 Xiaochang Zhang
Xiaochang Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 25 July 2024
Sec. Neuroplasticity and Development
Volume 17 - 2024 | https://doi.org/10.3389/fnmol.2024.1412964
This article is part of the Research Topic Come as You R(NA): Post-transcriptional Regulation Will Do the Rest View all 12 articles
Pediatric neurological disorders are frequently devastating and present unmet needs for effective medicine. The successful treatment of spinal muscular atrophy with splice-switching antisense oligonucleotides (SSO) indicates a feasible path to targeting neurological disorders by redirecting pre-mRNA splicing. One direct outcome is the development of SSOs to treat haploinsufficient disorders by targeting naturally occurring non-productive splice isoforms. The development of personalized SSO treatment further inspired the therapeutic exploration of rare diseases. This review will discuss the recent advances that utilize SSOs to treat pediatric neurological disorders.
Over the last two decades, causal variants for pediatric neurological disorders have been increasingly uncovered by high-throughput DNA sequencing. Many clinically comparable disease symptoms, such as developmental and epileptic encephalopathy (DEE), turn out to be caused by mutations in dozens of genes that have different biological functions and pathophysiology. Consequently, human diseases are increasingly classified based on their molecular causes and clinical presentations. Such accumulating genetic evidence offers unique opportunities to develop gene- or variant-specific treatments in addition to generic symptom-oriented drugs. Precision medicine strategies for neurological disorders, such as gene replacement therapy, genome editing, and splicing modulation, have been actively explored (Deverman et al., 2018; Nussbacher et al., 2019; Raguram et al., 2022). Antisense oligonucleotides (ASO) represent one type of such therapeutic means and have shown promising clinical outcomes for spinal muscular atrophy (SMA), Duchenne muscular dystrophy (DMD), and Amyotrophic Lateral Sclerosis (ALS), among other ongoing clinical and preclinical studies (Rinaldi and Wood, 2018).
ASOs are modified short nucleotides that bind to pre-mRNA through Watson-Crick base pairing (Kole et al., 2012). ASOs can be used as steric blockers to intervene in processes such as splicing and protein translation, or as gapmers to promote RNase H1-mediated target mRNA degradation. The nucleobases and the backbone are modified to resist nuclease degradation, enhance the target binding, and boost cellular intake. ASO modifications, such as 2’-O-methoxyethyl-modified (MOE) nucleotides with phosphorothioate (PS) backbone, have been clinically tested and proven to be generally tolerated (Egli and Manoharan, 2023). Various modifications have been developed to enhance efficacy and decrease toxicity. Splice-switching oligonucleotides (SSO) are a specific category of ASOs that bind to pre-mRNA as steric blockers and redirect splicing. SSOs have been successfully developed to treat SMA and DMD (Voit et al., 2014; Finkel et al., 2017). ASO gapmers have been recently approved by the FDA to treat SOD1 ALS. This review focuses on the progress of SSOs in targeting pediatric neurological conditions.
Most human protein-coding genes are split by introns, which are spliced out by the spliceosome (Berget et al., 1977; Chow et al., 1977). Introns are collectively defined by their 5′ splice donor site (5’SS), 3′ acceptor site (3’SS), the branchpoint adenosine, the poly-pyrimidine tract upstream of the 3’SS, and other regulatory sequences. Pre-mRNA splicing allows the reshuffle of different exons (Gilbert, 1978), and RNA-seq analyses showed that over 95% of intron-containing human genes undergo alternative splicing (AS) to generate multiple mRNA isoforms (Pan et al., 2008; Wang et al., 2008). Alternative splicing can lead to skipped exons (SE), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), mutually exclusive exons (MXE), and retained introns (RI) (Graveley, 2001). Alternative splicing can happen in species-, tissue- and cell-type-specific manners (Barbosa-Morais et al., 2012; Merkin et al., 2012; Feng et al., 2021). Alternative splicing is prevalent in the brain, and recent long-read sequencing analyses have uncovered coordinated splicing of distant exons (Gupta et al., 2018; Yang et al., 2023; Zhang et al., 2023). Alternative splicing is modulated by intronic and exonic cis-regulatory sequences and their associated RNA-binding proteins (Black, 2003; Wang and Burge, 2008; Barash et al., 2010; Xiong et al., 2015; Bao et al., 2019; Van Nostrand et al., 2020). The natural occurrence of alternative splicing and the identification of splicing enhancers/suppressors indicate that re-directing splicing holds its own dimension for gene regulation and therapeutic intervention.
About 10% of exonic human mutations are estimated to cause diseases by disrupting pre-mRNA splicing (Soemedi et al., 2017). While whole-exome sequencing detects exonic and splice site mutations for genetically defined disorders, integrating transcriptome and whole-genome analysis uncovers more causal intronic splicing mutations (Cummings et al., 2017; Kim et al., 2023). These splicing mutations frequently introduce aberrant splice sites that lead to loss-of-function or hypomorphic alleles. Disease-causing splicing variants can be suppressed to treat human diseases. Redirecting splicing can also lead to beneficial effects by (1) bypassing nonessential inframe exons that carry pathogenic mutations, (2) bypassing an additional exon to correct the reading frame, and (3) redirecting alternative splicing to promote functional isoform production.
SSOs bind to pre-mRNA through Watson-Crick base pairing and redirect pre-mRNA splicing (Kole et al., 2012; Centa and Hastings, 2022). The SSO binding sites are frequently splicing enhancers or suppressors, and the double-stranded SSO-pre-mRNA can block RNA–RNA or RNA-protein interactions that modulate splice site usage. Since the success of SSOs in treating DMD and SMA (Hua et al., 2011; Finkel et al., 2017), redirecting pre-mRNA splicing has been increasingly recognized as a powerful therapeutic strategy to treat neurological disorders (Hill and Meisler, 2021; Nikom and Zheng, 2023). The sequence flexibility and the clinically proven chemistry have made SSO a fast-growing platform for personalized medicine. The development of the SSO drug Milasen for a girl named Mila is inspirational, and the approach displayed promising progress toward previously undruggable targets and rare mutations (Kim et al., 2019, 2023). This review focuses on recently reported SSO strategies targeting pediatric neurological conditions and the value of genetic tools.
