 Yun-Yan He1,2†
Yun-Yan He1,2† Sheng Luo2†
Sheng Luo2† Liang Jin2,3
Liang Jin2,3 Peng-Yu Wang2Jie Xu2Hong-Liang Jiao4Hong-Jun Yan5Yao Wang5
Peng-Yu Wang2Jie Xu2Hong-Liang Jiao4Hong-Jun Yan5Yao Wang5 Qiong-Xiang Zhai6Jing-Jing Ji2Weng-Jun Zhang2
Qiong-Xiang Zhai6Jing-Jing Ji2Weng-Jun Zhang2 Peng Zhou2
Peng Zhou2 Hua Li5
Hua Li5 Wei-Ping Liao2Song Lan7*Lin Xu1* for the China Epilepsy Gene 1.0 Project
Wei-Ping Liao2Song Lan7*Lin Xu1* for the China Epilepsy Gene 1.0 Project- 1Department of Neurology, Women and Children’s Hospital, Qingdao University, Qingdao, China
- 2Department of Neurology, Institute of Neuroscience, Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, The Second Affiliated Hospital, Guangzhou Medical University, Guangzhou, China
- 3Department of Neurology, The Affiliated Nanhua Hospital, Hengyang Medical School, University of South China, Hengyang, China
- 4Department of Neurosurgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 5Epilepsy Center, Guangdong 999 Brain Hospital, Guangzhou, China
- 6Department of Pediatrics, Guangdong General Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
- 7Department of Neurology, Maoming People’s Hospital, Maoming, China
Background: The DLG3 gene encodes disks large membrane-associated guanylate kinase scaffold protein 3, which plays essential roles in the clustering of N-methyl-D-aspartate receptors (NMDARs) at excitatory synapses. Previously, DLG3 has been identified as the causative gene of X-linked intellectual developmental disorder—90 (XLID-90; OMIM# 300850). This study aims to explore the phenotypic spectrum of DLG3 and the genotype-phenotype correlation.
Methods: Trios-based whole-exome sequencing was performed in patients with epilepsy of unknown causes. To analyze the genotype-phenotype correlations, previously reported DLG3 variants were systematically reviewed.
Results: DLG3 variants were identified in seven unrelated cases with epilepsy. These variants had no hemizygous frequencies in controls. All variants were predicted to be damaging by silico tools and alter the hydrogen bonds with surrounding residues and/or protein stability. Four cases mainly presented with generalized seizures, including generalized tonic-clonic and myoclonic seizures, and the other three cases exhibited secondary generalized tonic-clonic seizures and focal seizures. Multifocal discharges were recorded in all cases during electroencephalography monitoring, including the four cases with generalized discharges initially but multifocal discharges after drug treating. Protein-protein interaction network analysis revealed that DLG3 interacts with 52 genes with high confidence, in which the majority of disease-causing genes were associated with a wide spectrum of neurodevelopmental disorder (NDD) and epilepsy. Three patients with variants locating outside functional domains all achieved seizure-free, while the four patients with variants locating in functional domains presented poor control of seizures. Analysis of previously reported cases revealed that patients with non-null variants presented higher percentages of epilepsy than those with null variants, suggesting a genotype-phenotype correlation.
Significance: This study suggested that DLG3 variants were associated with epilepsy with/without NDD, expanding the phenotypic spectrum of DLG3. The observed genotype-phenotype correlation potentially contributes to the understanding of the underlying mechanisms driving phenotypic variation.
1 Introduction
Neurodevelopmental disorder (NDD) is the most common neurological disease in children, including intellectual development disorder (ID), developmental speech or language disorder (DSD), autism spectrum disorder (ASD), developmental learning disorder (DLD), attention deficit hyperactivity disorder (ADHD), tic disorder (TD) and others (Zhu et al., 2023). Epilepsy is one of the most common comorbidities in patients with neurodevelopmental disorders, whereas up to 26% of individuals with NDDs have epilepsy (McGrother et al., 2006; Ali Rodriguez et al., 2018; Heyne et al., 2018; Turner et al., 2021). NDD is a vital risk factor for epileptogenesis, and frequent seizures can also lead to or worsen NDD (Pisella et al., 2019). Increasing evidence has highlighted the genetic overlap of both epilepsy and NDD (Shimizu et al., 2022). Previously, a series of genes have been identified as causative genes for both epilepsy and NDD, such as GRIN2A, GRIN2B, BCOR, FRMPD4, APC2, NEXMIF, SZT2, SHROOM4, BRWD3, KCNK4, and UNC79 (Liu et al., 2021; Bian et al., 2022; Li et al., 2022, 2023; Yan et al., 2022; Bayat et al., 2023; Jin et al., 2023; Luo et al., 2023; Tian et al., 2023; Wang et al., 2023; Ye et al., 2023). However, the majority of overlapping genetic etiologies for epilepsy and NDD remain undetermined.
The N-methyl-D-aspartate receptor (NMDAR) is one of the main excitatory receptors in the central nervous system, with essential roles in regulating neuroplasticity, excitatory neurotransmission, and the development of learning and memory (Chen et al., 2021). Genes encoding NMDAR subunits (such as GRIN1, GRIN2A, and GRIN2B) have been identified to be associated with broad-spectrum phenotypes, including epilepsies, epilepsies with NDD, and NDD without seizures (Endele et al., 2010; Charron et al., 2022). Similar to NMDAR genes, an increasing number of genes encoding NMDAR-associated proteins have been identified to be associated with a broad phenotypic spectrum, such as DLG4 and SYNGAP1 (Moutton et al., 2018; Agarwal et al., 2019). The phenotypic spectrum of other genes encoding NMDAR-associated proteins warrants further study.
The DLG3 gene (OMIM* 300189), located in Xq13.1, encodes disks large membrane-associated guanylate kinase scaffold protein 3. The DLG3 protein, also known as synapse-associated protein 102 (SAP102), is an NMDAR-associated protein with essential roles in clustering of NMDARs at excitatory synapses and regulating cell proliferation. It is highly expressed in the human brain, particularly in the cortex.1 Hemizygous knockout of DLG3 in mice led to abnormalities in the cortex and synapse morphology, impairment in spatial learning, and abnormal excitatory postsynaptic currents (Cuthbert et al., 2007). Previously, DLG3 has been identified as the causative gene of X-linked intellectual developmental disorder—90 (XLID-90; OMIM# 300850). It is unknown whether DLG3 is associated with epilepsy and shares a broad phenotypic spectrum similar to NMDAR/NMDAR-associated genes.
In this study, we performed trio-based whole-exome sequencing (WES) in a cohort of patients with childhood epilepsy without acquired causes. Six novel DLG3 variants were identified in seven unrelated cases with heterogeneous epilepsies, including three with epilepsy and four with epilepsy and ID. Previously reported DLG3 variants were systematically reviewed to explore the underlying mechanism of phenotypic heterogeneity. This study suggested that DLG3 may be associated with epilepsy without neurodevelopmental disability.
2 Materials and methods
2.1 Patients
The patients were recruited from multiple centers through the platform of China Epilepsy Project 1.0, including the Women and Children’s Hospital affiliated with Qingdao University, the Second Affiliated Hospital of Guangzhou Medical University, the First Affiliated Hospital of Zhengzhou University, Maoming People’s Hospital, Guangdong 999 Brain Hospital, and Guangdong Province People’s Hospital. Patients with acquired epilepsy were excluded, such as trauma, immunity, and infection. Detailed clinical information, including disease progression, prognosis, personal history, family history and results from general and neurological examinations, was collected from patients or their families.
2.2 Whole-exon sequencing
Blood samples of the probands were collected to extract genomic DNA. Whole-exon sequencing was performed using a NextSeq500 sequencing instrument (Illumina, San Diego, CA, USA) following the standard procedures previously described (Wang et al., 2018). The sequencing data were generated using massively parallel sequencing with an average depth of > 125x and > 98% coverage of the capture region on the chip, ensuring the acquisition of high-quality reads. These reads were mapped to the Genome Reference Consortium Human genome build 37 by Burrows-Wheeler alignment. Variants were called and qualified with the Genome Analysis Toolkit (DePristo et al., 2011). Sanger sequences were used to validate candidate variants.
2.3 Genetic analysis
To identify potentially pathogenic variants, an individualized analytical approach was employed for each case, following the methodology outlined in our previous study (Li et al., 2021; Wang et al., 2021). We screened DLG3 variants with the explainable origination for genetic diseases, including de novo and hemizygous mutations. All DLG3 variants identified in this study were annotated into the reference transcript NM_021120.4.
2.4 Literature review and analysis of genotype-phenotype correlation
The DLG3 variants and associated clinical information were systematically reviewed from the PubMed database and the Human Gene Mutation Database (HGMD) up to September 2023. Variants with undefined origination or unexplained origination for the occurrence of genetic diseases were excluded. Null variants that result in truncated protein were employed to identify variants, including canonical splice site variants frameshift, nonsense, and initiation codon (Richards et al., 2015). Other variants were classified into non-null variants, such as missense and intron variants.
