 Sholto de Wet
Sholto de Wet Rensu Theart2
Rensu Theart2 Ben Loos
Ben Loos
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 31 August 2023
Sec. Molecular Signalling and Pathways
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1225227
Neurodegenerative diseases are often characterized by hydrophobic inclusion bodies, and it may be the case that the aggregate-prone proteins that comprise these inclusion bodies are in fact the cause of neurotoxicity. Indeed, the appearance of protein aggregates leads to a proteostatic imbalance that causes various interruptions in physiological cellular processes, including lysosomal and mitochondrial dysfunction, as well as break down in calcium homeostasis. Oftentimes the approach to counteract proteotoxicity is taken to merely upregulate autophagy, measured by an increase in autophagosomes, without a deeper assessment of contributors toward effective turnover through autophagy. There are various ways in which autophagy is regulated ranging from the mammalian target of rapamycin (mTOR) to acetylation status of proteins. Healthy mitochondria and the intracellular energetic charge they preserve are key for the acidification status of lysosomes and thus ensuring effective clearance of components through the autophagy pathway. Both mitochondria and lysosomes have been shown to bear functional protein complexes that aid in the regulation of autophagy. Indeed, it may be the case that minimizing the proteins associated with the respective neurodegenerative pathology may be of greater importance than addressing molecularly their resulting inclusion bodies. It is in this context that this review will dissect the autophagy signaling pathway, its control and the manner in which it is molecularly and functionally connected with the mitochondrial and lysosomal system, as well as provide a summary of the role of autophagy dysfunction in driving neurodegenerative disease as a means to better position the potential of rapamycin-mediated bioactivities to control autophagy favorably.
Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) are progressive in nature and often consist of both inherited as well as sporadic forms (DiFiglia et al., 1997; Baba et al., 1998; Näslund et al., 2000; Meredith et al., 2002; Le and Appel, 2004; Li et al., 2004; Orr et al., 2008; Genuis and Kelln, 2015). Within the context of an increased aging population, as is the case today, the incidence of sporadic AD and PD cases has been seen to be much greater than those of the genetic variants (Alzheimer’s Association, 2019, 2022). The loss of cognitive function is an overlapping feature of each of these diseases. Additionally, each disease is histologically characterized according to the dysfunction of a particular protein that leads to the formation of inclusion bodies that consist primarily of these proteins within regions of the brain that correlate with the symptoms of the disease in question. For example, AD is characterized by the formation of senile plaques that present with β-amyloid deposition in the cerebral cortex (Braak and Braak, 1991), PD by the formation of Lewy bodies containing α-synuclein in the neurons of the substantia nigra (Spillantini et al., 1998) and HD by intranuclear inclusions containing of mutant huntingtin protein (Yu et al., 2003). Prior to the manifestation of a molecular pathology and symptoms these proteins form a necessary part of normal brain physiology, however, there is an increased production of their misfolded forms, which are aggregate-prone, leading to the eventual formation of inclusion bodies (Chiti and Dobson, 2009; Pechmann et al., 2009). Misfolded proteins have been shown to be a fact of life but are quickly cleared before they are produced to such an extent that they outnumber the presence of the native protein forms (Swart et al., 2014; Sweeney et al., 2017). This clearance is maintained by degradative systems such as the ubiquitin proteasomal system (UPS) and macroautophagy (Bence et al., 2001; Komatsu et al., 2006; Tanaka and Matsuda, 2014). However, it is known that the UPS becomes overwhelmed and ineffective when there is an overproduction of aggregated proteins, as is the case in neurodegenerative diseases, leading to a greater dependence of the system upon autophagy as a means of clearance (Figure 1). Although the components are well known in isolation, the interplay between protein cargo and its solubility state, mitochondrial health and lysosomal dysfunction appears central to the development of neurodegenerative diseases. In this review we will discuss the mechanisms in place for protein quality control; particularly macroautophagy, as well as the cellular dysfunction that impacts mitochondria and lysosomes which appear to be at the heart of neurodegenerative disease pathogenesis.
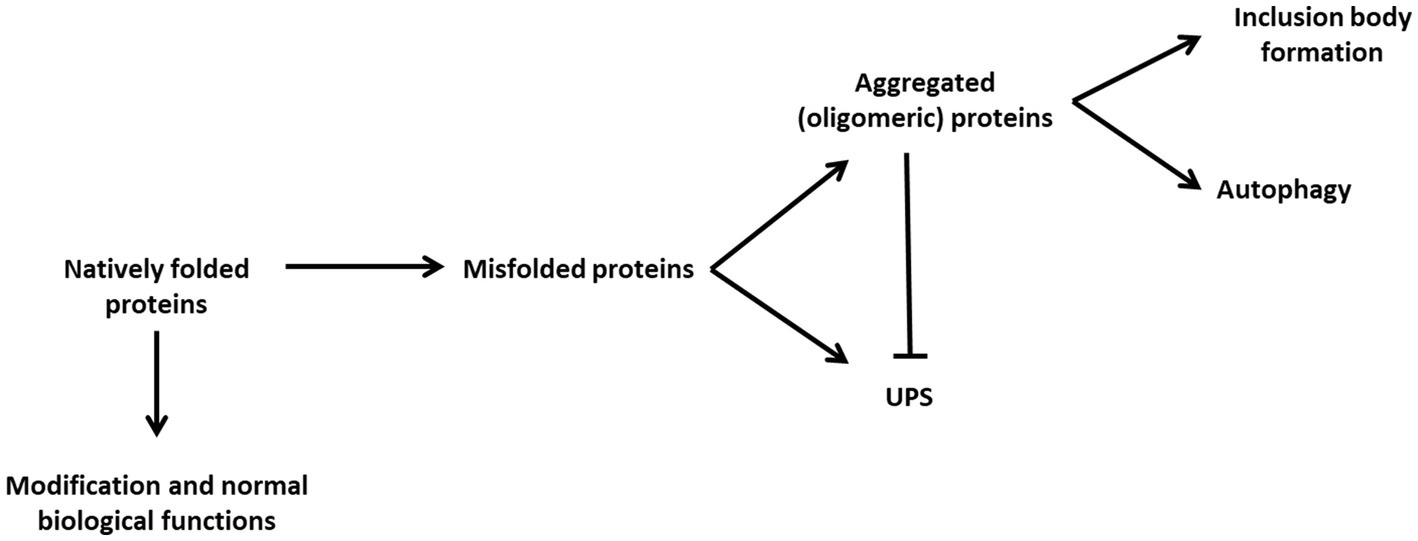
Figure 1. The proteostatic balance through cellular degradation strategies. Natively folded proteins usually undergo modification and processing contributing to normal biological function. However, natively folded proteins may suffer events resulting in their misfolding. The UPS is capable of degrading monomeric misfolded proteins, however, misfolded proteins may become aggregated and unable to be degraded by the UPS. As such autophagy becomes the primary means of maintaining proteostasis by degrading aggregated proteins. These proteins may continue to aggregate to the point at which they cause cellular stress and reduce the ability of autophagy to maintain the proteostasis of the cell. Large, aggregated protein clusters become insoluble and form deposits within the different regions of the cell, depending on the particular disease, rather than undergoing degradative events.