SSOs can promote either exon skipping or inclusion. With a focus on pediatric neurological conditions, recently reported SSO strategies generally fall into three main categories (Figure 1). The most straightforward application of SSOs would be suppressing an abnormal splice site introduced by a specific mutation – a variant-specific SSO (Figure 1A). SSOs have also been developed to skip a nonessential exon that is either inframe and carries deleterious mutations or correct the reading frame caused by frameshift mutations – an exon-specific SSO (Figure 1B). Lastly, SSOs can increase protein expression through a paralog and rescue recessive diseases or boost protein expression from the wild-type allele and rescue haploinsufficiency – such SSOs are independent of mutations and can be considered gene-specific SSOs (Figure 1C). While variant- and exon-specific SSOs play prominent roles in personalized medicine, a gene-specific SSO can be used to treat patients carrying mutations across the same gene. SSO-mediated therapy, or treatments for genetic disorders in general, would be more effective when used for sooner intervention in disease progression than later. Thus, early genetic diagnosis-aided treatment before the existence of irreversible disease presentations, as shown by a recent study (Kim et al., 2023), appears to be a promising practice.
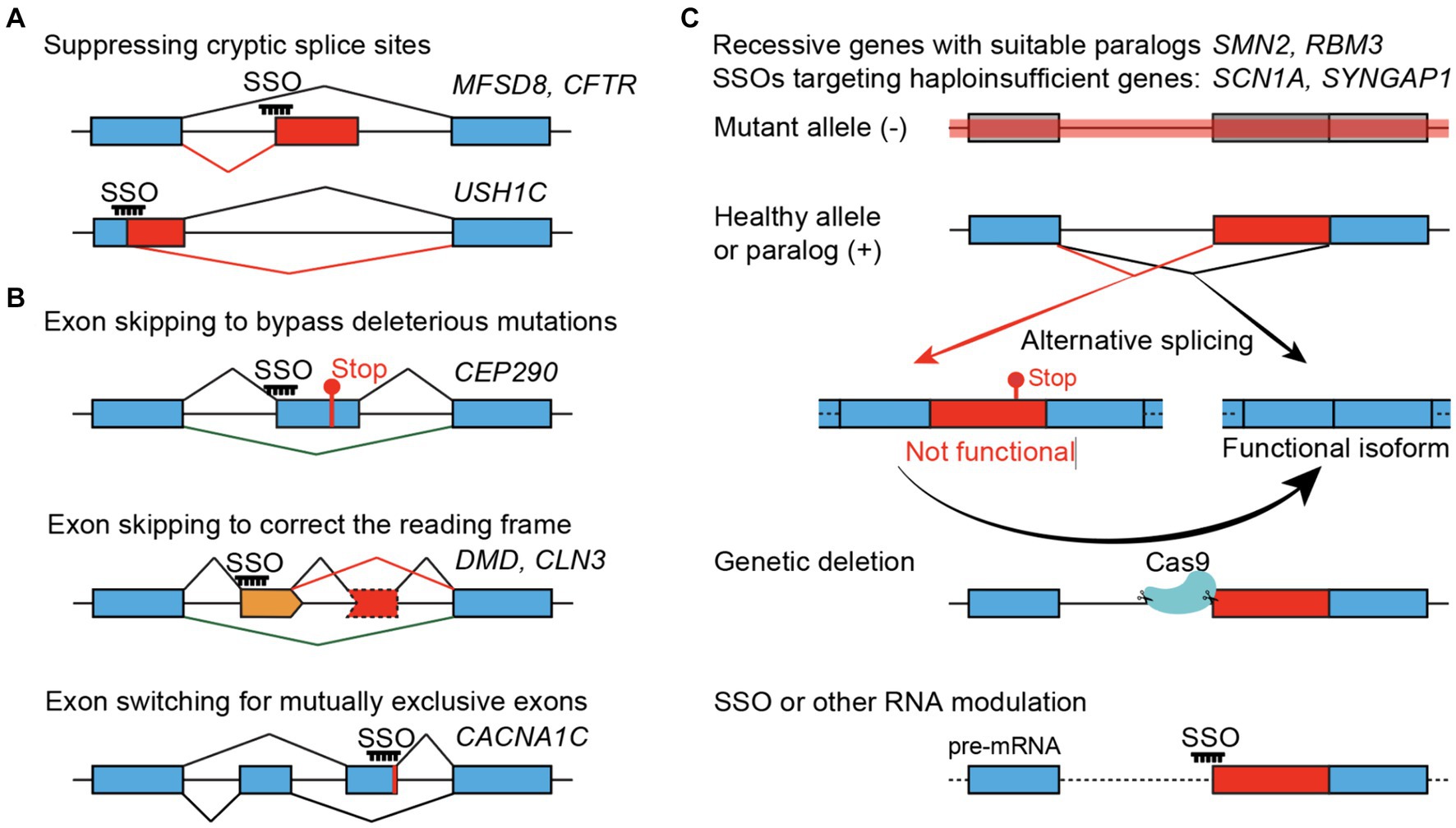
Figure 1. SSO-mediated therapeutic strategies. (A) Variant-specific SSOs suppress the gain of cryptic splice sites in the introns (top) or exons (bottom). (B) Exon-specific SSOs. Bypassing a non-essential exon that carries pathogenic mutations (top), skipping an additional non-essential exon (orange) to correct the translational reading frame (middle), or switching for a functional mutually exclusive exon (bottom). (C) Gene-specific SSOs treating recessive or haploinsufficient conditions by converting naturally occurring non-functional (or unstable) splice isoforms to functional isoforms, using SYNGAP1 as an example. Genetic suppression of non-productive splicing, mimicking the maximal and constant effect of an SSO, can provide in vivo evidence about the neurological and organismal functions of the non-productive isoform, to what extent the protein level can be restored, and whether it can rescue phenotypes associated with loss-of-function alleles.
Recessive diseases frequently involve loss-of-function alleles, and several SSO-based therapeutic strategies have been reported (Figure 1). SSO can promote the inclusion or exclusion of specific exons. Thus, it is straightforward to use SSOs to suppress undesired exons, such as abnormal/cryptic splice sites. SSOs can also block splicing silencers and promote exon inclusion to make functional proteins, such as the SMN2 case below.
Spinal muscular atrophy (SMA) is a motor neuron disorder caused by recessive loss-of-function mutations in SMN1 (Lefebvre et al., 1995). The loss of spinal cord motor neurons in SMA patients leads to muscle weakness and atrophy, and the disease presentations fall into different clinical categories based on the age of onset and the severity of symptoms. Type 1 SMA, with the disease onset by 6 months of age and an expected life shorter than 2 years, is the most severe form and affects about 50% of all cases. SMN2 is a hominid-specific duplication of SMN1, and increased SMN2 copy numbers are inversely correlated with SMA severity (Rochette et al., 2001; Calucho et al., 2018). Compared to SMN1, SMN2 carries a single synonymous C-to-T change in exon 7 that causes 90% of SMN2 mRNA to skip exon 7 and encode an unstable protein (Monani et al., 1999). The exon7-included SMN2 mRNA encodes an identical protein to SMN1. Multiple strategies, including SSOs and splicing modulatory small molecules, have been developed to promote SMN2 exon 7 inclusion and treat SMA (Hua et al., 2010). The FDA-approved SSO Spinraza/nusinersen consists of 18 2’-MOE nucleotides with a PS backbone, binds to SMN2 intron7, and promotes the inclusion of SMN2 exon7 (Hua et al., 2011). Nusinersen has been shown to significantly improve the motor conditions and life expectancy of SMA patients (Finkel et al., 2017; Mercuri et al., 2018).