2.5 Bioinformatic analyses
In order to evaluate the detrimental impact of candidate missense variants, protein modeling was conducted using the Iterative Threading ASSEmbly Refinement software (I-TASSER) (Yang and Zhang, 2015). PyMOL Molecular Graphics System (version 2.3.2; Schrödinger, LLC; New York, USA) was utilized to visualize and analyze the protein structure changes. The protein stability changes of each variant were predicted using the I-Mutant Suite server (Capriotti et al., 2005), which indicated the free energy change (ΔΔG). Negative ΔΔG values indicate a decrease in mutant protein stability. The VarSite web server was used to analyze amino acid and hydrophobicity changes (Laskowski et al., 2020).
2.6 Protein-protein interaction (PPI) network analysis
The protein-protein interaction (PPI) network of the DLG3 protein was analyzed using the STRING database (version: 12.0; University of Zurich, Zurich, Switzerland) (Szklarczyk et al., 2023). The interactive genes with a confidence score ≥ 0.7 were taken into analysis. The PPI networks were visualized by Cytoscape (version 3.10.1).
2.7 Statistical analysis
Statistical analyses were conducting using R (version 4.0.3). The two-tailed Fisher’s exact test was used to compare the differences between groups. P-value < 0.05 was considered statistically significant.
3 Results
3.1 Identification of DLG3 variants
Six DLG3 variants were identified in seven unrelated male individuals, including c.18C > G/p.His6Gln, c.128G > T/p.Gly43Val, c.463C > T/p.Pro155Ser, c.593G > A/p.Arg198Gln, c.1415G > A/p.Arg472His, and c.1998T > A/p.Asn666Lys (Figures 1A, B and Table 1). The variant c.128G > T/p.Gly43Val was recurrently identified in cases 2 and 3. All variants originated from their asymptomatic mothers, consistent with Mendelian X-linked recessive (XLR) inheritance.
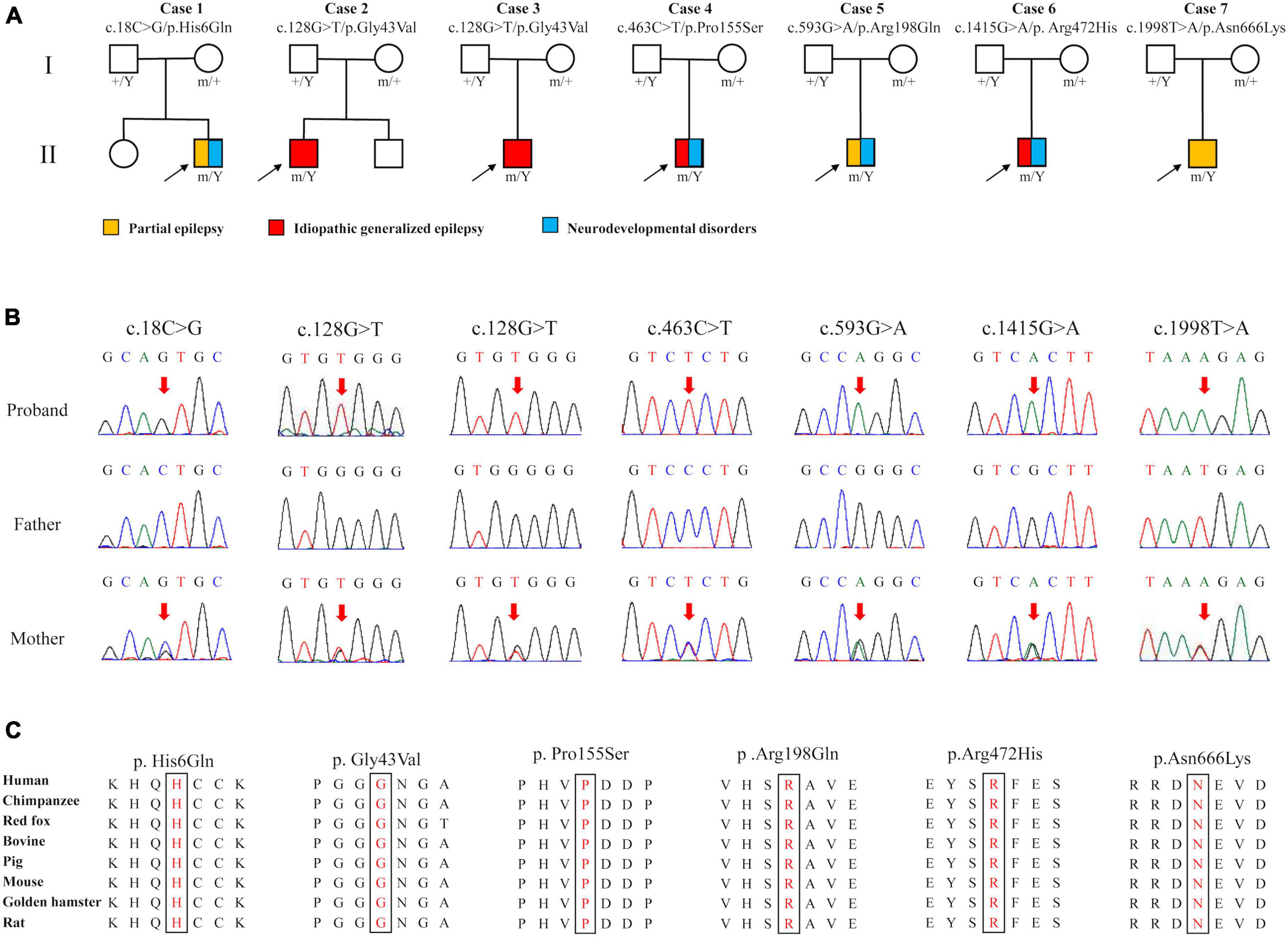
Figure 1. Genetic data of cases with DLG3 variants. (A) Pedigrees of the cases with DLG3 mutations and their corresponding phenotypes. (B) DNA sequencing chromatograms of the cases with DLG3 variants. Red arrows indicate the positions of the mutations. (C) The amino acid sequence alignment showed that the six identified missense variants are residues with high conservation in mammals.

Table 1. Clinical characteristics of individuals with DLG3 variants.
These variants were absent in the male controls of the gnomAD database (Table 2). The six variants were found to be located in residues that were highly conserved among mammals based on amino acid sequence alignment (Figure 1C). All variants were predicted to be “damaging” or “conserved” by more than five in silico tools (Table 2).
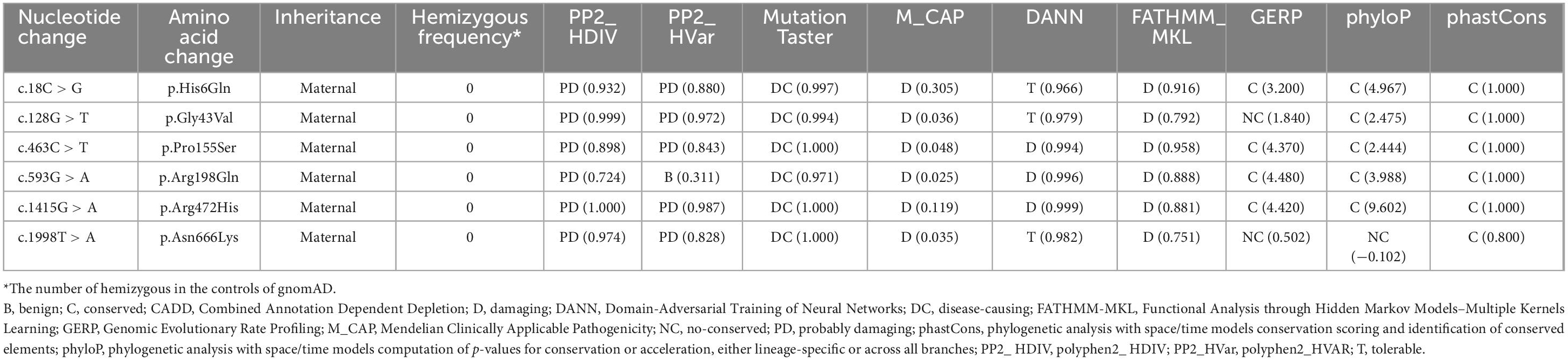
Table 2. Genetic characteristics of DLG3 mutations identified in this study.
The molecular effects of the missense variants were assessed through protein modeling and visualized using PyMOL. Among the six missense variants, three were predicted to alter hydrogen bonds with neighboring residues. The remaining three missense variants (p.His6Gln, p.Gly43Val, and p.Pro155Ser) were not predicted to alter hydrogen bonds with surrounding residues but were predicted to decrease the protein stability (Figures 2A, C). Four variants (p.His6Gln, p.Gly43Val, p.Pro155Ser, and p.Arg472His) were also predicted to cause hydrophobicity changes, based on the Fauchère and Pliska hydrophobicity scale (Figure 2B).
