 Anna Lozano-Ureña
Anna Lozano-Ureña José M. Frade
José M. Frade
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci., 22 December 2023
Sec. Molecular Signalling and Pathways
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1271820
This article is part of the Research TopicRole of Neurotrophins and Their Receptors in Brain Development, Neurological Disorders and Nervous System DiseasesView all 8 articles
Alterations in adult neurogenesis are a common hallmark of neurodegenerative diseases. Therefore, understanding the molecular mechanisms that control this process is an indispensable requirement for designing therapeutic interventions addressing neurodegeneration. Neurotrophins have been implicated in multiple functions including proliferation, survival, and differentiation of the neural stem cells (NSCs), thereby being good candidates for therapeutic intervention. Brain-derived neurotrophic factor (BDNF) belongs to the neurotrophin family and has been proven to promote neurogenesis in the subgranular zone. However, the effects of BDNF in the adult subventricular zone (SVZ) still remain unclear due to contradictory results. Using in vitro cultures of adult NSCs isolated from the mouse SVZ, we show that low concentrations of BDNF are able to promote self-renewal and proliferation in these cells by activating the tropomyosin-related kinase B (TrkB) receptor. However, higher concentrations of BDNF that can bind the p75 neurotrophin receptor (p75NTR) potentiate TrkB-dependent self-renewal and proliferation and promote differentiation of the adult NSCs, suggesting different molecular mechanisms in BDNF-promoting proliferation and differentiation. The use of an antagonist for p75NTR reduces the increment in NSC proliferation and commitment to the oligodendrocyte lineage. Our data support a fundamental role for both receptors, TrkB and p75NTR, in the regulation of NSC behavior.
Two main regions maintain the potential to generate new neurons in the adult mammalian brain: the subventricular zone (SVZ) in the wall of the lateral ventricles and the subgranular zone (SGZ) in the dentate gyrus of the hippocampus (Taupin and Gage, 2002; Chaker et al., 2016; Gonçalves et al., 2016). The neural stem cells (NSCs) are not only responsible for carrying out this process of neurogenesis but also contribute to generating new astrocytes and oligodendrocytes throughout life (Taupin and Gage, 2002; Sohn et al., 2015), thus becoming potential agents for brain repair. Under homeostatic conditions, a careful interchange between cellular and molecular processes in the microenvironment constantly regulates the activity of NSCs (Fuentealba et al., 2012). In order to avoid loss or excess of the stem cell (SC) population, self-renewal and proliferation must be acutely regulated in coordination with differentiation processes. Thus, the differentiation of NSCs requires an intermediate state in which these cells become committed [i.e., neural progenitor cells (NPCs)] although they still show proliferative potential (Llorente et al., 2022).
Brain-derived neurotrophic factor (BDNF) is the most widely distributed member of the neurotrophin (NT) family in the central nervous system (Leibrock et al., 1989), with important implications in neuronal survival and differentiation (Eide et al., 1993). BDNF interacts with two receptors, the tropomyosin-related kinase B (TrkB) receptor (Klein et al., 1989) and the p75 neurotrophin receptor (p75NTR), known to interact with all NTs (Rodriguez-Tebar et al., 1992). Classically, NTs promote survival, proliferation, and correct maturation by Trk receptor signaling through its associated kinase activity (Mitra et al., 1987), whereas p75NTR has been more involved in apoptosis (Frade et al., 1996) and in other cellular pathways depending on the intracellular complexes it constitutes (Roux and Barker, 2002). A recent study has begun to clarify the complexity of p75NTR signaling. This includes proteolytic processing through γ-secretase to release its intracellular domain (Vicario et al., 2015) that translocates to the nucleus (Parkhurst et al., 2010) and the conformational rearrangement of disulfide-linked receptor dimers (Klein et al., 1990) that allows the access of intracellular effectors to the receptor (Lin et al., 2015). BDNF, TrkB, and its truncated form TrkB.T1, known to lack the kinase domain (Klein et al., 1990), are all expressed in the murine SVZ (Vilar and Mira, 2016) as well as throughout the migratory pathway (Chiaramello et al., 2007). p75NTR is also expressed by cycling cells of the SVZ (Okano et al., 1996; Giuliani et al., 2004), including intermediate progenitors (Galvão et al., 2008). In addition, p75NTR can be detected in neuroblasts of the SVZ/RMS (Galvão et al., 2008), and genetic depletion of p75NTR reduces the migration capacity of the neuroprogenitors in the SVZ both in physiological conditions and after cortical injury (Young et al., 2007; Deshpande et al., 2022). The complexity of NT signaling is increased due to the known association of p75NTR with members of the Trk family (Hempstead et al., 1991; Zanin et al., 2019). This is also the case for BDNF as the treatment with BDNF in embryonic hippocampal neurons elicits the association of TrkB and p75NTR, facilitating the TrkB signaling and promoting neuronal survival and function (Zanin et al., 2019).
The activity of BDNF by the high-affinity binding to TrkB has been widely described in the hippocampal neurogenic niche (Bartkowska et al., 2007; Li et al., 2008; Vilar and Mira, 2016); however, its role in the NSCs located at the SVZ is not fully understood (Bath et al., 2012; Vilar and Mira, 2016). Although both BDNF receptors, TrkB and p75NTR, are present in the adult SVZ (Tervonen et al., 2006; Galvão et al., 2008; Bath et al., 2012; Vilar and Mira, 2016), the implication of these receptors in NSC decision-making remains to be established. BDNF/TrkB participates in the proliferation and differentiation of the neuroprogenitors, and in the survival and maturation of the new neurons (Berghuis et al., 2006; Bath et al., 2012; Chen et al., 2013). BDNF/p75NTR seems to regulate cell proliferation and migration of neuroblasts to the olfactory bulb (OB) (Snapyan et al., 2009; Bath et al., 2012; Deshpande et al., 2022).
Alterations in the niche environment as a consequence of stroke or neurodegenerative diseases, among others, drive a disorder in the amount of BDNF and its receptors (Holsinger et al., 2000; Jiao et al., 2016; Deshpande et al., 2022). These changes in BDNF concentration might imply the activation of different signaling pathways and, thus, the different context-dependent effects observed in previous studies (Bath et al., 2012). Investigating the function of BDNF and the molecular mechanisms implicated in the regulation of adult NSCs is essential to understand the potential contribution of adult NSCs to brain repair and as a therapeutic tool. Here, we analyzed the effect of low and high concentrations of BDNF in the self-renewal, proliferation, and differentiation capacity of NSCs isolated from the adult SVZ and the contribution of TrkB and p75NTR receptors in the adult NSCs response.
NSCs were obtained from mice with a C57BL6 background. Mice were maintained in a 12-h light/dark cycle with free access to food and water ad libitum according to the Animal Care and Ethics Committee of the CSIC. Adult NSCs were isolated from 3-month-old mice after cervical dislocation. The brains were dissected out, and both SVZs from each hemisphere were extracted and cut into small fragments. The pieces were incubated with 0.025% Trypsin-EDTA (Gibco; Cat #25300054) for 30 min at 37°C. The tissue was then transferred to Dulbecco's modified Eagle's medium (DMEM)/F12 (1:1 v/v; Life Technologies, Cat #21331020) and carefully triturated with a fire-polished Pasteur pipette to a single cell suspension. Isolated cells were collected by centrifugation, resuspended in the NSC medium based on DMEM/F12 containing 2 mM Glutamax (Gibco; Cat #35050038), 1X B27 without vitamin A (Gibco; Cat #11500446), 2X antibiotic-antimycotic (Gibco; Cat #15240062), 2 μg/ml heparin (Sigma; Cat #H3393), supplemented with 20 ng/ml epidermal growth factor (EGF; Peprotech, Cat #AF-100-15), and 10 ng/ml fibroblast growth factor (FGF; Peprotech; cat# 100-18B), and maintained in a 95% air−5% CO2 humidified atmosphere at 37°C (Bizy and Ferron, 2015; Belenguer et al., 2016). Neurospheres were allowed to develop for 7–10 days in these conditions. Each culture was generated using both SVZs from one adult mouse. Thus, each experimental point in the graphs represents the mean value of the replicates of a single independent animal. For culture expansion, primary neurospheres were disaggregated with Accutase (0.5 mM; Sigma; Cat #A6964) for 10 min at room temperature and washed with the NSC medium without mitogens to generate single-cell suspension. Then, 62.5 cells/μl were plated in the fresh mitogen-completed medium in a 95% air−5% CO2 humidified atmosphere at 37°C and maintained for 6–7 passages maximum. In order to determine the self-renewal capacity of the NSCs, secondary neurospheres were disaggregated, NSCs were plated at low density (5 cells/μl) in the fresh mitogen-completed medium, and the number of neurospheres was counted 5 days later. In the self-renewal experiment, four replicates for each culture were used, and the average value was estimated. All these experiments were repeated four times with different cultures. Images of the neurospheres were taken using the PAULA Smart Cell Imager (Leica), and the diameters of the spheres were estimated by ImageJ.
To estimate proliferation, 62.5 cells/μl were plated after Accutase disaggregation in the fresh mitogen-completed medium in a 95% air−5% CO2 humidified atmosphere at 37°C. After 3 days, neurospheres were plated onto cover glasses coated with 1X Matrigel (Corning, Cat #356234) for 15 min, allowing NSC attachment and fixed for staining with 2% paraformaldehyde (PFA) 0.1M phosphate buffer saline pH 7.4 (PBS) for 15 min at 37°C. For the differentiation assay, 80,000 cells/cm2 were seeded in Matrigel-coated coverslips and incubated for 2 days (2 DIV) in the NSC culture medium without EGF. The medium was then changed with the fresh medium without FGF and supplemented with 2% fetal bovine serum (FBS; Gibco; Cat #10438-026) for 5 more days (7 DIV) to allow terminal differentiation. Cultures were fixed for staining at 7 days of differentiation with 2% PFA 0.1M PBS for 15 min at 37°C. The BDNF treatment was performed by incubating the NSCs with either 10 ng/ml (low concentration) or 50 ng/ml (high concentration) of Recombinant Human/Murine/Rat BDNF (PeproTech; Cat #450-02) since the single cell suspension is plated. When indicated, NSC cultures were treated with 10 μM ANA-12 (MedChemExpress; Cat #HY-12497) (hereafter referred to as TrkB-i) or 10 μM THX-B (MedChemExpress; Cat #HY-137322) (hereafter referred to as p75-i) at the time of plating to inhibit TrkB or p75NTR, respectively. The specificity and selectivity of both antagonists have been previously evaluated (Bai et al., 2010; Cazorla et al., 2011). Control cultures were exposed to 1:1,000 of DMSO (Sigma; Cat.# D5879). In both proliferation and differentiation assays, 10 random images were taken with ~400 cells analyzed for each culture. These experiments were performed four times with independent cultures.