A straightforward application of SSOs would be suppressing abnormal/cryptic splice sites introduced by pathogenic mutations (Figure 1A). This strategy has been explored to treat multiple diseases, such as the USH1C Usher syndrome (Lentz et al., 2013). Autosomal recessive USH1C mutations cause type 1 Usher syndrome concerning congenital sensorineural deafness, vestibular dysfunction, and blindness (Verpy et al., 2000). The USH1C c.216G > A creates a cryptic 5′ splice site in exon 3, and an SSO covering the mutation and cryptic splice site significantly corrected the splicing error (Lentz et al., 2013). Remarkably, a single-dose SSO injection in the neonatal Ush1c c.216AA mice rescued abnormalities of cochlear hair cells, and vestibular and low-frequency hearing deficits, indicating strong therapeutic potential (Lentz et al., 2013).
While malfunctioning splicing can be suppressed, recessive mutations in the protein-coding regions may not be as straightforward to target with SSOs. In parallel to the nusinersen clinical trial, exon-skipping SSOs have been developed to treat Duchenne muscular dystrophy (DMD). DMD is an X-linked progressive muscle-wasting disease caused by loss-of-function mutations in the DMD/dystrophin gene. The dystrophin protein has 24 repeated spectrin-like domains, and truncated dystrophin proteins with fewer spectrin-like repeats were found in patients who showed much milder symptoms (England et al., 1990). Human genetics studies indicate that bypassing exons in the middle of dystrophin while preserving its N- and C-terminal domains can be beneficial (Matsuo, 1996). About half of DMD patients have deletion mutations in a hotspot region between exons 45–55 (Duan et al., 2021). Multiple SSOs have been successfully developed to skip exons such as 51, 53, or 45 (Exondys 51, Vyondys 53, and Amondys 45) to correct translational reading frames and produce partially functional dystrophin proteins (Roberts et al., 2023) (Figure 1B).
Exon-skipping SSOs have been explored for targeting other diseases, such as correcting the reading frame in CLN3 Batten’s disease (below), bypassing an inframe CEP290 exon 41 that carries pathogenic mutations for Jobert syndrome (Ramsbottom et al., 2018), and suppressing a cryptic splice site in CFTR cystic fibrosis (Michaels et al., 2020). The Hastings group developed an exon-skipping strategy in mice to target a mutant allele that causes CLN3, a form of Batten’s disease (Centa et al., 2020). Patients carrying recessive CLN3 mutations experience disease onset in early childhood and typically decease by 20–30 years of age (IBDC, 1995). A substantial portion of patients are affected by a deletion spanning exons 7 and 8 (Δex78), leading to a shift of the translational frame. SSOs have been reported to correct the reading frame by skipping exon 5 in cis (Δex578, Figure 1B). The SSO has been reported to robustly induce exon 5 skipping and improve motor coordination and survival in a Cln3 (Δex78) mouse model. The research group further created a Cln3 (Δex578) genetic model and showed that deleting exon 5 on top of Δex78 was beneficial in mice (Centa et al., 2023). These works suggest a promising exon-skipping strategy for CLN3 (Δex78) Batten’s disease.
The frontier of personalized medicine leaped forward with the N = 1 study on a child affected by CLN7, another form of Batten’s disease (Kim et al., 2019). CLN7 is a late-infantile-onset lysosomal storage disorder, and affected children would experience early normal development followed by function declines of the nervous system that lead to vision loss, drug-resistant epilepsy, progressive cerebral and cerebellar atrophy, and premature death (Topcu et al., 2004). CLN7 is caused by recessive mutations in MFSD8 (Siintola et al., 2007), but in the N = 1 case, clinical testing only identified one inherited MFSD8 allele (Kim et al., 2019). The Yu lab performed whole genome sequencing and identified an SVA-transposon insertion in MFSD8 intron 6, which promoted the inclusion of a cryptic 3′ splice site in SSOs targeting predicted splicing enhancers were screened to suppress abnormal splicing of MFSD8 (Figure 1A). The lead oligo milasen, an 18-nt SSO with 2’MOE modification and a PS backbone, was effective in patient cells and tolerated in rodents. Milasen was applied to the patient under an expanded-access protocol approved by the FDA and modeled after nusinersen. The N = 1 trial was shown to reduce seizure frequency and duration, suggesting a beneficial effect (Kim et al., 2019). This work paved the path for expedited genetic diagnosis and individualized drug development.
Important questions remain in SSO-mediated treatment. Given the diverse nature of pathogenic mutations, how can we identify targetable variants and design effective SSOs? A recent in-depth study of ataxia-telangiectasia (A-T) (Kim et al., 2023) began to address this question. A-T is an autosomal recessive disorder caused by the loss of the ATM gene required for DNA damage response and cell cycle progression (Savitsky et al., 1995). A-T patients typically show progressive cerebellar degeneration with early symptoms of ataxia, increased chance for cancer, and telangiectasias. A significant fraction of causal variants for A-T have been reported to cause abnormal splicing patterns (Teraoka et al., 1999), and SSOs (morpholino ASOs) have been developed to correct ATM splice variants (Du et al., 2007). A recent study reported whole-genome sequencing analyses of 235 A-T patients and classified plausible causal mutations depending on their molecular nature and potential for SSO treatment (Kim et al., 2023). Combining transcriptomic analyses and computation predictions, the authors estimated that 9 and 6% of the A-T patients carry “probable” and “possible” variants amenable to SSO targeting, respectively. The authors developed SSOs for two mutations and initiated clinical studies in A-T patients before disease onset. Thus, thorough genetics analysis estimated the SSO-targetable ratio to 9–15% in patients affected by rare diseases like A-T (Kim et al., 2023).
The variant- or exon-specific SSO strategies used in recessive disorders, such as suppressing cryptic splice sites and bypassing deleterious mutations in nonessential exons (Figures 1A,B), are also applicable to target the mutated alleles in dominant genetic disorders, especially for gain-of-function/activity alleles. Following the initial linkage analyses and cloning of inherited mutations, recent human genetics studies discovered widespread dominant mutations causal for neurodevelopmental disorders such as epilepsy and autism spectrum disorders (Helbig and Abou Tayoun, 2016, Satterstrom et al., 2020). For instance, over 1,400 SCN1A mutations have been reported as pathogenic in ClinVar (a public database to aggregate genetic variants and clinical findings), and a significant fraction of such mutations cause severe loss of function (frameshift, nonsense, splice site, and deletion). Furthermore, causal mutations for neurodevelopmental disorders have been reported in dozens to hundreds of genes. However, targeting such a vast number of mutated alleles using variant- or exon-specific SSOs presents a daunting task.