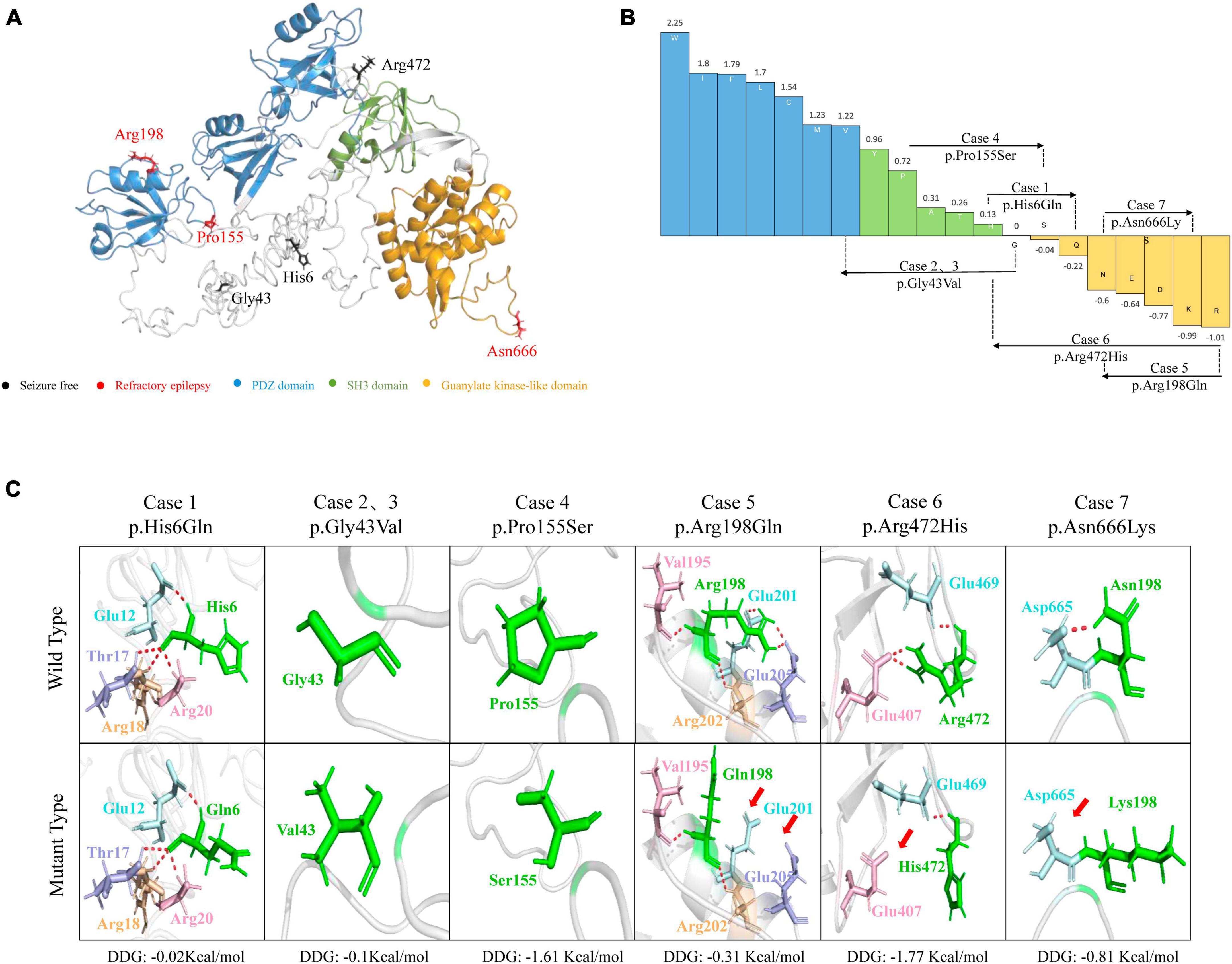
Figure 2. Schematic illustration of hydrogen bond changes, hydrophobicity changes and protein stability prediction. (A) Three-dimensional structure of DLG3 protein and localization of DLG3 variants of this study. (B) Fauchère and Pliska hydrophobicity scale exhibited the hydrophobicity of 20 amino acids. Abscissa: from left to right, hydrophobicity gradually decreased. Blue amino acids are hydrophobic, green amino acids are neutral, and yellow amino acids are hydrophilic. Amino acids with high positive values are more hydrophobic, whereas amino acids with low negative values are more hydrophilic. (C) Hydrogen bond changes and Gibbs free energy of folding (DDG) values of DLG3 variants. The residues where the mutations occurred are shown as green rods. The red dotted line represents hydrogen bonds. Arrows indicate the positions with hydrogen bond changes. Three of six variants were predicted to alter hydrogen bonds with surrounding residues. All variants were predicted to decrease protein stability.
No pathogenic or likely pathogenic variants in other epilepsy-related genes were identified in the seven patients (Supplementary Table 2) (Wang et al., 2017).
3.2 Clinical features of the cases with DLG3 variants
The detailed clinical features of the patients with DLG3 variants were summarized in Table 1. The patients were all diagnosed with epilepsy. The onset age of seizures ranged from 2 years old to 16 years old, with a median onset age of 4 years. Four cases mainly presented with generalized seizures (Cases 2, 3, 4 and 6), including generalized tonic-clonic and myoclonic seizures. Three cases (Cases 1, 5, and 7) exhibited secondary generalized tonic-clonic seizures and focal seizures. Seizure-free was achieved in cases 1, 2, 3, and 6. Multifocal discharges were recorded in all cases during EEG monitoring (Figures 3A–D). Generalized discharges were initially presented in cases 2, 3, 4 and 6, but multifocal discharges were also exhibited after being treated with antiseizure medications. MRI scans detected no structural abnormities of the brain, except case 4 with gray matter heterotopia (Figure 3E). Neurodevelopmental abnormalities were also exhibited in cases 1, 4, 5 and 6.
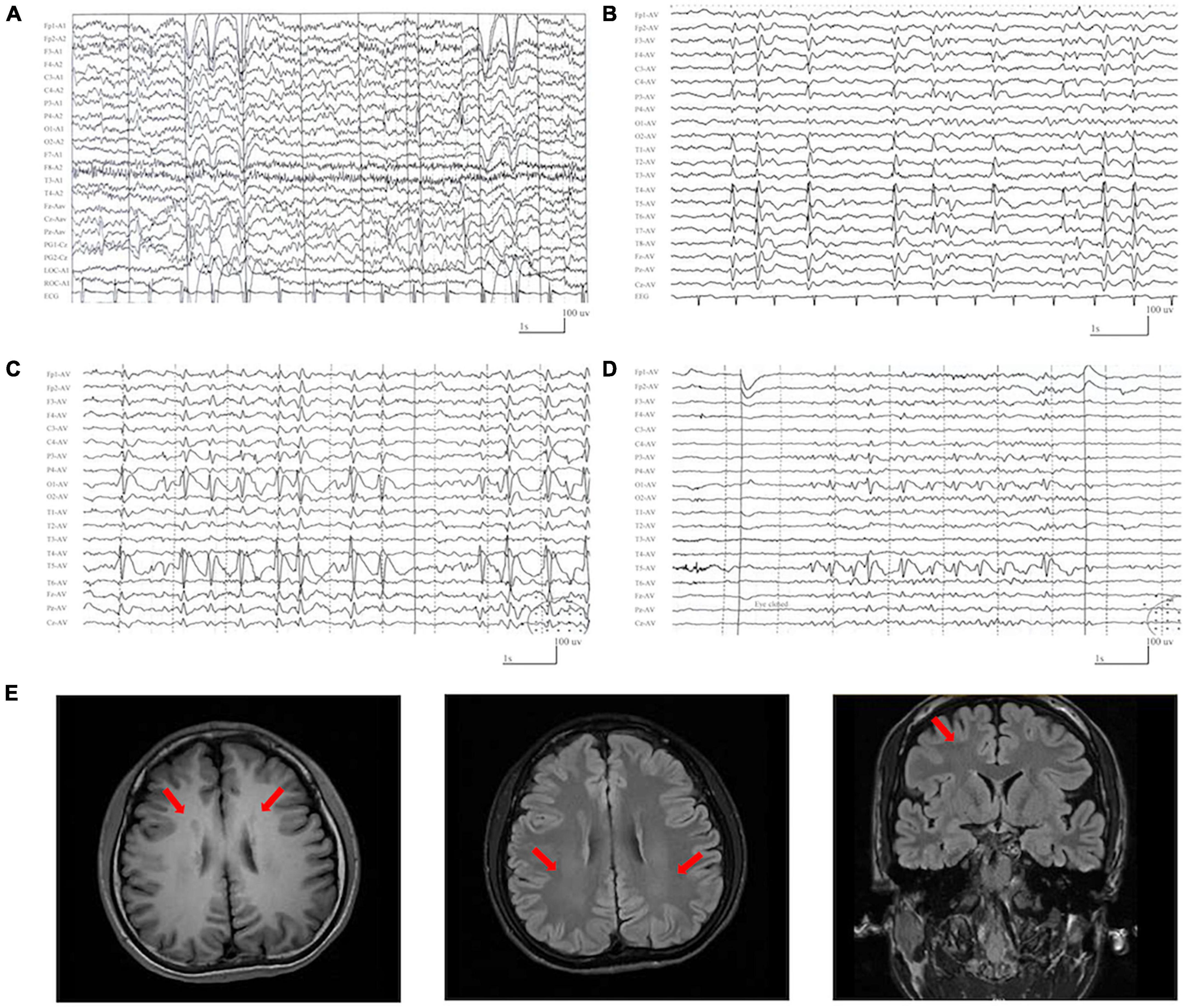
Figure 3. Representative EEG and magnetic resonance imaging (MRI) of the cases with DLG3 variants. (A) Interictal EEG of case 1 at 2 years of age showed bilateral and multifocal spike-slow waves. (B) Interictal EEG of case 6 at 7 years of age showed bilateral multifocal spikes and spikes-slow waves (obtained). (C,D) Interictal EEG of case 7 at 9 years of age showed spike-slow waves predominant at the left posterior head (C) and eye closure sensitivity (D). (E) The MRI of case 4 at the age of 16 years showed gray matter heterotopia.
