
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 10 January 2024
Sec. Molecular Signalling and Pathways
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1321090
This article is part of the Research Topic Sub-molecular Mechanism of Genetic Epilepsy, Volume II View all 5 articles
 Wen-Hui Liu1†
Wen-Hui Liu1† Sheng Luo1†
Sheng Luo1† Dong-Ming Zhang1Zi-Sheng Lin1Song Lan2Xin Li3
Dong-Ming Zhang1Zi-Sheng Lin1Song Lan2Xin Li3 Yi-Wu Shi1
Yi-Wu Shi1 Tao Su1
Tao Su1 Yong-Hong Yi1
Yong-Hong Yi1 Peng Zhou1*
Peng Zhou1* Bing-Mei Li1* for the China Epilepsy Gene 1.0 Project
Bing-Mei Li1* for the China Epilepsy Gene 1.0 ProjectBackground: The GABRA1 gene, encoding the GABRAR subunit α1, plays vital roles in inhibitory neurons. Previously, the GABRA1 gene has been identified to be associated with developmental and epileptic encephalopathy (DEE) and idiopathic generalized epilepsy (IGE). This study aims to explore the phenotypic spectrum of GABRA1 and molecular subregional effect analysis.
Methods: Trios-based whole-exome sequencing was performed in patients with epilepsy. Previously reported GABRA1 mutations were systematically reviewed to analyze the molecular subregional effects.
Results: De novo GABRA1 mutations were identified in six unrelated patients with heterogeneous epilepsy, including three missense mutations (p.His83Asn, p.Val207Phe, and p.Arg214Cys) and one frameshift mutation (p.Thr453Hisfs*47). The two missense mutations, p.His83Asn and p.Val207Phe, were predicted to decrease the protein stability but no hydrogen bond alteration, with which the two patients also presented with mild genetic epilepsy with febrile seizures plus and achieved seizure-free status by monotherapy. The missense variant p.Arg214Cys was predicted to decrease protein stability and destroy hydrogen bonds with surrounding residues, which was recurrently identified in three cases with severe DEE. The frameshift variant p.Thr453Hisfs*47 was located in the last fifth residue of the C-terminus and caused an extension of 47 amino acids, with which the patients presented with moderated epilepsy with generalized tonic-clonic seizures alone (GTCA) but achieved seizure-free status by four drugs. The four variants were not presented in gnomAD and were evaluated as “pathogenic/likely pathogenic” according to ACMG criteria. Analysis of all reported cases indicated that patients with mutations in the N-terminal extracellular region presented a significantly higher percentage of FS and DEE, and the patients with variants in the transmembrane region presented earlier seizure onset ages.
Significance: This study suggested that GABRA1 variants were potentially associated with a spectrum of epilepsies, including EFS+, DEE, and GTCA. Phenotypic severity may be associated with the damaging effect of variants. The molecular subregional effects help in understanding the underlying mechanism of phenotypic variation.
Gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the human brain (Sanacora et al., 2003). Functional GABA regulates neuronal excitability by interacting with neuronal receptors, including GABA A-type receptors (GABAAR) and GABA B-type receptors (GABABR). The GABAAR, a ligand-gated ion channel, is the primary inhibitory receptor of the central nervous system (Hannan et al., 2020; Papasergi-Scott et al., 2020; Feng et al., 2022). GABAAR is composed of various subunits, with various combinations. In the central nervous system, the most common components of GABAAR are two α subunits, two β subunits, and one γ subunit, forming a pentameric arrangement of αββαγ (Bai et al., 2019; Tomita, 2019). Dysregulation of GABAAR is the core mechanism of diverse central nervous disorders, especially epilepsies (Maljevic et al., 2019; Fu et al., 2022). Genes encoding the GABAAR subunit have been identified as causative genes of idiopathic generalized epilepsy (IGE) and/or developmental and epileptic encephalopathy (DEE), including GABRA1, GABRA2, GABRB3, GABRA5, GABRB1, GABRB2, GABRB3, and GABRG2 (OMIM, database).1
The GABRA1 gene (OMIM*137160), located at chromosomal locus 5q34, encodes the GABRAR subunit α 1. It forms the γ2β2α1β2α1 pentamer with GABRAR β2 subunits and GABRAR γ2 subunit, which is the most prevalent and functionally significant GABAAR subtype in the brain (Huang et al., 2001; Sanacora et al., 2003; Maljevic et al., 2019). The GABRA1 gene is highly expressed in the developing brain, especially in the frontal cortex, anterior cingulate cortex, hippocampus, and hypothalamus (GTEx database).2 In mice, knockout of gabra1 led to spontaneous seizures and pre-weaning lethality with incomplete penetrance (Vicini et al., 2001). In humans, the GABRA1 gene has been identified as the causative gene of developmental and epileptic encephalopathy-19 (DEE-19, OMIM* 615744) and the susceptibility genes of childhood absence epilepsy-4 (ECA-4, OMIM* 611136) and juvenile myoclonic epilepsy-5 (JME-5, OMIM* 611136). However, the phenotypic spectrum and genotype-phenotype correlation of GABRA1 remain undetermined.
In the present study, we performed GABRA1 trio-based whole exome sequencing (WES) in patients with epilepsy without acquired cause. Four de novo mutations in the GABRA1 variants were identified in six unrelated cases, including two with epilepsy with febrile seizures plus (EFS +), three with developmental and epileptic encephalopathy (DEE), and one with epilepsy with generalized tonic-clonic seizures alone (GTCA). To analyze the molecular subregional effects of GABRA1, we systematically reviewed all reported disease-causing variants and their associated phenotypes.
The patients of the study were recruited through the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University, the Second Hospital of Shandong University, and the Maoming People’s Hospital. Clinical data of the individuals were collected, including present age, gender, age at seizure onset, seizure course, detailed family history, general and neurological examination results, and effective antiseizure medications (ASMs). Magnetic resonance imaging (MRI) scans were conducted to detect structural abnormalities within the brain. A long-term (24-h) video electroencephalogram (EEG) was performed including an open–close eyes test, intermittent photic stimulation, hyperventilation, and sleeping recording. Epileptic seizures and epilepsies were diagnosed based on the classification guidelines developed by the Commission on Classification and Terminology of the Wirrell et al. (2022).
This study was approved by the ethics committee of The Second Affiliated Hospital of Guangzhou Medical University (2020-h5-49), and written informed consent was obtained from all patients and their parents.
Blood samples were collected from all probands, parents, and available family members to extract genomic DNA and perform trio-based whole-exome sequencing using the Illumina HiSeq 2000 system (BGI, Shenzhen, China). Detailed sequence procedures were described in previous studies (Wang et al., 2018; Cai et al., 2019; Shi et al., 2019).
To identify potential pathogenic variants, each case was analyzed as previously described (Chen et al., 2022; Li et al., 2022; Luo et al., 2022). Initially, polymorphic variants with a minor allele frequency (MAF) ≥ 0.005 in the gnomAD database were excluded from further analysis.3 Potentially disease-causing variants, including frameshift, nonsense, canonical splice site, initiation codon, in-frame, and missense variants were retained for further study. We screened GABRA1 mutations with potentially pathogenic inheritance patterns, including de novo, cosegregated, homozygous, and compound heterozygous, which demonstrate the genetic difference between the affected child and the asymptomatic parents in a given family (trio). All GABRA1 mutations identified in this study were annotated into the reference transcript NM_ 000806.5.
