 Matthew Tegowski1
Matthew Tegowski1
 Kate D. Meyer1,2*
Kate D. Meyer1,2*- 1Department of Biochemistry, Duke University School of Medicine, Durham, NC, United States
- 2Department of Neurobiology, Duke University School of Medicine, Durham, NC, United States
A major mechanism of post-transcriptional RNA regulation in cells is the addition of chemical modifications to RNA nucleosides, which contributes to nearly every aspect of the RNA life cycle. N6-methyladenosine (m6A) is a highly prevalent modification in cellular mRNAs and non-coding RNAs, and it plays important roles in the control of gene expression and cellular function. Within the brain, proper regulation of m6A is critical for neurodevelopment, learning and memory, and the response to injury, and m6A dysregulation has been implicated in a variety of neurological disorders. Thus, understanding m6A and how it is regulated in the brain is important for uncovering its roles in brain function and potentially identifying novel therapeutic pathways for human disease. Much of our knowledge of m6A has been driven by technical advances in the ability to map and quantify m6A sites. Here, we review current technologies for characterizing m6A and highlight emerging methods. We discuss the advantages and limitations of current tools as well as major challenges going forward, and we provide our perspective on how continued developments in this area can propel our understanding of m6A in the brain and its role in brain disease.
Introduction
RNAs contain over 170 distinct chemical modifications which play important roles in regulating RNA processing and function. Although most of these modifications occur in non-coding RNAs such as ribosomal RNA and tRNA, recent studies have revealed a diverse and dynamic “epitranscriptome” within cellular mRNAs as well. N6-methyladenosine (m6A) is the most abundant internal mRNA modification and is found in thousands of cellular mRNAs, in addition to a large number of non-coding RNAs. m6A plays important roles in several RNA processing events, including splicing, nuclear export, stability, and translation, making it a critical regulator of gene expression in cells (Murakami and Jaffrey, 2022; Flamand et al., 2023). Indeed, m6A contributes to a wide variety of physiological processes, including development, innate immunity, gametogenesis, and the cellular stress response. Additionally, and consistent with its importance for cellular function, m6A dysregulation has been implicated in a variety of human diseases, including several cancers (Yang et al., 2020; He and He, 2023). Thus, understanding m6A distribution, regulation, and function is critical for advancing our knowledge of human health and disease.
Within the brain, m6A levels are particularly abundant compared to other tissues (Meyer et al., 2012; Liu et al., 2020), and proper regulation of m6A is critical for processes such as neural stem cell function, brain development, learning and memory, response to stress, and neuronal signaling (Flamand and Meyer, 2019; Livneh et al., 2020).
Our current knowledge of m6A has been accelerated by technological advances which have enabled the identification of m6A sites transcriptome-wide. Additionally, emerging technologies for targeted m6A manipulation in select RNAs are promising tools that can enable functional studies of m6A in the brain and other tissues. Here, we review m6A detection and manipulation strategies and discuss major challenges that need to be overcome. We also provide our perspective on future directions and areas that are likely to drive the field forward.
m6A function and regulation in the brain
m6A is deposited in the nucleus co-transcriptionally by a large methyltransferase complex which includes METTL3 as the catalytic subunit and several additional accessory proteins including METTL14, WTAP, HAKAI, VIRMA, ZC3H13, and RBM15/15B (Shi et al., 2019; Zaccara et al., 2019; Flamand et al., 2023). Methylation occurs preferentially within the DRACH (D = A, G, U; R = A, G; H = A, C, U) consensus sequence, and recent studies have revealed that sequence specificity and gene architecture are the major determinants of methylation within cellular mRNAs (Garcia-Campos et al., 2019; Yang et al., 2022; He et al., 2023; Uzonyi et al., 2023). In addition, m6A can be removed by two eraser proteins, FTO and ALKBH5, which can contribute to dynamic regulation of m6A and gene expression under certain contexts (Shi et al., 2019; Flamand et al., 2023).
m6A has been shown to influence nearly every aspect of the RNA life cycle, including splicing, export, stability, localization, and translation (Shi et al., 2019; Zaccara et al., 2019; Flamand et al., 2023). However, the most well-established function of m6A in mRNAs is to recruit RNA degradation machinery through the binding of YTHDF proteins (Shi et al., 2019; Zaccara et al., 2019; Kontur et al., 2020; Zaccara and Jaffrey, 2020; Flamand et al., 2023). This m6A-dependent control of mRNA stability is critical for proper brain development, as this mechanism helps regulate the abundance of mRNAs that participate in neuronal stem cell function and cell cycle regulation (Yoon et al., 2017; Wang et al., 2018). m6A has also been shown to regulate mRNA metabolism in the brain in other ways, including promoting translation and nuclear export (Shi et al., 2019; Zaccara et al., 2019; Flamand et al., 2023). These functions are mediated by a variety of m6A reader proteins. For instance, YTHDF1 promotes methylated mRNA translation in neurons to control synaptic activity and learning and memory (Shi et al., 2018; Zou et al., 2023), and YTHDF2 promotes the differentiation of neural progenitors by degrading methylated transcripts (Li et al., 2018). The fragile X messenger ribonucleoprotein (FMRP) has been shown to preferentially bind methylated transcripts and facilitate their nuclear export (Edens et al., 2019). Additionally, our group identified RBM45 as a brain-enriched m6A reader protein that can impact splicing and regulate neuronal differentiation (Choi et al., 2022).
In addition to cortical development and neurogenesis, m6A also has important roles in regulating the function of mature neurons. Neurons are highly polarized cells, with complex dendritic processes that can make thousands of synaptic connections with other neurons. Proper synaptic function and plasticity requires the trafficking and local translation of mRNAs to synapses in an activity-dependent manner (Doyle and Kiebler, 2011; Holt et al., 2019; Roy et al., 2020). RNA localization is mediated by a variety of cis-acting elements, such as sequence and structure, which are bound by RNA-binding effector proteins (Doyle and Kiebler, 2011). m6A profiling of synaptic RNAs showed that several methylated transcripts are localized at synapses, suggesting that m6A could serve as an additional cis-acting element to control RNA localization in neurons (Merkurjev et al., 2018). Indeed, subsequent work from our group showed that hundreds of transcripts, including many that encode proteins important for synaptic maintenance and plasticity, are localized to distal processes in neurons in an m6A-dependent manner (Flamand and Meyer, 2022). We further showed that this is mediated through YTHDF proteins. However, why some methylated transcripts are degraded by YTHDF proteins while others are transported to distal processes is unknown, and it likely depends on other context-dependent factors, such as additional sequence and structural elements and interactions with other RBPs.