For immunocytochemical staining, fixed cells were permeabilized and blocked with PBS 0.2% Triton X-100 (Sigma; Cat.#X100) containing 10% normal goat serum and 1% glycine (Thermo Scientific; Cat #A13816.36) for 1 h at RT, incubated with primary antibodies, and prepared in the same blocking solution overnight at 4°C. Cells were washed three times with PBS 1X and incubated with secondary antibodies for 1 h at RT. Primary and secondary antibodies and dilutions used are listed in Tables 1, 2, respectively. DAPI (1 μg/ml) was used to counterstain DNA. The samples were washed three times with PBS 1X and mounted with the ImmunoSelect antifading mounting medium (Dianova; Cat #038447). Images were acquired at 20x or 40x magnification with a Leica SP5 confocal microscope. For fluorescence intensity quantification, maximal projection images were generated, and the mean gray intensities of p-TrkB, TrkB, and p75NTR were measured with ImageJ/Fiji software and recorded as arbitrary fluorescence units (a.u.). p-TrkB data were normalized to TrkB intensity.
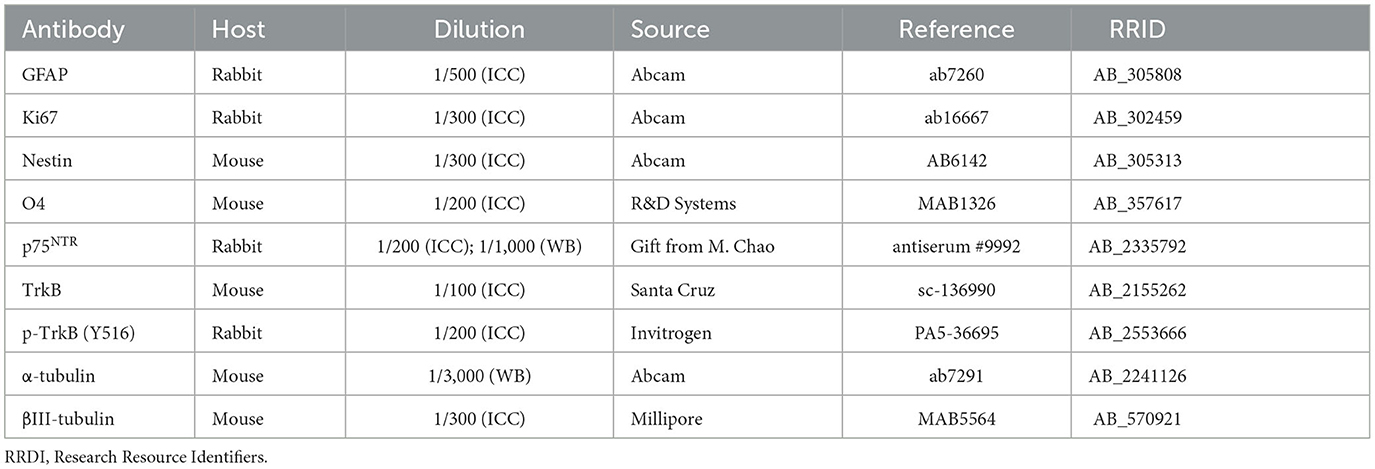
Table 1. List of primary antibodies for immunocytochemistry (ICC) and Western blot (WB).

Table 2. List of secondary antibodies for immunocytochemistry (ICC) and Western blot (WB).
Protein detection by Western blot was performed after protein extraction using 20 mM Tris pH 6.8 (Sigma; Cat.#10708976001) containing 1% Triton X-100 (Sigma), 0.5% sodium dodecyl sulfate (SDS) (Sigma; Cat.#L4509), 1 mM ethylene-dinitrilotetraacetic acid (EDTA) (Merck; Cat.#1.08418.0250), 10 mM β-mercaptoethanol (Sigma; Cat.#M-7154), and 1× cOmplete Mini, EDTA-free protease inhibitor (Roche; Cat.#11836170001). Total protein amount was determined by the Bradford Assay (BioRad; Cat.#500-0006). Proteins were denatured by heat (5 min at 100°C) and loaded in 4–20% precast polyacrylamide gels (BioRad; Cat.#4561095). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Merck; Cat.#IPFL00010) and immunoblots were carried out with primary antibodies (Table 1), incubated overnight, and secondary antibodies (Table 2) during 1 h. Antibodies were diluted in Intercept® Blocking Buffer (LI-COR; Cat.#927-60001). After antibodies incubation, the membranes were washed with Tris-buffered saline (TBS) containing 0.1% Tween 20 (Sigma; Cat.#P1379). Finally, protein bands were visualized using the Odyssey CLx Infrared Imaging System (LI-COR).
RNAs were extracted with the RNAeasy mini kit (Qiagen; Cat.# 74104) including DNase treatment, following the manufacturer's guidelines. For quantitative PCR (qPCR), 1 μg of total RNA was reverse transcribed using random primers and SuperScript IV Reverse Transcriptase (ThermoFisher Scientific; Cat# 15317696), following standard procedures. Thermocycling was performed in a final volume of 15 μl, containing 1 μl of cDNA sample (diluted 1:7), and the reverse transcribed RNA was amplified by PCR with appropriate primers from PrimePCR SYBR Green Assay (Cultek; Cat. PB20.11) (see Table 3). qPCR was used to measure gene expression levels normalized to Rpl27, the expression of which did not differ between the groups. qPCR reactions were performed in a 7500 real-time PCR equipment (Applied Biosystems). Raw data from this analysis is shown in Supplementary Table 1.
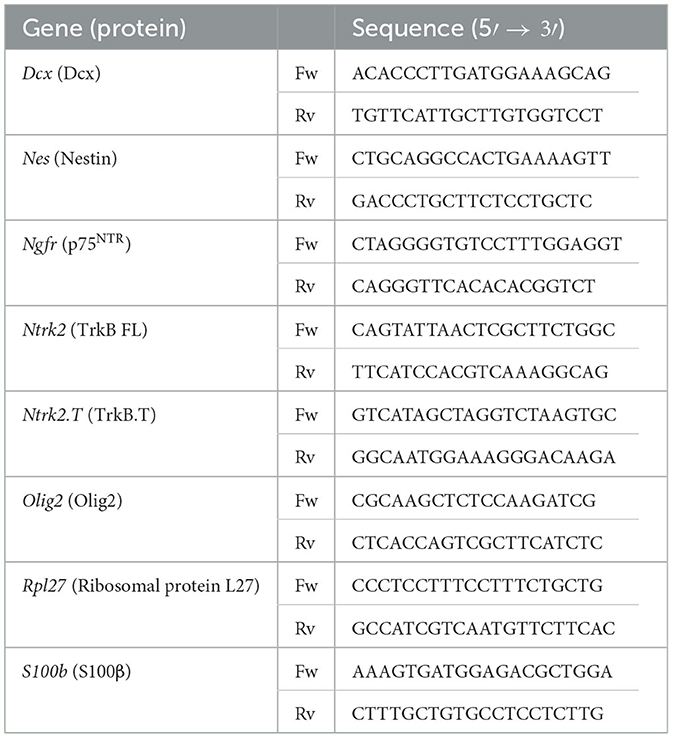
Table 3. List of primers for qPCR analysis.
All statistical tests were performed using the GraphPad Prism Software, version 7.00 for Windows. The significance of the differences between groups was evaluated by the two-tailed paired Student t-test or one-way ANOVA followed by a Tukey post-hoc test. The presence of outlier values was evaluated by Grubb's test. A p-value of < 0.05 was considered statistically significant. Data are presented as the mean ± standard error of the mean (SEM) and the number of independent cultures (n), and p-values are indicated in the figures.
BDNF has proved to act as a pro-neurogenic factor promoting the proliferation and differentiation of NSCs (Lee et al., 2002; Islam et al., 2009; Chen et al., 2013; Liu et al., 2014; Langhnoja et al., 2021). BDNF activity is mediated by high-affinity binding to the TrkB receptor (Naylor et al., 2002), and this neurotrophic factor is able to interact with low-affinity to p75NTR (Rodriguez-Tebar et al., 1990). Both receptors are expressed in the adult NSCs (Young et al., 2007; Islam et al., 2009; Bath et al., 2012; Faigle and Song, 2013; Vilar and Mira, 2016). A clear positive role for the TrkB pathway has been described in the function of BDNF on the embryonic or P0 NSC proliferation (Islam et al., 2009; Chen et al., 2013), and the proliferative role of p75NTR in the NSCs located in the adult SVZ (Young et al., 2007) remains to be established. To understand the mechanism behind BDNF's effects on the neurogenic population, adult NSCs were treated with two different doses of this neurotrophic factor (10 and 50 ng/ml). We chose these concentrations as the former mainly activates TrkB, while the latter also activates p75NTR since the Kd of the interaction of BDNF with p75NTR is approximately 10−9 M (~25 ng/ml) (Rodriguez-Tebar et al., 1990). First, self-renewal capacity was tested by determining the number of neurospheres after 5 days of NSCs cultured at low density with low (10 ng/ml) or high (50 ng/ml) concentrations of BDNF (Figure 1A). The presence of BDNF at 10 ng/ml in the NSC cultures significantly increased the number of neurospheres compared to untreated cultures, being this effect potentiated by the addition of BDNF at 50 ng/ml (Figure 1A). This suggests that p75NTR facilitates NSC self-renewal. Moreover, the diameter of these neurospheres was significantly higher in BDNF-treated NSCs (Figure 1B), suggesting an enhancement of NSC proliferation capacity. Both exposures to 10 and 50 ng/ml of BDNF showed a significant increment in the diameter of the neurospheres compared with untreated cultures, whereas no differences were detected between both concentrations of BDNF (Figure 1B). The proliferative capacity of adult NSCs was analyzed by measuring the percentage of positive cells for the cell cycle marker Ki67 (Figure 1C). Both concentrations of BDNF showed a significant increase in the proliferation ratio compared with untreated NSCs. Again, no differences in the percentage of Ki67+ cells were detected between 10 and 50 ng/ml treated cultures (Figure 1C), indicating that the lowest concentration of the neurotrophic factor was sufficient to activate the proliferation pathway.