The consequences of missense variants were predicted by multiple in silico tools with detailed scores acquired from the VarCards database (Li et al., 2018),4 including SIFT, VEST3, MetaSVM, MetaLR, M-CAP, CADD, DANN, Eigen, Fathmm-MKL, and REVEL. The Varsite web server was utilized to examine the hydrophobicity alterations of variants (Laskowski et al., 2020).5 MetaDome analyses the mutation tolerance at each position in a human protein. The conservation of mutated positions was evaluated through sequence alignment across different species.6
The structural impact of potential missense variants was evaluated by conducting protein modeling using the Iterative Threading ASSEmbly Refinement software (I-TASSER) (Yang and Zhang, 2015).7 The visualization and analysis of the protein’s three-dimensional structure were conducted using the PyMOL Molecular Graphics System (Version 2.3.2). To estimate the impact of protein stability of variants, the I-Mutant server was used to predict the free energy change value (DDG, kcal/mol) (Capriotti et al., 2005).8
To evaluate the relationship between genotype and phenotype, we conducted a comprehensive review of all previously reported GABRA1 mutations and associated clinical information via the PubMed database9 and the Human Gene Mutation Database (HGMD)10 until August 2023, likely our previously studies (Yan et al., 2022; Luo et al., 2023). Variants with undefined origination or unexplained origination for the occurrence of genetic diseases were excluded. The variants were visualized in the protein diagram. The number of variants with varied phenotypes in distinct functional domains was compared by Fisher’s exact test, to analyze the molecular subregional effects.
The statistical analyses were performed utilizing R software (version 4.0.3). Fisher’s exact test was used to evaluate the frequencies of GABRA1 mutations in the case-cohort and the control populations (2015). A P-value < 0.05 was considered to be statistically significant.
A total of four GABRA1 mutations were identified in six unrelated cases, including three missense mutations (c.247C > A/p.His83Asn, c.619G > T/p.Val207Phr, and c.640C > T/p.Arg214Cys) and one frameshift mutation (c.1356delC/p.Thr453Hisfs*47) (Figures 1A, B; Table 1). The mutations c.640C > T/p.Arg214Cys was recurrently identified in three cases, including cases 3, 4, and 5. The six variants were all of de novo, consistent with the autosomal dominant inheritance pattern.
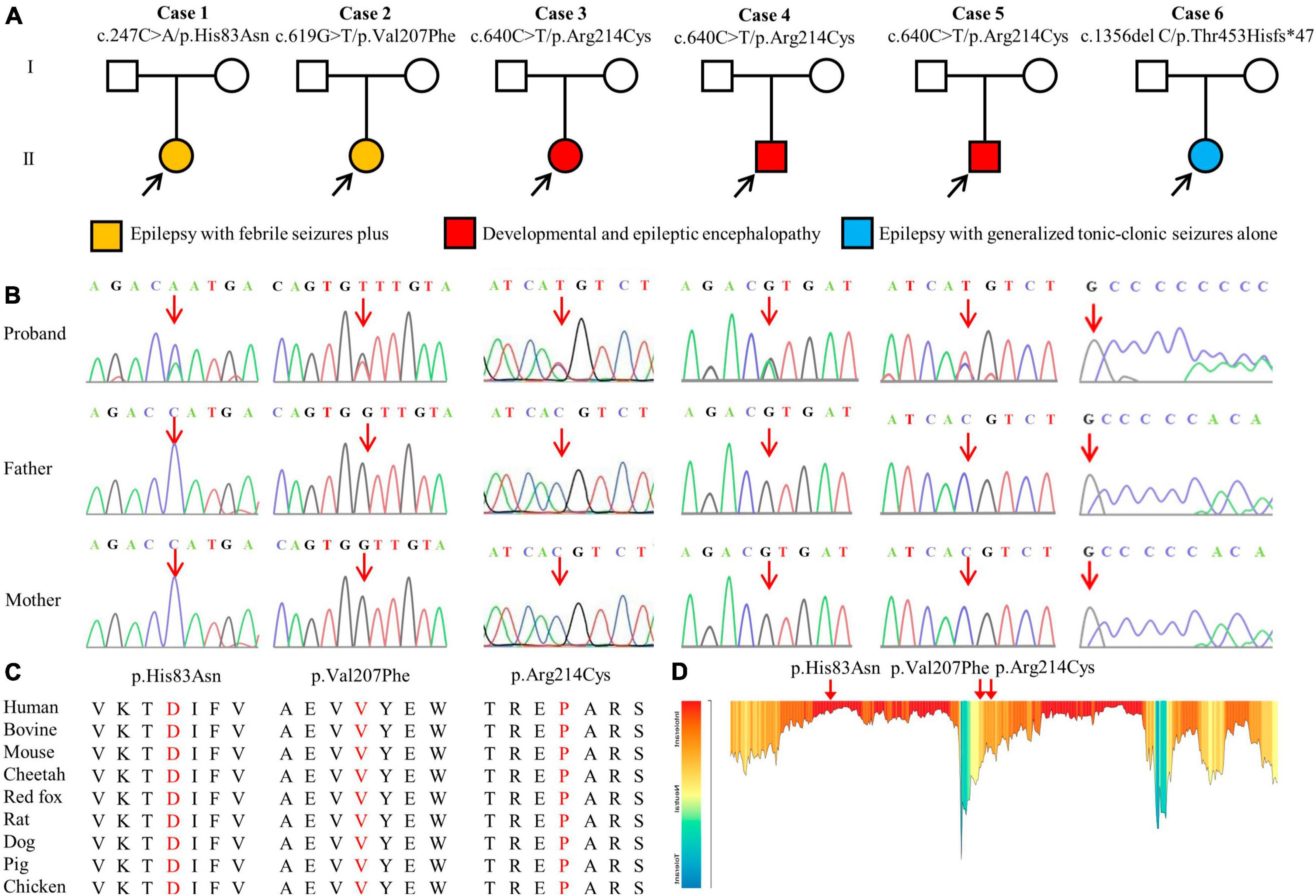
Figure 1. Genetic data of patients with GABRA1 mutations. (A) Pedigrees of the six de novo mutations and their corresponding phenotypes. (B) DNA sequence chromatogram of the GABRA1 mutations. Arrows indicate the positions of the mutations. (C) The amino acid sequence alignment of the three missense mutations shows that the residues are highly conserved across mammals. (D) The tolerance of three missense mutations of GABRA1 predicted by the MetaDome algorithm.