3.3 Location of DLG3 variants and molecular subregion effects
The SAP102 protein, contains three tandem amino-terminal PSD-95/Dlg/ZO-1 (PDZ) domains mediating protein-protein interactions, a src homology 3 (SH3) domain, and a C-terminal guanylate-kinase like (GK) domain (Kuwahara et al., 1999). The variants Pro155Ser and Arg198Gln were located in PDZ1 domain, and Asn666Lys was located in guanylate-kinase like domain, while other three variants (p.His6Gln, p.Gly43Val, and p.Arg472His) were located outside the functional domains (Figure 4).
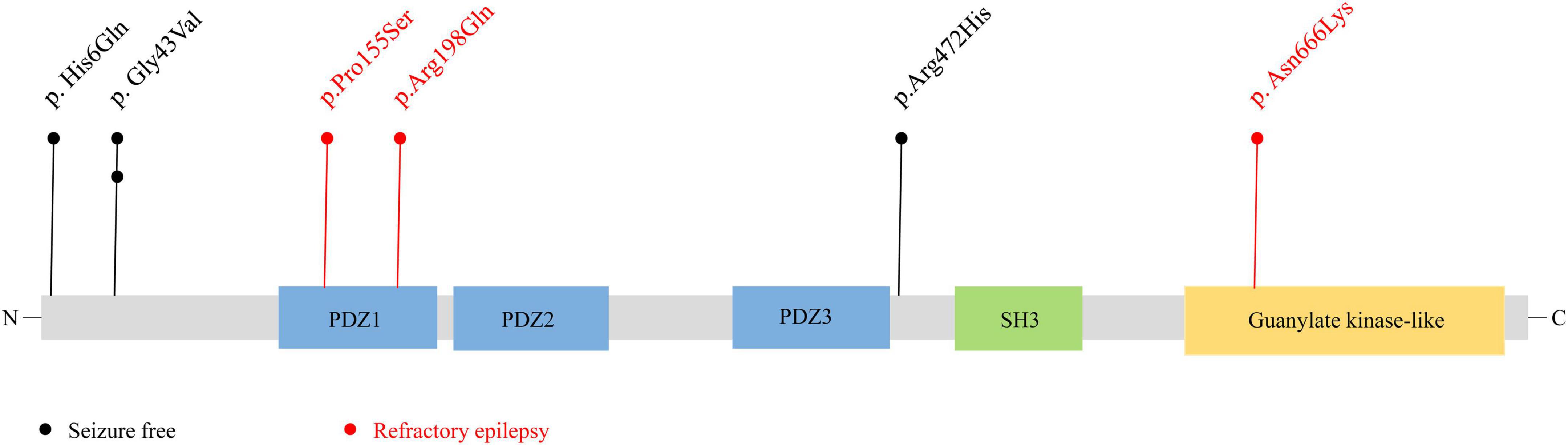
Figure 4. Schematic illustration of variant location. Schematic diagram of the DLG3 protein and the localization of the DLG3 variants identified in this study. Variants identified in patients with seizure-free are shown in black. Variants identified in patients with poor control of seizures are shown in red.
Previous studies showed that the location of the variants was associated with the variation of clinical phenotypes (Tang et al., 2020). It is noted that the three variants locating outside functional domains were all identified in patients achieving seizure-free (Cases 1, 2, 3 and 6), while other three variants locating in functional domains were all identified in the patients with poor control of seizures (Case 4, 5 and 7).
3.4 Genotype-phenotype correlation
The previously reported variants and associated clinical information were systemically reviewed by using the HGMD (version: HGMD Professional 2023.1) and PubMed databases. A total of 27 variants were identified in 46 patients, including thirteen missense variants, five nonsense variants, five canonical splicing site variants, one intron variant, and three frameshift variants (Figure 5A and Supplementary Table 1). The majority of DLG3 variants were of XLR pattern, and only four variants were of X-linked dominant (XLD) pattern. Further analysis showed that the XLR variants were all inherited from asymptomatic mothers, whereas XLD variants were all de novo (Figures 5B–D). The phenotype of male patients in the cases with XLD variants was more severe than that of female patients (Tarpey et al., 2004; Sandestig et al., 2020). To analyze the relationship between genotype and phenotype, the genotype was classified into null variants and non-null variants. Patients with non-null variants presented higher percentages of epilepsy with/without NDD than those with null variants (10/14 vs. 1/13; p = 0.001) (Figure 5E), potentially suggesting a genotype-phenotype correlation.
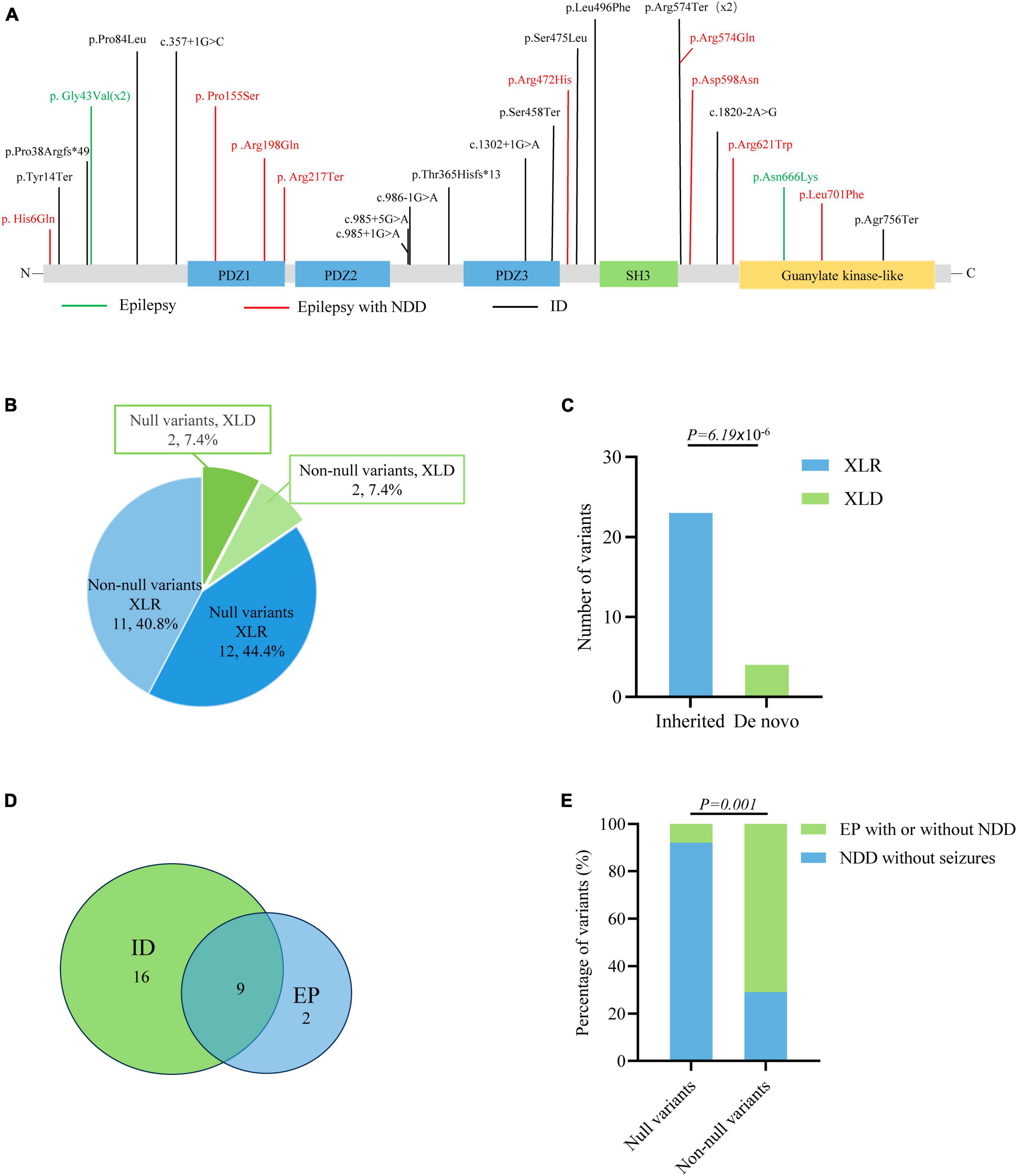
Figure 5. Genotype–phenotype correlation analysis of DLG3 variants. (A) Schematic diagram of the DLG3 protein and the localization of the DLG3 variants identified in this study and reported previously. Variants associated with intellectual disability (ID) are shown in black, variants associated with epilepsy are shown in green, and variants associated with epilepsy and neurodevelopmental disorders (NDDs) are shown in red. * Means termination codon. (B) Pie chart of the genotype distribution of DLG3 variants. (C) The stacked bar chart of inherited patterns and origination of the DLG3 variants (n = 27). (D) Venn diagram of the phenotypes of patients with DLG3 variants. A total of 27 variants were identified, including sixteen variants associated with intellectual disability, nine variants associated with both intellectual disability and epilepsy, and two variants associated with epilepsy. (E) The stacked bar chart of phenotypes of the variants with different genotypes. Variants with non-null variants presented higher percentages in patients with epilepsy with/without NDD than null variants (10/14 vs. 1/13; p = 0.001).