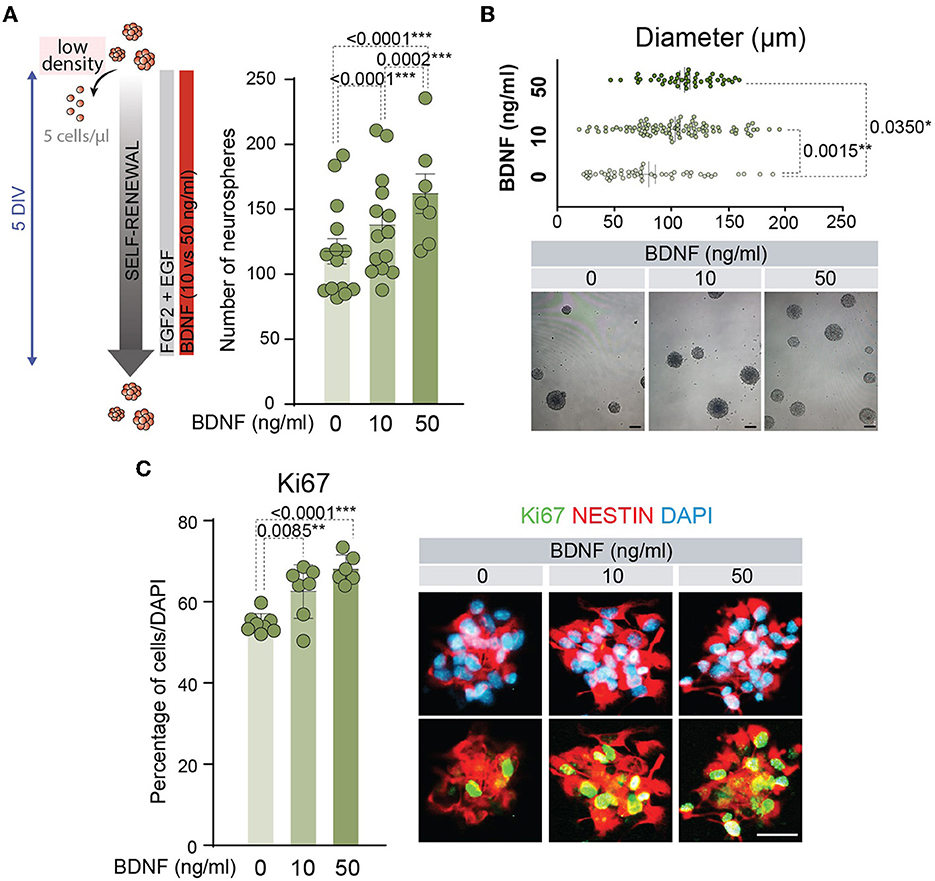
Figure 1. BDNF promotes NSC self-renewal and proliferation. (A) Schematic representation of the treatments for different concentrations of BDNF in adult NSCs in the self-renewal assay (left panel). Number of neurospheres after the culture of adult NSCs at low density (5 cells/μl) in the absence or presence of 10 or 50 ng/ml BDNF (right panel). (B) Diameter of the neurospheres in the self-renewal assay in the absence or presence of 10 or 50 ng/ml of BDNF (upper panel). Representative images of neurospheres formed in the absence or presence of BDNF treatments (lower panel). (C) Percentage of proliferative NSCs at high density (62.5 cells/μl), measured as the proportion of Ki67+ cells, in untreated or BDNF-treated cultures (10 or 50 ng/ml). Immunochemistry images for the proliferative marker Ki67 (green) and the neural precursor marker Nestin (red) in NSCs treated with different concentrations of BDNF are also shown. DAPI was used to counterstain DNA. All error bars show SEM. p-values and the number of samples (circles) are indicated. Only differences that are statistically significant are shown. Scale bars in (B) 100 μm; in (C) 20 μm.
Several studies have shown that BDNF exerts a positive effect on the differentiation of NSCs into neurons (Chen et al., 2013; Liu et al., 2014) and oligodendrocytes (Chen et al., 2013; Langhnoja et al., 2021). In accordance, the mRNA levels of relevant differentiation markers were analyzed by qPCR in cDNAs obtained from adult NSCs. This analysis indicated that the expression of the neuronal marker Dcx showed a tendency to increase and the oligodendrocyte marker Olig2 was significantly upregulated in the NSCs treated with 50 ng/ml of BDNF, suggesting that treatment with a high dose of BDNF predisposes NSCs toward a more committed state. Instead, the presence of 10 ng/ml of BDNF in the medium was not sufficient to increase the levels of mRNA of these lineage genes (Figure 2A). Neither the expression of the mRNA encoding the astrocytic marker S100β (S100b) nor the neural precursor gene Nestin (Nes) showed differences between untreated and BDNF-treated NSCs (Figures 2A, B). To test if the upregulation of the neuronal and oligodendrocytic genes in the adult NSCs after 50 ng/ml BDNF treatment drove an increment in the percentage of neurons and oligodendrocytes in differentiating conditions, the number of TUJ1+, O4+, and GFAP+ cells, representing neurons, oligodendrocytes, and astrocytes, respectively, were estimated after seven DIV in NSCs maintained in differentiation conditions. The percentage of neurons and oligodendrocytes were increased in the 50 ng/ml BDNF treated cultures, at the expense of astrocyte generation, which decreased in this condition compared with untreated cells (Figures 2C–F). Moreover, the treatment with the low dose of BDNF (10 ng/ml) did not alter the differentiation capacity of adult NSCs regarding untreated cultures (Figures 2C–F), thus requiring a higher concentration of BDNF to activate the differentiation pathway. These data, together with those from the proliferation analysis shown above, suggest different mechanisms for BDNF to promote proliferation or differentiation in a dose-dependent manner.
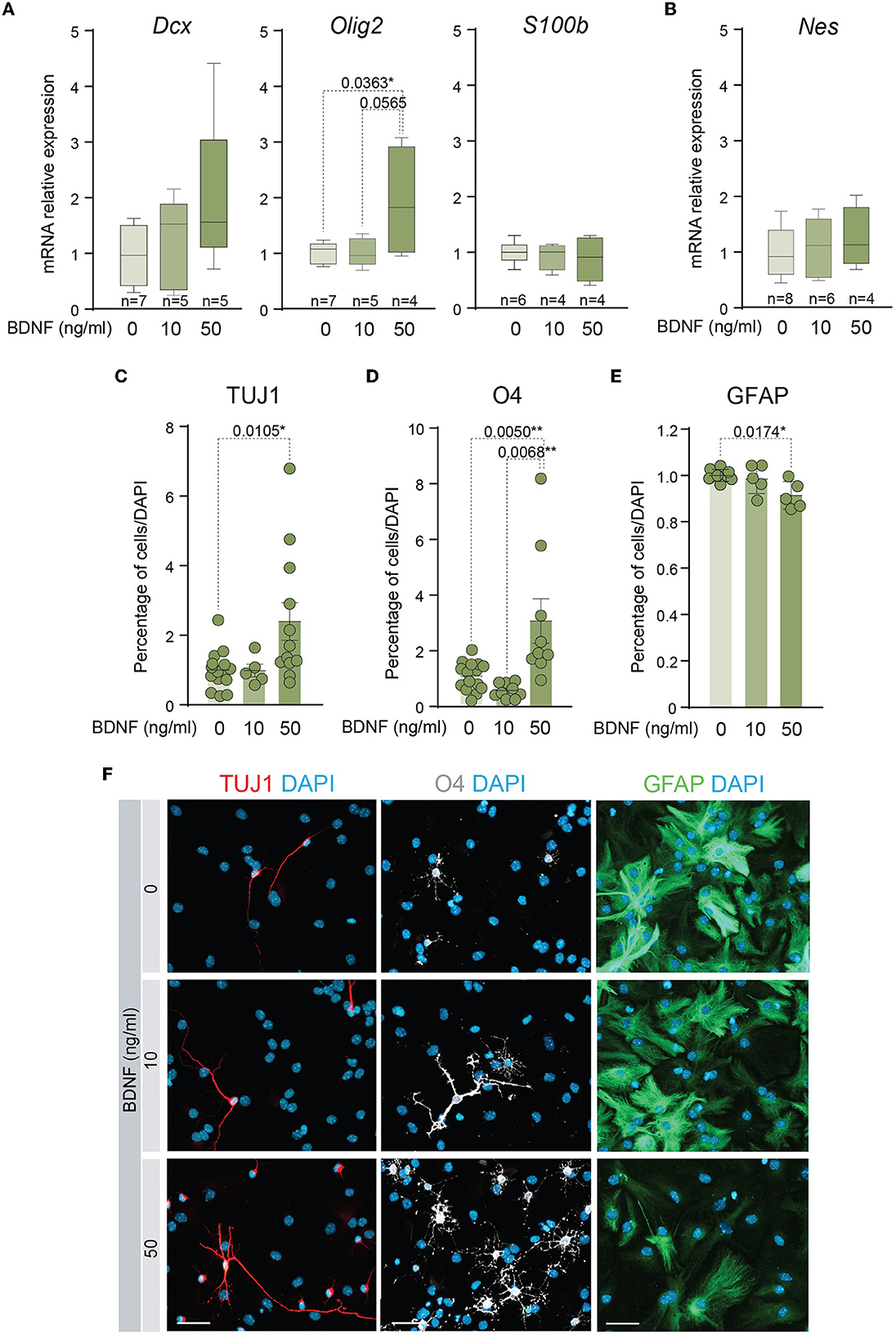
Figure 2. A higher dose of BDNF is required to favor neuronal and oligodendroglial differentiation. (A) Boxplots illustrating the expression of the neuronal marker Dcx, the oligodendrocytic marker Olig2, and the astrocytic marker S100β, in BDNF-treated NSCs (0, 10, or 50 ng/ml). Rpl27 was used as a housekeeping gene. (B) Boxplots illustrating the expression of Nestin in adult NSCs after being treated with 0, 10, or 50 ng/ml of BDNF. Rpl27 was used as a housekeeping gene. (C) Percentage of neurons, measured as TUJ1+ cells, after 7 days under differentiation-promoting condition in the absence or presence of 10 or 50 ng/ml of BDNF. (D) Percentage of oligodendrocytes, measured as O4+ cells, after 7 days under differentiation-promoting condition in the absence or presence of 10 or 50 ng/ml of BDNF. (E) Percentage of astrocytes, measured as GFAP+ cells, after 7 days under differentiation-promoting condition in the absence or presence of 10 or 50 ng/ml of BDNF. (F) Immunocytochemistry images for TUJ1 (red), O4 (gray), or GFAP (green) in NSCs after 7 DIV of differentiation in the absence or presence of 10 or 50 ng/ml of BDNF. DAPI was used to counterstain DNA. In (C–E), p-values and the number of samples (circles) are indicated, and all error bars show SEM. Only differences that are statistically significant are shown. Scale bars in (F) 30 μm.