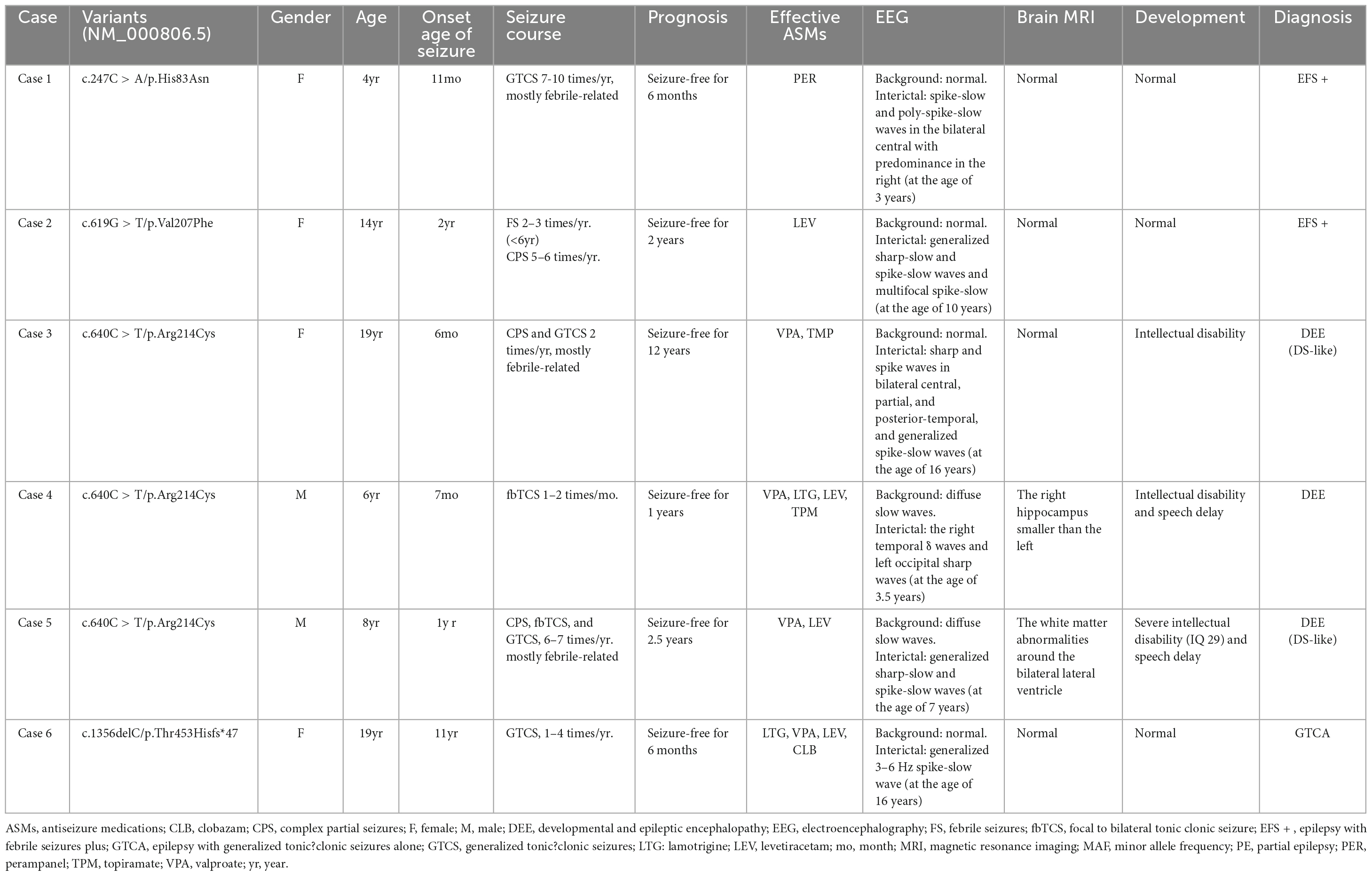
Table 1. Clinical features of the individuals with GABRA1 mutations.
The four de novo variants were not present in the population of the gnomAD database. Amino acid sequence alignment suggested that the variants were located in residues with high conservation across mammals (Figure 1C). The three variants were located in residues that are intolerant of missense variants, according to the MataDome prediction (Figure 1D). The missense mutations were all predicted to be “conserved” or “damaging” by multiple in silico algorithms. According to the ACMG guidelines, the frameshift mutation p. Thr453Hisfs*47 and the recurrently identified missense mutation p. Arg214Cys were evaluated as “pathogenic,” while the other two missense mutations, p.His83Asn and p.Val207Phe, were rated as “likely pathogenic” (Table 2).
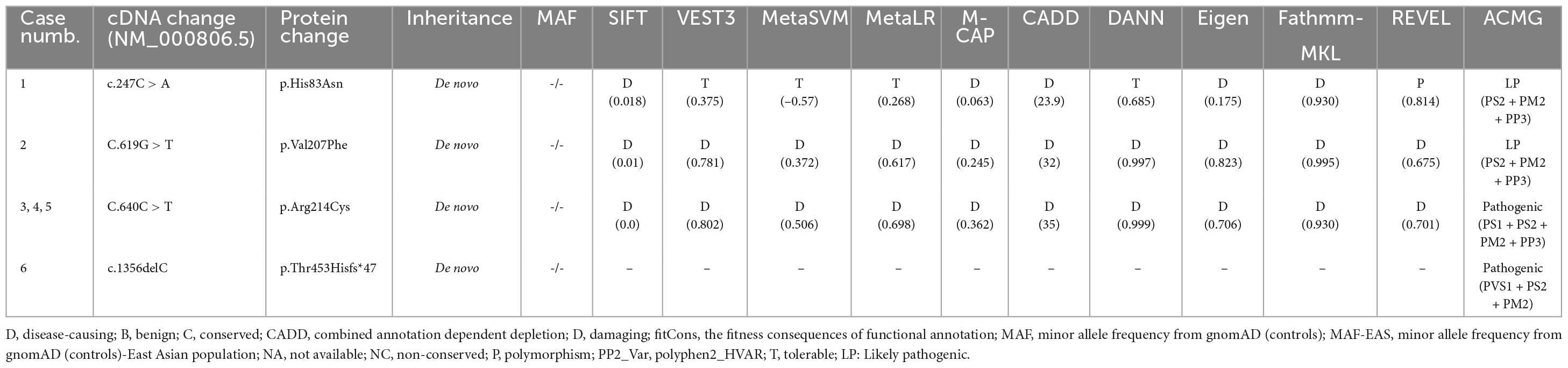
Table 2. Genetic characteristics of GABRA1 mutations.
Heterogeneous epilepsies were present in the six cases with GABRA1 mutations, including two with EFS+, three with DEE, and one with GTCA. Detailed clinical information was summarized in Table 1.
The two patients in cases 1 and 2 were diagnosed with EFS+, in which variants p.His83Asn and p.Val207Phe were identified, respectively. The two patients initially presented with febrile seizures (FS) featuring generalized tonic-clonic seizures (GTCS) at the age of 6 years old and subsequently afebrile GTCS. Seizures in the two patients were infrequent (yearly) and were controlled by monotherapy with perampanel (PER) or levetiracetam (LEV). Their interictal EEG recorded epileptic discharges, manifesting multifocal and generalized discharges in the Rolandic region (Figures 2A, B). The brain MRI scans were normal. The two patients showed normal neurodevelopment.