Interestingly, a recent study showed that mRNA stability is a major determinant of mRNA localization in neurons, with more stable transcripts being enriched in neurites (Loedige et al., 2023). The authors reported that neurite-enriched RNAs have lower levels of m6A, and they found that disrupting m6A or other factors that control RNA stability promotes neurite enrichment of neuronal transcripts. These studies examined m6A-mediated localization in primary cortical neurons, in contrast to hippocampal neurons used in our work, so it is possible that m6A may have unique roles in different neuronal subtypes. However, even within hippocampal neurons, we identified many transcripts with increased neurite localization following Mettl3 depletion in addition to the hundreds of transcripts that showed decreased neurite localization (Flamand and Meyer, 2022). Thus, the effects of m6A on RNA localization may be transcript-specific. Further studies will be necessary for defining the cell type-and transcript-dependent effects of m6A on RNA localization in the brain.
In addition to RNA localization, recent work has demonstrated that m6A promotes local, activity-dependent translation of mRNAs in hippocampal neurons. This process is mediated by YTHDF1, which is required in hippocampal neurons for proper learning and memory (Shi et al., 2018). Supporting these data, deletion of Mettl3 in the mouse hippocampus also leads to impaired learning and memory (Zhang et al., 2018). Furthermore, a mechanism by which YTHDF1 can promote activity-dependent translation in the hippocampus has been uncovered. Basal interactions between FMRP and YTHDF1 sequester YTHDF1. However, FMRP is phosphorylated upon neuronal activity, resulting the release of YTHDF1, allowing it to promote the translation of methylated transcripts (Zou et al., 2023). Altogether, m6A has been shown to regulate neuronal development and function by regulating RNA stability, localization, and translation.
Current methods and recent advances in m6A mapping
The first method for transcriptome-wide m6A mapping was developed in 2012 and involved using m6A antibodies to immunoprecipitate methylated RNAs followed by next-generation sequencing to identify the methylated targets (Dominissini et al., 2012; Meyer et al., 2012). This method, called MeRIP-seq or m6A-seq, has been widely used to globally profile m6A across a variety of tissues, cell types, and conditions, and it continues to be the predominant method used in most studies. Improvements to the technique have enabled single-nucleotide resolution m6A mapping (miCLIP and m6A-CLIP) (Linder et al., 2015; Ke et al., 2017), provided profiles of m6A within individual RNA isoforms (m6A-LAIC-seq) (Molinie et al., 2016), and reduced the RNA input requirements through more efficient library preparation (Zeng et al., 2018; Dierks et al., 2021).
Although widely used, antibody-based m6A mapping methods have their drawbacks. This includes cross-reactivity of m6A antibodies with m6Am, a chemically similar modification that is part of the 5′ cap structure. In addition, m6A site calling can be stochastic due to variability in antibody immunoprecipitation efficiency, and most studies lack sufficient replicate numbers to make accurate site calls (McIntyre et al., 2020). Furthermore, most global m6A mapping strategies lack the ability to quantify m6A stoichiometry. This has made studies of m6A dynamics difficult and has contributed to discrepancies in the literature regarding how m6A responds to cellular stress and other states.
Recently, two methods for simultaneous m6A mapping and quantification have overcome this problem. GLORI uses nitrous acid to deaminate unmodified A to I while leaving m6A unchanged. This results in unmodified A being read as G in sequencing reads, with m6A remaining as A (Liu et al., 2023). eTAM-seq similarly relies on exclusive deamination of unmodified A, but it does so through an evolved TadA8.20 enzyme which selectively targets unmodified A (Xiao et al., 2023). Both methods offer a simple approach for identifying m6A with nucleotide specificity, and they have the added advantage of being able to measure m6A stoichiometry transcriptome-wide. Further improvements to GLORI and eTAM-seq to limit RNA degradation will facilitate more widespread use of these methods and will help pave the way for their potential use in single-cell m6A mapping (below). Additionally, several other antibody-independent m6A profiling methods have been developed in recent years (reviewed in Owens et al., 2021). These approaches employ a variety of different strategies, including the use of methionine analogs to label m6A sites (m6A-label-seq) (Shu et al., 2020), chemical labeling of FTO-directed m6A demethylation intermediates (m6A-SEAL) (Wang et al., 2020), and treating RNA with modification-sensitive endoribonucleases (MAZTER-seq and m6A-REF-seq) (Garcia-Campos et al., 2019; Chen et al., 2022). Strategies for site-specific m6A quantification in RNAs of interest have also been developed, which serve as useful tools for investigating m6A within individual transcripts and/or validating the results of global m6A mapping for a subset of RNAs (Liu et al., 2013; Xiao et al., 2018; Castellanos-Rubio et al., 2019).
In addition to antibody-based, enzyme-assisted, and biochemical methods for m6A mapping, nanopore sequencing has emerged as a technology with great promise for profiling m6A and other RNA modifications. This direct RNA sequencing method involves driving RNAs through a protein nanopore and measuring the variations in ionic current that occur as different nucleotides pass through the pore (Garalde et al., 2018) (Figure 1). Chemical modifications in RNAs can alter the current intensity or dwell time of the RNA as it moves through the pore, and these unique signatures can then be used to detect the presence of modifications (Jain et al., 2022). Several studies have demonstrated the ability of nanopore technology to call m6A sites (Zhong et al., 2023). A key advantage of this approach is that native, full-length RNA molecules can be sequenced, therefore enabling a deeper understanding of m6A distribution within distinct transcript isoforms, the presence of m6A clusters in single RNA molecules, and potential co-occurrence of m6A with other modifications (Leger et al., 2021; Huang et al., 2024; Mateos et al., 2024).