3.5 Protein-protein interactive network analysis
Protein-protein interactions, the basis of cellular metabolism, are indispensable in all life activities. The interactive genes tend to be associated with similar phenotypes. We thus further investigated the interactive partner of DLG3 protein and their associated phenotypes to explore the underlying phenotypic spectrums of DLG3 gene. DLG3 protein is predicted to interact with 52 proteins with high confidence (minimum required interaction score ≥ 0.7, String database). The majority of disease-causing genes encoding the proteins interacting with the DLG3 protein are associated with neurological diseases, including five causative genes of both epilepsy and NDD (GRIN1, GRIN2A, GRIN2B, GRIK2, and NBEA), eleven causative genes of NDD with seizures (GRIA1, GRIA2, CASK, NRXN1, NLGN3, NEDD4L, SHANK3, GPSM2, DLG4, SYNGAP1, and UBE3A), and four causative genes of NDD without seizures (NLGN4X, CACNG2, SHANK2, and NLGN1) (Figure 6). These broad phenotypes of genes encoding proteins interacting with DLG3 protein provided possible clues for the association between DLG3 and epilepsy.
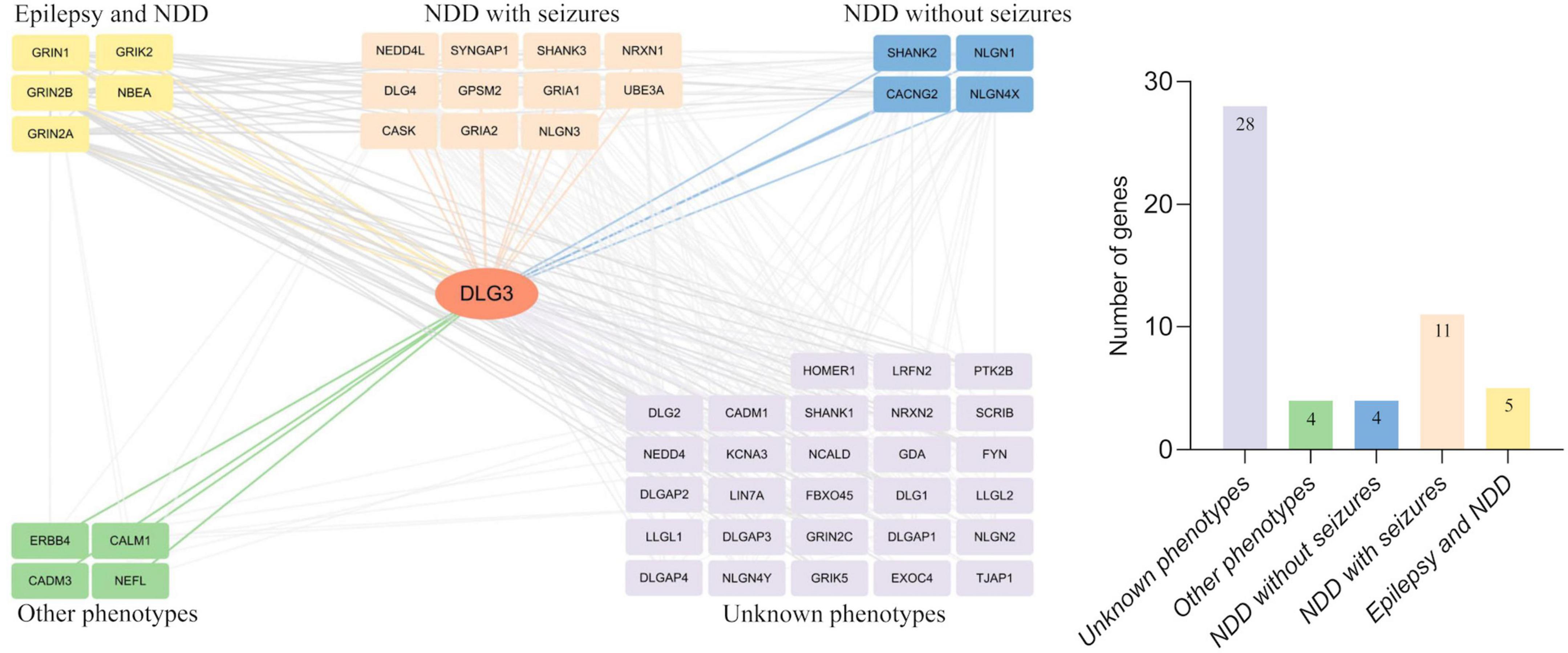
Figure 6. Analysis of genes interacting with DLG3. The DLG3 protein interacted with 52 proteins with high confidence [(left), minimum required interaction score ≥ 0.7, STRING database], including five genes identified to be associated with both epilepsy and neurodevelopmental disorder (NDD), eleven genes identified to be associated with NDD with seizures, four genes identified to be associated with NDD without seizures, four genes identified to be associated with other phenotypes, and 28 genes not identified to be associated with phenotypes (right).
4 Discussion
In this study, we identified six novel DLG3 variants in seven unrelated patients with heterozygous epilepsies, including three with only epilepsy and four with epilepsy and NDD. The variants were not presented as hemizygous states in controls. These variants were located in residues with high conservation and were predicted to be damaging by multiple in silico tools. Protein modeling showed that the variants altered the hydron bonding with surrounding residues and protein stability. Genotype-phenotype correlation analysis revealed that patients with non-null variants presented higher percentages of epilepsy with/without NDD than those with null variants. This study suggested that DLG3 variants were potentially associated with epilepsy with or without NDD. The genotype-phenotype correlation helps in understanding the underlying mechanism of phenotypic variation.
The DLG3 gene was highly conserved with homologs in Drosophila melanogaster, Mus musculus, and Homo sapiens. In Drosophila melanogaster, the majority of knockout DLG3 ortholog lines presented preadult lethality and abnormities in neuroanatomy neurophysiology (FlyBase ID: FBgn0001624). In Mus musculus, hemizygous knockout of DLG3 led to abnormalities in the cortex and synapse morphology, impairment in spatial learning, and abnormal excitatory postsynaptic currents (Cuthbert et al., 2007). In Homo sapiens, data from large-scale genome sequencing reveal that DLG3 is intolerant to loss-of-function (LOF) variants, with a probability of being LOF intolerant (pLi) of 1. The variants of the presented study were predicted to be “damaged” by diverse in silico tools and alter hydrogen bonding and/or polarity to disrupt protein stability, which may be associated with LOF. This evidence suggests that LOF may be the pathogenic mechanism for DLG3.