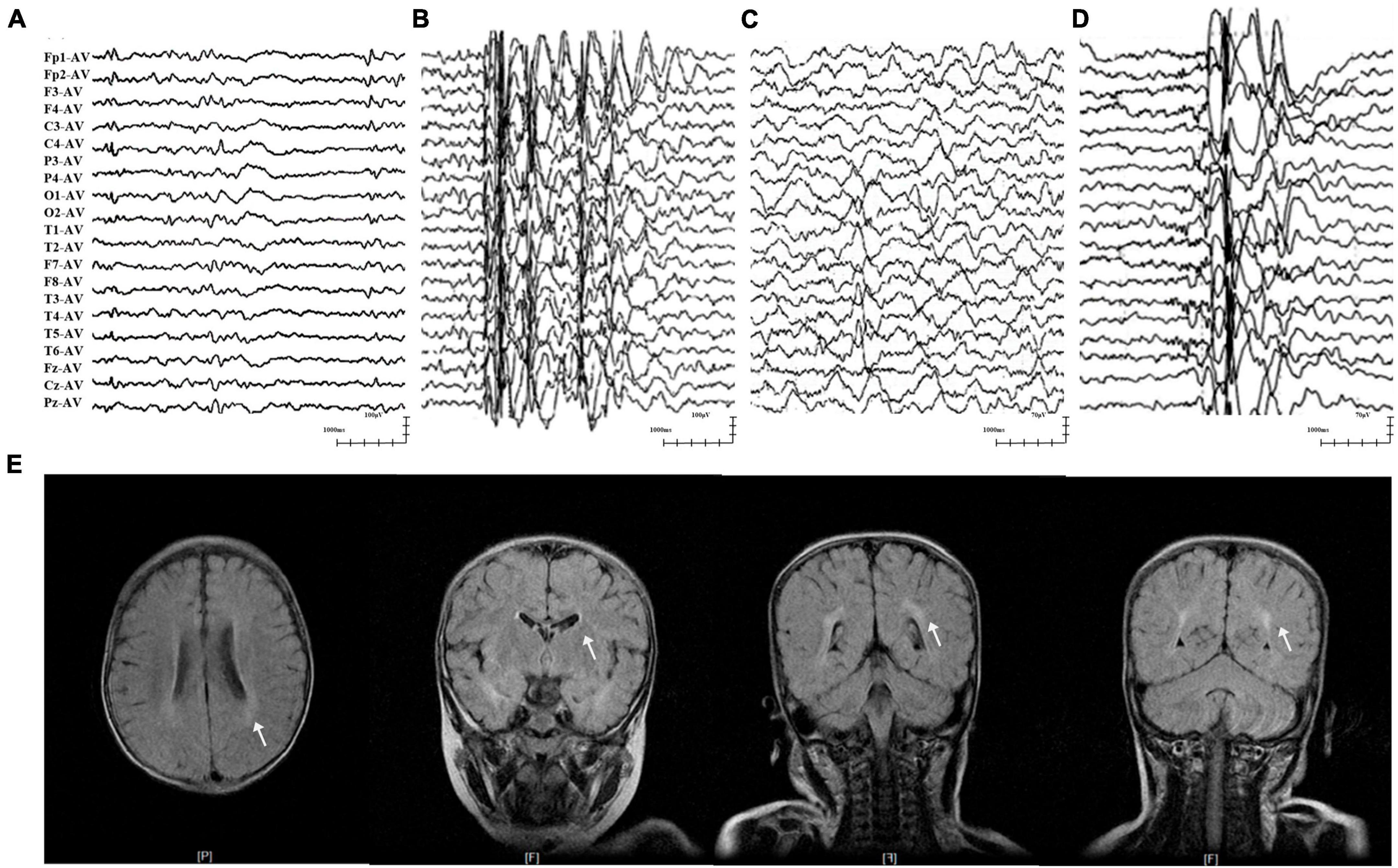
Figure 2. Representative EEG and MRI of patients with GABRA1 variants. (A) Interictal EEG of case 1 showed multifocal waves at the age of 3 years. (B) Interictal EEG of case 2 showed generalized spike-slow waves at the age of 10 years. (C) Interictal EEG of case 4 showed right temporal slow waves at the age of 3.5 years. (D) Interictal EEG of case 5 showed generalized spike-slow waves and irregular 0.5–3 slow activity bursts 0.5–2 s during sleep. (E) The MRI of case 5 showed an abnormal white matter signal around the posterior horn of the lateral ventricle.
The three patients with the identical variant p.Arg214Cys were all diagnosed with DEE, including two who presented with Dravet-like syndrome (Cases 3 and 5). The three patients all presented infancy-onset seizures with multiple seizure types, including GTCS, focal to bilateral tonic-clonic seizure (fbTCS), and focal seizure (Fos). Seizures in cases 3 and 5 were mostly febrile-related. Seizure-free status in the three patients was achieved by combined treatment including valproate (VPA). Their EEG recorded epileptic discharge with features of DEE, including interictal multifocal, bilateral, and/or generalized discharges and disuse slow waves in the background during the waking stage (Figures 2C, D). Brain MRI scans detected structural abnormalities, including a smaller right hippocampus in case 4 and white matter abnormalities around the bilateral lateral ventricle in case 5 (Figure 2E). The three patients all exhibited intellectual disability (ID), and two patients (cases 4 and 5) also presented speech delays.
The patient in case 6, who had the frameshift mutation p.Thr453Hisfs*47, was diagnosed with GTCA. She exhibited yearly GTCS since eleven-years-old. Seizure-free status was achieved for 6 months after treatment with four ASMs, including VPA, LEV, lamotrigine (LTG), and clobazam (CLB). EEG recording showed generalized 3–6 Hz spike-slow waves. Brain MRIs and neurodevelopment of the patient were normal.
The GABA receptor subunit alpha1 encoded by GABRA1 consists of one N-terminal extracellular domain, four transmembrane domains, three linkers between transmembrane domains, and one C-terminal extracellular domain (Macdonald et al., 2010). The three missense mutations identified in the present study were all located in the N-terminus extracellular domain, while the frameshift mutation was located in the latest fourth residue of the C-terminus extracellular domain (Figure 3A).