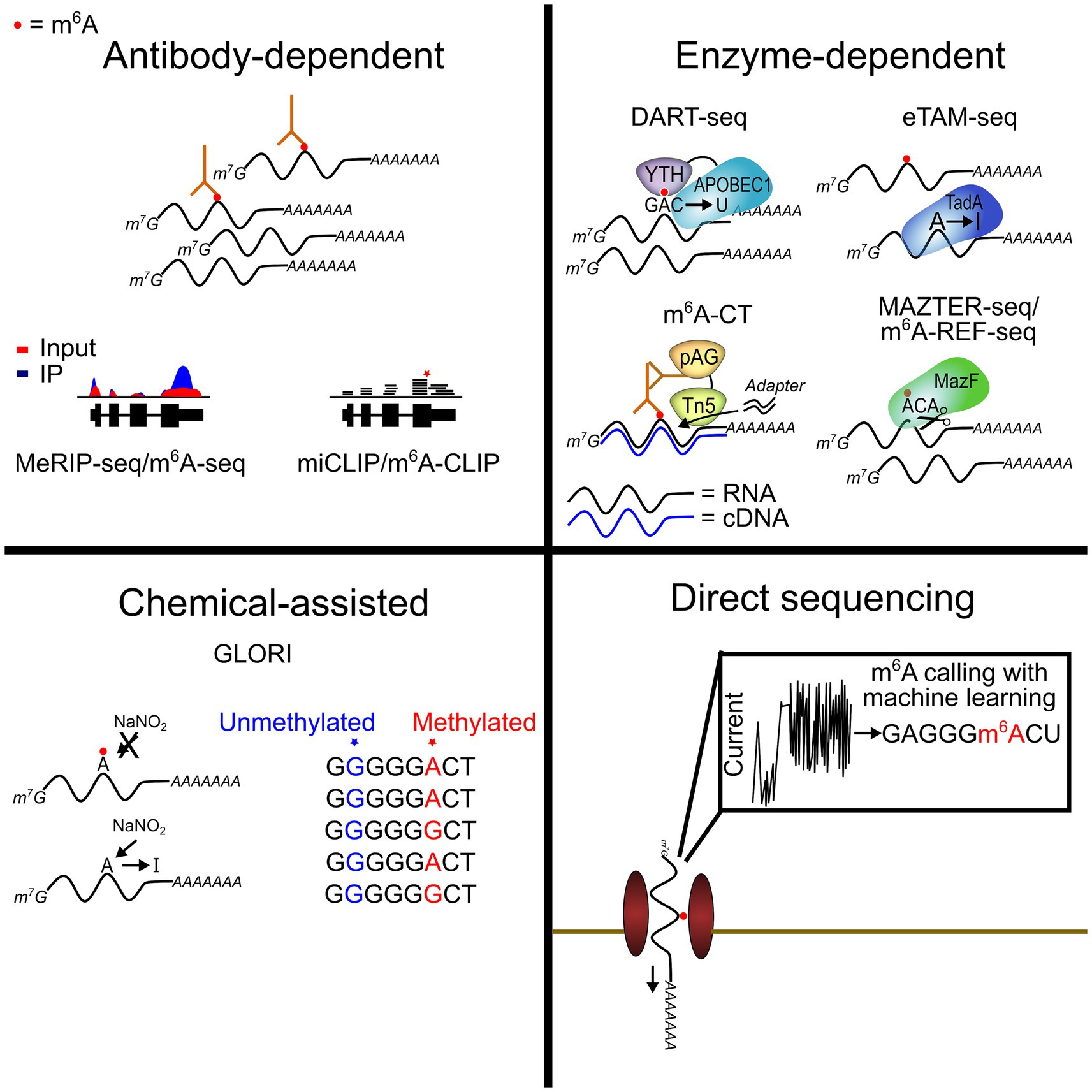
Figure 1. Current transcriptome-wide m6A profiling methods. Recent advances have provided new techniques for m6A profiling across the transcriptome. Some of these methods allow for highly quantitative detection of m6A sites at single-nucleotide resolution (GLORI and eTAM-seq). Others have facilitated the use of low-input samples, including single cells (DART-seq and m6A-CT).
Mapping m6A in single cells
The brain is a complex mixture of diverse cell types. However, all m6A profiling studies done in the brain thus far have used bulk tissue samples, which represent the cumulative m6A signal across different cell types and provide no information on the distribution or abundance of m6A within individual cells. By mapping methylated transcripts in single cells, the methylomes of all cell types can be elucidated, which would provide unprecedented insights into how m6A contributes to brain function and disease through influencing gene expression in distinct cell types.
Several approaches have recently been developed to achieve single-cell m6A profiling. Some studies have used m6A antibodies to perform a low-input MeRIP-seq from single cells (Li et al., 2023; Yao et al., 2023). These methods can identify methylated transcripts from individual cells, but they have some drawbacks. First, the high signal-to-noise ratio resulting from antibody enrichment complicates peak calling, especially when using low input samples. Second, these methods are generally not highly scalable and have profiled m6A in a few dozen cells at most (Li et al., 2023; Yao et al., 2023). The recent development of single-nucleus m6A-CUT&Tag (sn-m6A-CT) addresses these issues by coupling antibody-based methylated RNA enrichment with Tn5 transposase-mediated tagmentation (Hamashima et al., 2023). While this method still relies on m6A antibodies, it improves signal-to-noise relative to strategies based on immunoprecipitation alone and can be used in any cell type or tissue of interest. Furthermore, it is amenable to droplet-based library preparation methods, making it a truly high-throughput technique.