Proteostasis, that is the maintenance of protein homeostasis by controlling the abundance, conformation, binding interactions, and distribution of proteins by means of synthesis, degradation or modification is an integral part of normal metabolic functioning of a cell (Balch et al., 2008). Indeed, due to the important roles that proteins play in cellular processes such as signaling, the immune response, and structure, changes to proteostasis may result in cellular dysfunction and detrimental effects such as ER stress and autophagy dysfunction (Chen et al., 2001; Mcnaught et al., 2002; Chiti and Dobson, 2006; Hara et al., 2006). Therefore, there is a clear need for this balance to be carefully maintained for the sake of cell survival. Two degradative pathways exist and are used by the cell to ensure that proteostasis is maintained.
The first degradative pathway is the ubiquitin-proteasomal system (UPS) which is critical for the rapid degradation of short-lived, monomeric proteins (Kraft et al., 2010). The ubiquitin tagging system component is responsible for covalently binding cytoplasmic entities with ubiquitin, which acts as the recognition motif for the 26S subunit of the proteasome. Thus, the ubiquitination system provides the UPS with a high degree of specificity. It is however possible for the proteasome to become overwhelmed by substrates present (Bence et al., 2001; Mcnaught et al., 2002; Rideout et al., 2004; Pandey et al., 2007; Wang et al., 2020). This occurs when protein targets express irregular structures, resulting in misfolded conformations which are hydrophobic and aggregate-prone. As these proteins aggregate, they from larger clusters and are unable to pass through the proteasome (Johnston et al., 1998). In this instance, the proteostatic balance has been disrupted and the potential for the formation of insoluble structures such as aggresomes or inclusion bodies has been increased. This disruption leads to a proteostatic shift in which the system becomes more reliant upon autophagy as the means to degrade proteins.
Autophagy is the second key degradative pathway operating in cells. Three types of autophagy exist, namely microautophagy, macroautophagy and chaperone-mediated autophagy. Although each of these are well distinguished from one another, they share the function of delivering cytoplasmic components to lysosomes which contain acidic hydrolases; thereby aiding in degradation of these recruited components. Macroautophagy (hereafter autophagy) is the most well studied of its variants and morphologically distinct from the other forms due to the use of vesicular structures known as autophagosomes to shuttle cytoplasmic components to lysosomes, thereby facilitating degradation (Jahreiss et al., 2008; du Toit et al., 2018a). Autophagy has been shown to display pro-survival functions in cells following metabolic perturbation and cellular stress conditions (Mizushima et al., 2004, 2008; Ravikumar et al., 2010; Sumpter and Levine, 2010). Its degradative capabilities have been shown to extend to many cytoplasmic components, including proteins, mitochondria, peroxisomes and even micro-organisms.
In the past, autophagy was described as a non-selective form of degrading cytoplasmic components for the purpose of generating amino acids to be used in ATP production. Although this is true given nutrient-poor conditions (Sahani et al., 2014), it is now known that autophagy also controls selective cargo degradation given imbalanced homeostasis; including disruption in mitochondrial, lysosomal and ferritin homeostasis (Galluzzi et al., 2017). This selective targeting of cytoplasmic components as cargo is accomplished by proteins that act as receptors of ubiquitinated elements, effectively enabling the distinction between different types of disruptive components and targeting them to the autophagosome for subsequent degradation by lysosomal hydrolases (see section 6). Should the synthesis of misfolded proteins and aggregated proteins outweigh the synthesis and presence of fully functional proteins, proteostasis will be disrupted (Figure 1). Misfolded proteins have been shown to interact with other components of the cell such as mitochondria and the endoplasmic reticulum (ER), disrupting their function and further contributing to disrupted conditions (Lasagna-Reeves et al., 2011; Fang et al., 2019; Sharoar et al., 2019; Evans and Holzbaur, 2020). It becomes crucial therefore for the presence of these proteins to be minimized to preserve the viability of the cell.
A basal level of autophagy is constantly present within the cell and serves a “housekeeping” function; eliminating old or damaged cellular components that would otherwise disrupt homeostasis (Mizushima et al., 2004; Alers et al., 2012; Sarkar, 2013). However, in the event of a disruption in cellular homeostasis due to, for example, starvation or increased misfolded protein production, the rate of clearance through autophagy will need to increase (Yang and Klionsky, 2010; Kim et al., 2011).
Autophagy can be characterized as a step-by-step process (Figure 2) which begins with the initial synthesis of the phagophore which then matures into an autophagosome; the double-membrane vesicle that characterizes the pathway (Loos et al., 2014). Once it has sequestered cargo, the autophagosome is transported to and fuses with a lysosome, resulting in an autolysosome, which ensures the degradation of cargo. The term ‘autophagy flux’—the rate of protein degradation through the autophagy pathway—can be used to quantitatively describe the overall degradative activity and capacity of the pathway (Klionsky et al., 2016). Given conditions such as starvation or increased misfolded protein load, it becomes necessary for the autophagy flux to be increased. Indeed, given such conditions it has been observed that the cellular system will increase the number of autophagosomes thereby effectively increasing the autophagosome pool size available for cargo sequestration and delivery (Boland et al., 2008; du Toit et al., 2018b; de Wet et al., 2021).