For haploinsufficient mutations, the healthy allele offers another layer of therapeutic potential. Increasing protein expression from the healthy allele can potentially establish a gene-specific instead of a variant- or exon-specific solution. In principle, this is achievable by boosting transcription, suppressing mRNA degradation, promoting translation, or suppressing protein degradation. Strategies suppressing naturally occurring non-productive isoforms, boosting translation by recruiting ribosomes, degrading naturally occurring antisense transcripts, and targeted de-ubiquitination have been explored to treat haploinsufficiency (Meng et al., 2015; Han et al., 2020; Kanner et al., 2020; Lim et al., 2020; Cao et al., 2023; Dawicki-McKenna et al., 2023; Yang et al., 2023).
Abnormal translation termination caused by premature codons (PTCs) triggers nonsense-mediated mRNA decay (NMD) in eukaryotes (Kurosaki et al., 2019). Interestingly, naturally occurring alternative splicing can trigger NMD (AS-NMD), and AS-NMD has been shown to autoregulate the master splicing factor SR proteins (Lewis et al., 2003; Lareau et al., 2007; Leclair et al., 2020). Recent studies have reported that AS-NMD developmentally regulates hundreds of genes in the brain (Zheng et al., 2012; Eom et al., 2013; Yan et al., 2015). Abnormally elevated AS-NMD in SNRPB, FLNA, and SCN1A by human mutations have been reported to cause cerebro–costo–mandibular syndrome (Lynch et al., 2014), structural brain malformation (Zhang et al., 2016), and epilepsy in humans (Carvill et al., 2018). Thus, the naturally occurring non-productive alternative splicing in disease-associated genes can be targetable switches for gene regulation.
If the gene of interest naturally expresses an alternative and non-productive isoform, converting the non-productive splice isoform to a functional form would be a promising approach to upregulate gene expression. The TANGO (targeted augmentation of nuclear gene output) method was reported in 2020, with a focus on SCN1A (Han et al., 2020; Lim et al., 2020). De novo loss-of-function mutations in SCN1A are leading causes of DEE, especially the Dravet syndrome, which is characterized by intractable febrile seizures. Human genetic studies showed that a fraction of SCN1A mRNA contains exon 20 N that triggers nonsense-mediated decay, and if the inclusion is abnormally increased by human mutations, it causes Dravet syndrome (Carvill et al., 2018). Lim et al. started by looking for non-productive alternative splicing in human disease-associated genes, screened SSOs in cultured cells, and showed the efficacy of two SCN1A ASOs in mice (Lim et al., 2020). Zhou et al. further showed an in-depth screening of SCN1A ASOs, their effect in upregulating mRNA and protein expression in mice, and their striking effects in rescuing lethality in a Dravet syndrome mouse model (Han et al., 2020). Clinical trials of the SSO in Dravet patients are ongoing and appear promising. These studies suggest that targeting the non-productive isoform can be a promising therapeutic approach.
SYNGAP1 encodes the synaptic Ras GTPase-activating protein and is required for synaptic plasticity. Haploinsufficient SYNGAP1 mutations are the leading causes of intellectual disability, infantile epilepsy, and other neurological symptoms (Hamdan et al., 2009). Transcriptomic analysis of the developing mouse and human brains uncovered alternative 3′ splice sites of SYNGAP1 intron10 that lead to NMD (A3SS-NMD, Figure 1C) (Yang et al., 2023). PTBP1/2 proteins directly promote the A3SS-NMD and suppress SYNGAP1 protein expression (Yang et al., 2023). Deletion of the A3SS-NMD in mice lead to upregulated Syngap1 protein. Importantly, such upregulated protein significantly alleviated the LTP deficits in the hippocampus and the neuronal excitability phenotype in cortical neurons caused by a compound Syngap1 knockout allele (Yang et al., 2023). We further screened SSOs in human iPSCs, and the lead SSO effectively increased the functional SYNGAP1 isoform in iPSC-derived neurons and cerebral organoids (Yang et al., 2023). Interestingly, some of the lead SYNGAP1 SSOs identified in independent studies overlap with each other (Lim et al., 2020; Dawicki-McKenna et al., 2023; Yang et al., 2023), indicating the existence of a splicing enhancer for the A3SS-NMD.
Timothy syndrome, caused by dominant mutations in CACNA1C, is a multi-organ disorder characterized by congenital heart disease, lethal arrhythmias, cognitive deficits, and autism (Splawski et al., 2004). One recurrent p.G460R mutation occurs in the mutually exclusive exon 8A, promotes the exon 8A inclusion over exon 8, and leads to the loss of voltage-dependent channel inactivation (Panagiotakos et al., 2019). While CACNA1C exon 8 gradually replaces exon 8A during neural development, it was speculated as beneficial if the mutated exon 8A switched to exon 8 early in patients (Figure 1B). Indeed, the lead SSO was shown to increase CACNA1C exon 8 inclusion and rescue delayed channel inactivation and interneuron migration defects in cortical organoids (Chen et al., 2024). Furthermore, the authors transplanted cortical organoids carrying the p.G460R (exon 8A) mutation to athymic rats and showed the SSO treatment rescued molecular and functional defects (Chen et al., 2024). This study indicates that switching functionally equivalent but mutually exclusive exons can bypass deleterious effects and demonstrates the application of a human organoid-rat chimeric system.
Cultured cell lines, patient-derived fibroblasts, human iPSCs, and iPSC-derived neural cultures provide valuable tools for SSO screens, and the in vivo testing of SSO toxicity in rodents has become an integral process before clinical studies. However, identifying SSOs that work effectively in vivo remains a major challenge. For the N = 1 or extremely rare life-threatening variants, the limited time frame would not allow the establishment of proper genetic models or the thorough in vivo testing of SSO efficacy. For SSOs that target a specific gene, an exon, or a recurrent allele, the in vivo studies would provide valuable insights. This is exemplified by the development of nusinersen, where the SMA mouse models provide crucial tools to determine the efficacy of SSOs at the molecular and physiological levels (Monani et al., 2000; Hua et al., 2011). More recently, the Dravet mouse model (Scn1a knockout) was instrumental in demonstrating the efficacy of the SCN1A SSO in upregulating protein expression and rescuing lethality (Miller et al., 2014; Han et al., 2020). While the SCN1A lead SSO sequence is conserved and can be conveniently tested in mice, this would not necessarily be true for many other targets and SSOs. Mice carrying human genes of interest, through either BAC transgenic or humanized gene replacement, would be helpful tools to facilitate SSO studies. Recently, athymic rats carrying transplanted human cortical organoids have been reported as a new chimeric model to test the efficacy of SSOs (Chen et al., 2024).
