.2009.42.4.185
Shen, H., Yanas, A., Owens, M. C., Zhang, C., Fritsch, C., Fare, C. M., et al. (2022). Sexually dimorphic RNA helicases DDX3X and DDX3Y differentially regulate RNA metabolism through phase separation. Mol. Cell 82, 2588–2603.e9. doi: 10.1016/j.molcel.2022.04.022
Sheng, Y., Tsai-Morris, C. H., Gutti, R., Maeda, Y., and Dufau, M. L. (2006). Gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) is a transport protein involved in gene-specific mRNA export and protein translation during spermatogenesis. J. Biol. Chem. 281, 35048–35056. doi: 10.1074/jbc.M605086200
Shih, C. Y., Chen, Y. C., Lin, H. Y., and Chu, C. Y. (2023). RNA helicase DDX6 regulates A-to-I editing and neuronal differentiation in human cells. Int. J. Mol. Sci. 24:3197. doi: 10.3390/ijms24043197
Shiina, N., Shinkura, K., and Tokunaga, M. (2005). A novel RNA-binding protein in neuronal RNA granules: regulatory machinery for local translation. J. Neurosci. 25, 4420–4434. doi: 10.1523/JNEUROSCI.0382-05.2005
Siletti, K., Hodge, R., Mossi Albiach, A., Lee, K. W., Ding, S. L., Hu, L., et al. (2023). Transcriptomic diversity of cell types across the adult human brain. Science 382:eadd7046. doi: 10.1126/science.add7046
Snijders Blok, L., Madsen, E., Juusola, J., Gilissen, C., Baralle, D., Reijnders, M. R., et al. (2015). Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am. J. Hum. Genet. 97, 343–352. doi: 10.1016/j.ajhg.2015.07.004
Soto-Rifo, R., Rubilar, P. S., Limousin, T., de Breyne, S., Decimo, D., and Ohlmann, T. (2012). DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J. 31, 3745–3756. doi: 10.1038/emboj.2012.220
Steward, O., and Schuman, E. M. (2001). Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 24, 299–325. doi: 10.1146/annurev.neuro.24.1.299
Sun, C., Nold, A., Fusco, C. M., Rangaraju, V., Tchumatchenko, T., Heilemann, M., et al. (2021). The prevalence and specificity of local protein synthesis during neuronal synaptic plasticity. Sci. Adv. 7:eabj0790. doi: 10.1126/sciadv.abj0790
Tanner, N. K., Cordin, O., Banroques, J., Doere, M., and Linder, P. (2003). The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell 11, 127–138. doi: 10.1016/s1097-2765(03)00006-6
Tanner, N. K., and Linder, P. (2001). DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell 8, 251–262. doi: 10.1016/s1097-2765(01)00329-x
Valentin-Vega, Y. A., Wang, Y. D., Parker, M., Patmore, D. M., Kanagaraj, A., Moore, J., et al. (2016). Cancer-associated DDX3X mutations drive stress granule assembly and impair global translation. Sci. Rep. 6:25996. doi: 10.1038/srep25996
van der Lelij, P., Chrzanowska, K. H., Godthelp, B. C., Rooimans, M. A., Oostra, A. B., Stumm, M., et al. (2010). Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 86, 262–266. doi: 10.1016/j.ajhg.2010.01.008
Weber, R., Wohlbold, L., and Chang, C.-T. (2024). Human DDX6 regulates translation and decay of inefficiently translated mRNAs. Elife 13:RP92426. doi: 10.7554/eLife.92426
Weston, A., and Sommerville, J. (2006). Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res. 34, 3082–3094. doi: 10.1093/nar/gkl409
Xing, Z., Ma, W. K., and Tran, E. J. (2019). The DDX5/Dbp2 subfamily of DEAD-box RNA helicases. Wiley Interdiscip. Rev. RNA 10:e1519. doi: 10.1002/wrna.1519
Yamada, M., Nitta, Y., Uehara, T., Suzuki, H., Miya, F., Takenouchi, T., et al. (2023). Heterozygous loss-of-function DHX9 variants are associated with neurodevelopmental disorders: human genetic and experimental evidences. Eur. J. Med. Genet. 66:104804. doi: 10.1016/j.ejmg.2023.104804
Yan, X., Mouillet, J.-F., Ou, Q., and Sadovsky, Y. (2003). A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol. Cell. Biol. 23, 414–423. doi: 10.1128/MCB.23.1.414-423.2003
Yang, L., Liu, Q., Zhao, Y., Lin, N., Huang, Y., Wang, Q., et al. (2024). DExH-box helicase 9 modulates hippocampal synapses and regulates neuropathic pain. iScience 27:109016. doi: 10.1016/j.isci.2024.109016
Yang, S., Winstone, L., Mondal, S., and Wu, Y. (2023). Helicases in R-loop formation and resolution. J. Biol. Chem. 299:105307. doi: 10.1016/j.jbc.2023.105307
Zanini, I. M., Soneson, C., Lorenzi, L. E., and Azzalin, C. M. (2017). Human cactin interacts with DHX8 and SRRM2 to assure efficient pre-mRNA splicing and sister chromatid cohesion. J. Cell Sci. 130, 767–778. doi: 10.1242/jcs.194068
Zeitelhofer, M., Karra, D., Macchi, P., Tolino, M., Thomas, S., Schwarz, M., et al. (2008). Dynamic interaction between P-bodies and transport ribonucleoprotein particles in dendrites of mature hippocampal neurons. J. Neurosci. 28, 7555–7562. doi: 10.1523/JNEUROSCI.0104-08.2008
Keywords: stress granules, P-bodies, miRNA, translation, R-loop
Citation: Lederbauer J, Das S, Piton A, Lessel D and Kreienkamp H-J (2024) The role of DEAD- and DExH-box RNA helicases in neurodevelopmental disorders. Front. Mol. Neurosci. 17:1414949. doi: 10.3389/fnmol.2024.1414949
Edited by:
Oriane Mauger, Max Planck Institute of Psychiatry, GermanyReviewed by:
Karl Emory Bauer, Ludwig Maximilian University of Munich, GermanyCopyright © 2024 Lederbauer, Das, Piton, Lessel and Kreienkamp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davor Lessel, ZC5sZXNzZWxAc2Fsay5hdA==; Hans-Jürgen Kreienkamp, S3JlaWVua2FtcEB1a2UuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.