Antibody-independent strategies for single-cell m6A profiling have also been developed. In 2022, our group introduced single-cell DART-seq (scDART-seq), which installs a unique mutation signature adjacent to m6A sites (Meyer, 2019; Tegowski et al., 2022). This is achieved by expressing a fusion protein consisting of the m6A-binding YTH domain tethered to the cytidine deaminase APOBEC1 (Meyer, 2019). The YTH domain recruits the fusion protein to sites of methylation while APOBEC1 edits nearby cytidines to uridines, enabling m6A sites to be identified as C-to-T mutations in the sequencing data. This method is compatible with any scRNA-seq preparation method and does not require additional RNA processing steps, making it a highly scalable strategy which is straightforward to implement (Figure 1). However, one limitation of scDART-seq is that it requires expressing the APOBEC1-YTH protein in cells of interest. This is easy to do in many cultured cell types but can be more challenging for certain tissues. Furthermore, expression of APOBEC1-YTH could also influence cell biology if expression is prolonged (Tegowski et al., 2022).
Emerging technologies for studying m6A at the single-molecule level
Most methods for m6A mapping rely on short read sequencing. Although these techniques can reveal m6A sites, they are unable to describe how these sites are distributed on individual RNA molecules. For example, many RNAs have multiple m6A sites, but whether these sites co-occur on the same RNA molecules is unknown. In addition, the distribution of m6A within distinct transcript isoforms is often difficult to assess when only a short fragment of the parent RNA is sequenced. Exploring methylation at the single-molecule level can help address these important questions.
As discussed above, nanopore sequencing has emerged as a technology with great promise for profiling m6A and other RNA modifications. This strategy provides information on full-length RNA molecules, which enables greater insight into the presence of modifications in splice variants or other RNA isoforms. Additionally, since RNA molecules are sequenced directly, potential biases introduced during cDNA synthesis and PCR amplification steps are avoided. However, nanopore-based RNA modification sequencing has some limitations. First, identification of modification sites requires the use of machine learning algorithms trained on datasets to enable de novo modification site calls, or the use of modification-free control samples to enable modification detection by comparative analysis (Hendra et al., 2022; Jain et al., 2022). For m6A, several computational tools have been developed for identifying methylated sites from nanopore data, with substantial variations in called sites and estimated accuracy (Zhong et al., 2023). More fundamentally, training m6A calling algorithms requires a known “ground-truth,” which can be difficult to know with certainty in all model systems.
In addition to DRS technologies such as nanopore sequencing, other methods exist that enable m6A identification in individual RNA molecules. For instance, DART-seq has been used with PacBio sequencing, which has enabled the identification of m6A sites along the full length of individual mRNAs (Meyer, 2019). In theory, other methods that induce m6A-associated mutations, such as eTAM-seq and GLORI, could also be combined with long-read sequencing to explore m6A on single molecules. However, these approaches have not been widely used, and given the rapid developments in nanopore technology, DRS will likely emerge as the method of choice for single-molecule m6A mapping.
In addition to sequencing-based approaches, other methods have been developed that enable analysis of individual methylated RNA molecules in cells. m6AISH-PLA uses proximity ligation between an m6A-recognizing antibody and a sequence-specific oligo targeted to sequences flanking the m6A site of interest. After ligation, rolling circle amplification (RCA) amplifies an engineered sequence recognized by a fluorescent detection probe (Ren et al., 2021). This method allows for the visualization of single methylated molecules in situ, facilitating novel investigations into m6A-mediated RNA localization and trafficking. One drawback to this method is that it cannot simultaneously visualize unmethylated transcripts, which could lead to misinterpretations if both methylated and unmethylated RNAs are similarly trafficked. However, an adaptation of the DART-seq technology, termed DART-FISH, can detect methylated and unmethylated transcripts (Sheehan et al., 2023). By expressing the APOBEC1-YTH enzyme in cells, transcripts with m6A-dependent C-to-U mutations can be discriminated from unmodified transcripts using padlock probe hybridization followed by RCA and hybridization of detection probes. By using distinct padlock probes for the C and U variants adjacent to an m6A site of interest, the unmethylated and methylated copies of an individual transcript can be visualized simultaneously. Since m6A has been implicated in subcellular RNA localization, approaches such as these which enable in situ visualization of m6A-modified transcripts can be powerful approaches for understanding the role of m6A in RNA trafficking or partitioning to subcellular compartments such as stress granules (Anders et al., 2018; Fu and Zhuang, 2020; Khong et al., 2022; Ries et al., 2023).
Strategies for targeted m6A manipulation and m6A-dependent gene expression control
Several groups have developed tools for targeted addition or removal of m6A in cellular RNAs of interest. These methods involve fusing m6A methyltransferase or demethylase enzymes to catalytically inactive Cas proteins coupled with guide RNA (gRNA)-mediated targeting of specific transcripts. For instance, Wilson et al. fused METTL3/14 to dCas13 to achieve site-specific methylation of several cellular mRNAs, including GAPDH, FOXM1, and SOX2. In addition, they showed that targeted methylation of ACTB led to transcript degradation and that methylation of the BRD8 and ZNF638 transcripts impacted splicing, consistent with previous reports of m6A function in these mRNAs (Wilson et al., 2020). Li et al. showed that ALKBH5 tethered to dCas13b can remove m6A from oncogenic transcripts EGFR and MYC in the presence of transcript-targeting gRNAs, leading to decreased protein expression and reduced cell proliferation (Li et al., 2020). Tethering of dCas9 to m6A methyltransferases and demethylases has also been used to achieve targeted m6A writing and erasing, respectively (Liu et al., 2019). Collectively, these tools have utility not only for basic research into m6A function but also as a potential therapeutic strategy to overcome the effects of hyper or hypomethylation during disease. Current challenges include minimizing off-targeting effects to ensure transcript specificity and optimization of methylation and demethylation efficiency. However, the use of CRISPR/Cas-based technologies for targeting RNA has accelerated at a rapid pace, and as these and other methods continue to expand, we anticipate that the tools for manipulating m6A and other RNA modifications will also improve. Indeed, these methods have already been expanded to include light-activated m6A modification systems which add temporal specificity (Lan et al., 2021; Shi et al., 2022).