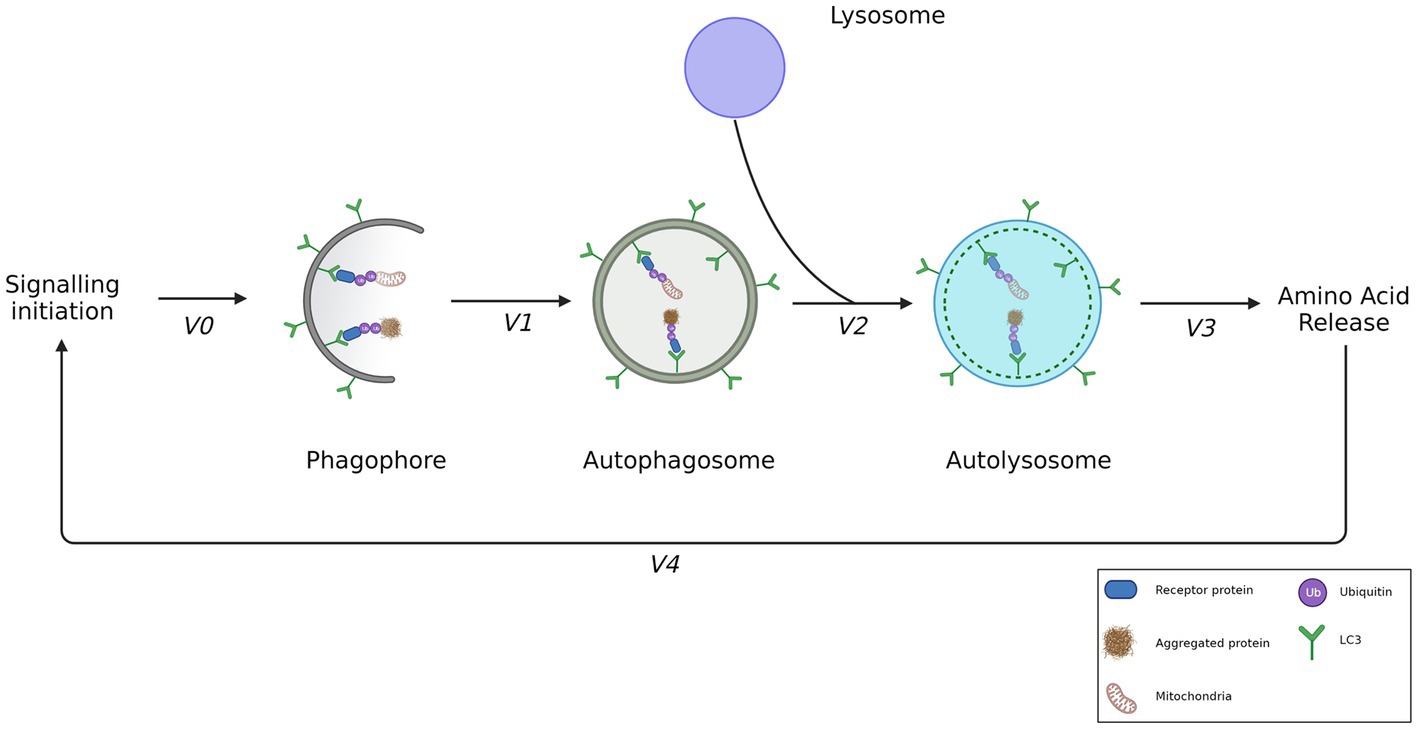
Figure 2. The step-wise rate of autophagosome flux. Signaling events result in the biogenesis of phagophores and lysosomes. Phagophores are decorated by LC3-II, enabling receipt of ubiquitinated cargo such as depolarized mitochondria or aggregated proteins. The formation rate of an autophagosome from a phagophore can be described by measuring V1, enclosing the recruited cargo. V2 however, is the best means of measuring autophagosome flux as it demonstrates the rate of autophagosome degradation by the formation of autolysosomes. V3 demonstrates the rate of amino acid release following the degradation of cargo. V4 shows the feedback of amino acids exerted on the initial signaling event. Adapted from Loos et al., (2014).
As the autophagy pathway involves the participation of many molecular role players, including autophagosomes, lysosomes and cargo, it becomes necessary to discern which of these are being used to measure and define autophagy flux. The term “autophagosome flux” has been proposed and is defined as the rate of flow along the vesicular pathway. The techniques used to measure it infers a distinction between the vesicular—that is, the molecular machinery that make up the autophagosome—and the cargo flux (Loos et al., 2014). This is of importance as not all proteins are degraded at the same rate nor does autophagy occur at the same rate in every tissue type. Indeed, whilst there may be indications of high autophagic activity—such as large autophagosome pool sizes— the inherent degradation rate of proteins may differ according to different tissue types or even brain regions (Mizushima et al., 2004; Lumkwana et al., 2017). Additionally, we have previously shown that an increase in autophagosome pool size does not necessarily infer an equal or proportional increase in the cargo receptor abundance and availability (de Wet et al., 2021). Hence, a distinction between the cargo clearance and autophagosome flux as separate entities may be necessary. To measure autophagosome flux one must consider the total number of autophagosomes within the cell at a time point and contrast this with the number of autolysosomes at the same time, in the absence and presence of an autophagosome/lysosome fusion inhibitor or lysosomal deacidifying agent, such Bafilomycin A1 or chloroquine (Li et al., 2013; du Toit et al., 2018a). In doing so, it becomes possible to measure the change in the number of autophagosomes, lysosomes and autolysosomes, respectively, that are present in the cell within a given time frame and thereby reveal their relationship in contributing toward the degradative potential of the cell. For this reason, autophagosome, lysosome and autolysosome pool sizes should be measured simultaneously to better understand the autophagosome flux, while measuring both the autophagosome and cargo flux may enable greater insight into the clearance capabilities of the pathway.
Indeed, the step-by-step nature of the autophagy pathway allows a greater number of parameters to be assessed to describe the degradative activity of the system. The following sub-sections will be used to discuss the control and stages of autophagy flux as separate entities such that it is clearer what factors may influence the degradative capacity of autophagy (Figure 2).
The autophagy pathway is well regulated such that the basal flux becomes rapidly increased given the introduction of a metabolic stressor or metabolic perturbation. The pathway that regulates the induction of autophagy is comprised of a complex network of proteins that integrate various factors and so impact autophagy flux (Figure 3). Since mitochondria require amino acids to preserve oxidative phosphorylation activity as well as glucogenic and ketogenic amino acids as additional substrates to drive ATP synthesis, the role of AMP-activated protein kinase (AMPK) has been closely tied to mitochondrial requirements and health (Hardie et al., 2012; Rabinovitch et al., 2017). Moreover, the ability of the AMPK machinery to sense ATP levels and hence energetic charge of the cell and to regulate autophagy accordingly demonstrates a close molecular link between mitochondrial health, the energy status of the cell and autophagy activity. Furthermore, the control of AMPK over autophagy induction becomes of great importance when one considers that autophagy is capable of providing amino acids that are used for mitochondrial respiration (Kim et al., 2011; Burman et al., 2017). When AMPK senses a lack of ATP, it becomes phosphorylated (Hardie et al., 2012; Rabinovitch et al., 2017) and subsequently phosphorylates the TSC2 (tuberous sclerosis complex) component of the TSC1/2 heterodimer, deactivating it. The result thereof is a downstream inhibition of the mammalian target of rapamycin complex 1 (mTOR). Due to the large degree of signaling-associated input it receives to regulate autophagy, mTOR is often referred to as the master regulator of autophagy (Wullschleger et al., 2006; Boyle and Randow, 2013). mTOR has been shown to receive and integrate stimuli from various signaling pathways in such a way that it aids in cellular repair and growth (Betz and Hall, 2013). AMPK and mTOR have been shown to regulate autophagy in accordance with the metabolic demand of the cell by phosphorylating components of the Ulk1/2-Atg13-FIP200 (ULK) complex: the initiator of autophagosome biogenesis (Alers et al., 2012). Indeed, given starvation conditions, AMPK becomes phosphorylated, subsequently phosphorylating mTOR such that autophagy is induced, as observed by the dephosphorylated state of the ULK complex (Kim et al., 2011).