The previous results of proliferation and differentiation of the adult NSCs in the presence of low or high doses of BDNF could be explained by the use of different signaling mechanisms to activate each cellular process. Precisely, BDNF presents high-affinity binding to TrkB (Naylor et al., 2002) and low-affinity binding to p75NTR (Rodriguez-Tebar et al., 1990), two receptors that are expressed by NSCs, showing a dynamic pattern of expression during proliferation and differentiation of these cells (Figure 3A). To understand if the different cellular response of BDNF in a dose-dependent manner could be due to the intervention of different receptors/pathways, adult NSCs were treated with 10 or 50 ng/ml of BDNF, and the gene expression of both receptors, Ntrk2 (TrkB) and Ngfr (p75NTR), was measured by qPCR (Figures 3B, C). To this aim, BDNF was added after neurosphere disaggregation, and the expression of these receptors was analyzed in the newly formed neurospheres after 5 days in the presence of the neurotrophin. The Ntrk2 gene encodes three receptor isoforms generated by alternative splicing, the full-length isoform (TrkB FL), and two truncated versions of the protein lacking the kinase domain, with TrkB.T1 being the most expressed in the NSCs from the SVZ (Islam et al., 2009; Vilar and Mira, 2016). Thus, the expression of the transcripts encoding both TrkB FL and TrkB.T1 (TrkB FL and TrkB.T1, respectively) was analyzed in adult NSCs grown in the absence or presence of 10 or 50 ng/ml BDNF (Figure 3B). The presence of BDNF in the culture medium resulted in a significant increment of both TrkB FL and TrkB.T1 expressions, regardless of the BDNF concentration (Figure 3B), suggesting that its expression is regulated by the activation of TrkB. As previously shown (Islam et al., 2009), the expression of TrkB.T1 was higher than that of TrkB FL (Figure 3B). In contrast to its mRNA levels, the expression of the TrkB protein using an antibody recognizing the extracellular domain (i.e., recognizing all TrkB isoforms) was not observed to show an increased response to BDNF (Figure 3D), suggesting that post-transcriptional mechanisms regulate TrkB protein expression. As expected, exposure of neurospheres to BDNF resulted in the increase of TrkB phosphorylation in Y516 (Figure 3D), a residue that becomes phosphorylated upon TrkB activation (Mazzaro et al., 2016). This activation of TrkB signaling in NSCs confirms previous published data suggesting TrkB activation in NSCs (Chen et al., 2017). Moreover, the application of the selective TrkB antagonist ANA-12 (TrkB-i) (Cazorla et al., 2011) to neurospheres treated with 10 ng/ml BDNF resulted in the reduction of Y516 TrkB phosphorylation to basal levels (Figure 3D). In contrast to TrkB FL and TrkB.T1 expressions, the expression of Ngfr was significantly upregulated in the NSC cultures only after high-dose exposure to BDNF (Figure 3C), indicating that the presence of high levels of BDNF promotes the activation of a signaling pathway resulting in the expression of Ngfr. The requirement for the dose of BDNF suggests that the upregulation of p75NTR is modulated by its own activation. To confirm this hypothesis, the expression of Ngfr was measured in NSCs treated with 50 ng/ml BDNF in the presence of TrkB-i or the selective p75NTR antagonist THX-B (Bai et al., 2010) (p75-i) (Figure 3E). The presence of TrkB-i did not change the Ngfr mRNA levels when NSCs were treated with 50 ng/ml of BDNF, and the expression of Ngfr was not upregulated after 50 ng/ml BDNF treatment in the presence of p75-i (Figure 3E), showing that the increment in the expression of the p75NTR receptor was regulated by the interaction of BDNF with this receptor. The increment in Ngfr mRNA at 50 ng/ml of BDNF treatment was confirmed at the protein level by Western blot (Figure 3F) and immunocytochemistry (Figure 3G), using a previously characterized antibody (Huber and Chao, 1995).
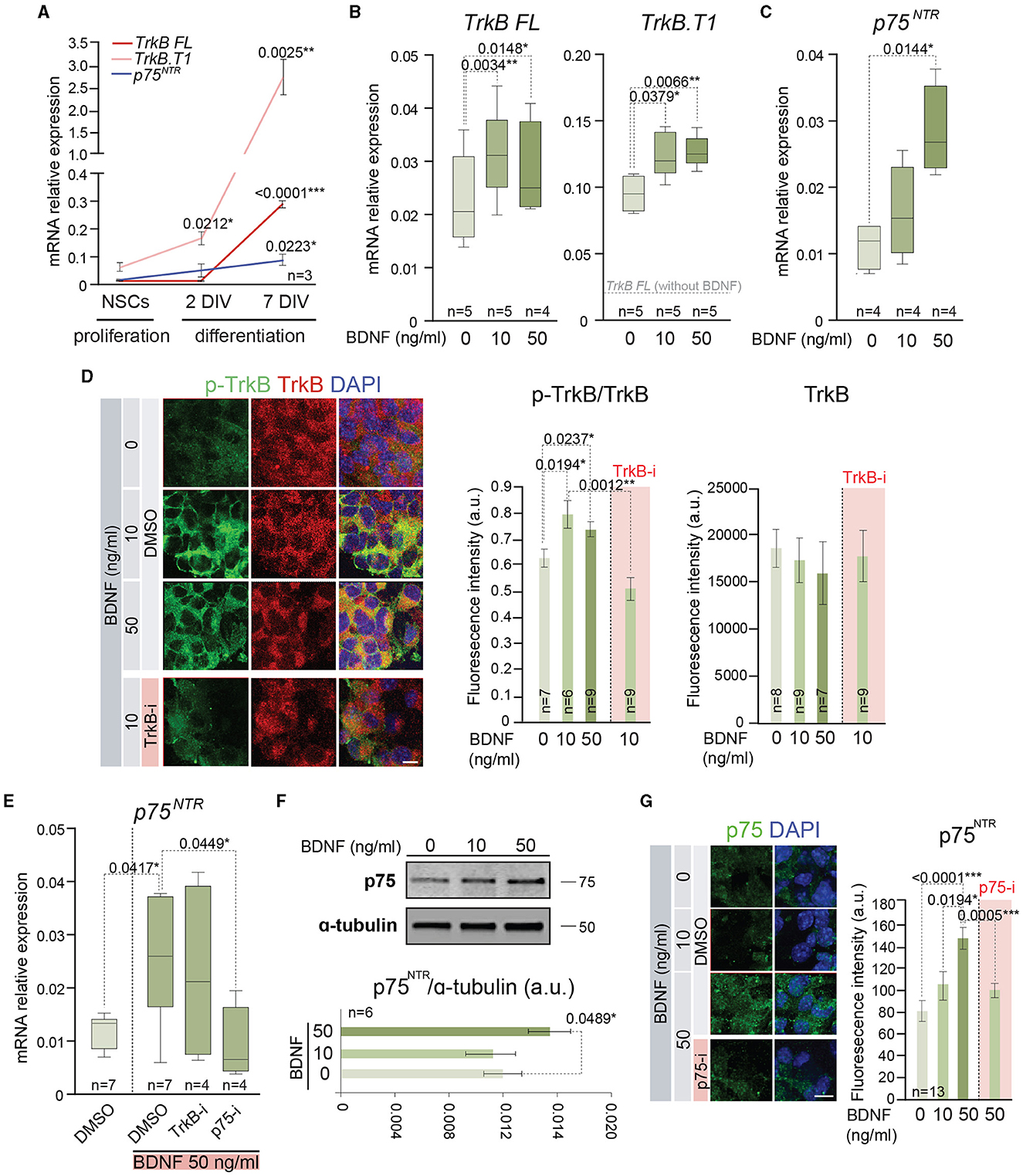
Figure 3. BDNF induces the expression of functional TrkB and p75NTR receptors. (A) Expression of mRNA encoding the full-length (TrkB FL) and the truncated (TrkB.T1) isoform of Ntrk2 gen and p75NTR (Ngfr) in adult NSCs maintained in the absence of exogenous BDNF in proliferation-promoting conditions and during the differentiation process [after 2 days in vitro (DIV) and 7 DIV]. (B) Boxplots illustrating the expression of TrkB FL and TrkB.T1 isoform of Ntrk2 gen in adult NSCs after being treated with 0, 10, or 50 ng/ml of BDNF. (C) Boxplots illustrating the expression of Ngfr (p75NTR) in BDNF-treated NSCs (0, 10, or 50 ng/ml). (D) Representative high-magnification images illustrating the immunostaining for p-TrkB in Y516 (green) and TrkB (red) in untreated or BDNF-treated (10 or 50 ng/ml) neurospheres as well as 10 ng/ml BDNF-treated neurospheres with the TrkB antagonist (TrkB-i). Vehicle: DMSO (left panel). Quantification of p-TrkB/TrkB and TrkB fluorescence intensity (in arbitrary units, a.u.) in these cultures (middle and right panels). (E) Boxplots illustrating the expression of p75NTR receptor, Ngfr, in adult NSCs in the absence or presence of 50 ng/ml of BDNF and treated with the antagonists TrkB-i or p75-i. DMSO was used as a control. (F) Immunoblot for p75NTR protein in NSC cultures treated with 0, 10, or 50 ng/ml of BDNF (upper panel). Quantification in the Western blot of p75NTR relative to α-tubulin protein (bottom panel). (G) Representative images illustrating the immunostaining for p75NTR in untreated or treated NSCs with 10 or 50 ng/ml of BDNF as well as 50 ng/ml BDNF-treated cells with the p75NTR antagonist (p75-i). Quantification of p75NTR fluorescence intensity in arbitrary units is shown as mean ± SEM (n = 13). In (A–C, E), Rpl27 was used as a housekeeping gene. DAPI was used to counterstain DNA in (D, G). In (A, D, F), error bars show SEM. In all panels, p-values and the number of samples are indicated. Only differences that are statistically significant are shown. Scale bars: 20 μm (D); 10 μm (G).