The tools above use targeted manipulation of m6A levels in specific RNAs to control the expression of genes of interest. This holds promise as a potential therapeutic strategy, since m6A dysregulation can lead to abnormal expression of specific genes to promote the pathogenesis of cancer and other diseases (Yang et al., 2020; Jiang et al., 2021; Delaunay et al., 2024). However, an alternative approach is to couple the presence of m6A with the expression of desired proteins. Recently, our group developed a genetically encoded m6A sensor system (GEMS), which couples mRNA methylation with expression of a protein of interest (Marayati et al., 2024). This is achieved by expressing a reporter mRNA together with APOBEC1-YTH in cells. The reporter mRNA contains an m6A sensor sequence that, when methylated, recruits APOBEC1-YTH to convert nearby cytidines to uridines, in turn generating one or more stop codons that block translation of a degradation tag after the coding sequence of the protein of interest. The result is stable protein production only when the mRNA is methylated. We used this system to achieve m6A-coupled expression of tumor suppressor proteins in cancer cells, which led to decreased cell proliferation and migration (Marayati et al., 2024). Although m6A-coupled protein expression technologies such as this still require further optimization, the ability to sense m6A in living cells offers an attractive platform both for methylation-sensitive protein expression as well as for studies of m6A dynamics in the brain and other tissues.
Discussion
Much of our understanding of m6A in the brain has been driven by recent advances in m6A mapping technologies. These tools have not only enabled the identification of methylated RNAs within the brain and other tissues but have also provided a deeper understanding of m6A dynamics and function. Although antibody-based methods have been the predominant method of choice for transcriptome-wide m6A mapping, newer approaches have emerged in the last few years which overcome many of the limitations of antibody-based approaches. For instance, GLORI and eTAM-seq offer not only nucleotide-resolution m6A mapping, but they also enable quantification of m6A stoichiometry. The ability to measure changes in m6A abundance is an important advance, since methods for reliable, sensitive quantification of m6A stoichiometry transcriptome-wide have been largely elusive, which has contributed to discrepancies regarding the dynamic nature of m6A. Although GLORI and eTAM-seq have great potential for becoming the new gold standard of m6A mapping and quantification, further refinements of these methods to improve sensitivity and reduce RNA degradation will be needed for their widespread adoption.
Direct RNA sequencing with nanopore technology also holds great promise for enabling m6A identification at the single-molecule level and within different transcript isoforms. Additionally, nanopore sequencing can potentially be used to identify multiple modifications within a single RNA molecule, which is an area that we currently have little knowledge about. However, achieving these goals will require improved throughput and accuracy, as well as establishment of consistent data analysis pipelines and appropriate training datasets. Nevertheless, rapid progress is being made in nanopore-based modification mapping, so we anticipate that this technology will become increasingly widespread in the coming years.
The ability to map m6A in single cells is an important step forward for deepening our understanding of m6A regulation and function. The recent development of scDART-seq (Tegowski et al., 2022), scm6A-seq (Yao et al., 2023), picoMeRIP-seq (Li et al., 2023), and single-nucleus m6A-CUT&Tag (sn-m6A-CT) (Hamashima et al., 2023) have been critical advances and have revealed new insights into m6A distribution and regulation within individual cells of a population. Applying single-cell m6A mapping methods to the brain will undoubtedly uncover new information about m6A dynamics and regulation within distinct brain cell types. In particular, our understanding of m6A function in non-neuronal cells is limited, so such studies will greatly facilitate future discoveries in this area.
Going forward, it will be important to further develop single-cell m6A mapping technologies to enable their widespread use across cell or tissue types of interest. Additionally, methods such as GLORI or eTAM-seq may be promising antibody-independent strategies for single-cell m6A mapping, but their sensitivity for low-input RNA must be further developed, and their propensity to induce RNA degradation must be addressed. Nevertheless, this is an exciting time for m6A mapping technology in single cells, with a few tools already available and further developments undoubtedly on the horizon. Having the ability to combine m6A mapping with other single-cell “omics” technologies will be very powerful for furthering our understanding of the interplay between m6A and other gene regulatory processes such as chromatin remodeling, transcription regulation, and RNA processing events.
In addition to technologies for mapping and quantifying m6A, there are emerging tools for targeted manipulation of m6A which can achieve selective methylation or demethylation of RNAs of interest. The ability to selectively add or remove m6A from RNAs is a useful tool for investigating m6A function. However, one consideration is that m6A sites cluster in RNAs, and recent studies have indicated that cellular RNAs contain many more m6A sites than previously thought (Tegowski et al., 2022; Liu et al., 2023). Thus, the effects of adding or removing a single m6A site may be compensated for through methylation of other nearby adenosines within a given region of methylation. This is also an important consideration when developing m6A targeting tools for therapeutic applications, as multiple m6A sites may exist at nearby positions in a transcript of interest. However, methylating or demethylating single sites has been shown to impact RNA expression in cells (Liu et al., 2019; Li et al., 2020; Wilson et al., 2020), suggesting that compensation by nearby m6A sites does not happen for all RNAs. It is also possible that the individual m6A sites that make up methylation “clusters” occur on different RNA molecules, which would make compensation by nearby adenosines less likely. Most m6A profiling strategies do not report the individual RNA molecules in which m6A sites reside, underscoring the need to develop better tools for single-molecule m6A mapping.
Going forward, it will be important for the field to address issues related to sensitivity and reproducibility of methods for studying m6A. Newer technologies such as GLORI and eTAM-seq that enable high-resolution m6A mapping as well as quantification can potentially enable better insights into m6A dynamics, since many m6A sites may be regulated by changes in abundance as opposed to strict gain or loss of methylation. Additionally, the recent development of tools for sensing m6A provide new opportunities for studying m6A dynamics in living cells, in contrast to other methods that require RNA isolation. Our understanding of how m6A is regulated within the brain during both healthy and disease states will undoubtedly be accelerated by the ability to map and quantify m6A within the brain and in specific cell types. Thus, further development of single-cell m6A profiling approaches will be important. Finally, nanopore sequencing or other methods that provide single-molecule information have the potential to provide deeper insights into roles of m6A in distinct transcript isoforms, as well as the possibility of multiple different modifications co-occurring on the same RNAs. We anticipate that continued development of these methods in the coming years will make them more widely used for studies of m6A in the brain.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
MT: Writing – original draft, Writing – review & editing. KM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (1DP1DA046584, 1R01MH118366, and 1RM1HG011563 to KM).