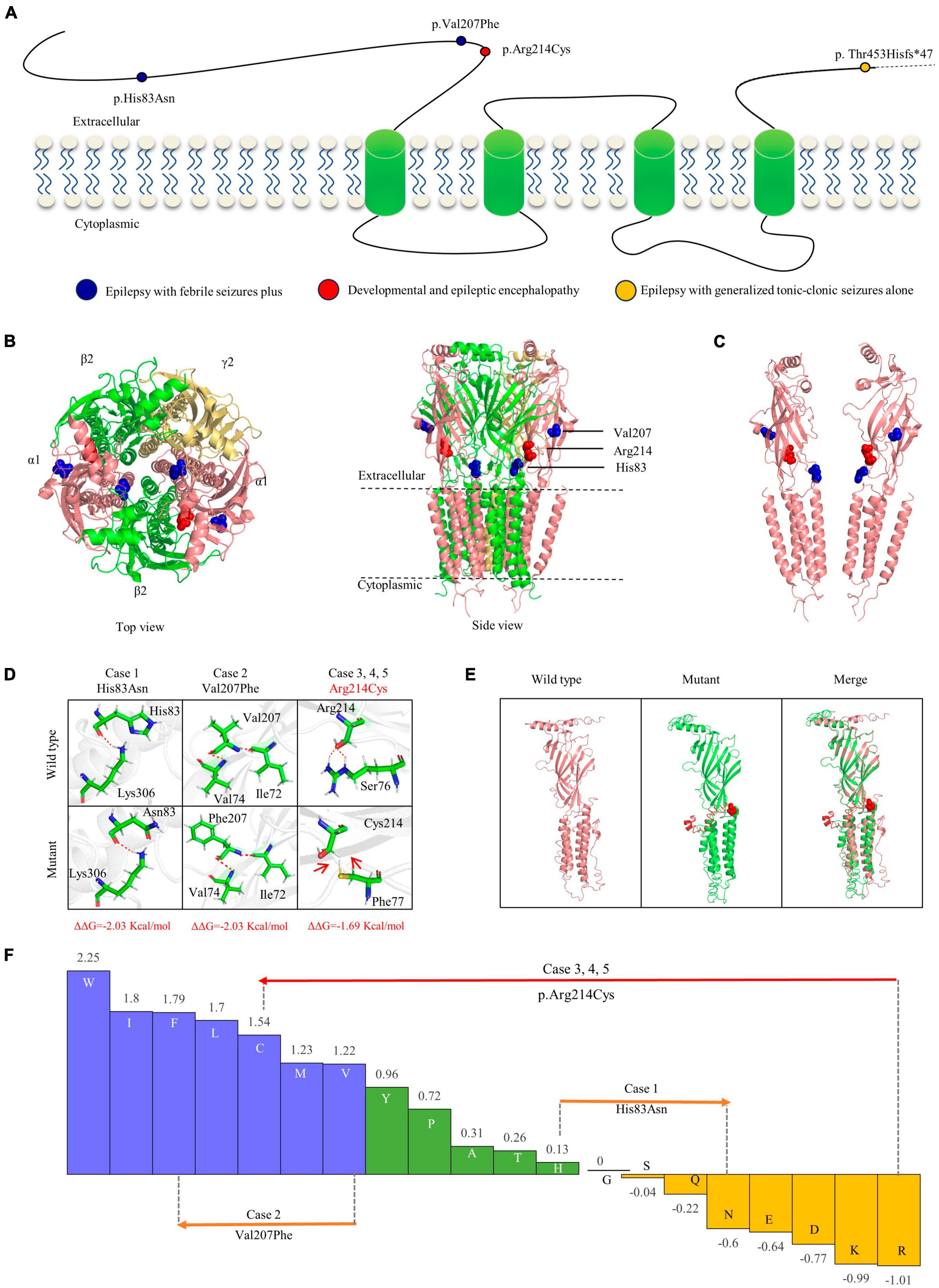
Figure 3. Schematic illustration of mutations and interactions with surrounding amino acids. (A) Schematic diagram of the GABRA1 protein and the localization of the GABRA1 mutations identified in the study. (B) The 3D structural model of GABAAR γ2β2α1β2α2 and the location of the GABRA1 mutations. (C) The location of the three missense mutations in the GABRA1 protein. (D) Hydrogen bond changes and DDG values of the three missense mutations. The red dotted line represents hydrogen bonds. (E) The structural alteration of the frameshift mutation. (F) Fauchère and Pliska hydrophobicity scale exhibited the hydrophobicity of 20 amino acids. Abscissa: from left to right, hydrophobicity gradually decreased. Blue amino acids are hydrophobic, green amino acids are neutral, and yellow amino acids are hydrophilic. Amino acids with high positive values are more hydrophobic, whereas amino acids with low negative values are more hydrophilic.
Protein modeling and single point mutation free energy change prediction were utilized to assess the structural impact of the identified mutations. The two missense mutations (p.His83Asn and p.Val207Phe) of the cases with mild EFS + were predicted to decrease the protein stability with ΔΔG >> 0.5 Kcal/mol but no hydrogen bond alteration (Figures 3B–E). The missense mutation p.Arg214Cys, which was recurrently identified in three severe DEE cases, was not only predicted to decrease protein stability but also destroy hydrogen bonds with surrounding residues. The frameshift mutation p.Thr453Hisfs*47 identified in the case with moderate GTCA was located in the last fifth residue of the C-terminus, causing an extension of 47 amino acids. The mutant protein was also predicted to cause abnormal spatial folding (Figure 3F).
A total of 25 GABRA1 mutations with explainable origins for the occurrence of genetic diseases have been identified in 47 patients (36 cases) so far, including two novel missense and the first frameshift mutation from the present study (Supplementary Table 1). The patients presented with heterogeneous epilepsies, including DEEs, IGEs, and EFS + (Figure 4A). Variable seizure types were exhibited in those patients, including GTCS, Fos, myoclonic seizures, FS, absence, fbTCS, spasms, status epilepticus, atonic, clonic, eyelid myoclonia, and tonic seizures. GTCS, Fos, and myoclonic seizures were the most common seizure types (Figure 4B). Distinct outcomes and drug responses were also presented in these patients, including ten patients with refractory seizures, eleven patients achieving seizure-free status, and six patients with seizure remission (Figure 4C). VPA, LEV, and LTG were the most commonly used drugs in patients with a positive drug response (Figure 4D).
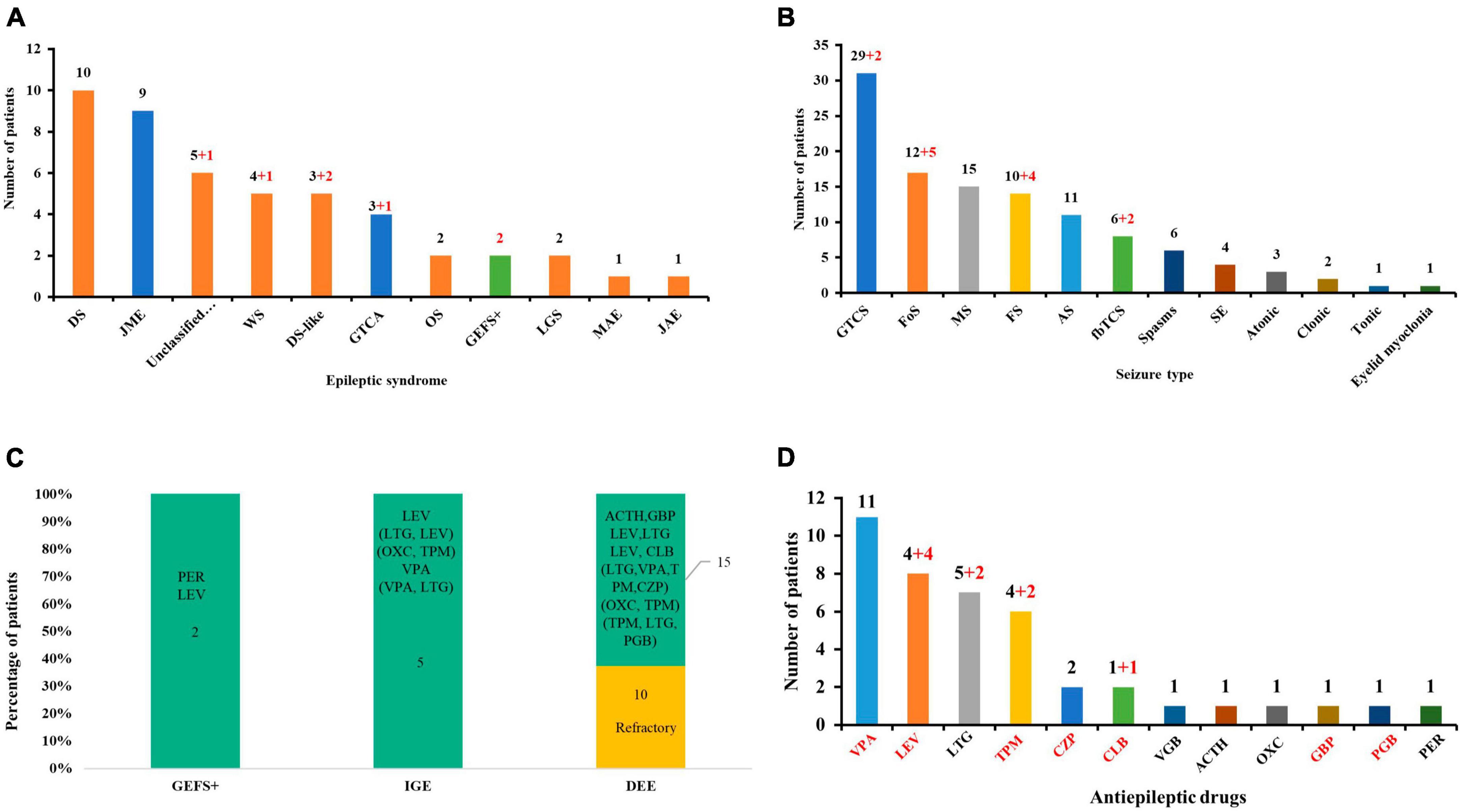
Figure 4. Clinical and genetic characteristics analysis of available GABRA1 mutations. (A) The epileptic syndrome of patients with GABRA1 variants. Red numbers indicate data from this study. The red column represents DEEs; the blue column represents IGEs; and the green column represents EFS+. (B) The seizure types of patients with GABRA1 variants. Red numbers indicate data from this study. (C) The stacked bar chart of the drug response of the patients with different phenotypes. The orange colors show that the percentage of patients was refractory, and the green colors show the percentage of patients with a positive response to drugs. (D) The effective ASMs used in patients with GABRA1 variants. Red numbers indicate data from this study.