The DLG3 protein is an important scaffold protein interacting with synaptic proteins, especially in excitatory synapses (Sheng and Hoogenraad, 2007; Murata and Constantine-Paton, 2013). Protein–protein interaction network analysis showed that DLG3 interacted with 52 genes, of which 24 genes have been identified to be associated with disease. The sixteen genes are associated with NDD with seizures or developmental and epileptic encephalopathy (Figure 6). With increasing disease-causing variants identified, sixteen genes (including GRIN1, GRIN2A, GRIN2B, GRIK2, NBEA, GRIA1, GRIA2, NEDD4L, CASK, NRXN1, NLGN3, SHANK3, GPSM2, DLG4, SYNGAP1, and UBE3A) have been identified to be associated with a wide phenotypic spectrum from mild epilepsy to severe DEE/NDD with seizures (Endele et al., 2010; Harrison et al., 2011; Moog et al., 2011; Tan et al., 2011; Doherty et al., 2012; Soorya et al., 2013; Broix et al., 2016; Lemke et al., 2016; Moutton et al., 2018; Mulhern et al., 2018; Agarwal et al., 2019; Salpietro et al., 2019; Liu et al., 2021; Stolz et al., 2021; Blakes et al., 2022; Ismail et al., 2022). The wide phenotypic spectrum of DLG3-interacting genes provides possible clues for the association between DLG3 and epilepsy. Previously, DLG3 variants have been reported to be associated with XLID-90 (Supplementary Table 1). This study identified DLG3 variants in three patients with mild epilepsy without NDD, broadening the phenotypic spectrum of DLG3.
The DLG3 protein, contains three PDZ domains, a SH3 domain, and a GK domain. The PDZ domains interact with a diverse range of membrane proteins, such as ionotropic glutamate receptors, cell-surface adhesion molecules (Wei et al., 2018). The SH3 and GK domains interact with cytoskeletal proteins and intracellular signaling complexes (Wei et al., 2018). In this study, the three patients with variants locating outside functional domains all achieved seizure-free, while the four patients with variants locating in functional domains presented poor control of seizures, suggesting a molecular sub-regional effect. Compared with those in functional domain, variants locating out the functional domain may cause mild damaging effect and subsequently favorable outcomes, which may be one of the explanations of phenotypic severity.
The DLG3 protein, predominantly distributed in the postsynaptic densities of excitatory synapses, plays vital roles in synaptic development and synaptic transmission (Sans et al., 2000; Cuthbert et al., 2007; Elias and Nicoll, 2007; Elias et al., 2008; Zheng et al., 2011; Wei et al., 2015). Synaptic abnormalities are one of the core processes in the occurrence of neurodevelopmental disorders and epilepsies (Blanpied and Ehlers, 2004). The genotype-phenotype correlation analysis showed that patients with non-destructive variants mainly exhibited epilepsy with or without NDD, whereas patients with destructive variants primarily presented with intellectual disability (Figure 5). It is possible that phenotypic heterogeneity is associated with the damaging effect of DLG3 variants (Figure 7). Variants of mild or moderate damage would cause subtle functional alteration of synapse with abnormal electrophysiological activity and subsequently epilepsies and/or intellectual disability; while variants of complete loss of function would lead to decreased synaptic conduction, subsequently intellectual disability. However, the functional alternations of all identified DLG3 variants, including those previously reported variants, were not experimentally validated. The association between phenotypic heterogeneity and detailed mechanisms of variants, such as gain of function and dominant-negative effects, warrants functional studies.
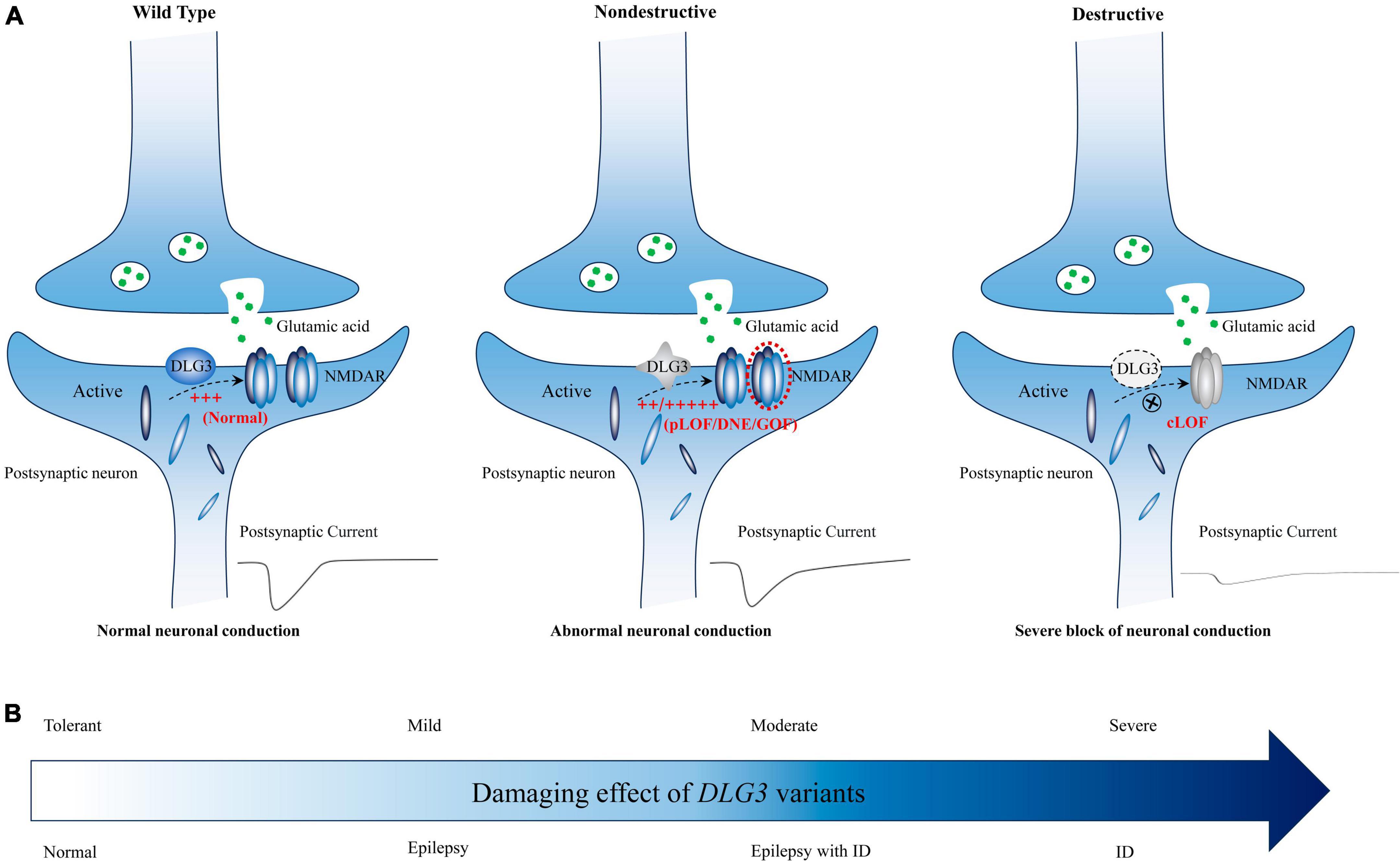
Figure 7. Schematic diagram of the possible association between DLG3 variants on synaptic damage and related phenotypes. (A) Possible synaptic damage and postsynaptic current alteration of various DLG3 variants. cLOF, complete loss of function; DNE, dominant-negative effects; GOF, gain of function; NMDARE, N-methyl-D-aspartate receptors; pLOF, partial loss of function. (B) The possible association between phenotypes and synaptic damage. Variants of mild or moderate damage would cause subtle functional alteration of synapse with abnormal electrophysiological activity and subsequently epilepsies and/or intellectual disability; while variants of complete loss of function would lead to decreased synaptic conduction, subsequently intellectual disability.
The DLG3 protein mediates NMDA receptor trafficking and contributes to NMDA receptor clustering and anchoring in the PSD (Lau et al., 1996; Nagano et al., 1998). Dysregulation of NMDAR subunits is one of the important mechanisms of partial epilepsy (Xu and Luo, 2018). In this study, the cases with DLG3 variants all presented with multifocal discharges on EEG, in which four cases exhibited generalized discharges initially but also multifocal discharges after being treated with antiseizure medications. The phenotype of multifocal discharges potentially reflects the functional impact of DLG3 variants in mediating NMDAR trafficking and may be one of the core features of patients with DLG3 variants. However, the detailed functional alterations of these variants remain to be functionally validated.
In this study, gray matter heterotopia (GMH) was presented in the patient of case 4, which was distinguished from the other six cases. Abnormities of brain structure were also not presented in the previously reported cases (Supplementary Table 1). The DLG3 variants may partially contribute to the phenotype of these cases but not gray matter heterotopia. However, Mendelian variants associated with cortical malformations were not identified in this case (Supplementary Table 2). It is unknown whether other factors are involved in the pathogenicity, which warrants further verification.
In summary, this study suggested that DLG3 variants were associated with epilepsy with/without NDD, expanding the phenotypic spectrum of DLG3. The observed genotype-phenotype correlation contributes to our understanding of the underlying mechanisms of phenotypic variation.