Acknowledgments
We thank Dr. Clémence Bernard, Dr. Michael Kiebler, and Dr. Oriane Mauger for giving us the opportunity to write this perspective review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anders, M., Chelysheva, I., Goebel, I., Trenkner, T., Zhou, J., Mao, Y., et al. (2018). Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Sci. Alliance 1:e201800113. doi: 10.26508/lsa.201800113
Castellanos-Rubio, A., Santin, I., Olazagoitia-Garmendia, A., Romero-Garmendia, I., Jauregi-Miguel, A., Legarda, M., et al. (2019). A novel RT-QPCR-based assay for the relative quantification of residue specific m6A RNA methylation. Sci. Rep. 9, 1–7. doi: 10.1038/s41598-019-40018-6
Chen, H.-X., Zhang, Z., Ma, D.-Z., Chen, L.-Q., and Luo, G.-Z. (2022). Mapping single-nucleotide m6A by m6A-REF-seq. Methods 203, 392–398. doi: 10.1016/j.ymeth.2021.06.013
Choi, S. H., Flamand, M. N., Liu, B., Zhu, H., Hu, M., Wang, M., et al. (2022). RBM45 is an m6A-binding protein that affects neuronal differentiation and the splicing of a subset of mRNAs. Cell Rep. 40:111293. doi: 10.1016/j.celrep.2022.111293
Delaunay, S., Helm, M., and Frye, M. (2024). RNA modifications in physiology and disease: towards clinical applications. Nat. Rev. Genet. 25, 104–122. doi: 10.1038/s41576-023-00645-2
Dierks, D., Garcia-Campos, M. A., Uzonyi, A., Safra, M., Edelheit, S., Rossi, A., et al. (2021). Multiplexed profiling facilitates robust m6A quantification at site, gene and sample resolution. Nat. Methods 18, 1060–1067. doi: 10.1038/s41592-021-01242-z
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. doi: 10.1038/nature11112
Doyle, M., and Kiebler, M. A. (2011). Mechanisms of dendritic mRNA transport and its role in synaptic tagging. EMBO J. 30, 3540–3552. doi: 10.1038/emboj.2011.278
Edens, B. M., Vissers, C., Su, J., Arumugam, S., Xu, Z., Shi, H., et al. (2019). FMRP modulates neural differentiation through m6A-dependent mRNA nuclear export. Cell Rep. 28, 845–854.e5. doi: 10.1016/J.CELREP.2019.06.072
Flamand, M. N., and Meyer, K. D. (2019). The epitranscriptome and synaptic plasticity. Curr. Opin. Neurobiol. 59, 41–48. doi: 10.1016/J.CONB.2019.04.007
Flamand, M. N., and Meyer, K. D. (2022). m6A and YTHDF proteins contribute to the localization of select neuronal mRNAs. Nucleic Acids Res. 50, 4464–4483. doi: 10.1093/nar/gkac251
Flamand, M. N., Tegowski, M., and Meyer, K. D. (2023). The proteins of mRNA modification: writers, readers, and erasers. Annu. Rev. Biochem. 92, 145–173. doi: 10.1146/annurev-biochem-052521-035330
Fu, Y., and Zhuang, X. (2020). m6A-binding YTHDF proteins promote stress granule formation. Nat. Chem. Biol. 16, 955–963. doi: 10.1038/s41589-020-0524-y
Garalde, D. R., Snell, E. A., Jachimowicz, D., Sipos, B., Lloyd, J. H., Bruce, M., et al. (2018). Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206. doi: 10.1038/nmeth.4577
Garcia-Campos, M. A., Edelheit, S., Toth, U., Safra, M., Shachar, R., Viukov, S., et al. (2019). Deciphering the “m6A code” via antibody-independent quantitative profiling. Cell 178, 731–747.e16. doi: 10.1016/J.CELL.2019.06.013
Hamashima, K., Wong, K. W., Sam, T. W., Teo, J. H. J., Taneja, R., Le, M. T. N., et al. (2023). Single-nucleus multiomic mapping of m6A methylomes and transcriptomes in native populations of cells with sn-m6A-CT. Mol. Cell 83, 3205–3216.e5. doi: 10.1016/j.molcel.2023.08.010
He, P. C., and He, C. (2023). mRNA accessibility within mRNPs as a determinant of gene expression. Trends Biochem. Sci. 49, 199–207. doi: 10.1016/j.tibs.2023.11.003
He, P. C., Wei, J., Dou, X., Harada, B. T., Zhang, Z., Ge, R., et al. (2023). Exon architecture controls mRNA m6A suppression and gene expression. Science 379, 677–682. doi: 10.1126/science.abj9090
Hendra, C., Pratanwanich, P. N., Wan, Y. K., Goh, W. S. S., Thiery, A., and Göke, J. (2022). Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat. Methods 19, 1590–1598. doi: 10.1038/s41592-022-01666-1
Holt, C. E., Martin, K. C., and Schuman, E. M. (2019). Local translation in neurons: visualization and function. Nat. Struct. Mol. Biol. 26, 557–566. doi: 10.1038/s41594-019-0263-5
Huang, S., Wylder, A. C., and Pan, T. (2024). Simultaneous nanopore profiling of mRNA m6A and pseudouridine reveals translation coordination. Nat. Biotechnol. 42, 1–5. doi: 10.1038/s41587-024-02135-0
Jain, M., Abu-Shumays, R., Olsen, H. E., and Akeson, M. (2022). Advances in nanopore direct RNA sequencing. Nat. Methods 19, 1160–1164. doi: 10.1038/s41592-022-01633-w
Jiang, X., Liu, B., Nie, Z., Duan, L., Xiong, Q., Jin, Z., et al. (2021). The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 6, 1–16. doi: 10.1038/s41392-020-00450-x
Ke, S., Pandya-Jones, A., Saito, Y., Fak, J. J., Vågbø, C. B., Geula, S., et al. (2017). m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006. doi: 10.1101/gad.301036.117