A previous study indicated that phenotypic heterogeneity was associated with molecular subregional effects (Liao et al., 2022). The location of variants and associated phenotypes were thus visualized for analysis. It is suggested that the patients with variants in the transmembrane region all presented with severe DDE, showing a significantly higher percentage of DEE than those with mutations in other regions (Figure 5A). The patients with variants in the transmembrane region also exhibited earlier seizure onset ages (less than 6 months old) than those with variants in other regions (P = 4.0 × 10–2) (Figure 5B). The patients with mutations in the N-terminal extracellular region presented a significantly higher percentage of FS than those with mutations in other regions (P = 1.0 × 10–2) (Figure 5C). These findings suggested a potential molecular subregional effect.
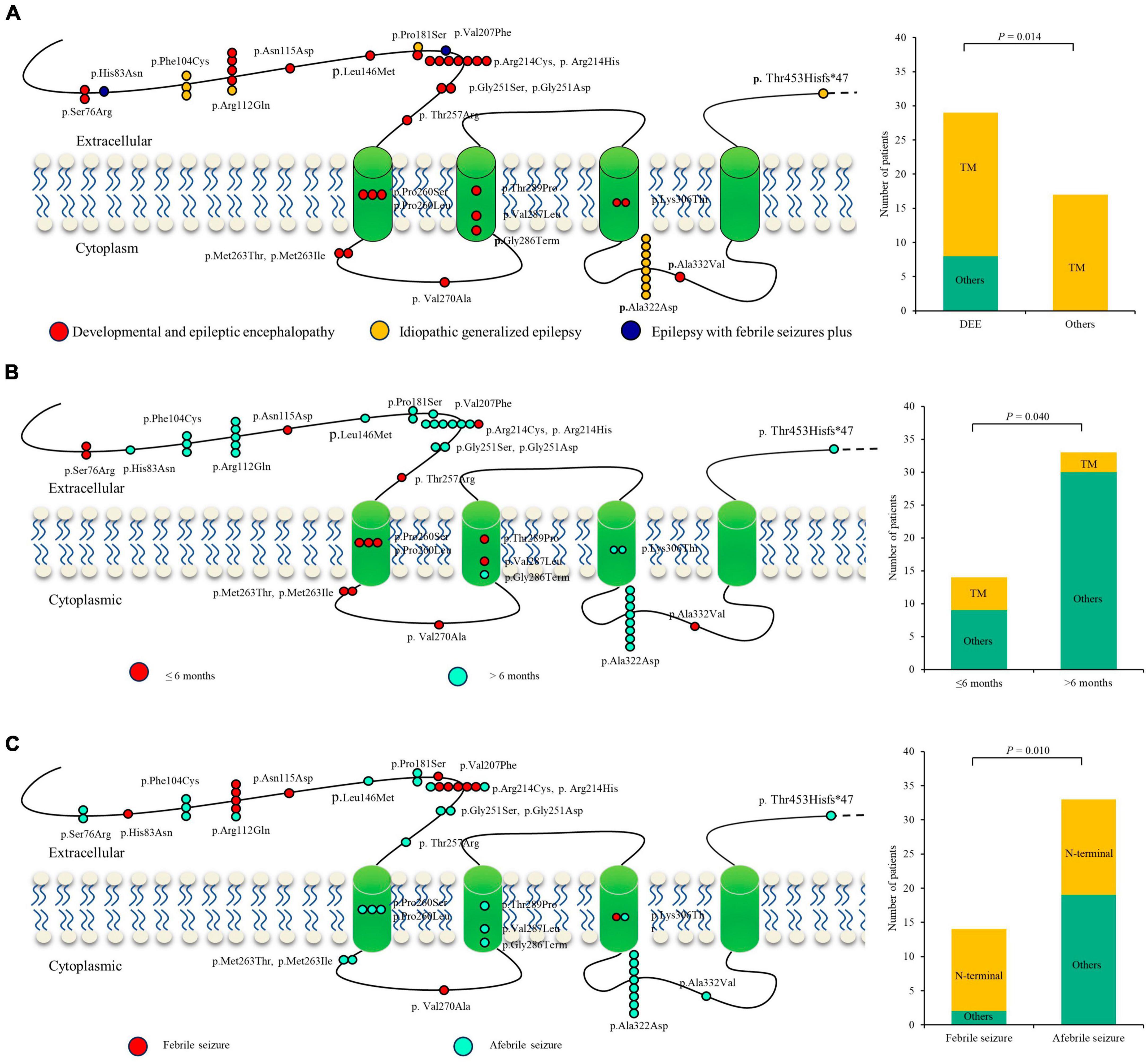
Figure 5. Molecular subregional effects analysis of GABRA1 mutations. (A) The locations of GABRA1 variants associated with various epilepsy syndromes, including DEEs, IGEs, and EFS+. The patients with variants in the transmembrane region all presented with severe DDE, showing a significantly higher percentage of DEE than those with mutations in other regions. TM, transmember. (B) The locations of GABRA1 variants associated with varied onset ages of seizures, including early onset (younger than 6 months) and late onset (older than 6 months). The patients with variants in the transmembrane region also exhibited earlier seizure onset ages (less than 6 months old) than those with variants in other regions. (C) The locations of GABRA1 variants associated with febrile seizure (FS) and afebrile seizure. The patients with mutations in the N-terminal extracellular region presented a significantly higher percentage of FS than those with mutations in other regions.