The expression data of TrkB and p75NTR in proliferating and differentiating conditions suggest that both receptors are involved in NSC behavior. To determine the implications of TrkB and p75NTR in these processes, NSCs were treated with TrkB-i and p75-i, respectively (Figure 4A). NSCs were cultured at low density to evaluate self-renewal capacity in the absence or presence of 10 or 50 ng/ml of BDNF as above, using 10 μM of TrkB-i or 10 μM of p75-i to inhibit TrkB or p75NTR specifically (Figure 4B). Control NSCs were treated with DMSO. The presence of TrkB-i in the medium revealed that the TrkB pathway is essential for NSCs to self-renew, independently of the presence of exogenous BDNF, a finding consistent with the expression of Bdnf by the adult NSCs (Figure 4C). Blocking this receptor significantly decreased the number of neurospheres in 0, 10, and 50 ng/ml of BDNF treatments (Figure 4B). These data were consistent with previous results showing a decrease of newly born neurons in the OB of TrkB heterozygous mice (Bath et al., 2008). In contrast, treatment of NSCs with p75-i in the absence of BDNF showed no effect on the self-renewal capacity of the NSCs (Figure 4B). The presence of 10 ng/ml of BDNF jointly with this antagonist did not alter this ability either (Figure 4B) indicating that lower concentrations of BDNF act through the TrkB pathway. However, treatment with 50 ng/ml of BDNF in the presence of the p75NTR antagonist resulted in a decrease in the number of neurospheres (Figure 4B), indicating that the higher concentration of BDNF activated a TrkB/p75NTR-dependent pathway that becomes necessary to control NSC self-renewal. Previous studies demonstrated that TrkA formed complexes with p75NTR, increasing the affinity and selectivity of NGF binding (Hempstead et al., 1991). Another study showed that BDNF induces TrkB association with p75NTR in embryonic hippocampal neurons after TrkB activation (Zanin et al., 2019). Importantly, this latter study demonstrated that p75NTR is necessary for optimal TrkB signaling and function through the PI3K pathway in embryonic neurons (Zanin et al., 2019). In contrast to these studies, where p75NTR optimizes the signaling capacity of the Trk family receptors, our observation suggests that a novel functional interaction between p75NTR and TrkB exists in the adult NSCs as the blockade of p75NTR prevents TrkB function.
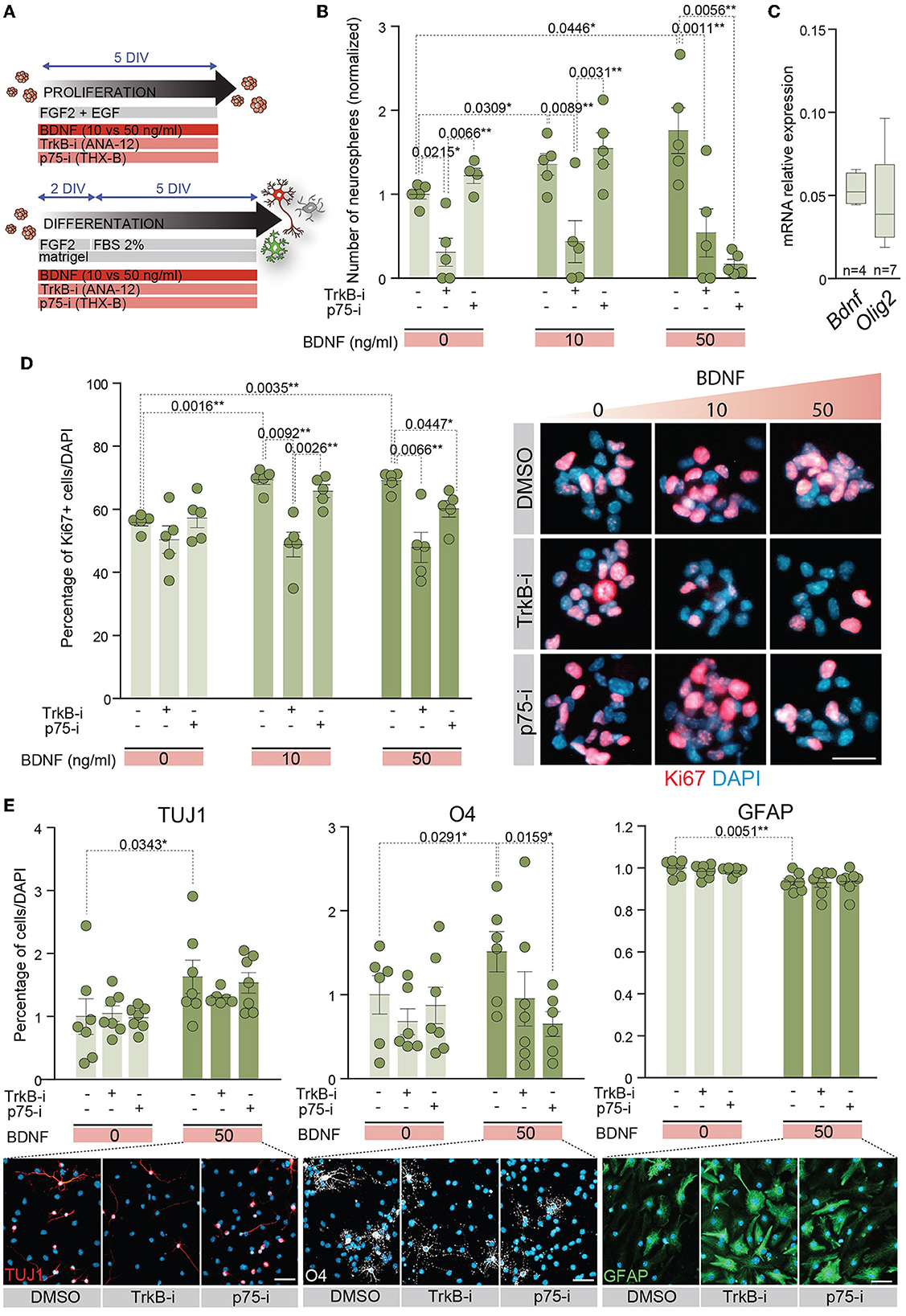
Figure 4. p75NTR regulates adult NSC proliferation and differentiation in a higher dose of BDNF context. (A) Schematic representation of the treatments for TrkB and p75NTR inhibition in adult NSCs in proliferation or differentiation conditions. (B) Number of neurospheres in 0, 10, or 50 ng/ml BDNF treatments in the presence of the TrkB antagonist, TrkB-i, or the p75 antagonist, p75-i. As a control, DMSO was added to the cultures without antagonists. (C) Boxplots illustrating the expression of Bdnf and Olig2 by qPCR in untreated adult NSCs. Olig2 expression is shown as a positive control of a neural expressed gene. Rpl27 was used as a housekeeping gene. (D) Percentage of Ki67 positive cells in NSCs treated with 0, 10, or 50 ng/ml of BDNF in the presence of the antagonists TrkB-i or p75-i (left panel). Immunocytochemistry images for Ki67 (red) in these conditions (right panel). (E) Percentage of TUJ1+ neurons, O4+ oligodendrocytes, and GFAP+ astrocytes after 7 DIV under differentiation-promoting conditions in 50 ng/ml BDNF-treated or untreated NSCs in the presence of TrkB-i or p75-i antagonists (upper panels). As a control, DMSO was added to the cultures without antagonists. Immunocytochemistry images for the lineage markers TUJ1 (red), O4 (gray), and GFAP (green) in these conditions are also shown (lower panels). DAPI was used to counterstain DNA. Error bars show SEM. p-values and the number of samples (circles) are indicated. Only differences that are statistically significant are shown. Scale bars in (D) 20 μm; in (E) 30 μm.