Khong, A., Matheny, T., Huynh, T. N., Babl, V., and Parker, R. (2022). Limited effects of m6A modification on mRNA partitioning into stress granules. Nat. Commun. 13:3735. doi: 10.1038/s41467-022-31358-5
Kontur, C., Jeong, M., Cifuentes, D., and Giraldez, A. J. (2020). Ythdf m6A readers function redundantly during zebrafish development. Cell Rep. 33:108598. doi: 10.1016/j.celrep.2020.108598
Lan, L., Sun, Y.-J., Jin, X.-Y., Xie, L.-J., Liu, L., and Cheng, L. (2021). A light-controllable chemical modulation of m6A RNA methylation. Angew. Chem. Int. Ed. 60, 18116–18121. doi: 10.1002/anie.202103854
Leger, A., Amaral, P. P., Pandolfini, L., Capitanchik, C., Capraro, F., Miano, V., et al. (2021). RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 12:7198. doi: 10.1038/s41467-021-27393-3
Li, J., Chen, Z., Chen, F., Xie, G., Ling, Y., Peng, Y., et al. (2020). Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res. 48, 5684–5694. doi: 10.1093/nar/gkaa269
Li, Y., Wang, Y., Vera-Rodriguez, M., Lindeman, L. C., Skuggen, L. E., Rasmussen, E. M. K., et al. (2023). Single-cell m6A mapping in vivo using picoMeRIP–seq. Nat. Biotechnol. 41, 1–6. doi: 10.1038/s41587-023-01831-7
Li, M., Zhao, X., Wang, W., Shi, H., Pan, Q., Lu, Z., et al. (2018). Ythdf2-mediated m6A mRNA clearance modulates neural development in mice. Genome Biol. 19, 1–16. doi: 10.1186/S13059-018-1436-Y/TABLES/1
Linder, B., Grozhik, A. V., Olarerin-George, A. O., Meydan, C., Mason, C. E., and Jaffrey, S. R. (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. doi: 10.1038/nmeth.3453
Liu, J., Li, K., Cai, J., Zhang, M., Zhang, X., Xiong, X., et al. (2020). Landscape and regulation of m6A and m6Am Methylome across human and mouse tissues. Mol. Cell 77, 426–440.e6. doi: 10.1016/J.MOLCEL.2019.09.032
Liu, N., Parisien, M., Dai, Q., Zheng, G., He, C., and Pan, T. (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. doi: 10.1261/rna.041178.113
Liu, C., Sun, H., Yi, Y., Shen, W., Li, K., Xiao, Y., et al. (2023). Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nat. Biotechnol. 41, 355–366. doi: 10.1038/s41587-022-01487-9
Liu, X.-M., Zhou, J., Mao, Y., Ji, Q., and Qian, S.-B. (2019). Programmable RNA N6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol. 15, 865–871. doi: 10.1038/s41589-019-0327-1
Livneh, I., Moshitch-Moshkovitz, S., Amariglio, N., Rechavi, G., and Dominissini, D. (2020). The m6A epitranscriptome: transcriptome plasticity in brain development and function. Nat. Rev. Neurosci. 21, 36–51. doi: 10.1038/s41583-019-0244-z
Loedige, I., Baranovskii, A., Mendonsa, S., Dantsuji, S., Popitsch, N., Breimann, L., et al. (2023). mRNA stability and m6A are major determinants of subcellular mRNA localization in neurons. Mol. Cell 83, 2709–2725.e10. doi: 10.1016/j.molcel.2023.06.021
Marayati, B. F., Thompson, M. G., Holley, C. L., Horner, S. M., and Meyer, K. D. (2024). Programmable protein expression using a genetically encoded m6A sensor. Nat. Biotechnol. 42, 1–12. doi: 10.1038/s41587-023-01978-3
Mateos, P. A., Sethi, A. J., Ravindran, A., Srivastava, A., Kanchi, M., Mahmud, S., et al. (2024). Simultaneous identification of m6A and m5C reveals coordinated RNA modification at single-molecule resolution. bioRxiv. doi: 10.1101/2022.03.14.484124
McIntyre, A. B. R., Gokhale, N. S., Cerchietti, L., Jaffrey, S. R., Horner, S. M., and Mason, C. E. (2020). Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci. Rep. 10:6590. doi: 10.1038/s41598-020-63355-3
Merkurjev, D., Hong, W.-T., Iida, K., Oomoto, I., Goldie, B. J., Yamaguti, H., et al. (2018). Synaptic N6-methyladenosine (m6A) epitranscriptome reveals functional partitioning of localized transcripts. Nat. Neurosci. 21, 1004–1014. doi: 10.1038/s41593-018-0173-6
Meyer, K. D. (2019). DART-seq: an antibody-free method for global m6A detection. Nat. Methods 16, 1275–1280. doi: 10.1038/s41592-019-0570-0
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/J.CELL.2012.05.003
Molinie, B., Wang, J., Lim, K. S., Hillebrand, R., Lu, Z. X., Van Wittenberghe, N., et al. (2016). M6 A-LAIC-seq reveals the census and complexity of the m6 a epitranscriptome. Nat. Methods 13, 692–698. doi: 10.1038/nmeth.3898
Murakami, S., and Jaffrey, S. R. (2022). Hidden codes in mRNA: control of gene expression by m6A. Mol. Cell 82, 2236–2251. doi: 10.1016/J.MOLCEL.2022.05.029
Owens, M. C., Zhang, C., and Liu, K. F. (2021). Recent technical advances in the study of nucleic acid modifications. Mol. Cell 81, 4116–4136. doi: 10.1016/j.molcel.2021.07.036
Ren, X., Deng, R., Zhang, K., Sun, Y., Li, Y., and Li, J. (2021). Single-cell imaging of m6A modified RNA using m6A-specific in situ hybridization mediated proximity ligation assay (m6AISH-PLA). Angew. Chem. Int. Ed. 60, 22646–22651. doi: 10.1002/anie.202109118
Ries, R. J., Pickering, B. F., Poh, H. X., Namkoong, S., and Jaffrey, S. R. (2023). m6A governs length-dependent enrichment of mRNAs in stress granules. Nat. Struct. Mol. Biol. 30, 1525–1535. doi: 10.1038/s41594-023-01089-2