In addition to testing SSOs in models of human diseases, the feasibility of SSO strategies can also be genetically tested for the desired splicing changes. This has been demonstrated by genetically deleting exon 5 in the CLN3 (Δex78) Batten’s disease model, where the CLN3 (Δex578) allele has been shown to restore the reading frame and suppress the sensorimotor deficits (Centa et al., 2023) (Figure 1B, bottom). The heterozygous deletion of Syngap1 A3SS-NMD has been recently shown to rescue haploinsufficiency in mice (Yang et al., 2023) (Figure 1C). These mouse genetic studies are critical to addressing questions that are otherwise hard to tackle: (1) Can the exon-skipping or NMD-suppression strategies yield the desired molecular and physiological outcome. For instance, when and how much protein upregulation can be achieved in vivo when the NMD exon is completely blocked. (2) Whether the splicing manipulation is deleterious for animal development. For the exon-skipping strategy, it is essential to know that the truncated protein would not gain toxicity or have more harmful effects than the otherwise loss-of-function allele. To treat haploinsufficiency by suppressing AS-NMD, it is crucial to understand the developmental function of the AS-NMD exons, which can be essential for brain development and functions. For example, deletion of the Bak1 AS-NMD exon in mice induces abnormal neuronal loss and perinatal lethality (Lin et al., 2020). Homozygous deletion of A3SS-NMD exon in mouse Syngap1 led to deficits in long-term potentiation (Yang et al., 2023). Furthermore, CRISPR screens in cell lines have reported that AS-NMD exons can modulate cell proliferation and survival (Thomas et al., 2020). Thus, AS-NMD exons can be essential, and completely blocking AS-NMD may have undesired consequences. (3) Whether the genetic manipulation, mimicking the maximum effect of SSO treatment, can rescue or alleviate phenotypes in mouse models of human diseases.
For developmental and progressive disorders, it is important to have an early genetic diagnosis for targeted therapy. The unprecedented identification of causal variants with exome, genome, and transcriptome analyses has set the stage for precision medicine. Genetic diagnosis takes only days to weeks and saves precious time for therapeutic development. The flexible yet specific targeting by SSOs and the clinically proven chemistry make it possible to target a particular gene, an exon, or even a unique mutation. This is achieved by suppressing cryptic splice sites, skipping specific exons, or boosting gene expression by redirecting naturally occurring alternative splicing. In addition to early-onset neurological disorders, SSOs have also been designed to target models of aging and neurodegenerative disorders (Chang et al., 2018; Korecka et al., 2019; Nikom and Zheng, 2023; Preussner et al., 2023). Furthermore, splice-modulatory small molecules are rising to treat neurological disorders such as SMA and Huntington’s disease (Palacino et al., 2015; Ratni et al., 2018; Bhattacharyya et al., 2021; Tang et al., 2021; Krach et al., 2022).
Naturally occurring alternative splicing events are potentially amenable to treating neurological disorders through different mechanisms: (1) Redirecting alternative splicing to promote the “healthier” allele. This has been demonstrated by nusinersen, which promotes SMN2 exon7 inclusion to make a stable protein. Most alternative exons (SE, A5SS, A3SS, MXE) are inframe, and pathogenic mutations within such exons can be potentially bypassed by enhancing alternative exon usage. (2) Treating haploinsufficiency by converting unproductive isoforms to functional forms. Suppression of Scn1a exon20N-NMD and Syngap1 A3SS-NMD has been shown to alleviate haploinsufficiency in mice (Han et al., 2020; Yang et al., 2023). In SCN1A, FLNA, and SNRPB cases, deleterious mutations have been reported to increase AS-NMD and cause neurodevelopmental disorders (Lynch et al., 2014; Zhang et al., 2016; Carvill et al., 2018). Such human mutations may provide insights into how AS-NMD exons are regulated. In addition to AS-NMD, retained introns can be dynamically regulated and frequently prevent the host transcript from making functional proteins (Braunschweig et al., 2014; Mauger et al., 2016). Promoting intron excision may be another way to boost protein expression. Recent studies of nascent RNAs led to the estimation that ~15% of human protein-coding transcripts are degraded through AS-NMD, suggesting a large space for gene regulation (Fair et al., 2023). Deeper transcriptomic analyses and a better understanding of the splicing code will provide new insights into splice isoform regulation and enhance the discovery of SSO targets (Gandal et al., 2018; Li et al., 2018; Bao et al., 2019; Fair et al., 2023).
The gene- and exon-specific SSOs can be applied to conceivably many patients carrying mutations in the same gene or exon, and such SSOs have been going through clinical trials to determine their toxicity and efficacy. In contrast, variant-specific SSOs are enthusiastically pursued for personalized medicine or treating extremely rare cases (Kim et al., 2019; Crooke, 2022; Aartsma-Rus et al., 2023). Such N = 1 therapy presents new challenges and necessitates new guidelines for the SSO design and preclinical testing. An emerging question is what diseases, genes, and pathogenic variants are treatable by SSOs or ASOs in general. SSOs have been estimated to target 9–15% of A-T patients (Kim et al., 2023) and a higher ratio for DMD patients (Bladen et al., 2015). A much broader group of genes and about half of the pathogenic variants have been considered druggable with ASOs and other gene-regulatory mechanisms (Mittal et al., 2022). The active research and collaborative efforts in the field are drawing a promising future for SSO therapy.
XZ: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. XZ was supported by grants from the National Institutes of Health (DP2-GM137423 and R01-MH130594).