Data availability statement
The data presented in the study are deposited in the NCBI-GenBank database, accession numbers OR818645 and OR818665.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Women and Children’s Hospital Affiliated with Qingdao University. Written informed consent to participate in this study was provided by the participant’s legal guardian/next of kin. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
Y-YH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. ShL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LJ: Software, Writing – review & editing. P-YW: Methodology, Writing – review & editing. JX: Resources, Writing – review & editing. H-LJ: Resources, Writing – review & editing. H-JY: Resources, Writing – review & editing. YW: Resources, Writing – review & editing. Q-XZ: Resources, Writing – review & editing. J-JJ: Resources, Writing – review & editing. W-JZ: Resources, Writing – review & editing. PZ: Resources, Writing – review & editing. HL: Resources, Writing – review & editing. W-PL: Writing – review & editing. SoL: Resources, Writing – review & editing. LX: Writing – review & editing, Funding acquisition, Methodology, Resources.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Talent Training Fund of the Women and Children’s Hospital Affiliated with Qingdao University for LX. The funders had no role in the study design, data collection and analysis, and decision to publish or prepare the manuscript.
Acknowledgments
The help of patients and clinicians participating in this work is greatly appreciated.
Web resources
OMIM: https://omim.org/.
GTEx database: https://www.gtexportal.org/.
PubMed database: http://www.ncbi.nlm.nih.gov/pubmed/.
Human Gene Mutation Database (HGMD): http://www.hgmd.cf.ac.uk/ac/index.php.
Iterative Threading ASSEmbly Refinement (I-TASSER): https://zhanglab.ccmb.med.umich.edu/I-TASSER/.
VarSite database: VarSite home page (ebi.ac.uk).
I-Mutant Suite server: https://folding.biofold.org/i-mutant/i-mutant2.0.html.
STRING database: https://string-db.org/.
FlyBase database: http://flybase.org/.
Mouse Genome Informatics (MGI): http://www.informatics.jax.org/.
Genome Aggregation Database (GnomAD): https://gnomad.broadinstitute.org/.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1290919/full#supplementary-material
Footnotes
References
Agarwal, M., Johnston, M. V., and Stafstrom, C. E. (2019). SYNGAP1 mutations: Clinical, genetic, and pathophysiological features. Int. J. Dev. Neurosci 78, 65–76. doi: 10.1016/j.ijdevneu.2019.08.003
Ali Rodriguez, R., Joya, C., and Hines, R. M. (2018). Common ribs of inhibitory synaptic dysfunction in the umbrella of neurodevelopmental disorders. Front. Mol. Neurosci. 11:132. doi: 10.3389/fnmol.2018.00132
Bayat, A., Liu, Z., Luo, S., Fenger, C. D., Hojte, A. F., Isidor, B., et al. (2023). A new neurodevelopmental disorder linked to heterozygous variants in UNC79. Genet. Med. 25:100894. doi: 10.1016/j.gim.2023.100894
Bian, W. J., Li, Z. J., Wang, J., Luo, S., Li, B. M., Gao, L. D., et al. (2022). SHROOM4 variants are associated with X-linked epilepsy with features of generalized seizures or generalized discharges. Front. Mol. Neurosci. 15:862480. doi: 10.3389/fnmol.2022.862480
Blakes, A. J. M., English, J., Banka, S., and Basu, H. (2022). A homozygous GRIN1 null variant causes a more severe phenotype of early infantile epileptic encephalopathy. Am. J. Med. Genet. A 188, 595–599. doi: 10.1002/ajmg.a.62528
Blanpied, T. A., and Ehlers, M. D. (2004). Microanatomy of dendritic spines: Emerging principles of synaptic pathology in psychiatric and neurological disease. Biol. Psychiatry 55, 1121–1127. doi: 10.1016/j.biopsych.2003.10.006
Broix, L., Jagline, H., Ivanova, E., Schmucker, S., Drouot, N., Clayton-Smith, J., et al. (2016). Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia. Nat. Genet. 48, 1349–1358. doi: 10.1038/ng.3676
Capriotti, E., Fariselli, P., and Casadio, R. I. (2005). -Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310. doi: 10.1093/nar/gki375
Charron, J. G., Hernandez, A., Bilinovich, S. M., Vogt, D. L., Bedinger, L. A., Seaver, L. H., et al. (2022). N-methyl-d-aspartate (n.d.) receptor genetics: The power of paralog homology and protein dynamics in defining dominant genetic variants. Am. J. Med. Genet. A 188, 556–568. doi: 10.1002/ajmg.a.62554
Chen, S., Xu, D., Fan, L., Fang, Z., Wang, X., and Li, M. (2021). Roles of N-Methyl-D-aspartate receptors (NMDARs) in Epilepsy. Front. Mol. Neurosci. 14:797253. doi: 10.3389/fnmol.2021.797253
Cuthbert, P. C., Stanford, L. E., Coba, M. P., Ainge, J. A., Fink, A. E., Opazo, P., et al. (2007). Synapse-associated protein 102/dlgh3 couples the NMDA receptor to specific plasticity pathways and learning strategies. J. Neurosci. 27, 2673–2682. doi: 10.1523/JNEUROSCI.4457-06.2007
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498.
Doherty, D., Chudley, A. E., Coghlan, G., Ishak, G. E., Innes, A. M., Lemire, E. G., et al. (2012). GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome. Am. J. Hum. Genet. 90, 1088–1093.
Elias, G. M., Elias, L. A., Apostolides, P. F., Kriegstein, A. R., and Nicoll, R. A. (2008). Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc. Natl. Acad. Sci. U. S. A. 105, 20953–20958. doi: 10.1073/pnas.0811025106
Elias, G. M., and Nicoll, R. A. (2007). Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 17, 343–352.
Endele, S., Rosenberger, G., Geider, K., Popp, B., Tamer, C., Stefanova, I., et al. (2010). Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 42, 1021–1026. doi: 10.1038/ng.677
Harrison, V., Connell, L., Hayesmoore, J., McParland, J., Pike, M. G., and Blair, E. (2011). Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am. J. Med. Genet. A 155A, 2826–2831. doi: 10.1002/ajmg.a.34255
Heyne, H. O., Singh, T., Stamberger, H., Abou Jamra, R., Caglayan, H., Craiu, D., et al. (2018). De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 50, 1048–1053.
Ismail, V., Zachariassen, L. G., Godwin, A., Sahakian, M., Ellard, S., Stals, K. L., et al. (2022). Identification and functional evaluation of GRIA1 missense and truncation variants in individuals with ID: An emerging neurodevelopmental syndrome. Am. J. Hum. Genet. 109, 1217–1241. doi: 10.1016/j.ajhg.2022.05.009
Jin, L., Li, Y., Luo, S., Peng, Q., Zhai, Q. X., Zhai, J. X., et al. (2023). Recessive APC2 missense variants associated with epilepsies without neurodevelopmental disorders. Seizure 111, 172–177. doi: 10.1016/j.seizure.2023.08.008
Kuwahara, H., Araki, N., Makino, K., Masuko, N., Honda, S., Kaibuchi, K., et al. (1999). A novel NE-dlg/SAP102-associated protein, p51-nedasin, related to the amidohydrolase superfamily, interferes with the association between NE-dlg/SAP102 and N-methyl-D-aspartate receptor. J. Biol. Chem. 274, 32204–32214. doi: 10.1074/jbc.274.45.32204
Laskowski, R. A., Stephenson, J. D., Sillitoe, I., Orengo, C. A., and Thornton, J. M. (2020). VarSite: Disease variants and protein structure. Protein Sci. 29, 111–119.
Lau, L. F., Mammen, A., Ehlers, M. D., Kindler, S., Chung, W. J., Garner, C. C., et al. (1996). Interaction of the N-methyl-D-aspartate receptor complex with a novel synapse-associated protein, SAP102. J. Biol. Chem. 271, 21622–21628. doi: 10.1074/jbc.271.35.21622
Lemke, J. R., Geider, K., Helbig, K. L., Heyne, H. O., Schutz, H., Hentschel, J., et al. (2016). Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy. Neurology 86, 2171–2178. doi: 10.1212/WNL.0000000000002740
Li, J., Lin, S. M., Qiao, J. D., Liu, X. R., Wang, J., Jiang, M., et al. (2021). CELSR3 variants are associated with febrile seizures and epilepsy with antecedent febrile seizures. CNS Neurosci. Ther. 28, 382–389. doi: 10.1111/cns.13781
Li, R. K., Li, H., Tian, M. Q., Li, Y., Luo, S., Liang, X. Y., et al. (2023). Investigation of FRMPD4 variants associated with X-linked epilepsy. Seizure [Online ahead of print.]. doi: 10.1016/j.seizure.2023.05.014
Li, X., Bian, W. J., Liu, X. R., Wang, J., Luo, S., Li, B. M., et al. (2022). BCOR variants are associated with X-linked recessive partial epilepsy. Epilepsy Res. 187:107036. doi: 10.1016/j.eplepsyres.2022.107036
Liu, X. R., Xu, X. X., Lin, S. M., Fan, C. Y., Ye, T. T., Tang, B., et al. (2021). GRIN2A variants associated with idiopathic generalized epilepsies. Front. Mol. Neurosci. 14:720984. doi: 10.3389/fnmol.2021.720984
Luo, S., Ye, X. G., Jin, L., Li, H., He, Y. Y., Guan, B. Z., et al. (2023). SZT2 variants associated with partial epilepsy or epileptic encephalopathy and the genotype-phenotype correlation. Front. Mol. Neurosci. 16:1162408. doi: 10.3389/fnmol.2023.1162408
McGrother, C. W., Bhaumik, S., Thorp, C. F., Hauck, A., Branford, D., and Watson, J. M. (2006). Epilepsy in adults with intellectual disabilities: Prevalence, associations and service implications. Seizure 15, 376–386.