Roy, R., Shiina, N., and Wang, D. O. (2020). More dynamic, more quantitative, unexpectedly intricate: advanced understanding on synaptic RNA localization in learning and memory. Neurobiol. Learn. Mem. 168:107149. doi: 10.1016/j.nlm.2019.107149
Sheehan, C. J., Marayati, B. F., Bhatia, J., and Meyer, K. D. (2023). In situ visualization of m6A sites in cellular mRNAs. Nucleic Acids Res. 51:e101. doi: 10.1093/nar/gkad787
Shi, H., Wei, J., and He, C. (2019). Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650. doi: 10.1016/j.molcel.2019.04.025
Shi, H., Xu, Y., Tian, N., Yang, M., and Liang, F.-S. (2022). Inducible and reversible RNA N6-methyladenosine editing. Nat Commun. 13:1958. doi: 10.1038/s41467-022-29665-y
Shi, H., Zhang, X., Weng, Y.-L., Lu, Z., Liu, Y., Lu, Z., et al. (2018). m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. doi: 10.1038/s41586-018-0666-1
Shu, X., Cao, J., Cheng, M., Xiang, S., Gao, M., Li, T., et al. (2020). A metabolic labeling method detects m6A transcriptome-wide at single base resolution. Nat. Chem. Biol. 16, 887–895. doi: 10.1038/s41589-020-0526-9
Tegowski, M., Flamand, M. N., and Meyer, K. D. (2022). scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells. Mol. Cell 82, 868–878.e10. doi: 10.1016/J.MOLCEL.2021.12.038
Uzonyi, A., Dierks, D., Nir, R., Kwon, O. S., Toth, U., Barbosa, I., et al. (2023). Exclusion of m6A from splice-site proximal regions by the exon junction complex dictates m6A topologies and mRNA stability. Mol. Cell 83, 237–251.e7. doi: 10.1016/j.molcel.2022.12.026
Wang, Y., Li, Y., Yue, M., Wang, J., Kumar, S., Wechsler-Reya, R. J., et al. (2018). N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 21, 195–206. doi: 10.1038/s41593-017-0057-1
Wang, Y., Xiao, Y., Dong, S., Yu, Q., and Jia, G. (2020). Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol. 16, 896–903. doi: 10.1038/s41589-020-0525-x
Wilson, C., Chen, P. J., Miao, Z., and Liu, D. R. (2020). Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 38, 1431–1440. doi: 10.1038/s41587-020-0572-6
Xiao, Y.-L., Liu, S., Ge, R., Wu, Y., He, C., Chen, M., et al. (2023). Transcriptome-wide profiling and quantification of N6-methyladenosine by enzyme-assisted adenosine deamination. Nat. Biotechnol. 41, 993–1003. doi: 10.1038/s41587-022-01587-6
Xiao, Y., Wang, Y., Tang, Q., Wei, L., Zhang, X., and Jia, G. (2018). An elongation-and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N6-Methyladenosine modification. Angew. Chem. Int. Ed. 57, 15995–16000. doi: 10.1002/anie.201807942
Yang, C., Hu, Y., Zhou, B., Bao, Y., Li, Z., Gong, C., et al. (2020). The role of m6A modification in physiology and disease. Cell Death Dis. 11, 960–916. doi: 10.1038/s41419-020-03143-z
Yang, X., Triboulet, R., Liu, Q., Sendinc, E., and Gregory, R. I. (2022). Exon junction complex shapes the m6A epitranscriptome. Nat. Commun. 13:7904. doi: 10.1038/s41467-022-35643-1
Yao, H., Gao, C.-C., Zhang, D., Xu, J., Song, G., Fan, X., et al. (2023). scm6A-seq reveals single-cell landscapes of the dynamic m6A during oocyte maturation and early embryonic development. Nat. Commun. 14:315. doi: 10.1038/s41467-023-35958-7
Yoon, K.-J., Ringeling, F. R., Vissers, C., Jacob, F., Pokrass, M., Jimenez-Cyrus, D., et al. (2017). Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 171, 877–889.e17. doi: 10.1016/J.CELL.2017.09.003
Zaccara, S., and Jaffrey, S. R. (2020). A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 181, 1582–1595.e18. doi: 10.1016/J.CELL.2020.05.012
Zaccara, S., Ries, R. J., and Jaffrey, S. R. (2019). Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624. doi: 10.1038/s41580-019-0168-5
Zeng, Y., Wang, S., Gao, S., Soares, F., Ahmed, M., Guo, H., et al. (2018). Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol. 16:e2006092. doi: 10.1371/journal.pbio.2006092
Zhang, Z., Wang, M., Xie, D., Huang, Z., Zhang, L., Yang, Y., et al. (2018). METTL3-mediated N6-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 28, 1050–1061. doi: 10.1038/s41422-018-0092-9
Zhong, Z.-D., Xie, Y.-Y., Chen, H.-X., Lan, Y.-L., Liu, X.-H., Ji, J.-Y., et al. (2023). Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing. Nat. Commun. 14:1906. doi: 10.1038/s41467-023-37596-5
Keywords: RNA, m6A, epitranscriptome, methods, brain
Citation: Tegowski M and Meyer KD (2024) Studying m6A in the brain: a perspective on current methods, challenges, and future directions. Front. Mol. Neurosci. 17:1393973. doi: 10.3389/fnmol.2024.1393973
Edited by:
Clémence Bernard, University of Exeter, United KingdomReviewed by:
Gary Patrick Brennan, University College Dublin, IrelandDan Ohtan Wang, New York University Abu Dhabi, United Arab Emirates
Copyright © 2024 Tegowski and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kate D. Meyer, a2F0ZS5tZXllckBkdWtlLmVkdQ==