The author would like to thank Oriane Mauger, Clémence Bernard, and Michael Kiebler for the opportunity to contribute this review; and thank Runwei Yang and other colleagues for critically reading this manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aartsma-Rus, A., Garanto, A., van Roon-Mom, W., McConnell, E. M., Suslovitch, V., Yan, W. X., et al. (2023). Consensus guidelines for the design and in vitro preclinical efficacy testing N-of-1 exon skipping antisense oligonucleotides. Nucleic Acid Ther. 33, 17–25. doi: 10.1089/nat.2022.0060
Bao, S., Moakley, D. F., and Zhang, C. (2019). The splicing code Goes deep. Cell 176, 414–416. doi: 10.1016/j.cell.2019.01.013
Barash, Y., Calarco, J. A., Gao, W., Pan, Q., Wang, X., Shai, O., et al. (2010). Deciphering the splicing code. Nature 465, 53–59. doi: 10.1038/nature09000
Barbosa-Morais, N. L., Irimia, M., Pan, Q., Xiong, H. Y., Gueroussov, S., Lee, L. J., et al. (2012). The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593. doi: 10.1126/science.1230612
Berget, S. M., Moore, C., and Sharp, P. A. (1977). Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 74, 3171–3175. doi: 10.1073/pnas.74.8.3171
Bhattacharyya, A., Trotta, C. R., Narasimhan, J., Wiedinger, K. J., Li, W., Effenberger, K. A., et al. (2021). Small molecule splicing modifiers with systemic HTT-lowering activity. Nat. Commun. 12:7299. doi: 10.1038/s41467-021-27157-z
Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336. doi: 10.1146/annurev.biochem.72.121801.161720
Bladen, C. L., Salgado, D., Monges, S., Foncuberta, M. E., Kekou, K., Kosma, K., et al. (2015). The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 36, 395–402. doi: 10.1002/humu.22758
Braunschweig, U., Barbosa-Morais, N. L., Pan, Q., Nachman, E. N., Alipanahi, B., Gonatopoulos-Pournatzis, T., et al. (2014). Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 24, 1774–1786. doi: 10.1101/gr.177790.114
Calucho, M., Bernal, S., Alias, L., March, F., Vencesla, A., Rodriguez-Alvarez, F. J., et al. (2018). Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 28, 208–215. doi: 10.1016/j.nmd.2018.01.003
Cao, Y., Liu, H., Lu, S. S., Jones, K. A., Govind, A. P., Jeyifous, O., et al. (2023). RNA-based translation activators for targeted gene upregulation. Nat. Commun. 14:6827. doi: 10.1038/s41467-023-42252-z
Carvill, G. L., Engel, K. L., Ramamurthy, A., Cochran, J. N., Roovers, J., Stamberger, H., et al. (2018). Aberrant inclusion of a poison exon causes Dravet syndrome and related SCN1A-associated genetic epilepsies. Am. J. Hum. Genet. 103, 1022–1029. doi: 10.1016/j.ajhg.2018.10.023
Centa, J. L., and Hastings, M. L. (2022). Targeting alternative splicing for therapeutic interventions. Methods Mol. Biol. 2537, 21–36. doi: 10.1007/978-1-0716-2521-7_2
Centa, J. L., Jodelka, F. M., Hinrich, A. J., Johnson, T. B., Ochaba, J., Jackson, M., et al. (2020). Therapeutic efficacy of antisense oligonucleotides in mouse models of CLN3 batten disease. Nat. Med. 26, 1444–1451. doi: 10.1038/s41591-020-0986-1
Centa, J. L., Stratton, M. P., Pratt, M. A., Osterlund Oltmanns, J. R., Wallace, D. G., Miller, S. A., et al. (2023). Protracted CLN3 batten disease in mice that genetically model an exon-skipping therapeutic approach. Mol. Ther. Nucleic Acids 33, 15–27. doi: 10.1016/j.omtn.2023.05.025
Chang, J. L., Hinrich, A. J., Roman, B., Norrbom, M., Rigo, F., Marr, R. A., et al. (2018). Targeting amyloid-beta precursor protein, APP, splicing with antisense oligonucleotides reduces toxic amyloid-beta production. Mol. Ther. 26, 1539–1551. doi: 10.1016/j.ymthe.2018.02.029
Chen, X., Birey, F., Li, M. Y., Revah, O., Levy, R., Thete, M. V., et al. (2024). Antisense oligonucleotide therapeutic approach for Timothy syndrome. Nature 628, 818–825. doi: 10.1038/s41586-024-07310-6
Chow, L. T., Gelinas, R. E., Broker, T. R., and Roberts, R. J. (1977). An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12, 1–8. doi: 10.1016/0092-8674(77)90180-5
Crooke, S. T. (2022). Addressing the needs of patients with ultra-rare mutations one patient at a time: the n-lorem approach. Nucleic Acid Ther. 32, 95–100. doi: 10.1089/nat.2021.0046
Cummings, B. B., Marshall, J. L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A. R., et al. (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 9:5209. doi: 10.1126/scitranslmed.aal5209
Dawicki-McKenna, J. M., Felix, A. J., Waxman, E. A., Cheng, C., Amado, D. A., Ranum, P. T., et al. (2023). Mapping PTBP2 binding in human brain identifies SYNGAP1 as a target for therapeutic splice switching. Nat. Commun. 14:2628. doi: 10.1038/s41467-023-38273-3
Deverman, B. E., Ravina, B. M., Bankiewicz, K. S., Paul, S. M., and Sah, D. W. Y. (2018). Gene therapy for neurological disorders: progress and prospects. Nat. Rev. Drug Discov. 17, 641–659. doi: 10.1038/nrd.2018.110
Du, L., Pollard, J. M., and Gatti, R. A. (2007). Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proc. Natl. Acad. Sci. USA 104, 6007–6012. doi: 10.1073/pnas.0608616104
Duan, D., Goemans, N., Takeda, S., Mercuri, E., and Aartsma-Rus, A. (2021). Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 7:13. doi: 10.1038/s41572-021-00248-3
Egli, M., and Manoharan, M. (2023). Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 51, 2529–2573. doi: 10.1093/nar/gkad067
England, S. B., Nicholson, L. V., Johnson, M. A., Forrest, S. M., Love, D. R., Zubrzycka-Gaarn, E. E., et al. (1990). Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343, 180–182. doi: 10.1038/343180a0
Eom, T., Zhang, C., Wang, H., Lay, K., Fak, J., Noebels, J. L., et al. (2013). NOVA-dependent regulation of cryptic NMD exons controls synaptic protein levels after seizure. eLife 2:e00178. doi: 10.7554/eLife.00178
Fair, B., Najar, C. B. A., Zhao, J., Lozano, S., Reilly, A., Mossian, G., et al. (2023). Global impact of aberrant splicing on human gene expression levels. bioRxiv.