In this study, we identified novel GABRA1 variants in six unrelated cases, including one frameshift and three missense mutations. The mutations were of de novo, had no frequencies in controls, and were predicted to alter protein stability. According to the ACMG criteria, the four variants were all evaluated to be “pathogenic” or “likely pathogenic.” The six patients presented with heterogeneous epilepsies, including two with EFS+, three with DEE, and one with GTCA. Analysis of all reported cases indicated that patients with mutations in the N-terminal extracellular region presented a significantly higher percentage of FS, and patients with variants in the transmembrane region presented earlier seizure onset ages, indicating a potential molecular subregional effect. This study suggested that GABRA1 is associated with a spectrum of epilepsies, including severe DEE, IGEs, and EFS+. The molecular subregional effects analysis provided novel insight into the phenotypic heterogeneity of GABRA1.
The GABRA1 gene is highly conserved across various species, including zebrafish, mice, and humans. In zebrafish, knockout of GABRA1 causes epileptic seizures triggered by light (Samarut et al., 2018). In mice, knockout of GABRA1 led to spontaneous seizures and pre-weaning lethality with incomplete penetrance (Vicini et al., 2001). In humans, genomic data reveal that GABRA1 is intolerant to loss-of-function (LOF) variants with the pLI (the probability of being LOF intolerant) index of 0.91 and LOEUF (LOF observed/expected upper bound fraction) index of 0.91 (Karczewski et al., 2020). In this study, we identified one frameshift and three missense mutations of GABRA1 in six unrelated cases. The frameshift mutation (p.Thr453Hisfs*47) was predicted to cause a truncated protein, which is associated with LOF. The three missense variants were all predicted to be damaged by multiple in silico tools and decrease protein stability, which may also be associated with LOF. These findings suggested that LOF may be the epileptogenic mechanism of GABRA1.
Abnormalities of GABAAR were one of the most essential mechanisms of the occurrence of multiple epilepsies, particularly IGEs, DEEs, and EFS+. The GABRAR subunit α1 encoded by the GABRA1 gene is the core subunit of the GABAAR γ2β2α1β2α1 pentamer, the most prevalent and functionally significant subtype in the brain. Previously, the GABRA1 gene has been identified as one of the causative genes of DEE and the susceptibility gene of IGEs, including ECA-4 and EJM-5. In this study, we identified six novel cases with GABRA1 mutations, including two cases with EFS + and one case with the IGE subtype of GTCA, expanding the phenotypic spectrum of GABRA1.
The two missense variants identified in cases 1 and 2 were predicted to decrease the protein stability but no hydrogen binding alteration, indicating mild damaging effects; the two cases exhibited the mild phenotype of EFS + and achieved seizure-free status by monotherapy. The variant p.Arg214Cys recurrently identified in cases 3, 4, and 5 was predicted to destroy hydrogen bonds with surrounding residues and decrease the protein stability, suggesting a severely damaging effect; the three patients also presented with the severe phenotype of DEE. The frameshift variant p.Thr453Hisfs*47 was located in the C-terminus, which only led to a frameshift of four amino acids and an additional extension of 47 amino acids and may reflect moderate damaging effects; the patient also exhibited a moderate phenotype manifesting mild GTCA but achieving seizure-free status by four drugs. This finding suggests that phenotypic severity is possibly associated with the damaging effects of variants.
Previous studies have indicated that molecular subregional effects may also be associated with phenotypic heterogeneity (Liao et al., 2022). This study suggested that the patients with variants in the transmembrane region tended to present with severe DEE and earlier seizure onset ages, while patients with variants in the N-terminal extracellular region appeared to exhibit FS. These findings suggested a potential molecular subregional effect, which warrants a larger case-cohort study to further determine.
The three patients (Cases 3, 4, and 5) with the identical variant p.Arg214Cys presented with comparable phenotypic severity but subtle phenotypic variation, including two with Dravet-like syndrome and one with DEE without FS. The three previously reported cases with the identical variant p.Arg214Cys also presented phenotypic heterogeneity, including two with Dravet syndrome and one with Lennox-Gastaut syndrome (Johannesen et al., 2016; Cai et al., 2019). An analogous situation was also presented in other hotspot mutations, including p.Ser76Arg, p.Phe104Cys, p.Arg112Gln, p.Pro181Ser, p.Arg214His, p.Arg214Cys, p.Pro260Leu, and p.Lys306Thr (Supplementary Table 1). It is unknown whether modifier genes or polygenic interactions also partially contribute to phenotypic heterogeneity.
In this study, seizure-free status was achieved in all patients, in which five of six patients were treated with VPA and/or LEV. Analysis of previously reported patients with GABRA1 mutations also showed that VPA and LEV were the most commonly used ASMs in patients with positive drug responses. Previous studies suggested that the GABAergic system was one of the main targets of VPA and LEV, which may be a possible explanation for the positive effect of the two drugs in patients with GABRA1 mutations (Deshpande and Delorenzo, 2014; Romoli et al., 2019). Early genetic diagnosis may help in the clinical management of patients with GABRA1 mutations.
In conclusion, this study suggested that GABRA1 variants were potentially associated with a spectrum of epilepsies, including EFS+, DEE, and GTCA. Phenotypic severity may be associated with the damaging effect of variants. The molecular subregional effects help in understanding the underlying mechanism of phenotypic variation. Early use of VPA, LEV, and/or LTG may be the optimal treatment option for patients with GABRA1 mutations if early genetic diagnosis is acquired.
The data presented in the study are deposited in the NCBI-genebank repository, accession number OR818666-OR818683.
The studies involving humans were approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
W-HL: Writing—original draft. SLuo: Conceptualization, Formal analysis, Supervision, Writing—review and editing. D-MZ: Resources, Validation, Writing—review and editing. Z-SL: Writing—review and editing. SLan: Writing—review and editing. XL: Writing—review and editing. Y-WS: Writing—review and editing. TS: Writing—review and editing. Y-HY: Writing—review and editing. PZ: Writing—review and editing. B-ML: Writing—review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant Nos. 82171439 and 82271505), the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2021A1515010986), the Guangzhou City School Joint Funding Project Fund (Grant No. 2023A03J0412), the Science and Technology Project of Guangzhou (Grant Number 202201020106), the Multi-center Clinical Research Fund Project of the Second Affiliated Hospital of Guangzhou Medical University (2021-LCYJ-DZX-02), and the Scientific Research Project of Guangzhou Education Bureau (Grant Number 202235395).