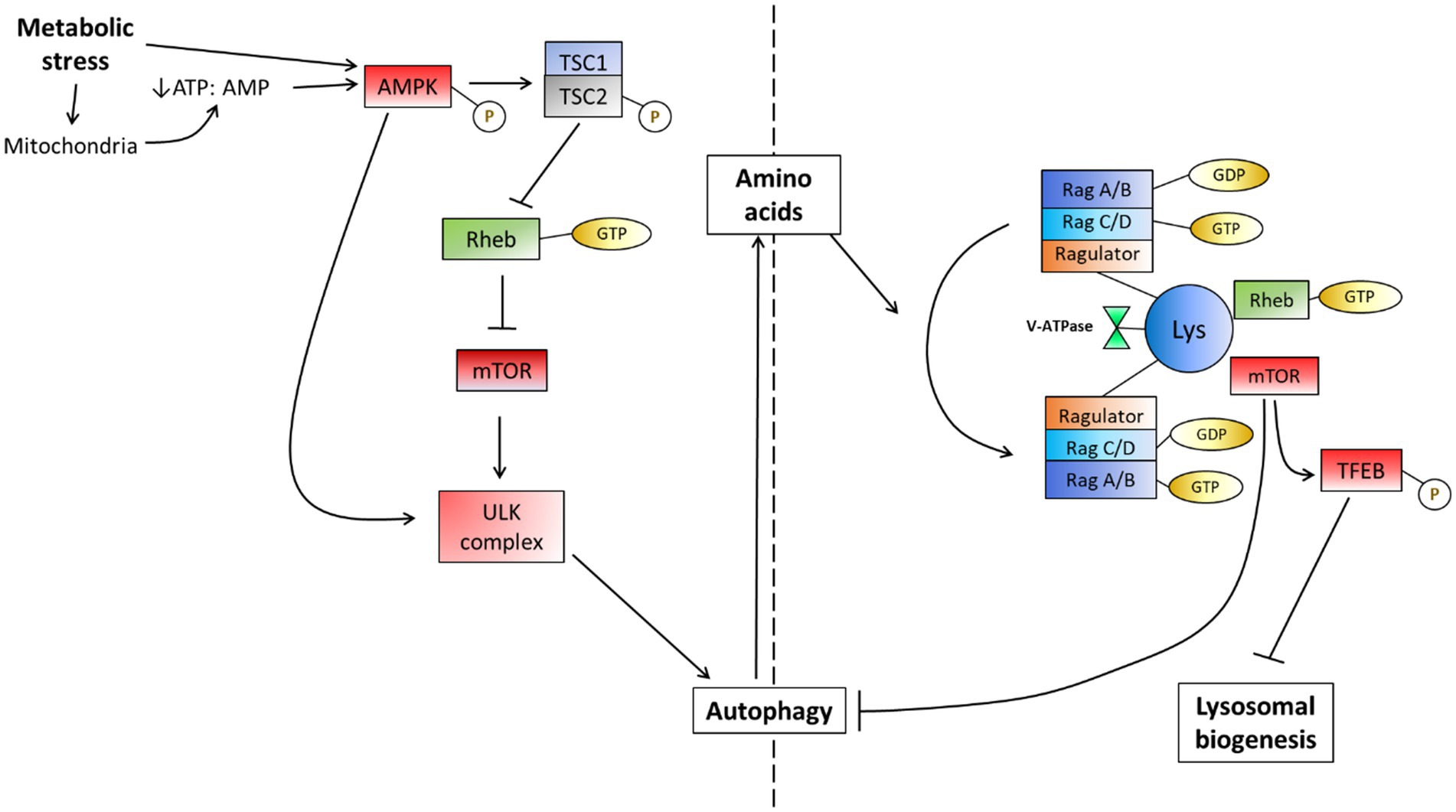
Figure 3. The mTOR-dependent response to metabolic stress in the form of starvation. Metabolic perturbations encompass many potential factors which often lead to a response directed toward the mitochondria. This may cause the depletion of ATP due to its actions on mitochondria or may be directly sensed by AMPK to induce autophagy by inhibiting mTOR. AMPK may also directly dephosphorylate the ULK complex to induce autophagy independently of mTOR, however, this often occurs at lower levels. The availability of amino acids is additionally sensed by the v-ATPase on the lysosome membrane and causes a change in the Rag complex activation, recruiting mTOR and thereby driving an inhibition reaction on autophagy when amino acids become available [Adapted from Sarkar (2013)].
In addition to the regulation of the ULK complex by AMPK and mTOR, there appears to be a molecular link between the overall abundance of lysosomes per cell and the regulation of autophagosome synthesis initiation (Sancak et al., 2010; Yu et al., 2010; Martina et al., 2012). Studies have shown that transcription factor EB (TFEB); the master regulator of lysosomal biogenesis, plays an important role in regulating the expression levels of autophagy genes. Given starvation conditions, TFEB translocates from the cytoplasm to the nucleus to induce lysosome biogenesis. Additionally, it has been observed that the overexpression of TFEB results in an increased number of autophagosomes, demonstrating a direct autophagy inducing role (Sardiello et al., 2009; Settembre et al., 2011). mTOR has been shown to regulate TFEB activity to avoid excessive levels of autophagy (Martina et al., 2012). To achieve this, mTOR must first be recruited to the lysosome. Lysosome membranes are decorated by amino acid-sensing complexes that consist of Ras-related GTPase-binding proteins (Rags) and Ragulators (Eunjung et al., 2008). Rags exist as heterodimers consisting of either RagA or RagB that is bound with either RagC or RagD; forming a trimeric complex that is collectively known as the Ragulator, and it links the Rags with v-ATPase. Low concentrations of amino acids activate v-ATPase, resulting the enhanced acidification of lysosomes through the hydrolysis of ATP (Sancak et al., 2008; Bordi et al., 2016). When amino acids are sensed, the Rag complex will be activated and causes the translocation of mTOR to the lysosome membrane. Rheb GTPase is a key activator of mTOR activity and its localization to the lysosome membrane is a key regulator of mTOR activity (Tee et al., 2003; Eunjung et al., 2008). Both the AMPK and Rag machinery hence sense the energetic state of the cell and regulate autophagy accordingly, demonstrating the importance of mitochondria and lysosomes in the control of autophagy induction. mTOR is able to carry out its inhibitory actions on TFEB and therefore lysosome and autophagosome biogenesis when Rheb is bound with GTP (Zoncu et al., 2011).
Evidently, mTOR is capable of regulating autophagy through many avenues and has an impact on both autophagosome and lysosome pool sizes. This lends greater control over the degradation of cytoplasmic components, thereby contributing toward effective control over basal autophagy flux in response to metabolic stress. It becomes clear however, that mTOR is not the sole regulator of the autophagy pathway. Rather, the interplay between mTOR, AMPK, TFEB and the Rag complex are key to autophagy signaling and regulation. Indeed, in the case of Alzheimer’s disease, reduced removal of aggregated amyloid-β results in disrupted proteostasis and the manifestation of the neurodegenerative symptoms of Alzheimer’s disease (see section 8.1) (Haass and Selkoe, 2007). Additionally, excessive levels of autophagy flux have been shown to be harmful, with the induction of a specific autophagy-dependent cell death termed autosis, which is triggered when there is a high demand for membrane material to generate autophagosomes, thus resulting in damage to the ER and mitochondria (Kriel and Loos, 2019).
The activity of mTOR, along with the energy sensing activity of AMPK and the lysosome-associated machinery, allows for the fine-tuning of the autophagy pathway response according to the metabolic needs of the cell. These interactions represent the mTOR-dependent means of autophagosome biosynthesis signaling. There are however mechanisms of inducing autophagy that are independent of mTOR activity. For example, the recruitment of Beclin-1 is crucial as a molecular machinery component during the formation of the phagophore; the pre-autophagosome membrane (Kihara et al., 2001; Pattingre et al., 2005; Russell et al., 2013). Additionally, there is a close interaction between the inositol signaling pathway and calcium presence and abundance (Sarkar et al., 2005; Vicencio et al., 2009) as both work to regulate autophagy induction independent of mTOR. Furthermore, the regulation of acetylation, the post-translational modification that involves the transfer of an acetyl group, has gained increasingly attention due to its role in controlling autophagy flux (Xie et al., 2010). Indeed, components such as sirtuin-1 (Sirt1); a NAD-dependent deacetylase capable of sensing metabolic stress (Haigis and Sinclair, 2010), and E1A-binding protein p300 (EP300); an acetyltransferase (Lee and Finkel, 2009) are two agents that influence the acetylation status of proteins. Both aid in regulating the recruitment of core autophagosome machinery components such as Atg5, Atg7 and LC3 to the phagophore and thus control autophagosome elongation (Bánréti et al., 2013; Pietrocola et al., 2015; Pineda-Ramírez et al., 2020). Indeed, substances such as spermidine, curcumin and resveratrol have been shown to cause changes in autophagy signaling that lead to the increase of autophagy flux by increasing autophagosome biogenesis (Chung et al., 2010; Morselli et al., 2011; Ai et al., 2015; Pietrocola et al., 2015).
The molecular events necessary following the signaling event that led to the generation of a mature autophagosome that contains sequestered cytoplasmic cargo and is functionally able to interact with a lysosome for degradation, is highly complex and comprises of multiple steps (Figure 4). This section will address the autophagosome machinery, that is, the core proteins recruited to the phagophore membrane, leading to the formation of a mature autophagosome. Autophagosome biosynthesis is marked by the initial translocation of the ULK complex to the ER, the site of autophagosome initiation (Koyama-Honda et al., 2013). Once present on the ER membrane, the ULK complex recruits the PI3K complex III (PI3KC3) containing Atg14L (Russell et al., 2013). Once recruited to the ER membrane, PI3KC3 phosphorylates phosphatidylinositol (PtdIns)—a lipid present on the membranes of many organelles, including the ER—to produce PI(3)P (phosphatidylinositol-3-phosphate). In parallel with this event, there is recruitment of proteins that specifically bind to PI(3)P, such as DFCP1 (double FYVE domain-containing protein 1) and WIPI (WD-repeat-interacting phosphoinositide protein). The exact role of these proteins remains unclear; however, it seems clear that their recruitment precedes that of the autophagy-related proteins (Axe et al., 2008; Koyama-Honda et al., 2013; Amaya et al., 2015). Additionally, these proteins contain a FYVE domain, which enables them to bind with the membrane of the ER and possibly the Golgi apparatus and therefore the vesicle derivatives of each. As such, PI(3)P-binding is considered to form a scaffolding matrix on the phagophore that facilitates the recruitment of autophagy-related (Atg) proteins to the phagophore after the nucleation event.
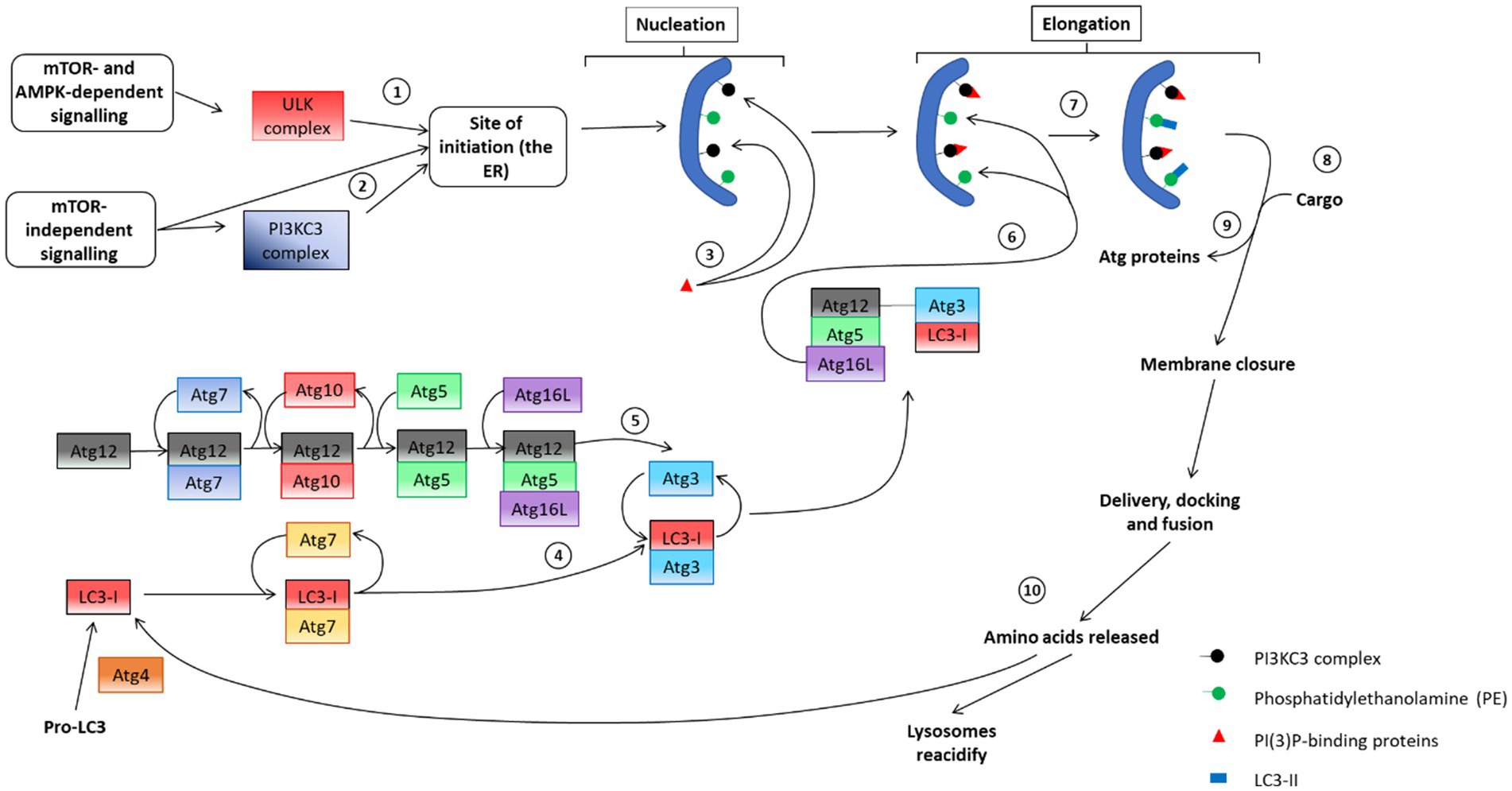
Figure 4. An overview of the autophagosome pathway, including the molecular machinery of each step. (1) Initial mTOR dependent recruitment of the ULK complex to the ER to facilitate the (2) subsequent recruitment of the PI3KC3 complex to the ER to enable phagophore nucleation. (3) PI(3)P-binding proteins aid in the nucleation step and assist in the recruitment of cargo. (4) and (5) depicts the two ubiquitin-like conjugation systems that are in place to assist in phagophore elongation. These proteins engage in the recruitment of the other components to the phagophore membrane. (6) LC3-I interacts with PE on the phagophore membrane, causing a lipidation reaction that produces LC3-II, aiding in the (7) recruitment of cargo to the membrane. (8) Atg proteins dissociate from the autophagosome membrane and aid in membrane closure. Following closure, autophagosomes are considered mature and interact with microtubules to facilitate delivery, docking and fusion with lysosomes to undergo degradation of sequestered cargo. Following degradation, the autophagy system must be reset in preparation for subsequent autophagy events. To this end (9) lysosomes are reacidified and LC3 is recycled under care of the Atg4 protein [adapted from Sarkar (2013)].
Before an autophagosome can sequester and deliver cargo, it requires modification from two ubiquitin-like conjugation systems to facilitate its elongation (Figure 4). First, Atg12 is conjugated to Atg5, which occurs due to a reaction with Atg7 and then with Atg10. Next, Atg16L is non-covalently conjugated to the Atg12-Atg5 complex, resulting in the formation of the Atg12-5-16 L complex (Mizushima et al., 2003; Tanida et al., 2004). The complex is then targeted toward phosphatidylethanolamine (PE), another membrane lipid located on the pre-autophagosome membrane (Mizushima et al., 1999).
The second conjugation system involves the transfer of microtubule-associated protein 1 light chain 3 (LC3). This process begins with the cleavage of pro-LC3 by Atg4 resulting in LC3-I. LC3-I is subsequently conjugated with Atg7 and then with Atg3. The interaction between Atg12 and Atg5 enables Atg12 to sequester Atg3, removing it and bringing LC3-I into proximity with PE. The reaction between LC3-I and PE results in the lipid-bound LC3-II (Fujita et al., 2008b). The presence of LC3-II marks the end of autophagosomal elongation and autophagosomes are now prepared for cargo sequestration (section 6). Once cargo has been sequestered, Atg proteins dissociate from the autophagosome membrane. This dissociation is thought to enable the closure of the autophagosome (Cebollero et al., 2012). LC3-II remains as part of the autophagosome membrane for the duration of its lifetime and is used as a marker of mature autophagosomes when assessing autophagosome abundance or autophagosome puncta counts (Pankiv et al., 2007; Boland et al., 2008; Jahreiss et al., 2008; du Toit et al., 2018a). Additionally, LC3-II is useful in enabling interaction between autophagosomes and microtubules, acting as an anchor point for molecular motors; proteins that drive the movement of the autophagosome (Fass et al., 2006; Jahreiss et al., 2008; Pankiv et al., 2010). Microtubules play an important role in the maturation of autophagosomes as well as their delivery, docking and fusion with lysosomes (Shen and Mizushima, 2014). The role of microtubule-dependent delivery in the autophagy process will be further discussed in section 7.
After lysosomal-dependent degradation, amino acids are released into the cytosol where they can be used for aerobic respiration by mitochondria (Scherz-Shouval and Elazar, 2007; Rabinowitz and White, 2010). LC3-II is delipidated to LC3-I by Atg4, thereby replenishing the cytoplasmic pool for future autophagy events (Fujita et al., 2008a; Kaizuka et al., 2016). The proteins involved in ensuring functional autophagosome generation and providing autophagosomes with the ability to successfully sequester cargo and interact with microtubules to enable their delivery to lysosomes demonstrate a high level of complexity. The molecular machinery necessary for autophagosome synthesis is of key interest and studies are ongoing to elucidate the precise role of many of these protein during autophagy enhancement and cargo sequestration.
Once elongated, the autophagosome is ready to receive cargo that is to be delivered to the lysosome for degradation. This step has received major attention as the autophagosome may either sequester cytoplasmic components as cargo in a non-selective, bulk manner (Mizushima, 2007), or in a highly selective manner (Kirkin et al., 2009; Zhang and Ney, 2009; Deosaran et al., 2013). The understanding of the specificity of autophagy has created a need amongst researchers to differentiate between several subtypes of autophagy according to the specific cargo targets in the context of metabolic or homeostatic perturbation (Galluzzi et al., 2017). The exact mechanism by which cargo recognition is regulated is not clear, however ubiquitinated cargo appears to be an overlapping feature (Kim et al., 2008) which is exploited by proteins bearing a ubiquitin binding area (UBA) domain as well as a LC3-interacting region (LIR). In this way, these receptor proteins, sometimes termed adaptors, are able to bind ubiquitinated components as well as LC3-II, thereby linking cargo with autophagosome machinery for effective cargo sequestration (Pankiv et al., 2007; Fujita et al., 2008a; Figure 2).
One such receptor protein is p62/SQSTM1 (hereafter p62). Due to its various functional domains, p62 has been shown to play an important role in many physiological processes such as cellular signaling pathways, inclusion body formation and tumorigenesis (Moscat and Diaz-Meco, 2009; Lim et al., 2015). In the context of cargo degradation, p62 has been shown to be present in autophagy progression and is therefore useful as a marker of autophagy activity (Ravikumar et al., 2005). Additionally, p62 has been shown to bear a Phox and Bem1 (PB1) domain which enables it to oligomerize with other p62 receptors. In so doing, p62 can self-aggregate along with its cargo to further enhance degradation (Kirkin et al., 2009). Although p62 appears to be involved in many forms of autophagy, its importance lies in its function to serve as a receptor that is primarily associated with aggrephagy; the autophagy-dependent degradation of protein aggregates (Kopito, 2000; Bjørkøy et al., 2005; Caccamo et al., 2017). This role is vital in the context of neurodegenerative diseases in which harmful misfolded proteins are generated and may lead to the formation of inclusion bodies if left unchecked (Komatsu et al., 2007; Salminen et al., 2012; Sarkar et al., 2014).
Another receptor protein, neighbor of BRCA1 gene 1 (NBR1), has been shown play several roles in maintaining cell survival (Mardakheh et al., 2010; Kim et al., 2019; Marsh et al., 2020), including the autophagy-dependent degradation of peroxisomes, known as pexophagy (Deosaran et al., 2013). Furthermore, NBR1 has also been shown to play an important role in aggrephagy as a compensatory receptor given the absence of p62 (Kirkin et al., 2009). Evidence has shown that NBR1 may be cleared through the endosomal rather than the autophagosomal pathway (Mardakheh et al., 2010), however, given that NBR1 bears the LIR and UBA domains necessary for autophagy-associated degradation, it may be plausible that NBR1 is preferentially degraded through the endosomal pathway, but has increased involvement in the autophagy-lysosomal pathway as p62 availability decreases.
Mitochondria are vital in the maintenance of the cell survival. Mitochondria are spread out through the cell as highly dynamic network; undergoing fission and fusion reactions as necessary. Fusing to facilitate efficient oxidative phosphorylation as well as protecting and maintaining mitochondrial DNA and undergoing fission to distribute mitochondria across the cell and segmenting depolarized portions to ensure their subsequent clearance (Parone et al., 2008; Twig et al., 2008; Wang et al., 2009; Perciavalle et al., 2012; Rana et al., 2017). To this end, mitophagy; the autophagy-dependent degradation of mitochondria, is yet another key autophagy subtype. Indeed, failure to eliminate dysfunctional/depolarized mitochondria results in the continued production of ROS, therefore threatening cell viability (Quintanilla et al., 2009; Wu et al., 2011). Several mitophagy receptors have been identified, presumably demonstrating the importance of mitochondrial health and turnover. These include receptors such as BNIP3 and NIX, optineurin (OPTN), optic atrophy 1 (OPA1) and the PINK/Parkin system (Zhang and Ney, 2009; Shi et al., 2014; Marceau et al., 2015; Diot et al., 2018; Padman et al., 2019; Evans and Holzbaur, 2020). The variety of identified receptors for mitophagy is a true testament to the importance of maintaining a healthy mitochondrial network and minimizing oxidative stress produced by dysfunctional mitochondria.
Of the various subtypes of autophagy that exist, aggrephagy is the one most often studied and best characterized. This is due to its critical role in re-establishing disrupted proteostasis, especially when the activity of the UPS has been impeded. Other types of autophagy, such as lysophagy, reticulophagy, xenophagy and several others have also been described along with their potential receptors (reviewed in Galluzzi et al., 2017). It is important to note that the selective nature of autophagy has only recently been identified and investigations into the exact mechanism through which cargo is sequestered are ongoing.
At a molecular level, neurodegenerative diseases are characterized by the increased presence of soluble oligomers, dysfunctional mitochondria, deacidified lysosomes and ER stress (Figure 5). Which proteinaceous cargo is problematic is what defines the neurodegenerative disease and will be discussed in subsequent sections. Due to the conformation taken on by misfolded protein monomers (Hamano et al., 2008; Chiti and Dobson, 2009), these proteins become increasingly aggregated and the resulting oligomers cause a proteostatic imbalance (Tanaka and Matsuda, 2014; Martínez et al., 2017). Additionally, due to the extent to which these proteins are aggregated, they become unable to pass through the proteasome component of the UPS. Basal autophagy has been shown to be upregulated in response to the accumulation of proteinaceous components, demonstrating a shift in protein degradation that is reliant upon autophagy (Nixon, 2007; Lim et al., 2015).
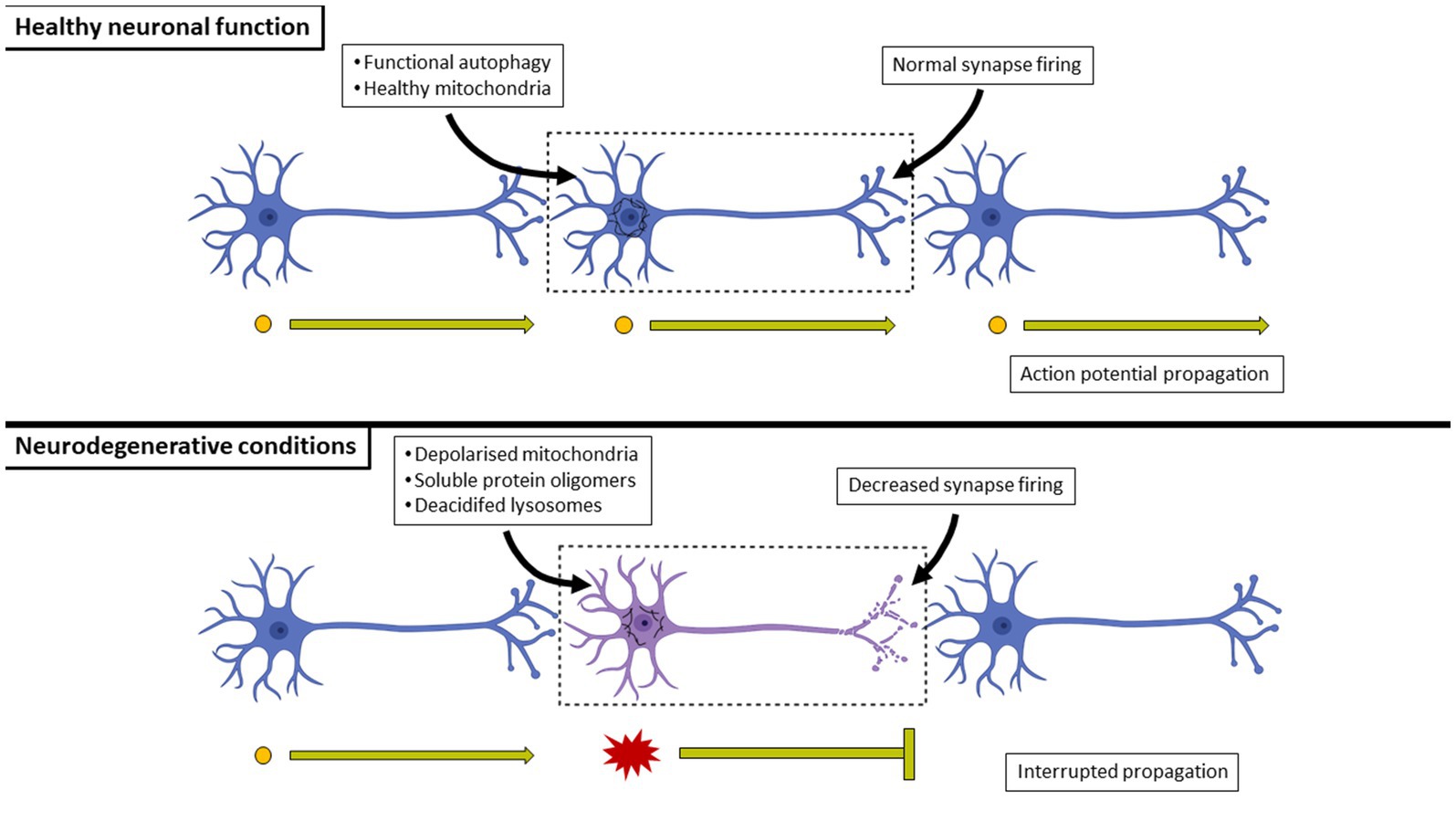
Figure 5. A comparison between healthy and neurodegenerating neuronal connections. In healthy neurons, basal autophagy is at such (typically very high) level that harmful misfolded proteins are minimized and eliminated, preventing the formation of aggregates and the co-localization with mitochondria which would otherwise impair ATP generation. In contrast, the over-production of harmful misfolded proteins will result in their aggregation. Their overproduction may occur at a rate greater than that of the cell’s autophagy flux, effectively leading to their harmful activities of aggregating to the point of forming insoluble inclusion bodies, impairing mitochondrial respiration and leading to a decrease in synaptic activity of the cell. Neurons eventually undergo cell death as a result, effectively breaking circuits.