Feng, H., Moakley, D. F., Chen, S., McKenzie, M. G., Menon, V., and Zhang, C. (2021). Complexity and graded regulation of neuronal cell-type-specific alternative splicing revealed by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 118, 1–12. doi: 10.1073/pnas.2013056118
Finkel, R. S., Mercuri, E., Darras, B. T., Connolly, A. M., Kuntz, N. L., Kirschner, J., et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732. doi: 10.1056/NEJMoa1702752
Gandal, M. J., Zhang, P., Hadjimichael, E., Walker, R. L., Chen, C., Liu, S., et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, 1–15. doi: 10.1126/science.aat8127
Graveley, B. R. (2001). Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 17, 100–107. doi: 10.1016/S0168-9525(00)02176-4
Gupta, I., Collier, P. G., Haase, B., Mahfouz, A., Joglekar, A., Floyd, T., et al. (2018). Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 36, 1197–1202. doi: 10.1038/nbt.4259
Hamdan, F. F., Gauthier, J., Spiegelman, D., Noreau, A., Yang, Y., Pellerin, S., et al. (2009). Synapse to disease, mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 360, 599–605. doi: 10.1056/NEJMoa0805392
Han, Z., Chen, C., Christiansen, A., Ji, S., Lin, Q., Anumonwo, C., et al. (2020). Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 12, 1–14. doi: 10.1126/scitranslmed.aaz6100
Helbig, I., and Abou Tayoun, A. N. (2016). Understanding genotypes and phenotypes in epileptic encephalopathies. Mol. Syndromol 7, 172–181. doi: 10.1159/000448530
Hill, S. F., and Meisler, M. H. (2021). Antisense oligonucleotide therapy for neurodevelopmental disorders. Dev. Neurosci. 43, 247–252. doi: 10.1159/000517686
Hua, Y., Sahashi, K., Hung, G., Rigo, F., Passini, M. A., Bennett, C. F., et al. (2010). Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 24, 1634–1644. doi: 10.1101/gad.1941310
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F., et al. (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. doi: 10.1038/nature10485
IBDC (1995). Isolation of a novel gene underlying batten disease, CLN3. The international batten disease consortium. Cell 82, 949–957.
Kanner, S. A., Shuja, Z., Choudhury, P., Jain, A., and Colecraft, H. M. (2020). Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 17, 1245–1253. doi: 10.1038/s41592-020-00992-6
Kim, J., Hu, C., Moufawad El Achkar, C., Black, L. E., Douville, J., Larson, A., et al. (2019). Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 381, 1644–1652. doi: 10.1056/NEJMoa1813279
Kim, J., Woo, S., de Gusmao, C. M., Zhao, B., Chin, D. H., DiDonato, R. L., et al. (2023). A framework for individualized splice-switching oligonucleotide therapy. Nature 619, 828–836. doi: 10.1038/s41586-023-06277-0
Kole, R., Krainer, A. R., and Altman, S. (2012). RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 11, 125–140. doi: 10.1038/nrd3625
Korecka, J. A., Thomas, R., Christensen, D. P., Hinrich, A. J., Ferrari, E. J., Levy, S. A., et al. (2019). Mitochondrial clearance and maturation of autophagosomes are compromised in LRRK2 G2019S familial Parkinson's disease patient fibroblasts. Hum. Mol. Genet. 28, 3232–3243. doi: 10.1093/hmg/ddz126
Krach, F., Stemick, J., Boerstler, T., Weiss, A., Lingos, I., Reischl, S., et al. (2022). An alternative splicing modulator decreases mutant HTT and improves the molecular fingerprint in Huntington's disease patient neurons. Nat. Commun. 13:6797. doi: 10.1038/s41467-022-34419-x
Kurosaki, T., Popp, M. W., and Maquat, L. E. (2019). Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 20, 406–420. doi: 10.1038/s41580-019-0126-2
Lareau, L. F., Inada, M., Green, R. E., Wengrod, J. C., and Brenner, S. E. (2007). Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446, 926–929. doi: 10.1038/nature05676
Leclair, N. K., Brugiolo, M., Urbanski, L., Lawson, S. C., Thakar, K., Yurieva, M., et al. (2020). Poison exon splicing regulates a coordinated network of SR protein Expression during differentiation and tumorigenesis. Mol. Cell 80, 648–665.e9. doi: 10.1016/j.molcel.2020.10.019
Lefebvre, S., Burglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lentz, J. J., Jodelka, F. M., Hinrich, A. J., McCaffrey, K. E., Farris, H. E., Spalitta, M. J., et al. (2013). Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 19, 345–350. doi: 10.1038/nm.3106
Lewis, B. P., Green, R. E., and Brenner, S. E. (2003). Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA 100, 189–192. doi: 10.1073/pnas.0136770100
Li, M., Santpere, G., Imamura Kawasawa, Y., Evgrafov, O. V., Gulden, F. O., Pochareddy, S., et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, 1–14. doi: 10.1126/science.aat7615
Lim, K. H., Han, Z., Jeon, H. Y., Kach, J., Jing, E., Weyn-Vanhentenryck, S., et al. (2020). Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 11:3501. doi: 10.1038/s41467-020-17093-9
Lin, L., Zhang, M., Stoilov, P., Chen, L., and Zheng, S. (2020). Developmental attenuation of neuronal apoptosis by neural-specific splicing of Bak1 microexon. Neuron 107, 1180–1196.e8. doi: 10.1016/j.neuron.2020.06.036
Lynch, D. C., Revil, T., Schwartzentruber, J., Bhoj, E. J., Innes, A. M., Lamont, R. E., et al. (2014). Disrupted auto-regulation of the spliceosomal gene SNRPB causes cerebro-costo-mandibular syndrome. Nat. Commun. 5:4483. doi: 10.1038/ncomms5483
Matsuo, M. (1996). Duchenne/Becker muscular dystrophy: from molecular diagnosis to gene therapy. Brain and Development 18, 167–172. doi: 10.1016/0387-7604(96)00007-1
Mauger, O., Lemoine, F., and Scheiffele, P. (2016). Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron 92, 1266–1278. doi: 10.1016/j.neuron.2016.11.032
Meng, L., Ward, A. J., Chun, S., Bennett, C. F., Beaudet, A. L., and Rigo, F. (2015). Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412. doi: 10.1038/nature13975
Mercuri, E., Darras, B. T., Chiriboga, C. A., Day, J. W., Campbell, C., Connolly, A. M., et al. (2018). Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635. doi: 10.1056/NEJMoa1710504
Merkin, J., Russell, C., Chen, P., and Burge, C. B. (2012). Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338, 1593–1599. doi: 10.1126/science.1228186
Michaels, W. E., Bridges, R. J., and Hastings, M. L. (2020). Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucleic Acids Res. 48, 7454–7467. doi: 10.1093/nar/gkaa490
Miller, A. R., Hawkins, N. A., McCollom, C. E., and Kearney, J. A. (2014). Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 13, 163–172. doi: 10.1111/gbb.12099
Mittal, S., Tang, I., and Gleeson, J. G. (2022). Evaluating human mutation databases for "treatability" using patient-customized therapy. Med. J. 3, 740–759. doi: 10.1016/j.medj.2022.08.006