The help of patients and clinicians participating in this work is greatly appreciated.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1321090/full#supplementary-material
Bai, Y.-F., Chiu, M., Chan, E. S., Axerio-Cilies, P., Lu, J., Huh, L., et al. (2019). Pathophysiology of and therapeutic options for a GABRA1 variant linked to epileptic encephalopathy. Mol. Brain 12, 1–17. doi: 10.1186/s13041-019-0513-9
Cai, K., Wang, J., Eissman, J., Wang, J., Nwosu, G., Shen, W., et al. (2019). A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp. Neurol. 320:112973. doi: 10.1016/j.expneurol.2019.112973
Capriotti, E., Fariselli, P., and Casadio, R. (2005). I-Mutant2. 0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310. doi: 10.1093/nar/gki375
Chen, Z., Luo, S., Liu, Z. G., Deng, Y. C., He, S. L., Liu, X. R., et al. (2022). CELSR1 variants are associated with partial epilepsy of childhood. Am. J. Med. Genet. B Neuropsychiatr. Genet. 189, 247–256. doi: 10.1002/ajmg.b.32916
Deshpande, L. S., and Delorenzo, R. J. (2014). Mechanisms of levetiracetam in the control of status epilepticus and epilepsy. Front. Neurol. 5:11. doi: 10.3389/fneur.2014.00011
Feng, Y., Wei, Z.-H., Liu, C., Li, G.-Y., Qiao, X.-Z., Gan, Y.-J., et al. (2022). Genetic variations in GABA metabolism and epilepsy. Seizure 101, 22–29. doi: 10.1016/j.seizure.2022.07.007
Fu, X., Wang, Y.-J., Kang, J.-Q., and Mu, T.-W. (2022). “GABAA receptor variants in epilepsy,” in Epilepsy, ed. S. Czuczwar (Brisbane, AU: Exon Publications).
Hannan, S., Minere, M., Harris, J., Izquierdo, P., Thomas, P., Tench, B., et al. (2020). GABAAR isoform and subunit structural motifs determine synaptic and extrasynaptic receptor localisation. Neuropharmacology 169:107540. doi: 10.1016/j.neuropharm.2019.02.022
Huang, R.-Q., Bell-Horner, C. L., Dibas, M. I., Covey, D. F., Drewe, J. A., and Dillon, G. H. (2001). Pentylenetetrazole-induced inhibition of recombinant γ-aminobutyric acid type A (GABAA) receptors: mechanism and site of action. J. Pharmacol. Exp. Therap. 298, 986–995.
Johannesen, K., Marini, C., Pfeffer, S., MøLLER, R. S., Dorn, T., Niturad, C. E., et al. (2016). Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology 87, 1140–1151.
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., ALFöLDI, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443.
Laskowski, R. A., Stephenson, J. D., Sillitoe, I., Orengo, C. A., and Thornton, J. M. (2020). VarSite: Disease variants and protein structure. Protein Sci. 29, 111–119. doi: 10.1002/pro.3746
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018). VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46, D1039–D1048.
Li, X., Bian, W. J., Liu, X. R., Wang, J., Luo, S., Li, B. M., et al. (2022). BCOR variants are associated with X-linked recessive partial epilepsy. Epilepsy Res. 187, 107036. doi: 10.1016/j.eplepsyres.2022.107036
Liao, W. P., Chen, Q., Jiang, Y. W., Luo, S., and Liu, X. R. (2022). Editorial: Sub-molecular mechanism of genetic epilepsy. Front. Mol. Neurosci. 15:958747. doi: 10.3389/fnmol.2022.958747
Luo, S., Liu, Z. G., Wang, J., Luo, J. X., Ye, X. G., Li, X., et al. (2022). Recessive LAMA5 Variants Associated With Partial Epilepsy and Spasms in Infancy. Front. Mol. Neurosci. 15:825390. doi: 10.3389/fnmol.2022.825390
Luo, S., Ye, X. G., Jin, L., Li, H., He, Y. Y., Guan, B. Z., et al. (2023). SZT2 variants associated with partial epilepsy or epileptic encephalopathy and the genotype-phenotype correlation. Front. Mol. Neurosci. 16:1162408. doi: 10.3389/fnmol.2023.1162408
Macdonald, R. L., Kang, J. Q., and Gallagher, M. J. (2010). Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 588, 1861–1869.
Maljevic, S., Møller, R. S., Reid, C. A., PéREZ-Palma, E., Lal, D., May, P., et al. (2019). Spectrum of GABAA receptor variants in epilepsy. Curr. Opin. Neurol. 32, 183–190.
Papasergi-Scott, M. M., Robertson, M. J., Seven, A. B., Panova, O., Mathiesen, J. M., and Skiniotis, G. (2020). Structures of metabotropic GABAB receptor. Nature 584, 310–314.
Romoli, M., Mazzocchetti, P., D’Alonzo, R., Siliquini, S., Rinaldi, V. E., Verrotti, A., et al. (2019). Valproic acid and epilepsy: from molecular mechanisms to clinical evidences. Curr. Neuropharmacol. 17, 926–946.
Samarut, É, Swaminathan, A., RICHé, R., Liao, M., Hassan-Abdi, R., Renault, S., et al. (2018). γ−Aminobutyric acid receptor alpha 1 subunit loss of function causes genetic generalized epilepsy by impairing inhibitory network neurodevelopment. Epilepsia 59, 2061–2074. doi: 10.1111/epi.14576
Sanacora, G., Mason, G. F., Rothman, D. L., Hyder, F., Ciarcia, J. J., Ostroff, R. B., et al. (2003). Increased cortical GABA concentrations in depressed patients receiving ECT. Am. J. Psychiatry 160, 577–579.
Shi, Y.-W., Zhang, Q., Cai, K., Poliquin, S., Shen, W., Winters, N., et al. (2019). Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 142, 3028–3044. doi: 10.1093/brain/awz250
Tomita, S. (2019). Molecular constituents and localization of the ionotropic GABA receptor complex in vivo. Curr. Opin. Neurobiol. 57, 81–86.
Vicini, S., Ferguson, C., Prybylowski, K., Kralic, J., Morrow, A. L., and Homanics, G. E. (2001). GABAA receptor α1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J. Neurosci. 21, 3009–3016. doi: 10.1523/JNEUROSCI.21-09-03009.2001
Wang, J.-Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X.-R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Wirrell, E., Tinuper, P., Perucca, E., and Moshé, S. L. (2022). Introduction to the epilepsy syndrome papers. Wiley Online Library.
Yan, H. J., He, Y. Y., Jin, L., Guo, Q., Zhou, J. H., and Luo, S. (2022). Expanding the phenotypic spectrum of KCNK4: From syndromic neurodevelopmental disorder to rolandic epilepsy. Front. Mol. Neurosci. 15:1081097. doi: 10.3389/fnmol.2022.1081097
Keywords: GABRA1 gene, epilepsy, variants, GABAAR, molecular subregional effect
Citation: Liu W-H, Luo S, Zhang D-M, Lin Z-S, Lan S, Li X, Shi Y-W, Su T, Yi Y-H, Zhou P and Li B-M (2024) De novo GABRA1 variants in childhood epilepsies and the molecular subregional effects. Front. Mol. Neurosci. 16:1321090. doi: 10.3389/fnmol.2023.1321090
Received: 13 October 2023; Accepted: 13 December 2023;
Published: 10 January 2024.
Edited by:
Qian Chen, Massachusetts Institute of Technology, United StatesReviewed by:
Yunjian Zhang, Fudan University, ChinaCopyright © 2024 Liu, Luo, Zhang, Lin, Lan, Li, Shi, Su, Yi, Zhou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing-Mei Li, enN1bGJtQGhvdG1haWwuY29t; Peng Zhou, emhvdXBlbmdsYW9kaWFvQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.