The proliferation capacity of adult NSCs was also analyzed in the presence of the receptor antagonists (Figure 4D). NSCs were plated in proliferation-promoting conditions and treated with different doses of BDNF. The percentage of proliferating cells was determined by the number of Ki67+ cells. The treatment with either antagonist in the absence of exogenous BDNF showed no alterations in the percentage of proliferative NSCs (Figure 4D). The presence of the TrkB-i in NSCs treated with low or high concentrations of BDNF prevented the increase in the percentage of Ki67+ cells induced by this neurotrophin, reaching the untreated culture levels (Figure 4D). However, the presence of the p75-i decreased the Ki67 percentage to untreated culture levels only in NSCs treated with 50 ng/ml BDNF (Figure 4D), demonstrating activation of the p75NTR pathway when BDNF levels are high, leading to increased proliferation.
Since BDNF-mediated differentiation requires high levels of BDNF (Figures 2C–F), we investigated whether the p75NTR activation observed under proliferative conditions was also required to achieve terminal differentiation of adult NSCs. Thus, NSCs were differentiated in the presence of a high concentration of BDNF and either of the antagonists TrkB-i and p75-i (Figure 4E). In the absence of BDNF, no alterations were detected in the percentage of neurons, oligodendrocytes, and astrocytes after the receptor blockage (Figure 4E). The differentiation of NSCs with 50 ng/ml of BDNF increased the percentage of neurons and oligodendrocytes at the expense of astrocytes, as previously demonstrated. However, only p75NTR inhibition with p75-i was able to rescue the proportion of oligodendrocytes observed in the control cultures with statistical significance (Figure 4E). No statistically significant alterations were observed in the percentage of astrocytes and neurons with the TrkB-i and p75-i antagonists. However, a decrease in the proportion of oligodendrocytes with TrkB-i antagonist in 50 ng/ml BDNF-treated cultures was detected, not reaching statistical significance (Figure 4E).
We have shown in this study that BDNF facilitates self-renewal and cell cycle progression in NSCs isolated from the SVZ of adult mice. These processes are mediated by the TrkB/TrkB.T1 receptors as they can be blocked by ANA-12 (TrkB-i), an inhibitor that interacts with the binding domain of BDNF in the extracellular domain of these receptors (Cazorla et al., 2011). Interestingly, both self-renewal and cell cycle progression become dependent on p75NTR when the concentration of BDNF is high enough to activate this latter receptor. Under this condition, BDNF does not exert proliferative effects if the p75NTR function is pharmacologically blocked. In addition, we have demonstrated that BDNF induces the differentiation of NSCs into oligodendrocytes through a p75NTR-dependent mechanism as it requires a BDNF concentration above its Kd for the binding to p75NTR and can be pharmacologically blocked with a p75NTR-specific inhibitor. We have also shown that BDNF triggers neuronal differentiation when applied at a high dose (Figure 5).

Figure 5. Scheme showing how BDNF regulates self-renewal, proliferation, and differentiation of adult NSCs by TrkB and p75NTR receptors. In homeostasis conditions (left), NSCs require TrkB signaling to maintain their self-renewal capacity. Inhibition of this receptor by a specific TrkB antagonist (TrkB-i) reduces the formation of neurospheres. Exposure to low concentrations of BDNF (10 ng/ml) increases the self-renewal and proliferative activity of the adult NSCs, which is prevented by the TrkB antagonist. Exposure to high concentrations of BDNF (50 ng/ml) (right) induces the upregulation of p75NTR, thus becoming self-renewal and proliferation dependent on p75NTR. The high concentration of BDNF also promotes NSC commitment to neuronal and oligodendroglial lineage. This increment of oligodendrocytes is prevented by a p75NTR-specific antagonist (p75-i). Thus, in an exogenous BDNF context, TrkB regulates self-renewal and proliferation in NSCs isolated from the adult SVZ, whereas p75NTR is implicated in self-renewal, proliferation, and differentiation in the presence of a high dose of the neurotrophin.
Our results indicate that BDNF is required for NSC self-renewal. This effect is dose-dependent since a significantly higher number of neurospheres can be observed in the presence of 50 ng/ml BDNF when compared to 10 ng/ml. This facilitation has been previously described for the BDNF-dependent survival of rat hippocampal neurons (Zanin et al., 2019). The mechanism by which 50 ng/ml BDNF potentiates NSC self-renewal might depend on the observed upregulation of p75NTR expression at this high BDNF concentration. This increase in p75NTR at high BDNF dose is reminiscent of the effect of NGF in astrocytes, which can also upregulate p75NTR expression in these cells (Kumar et al., 1993). In the adult SVZ, the p75NTR-positive population contains all of the neurosphere-producing precursor cells (Young et al., 2007). Therefore, we suggest that the observed increase of p75NTR in our cultures when treated with 50 ng/ml BDNF is likely due to the activation of p75NTR and the upregulation of its levels in all the neurosphere-constituting cells. In fact, the upregulation of the mRNA and protein p75NTR levels was blocked in the presence of p75-i, indicating activation of this receptor after high-dose BDNF treatment to promote its own expression.
In this study, we have demonstrated that BDNF facilitates the proliferation of adult mouse NSCs in vitro. This observation is consistent with the finding that BDNF stimulates the proliferation of newborn NSCs (Chen et al., 2013), human iPSCs-derived NPCs (Pansri et al., 2021), and embryonic neural precursors (Bartkowska et al., 2007). The stimulation of proliferation triggered by BDNF likely depends on TrkB since the use of TrkB-i prevents it. This is consistent with the known activation by TrkB of the Ras–Raf–MEK–ERK signaling pathway (Reichardt, 2006), which favors cell cycle progression when ERK translocates to the nucleus and phosphorylate transcription factor substrates that are responsible for the mitogenic response (Mebratu and Tesfaigzi, 2009). This is consistent with our observation that BDNF induces the phosphorylation of TrkB in Y516, a residue known to participate in the latter signaling pathway (Fan et al., 2020). Nevertheless, TrkB.T1 may also participate in the facilitation of NSC proliferation by BDNF as the truncated form of TrkB has been suggested to induce BDNF-dependent proliferative effects on both embryonic NSCs (Islam et al., 2009) and embryonic neural progenitors (Tervonen et al., 2006).
Our results indicate that the intrinsic ability of TrkB to confer both self-renewal and proliferative capacity to NSCs (i.e., the proliferative capacity that would be observed upon pharmacological inhibition of p75NTR) becomes unexpectedly abolished when BDNF is added at 50 ng/ml. We explain this result in terms of the differential capacity of BDNF to activate p75NTR depending on its concentration (Rodriguez-Tebar et al., 1990). We propose that the activation of p75NTR with 50 ng/ml BDNF seems to permanently modify the proliferative signaling of TrkB. We refer to this effect as “co-receptor dependence for TrkB signaling.” The mechanism of acquisition of this novel co-receptor dependence is currently unknown. However, it should not derive from a different mode of TrkB activation by the higher BDNF concentration since the binding capacity of BDNF to the high-affinity TrkB receptor has already reached a plateau at the range of 10–50 ng/ml (Rodriguez-Tebar et al., 1990). This co-receptor dependence for TrkB signaling might be physiologically relevant in vivo, in neurogenic regions where local enrichment of BDNF results in the upregulation of p75NTR and the modulation of TrkB/p75NTR signaling. Our results are consistent with previous studies in postnatal hippocampal NSCs demonstrating the implication of p75NTR in the proliferation capacity of these cells since the p75NTR-ligand proNGF inhibits proliferation of the NCSs (Guo et al., 2013). As proNGF cannot interact with TrkB, it likely prevents the functional interaction of p75NTR with the latter in response to endogenously produced BDNF. This effect was also abolished in p75NTR knock-out mice (Guo et al., 2013), thus providing genetic evidence that this receptor is involved in the proliferation of NSCs.
Our results also indicate that the pharmacological inhibition of TrkB results in a dramatic reduction in the number of neurospheres even in the absence of added BDNF, suggesting that low levels of this neurotrophin may be released by the NSCs facilitating their self-renewal. Indeed, previous studies have shown BDNF expression in the SVZ (Galvão et al., 2008) and embryonic NSCs (Blurton-Jones et al., 2009). We have shown that Bdnf-specific mRNA is transcribed by adult NSCs, a finding consistent with a previous study by Goldberg et al. (2015). In contrast, TrkB inhibition in the Ki67 proliferation assay without exogenous BDNF does not lead to a significant reduction in cell cycle progression. The main difference between both results is the density of the NSCs that were used. In the proliferative assay, high NSC density was employed, while in the self-renewal assay, NSCs were plated at low density. Therefore, one explanation for this discrepancy may derive from a hypothetical capacity of TrkB to stimulate either the expression or function of the cell adhesion molecules involved in the generation of the neurospheres (Zhou et al., 1997). Consequently, NSCs would not adhere to each other to generate multicellular structures in the presence of TrkB-i.
In this study, we have demonstrated that BDNF induces the differentiation of adult NSCs in vitro into oligodendrocytes and neurons, as previously shown to take place in newborn NSCs (Chen et al., 2013; Langhnoja et al., 2021). This is consistent with the capacity of BDNF to promote the progression of oligodendrocyte lineage and to enhance myelination through the p75NTR receptor (Cosgaya et al., 2002). The studies by Chen et al. (2013) and Langhnoja et al. (2021) mentioned above did not compare the roles of TrkB and p75NTR in this process. Nevertheless, we note that high concentrations of BDNF were used by these authors to detect a potent differentiative effect on newborn NSCs (25 and 50 ng/ml BDNF, respectively). We therefore decided to explore which BDNF receptor is responsible for the differentiative effect of BDNF. Our results indicate that BDNF induces differentiation through p75NTR-dependent signaling based on two lines of evidence. On the one hand, this effect could not be observed with 10 ng/ml BDNF, a concentration that is insufficient to activate p75NTR (Rodriguez-Tebar et al., 1990). On the other hand, the use of p75-i, in contrast to TrkB-i, significantly blocked BDNF-dependent oligodendrocyte differentiation. These results agree with the known inhibition of oligodendrogenesis in a p75NTR-dependent manner since this process was blocked in the presence of proNGF and p75NTR knock-out mice (Guo et al., 2013).
We have observed that the p75NTR-specific inhibitor was not able to prevent neuronal differentiation in vitro, which is consistent with the observation that p75NTR null mice had nearly identical levels of surviving BrdU-positive cells in the OB relative to wild-type mice 28 days after DNA labeling with this nucleotide analog (Bath et al., 2008). This contrasts with our observation that 50 ng/ml BDNF, but not 10 ng/ml BDNF, is required to induce neuronal differentiation in our cultures. In this regard, we note that a great statistical error can be observed in the increase of TUJ1-positive cells when the NSCs are treated with 50 ng/ml BDNF under differentiative conditions (Figures 2C, 4E). Therefore, it cannot be strongly concluded that BDNF triggers a clear effect on neuronal differentiation through p75NTR.
Our results are consistent with the observation that intraventricular administration of BDNF increases the number of newly generated neurons in the adult rat olfactory bulb (Zigova et al., 1998; Benraiss et al., 2001; Henry et al., 2007). They are also consistent with the reduction in the number of newborn neurons that is observed in the OB of mice lacking one copy of the Bdnf gene (Bath et al., 2008). They are also consistent with the claim that TrkB is not essential for adult SVZ neurogenesis (Galvão et al., 2008). Mechanistically, the observation that neurotrophin binding to p75NTR modulates Rho activity and axonal outgrowth (Yamashita et al., 1999) and that developmental biology is one of the enriched pathways associated with p75NTR function (Sajanti et al., 2020) may explain the differentiative effect of BDNF-dependent activation of p75NTR in adult NSCs.
Taken together, our results provide the basis to understand the role of BDNF in the homeostasis of SVZ-derived adult NSCs and the implications of this relevant neurotrophin in pathological conditions as we have clarified the differential contribution of TrkB and p75NTR to BDNF-dependent self-renewal, proliferation, and differentiation of adult NSCs. Furthermore, our results reveal an undescribed mechanism based on a co-receptor dependence for TrkB signaling in the regulation of self-renewal and proliferation of adult NSCs that may be a clue to understand BDNF effects in the neurogenic niche.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
The animal study was approved by Comité de Ética (Consejo Superior de Investigaciones Científicas). The study was conducted in accordance with the local legislation and institutional requirements.
AL-U: Conceptualization, Formal analysis, Methodology, Writing–original draft, Writing–review & editing. JF: Conceptualization, Funding acquisition, Supervision, Writing–review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Ministerio de Ciencia e Innovación, grant number PID2021-128473OB-I00, supported by MCIN/AEI/10.13039/501100011033 and ERDF A way of making Europe. AL-U was contracted by PTI+ Neuroaging (CSIC) and currently holds a Juan de la Cierva Formación 2021 contract from Ministerio de Ciencia e Innovación (FJC2021-046729-I).
The authors thank M. Chao (New York University) for providing #9992 antibody against the intracellular domain of p75NTR, and A. Fraj, A. M. Llabrés-Mas, A. Garrido-García, and V. Cano-Daganzo for their technical help.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
One author (JF) declared that he was an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1271820/full#supplementary-material
Bai, Y., Dergham, P., Nedev, H., Xu, J., Galan, A., Rivera, J. C., et al. (2010). Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J. Biol. Chem. 285, 39392–39400. doi: 10.1074/jbc.M110.147801
Bartkowska, K., Paquin, A., Gauthier, A. S., Kaplan, D. R., and Miller, F. D. (2007). Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 134, 4369–4380. doi: 10.1242/dev.008227
Bath, K. G., Akins, M. R., and Lee, F. S. (2012). BDNF control of adult SVZ neurogenesis. Dev. Psychobiol. 54, 578–589. doi: 10.1002/dev.20546
Bath, K. G., Mandairon, N., Jing, D., Rajagopal, R., Kapoor, R., Chen, Z. Y., et al. (2008). Variant brain-derived neurotrophic factor (Val66Met) alters adult olfactory bulb neurogenesis and spontaneous olfactory discrimination. J. Neurosci. 28, 2383–2393. doi: 10.1523/JNEUROSCI.4387-07.2008
Belenguer, G., Domingo-Muelas, A., Ferron, S. R., Morante-Redolat, J. M., and Farinas, I. (2016). Isolation, culture and analysis of adult subependymal neural stem cells. Differentiation 91, 28–41. doi: 10.1016/j.diff.2016.01.005
Benraiss, A., Chmielnicki, E., Lerner, K., Roh, D., and Goldman, S. A. (2001). Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J. Neurosci. 21, 6718–6731. doi: 10.1523/JNEUROSCI.21-17-06718.2001
Berghuis, P., Agerman, K., Dobszay, M. B., Minichiello, L., Harkany, T., and Ernfors, P. (2006). Brain-derived neurotrophic factor selectively regulates dendritogenesis of parvalbumin-containing interneurons in the main olfactory bulb through the PLCgamma pathway. J. Neurobiol. 66, 1437–1451. doi: 10.1002/neu.20319
Bizy, A., and Ferron, S. R. (2015). Isolation, long-term expansion, and differentiation of murine neural stem cells. Methods Mol. Biol. 1212, 103–112. doi: 10.1007/7651_2014_91
Blurton-Jones, M., Kitazawa, M., Martinez-Coria, H., Castello, N. A., Muller, F. J., Loring, J. F., et al. (2009). Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 106, 13594–13599. doi: 10.1073/pnas.0901402106
Cazorla, M., Premont, J., Mann, A., Girard, N., Kellendonk, C., and Rognan, D. (2011). Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest. 121, 1846–1857. doi: 10.1172/JCI43992
Chaker, Z., Codega, P., and Doetsch, F. (2016). A mosaic world: puzzles revealed by adult neural stem cell heterogeneity. Wiley Interdiscip. Rev. Dev. Biol. 5, 640–658. doi: 10.1002/wdev.248
Chen, B. Y., Wang, X., Wang, Z. Y., Wang, Y. Z., Chen, L. W., and Luo, Z. J. (2013). Brain-derived neurotrophic factor stimulates proliferation and differentiation of neural stem cells, possibly by triggering the Wnt/beta-catenin signaling pathway. J. Neurosci. Res. 91, 30–41. doi: 10.1002/jnr.23138
Chen, T., Wu, Y., Wang, Y., Zhu, J., Chu, H., Kong, L., et al. (2017). Brain-derived neurotrophic factor increases synaptic protein levels via the MAPK/Erk signaling pathway and Nrf2/Trx axis following the transplantation of neural stem cells in a rat model of traumatic brain injury. Neurochem. Res. 42, 3073–3083. doi: 10.1007/s11064-017-2340-7
Chiaramello, S., Dalmasso, G., Bezin, L., Marcel, D., Jourdan, F., Peretto, P., et al. (2007). BDNF/ TrkB interaction regulates migration of SVZ precursor cells via PI3-K and MAP-K signalling pathways. Eur. J. Neurosci. 26, 1780–1790. doi: 10.1111/j.1460-9568.2007.05818.x
Cosgaya, J. M., Chan, J. R., and Shooter, E. M. (2002). The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298, 1245–1248. doi: 10.1126/science.1076595
Deshpande, S. S., Malik, S. C., Conforti, P., Lin, J. D., Chu, Y. H., Nath, S., et al. (2022). P75 neurotrophin receptor controls subventricular zone neural stem cell migration after stroke. Cell Tissue Res. 387, 415–431. doi: 10.1007/s00441-021-03539-z
Eide, F. F., Lowenstein, D. H., and Reichardt, L. F. (1993). Neurotrophins and their receptors–current concepts and implications for neurologic disease. Exp. Neurol. 121, 200–214. doi: 10.1006/exnr.1993.1087
Faigle, R., and Song, H. (2013). Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim. Biophys. Acta. 1830, 2435–2448. doi: 10.1016/j.bbagen.2012.09.002
Fan, C. H., Lin, C. W., Huang, H. J., Lee-Chen, G. J., Sun, Y. C., Lin, W., et al. (2020). LMDS-1, a potential TrkB receptor agonist provides a safe and neurotrophic effect for early-phase Alzheimer's disease. Psychopharmacol. 237, 3173–3190. doi: 10.1007/s00213-020-05602-z
Frade, J. M., Rodriguez-Tebar, A., and Barde, Y. A. (1996). Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383, 166–168. doi: 10.1038/383166a0
Fuentealba, L. C., Obernier, K., and Alvarez-Buylla, A. (2012). Adult neural stem cells bridge their niche. Cell Stem Cell 10, 698–708. doi: 10.1016/j.stem.2012.05.012
Galvão, R. P., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (2008). Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J. Neurosci. 28, 13368–13383. doi: 10.1523/JNEUROSCI.2918-08.2008
Giuliani, A., D'Intino, G., Paradisi, M., Giardino, L., and Calza, L. (2004). p75(NTR)-immunoreactivity in the subventricular zone of adult male rats: expression by cycling cells. J. Mol. Histol. 35, 749–758. doi: 10.1007/s10735-004-9609-2
Goldberg, N. R. S., Caesar, J., Park, A., Sedgh, S., Finogenov, G., Masliah, E., et al. (2015). Neural stem cells rescue cognitive and motor dysfunction in a transgenic model of dementia with lewy bodies through a BDNF-dependent mechanism. Stem Cell Rep. 5, 791–804. doi: 10.1016/j.stemcr.2015.09.008
Gonçalves, J. T., Schafer, S. T., and Gage, F. H. (2016). Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell 167, 897–914. doi: 10.1016/j.cell.2016.10.021
Guo, J., Wang, J., Liang, C., Yan, J., Wnag, Y., Liu, G., et al. (2013). proNGF inhibits proliferation and oligoendrogenesis of postnatal hippocampal neural stem/progenitor cells through p75NTR in vitro. Stem Cell Res. 11, 874–887. doi: 10.1016/j.scr.2013.05.004
Hempstead, B. L., Martin-Zanca, D., Kaplan, D. R., Parada, L. F., and Chao, M. V. (1991). High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350, 678–683. doi: 10.1038/350678a0
Henry, R. A., Hughes, S. M., and Connor, B. (2007). AAV-mediated delivery of BDNF augments neurogenesis in the normal and quinolinic acid-lesioned adult rat brain. Eur. J. Neurosci. 25, 3513–3525. doi: 10.1111/j.1460-9568.2007.05625.x
Holsinger, R. M., Schnarr, J., Henry, P., Castelo, V. T., and Fahnestock, M. (2000). Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res. Mol. Brain Res. 76, 347–354. doi: 10.1016/S0169-328X(00)00023-1
Huber, L. J., and Chao, M. V. (1995). Mesenchymal and neuronal cell expression of the p75 neurotrophin receptor gene occur by different mechanisms. Dev. Biol. 167, 227–238. doi: 10.1006/dbio.1995.1019
Islam, O., Loo, T. X., and Heese, K. (2009). Brain-derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK-B receptor, MAP kinase, AKT, and STAT-3 signaling pathways. Curr. Neurovasc. Res. 6, 42–53. doi: 10.2174/156720209787466028
Jiao, S. S., Shen, L. L., Zhu, C., Bu, X. L., Liu, Y. H., Liu, C. H., et al. (2016). Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer's disease. Transl. Psychiatry 6, e907. doi: 10.1038/tp.2016.186
Klein, R., Conway, D., Parada, L. F., and Barbacid, M. (1990). The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61, 647–656. doi: 10.1016/0092-8674(90)90476-U
Klein, R., Parada, L. F., Coulier, F., and Barbacid, M. (1989). trkB, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J. 8, 3701–3709. doi: 10.1002/j.1460-2075.1989.tb08545.x
Kumar, S., Pena, L. A., and de Vellis, J. (1993). CNS glial cells express neurotrophin receptors whose levels are regulated by NGF. Brain Res. Mol. Brain Res. 17, 163–168. doi: 10.1016/0169-328X(93)90086-5
Langhnoja, J., Buch, L., and Pillai, P. (2021). Potential role of NGF, BDNF, and their receptors in oligodendrocytes differentiation from neural stem cell: an in vitro study. Cell Biol. Int. 45, 432–446. doi: 10.1002/cbin.11500
Lee, J., Duan, W., and Mattson, M. P. (2002). Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 82, 1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x
Leibrock, J., Lottspeich, F., Hohn, A., Hofer, M., Hengerer, B., Masiakowski, P., et al. (1989). Molecular cloning and expression of brain-derived neurotrophic factor. Nature 341, 149–152. doi: 10.1038/341149a0
Li, Y., Luikart, B. W., Birnbaum, S., Chen, J., Kwon, C. H., Kernie, S. G., et al. (2008). TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59, 399–412. doi: 10.1016/j.neuron.2008.06.023
Lin, Z., Tann, J. Y., Goh, E. T., Kelly, C., Lim, K. B., Gao, J. F., et al. (2015). Structural basis of death domain signaling in the p75 neurotrophin receptor. Elife 4, e11692. doi: 10.7554/eLife.11692.023
Liu, F., Xuan, A., Chen, Y., Zhang, J., Xu, L., Yan, Q., et al. (2014). Combined effect of nerve growth factor and brain-derived neurotrophic factor on neuronal differentiation of neural stem cells and the potential molecular mechanisms. Mol. Med. Rep. 10, 1739–1745. doi: 10.3892/mmr.2014.2393
Llorente, V., Velarde, P., Desco, M., and Gomez-Gaviro, M. V. (2022). Current understanding of the neural stem cell niches. Cells 11:3002. doi: 10.3390/cells11193002
Mazzaro, N., Barini, E., Spillantini, M. G., Goedert, M., Medini, P., and Gasparini, L. (2016). Tau-driven neuronal and neurotrophic dysfunction in a mouse model of early tauopathy. J. Neurosci. 36, 2086–2100. doi: 10.1523/JNEUROSCI.0774-15.2016
Mebratu, Y., and Tesfaigzi, Y. (2009). How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle 8, 1168–1175. doi: 10.4161/cc.8.8.8147
Mitra, G., Martin-Zanca, D., and Barbacid, M. (1987). Identification and biochemical characterization of p70TRK, product of the human TRK oncogene. Proc. Natl. Acad. Sci. USA 84, 6707–6711. doi: 10.1073/pnas.84.19.6707
Naylor, R. L., Robertson, A. G., Allen, S. J., Sessions, R. B., Clarke, A. R., Mason, G. G., et al. (2002). A discrete domain of the human TrkB receptor defines the binding sites for BDNF and NT-4. Biochem. Biophys. Res. Commun. 291, 501–507. doi: 10.1006/bbrc.2002.6468
Okano, H. J., Pfaff, D. W., and Gibbs, R. B. (1996). Expression of EGFR-, p75NGFR-, and PSTAIR (cdc2)-like immunoreactivity by proliferating cells in the adult rat hippocampal formation and forebrain. Dev. Neurosci. 18, 199–209. doi: 10.1159/000111408
Pansri, P., Phanthong, P., Suthprasertporn, N., Kitiyanant, Y., Tubsuwan, A., Dinnyes, A., et al. (2021). Brain-derived neurotrophic factor increases cell number of neural progenitor cells derived from human induced pluripotent stem cells. PeerJ. 9, e11388. doi: 10.7717/peerj.11388
Parkhurst, C. N., Zampieri, N., and Chao, M. V. (2010). Nuclear localization of the p75 neurotrophin receptor intracellular domain. J. Biol. Chem. 285, 5361–5368. doi: 10.1074/jbc.M109.045054
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond,. B,. Biol. Sci. 361, 1545–1564. doi: 10.1098/rstb.2006.1894
Rodriguez-Tebar, A., Dechant, G., and Barde, Y. A. (1990). Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron 4, 487–492. doi: 10.1016/0896-6273(90)90107-Q
Rodriguez-Tebar, A., Dechant, G., Gotz, R., and Barde, Y. A. (1992). Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO J. 11, 917–922. doi: 10.1002/j.1460-2075.1992.tb05130.x
Roux, P. P., and Barker, P. A. (2002). Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 67, 203–233. doi: 10.1016/S0301-0082(02)00016-3
Sajanti, A., Lyne, S. B., Girard, R., Frantzen, J., Rantamaki, T., Heino, I., et al. (2020). A comprehensive p75 neurotrophin receptor gene network and pathway analyses identifying new target genes. Sci. Rep. 10, 14984. doi: 10.1038/s41598-020-72061-z
Snapyan, M., Lemasson, M., Brill, M. S., Blais, M., Massouh, M., Ninkovic, J., et al. (2009). Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J. Neurosci. 29, 4172–4188. doi: 10.1523/JNEUROSCI.4956-08.2009
Sohn, J., Orosco, L., Guo, F., Chung, S. H., Bannerman, P., Mills Ko, E., et al. (2015). The subventricular zone continues to generate corpus callosum and rostral migratory stream astroglia in normal adult mice. J. Neurosci. 35, 3756–3763. doi: 10.1523/JNEUROSCI.3454-14.2015
Taupin, P., and Gage, F. H. (2002). Adult neurogenesis and neural stem cells of the central nervous system in mammals. J. Neurosci. Res. 69, 745–749. doi: 10.1002/jnr.10378
Tervonen, T. A., Ajamian, F., De Wit, J., Verhaagen, J., Castren, E., and Castren, M. (2006). Overexpression of a truncated TrkB isoform increases the proliferation of neural progenitors. Eur. J. Neurosci. 24, 1277–1285. doi: 10.1111/j.1460-9568.2006.05010.x
Vicario, A., Kisiswa, L., Tann, J. Y., Kelly, C. E., and Ibanez, C. F. (2015). Neuron-type-specific signaling by the p75NTR death receptor is regulated by differential proteolytic cleavage. J. Cell Sci. 128, 1507–1517. doi: 10.1242/jcs.161745
Vilar, M., and Mira, H. (2016). Regulation of neurogenesis by neurotrophins during adulthood: expected and unexpected roles. Front. Neurosci. 10:26. doi: 10.3389/fnins.2016.00026
Yamashita, T., Tucker, K. L., and Barde, Y. A. (1999). Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24, 585–593. doi: 10.1016/S0896-6273(00)81114-9
Young, K. M., Merson, T. D., Sotthibundhu, A., Coulson, E. J., and Bartlett, P. F. (2007). p75 neurotrophin receptor expression defines a population of BDNF-responsive neurogenic precursor cells. J. Neurosci. 27, 5146–5155. doi: 10.1523/JNEUROSCI.0654-07.2007
Zanin, J. P., Montroull, L. E., Volosin, M., and Friedman, W. J. (2019). The p75 neurotrophin receptor facilitates TrkB signaling and function in rat hippocampal neurons. Front. Cell. Neurosci. 13:485. doi: 10.3389/fncel.2019.00485
Zhou, H., Welcher, A. A., and Shooter, E. M. (1997). BDNF/NT4-5 receptor TrkB and cadherin participate in cell-cell adhesion. J. Neurosci. Res. 49, 281–291.
Keywords: SVZ, self-renewal, neurosphere, neurotrophin, TrkB.T1, oligodendrocyte, neuron, co-receptor dependence
Citation: Lozano-Ureña A and Frade JM (2023) Differential contribution of TrkB and p75NTR to BDNF-dependent self-renewal, proliferation, and differentiation of adult neural stem cells. Front. Mol. Neurosci. 16:1271820. doi: 10.3389/fnmol.2023.1271820
Received: 03 August 2023; Accepted: 01 December 2023;
Published: 22 December 2023.
Edited by:
Catarina Osorio, Erasmus Medical Center, NetherlandsReviewed by:
Barbara Hausott, Innsbruck Medical University, AustriaCopyright © 2023 Lozano-Ureña and Frade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Frade, ZnJhZGVAY2FqYWwuY3NpYy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.