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H., et al. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/hmg/8.7.1177
Monani, U. R., Sendtner, M., Coovert, D. D., Parsons, D. W., Andreassi, C., Le, T. T., et al. (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in SMN(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339. doi: 10.1093/hmg/9.3.333
Nikom, D., and Zheng, S. (2023). Alternative splicing in neurodegenerative disease and the promise of RNA therapies. Nat. Rev. Neurosci. 24, 457–473. doi: 10.1038/s41583-023-00717-6
Nussbacher, J. K., Tabet, R., Yeo, G. W., and Lagier-Tourenne, C. (2019). Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 102, 294–320. doi: 10.1016/j.neuron.2019.03.014
Palacino, J., Swalley, S. E., Song, C., Cheung, A. K., Shu, L., Zhang, X., et al. (2015). SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 11, 511–517. doi: 10.1038/nchembio.1837
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. doi: 10.1038/ng.259
Panagiotakos, G., Haveles, C., Arjun, A., Petrova, R., Rana, A., Portmann, T., et al. (2019). Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. eLife 8:1037. doi: 10.7554/eLife.51037
Preussner, M., Smith, H. L., Hughes, D., Zhang, M., Emmerichs, A. K., Scalzitti, S., et al. (2023). ASO targeting RBM3 temperature-controlled poison exon splicing prevents neurodegeneration in vivo. EMBO Mol. Med. 15:e17157. doi: 10.15252/emmm.202217157
Raguram, A., Banskota, S., and Liu, D. R. (2022). Therapeutic in vivo delivery of gene editing agents. Cell 185, 2806–2827. doi: 10.1016/j.cell.2022.03.045
Ramsbottom, S. A., Molinari, E., Srivastava, S., Silberman, F., Henry, C., Alkanderi, S., et al. (2018). Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model. Proc. Natl. Acad. Sci. USA 115, 12489–12494. doi: 10.1073/pnas.1809432115
Ratni, H., Ebeling, M., Baird, J., Bendels, S., Bylund, J., Chen, K. S., et al. (2018). Discovery of Risdiplam, a selective survival of motor Neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 61, 6501–6517. doi: 10.1021/acs.jmedchem.8b00741
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21. doi: 10.1038/nrneurol.2017.148
Roberts, T. C., Wood, M. J. A., and Davies, K. E. (2023). Therapeutic approaches for Duchenne muscular dystrophy. Nat. Rev. Drug Discov. 22, 917–934. doi: 10.1038/s41573-023-00775-6
Rochette, C. F., Gilbert, N., and Simard, L. R. (2001). SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 108, 255–266. doi: 10.1007/s004390100473
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., de Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. doi: 10.1016/j.cell.2019.12.036
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753. doi: 10.1126/science.7792600
Siintola, E., Topcu, M., Aula, N., Lohi, H., Minassian, B. A., Paterson, A. D., et al. (2007). The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 81, 136–146. doi: 10.1086/518902
Soemedi, R., Cygan, K. J., Rhine, C. L., Wang, J., Bulacan, C., Yang, J., et al. (2017). Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 49, 848–855. doi: 10.1038/ng.3837
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. doi: 10.1016/j.cell.2004.09.011
Tang, Z., Zhao, J., Pearson, Z. J., Boskovic, Z. V., and Wang, J. (2021). RNA-targeting splicing modifiers: drug development and screening assays. Molecules 26:2263. doi: 10.3390/molecules26082263
Teraoka, S. N., Telatar, M., Becker-Catania, S., Liang, T., Onengut, S., Tolun, A., et al. (1999). Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am. J. Hum. Genet. 64, 1617–1631. doi: 10.1086/302418
Thomas, J. D., Polaski, J. T., Feng, Q., De Neef, E. J., Hoppe, E. R., McSharry, M. V., et al. (2020). RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet. 52, 84–94. doi: 10.1038/s41588-019-0555-z
Topcu, M., Tan, H., Yalnizoglu, D., Usubutun, A., Saatci, I., Aynaci, M., et al. (2004). Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological and histopathologic studies. Turk. J. Pediatr. 46, 1–10
Van Nostrand, E. L., Freese, P., Pratt, G. A., Wang, X., Wei, X., Xiao, R., et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. doi: 10.1038/s41586-020-2077-3
Verpy, E., Leibovici, M., Zwaenepoel, I., Liu, X. Z., Gal, A., Salem, N., et al. (2000). A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies usher syndrome type 1C. Nat. Genet. 26, 51–55. doi: 10.1038/79171
Voit, T., Topaloglu, H., Straub, V., Muntoni, F., Deconinck, N., Campion, G., et al. (2014). Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 13, 987–996. doi: 10.1016/S1474-4422(14)70195-4
Wang, Z., and Burge, C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813. doi: 10.1261/rna.876308
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K., et al. (2015). The human splicing code reveals new insights into the genetic determinants of disease. Science 347:1254806. doi: 10.1126/science.1254806
Yan, Q., Weyn-Vanhentenryck, S. M., Wu, J., Sloan, S. A., Zhang, Y., Chen, K., et al. (2015). Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc. Natl. Acad. Sci. USA 112, 3445–3450. doi: 10.1073/pnas.1502849112
Yang, R., Feng, X., Arias-Cavieres, A., Mitchell, R. M., Polo, A., Hu, K., et al. (2023). Upregulation of SYNGAP1 expression in mice and human neurons by redirecting alternative splicing. Neuron 111, 1637–1650.e5. doi: 10.1016/j.neuron.2023.02.021
Yang, Y., Yang, R., Kang, B., Qian, S., He, X., and Zhang, X. (2023). Single-cell long-read sequencing in human cerebral organoids uncovers cell-type-specific and autism-associated exons. Cell Rep. 42:113335. doi: 10.1016/j.celrep.2023.113335
Zhang, Z., Bae, B., Cuddleston, W. H., and Miura, P. (2023). Coordination of alternative splicing and alternative polyadenylation revealed by targeted long read sequencing. Nat. Commun. 14:5506. doi: 10.1038/s41467-023-41207-8
Zhang, X., Chen, M. H., Wu, X., Kodani, A., Fan, J., Doan, R., et al. (2016). Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166, 1147–1162.e15. doi: 10.1016/j.cell.2016.07.025
Keywords: ASO, SSO, neurodevelopmental disorder, epilepsy, autism, alternative splicing, nonsense-mediated mRNA decay, Syngap1
Citation: Zhang X (2024) Splice-switching antisense oligonucleotides for pediatric neurological disorders. Front. Mol. Neurosci. 17:1412964. doi: 10.3389/fnmol.2024.1412964
Edited by:
Oriane Mauger, Max Planck Institute of Psychiatry, GermanyReviewed by:
Florian Heyd, Free University of Berlin, GermanyCopyright © 2024 Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaochang Zhang, eGN6aGFuZ0B1Y2hpY2Fnby5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.