Moog, U., Kutsche, K., Kortum, F., Chilian, B., Bierhals, T., Apeshiotis, N., et al. (2011). Phenotypic spectrum associated with CASK loss-of-function mutations. J. Med. Genet. 48, 741–751.
Moutton, S., Bruel, A. L., Assoum, M., Chevarin, M., Sarrazin, E., Goizet, C., et al. (2018). Truncating variants of the DLG4 gene are responsible for intellectual disability with marfanoid features. Clin. Genet. 93, 1172–1178. doi: 10.1111/cge.13243
Mulhern, M. S., Stumpel, C., Stong, N., Brunner, H. G., Bier, L., Lippa, N., et al. (2018). NBEA: Developmental disease gene with early generalized epilepsy phenotypes. Ann. Neurol. 84, 788–795. doi: 10.1002/ana.25350
Murata, Y., and Constantine-Paton, M. (2013). Postsynaptic density scaffold SAP102 regulates cortical synapse development through EphB and PAK signaling pathway. J. Neurosci. 33, 5040–5052. doi: 10.1523/JNEUROSCI.2896-12.2013
Nagano, T., Jourdi, H., and Nawa, H. (1998). Emerging roles of Dlg-like PDZ proteins in the organization of the NMDA-type glutamatergic synapse. J. Biochem. 124, 869–875. doi: 10.1093/oxfordjournals.jbchem.a022200
Pisella, L. I., Gaiarsa, J. L., Diabira, D., Zhang, J., Khalilov, I., Duan, J., et al. (2019). Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 12:eaay0300. doi: 10.1126/scisignal.aay0300
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424.
Salpietro, V., Dixon, C. L., Guo, H., Bello, O. D., Vandrovcova, J., Efthymiou, S., et al. (2019). AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 10:3094.
Sandestig, A., Green, A., Aronsson, J., Ellnebo, K., Stefanova, M., and Novel, A. (2020). DLG3 mutation expanding the phenotype of X-linked intellectual disability caused by DLG3 nonsense variants. Mol. Syndromol. 10, 281–285. doi: 10.1159/000502601
Sans, N., Petralia, R. S., Wang, Y. X., Blahos, J. II, Hell, J. W., Wenthold, R. J., et al. (2000). change in NMDA receptor-associated proteins at hippocampal synapses. J. Neurosci. 20, 1260–1271.
Sheng, M., and Hoogenraad, C. C. (2007). The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 76, 823–847.
Shimizu, H., Morimoto, Y., Yamamoto, N., Tayama, T., Ozawa, H., and Imamura, A. (2022). Overlap Between Epilepsy and Neurodevelopmental Disorders: Insights from Clinical and Genetic Studies, ed. S. J. Czuczwar (Brisbane, AU: Epilepsy).
Soorya, L., Kolevzon, A., Zweifach, J., Lim, T., Dobry, Y., Schwartz, L., et al. (2013). Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 4:18. doi: 10.1186/2040-2392-4-18
Stolz, J. R., Foote, K. M., Veenstra-Knol, H. E., Pfundt, R., Ten Broeke, S. W., de Leeuw, N., et al. (2021). Clustered mutations in the GRIK2 kainate receptor subunit gene underlie diverse neurodevelopmental disorders. Am. J. Hum. Genet. 108, 1692–1709. doi: 10.1016/j.ajhg.2021.07.007
Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., et al. (2023). The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. doi: 10.1093/nar/gkac1000
Tan, W. H., Bacino, C. A., Skinner, S. A., Anselm, I., Barbieri-Welge, R., Bauer-Carlin, A., et al. (2011). Angelman syndrome: Mutations influence features in early childhood. Am. J. Med. Genet. A 155A, 81–90. doi: 10.1002/ajmg.a.33775
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2020). Optimization of in silico tools for predicting genetic variants: Individualizing for genes with molecular sub-regional stratification. Brief Bioinform. 21, 1776–1786. doi: 10.1093/bib/bbz115
Tarpey, P., Parnau, J., Blow, M., Woffendin, H., Bignell, G., Cox, C., et al. (2004). Mutations in the DLG3 gene cause nonsyndromic X-linked mental retardation. Am. J. Hum. Genet. 75, 318–324. doi: 10.1086/422703
Tian, M. Q., Liu, X. R., Lin, S. M., Wang, J., Luo, S., Gao, L. D., et al. (2023). Variants in BRWD3 associated with X-linked partial epilepsy without intellectual disability. CNS Neurosci. Ther. 29, 727–735. doi: 10.1111/cns.14057
Turner, T. J., Zourray, C., Schorge, S., and Lignani, G. (2021). Recent advances in gene therapy for neurodevelopmental disorders with epilepsy. J. Neurochem. 157, 229–262. doi: 10.1111/jnc.15168
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wang, J., Qiao, J. D., Liu, X. R., Liu, D. T., Chen, Y. H., Wu, Y., et al. (2021). UNC13B variants associated with partial epilepsy with favourable outcome. Brain 144, 3050–3060. doi: 10.1093/brain/awab164
Wang, J. Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X. R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: Toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Wang, X., Mei, D., Gou, L., Zhao, S., Gao, C., Guo, J., et al. (2023). Functional evaluation of a novel GRIN2B missense variant associated with epilepsy and intellectual disability. Neuroscience 526, 107–120. doi: 10.1016/j.neuroscience.2023.06.018
Wei, Z., Behrman, B., Wu, W. H., and Chen, B. S. (2015). Subunit-specific regulation of N-methyl-D-aspartate (n.d.) receptor trafficking by SAP102 protein splice variants. J. Biol. Chem. 290, 5105–5116. doi: 10.1074/jbc.M114.599969
Wei, Z., Wu, G., and Chen, B. S. (2018). Regulation of SAP102 synaptic targeting by phosphorylation. Mol. Neurobiol. 55, 6215–6226. doi: 10.1007/s12035-017-0836-4
Xu, X. X., and Luo, J. H. (2018). Mutations of N-Methyl-D-aspartate receptor subunits in epilepsy. Neurosci. Bull. 34, 549–565. doi: 10.1007/s12264-017-0191-5
Yan, H. J., He, Y. Y., Jin, L., Guo, Q., Zhou, J. H., and Luo, S. (2022). Expanding the phenotypic spectrum of KCNK4: From syndromic neurodevelopmental disorder to rolandic epilepsy. Front. Mol. Neurosci. 15:1081097. doi: 10.3389/fnmol.2022.1081097
Yang, J., and Zhang, Y. I. (2015). -TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi: 10.1093/nar/gkv342
Ye, Z. L., Yan, H. J., Guo, Q. H., Zhang, S. Q., Luo, S., Lian, Y. J., et al. (2023). NEXMIF variants are associated with epilepsy with or without intellectual disability. Seizure [Online ahead of print]. doi: 10.1016/j.seizure.2023.08.012
Zheng, C. Y., Seabold, G. K., Horak, M., and Petralia, R. S. (2011). MAGUKs, synaptic development, and synaptic plasticity. Neuroscientist 17, 493–512. doi: 10.1177/1073858410386384
Keywords: DLG3 gene, epilepsy, neurodevelopmental disorder, variants, Genotype-phenotype correlation
Citation: He Y-Y, Luo S, Jin L, Wang P-Y, Xu J, Jiao H-L, Yan H-J, Wang Y, Zhai Q-X, Ji J-J, Zhang W-J, Zhou P, Li H, Liao W-P, Lan S and Xu L (2024) DLG3 variants caused X-linked epilepsy with/without neurodevelopmental disorders and the genotype-phenotype correlation. Front. Mol. Neurosci. 16:1290919. doi: 10.3389/fnmol.2023.1290919
Received: 11 October 2023; Accepted: 28 November 2023;
Published: 05 January 2024.
Edited by:
Michel J. Simonneau, École Normale Supérieure Paris-Saclay, FranceReviewed by:
Antonio Falace, Pediatric Neurology and Muscular Diseases Unit IRCCS Istituto Giannina Gaslini, ItalyAmanda Morato Do Canto, State University of Campinas, Brazil
Copyright © 2024 He, Luo, Jin, Wang, Xu, Jiao, Yan, Wang, Zhai, Ji, Zhang, Zhou, Li, Liao, Lan and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Xu, bHlubmV4dTIwMDBAaG90bWFpbC5jb20=; Song Lan, ZG9jdG9yLmxhbnNvbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship