
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 28 September 2023
Sec. Brain Disease Mechanisms
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1244133
This article is part of the Research Topic The Legacy of Dr. Rita Levi-Montalcini: Advances in Neurotrophic Factors in Brain Disease Development and Treatment View all 7 articles
 Daniele Narducci1
Daniele Narducci1 Despoina Charou2,3
Despoina Charou2,3 Thanasis Rogdakis2,3
Thanasis Rogdakis2,3 Ioanna Zota2,3
Ioanna Zota2,3 Vivi Bafiti1
Vivi Bafiti1 Maria Zervou1
Maria Zervou1 Theodora Katsila1
Theodora Katsila1 Achille Gravanis2,3
Achille Gravanis2,3 Kyriakos C. Prousis1
Kyriakos C. Prousis1 Ioannis Charalampopoulos2,3
Ioannis Charalampopoulos2,3 Theodora Calogeropoulou1*
Theodora Calogeropoulou1*Introduction: The neurotrophin system plays a pivotal role in the development, morphology, and survival of the nervous system, and its dysregulation has been manifested in numerous neurodegenerative and neuroinflammatory diseases. Neurotrophins NGF and BDNF are major growth factors that prevent neuronal death and synaptic loss through binding with high affinity to their specific tropomyosin-related kinase receptors namely, TrkA and TrkB, respectively. The poor pharmacokinetic properties prohibit the use of neurotrophins as therapeutic agents. Our group has previously synthesized BNN27, a prototype small molecule based on dehydroepiandrosterone, mimicking NGF through the activation of the TrkA receptor.
Methods: To obtain a better understanding of the stereo-electronic requirements for selective activation of TrkA and TrkB receptors, 27 new dehydroepiandrosterone derivatives bearing a C17-spiro-dihydropyran or cyclobutyl moiety were synthesized. The new compounds were evaluated for their ability (a) to selectively activate the TrkA receptor and its downstream signaling kinases Akt and Erk1/2 in PC12 cells, protecting these cells from serum deprivation-induced cell death, and (b) to induce phosphorylation of TrkB and to promote cell survival under serum deprivation conditions in NIH3T3 cells stable transfected with the TrkB receptor and primary cortical astrocytes. In addition the metabolic stability and CYP-mediated reaction was assessed.
Results: Among the novel derivatives, six were able to selectively protect PC12 cells through interaction with the TrkA receptor and five more to selectively protect TrkB-expressing cells via interaction with the TrkB receptor. In particular, compound ENT-A025 strongly induces TrkA and Erk1/2 phosphorylation, comparable to NGF, and can protect PC12 cells against serum deprivation-induced cell death. Furthermore, ENT-A065, ENT-A066, ENT-A068, ENT-A069, and ENT-A070 showed promising pro-survival effects in the PC12 cell line. Concerning TrkB agonists, ENT-A009 and ENT-A055 were able to induce phosphorylation of TrkB and reduce cell death levels in NIH3T3-TrkB cells. In addition, ENT-A076, ENT-A087, and ENT-A088 possessed antiapoptotic activity in NIH-3T3-TrkB cells exclusively mediated through the TrkB receptor. The metabolic stability and CYP-mediated reaction phenotyping of the potent analogs did not reveal any major liabilities.
Discussion: We have identified small molecule selective agonists of TrkA and TrkB receptors as promising lead neurotrophin mimetics for the development of potential therapeutics against neurodegenerative conditions.
Traced back to over 600 million years ago (Hallböök et al., 2006), the neurotrophin (NT) system plays a pivotal role in the development, morphology, and survival of the nervous system (Shahnaz et al., 2015). In addition, it influences a wide range of functions, among which the most studied are related to memory (Mitre et al., 2016), the perception of fear (Penzo et al., 2015), pain (Allen and Dawbarn, 2006), hunger (Ceren et al., 2015), and taste (Fei and Krimm, 2013), and even insulin secretion and receptivity (Baeza-Raja et al., 2012; Houtz et al., 2016).
Over the years, various secreted growth factors belonging to the neurotrophins family were discovered the most important being the nerve growth factor (NGF), the brain-derived neurotrophic factor (BDNF), and neurotrophin 3 (NT-3). To produce their beneficial effects, neurotrophins bind with high affinity to a specific tropomyosin receptor kinase (Trk), namely, NGF and NT-3 (with less affinity than NGF) to TrkA, BDNF to TrkB, and NT-3 to TrkC. The last member of the neurotrophin system is the 75 kDa pan-neurotrophin receptor (p75NTR), bound with low affinity by all neurotrophins and with high affinity by their precursors: the proneurotrophins (pro-NTs) (Chao et al., 2006; Mitre et al., 2016). The correct functioning of the neurotrophin system was famously schematized by Bai et al. (2005) with a Yin and Yang representation, to remark how the homeostasis relies on a finely regulated balance between NT/Trk-stimulated survival and pro-NT/p75-stimulated apoptosis. Dysregulations in the aforementioned equilibrium of the neurotrophin system were observed in numerous neurodegenerative and neuroinflammatory diseases. Among the most famous, Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS) are highlighted (Meldolesi, 2017).
Following this general understanding, activation of the Trks holds the potential to revolutionize the treatment of neurodegeneration from being merely palliative to disease-modifying. Alas, despite numerous past and ongoing attempts, neurotrophins themselves are proving to be an unviable therapeutic modality because of the typical pharmacokinetic issues that characterize peptide-based medications. These include short half-life, poor oral bioavailability, and low ability to cross the blood–brain barrier (Pardridge, 2002), accompanied by topical hyperalgesia, undesirably high spike-like potency, and counterproductive p75NTR activation (Josephy-Hernandez et al., 2017).
A variety of strategies have been conceived to target the neurotrophin receptors and were recently reviewed (Saragovi et al., 2019; Gudasheva et al., 2021; Jeyaram et al., 2023; Rahman et al., 2023). The current study focuses specifically on new small molecule neurotrophin mimetics capable of achieving selective Trk receptor activation circumventing the limitations associated with the direct use of neurotrophins in therapy, thus paving the way for more effective and tailored neuroprotection strategies. In this context, the endogenous neurosteroid dehydroepiandrosterone (DHEA) acts as the stepstone of our study as it can bind to all the neurotrophin receptors stimulating neurogenesis, neuronal survival, and neurite outgrowth and reduce neuroinflammation (Baulieu, 1996; Lazaridis et al., 2011; Pediaditakis et al., 2015; Alexaki et al., 2018; Yilmaz et al., 2019).
Capitalizing on these beneficial properties, Calogeropoulou et al. (2009) designed and synthesized a series of modified DHEA derivatives aiming to enhance the potency and selectivity of the neurotrophin receptors while hindering metabolism into androgens and estrogens. Among these, the C17-spiro-epoxy analog BNN27 emerged as the lead compound. BNN27 is a selective TrkA and p75NTR agonist (Pediaditakis et al., 2016a,b) and can exert neuroprotective and anti-inflammatory activity in several in vitro and in vivo studies (Glajch et al., 2016; Bonetto et al., 2017; Pitsikas and Gravanis, 2017; Ibán-Arias et al., 2018; Tsika et al., 2021). Furthermore, BNN27 does not bind to hormonal receptors and is not metabolically converted to estrogens or androgens (Calogeropoulou et al., 2009; Bennett et al., 2016). However, it was shown that BNN27 undergoes rapid hydroxylation and subsequent excretion during hepatic catabolism, resulting in an elimination constant (Kel) of 0.465 h−1 and a mean residence time (MRT) of 2.154 h in murine models (Bennett et al., 2016; Tsika et al., 2021). To address the metabolic stability issue of BNN27 while maintaining potency and enhancing selectivity toward TrkA or TrkB receptors, our initial research efforts sought to replace the C-17 spiro epoxide moiety with the more stable cyclopropyl group with promising results (Rogdakis et al., 2022; Yilmaz et al., 2022).
In the current study, we set out to investigate the presence of larger spirocyclic moieties, in particular, cyclobutyl and dihydropyran rings, attached to DHEA at the C17 position (Figure 1). In turn, these rings were decorated with carefully selected substituents to enrich the structure–activity relationships for neurotrophin mimetic activity. Additionally, we synthesized one derivative featuring a C17-spiro-tetrahydropyran ring and one with a C17-spiro-oxepan-3-one moiety.
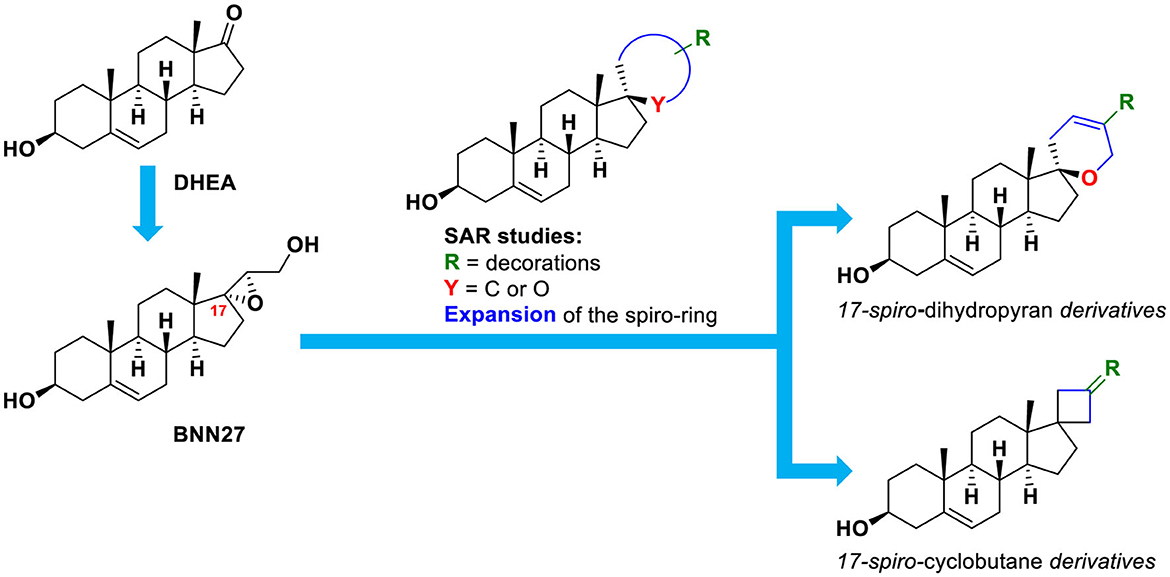
Figure 1. Conceptual representation of the design of the compounds of the present study.
A solution of tetrabutylammonium fluoride (TBAF) (1.0 M in THF, 1.3 mL, 1.3 mmol) was added to a solution of compound 4 (102 mg, 0.18 mmol) in anhydrous tetrahydrofuran (2 mL), and the reaction was stirred at 25°C for 48 h. The reaction mixture was diluted with dichloromethane and the organic phase was washed with water and brine, dried over Na2SO4, and the solvent was removed in vacuo. The residue was purified by FCC (elution solvent: petroleum ether 40–60°C/diethyl ether: 9/1) to afford ENT-A002 as a white crystalline solid (52 mg, 83% yield). Rf: 0.08 (petroleum ether 40–60°C/diethyl ether: 8/2); mp: 119–121°C; 1H NMR (600 MHz, CDCl3): δ 5.75–5.74 (m, 1H), 5.69–5.67 (m, 1H), 5.33 (d, J = 4.2 Hz, 1H), 4.21–4.14 (m, 2H), 3.52–3.47 (m, 1H), 2.36 (d, J = 16.9 Hz, 1H), 2.28 (dd, J = 3.0, 12.7 Hz, 1H), 2.23–2.20 (m, 1H), 2.00–1.05 (m, 18H), 1.01 (s, 3H), 0.87 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 140.8, 125.8, 123.3, 121.3, 83.0, 71.6, 63.2, 51.0, 50.0, 45.8, 42.2, 37.2, 36.5, 33.2, 32.9, 32.4, 31.7, 31.5, 31.2, 23.3, 20.8, 19.3, 13.6; HR-MS (APCI+): calcd for C23H35O2 [M+H]+ 343.2632, found 343.2623; calcd for C23H33O [M-H2O+H]+ 325.2526, found 325.2520.
HF·pyridine complex (1.3 mL, 14.43 mmol) was added to a solution of compound 6 (0.505 g, 1.00 mmol) in anhydrous dichloromethane (33.0 mL) at 0°C, and the reaction was stirred at the same temperature for 40 min. The reaction was quenched with water and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by FCC (ethyl acetate/petroleum ether 35-60°C: 4/6) to afford ENT-A025, as a white solid (0.39 g, yield quantitative). Rf: 0.30 (ethyl acetate/petroleum ether 35-60°C: 4/6); mp: 128–130°C; 46.05° (c = 0.00160 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.88 (bs, 1H), 5.36–5.33 (m, 1H), 4.25 (ABq, JAB = 16.4 Hz, 2H), 3.99 (s, 2H), 3.55–3.48 (m, 1H), 2.43–0.86 (m, 22H), 1.02 (s, 3H), 0.92 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 141.0, 133.0, 124.4, 121.5, 83.2, 71.9, 63.8, 51.5, 50.2, 45.9, 45.8, 42.4, 37.4, 36.7, 33.7, 33.3, 32.7, 31.8, 31.5, 29.8, 23.4, 21.0, 19.5, 13.7. HR-MS (APCI+): m/z calculated for C24H3635ClO2 391.2398 [M+H]+, found 391.2396; HPLC: 100% purity, RT = 27.42 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 20% ethyl acetate −80% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
A solution of ENT-A025 (20.0 mg, 0.06 mmol) and KCN (10.6 mg, 0.16 mmol) in a mixture of EtOH/water/dichloromethane: 2/1/1(1.6 mL) was vigorously stirred at 25°C for 12 h. Subsequently, the reaction was stirred at 40°C for 5 h, and then KCN (10.6 mg, 0.16 mmol) was added and the reaction was stirred at 25°C for an additional 12 h. The reaction mixture was evaporated under reduced pressure, and the residue was partitioned between aqueous saturated sodium thiosulphate solution and dichloromethane. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by FCC (elution solvent: dichloromethane/MeOH: 99/1) to afford ENT-A033, as a white solid (16 mg, 70% yield). Rf: 0.50 (dichloromethane/MeOH: 96/4); mp: 173–175°C; (c = 0.00128 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.91–5.89 (m, 1H), 5.37–5.33 (m, 1H), 4.13 and 4.10 (two d, ABq, JAB = 17.6 Hz, 2H), 3.56–3.48 (m, 1H), 2.98 (s, 2H), 2.43–0.83 (m, 21H), 1.02 (s, 3H), 0.91 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 141.0, 125.7, 123.3, 121.5, 116.8, 83.4, 71.9, 64.7, 51.4, 50.2, 45.9, 42.4, 37.4, 33.4, 33.2, 32.7, 31.8, 31.8, 31.3, 29.8, 23.5, 21.4, 21.0, 19.6, 13.7; HR-MS (APCI+): m/z calculated for C25H36NO2 382.2741 [M+H]+, found 382.2737; HPLC: 100% purity, RT = 36.47 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 20% ethyl acetate-−80% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Diethylamine (14.5 μL, 0.14 mmol) was added to a solution of ENT-A025 (27.3 mg, 0.07 mmol) in anhydrous dichloromethane (0.4 mL), and the reaction was stirred at 25°C for 48 h. To the reaction mixture, saturated aqueous ammonium chloride solution was added and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by FCC (elution solvent: dichloromethane/MeOH: 9/1) to afford ENT-A076 as a white solid (27 mg, 90% yield). Rf: 0.21 (dichloromethane/MeOH: 9/1); mp: 144°C, decomposition; 46.15° (c = 0.00130 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.67 (bs, 1H), 5.36–5.32 (m, 1H), 4.18 and 4.15 (two d, ABq, JAB = 17.3 Hz, 2H), 3.55–3.47 (m, 1H), 2.97 (s, 2H), 2.55–2.49 (m, 4H), 2.39–0.86 (m, 25H), 1.02 (s, 3H), 0.90 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 141.0, 134.1, 121.5, 121.5, 83.3, 71.9, 65.4, 56.6, 51.4, 50.3, 46.5, 45.9, 42.4, 37.5, 36.7, 33.5, 33.2, 32.7, 31.9, 31.8, 31.5, 23.5, 21.1, 19.6, 13.8, 11.3. HR-MS (APCI+): m/z calculated for C28H46O2N 428.3523 [M+H]+, found 428.3521; HPLC: 100% purity, RT = 14.23 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 15% chloroform-−85% acetone, isocratic, flow rate 2.1 mL/min at 25°C, and injection volume 20 μL.
A mixture of azide 9 (55.7 mg, 0.14 mmol), propargyl alcohol (34.0 μL, 0.42 mmol), CuSO4·5H2O (10.4 mg, 0.04 mmol), and sodium ascorbate (16.6 mg, 0.08 mmol) in dichloromethane/water/tBuOH (1/1/1, 3.0 mL) was stirred at 25°C for 12 h. Subsequently, the reaction mixture was extracted with dichloromethane and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: dichloromethane/MeOH: 96/4) to afford ENT-A037, as a white solid (31 mg, 49%). Rf: 0.28 (dichloromethane/MeOH: 95/5); mp: 202–204°C; (c = 0.00145 g/mL, CHCl3); 1H NMR (300 MHz, CD3OD): δ 7.87 (s, 1H), 5.88 (bs, 1H), 5.38–5.30 (m, 1H), 4.93 (bs, 2H), 4.67 (s, 2H), 4.04 (bs, 2H), 3.45–3.34 (m, 1H), 2.42 (d, J = 17.3 Hz, 1H), 2.26–0.86 (m, 20H), 1.03 (s, 3H), 0.88 (s, 3H); 13C NMR (75 MHz, CD3OD): 149.4, 142.3, 132.6, 125.5, 124.1, 122.2, 84.5, 72.4, 64.4, 56.5, 53.3, 52.5, 51.6, 47.0, 43.0, 38.6, 37.7, 34.4, 34.2, 33.9, 32.8, 32.3, 32.2, 24.3, 22.0, 19.8, 14.0; HR-MS (APCI+): m/z calculated for C27H40N3O3 454.3064 [M+H]+, found 454.3063; HPLC: 100% purity, RT = 41.36 min. Column: Agilent Eclipse XDB-C8 (4.6 × 150 mm, 5 μm), Method: eluting with 10% water-−90% methanol, isocratic, flow rate 1.0 mL/min at 10°C, and injection volume 20 μL.
A mixture of azide 9 (13.7 mg, 0.04 mmol), N,N-dimethyl-propargyl amine (12.0 μL, 0.11 mmol), CuSO4·5H2O (2.6 mg, 0.01 mmol), and sodium ascorbate (4.2 mg, 0.02 mmol) in dichloromethane/water/tBuOH (1/1/1, 1.0 mL) was stirred at 25°C for 12 h. Subsequently, the reaction mixture was extracted with dichloromethane and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: dichloromethane/MeOH: 93/7) to afford ENT-A046, as a white solid (17 mg, quantitative yield). Rf: 0.37 (dichloromethane/MeOH: 9/1); mp: 182°C, decomposition; (c = 0.00127 g/mL, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.60 (s, 1H), 5.83 (bs, 1H), 5.37–5.30 (m, 1H), 4.85 (s, 2H), 4.02 (bs, 2H), 3.70 (bs, 2H), 3.59–3.44 (m, 1H), 2.43–0.83 (m, 24H), 1.01 (s, 3H), 0.86 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 144.5, 141.0, 131.1, 124.9, 122.9, 121.4, 83.3, 71.8, 63.5, 54.2, 52.9, 51.4, 50.2, 45.9, 44.8, 42.4, 37.4, 36.7, 33.3, 33.2, 32.6, 31.8, 31.3, 29.8, 23.4, 21.0, 19.5, 13.7. HR-MS (APCI+): m/z calculated for C29H45N4O3 481.3537 [M+H]+, found 481.3541; HPLC: 100% purity, RT = 29.7 min. Column: Fortis C18 (10 × 250 mm, 10 μm), Method: eluting with 8% water−92% methanol, isocratic, flow rate 1.0 mL/min at 25°C, and injection volume 20 μL.
A mixture of azide 9 (24.0 mg, 0.06 mmol), prop-2-yn-1-yl-cyclohexane (26.0 μL, 0.18 mmol), CuSO4·5H2O (4.5 mg, 0.02 mmol), and sodium ascorbate (7.1 mg, 0.04 mmol) in dichloromethane/water/tBuOH (1/1/1, 1.0 mL) was stirred at 25°C for 12 h. Subsequently, the reaction mixture was extracted with dichloromethane and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 6/4) to afford ENT-A047, as a white solid (19 mg, 61% yield). Rf: 0.22 (petroleum ether 35–60°C/ethyl acetate: 1/1); mp: 181°C, decomposition; (c = 0.00137 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 7.22 (s, 1H), 5.82 (bs, 1H), 5.36–5.32 (m, 1H), 4.82 (s, 2H), 4.03 and 3.99 (two d, ABq, JAB = 17.8 Hz, 2H), 3.55–3.47 (m, 1H), 2.57 (d, J = 7.0 Hz, 2H), 2.44–0.83 (m, 33H), 1.01 (s, 3H), 0.87 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 140.8, 131.2, 124.4, 121.3, 120.8, 83.1, 71.7, 63.5, 52.6, 51.3, 50.1, 45.8, 42.2, 38.0, 37.3, 36.5, 33.4, 33.4, 33.1, 33.0, 33.0, 32.5, 31.7, 31.6, 31.2, 29.7, 26.4, 26.1, 23.3, 20.8, 19.4, 13.5. HR-MS (APCI+): m/z calculated for C33H50N3O2 520.3898 [M+H]+, found 520.3898; HPLC: 100% purity, RT = 32.74 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 35% ethyl acetate −65% toluene, isocratic, flow rate 2.1 mL/min at 25°C, and injection volume 20 μL.
A mixture of azide 9 (24.0 mg, 0.06 mmol), 3-phenyl-1-propyne (22.0 μL, 0.18 mmol), CuSO4·5H2O (4.5 mg, 0.02 mmol), and sodium ascorbate (7.1 mg, 0.04 mmol) in dichloromethane/water/tBuOH (1/1/1, 1.0 mL) was stirred at 25°C for 12 h. Subsequently, the reaction mixture was extracted with dichloromethane and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 7/3) to afford ENT-A055, as a white solid (22 mg, 71% yield). Rf: 0.51 (petroleum ether 35–60°C/ethyl acetate: 4/6); mp: 201–203°C; (c = 0.00125 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 7.34–7.22 (m, 5H), 7.14 (s, 1H), 5.81 (bs, 1H), 5.37–5.33 (m, 1H), 4.81 (s, 2H), 4.82 and 4.80 (two d, ABq, JAB = 15.4 Hz, 2H), 4.11 and 4.08 (two d, ABq, JAB = 16.6 Hz, 2H), 4.02 and 3.99 (two d, ABq, JAB = 17.5 Hz, 2H), 3.57–3.49 (m, 1H), 2.43–0.85 (m, 21H), 1.03 (s, 3H), 0.87 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 148.2, 141.0, 139.1, 131.2, 128.8, 128.8, 126.6, 124.7, 121.4, 121.2, 83.3, 71.8, 63.6, 52.9, 51.4, 50.2, 45.9, 42.4, 37.4, 36.7, 33.5, 33.2, 32.6, 32.4, 31.8, 31.8, 31.3, 23.4, 21.0, 19.5, 13.6. HR-MS (APCI+): m/z calculated for C33H44N3O2 514.3428 [M+H]+, found 514.3432; HPLC: 100% purity, RT = 24.79 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 35% ethyl acetate-−65% toluene, isocratic, flow rate 2.1 mL/min at 25°C, and injection volume 20 μL.
Step 1. Triethyl phosphonoacetate (0.11 mL, 0.52 mmol) was added to a suspension of NaH (20.8 mg, 0.52 mmol) in anhydrous tetrahydrofuran (0.4 mL) at 0°C, and the reaction was stirred at 25°C for 30 min. The reaction mixture was cooled to 0°C and a solution of aldehyde 10 (64.0 mg, 0.13 mmol) in anhydrous tetrahydrofuran (1.6 mL) was added and the mixture was stirred at the same temperature for 1 h. The reaction was quenched, at the same temperature, by the dropwise addition of brine and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The product was used without further purification in the next step (32 mg, 54% yield). Rf: 0.13 (petroleum ether 35–60°C/ethyl acetate: 85/15).
Step 2. HF·pyridine complex (0.2 mL, 2.22 mmol) was added dropwise to a solution of the crude product from step 1 (32 mg, 0.06 mmol) in anhydrous dichloromethane (2.4 mL) at 0°C, and the reaction was stirred at 0°C for 40 min. The reaction was quenched at 0°C with water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: petroleum ether 35–60°C/ethyl acetate: 85/15) to afford ENT-A007, as a white solid (20.0 mg, 35% yield over two steps). Rf: 0.47 (petroleum ether 35–60°C/ethyl acetate: 6/4); mp: 161−163°C; (c = 0.00135 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 7.20 (d, J = 16.2 Hz, 1H), 6.22 (bs, 1H), 5.58 (d, J = 16.2 Hz, 1H), 5.38–5.30 (m, 1H), 4.38 and 4.30 (two d, ABq, JAB = 16.5 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.60–3.43 (m, 1H), 2.54 (d, J = 18.5 Hz, 1H), 2.32–0.82 (m, 20H), 1.30 (t, J = 6.9 Hz, 3H), 1.01 (s, 3H), 0.92 (s, 3H). 13C NMR (75 MHz, CDCl3): δ δ 167.3, 143.2, 141.0, 134.5, 133.2, 121.4, 115.1, 83.5, 71.8, 62.6, 60.5, 51.5, 50.1, 42.3, 37.4, 36.7, 33.8, 33.3, 32.7, 32.6, 31.8, 31.7, 29.8, 23.4, 21.0, 19.5, 14.4, 13.6; HR-MS (APCI−): m/z calculated for C28H39O4 439.2854 [M-H]−, found m/z 439.2848; HPLC: 100% purity, RT = 29.99 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 20% ethyl acetate −80% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Step 1. Diethyl (cyanomethyl) phosphonate (30.0 μL, 0.28 mmol) was added to a suspension of NaH (11.2 mg, 0.28 mmol) in anhydrous tetrahydrofuran (0.2 mL) at 0°C, and the reaction was stirred at 25°C for 30 min. The reaction was cooled to 0°C and a solution of aldehyde 10 (34.0 mg, 0.07 mmol) in anhydrous tetrahydrofuran (0.7 mL) was added and the mixture was stirred at the same temperature for 1 h. The reaction was quenched, at the same temperature, by the dropwise addition of brine and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The product was used without further purification in the next step (27 mg, 71% yield). Rf: 0.13 (petroleum ether 35–60°C/Et2O: 94/6).
Step 2. HF·pyridine complex (60.0 μL, 0.67 mmol) was added dropwise to a solution of the crude product from step 1 (27.0 mg, 0.05 mmol) in anhydrous dichloromethane (1.7 mL) at 0°Cof, and the reaction was stirred at 0°C for 40 min. The reaction was quenched at 0°C with water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: petroleum ether 35–60°C/ethyl acetate: 80/20) to afford ENT-A009, as a white solid (17.0 mg, 64% yield over two steps). Rf: 0.53 (petroleum ether 35–60°C/ethyl acetate: 6/4); mp: 205°C, decomposition; = + 59.26° (c = 0.00135 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 6.91 (d, J = 16.7 Hz, 1H), 6.23 (bs, 1H), 5.38–5.30 (m, 1H), 5.00 (d, J = 16.8 Hz, 1H), 4.32 and 4.25 (two d, ABq, JAB = 17.4 Hz, 2H), 3.60–3.43 (m, 1H), 2.55 (d, J = 18.8 Hz, 1H), 2.32–0.83 (m, 20H), 1.02 (s, 3H), 0.92 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 149.0, 141.0, 135.9, 133.1, 121.4, 118.4, 93.3, 83.5, 71.9, 61.8, 51.5, 50.2, 46.0, 42.4, 37.4, 36.7, 33.7, 33.3, 32.7, 31.89, 31.77, 29.9, 23.5, 21.0, 19.6, 13.7. HR-MS (APCI−): m/z calculated for C26H3O2N 392.2595 [M-H]−, found 392.2587; HPLC: 100% purity, RT = 34.89 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 30% ethyl acetate-−70% cyclohexane, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Step 1. Potassium tert-butoxide (53.9 mg, 0.48 mmol) was added to a solution of (4-bromobutyl)-triphenyl-phosphonium bromide (114.8 mg, 0.24 mmol) in anhydrous tetrahydrofuran (0.36 mL) at 0°C, and the reaction was stirred at 25°C for 30 min. The reaction was cooled to 0°C, a solution of aldehyde 10 (30.0 mg, 0.06 mmol) in anhydrous tetrahydrofuran (0.36 mL) was added, and the mixture was stirred at the same temperature for 1 h. The reaction was quenched, at the same temperature, by the dropwise addition of brine and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The mixture was filtered through a silica pad (elution solvent: petroleum ether 35–60°C/ethyl acetate: 97/3) to remove triphenylphosphine oxide and the solvent evaporated in vacuo. The crude product was used without further purification in the next step (20 mg, 64% yield). Rf: 0.53 (petroleum ether 35–60°C/ethyl acetate: 94/6).
Step 2. HF·pyridine complex (52.0 μL, 0.58 mmol) was added dropwise to a solution of the crude product from step 1 (20.0 mg, 0.04 mmol) in anhydrous dichloromethane (1.3 mL) at 0°C, and the reaction was stirred at 0°C for 40 min. The reaction was quenched at 0°C with water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 7/3) to afford ENT-A065, as a white solid (17.0 mg, 64% yield over two steps). Rf: 0.23 (petroleum ether 35–60°C/ethyl acetate: 7/3); mp: 164–166°C; 6.58° (c = 0.00152 g/mL, CHCl3); NMR (600 MHz, CDCl3): δ 5.54 (bs, 1H), 5.52 (bs, 1H), 5.37–5.32 (m, 1H), 4.37 and 4.30 (two d, ABq, JAB = 16.0 Hz, 2H), 3.56–3.47 (m, 1H), 2.89–2.79 (m, 2H), 2.72 (t, J = 8.1 Hz, 2H), 2.47–0.85 (m, 23H), 1.02 (s, 3H), 0.91 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 141.4, 141.0, 135.0, 121.6, 120.77, 120.5, 83.1, 71.9, 64.5, 51.4, 50.3, 45.9, 42.4, 37.4, 36.7, 33.4, 33.2, 32.7, 32.4, 32.3, 31.9, 31.8, 31.8, 23.5, 21.0, 19.6, 17.4, 13.8. HR-MS (APCI+): m/z calculated for C28H41O2 409.3101 [M+H]+, found 409.3101; HPLC: 100% purity, RT = 34.89 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 20% ethyl acetate −80% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Step 1. NaHMDS (1M in tetrahydrofuran; 0.28 mL, 0.28 mmol) was added to a solution of (bromomethyl)-triphenyl-phosphonium bromide (112.1 mg, 0.28 mmol) in anhydrous tetrahydrofuran (0.42 mL) at 0°C, and the reaction was stirred at 25°C for 30 min. The reaction was cooled to 0°C, a solution of aldehyde 10 (33 mg, 0.07 mmol) in anhydrous tetrahydrofuran (0.42 mL) was added, and the mixture was stirred at the same temperature for 14 h. The reaction was quenched, at the same temperature, by the dropwise addition of brine and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The mixture was filtered through a silica pad (elution solvent: petroleum ether 35–60°C/ethyl acetate: 94/6) to remove triphenylphosphine oxide and the solvent evaporated in vacuo. The crude product was used as such in the next step (28 mg, 72% yield). Rf: 0.63 (petroleum ether 35–60°C/ethyl acetate: 94/6).
Step 2. HF·pyridine complex (65.0 μL, 0.72 mmol) was added dropwise to a solution of the crude product from step 1 (28.0 mg, 0.05 mmol) in anhydrous dichloromethane (1.5 mL) at 0°C, and the reaction was stirred at 0°C for 40 min. The reaction was quenched at 0°C with water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 7/3) to afford ENT-A066 (E,Z mixture 6:4), as a white solid (21.0 mg, 93% yield). Rf: 0.27 (petroleum ether 35–60°C/ethyl acetate: 7/3); mp: 68–70°C; [a]D24 = + 30.30° (c = 0.00165 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 6.63 (d, J = 14.2 Hz) and 6.50 (d, J = 8.1 Hz) (1H), 6.05 and 5.82 (two bs, 1H), 6.04–5.97 (m, 1H), 5.37–5.32 (m, 1H), 4.66 and 4.58 (two d, ABq, JAB = 16.5 Hz) and 4.31 and 4.26 (two d, ABq, JAB = 15.4 Hz) (2H), 3.56–3.48 (m, 1H), 2.47–0.86 (m, 21H), 1.02 and 1.02 (two s, 3H), 0.93 and 0.91 (two s, 3H); 13C NMR (151 MHz, CDCl3): δ 141.0, 136.3, 133.4, 133.3, 131.6, 129.0, 126.6, 121.5, 103.2, 102.8, 83.5, 82.8, 71.9, 64.7, 62.4, 51.4, 50.2, 45.9, 42.4, 37.4, 36.7, 33.7, 33.5, 33.3, 33.2, 32.7, 32.2, 32.1, 31.8, 31.8, 29.8, 23.5, 21.0, 19.5, 13.7. HR-MS (APCI+): m/z calculated for C25H36O2Br 447.1893 [M+H]+, found 447.1892; HPLC: 100% purity, RT = 27.36 min (E) and 28.47 min (Z). Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 25% ethyl acetate-−75% cyclohexane, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Step 1. Potassium tert-butoxide (26.9 mg, 0.24 mmol) was added to a solution of ethyl triphenyl-phosphonium bromide (89.1 mg, 0.24 mmol) in anhydrous tetrahydrofuran (0.42 mL) at 0°C, and the reaction was stirred at 25°C for 30 min. The reaction was cooled to 0°C, a solution of aldehyde 10 (50.0 mg, 0.10 mmol) in anhydrous tetrahydrofuran (0.42 mL) was added, and the mixture was stirred at the same temperature for 14 h. The reaction was quenched, at the same temperature, by the dropwise addition of brine and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The mixture was filtered through a silica pad (elution solvent: petroleum ether 35–60°C/ethyl acetate: 94/6) to remove triphenylphosphine oxide and the solvent evaporated in vacuo. The crude product was used as such in the next step (28 mg, 94% yield). Rf: 0.63 in petroleum ether 35–60°C/ethyl acetate: 94/6).
Step 2. HF·pyridine complex (78.0 μL, 0.87 mmol) was added to a solution of the crude product from step 1 (28 mg, 0.06 mmol) in anhydrous dichloromethane (1.8 mL) at 0°C, and the reaction was stirred at 0°C for 40 min. The reaction was quenched at 0°C with water and extracted with dichloromethane. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 7/3) to afford ENT-A068 (E, Z mixture), as a white solid (20 mg, 87% over two steps); Rf: 0.22 (petroleum ether 35–60°C/ethyl acetate: 7/3); mp: 103–105°C; (c = 0.00138 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.95 (d, J = 16.0 Hz, 0.4H), 5.68–5.63 (m, 1.6H), 5.50–5.45 (m, 0.6 H), 5.40–5.34 (m, 1.4H), 4.36–4.26 (m, 2H), 3.56–3.49 (m, 1H), 2.48–1.19 (m, 19H), 1.76 (d, J = 7.2 Hz) and 1.73 (d, J = 6.6 Hz) (3H), 1.024 and 1.023 (two s, 3H), and 0.92 (s, 3H). 13C NMR (151 MHz, CDCl3): 13C NMR (151 MHz, CDCl3): δ 141.0, 134.4, 134.1, 130.4, 127.8, 125.3, 123.2, 122.3, 122.0, 121.5, 83.6, 83.0, 71.9, 65.7, 63.3, 51.4, 50.3, 45.9, 42.4, 37.4, 36.7, 33.7, 33.5, 33.3, 33.2, 32.7, 31.9, 31.8, 31.7, 23.5, 21.0, 19.6, 18.6, 15.7, 14.3, 13.8; HR-MS (APCI+): m/z calculated for C26H39O2 383.2945 [M+H]+, found 383.2941; HPLC: 100% purity, RT = 25.17 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 25% ethyl acetate −75% cyclohexane, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Methyl magnesium bromide (3M in Et2O, 40.5 μL, 0.12 mmol) was added to a solution of ENT-A025 (31.7 mg, 0.081 mmol) in anhydrous tetrahydrofuran (0.81 mL) at 0°C, and the resulting solution was stirred at 25°C for 12 h. The reaction was quenched with a saturated aqueous solution of ammonium chloride and concentrated under reduced pressure. The residue was solubilized in ethyl acetate and washed with brine. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by FCC (elution system: hexane/ethyl acetate: 7/3) to obtain ENT-A070 as a white solid (27 mg, 90% yield). Rf: 0.30 (petroleum ether 35–60°C/ethyl acetate: 6/4); mp: 137–139°C; 5.26° (c = 0.00129 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.42 (bs, 1H), 5.35–5.31 (m, 1H), 4.08 and 4.04 (two d, ABq, JAB = 18.4 Hz, 2H), 3.54–3.46 (m, 1H), 2.36–0.85 (m, 23H), 1.01 (s, 3H), 0.98 (t, J = 7.4 Hz, 3H), 0.89 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 140.8, 137.5, 121.4, 115.9, 83.1, 71.7, 65.6, 51.2, 50.09, 45.1, 42.2, 37.3, 36.5, 33.3, 33.1, 32.5, 31.7, 31.6, 31.1, 25.3, 23.30, 20.9, 19.4, 13.6, 12.1; HR-MS (APCI+): m/z calculated for C25H39O2 371.2945 [M+H]+, found 371.2947; HPLC: 96.5% purity, RT = 27.36 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 25% ethyl acetate −75% cyclohexane, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
A round-bottom flask was loaded with ENT-A070 (20 mg, 0.03 mmol) and tetrahydrofuran (0.5 mL). The solution was reduced using H-Cube Mini+ over 5% PtS/C, at 25°C, hydrogen pressure 1 atm, and flow rate 1 mL/min. The reaction mixture was circulated three times through the 5% PtS/C cartridge until the reaction was complete (checked by 1H-NMR). The reaction was concentrated in vacuo and the residue was purified by FCC (elution solvent: ethyl acetate/hexane/ethyl acetate: 7/3) to afford ENT-A069 (mixture of C5′ epimers), as a white solid (15 mg, 70% yield). Rf: 0.54 (petroleum ether 35–60°C/ethyl acetate: 6/4); mp: 107–109°C; 40.65° (c = 0.00123 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.22–5.18 (m, 1H), 3.60 (dd, J = 12.2, 4.2 Hz) and 2.93 (t, J = 11.3 Hz) (1H), 3.49 (dd, J = 11.5, 3.2 Hz) and 3.24 (dd, J = 11.5, 5.9 Hz) (1H), 3.44–3.31 (m, 1H), 2.18–0.67 (m, 26H), 0.88 (s, 3H), 0.77–0.71 (m, 3H), 0.74 and 0.70 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 141.0, 121.6, 84.8, 84.7, 71.9, 69.4, 68.2, 51.4, 50.4, 50.2, 46.6, 46.2, 42.4, 38.0, 37.4, 37.4, 36.7, 36.7, 36.3, 35.8, 35.3, 32.7, 32.6, 32.1, 32.0, 31.9, 31.8, 30.4, 30.0, 26.5, 26.0, 25.5, 24.2, 24.0, 23.7, 21.2, 20.9, 19.5, 14.2, 13.9, 11.9, 11.4; HR-MS (APCI+): calculated for C25H41O2 m/z 373.3101 [M+H]+, found m/z 373.3100; HPLC: 100% purity, RT = 24.99 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 12% ethyl acetate −88% toluene, isocratic, flow rate 2.1 mL/min at 25°C, and injection volume 20 μL.
Potassium carbonate (611 mg, 4.42 mmol) was added to a solution of compound 13 (480 mg, 1.34 mmol) in a mixture of methanol/water (54/1, 547 mL). The resulting solution was stirred at 25°C for 4 h. Upon completion of the reaction, the reaction mixture was concentrated under reduced pressure, and the residue was extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 8/2) to afford ENT-A076, as a white crystalline solid (440 mg, quantitative yield). Rf: 0.13 (petroleum ether 35–60°C/ethyl acetate: 8/2); mp: 183–185°C; 130.77° (c = 0.00130 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.34–5.30 (m, 1H), 3.53–3.45 (m, 1H), 3.08 (d, J = 17.3 Hz, 1H), 3.00 (d, J = 17.3 Hz, 1H), 2.64 (dd, J = 17.3, 3.4 Hz, 1H), 2.43 (dd, J = 17.3, 3.4 Hz, 1H), 2.29–0.75 (m, 19H), 1.00 (s, 3H), 0.77 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 208.3, 140.9, 121.4, 71.7, 54.3, 52.6, 51.4, 50.0, 43.4, 42.8, 42.3, 37.5, 37.4, 36.6, 33.0, 32.0, 31.9, 31.6, 24.7, 20.6, 19.5, 14.6. HR-MS (APCI+): m/z calculated for C22H33O2 329.2475 [M+H]+, found 329.2474; HR-MS (APCI+): m/z calculated for C22H33O2 329.2475 [M+H]+, found 329.2474; HPLC: 100% purity, RT = 31.56 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 20% ethyl acetate −80% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Diethyl (cyanomethyl) phosphonate (64.7 μL, 0.40 mmol) was added to a suspension of NaH (60% dispersion in mineral oil, 16 mg, 0.40 mmol) in anhydrous tetrahydrofuran (0.3 mL) at 0°C, and the reaction mixture was stirred at 25°C for 30 min. The reaction was cooled to 0°C, a solution of ENT-A076 (38 mg, 0.10 mmol) in anhydrous tetrahydrofuran (1.0 mL) was added, and the mixture was stirred at 25°C for 12 h. The reaction was quenched, at 0°C, by the dropwise addition of brine, and the mixture was concentrated in vacuo. The residue was extracted with ethyl acetate, and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 8/2) to obtain ENT-A077 (E, Z mixture 1/1), a white crystalline solid (25 mg, 71% yield). Rf: 0.05 (petroleum ether 35–60°C/ethyl acetate: 8/2); mp: 161–163°C; (c = 0.00141 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.36–5.32 (m, 1H), 5.15 and 5.13 (two s, 1H), 3.56–3.50 (m, 1H), 2.99 (d, J = 16.6 Hz) and 2.78 (d, J = 16.6 Hz) (1H), 2.89 (t, J = 17.4 Hz, 1H), 2.64–0.86 (m, 21H), 1.01 (s, 3H), 0.69 and 0.67 (two s, 3H). 13C NMR (75 MHz, CDCl3): δ 167.9, 140.9, 121.6, 116.7, 110.2, 91.6, 71.9, 51.9, 50.1, 48.2, 43.4, 42.4, 41.3, 38.0, 37.8, 37.4, 37.3, 36.7, 32.9, 32.0, 31.7, 29.8, 24.6, 20.7, 19.6, 13.8. HR-MS (APCI+): m/z calculated for C24H34ON 352.2635 [M+H]+, found 352.2635; HPLC: 100% purity, RT = 31.56 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 30% ethyl acetate −70% cyclohexane, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
Triethyl phosphonoacetate (1.0 mL, 5.08 mmol) was added to a suspension of NaH (60% dispersion in mineral oil, 203 mg, 0.51 mmol) in anhydrous tetrahydrofuran (3.8 mL) at 0°C, and the reaction mixture was stirred at 25°C for 30 min. The solution was cooled to 0°C, a solution of ENT-A076 (469 mg, 1.27 mmol) in anhydrous tetrahydrofuran (12.7 mL) was added dropwise, and the mixture was stirred at 25°C for 12 h. The reaction was quenched, at 0°C, by the dropwise addition of brine, and the mixture was concentrated under reduced pressure. To the residue was added ethyl acetate, and the organic phase was washed with brine and dried over anhydrous Na2SO4 and was concentrated in vacuo. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 7/3) to obtain ENT-A079 (E,Z mixture 1:1) as a solid (122 mg, 32% yield). Rf: 0.14 (petroleum ether 35–60°C/ethyl acetate: 7/3); 1H NMR (300 MHz, CDCl3): δ 5.68–5.58 (m, 1H), 5.39–5.31 (m, 1H), 4.21–4.05 (m, 2H), 3.59–3.46 (m, 1H), 3.19–0.93 (m, 23H), 1.33–1.19 (m, 3H), 1.02 (s, 3H), and 0.70 and 0.67 (two s, 3H).
NaOH (70 mg, 1.75 mmol) was added to a mixture of ENT-A079 (E/Z mixture 1:1) (99 mg, 0.25 mmol) in a mixture of EtOH/water (2.5 mL/0.75 mL), and the resulting mixture was refluxed for 4 h. The reaction was cooled to room temperature, and the mixture was concentrated under reduced pressure. To the residue was added dropwise 10% HClaq with stirring until a pale-yellow precipitate formed, which was filtered, and the solid was washed with hexane and Et2O, resulting in a white solid. The solid was then dissolved in tetrahydrofuran, and the solution was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: ethyl acetate) to obtain ENT-A080 (E,Z mixture 1:1), as a white crystalline solid (87 mg, 94% yield). Rf: 0.58 (ethyl acetate); mp: 221°C, decomposition; 73.53° (c = 0.00136 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.66 and 5.64 (two s, 1H), 5.39–5.32 (m, 1H), 3.58–3.50 (m, 1H), 3.19–0.62 (m, 23H), 1.02 (s, 3H), 0.69 and 0.68 (two s, 3H). 13C NMR (151 MHz, CDCl3): δ 171.4, 166.9, 140.7, 121.5, 111.9, 71.7, 51.5, 50.0, 48.8, 43.2, 42.6, 42.2, 37.8, 37.3, 36.6, 32.7, 32.0, 31.8, 31.6, 29.7, 29.4, 24.5, 22.7, 20.6, 19.4, 14.1, 13.7. HR-MS (APCI−): m/z calculated for C24H33O3 369.2435 [M-H]−, found m/z 369.2421; HPLC: 100% purity, RT = 18.88 min and 19.92 min (each peak corresponding to one geometrical isomer). Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 50% ethyl acetate −50% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
NaOH (50 mg, 1.25 mmol) was added to a solution of ENT-A079 (E,Z mixture 1:1) (459 mg, 1.24 mmol) in MeOH/water: (54/1; 10.6 mL/0.2 mL). The mixture was stirred at 25°C for 12 h. The reaction mixture was concentrated at reduced pressure and to the residue was added dropwise with stirring 10% HClaq until a pale-yellow precipitate formed which was filtered, and the solid was washed with hexane and Et2O, affording a white solid. The solid was then dissolved in tetrahydrofuran, and the solution was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by FCC (elution solvent: hexane/ethyl acetate: 7/3) to obtain ENT-A080 (E, Z mixture 1:1, 144 mg, 31% yield), ENT-A087 (E,Z mixture 1:1; 100 mg, 21% yield), and ENT-A088 (mixture of the two C17 epimers in a 7/3 ratio; 33 mg, 7% yield), as white solids.
ENT-A087 (E, Z mixture 1:1) Rf: 0.12 (petroleum ether 35-60°C/ethyl acetate: 8/2); mp: 87–89°C; (c = 0,00126 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.63 and 5.61 (two s, 1H), 5.34–5.30 (m, 1H), 3.66 and 3.65 (two s, 3H), 3.54–3.45 (m, 1H), 3.14–0.87 (m, 23H), 0.99 (s, 3H), 0.67 and 0.65 (two s, 3H). 13C NMR (75 MHz, CDCl3): δ 167.2, 163.6, 140.9, 121.5, 112.2, 71.7, 71.7, 51.8, 51.0, 50.1, 49.0, 43.3, 42.3, 41.0, 38.8, 37.6, 37.4, 36.6, 32.8, 32.0, 31.6, 24.6, 24.6, 20.7, 19.5, 13.7. HR-MS (APCI+): m/z calculated for C25H37O3 [M+H]+ 385.2737, found 385.2735; HPLC: 100% purity, RT = 35.65 min and 38.66 min (each peak corresponding to one geometrical isomer). Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 25% ethyl acetate −75% cyclohexane, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
ENT-A088 (17R,S) (3S)-3-hydroxy-spiro[5-androsten-17,3′-oxepan]-7′-one Rf: 0.28 petroleum ether 35–60°C/ethyl acetate: 6/4); mp: 181–183°C; (c = 0.00121 g/mL, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.36–5.32 (m, 1H), 4.30 (d, J = 8.8 Hz) and 4.23 (d, J = 9.0 Hz) (1H), 4.05 (d, J = 9.0 Hz) and 3.88 (d, J = 8.8 Hz, 1H), 3.51 (s, 1H), 2.59–0.75 (m, 25H), 1.01 (s, 3H), and 0.78 and 0.76 (two s, 3H). 13C NMR (75 MHz, CDCl3): δ 177.1, 140.9, 121.3, 76.0, 75.0, 71.7, 60.5, 53.3, 53.1, 52.7, 50.0, 43.6, 42.3, 38.4, 37.4, 36.7, 36.1, 35.9, 34.8, 32.6, 32.5, 32.3, 32.1, 32.0, 31.9, 31.7, 24.5, 24.3, 20.6, 19.5, 16.1, 14.7, 14.3; HPLC: 100% purity, RT = 35.29 min and 37.43 min. Column: Kromasil 100-10-SIL (10 × 250 mm, 10 μm), Method: eluting with 25% ethyl acetate −75% toluene, isocratic, flow rate 1.5 mL/min at 25°C, and injection volume 20 μL.
PC12 cells were obtained from LGC Promochem and cultured under specified conditions. Cells were grown in DMEM medium (Thermo Fischer Scientific, cat# 10566016) containing 10% horse serum (Thermo Fischer Scientific, cat# 16050122), 5% fetal bovine serum (Thermo Fischer Scientific, cat# 10270106), 100 units/mL penicillin, and 0.1 mg/mL streptomycin (Thermo Fischer Scientific, cat# 15140122) at 5% CO2 and 37°C. Cells were used between passages 5 and 20.
For immunoprecipitation experiments, PC12 cells were used when they were at 70–80% confluent. Cells were starved from serum for 4 h and subsequently treated with 100 ng/mL NGF (Alomone Labs, cat# N-100) or 500 nM of each compound for 30 min. Cells were then lysed in Pierce™ IP Lysis Buffer (Thermo Fischer Scientific, cat# 87787) containing proteases (Sigma-Aldrich, cat# 539131) and phosphatase inhibitors (Millipore, cat# 524629). Lysates were then immunoprecipitated overnight at 4°C with TrkA antibody (1:100, Millipore, cat# 06-574) followed by 4 h incubation with protein G-plus agarose beads (Santa Cruz Biotechnology, cat# sc-2002). Beads were then collected, washed three times with lysis buffer, resuspended in SDS loading buffer, and subjected to Western blot against phosphorylated Tyrosine (1:1000, R&D systems, cat# BAM1676). Whole-cell lysates were subjected to Western blot against TrkA (1:1,000, Millipore, cat# 06-574), phosphorylated Akt (1:1,000, Cell Signaling, cat# 9271S), phosphorylated Erk1/2 (1:1,000, Cell Signaling, cat# 9101), total Akt (1:1,000, Cell Signaling, cat# 4,691), and total Erk1/2 (1:1000, Cell Signaling, cat# 9,194).
CellTox assay (G8742, Promega, Leiden, Belgium) was used to assess the survival of PC12 cells under serum deprivation conditions. PC12 cells were plated in 96-well plates, starved from serum for 4 h, and subsequently treated with NGF (100 ng/mL) or compound (500 nM) for 24 h. CellTox assay reagents and Hoescht (1:10,000, H3570, Invitrogen, Massachusetts, USA) were then added to each well for 30 min, and then, cells were imaged with a Zeiss AXIO Vert A1 fluorescent microscope. CellTox positive cells were normalized to the total number of cells for each image.
We tested compounds on three cellular systems, primary cortical astrocytes from 2-day-old mice (as a natively TrkB-expressing neuronal population), and NIH-3T3 fibroblasts stable transfected with TrkB and NIH-3T3 naïve cells. Cells were grown with high-glucose DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, and 100 units/mL penicillin at 37°C in a humidified 5% CO2 atmosphere. Phosphorylation results presented here are in primary astrocytes, and cell toxicity assay results are in NIH-3T3 stable transfected cells with TrkB.
Cells were plated in 12 well plates, at a density of 100,000 cells per well, with 6 h of serum deprivation carried out on the following day. To assess the effect of compounds on TrkB phosphorylation, 20-min treatments were applied, using either BDNF at 500 ng/mL (Peprotech 450-02) (positive control) or compounds at 1 μM.
Cell lysis for 10 min was carried out on ice using the Pierce IP Lysis Buffer by Thermo Scientific and a phospho-protease inhibitor cocktail by Millipore. After adding loading buffer (5 × Laemni), 25 μg from each protein sample were incubated for 5 min at 95°C and subjected to SDS-PAGE. Proteins were transferred to a nitrocellulose membrane at 350 mA for 2 h. Membrane blocking was performed using 5% bovine serum albumin (BSA) at room temperature for 1 h, before adding the primary antibodies to the blocking solution at 4°C overnight. HRP-conjugated secondary antibodies were used for the detection of chemiluminescence with ECL solution. Primary antibodies: Phospho-TrkB (Tyr816) Millipore # ABN1381, and Anti-TrkB Abcam #ab33655.
NIH-3T3 stable transfected cells with TrkB were plated in 96-well plates at a density of 10,000 cells/well, before carrying out serum starvation for 24 h. Cells were then treated for 24 h with either BDNF at 500 ng/mL (control) or the compounds at 1 μM.
For the cell toxicity assay, we used CellTox™ Green Dye (2,000 × , Promega) for the visualization of dead cells and Hoechst 33,342 solution (10,000 × ) for total cells. A ZEISS Axio Vert.A1 microscope was used for image capture, and the ImageJ software (https://imagej.nih.gov/ij/) was used for analysis. The number of CellTox positive cells was normalized against the total number of cells observed in each image.
Mixed glial cultures were isolated from the cortex of C57B/6 pups at post-natal day 2 (P2). Cells were plated in a medium containing high-glucose DMEM, 200 U/mL penicillin, 200 μg/mL streptomycin, and 10% fetal bovine serum (FBS). When cells reach 100% confluency (7–8 days), the anti-mitotic agent Ara-C was added in the media at a final concentration of 10 μM, for 3 to 4 days to target the highly proliferative microglial cells. Ara-C was removed, and primary astrocytes (97% purity) were cultured at 5% CO2 and 37°C.
Cells were plated at 70–80% confluency. The next day, they were deprived of serum for 6 h and subsequently treated with 500 ng/mL BDNF or 1 μM of compounds for 20 min. Cells were then lysed in Pierce™ IP Lysis Buffer (87788, Thermo Fischer Scientific, Rockford, USA), containing proteases (539138, Calbiochem, Darmstadt, Germany), and phosphatase inhibitors (524629, Calbiochem, Darmstadt, Germany). Lysates were then suspended in an SDS loading buffer and subjected to Western blot against phosphorylated TrkB (1:1,000, ABN1381, Sigma-Aldrich, St. Louis, MO, USA) and total TrkB (1:1,000, 07-225-I, Sigma-Aldrich, St. Louis, MO, USA).
Total RNA was extracted from cells using TRIzol reagent (15596026, Thermo Fisher, Waltham, MA, USA), and cDNA was synthesized using the High-Capacity cDNA Reverse Transcription kit (4368814, Thermo Fisher, Waltham, MA, USA) according to the supplier protocols. For qPCR experiments run with SYBR green dye, for 20 s at 95°C, followed by 40 cycles of 95°C for 3 s and 60°C for 30 s on a StepOne Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). B-Actin was used as a housekeeping gene to normalize the gene expression levels. Data were collected and analyzed using the StepOne Software v2.3 (Thermo Fischer Scientific, Waltham, MA, USA).
The data are presented as the mean ± standard error of the mean (SEM). Statistical analysis was conducted using GraphPad Prism 7 software (GraphPad Software Inc., San Diego, CA, USA). Student's t-test was used for the comparisons. A p-value of < 0.05 was considered to mark statistical significance. In cases where fold change values were utilized, statistical analysis was performed on the portion of each case normalized to the DMSO condition. A p-value of < 0.05 was considered statistically significant.
The physicochemical descriptors and ADME properties of the compounds were predicted through the application of the Qikprop module of the Schrödinger platform (Schrodinger Release 2020-3, 2020). Before Qikprop calculations, the compounds were imported in the Maestro platform of Schrodinger software as .smi files (Schrödinger Release 2020-3, 2020a), converted to 3D form, and optimized through the energy minimization module of MacroModel (Schrödinger Release 2020-3, 2020b) and Ligprep module (Schrödinger Release 2020-3, 2020c) by applying OPLS_2005 force field at pH = 7.0 ± 0.5.
For metabolic stability assessment, pooled human liver microsomes were employed at 0.5 mg/mL. Incubation conditions were optimized to ensure linear metabolite formation with respect to protein concentration and reaction time. All test compounds were assessed at 1 μM based on our ADMET standard operating procedures and protocols, also published previously (1 μM was selected based on our ADMET standard operating procedures and protocols also published previously) (Rogdakis et al., 2022; Yilmaz et al., 2022). Approximately 1 mM of NADPH served as a cofactor. Reactions took place in triplicates at 37°C. Negative and positive controls (low vs. rapid clearance) were included for low vs. rapid clearance. After 60 min, reactions were terminated. To profile the depletion of test compounds and hence determine the residual (%) of time zero, readouts were recorded by Lionheart FX (Agilent BioTek).
Human cytochrome P450 (CYP450) isoenzymes were expressed in Baculosomes®, purchased from Thermo Fisher Scientific (Waltham, MA, USA). All reagents were handled and prepared according to the manufacturer's protocol. Test compounds were assessed at 1 μM for their effects on CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Approximately 1 μM was selected based on our ADMET standard operating procedures and protocols also published previously (Rogdakis et al., 2022; Yilmaz et al., 2022). CYP450-enzymatic activity was determined based on the kinetic model for each CYP450 isoform (Cohen et al., 2003; Veith et al., 2009). Reactions took place in triplicates at 20°C. For this, NADP+ (10 mM in 100 mM potassium phosphate, pH 8.0) was converted into NADPH by the regeneration system present (glucose-6-phosphate at 333 mM and glucose-6-phosphate-dehydrogenase at 30 U/mL in 100 mM potassium phosphate, pH 8.0). Following the addition of the fluorescent substrate, signal monitoring over time took place immediately (<2 min) at suitable excitation and emission wavelengths (Agilent BioTek Lionheart FX). CYP450 inhibition (%) was determined based on the reaction rates (fluorescence intensity changes per unit time). In total, n = 60 measurements per minute were acquired (t = 60 min).
X is the rate observed in the presence of the test compound.
A is the rate observed in the presence of the negative (solvent, DMSO) control.
The first series of derivatives involves the modification of the DHEA scaffold by a C17-spiro-2H-dihydropyran ring variously decorated at the C5′ position. Initially, the unsubstituted derivative ENT-A002 was prepared from DHEA. Quantitative protection of the 3β-hydroxy group of DHEA as the tert-butyldiphenylsilyl ether (1a) was achieved using tert-butyldiphenylsilyl chloride in the presence of imidazole as the base and iodine. The steric hindrance imposed by the axial C18 methyl group on the C17-ketone (Xun et al., 1994) resulted in the exclusive addition of allylmagnesium bromide from the equatorial face of compound 1a with a conversion of 97%. The resulting tertiary alcohol (2a) was then deprotonated with NaH and alkylated by allyl bromide, yielding compound 3 in 97% yield. The desired (2H)dihydropyran ring was formed via a ring-closing metathesis reaction using diene 3 and Grubbs 2nd generation catalyst affording derivative 4 in 94% yield. Finally, the deprotection of the C3-alcohol of 4 was achieved using tetra-n-butylammonium fluoride (TBAF) to give compound ENT-A002 an 83% yield (Figure 2).
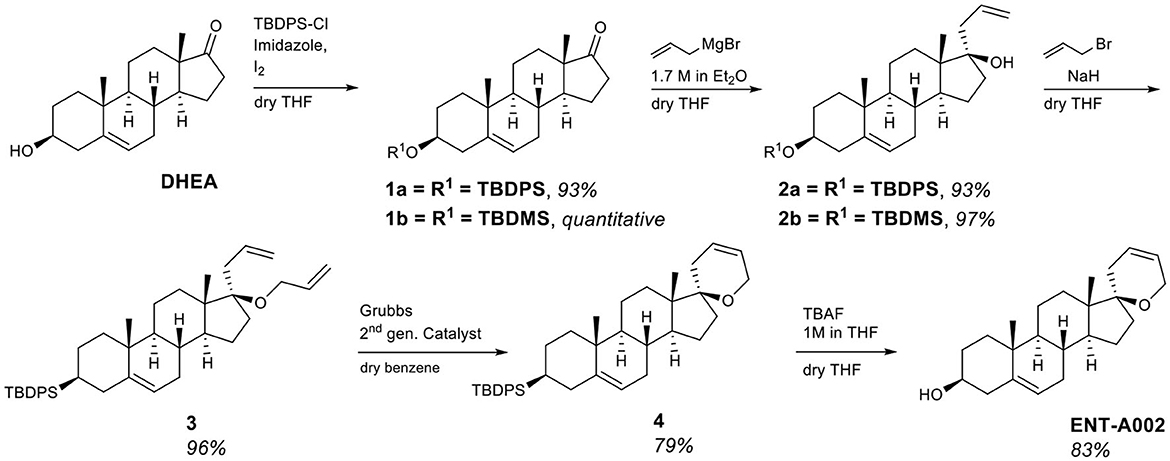
Figure 2. Synthesis of ENT-A002.
Compound ENT-A002 represents the unsubstituted canvas of this series. To explore the stereo-electronic requirements for selectivity and agonism on the neurotrophin Trk receptors, we incorporated a chloromethyl handle, on the C17-spiro-(2H)dihydropyran moiety (compound 6) that could further elaborate a variety of functionalities. Our synthetic strategy involved the protection of the C3-alcohol of DHEA with tert-butyldimethylchlorosilane to afford compound 1b in quantitative yield that was in turn transformed to compound 2b upon the addition of allyl magnesium bromide (Figure 2). Alkylation of the tertiary alcohol of compound 2b using 3-chloro-2-chloromethyl-propene and NaH as a base afforded the chloro-substituted diene 5 in 82% yield (Figure 3). Subsequently, a ring-closing metathesis reaction employing Hoveyda-Grubbs 2nd generation catalyst resulted in the formation of compound 6 in 79% yield (Figure 3). Deprotection of compound 6 using HF·pyridine complex gave the C5′-chloromethyl derivative ENT-A025. Alternatively, by treating compound 6 with TBAF, the fluorinated congener of ENT-A025 compound ENT-A034 was obtained in quantitative yield (Figure 3).
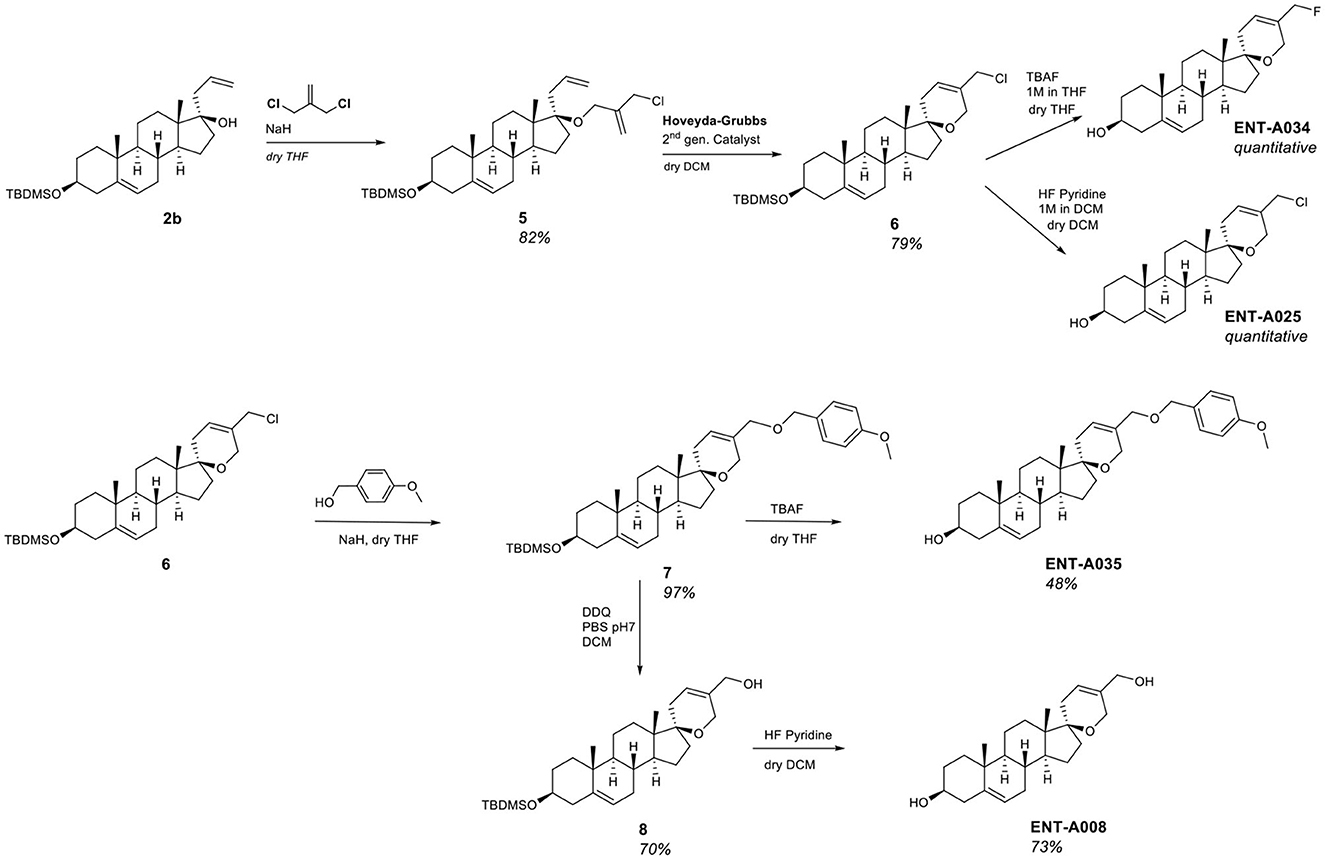
Figure 3. Synthesis of the key intermediate 6 and compounds ENT-A008, ENT-A025, ENT-A034, and ENT-A035.
Alkylation of compound 6 with para-methoxybenzyl alcohol in the presence of NaH resulted in the corresponding para-methoxybenzyl ether derivative 7 in 97% yield which enabled the obtainment of two derivatives, namely ENT-A035 and ENT-A008. Deprotection of the 3β-hydroxy group of 7 using TBAF afforded ENT-A035 in 48% yield. In turn, a reaction of 7 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), yielded the C5′-hydroxymethyl derivative 8 in 70% yield, which was then treated with HF·pyridine complex to afford ENT-A008 in 73% yield (Figure 3).
The thio analog ENT-A026 was obtained in quantitative yield through a two-step reaction. Initially, the chloromethyl group in ENT-A025 was transformed to iodomethyl using NaI in acetone that was reacted with methyl thioglycolate in dichloromethane to give ENT-A026 (Figure 4). In our subsequent investigation, we incorporated nitrogen-bearing substituents at C5′. First, we successfully introduced a cyanomethyl group in 70% yield by stirring ENT-A025 with KCN and catalytic KI. Then, the chloro group in ENT-A025 was replaced by various dialkylamino groups. In particular, the reaction of ENT-A025 with diethanolamine gave ENT-A036 in 28% yield, while the reaction with morpholine afforded ENT-A056 in 62% yield. Furthermore, the nucleophilic substitution of ENT-A025 by diethylamine gave ENT-A075 in 70% yield (Figure 4).
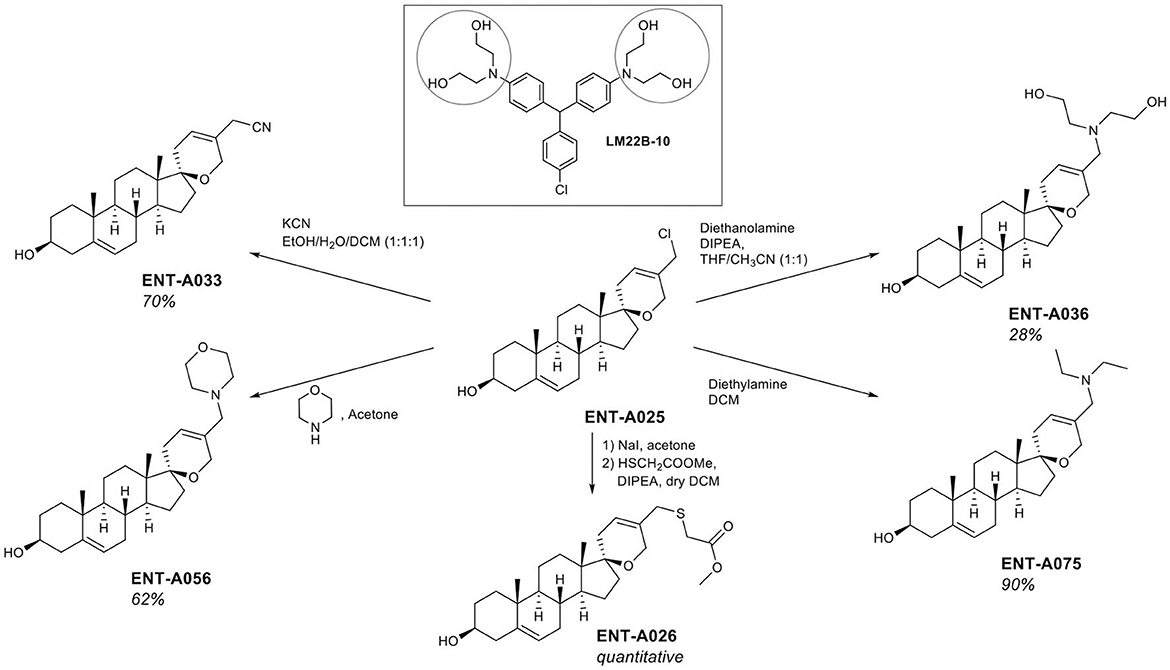
Figure 4. Synthesis of compounds ENT-A026, ENT-A036, ENT-A056, ENT-A075, and ENT-A033 and structure of LM22B-10.
To complement the nitrogen-containing derivatives, we introduced a 1,2,3-triazole moiety substituted by groups chosen for their ability to engage in hydrogen bonding or π-stacking interactions (Figure 5). Treatment of ENT-A025 with NaN3 gave the azido derivative 9. Click reaction of 9 with various substituted alkynes afforded the desired 1,2,3-triazolyl derivatives. Thus, the reaction of 9 with propargyl alcohol gave ENT-A035 in 35% yield, with N,N-dimethyl-propargyl amine afforded ENT-A046 in quantitative yield, with prop-2-yn-1-yl-cyclohexane yielded ENT-A047 in 67% yield and with 3-phenyl-1-propyne analog ENT-A055 in 71% yield (Figure 5).
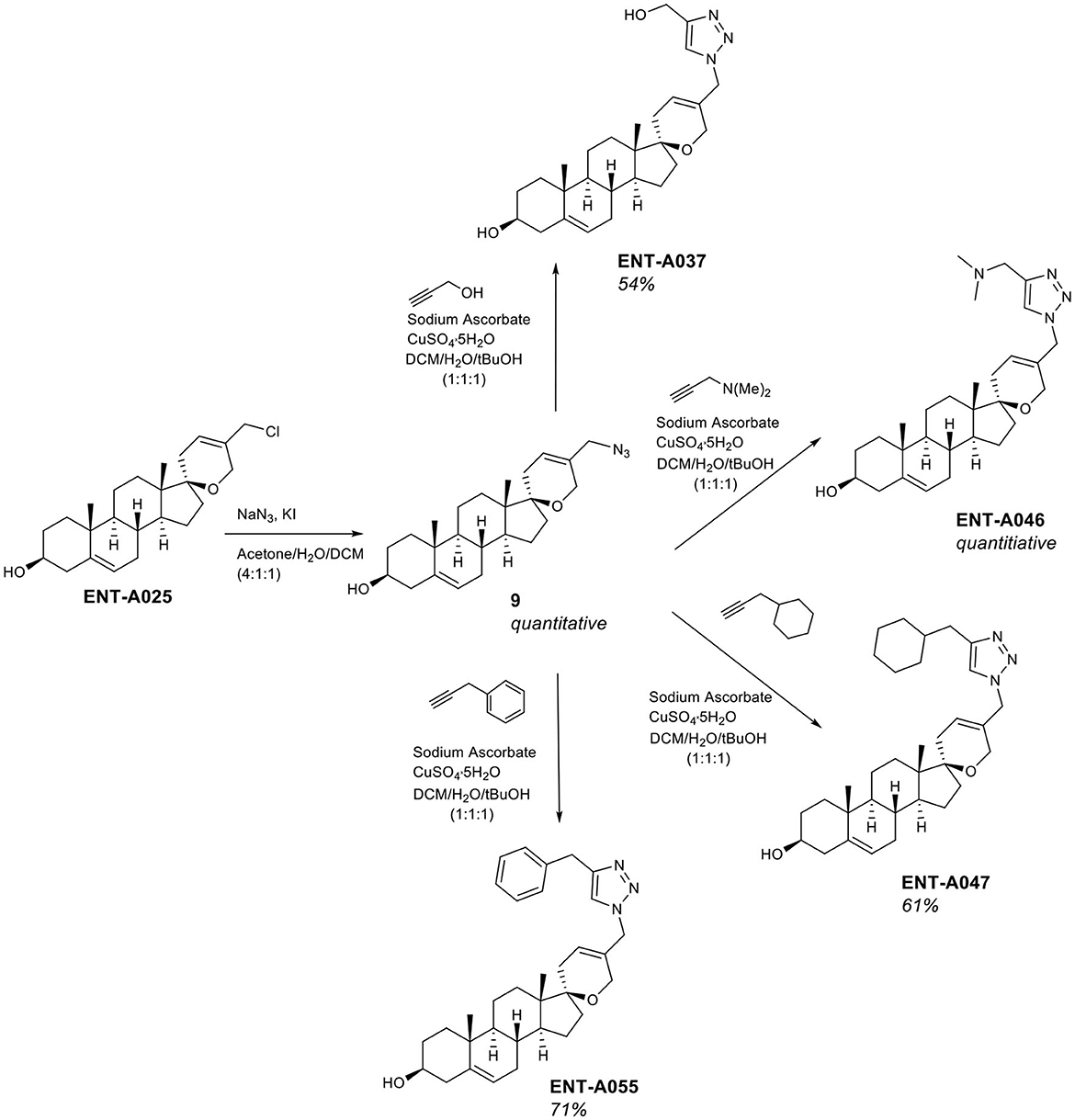
Figure 5. Synthesis of the 1,2,3-triazolyl-substituted derivatives ENT-A037, ENT-A046, ENT-A047, and ENT-A055.
To introduce unsaturated groups at C5′, the intermediate aldehyde derivative was required. Thus, compound 6 was fully converted to aldehyde 10 in a two-step one-pot reaction that involved the initial exchange of the chlorine atom with iodine, followed by oxidation using N-methyl-morpholine N-oxide (NMO) (Figure 6). Aldehyde 10 served as the starting material for generating a series of unsaturated derivatives through Horner-Emmons or Wittig reactions followed by deprotection of the C3-hydroxyl group (Figure 6). For the synthesis of ENT-A007 and ENT-A009, aldehyde 10 was treated with NaH and triethyl phosphonoacetate or diethyl (cyanomethyl)-phosphonate, respectively. Furthermore, ENT-A065 and ENT-A068 were prepared from 10 by employing (4-bromobutyl)-triphenyl-phosphonium bromide or ethyl triphenyl-phosphonium bromide in the presence of potassium tert-butoxide (tBuOK) as the base, respectively. Similarly, the use of (bromomethyl)-triphenyl-phosphonium bromide with sodium bis(trimethylsilyl)amide (NaHMDS) led to the formation of the ENT-A066 from 10. The resulting C3-protected intermediates were directly subjected to deprotection with HF·pyridine complex to afford the corresponding final products ENT-A007, ENT-A009, ENT-A065, ENT-A066, and ENT-A068 in 70 and 71% quantitative, 98 and 87% yield, respectively (Figure 6).
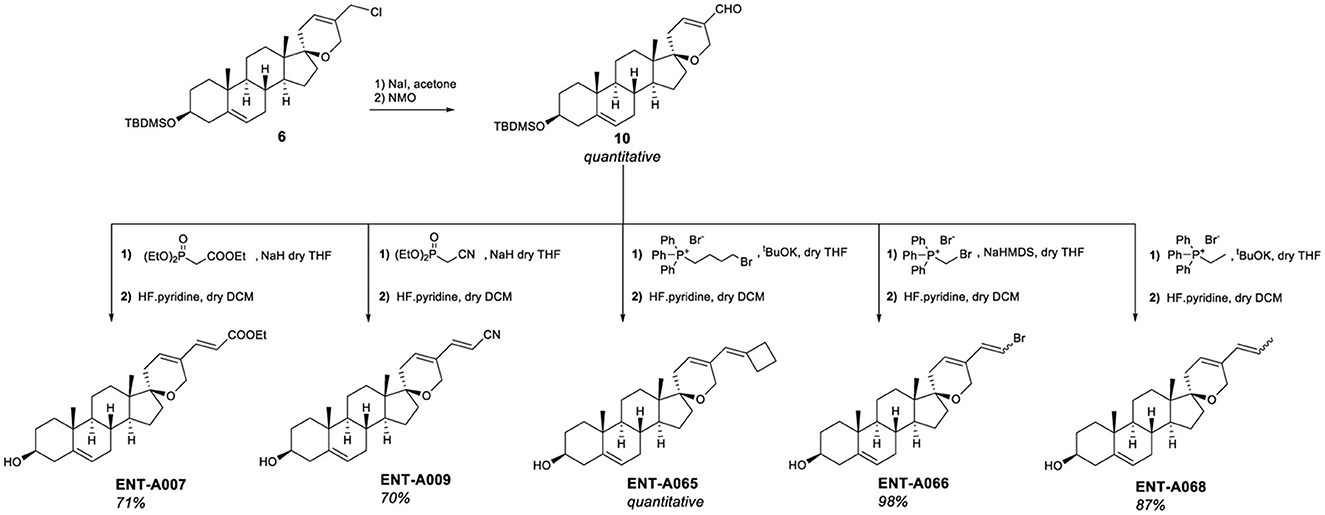
Figure 6. Synthesis of the unsaturated derivatives ENT-A007, ENT-A009, ENT-A065, ENT-A066, and ENT-A068.
Finally, our library of decorated C17-spiro-dihydropyran DHEA derivatives was complemented by two alkyl-substituted analogs. Grignard addition of methyl magnesium bromide to ENT-A025 gave the C5′ ethyl-substituted analog ENT-A070 in 90% yield. Finally, the double bond of the (2H)dihydropyran ring was selectively hydrogenated in ENT-A070 in 70% yield, using a flow-through hydrogenator (H-Cube Mini Plus™) and 5% PtS/C as catalyst to afford ENT-A069 (Figure 7).
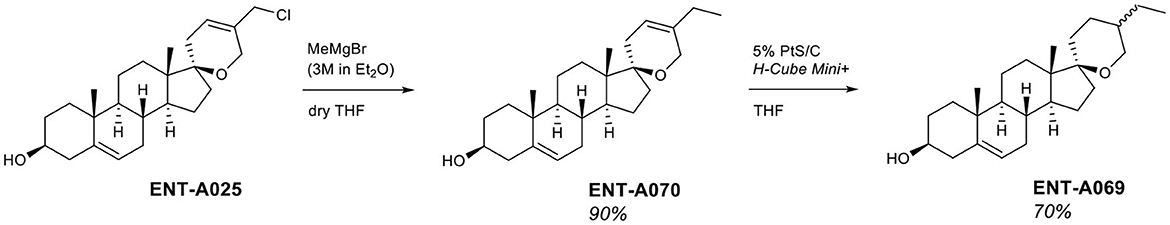
Figure 7. Synthesis of C5′-alkyl-substituted derivatives ENT-A070 and ENT-A069.
The second series of our study involves C17-spiro-cyclobutane DHEA derivatives (Figure 8). Initially, the C17-spiro-cyclobutanone DHEA analog was prepared using a reaction described by Zdzislaw and Blaszczyk (1990) (Figure 8). Wittig reaction of DHEA using methyltriphenylphosphonium bromide and tBuOK as base afforded quantitatively the C17-methylene derivative 11. Acetylation of the C3-alcohol using acetic anhydride gave 12 in quantitative yield which was then subjected to a two-step reaction for the formation of the 17-spiro-cyclobutanone derivative 13. This step proved to be particularly challenging, but we were able to increase the previously reported overall yield from 31.5 to 50% for the same substrate. The first step involved the dropwise addition of a solution of ClCOCCl3 and POCl3 to a mixture of activated zinc dust and compound 12. Due to the instability of the resulting intermediate, the crude product was carried forward to the next step. Thus, the mixture was dissolved in acetic acid, supplemented with zinc dust, and was refluxed overnight to give 13 (Figure 8). Quantitative deprotection of the C3-alcohol in 13 was effected using potassium carbonate in methanol/water, affording ENT-A076 (Figure 8). The Horner-Emmons reaction of 13 with cyanomethyl diethylphosphonate or carboxyethyl diethylphosphonate gave ENT-A077 and ENT-A079, respectively (Figure 8). However, despite our extensive efforts, ENT-A079 proved to be challenging. Not only it was obtained in low yield but also its purification was proven tedious. Thus, the ester group of ENT-A079 was hydrolyzed using NaOH in different solvent mixtures to afford acid ENT-A080 or the methyl ester ENT-A087 or the rearrangement spiro-oxepan-3-one derivative ENT-A088 (Figure 8). The respective yields were 94, 21, and 7%.
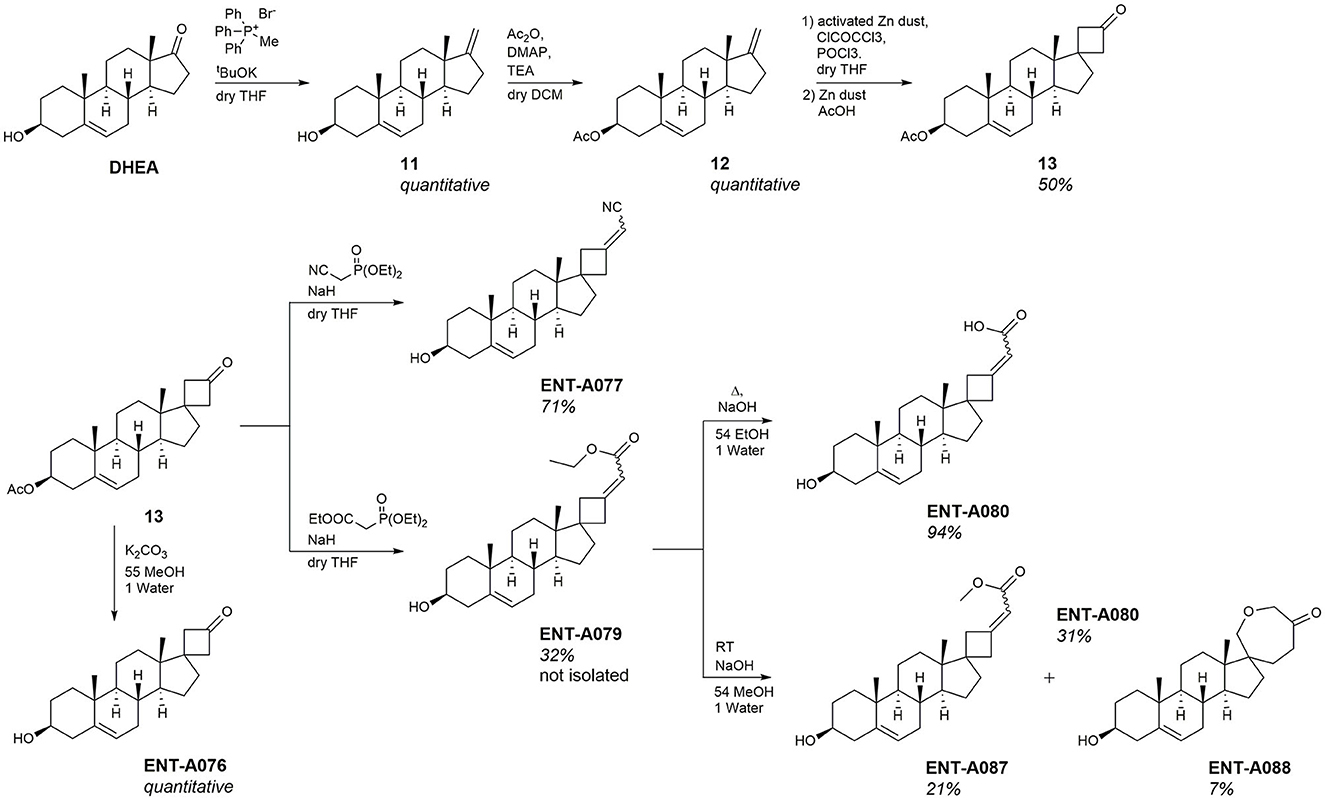
Figure 8. Synthesis of the key intermediate 13 and final compounds ENT-A076, ENT-A077, ENT-A079, ENT-A080, ENT-A087, and ENT-A088.
Initially, we tested compounds ENT-A002, ENT-A007, ENT-A009, ENTA025, ENT-A026, ENT-A033, ENT-A034, ENT-A035, ENT-A036, ENT-A037, ENT-A046, ENT-A047, ENT-A055, and ENT-A056 for their ability to protect PC12 cells from serum deprivation-induced cell death as well as for their ability to activate TrkA downstream pro-survival signaling kinases Akt and Erk1/2 (Figures 9A–C). We opted to employ PC12 cells, which express receptors TrkA and p75 and have been routinely used for investigating NGF-TrkA signaling. PC12 cells were subjected to a period of nutrient deprivation (without fetal bovine serum and horse serum in their culture medium) for 4 h. Following this, NGF (100 ng/mL) or one of the compounds (500 nM or 1 μM) was administered and allowed to interact with the cells for 24 h. Following that, CellTox and Hoescht dyes were added and cells were imaged to assess cell death. Control cases involved cells cultured in full medium (serum), cells under starvation conditions (serum-free), and cells under starvation conditions treated with DMSO (SF+DMSO). From this series, only the C5′-chloromethyl derivative ENT-A025 was identified to strongly induce TrkA and Erk1/2 phosphorylation, comparable to NGF action (Figures 9D–F) and to protect PC12 cells against serum deprivation-induced cell death (Figures 9G, H). Subsequently, compounds ENT-A065, ENT-A066, ENT-A068, ENT-A069, and ENT-A070 were evaluated, and derivatives ENT-A066, ENT-A068, ENT-A069, and ENT-A070 were found to promote PC12 cell survival during stress conditions (Figures 10A, B).
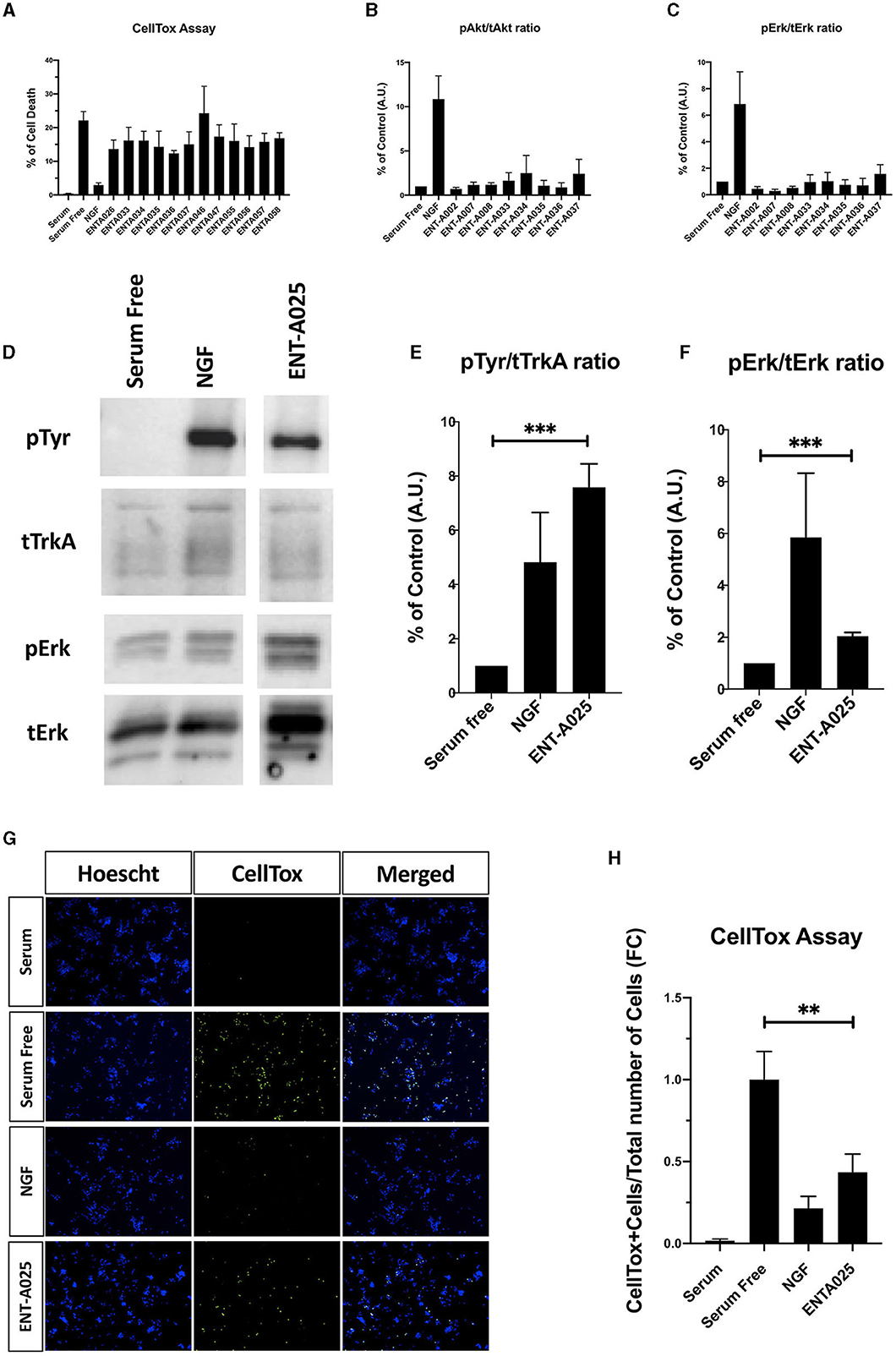
Figure 9. (A–C) Screening and identification of compounds for their ability to activate TrkA and its downstream signaling kinases as well as to promote PC12 cell survival. (A) Quantification of CellTox assay on PC12 cells starved from serum for 4 h and treated with compound (500 nM) or NGF (100 ng/mL) for 24 h, n = 3–6 independent experiments. (B, C) Quantification of phosphorylation of TrkA downstream signaling kinases Akt and ERK1/2 on PC12 cells treated with NGF (100 ng/mL) or compound (500 nM) for 30 min, n = 2–3 independent experiments for AKT, n = 3–4 independent experiments for ERK1/2. (D–H) ENT-A025 strongly activates TrkA, its downstream signaling kinase ERK1/2, and protects PC12 cells from cell death. (D–F) Western Blot and quantification of TrkA and ERK1/2 phosphorylation on PC12 cells treated with ENT-A025 (500 nM) or NGF (100 ng/mL). Data are shown as ±SEM, ***p < 0.001; Student's t-test against negative control, n = 4 independent experiments. (G, H) CellTox assay and quantification of PC12 cells starved from serum and treated with ENT-A025 (500 nM) or NGF (100 ng/mL). Data are shown as ±SEM, **p < 0.01; One-way ANOVA, multiple comparisons, Turkey's test correction, n = 5 independent experiments.
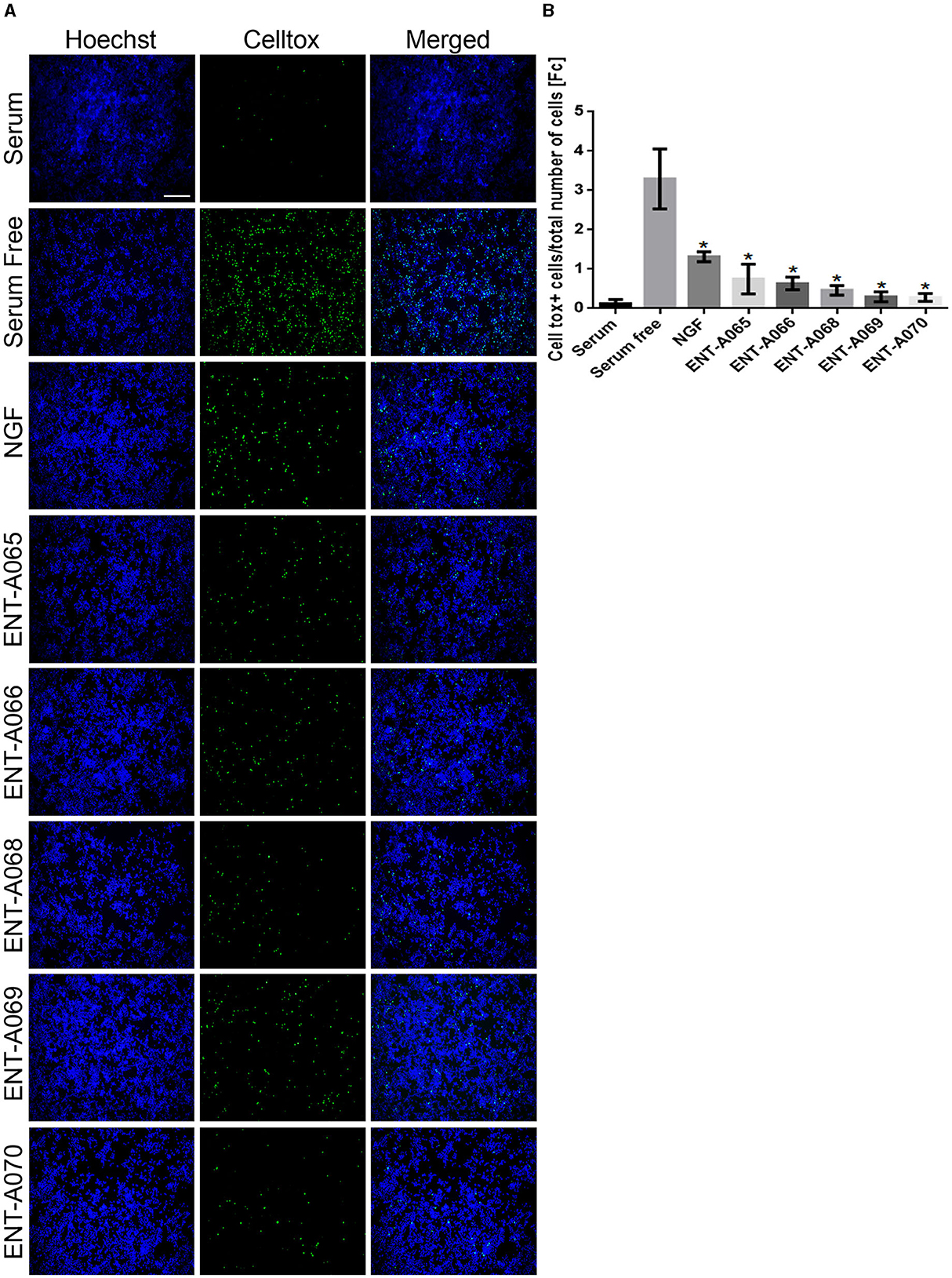
Figure 10. ENT-A065, ENT-A066, ENT-A068, ENT-A069, and ENT-A070 protect PC12 cells from serum deprivation-induced death. (A) PC12 cells were starved of serum and treated with each compound (1 μM) or NGF (100 ng/mL). CellTox dye for dead cells (green) and Hoechst for the total number of cells (blue) were used. (B) Quantification of CellTox+ cells showed that compounds ENT-A065, ENT-A066, ENT-A068, ENT-A069, and ENT-A070 promote cell survival as well as NGF does under stress conditions. Data are the average for 3 independent experiments, presented as mean ± SEM. Statistical analyses were performed by unpaired t-test; *p < 0.05.
The NIH-3T3 TrkB cell line, which is stable and transfected to specifically express the TrkB receptor, was used for the screening of the new compounds for their ability to protect cells from serum deprivation-induced apoptosis through the activation of the TrkB receptor. Cell death was induced by serum deprivation for 24 h and then BDNF (500 ng/mL), the native neurotrophin ligand of the TrkB receptor, or compounds ENT-A007, ENT-A009, ENTA025, ENT-A026, ENT-A033, ENT-A034, ENT-A035, ENT-A036, ENT-A037, ENT-A046, ENT-A047, ENT-A055, and ENT-A056 (1 μM) were added to the media for 24 h. The compounds ENT-A007, ENT-A009, ENT-A046, and ENT-A055 showed the ability to significantly reduce cell death levels compared to the serum-free control, comparable to the BDNF action (Figure 11).

Figure 11. CellTox, Cell Toxicity Assay in the NIH-3T3 TrkB-expressing cell line. Quantification and representative images of NIH-3T3 cells, untreated and treated with BDNF (500 ng/mL) or compounds ENT-A007, ENT-A009, ENT-A046, and ENT-A055 (1 μM) for 24 h, after 24 h of serum starvation, n = 3–5 independent experiments, error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Subsequently, the compounds that protected NIH3T3-TrkB cells from cell death were evaluated for their ability to phosphorylate TrkB. Mouse astrocytes were employed as a primary neuronal population that endogenously expresses TrkB, to assess more physiologically if the compounds have similar effects in a primary cell line that is dependent on TrkB signaling. Among derivatives ENT-A007, ENT-A009, ENT-A046, and ENT-A055 that showed cell protection, only ENT-A009 and ENT-A055 successfully increased phosphorylation levels of the TrkB receptor compared to the untreated control (Figure 12), in a comparable manner to BDNF.
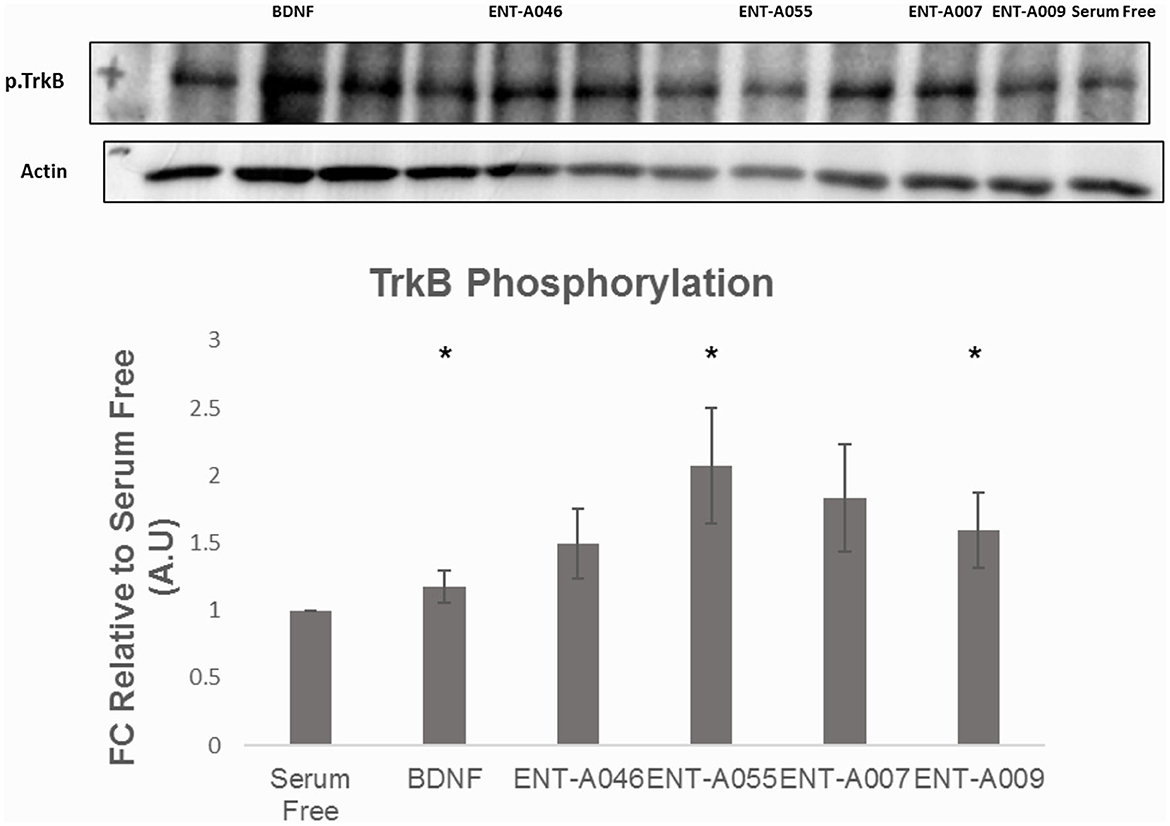
Figure 12. Phosphorylation assay in primary astrocytes. Quantification and representative images [(top), phospho-TrkB, and β-Actin] for the Western Blot Assay, n = 3 independent experiments. Cells were treated with BDNF (500 ng/mL) or compounds (1 μM) for 20 min, after 6 h of serum starvation. Error bars represent SEM. *p < 0.05.
Subsequently, compounds ENT-A075, ENT-A076, ENT-A077, ENT-A080, ENT-A087, and ENT-A088 were initially screened in the NIH3T3-TrkB stable transfected cells for their ability to promote cell survival under serum deprivation conditions as described above. Based on our findings, we can conclude that compounds ENT-A076, ENT-A087, and ENT-A088 demonstrated the ability to promote cell survival during serum starvation as well as BDNF (Figure 13).
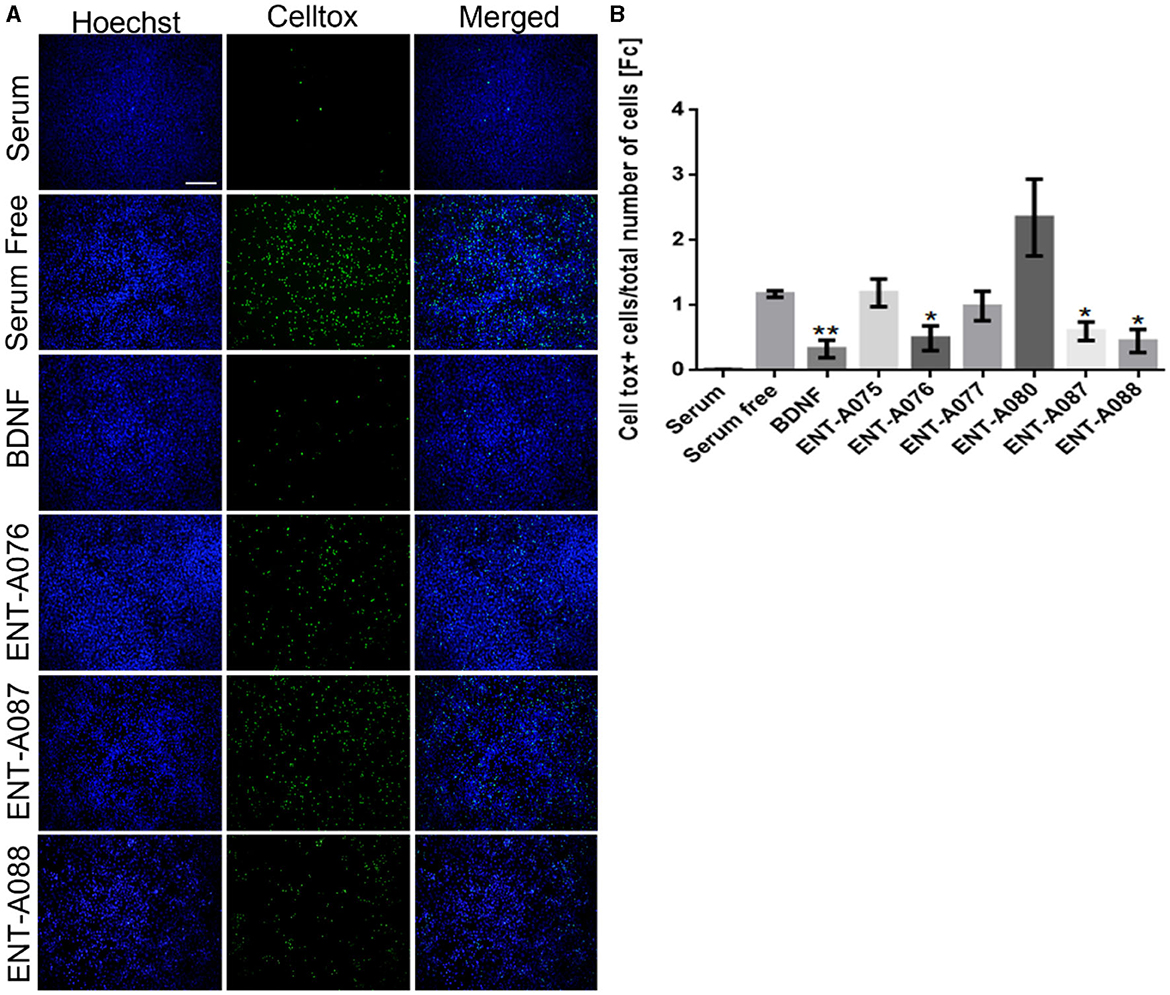
Figure 13. ENT-A076, ENT-A087, and ENT-A088 protect NIH3T3 TrkB stable transfected cells from serum deprivation-induced death. (A) NIH3T3 TrkB stable transfected cells were starved of serum and treated with each compound (1 μM) or BDNF (500 ng/mL). CellTox dye for dead cells (green) and Hoechst for the total number of cells (blue) were used. (B) Quantification of CellTox+ cells showed that compounds ENT-A076, ENT-A087, and ENT-A088 promote cell survival as well as BDNF under stress conditions. Data are the average for 3 independent experiments, presented as mean ± SEM. Statistical analyses were performed by unpaired t-test; *p < 0.05; **p < 0.01.
To confirm the involvement of the TrkB receptor in the observed cell survival effects by compounds ENT-A076, ENT-A087, and ENT-A088, NIH3T3 cells that do not naturally express any neurotrophin receptors were employed. These cells were treated with the compounds, or BDNF, for 24 h under starvation conditions. Control conditions included cells cultured with serum, serum-free conditions, and serum-free conditions supplemented with DMSO. The cells were subjected to a period of 18 h of nutrient deprivation before the treatments. However, neither the compounds nor BDNF were able to rescue the cells from cell death. Therefore, we infer that the involvement of the TrkB receptor is indeed essential for the cell-protective effects of ENT-A076, ENT-A087, and ENT-A088 (Figures 14A, B).
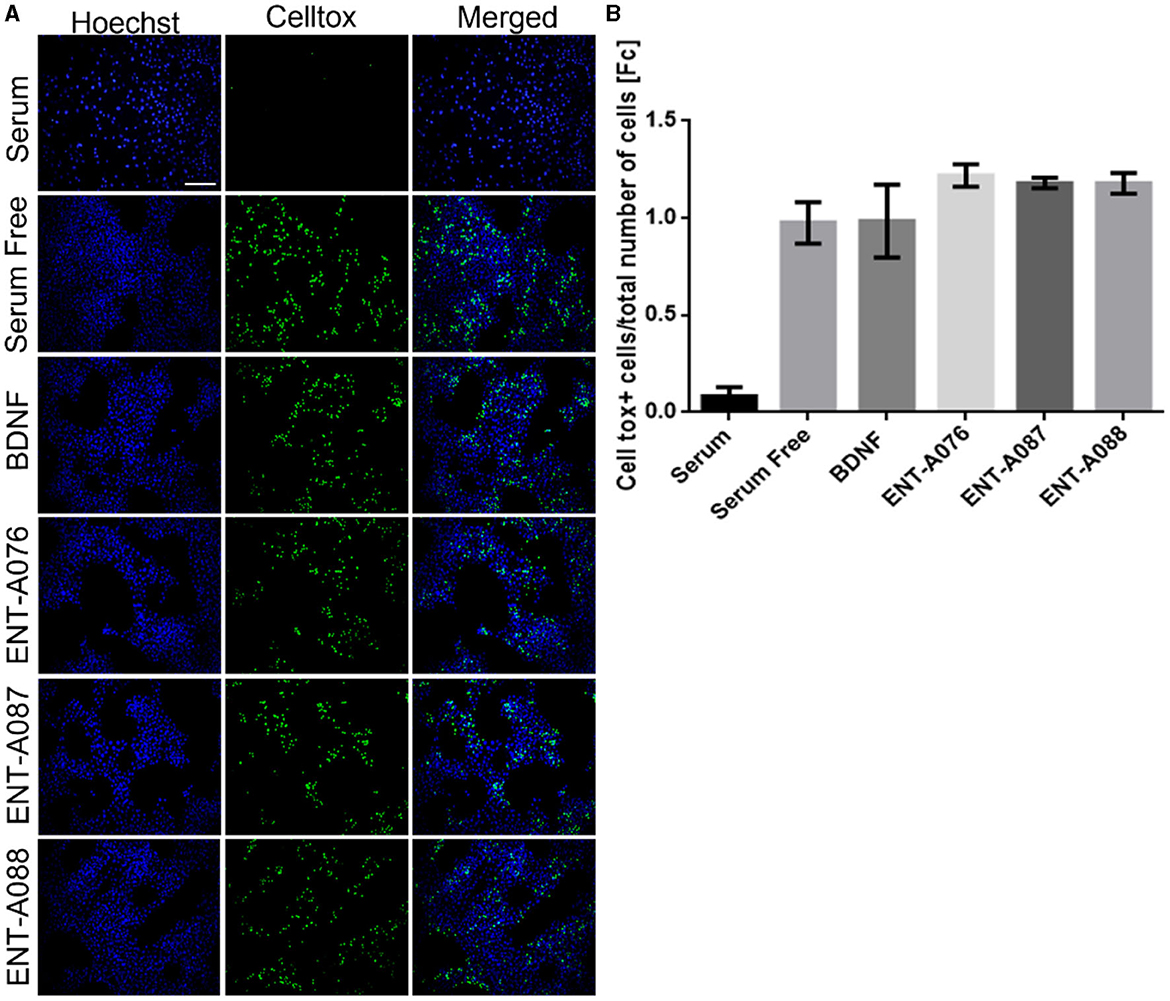
Figure 14. TrkB receptor is involved in the cell-protective effects of compounds ENT-A076, ENT-A087, and ENT-A088. (A) NIH3T3 naive cells were starved of serum and treated with each compound (1 μM) or BDNF (500 ng/mL). CellTox dye for dead cells (green) and Hoechst for the total number of cells (blue) were used. (B) Quantification of CellTox+ cells showed that neither BDNF nor compound administration was not capable of saving cells from death. Thus, we can assume that the cell survival effect is exclusively mediated through the activation of the TrkB receptor. Data are the average for 3 independent experiments, presented as mean ± SEM. Statistical analysis was performed by unpaired t-test.
The predicted physicochemical profile and drug-like properties of the compounds were satisfactory, displaying in general values within the 95% range of the known drugs except the limited aqueous solubility for a number of compounds. Moreover, the compounds ENT-A035, ENT-A0047, ENT-A065, and ENT-A055 exceed the lipophilicity range of values with potential impact in their distribution. The number of potential metabolites emerged slightly increased for the compounds ENT-A035, ENT-A036, ENT-A046, and ENT-A056. Regarding the potential reactivity of the compounds, the presence of unhindered ester functionality as in the compounds ENT-A026 and ENT-A088 or the carbonyl group in the 4-ring system has been highlighted (Supplementary Table S1).
The susceptibility of a test compound to biotransformation is defined as metabolic stability. For the ranking of test compounds, several approaches can be followed including parent structure loss during metabolic reactions or their intrinsic clearance (CLint) and in vitro half-life (t1/2) values (Masimirembwa et al., 2003; Barter et al., 2007). Herein, we chose the former.
Parent structure loss is classified as very slow (<5%), slow (5–19%), moderate (20–50%), fast (50–80%), or very fast (>80%). Such categories have been defined according to set criteria, namely, high metabolism (t1/2 value of <30 min), moderate metabolism (30 min <t1/2 value of <60 min), and low metabolism (t1/2 value of >60 min) (EMA, 2012, 2013; FDA, 2020).
ENT-A055, ENT-A058, ENT-A066, ENT-A070, ENT-A087, and ENT-A088 were very slowly depleted showing >95% residual of time zero at t = 60 min, except for ENT-A025 (92%), ENT-A069 (95%), and ENT-A076 (95%) that exhibit slow depletion (Figure 15A). Thus, all test compounds may correspond to low or medium intrinsic clearance classification bands.
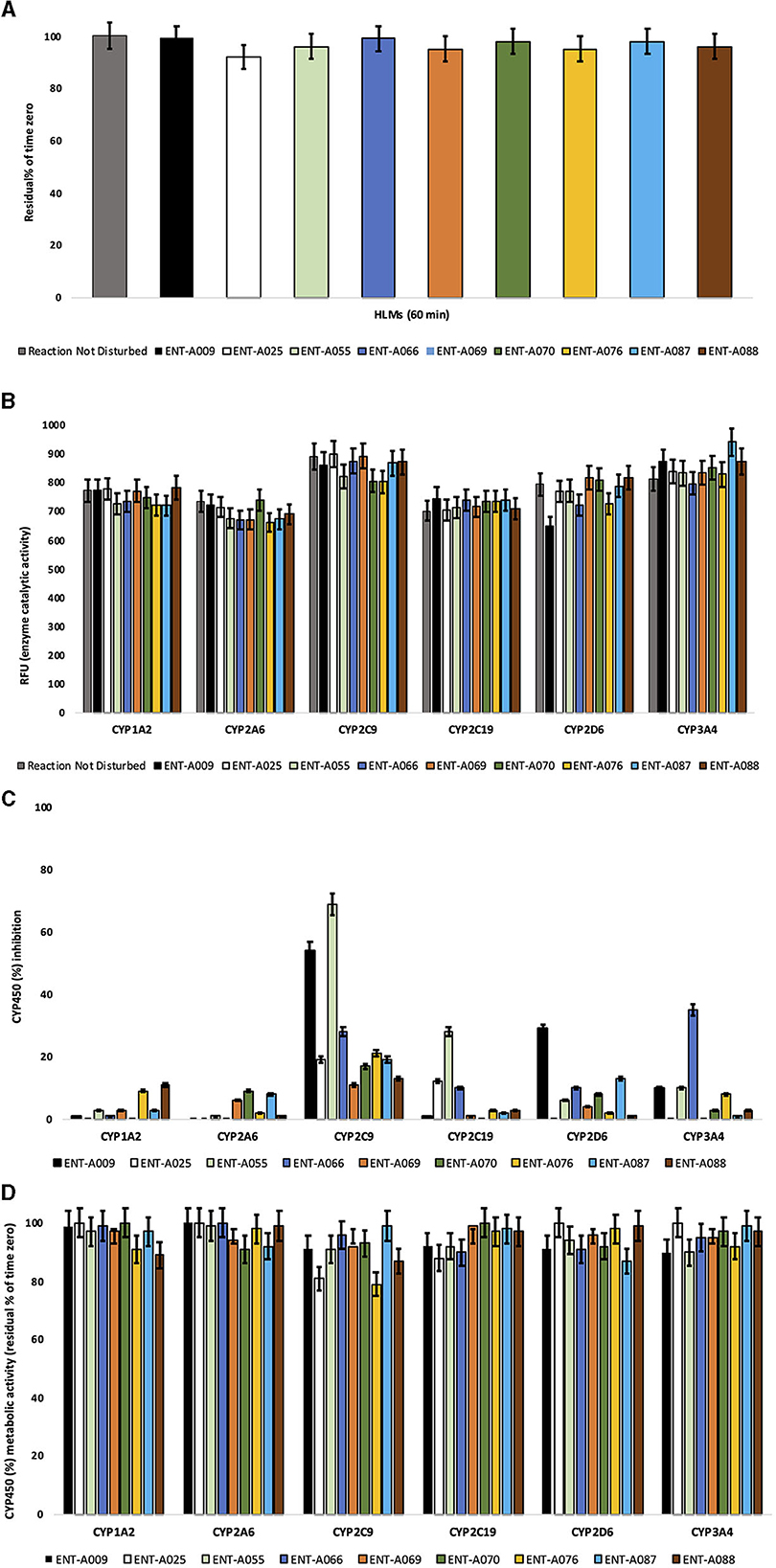
Figure 15. (A) Microsomal stability of test compounds at 1 μM upon incubation with pooled human liver microsomes (60 min). (B) Enzyme (catalytic) activity of the CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 isoenzymes upon the administration of test compounds at 1 μM (60 min). RFU, relative fluorescence units. Reaction Not Disturbed, reaction without test compounds. (C) CYP450 (%) inhibition of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 isoenzymes upon the administration of test compounds at 1 μM (60 min). (D) CYP450 (%) metabolic activity of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 isoenzymes upon the administration of test compounds at 1 μM (60 min).
Human CYP450 enzymes are crucial for xenobiotic biodegradation, metabolism, and toxicity as well as xenobiotic–host and/or xenobiotic–xenobiotic interactions (McGinnity et al., 2000). Upon linear velocity conditions in vitro, the depletion rate of test compounds may be extrapolated to (a) in vivo hepatic clearance, (b) extraction ratio, and (c) the effect of hepatic first-pass metabolism on total oral bioavailability. Biodegradation, metabolic, and toxicity liabilities can be identified early on and thus inform structure–activity relationships (SAR) (EMA, 2012, 2013; FDA, 2020).
For this, the activity of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 was assessed after the administration of test compounds at 1 μM to determine (a) the oxidative (CYP-mediated) metabolic stability profile in question and (b) the enzyme metabolizing isoforms responsible (the test system consists of recombinant human CYP450 and CYP450 reductase; cytochrome b5 may also be present). Herein, no concentration-dependent effects were reported. CYP450 substrates can alter enzyme activity by blocking the enzyme's active site, changing enzyme conformation, and disrupting enzyme structure and/or functioning (Pelkonen and Turpeinen, 2007; Zanger and Schwab, 2013). No test compound decreased the enzyme (catalytic) activity of the CYP450 system tested herein. CYP450 enzyme inhibition may lead to unexpectedly high exposure to co-administered xenobiotics and hence increase the risk for adverse effects. No product inhibition or mechanism-based inactivation of the CYP450 isoenzymes in question was obtained. Test compounds with poor solubility can show artificially low CYP450 inhibition and thus, chemical entities with potential drug–drug interaction toxicities may be overlooked. No solubility issues were observed. All test compounds were weak or moderate inhibitors of the CYP450 system. ENT-A055 and ENT-A009 showed moderate-to-strong inhibition for CYP2C9. Findings are summarized in Figures 15B–D.
Neurotrophins belong to a family of secreted proteins, widely expressed in the peripheral and the central nervous system, that majorly support neuronal survival, synaptic plasticity, and neurogenesis. The predominant neurotrophins in adult humans are nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) acting through their high-affinity receptors: TrkA and TrkB, respectively. Their fine regulation governs fundamental functions of the brain, such as memory and learning. Alterations of BDNF/TrkB signaling in the cortical and hippocampal area, in fact, were indicated as a hallmark of numerous neurodegenerative diseases, and a Trk agonist-mediated restoration holds the promise of a disease-modifying treatment (reviewed by Longo and Massa, 2013).
In accordance with the unmet need for neurotrophin receptor manipulation, we have now synthesized a library of 27 new C17-spiro DHEA derivatives in a quest to decipher the stereo-electronic requirements for selective activation of neurotrophin receptors TrkA or TrkB. Building on the discovery of the prototype BNN27, a selective TrkA agonist (Calogeropoulou et al., 2009), we expanded our seminal results with two series of derivatives substituted at C17 by six- or four-membered spirocyclic moieties. All the reported compounds showed increased metabolic stability as a result of the ring expansion of BNN27′s C17-spiro-oxirane moiety. The first series encompasses C17-spiro-dihydro(2H)pyran derivatives decorated at C5′ by a variety of pharmacophoric groups. In particular, our in-depth investigations began with the synthesis of the C5′-unsubstituted analog ENT-A002. Evaluation of the antiapoptotic activity in PC12 and NIH3T3-TrkB cells did not show any protective effects. Thus, we embarked on decorating the C5′ position of ENT-A002 with different substituents aiming to restore the neuroprotective effect of BNN27. Thus, we initially introduced an electrophilic handle via a chloromethyl substituent (compound ENT-A025) that in turn could enable the further derivatization to multiple analogs, thus expanding the diversity of our compound library. Gratifyingly, ENT-A025 strongly activates TrkA and its downstream signaling kinase ERK1/2 while protecting PC12 cells from cell death (Figure 9). Conversely, it cannot protect NIH3T3-TrkB cells from serum deprivation (Figure 11). Thus, the previous findings suggest ENT-A025 is a selective TrkA agonist, capable of mimicking some of the actions of NGF. Aiming to map the structural requirements for neurotrophin mimetic activity, we further derivatized ENT-A025 at the C5′ position and generated its fluoro congener compound ENT-A034, the hydroxymethyl analog ENT-A008, the (p-methoxybenzyloxy)methyl derivative compound ENT-A035, the C5′-methyl(thiomethylacetate)-substituted analog ENT-A026, and the cyanomethyl derivative ENT-A033. Unfortunately, none of these compounds provided any protective effects in PC12 (Figure 9) or NIH3T3-TrkB cells (data not shown). Interestingly, ENT-A008 bears the hydroxymethyl substituent of our prototype compound BNN27, indicating that the larger six-membered C17-spiro substituent compromised the activity. Continuing our structure–activity studies, we designed a small group of analogs borrowing structural features from the neurotrophin mimetic compounds reported by the groups of Longo and Massa (2013), in particular, LM22B-10 (Yang et al., 2016). This compound, characterized by three diethanolamino moieties, binds and activates TrkB and TrkC receptors to promote cell survival and induce neurite outgrowth. Thus, we incorporated the diethanolamino functionality on the C5′ position of ENT-A025, resulting in ENT-A036 (Scheme 4). Inspired by the potential of this pharmacophore, we envisaged two additional analogs modulating the nature of the amino group. In the first (ENT-A075), the two alcohol groups were replaced by hydrogens, and in the second (ENT-A056), they were etherized into a morpholine. Interestingly, only the diethylamino derivative ENT-A075 showed potent antiapoptotic activity in NIH3T3-TrkB cells (Figure 13). Subsequently, we introduced the well-known five-membered heterocyclic 1,2,3-triazole group at C5′ of ENT-A025. This pharmacophore moiety is considered an amide group bioisostere and has been employed in hit-to-lead optimization campaigns resulting in bioactive compounds encompassing an array of pharmacological actions (Kumar and Kavitha, 2013; Bonandi et al., 2017). Four 1,2,3-triazolyl derivatives were prepared substituted by hydroxymethyl (ENT-A037), N,N-dimethylaminomethyl (ENT-A046), or cyclohexylmethyl (ENT-A047) or benzyl (ENT-A055) groups on the heterocyclic ring. These carefully selected substituents could enable potential hydrogen bonding, π-stacking, or hydrophobic interactions with the neurotrophin receptors. We were very excited to identify ENT-A046 and ENT-A055 capable of protecting NIH3T3-TrkB cells from serum deprivation comparably to the natural ligand BDNF (Figure 11) without showing any antiapoptotic activity in PC12 cells (Figure 9A). However, when tested for their ability to phosphorylate TrkB in mouse astrocytes, only the benzyl-substituted 1,2,3-triazole containing derivative ENT-A055 successfully increased phosphorylation levels of the TrkB receptor compared to the untreated control (Figure 12). Thus, ENT-A055 is a selective TrkB agonist mimicking some of the protective effects of BDNF. These differential effects by ENT-A046 and ENT-A055 could reflect structurally induced differences in various signaling pathways and cellular phenotypes between the two molecules. The (2H)dihydropyran series was finally complemented by a group of compounds bearing a vinyl group at C5′. Compounds ENT-A007 and ENT-A009 were substituted by α,β-unsaturated moieties as nucleophilic warheads, ENT-A066 a bromovinyl substituent, while ENT-A065 and ENT-A068 as olefinic groups (cyclobutylmethylene or allyl, respectively). From this group, compounds ENT-A065, ENT-A066, and ENT-A068 were found to promote PC12 cell survival as well as NGF under stress conditions (Figure 10). In contrast, compound ENT-A009 was found to be a selective TrkB agonist, protecting NIH3T3-TrkB cells from cell death (Figure 11) and inducing TrkB phosphorylation comparably to BDNF (Figure 12). Interestingly, derivative ENT-A066 bears a C5′-bromovinyl substituent that is also present in our previously reported selective TrkA agonist ENT-A013 which mimics NGF activity in cell lines and primary neuronal populations and exhibits neuroprotective and anti-amyloid properties (Rogdakis et al., 2022). Thus, this strongly indicates that the bromovinyl moiety confers TrkA selectivity, irrespective of the size of the C17-spirocyclic moiety on the DHEA steroidal scaffold. Since compound ENT-A068 was a potent TrkA agonist, we synthesized two saturated derivatives replacing the chloro substituent in ENT-A025 with a methyl group and subsequently reducing the endocyclic double bond of the (2H)dihydropyran group (compounds ENT-A070 and ENT-A069, respectively). This would enable us to identify the contribution of the unsaturations on the neurotrophin mimetic activity. Both derivatives were able to protect PC12 cells from serum deprivation-induced death, albeit the (2H)-dihydropyran derivative (ENT-A070) being more potent (Figure 10). Thus, the presence of the endocyclic double bond is beneficial for the NGF mimetic activity.
Based on our previous results (Yilmaz et al., 2022) that we identified compound ENT-A010 bearing a cyclopropyl group conjugated to an α,β-unsaturated ester as a dual agonist of TrkA and TrkB receptors, we set out to prepare a small series of cyclobutyl derivatives to clarify the optimum size of the C17-spirocyclic moiety. Thus, the C17-cyclobutanone analog (ENT-A076), the α,β-unsaturated cyano (ENT-A077), α,β-unsaturated ethyl ester (ENT-A079), α,β-unsaturated methyl ester (ENT-A087), and α,β-unsaturated acid (ENT-A080) derivatives were prepared. As a side product during the synthesis of ENT-A087, the spiro-oxepan-3-one derivative ENT-A088 was formed via a rearrangement reaction. Excitingly, ENT-A076, ENT-A087, and ENT-A088 were shown to possess antiapoptotic activity in NIH-3T3-TrkB cells exclusively mediated through the TrkB receptor (Figures 13, 14). To get an insight into the drug-likeness of the new compounds, we predicted the physicochemical profile of the new synthetic compounds and were found to display values within the 95% range of the known drugs. As a drawback, a number of derivatives were predicted to have limited aqueous solubility. Finally, we performed an isozyme-specific CYP450 study to delineate the interactions among the active compounds with human CYP450 isoenzymes that account for most xenobiotic biodegradation, metabolism, and toxicity, as well as xenobiotic–xenobiotic and/or xenobiotic–host interactions. All the tested compounds were weak or moderate inhibitors of the CYP450 system. However, ENT-A007 and ENT-A009 showed moderate-to-strong inhibition for CYP2C9. Furthermore, none of the compounds tested showed biodegradation or liver metabolism (≥95% residual of time zero at t = 60 min) or safety issues.
In conclusion, this study focused on designing and synthesizing novel small neurotrophin mimetics to target neurotrophin receptors and combat neurodegeneration. A total of two series of DHEA derivatives, comprising 27 compounds, were synthesized and evaluated in vitro for their ability to stimulate survival in neurotoxicity models involving TrkA or TrkB receptors.
Among the compounds tested, ENT-A025 strongly induces TrkA and Erk1/2 phosphorylation, comparable to NGF, and can protect PC12 cells against serum deprivation-induced cell death. Furthermore, compounds ENT-A065, ENT-A066, ENT-A068, ENT-A069, and ENT-A070 have shown promising pro-survival effects in the PC12 cell line. These C17-spiro-dihydropyran derivatives will provide valuable insights into the structure–activity relationship (SAR) and identification of the most potent steroidal-based derivative for TrkA activation. In contrast, the SAR of the spiro-dihydropyran compounds showing pro-survival effects on TrkB is more complex. Compound ENT-A009, bearing a C5′-α,β-unsaturated cyano moiety, ENT-A075 with a C5′-diethylaminomethyl group, and ENT-A055 featuring a larger decoration by a benzyl-substituted 1,2,3-triazole group at C5′ position suggest a higher tolerability for the TrkB binding site(s) for these mimetics. Interestingly, the C17-spirocyclobutyl derivatives ENT-A076 and ENT-A087 exhibited potent antiapoptotic activity mediated through TrkB despite their smaller spiro-ring and decorations. Lastly, ENT-A088 holds promise in exploring even larger spirocyclic moieties for TrkB receptor activation.
Overall, these findings provide valuable insights into the optimal stereo-electronic requirements for neurotrophin receptor agonism, offering potential avenues for mapping and dissecting the structural properties of neurotrophin receptors with their agonists and thus selectively control their signaling, to develop effective therapeutic interventions against neurodegenerative diseases.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
TC, IC, AG, and KP designed and supervised the study. DN performed the synthesis and characterization of the new compounds. DC, TR, and IZ performed the screening experiments. TK and VB performed the in vitro ADMET studies. MZ performed the prediction of the drug-like properties of the new compounds. DN, DC, TR, IZ, VB, TK, AG, KP, IC, and TC interpreted the data and wrote the manuscript. All authors have read and approved the manuscript.
This research was funded by the European Union's Horizon 2020 research and innovation programme Euroneurotrophin under the Marie Skłodowska-Curie grant agreement no. 765704.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1244133/full#supplementary-material
Alexaki, V. I., Fodelianaki, G., Neuwirth, A., Mund, C., Kourgiantaki, A., Ieronimaki, E., et al. (2018). DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol. Psychiat. 23, 1410–1420, doi: 10.1038/mp.2017.167
Allen, S. J., and Dawbarn, D. (2006). Clinical relevance of the neurotrophins and their receptors. Clin. Sci. 110, 175–191. doi: 10.1042/CS20050161
Baeza-Raja, B., Li, P., Le Moan, N., Sachs, B. D., Schachtrup, C., Davalos, D., et al. (2012). P75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. PNAS 109, 5838–5843. doi: 10.1073/pnas.1103638109
Bai, L., Pang, P. T., and Woo, N. H. (2005). The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 6, 603–614. doi: 10.1038/nrn1726
Barter, Z. E., Bayliss, M. K., Beaune, P. H., Boobis, A. R., Carlie, D. J., Edwards, R. J., et al. (2007). Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug. Meta. 8 33–45. doi: 10.2174/138920007779315053
Baulieu, E. E. (1996). Dehydroepiandrosterone (DHEA): a fountain of youth? J. Clin. Endocrinol. Metab. 81, 3147–3151. doi: 10.1210/jcem.81.9.8784058
Bennett, J. P., O'Brien, L. C., and Brohawn, D. G. (2016). Pharmacological properties of microneurotrophin drugs developed for treatment of amyotrophic lateral sclerosis. Biochem. Pharmacol. 117, 68–77. doi: 10.1016/j.bcp.2016.08.001
Bonandi, E., Christodoulou, M. S., Fumagalli, G., Perdicchia, D., Rastelli, G., and Passarella, D. (2017). The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today. 22, 1572–1581. doi: 10.1016/j.drudis.2017.05.014
Bonetto, G., Charalampopoulos, I., Gravanis, A., and Karagogeos, D. (2017). The novel synthetic microneurotrophin BNN27 protects mature oligodendrocytes against cuprizone-induced death, through the NGF receptor TrkA. Glia 65, 1376–1394. doi: 10.1002/glia.23170
Calogeropoulou, T., Avlonitis, N., Minas, V., Alexi, X., Pantzou, A., Charalampopoulos, I., et al. (2009). Novel dehydroepiandrosterone derivatives with antiapoptotic, neuroprotective activity. J. Med. Chem. 52, 6569–6587. doi: 10.1021/jm900468p
Ceren, O., Zimmer, D. J., De Jonghe, B. C., Kalb, R. G., and Bence, K. C. (2015). Ablation of intact hypothalamic and/or hindbrain TrkB signaling leads to perturbations in energy balance. Mol. Metab. 4, 867–880. doi: 10.1016/j.molmet.2015.08.002
Chao, M. Y., Rithwick, R., and Lee, F. S. (2006). Neurotrophin signalling in health and disease. Clin. Sci. 110, 167–173. doi: 10.1042/CS20050163
Cohen, L. H., Remley, M. J., Raunig, D., and Vaz, A. D. N. (2003). In vitro drug interactions of cytochrome P450: an evaluation of fluorogenic to conventional substrates. Drug Metab. Dispos. 31, 1005–1015. doi: 10.1124/dmd.31.8.1005
Fei, D., and Krimm, R. F. (2013). Taste neurons consist of both a large TrkB-receptor-dependent and a small TrkB-receptor-independent subpopulation. PLoS ONE 8, 83460. doi: 10.1371/journal.pone.0083460
Glajch, K. E., Ferraiuolo, L., Mueller, K. A., Stopford, M. J., Prabhkar, V., Gravanis, A., et al. (2016). MicroNeurotrophins improve survival in motor neuron-astrocyte co-cultures but do not improve disease phenotypes in a mutant SOD1 mouse model of amyotrophic lateral sclerosis. PLoS ONE 11, 1–24. doi: 10.1371/journal.pone.0164103
Gudasheva, T. A., Povarnina, P. Y., Tarasiuk, A. V., and Seredenin, S. B. (2021). Low-molecular mimetics of nerve growth factor and brain-derived neurotrophic factor: Design and pharmacological properties. Med. Res. Rev. 41, 2746–2774. doi: 10.1002/med.21721
Hallböök, F., Wilson, K., Thorndyke, M., and Olinski, R. P. (2006). Formation and evolution of the chordate neurotrophin and Trk receptor genes. Brain Behav. Evol. 68, 133–144. doi: 10.1159/000094083
Houtz, J., Borden, P., Ceasrine, A., Minichiello, L., and Kuruvilla, R. (2016). Neurotrophin signaling is required for glucose-induced insulin secretion. Dev. Cell 39, 329–345. doi: 10.1016/j.devcel.2016.10.003
Ibán-Arias, R., Lisa, S., Mastrodimou, N., Kokona, D., Koulakis, E., Iordanidou, P., et al. (2018). The synthetic microneurotrophin BNN27 affects retinal function in rats with streptozotocin-induced diabetes. Diabetes. 67, 321–333. doi: 10.2337/db17-0391
Jeyaram, B. J., Palathoti, N., Dhanasekaran, M., Sivasamy, R., Ponnusankar, S., Dhanabal, S. P., et al. (2023). Neurotrophin mimetics and tropomyosin kinase receptors: a futuristic pharmacological tool for Parkinson's. Neurol. Sci. 44, 2265–2275 doi: 10.1007/s10072-023-06684-1
Josephy-Hernandez, S., Sean, J., Pirvulescu, I., Aboulkassim, T., and Saragovi, H. U. (2017). Neurotrophin receptor agonists and antagonists as therapeutic agents: an evolving paradigm. Neurobiol. Dis. 97, 139–155. doi: 10.1016/j.nbd.2016.08.004
Kumar, S. S., and Kavitha, H. P. (2013). Synthesis and biological applications of triazole derivatives-a review. Mini-Rev. Org. Chem. 10, 40–65. doi: 10.2174/1570193X11310010004
Lazaridis, I., Charalampopoulos, I., Alexaki, V. I., Avlonitis, N., Pediaditakis, I., Efstathopoulos, P., et al. (2011). Neurosteroid dehydroepiandrosterone interacts with nerve growth factor (NGF) receptors, preventing neuronal apoptosis. PLoS Biol. 9, e1001051. doi: 10.1371/journal.pbio.1001051
Longo, F. M., and Massa, S. M. (2013). Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 12, 507–525. doi: 10.1038/nrd4024
Masimirembwa, C. M., Bredberg, U., and Andersson, T. B. (2003). Metabolic stability for drug discovery and development: pharmacokinetic and biochemical challenges. Clin. Pharmacokinet. 42, 515–528. doi: 10.2165/00003088-200342060-00002
McGinnity, D. F., Parker, A. J., Soars, M., and Riley, R. J. (2000). Automated definition of the enzymology of drug oxidation by the major human drug metabolizing cytochrome P450s. Drug Metab. Dispos. 28, 1327–1334.
Meldolesi, J. (2017). Neurotrophin receptors in the pathogenesis, diagnosis and therapy of neurodegenerative diseases. Pharmacol. Res. 121, 129–137. doi: 10.1016/j.phrs.2017.04.024
Mitre, M., Mariga, A., and Chao, M. V. (2016). Neurotrophin signalling: novel insights into mechanisms and pathophysiology. Clin. Sci. 131, 13–23. doi: 10.1042/CS20160044
Pardridge, W. M. (2002). Neurotrophins, neuroprotection and the blood-brain barrier. Curr. Opin. Investig. Drugs 3, 1753–1757.
Pediaditakis, I., Efstathopoulos, P., Prousis, K. C., Zervou, M., Arévalo, J. C., Alexaki, V. I., et al. (2016a). Selective and differential interactions of BNN27, a Novel C17-spiroepoxy steroid derivative, with trka receptors, regulating neuronal survival and differentiation. Neuropharmacol. 111, 266–282. doi: 10.1016/j.neuropharm.2016.09.007
Pediaditakis, I., Iliopoulos, I., Theologidis, I., Delivanoglou, N., Margioris, A. N., Charalampopoulos, I., et al. (2015). Dehydroepiandrosterone: an ancestral ligand of neurotrophin receptors. Endocrinol. 156,16–23. doi: 10.1210/en.2014-1596
Pediaditakis, I., Kourgiantaki, A., Prousis, K. C., Potamitis, C., Xanthopoulos, K. P., Zervou, M., et al. (2016b). BNN27, a 17-spiroepoxy steroid derivative, interacts with and activates p75 neurotrophin receptor, rescuing cerebellar granule neurons from apoptosis. Front. Pharmacol 7, 512. doi: 10.3389/fphar.2016.00512
Pelkonen, O., and Turpeinen, M. (2007). In vitro–in vivo extrapolation of hepatic clearance: biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 3, 1066–1089. doi: 10.1080/00498250701620726
Penzo, M. A., Robert, V., Tucciarone, J., De Bundel, D., Wang, M., Van Aelst, L., et al. (2015). The paraventricular thalamus controls a central amygdala fear circuit. Nature 519 455–459. doi: 10.1038/nature13978
Pitsikas, N., and Gravanis, A. (2017). Neurobiology of Learning and Memory The novel dehydroepiandrosterone (DHEA) derivative BNN27 counteracts delay-dependent and scopolamine-induced recognition memory deficits in rats. Neurobiol. Learn. Mem. 140, 145–153. doi: 10.1016/j.nlm.2017.03.004
Rahman, M. M., Islam, M. R., Supti, F. A., Dhar, P. S., Shohag, S., Ferdous, J., et al. (2023). Exploring the therapeutic effect of neurotrophins and neuropeptides in neurodegenerative diseases: at a glance. Mol. Neurobiol. 60, 4206–4231. doi: 10.1007/s12035-023-03328-5
Rogdakis, T., Charou, D., Latorrata, A., Papadimitriou, E., Tsengenes, A., Athanasiou, C., et al. (2022). Development and biological characterization of a novel selective TrkA agonist with neuroprotective properties against amyloid toxicity. Biomedicines. 10, 614. doi: 10.3390/biomedicines10030614
Saragovi, H. U., Galan, A., and Levin, L. A. (2019). Neuroprotection: Pro-Survival and Anti-Neurotoxic Mechanisms as Therapeutic Strategies in Neurodegeneration. Front. Cell. Neurosci. 13, 231. doi: 10.3389/fncel.2019.00231
Shahnaz, R., Nazem, G., Mardani, M., Esfandiari, E., Esfahani, S., and Salehi, H. (2015). Neurotrophic factors and their effects in the treatment of multiple sclerosis. Adv. Biomed. Res. 4, 53. doi: 10.4103/2277-9175.151570
Tsika, C., Tzatzarakis, M. N., Antimisiaris, S. G., Tsoka, P., Efstathopoulos, P., Charalampopoulos, I., et al. (2021). Quantification of BNN27, a novel neuroprotective 17-spiroepoxy dehydroepiandrosterone derivative in the blood and retina of rodents, after single intraperitoneal administration. Pharmacol. Res. Perspect. 9, e00724. doi: 10.1002/prp2.724
Veith, H., Southall, N., Huang, R., James, T., Fayne, D., Artemenko, N., et al. (2009). Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 27, 1050–1055. doi: 10.1038/nbt.1581
Xun, L., Singh, S. M., and Labrie, F. (1994). Highly efficient nucleophilic addition of Alkyl grignard reagents to 17-Ketosteroids in the presence of Cerium(III) Chloride: Synthesis of 17α-Propyl-17β-Hydroxy-4-Androsten-3-One, an androgen receptor antagonist. Tetrahedron Lett. 35, 1157–1160. doi: 10.1016/0040-4039(94)88011-5
Yang, T., Massa, S. M., Tran, K. C., Simmons, D. A., Rajadas, J., Zeng, A. Y., et al. (2016). A small molecule TrkB/TrkC neurotrophin receptor co-activator with distinctive effects on neuronal survival and process outgrowth. Neuropharmacol. 110, 343–361. doi: 10.1016/j.neuropharm.2016.06.015
Yilmaz, C., Karali, K., Fodelianaki, G., Gravanis, A., Chavakis, T., Charalampopoulos, I., et al. (2019). Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 55, 788. doi: 10.1016/j.yfrne.2019.100788
Yilmaz, C., Rogdakis, T., Latorrata, A., Thanou, E., Karadima, E., Papadimitriou, E., et al. (2022). ENT-A010, a novel steroid derivative, displays neuroprotective functions and modulates microglial responses. Biomolecules. 9, 424. doi: 10.3390/biom12030424
Zanger, U. M., and Schwab, M. (2013). Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141. doi: 10.1016/j.pharmthera.2012.12.007
Keywords: neurotrophin mimetics, TrkA, TrkB, receptor agonists, signaling, neuroprotection, neurodegeneration, DHEA derivatives
Citation: Narducci D, Charou D, Rogdakis T, Zota I, Bafiti V, Zervou M, Katsila T, Gravanis A, Prousis KC, Charalampopoulos I and Calogeropoulou T (2023) A quest for the stereo-electronic requirements for selective agonism for the neurotrophin receptors TrkA and TrkB in 17-spirocyclic-dehydroepiandrosterone derivatives. Front. Mol. Neurosci. 16:1244133. doi: 10.3389/fnmol.2023.1244133
Received: 21 June 2023; Accepted: 31 August 2023;
Published: 28 September 2023.
Edited by:
Cleide Dos Santos Souza, The University of Sheffield, United KingdomReviewed by:
Lilian Kisiswa, Aarhus University, DenmarkCopyright © 2023 Narducci, Charou, Rogdakis, Zota, Bafiti, Zervou, Katsila, Gravanis, Prousis, Charalampopoulos and Calogeropoulou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theodora Calogeropoulou, dGNhbG9nQGVpZS5ncg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.