The ER is crucial in maintaining proteostasis and a disruption of that proteostasis, due to the overproduction of misfolded proteins, has been shown to induce ER stress; a major contributor toward the overall cellular stress that results in increased cytoplasmic calcium (Ca2+) (Goussakov et al., 2010; Hetz and Mollereau, 2014; Villegas et al., 2014; Kaushik and Cuervo, 2015; Martínez et al., 2017). Furthermore, the misfolded proteins associated with neurodegenerative diseases have been shown to co-localize with and disrupt the electron transport chain complexes of mitochondria, thereby disrupting neuronal bioenergetics (Manczak et al., 2006; Chinta et al., 2010; Lasagna-Reeves et al., 2011; Shirendeb et al., 2011). This mitochondrial disruption carries a knock-on effect as the lack of ATP generation leads to a decrease in the acidified lysosomal pool size. The loss of lysosomal acidity results in an increase in the number of autophagosomes and a decrease in the numbers of autolysosomes in the neuron as the disease progresses (Lee et al., 2011; Burbulla et al., 2017). The combined effects of increased intracellular Ca2+ along with dysfunctional mitochondria lead to an increased production of ROS within the cell, priming it toward death (Kishida and Klann, 2007; Loos et al., 2013). Subsequently, abundant cell death causes interruptions between neuronal connections within specific regions of the brain, thereby contributing to clinical symptoms that manifest in patients with the disease. Additionally, many neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease, have both genetic as well as non-genetic origins, indicating that these diseases may have familial roots or be a result of lifestyle, environment or aging (Andersen, 2003; Winklhofer and Haass, 2010; Genuis and Kelln, 2015; Kell and Pretorius, 2015; Xu et al., 2016; Colacurcio and Nixon, 2017; An et al., 2018). In short, the molecular pathway through which neurodegenerative diseases are manifested in patients is no simple matter and should be approached in a genetic as well as physiological and biochemical manner to reach a consensus on the best possible treatment approach.
Aggregated proteins continue to accumulate throughout the progression of the disease as the pool size of acidified lysosomes continues to decrease. As a result, there is disrupted cellular energetics and death that leads to decreased synaptic firing and consequently, disrupted neuronal networks (Figure 5). The respective insoluble inclusion bodies are used as histological markers of neurodegenerative disease, especially at late stages where autophagy function has been mostly lost due to the loss of acidified lysosomes and neurons have begun to undergo cell death. The region of the brain impacted by the build-up of these insoluble protein aggregates varies according to the particular disease. However, there are often overlapping clinical symptoms, such as dementia or decreased fine motor capabilities that are common between diseases.
Alzheimer’s disease (AD) is histologically identified by the presence of amyloid-β (Aβ) inclusion bodies in the cerebral cortex (Braak and Braak, 1991). Additionally, brains of AD patients are often shown to contain neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau filaments primarily within the hippocampal regions (Polito et al., 2014). The resulting clinical symptoms include memory loss, spatiotemporal confusion and mood changes (Alzheimer’s Association, 2019).
The amyloid precursor protein (APP) is a single-pass transmembrane protein that is found at high levels in the brain (Kamenetz et al., 2003). The exact physiological function of APP remains unclear, however its overexpression in cell models suggests that it aids in cell growth and survival as well as contributing toward the expression levels of dendritic spines (Oh et al., 2009; Lee K. J. et al., 2010), whereas knockout models in mice indicate that it plays a role in development and memory (Senechal et al., 2008). APP is most often located on the plasma membrane of neurons where it undergoes two splicing events as part of its normal processing, first by α-secretase and then by γ-secretase (O’Brien and Wong, 2011) to yield Aβ40. Alternatively, APP may be internalized into an early endosome, where it is cleaved at its β-site by β-site-APP-cleaving enzyme 1 (BACE1) and C-terminal product of β-secretase cleavage (βCTF) (Huse et al., 2000). The resulting protein is shorter than the product of α-secretase cleavage and subsequent cleavage by γ-secretase yields Aβ at a length of 42 (Aβ42) rather than 40 (Aβ40) amino acids (Nixon, 2007). Aβ generation is a normal part of brain metabolism as Aβ is generated in low concentrations at normal physiological conditions. Indeed Abramov et al. (2009) have shown that Aβ has a regulatory role in the release of presynaptic neurotransmitters at the hippocampal region. Importantly, the hydrophobic nature of Aβ42 makes it more prone to aggregation than Aβ40 (Iwatsubo et al., 1994; Xiao et al., 2015). The Aβ42 monomers and oligomers are found in senile plaques that characterize cerebral degeneration in AD (Mucke et al., 2000; Nilsson et al., 2013). Soluble Aβ42 oligomers have been shown to cause neuronal damage in two ways. Firstly, soluble Aβ42 can interact with the outer membrane of mitochondria, thereby leading to its co-localization with the electron transport chain (ETC) machinery (Manczak et al., 2006; Hansson Petersen et al., 2008; Cha et al., 2012; Lee J. H. et al., 2022; Lee S. E. et al., 2022). This interaction disrupts mitochondrial energy metabolism and leads to elevated levels of ROS production. Due to the sensitivity of mitochondria to increased oxidative stress, the increased cytoplasmic ROS produced from Aβ-associated mitochondria will trigger a cascading effect that negatively impacts neighboring non-Aβ-associated mitochondria, effectively stressing the cell if not properly resolved though, for example, mitophagy (D'Amelio et al., 2011; Zündorf and Reiser, 2011; Frank et al., 2012; Rao et al., 2014).
Secondly, the increased production of Aβ42 relative to Aβ40 results in a proteostatic imbalance (Yu et al., 2009). This imbalance appears to be linked with dysfunctional Ca2+ regulation (Takuma et al., 2005; Kloda et al., 2007) and an increased concentration of intracellular Ca2+ is known to cause a release of caspase co-factors from the mitochondria, thereby priming and sensitizing the cell toward cell death by apoptosis (Pinton et al., 2008; Affaticati et al., 2011). Autophagy responds to increased Aβ42 levels as a means of minimizing the harmful effects brought about by the aggregates (Boland et al., 2008; Klionsky et al., 2016; Caccamo et al., 2017). As the Aβ42 yield increases, there is a decreased ATP output by mitochondria due to ETC interference. As lysosomes need ATP to reacidify after degrading cargo, there is a gradual decrease in the number of acidified lysosomes as oligomers are continually produced (Nixon et al., 2005; Yu et al., 2010; Martina et al., 2012; Lee J. H. et al., 2022). Indeed, lysosomal acidification is crucial in the elimination of toxic oligomers, and a loss thereof has detrimental consequences (Lee et al., 2011). As a result, autophagy flux becomes gradually diminished and misfolded proteins continue to aggregate further. Additionally, this diminishment of autophagy activity will lead to the accumulation of dysfunctional mitochondria due to decreased mitophagy (Komatsu et al., 2006; Martín-Maestro et al., 2017; Lee J. H. et al., 2022; Lee S. E. et al., 2022). Failure to clear these dysfunctional mitochondria will lead to the continued production of ROS and subsequent cytotoxicity (Wang et al., 2009).
AD can be classified according to the age of onset. Early-onset AD (EOAD) occurs in patients younger than 65 years, sometimes as young as 30 years, due to mutations in the gene encoding APP as well as the genes encoding for presenilin 1 (PSEN1) or PSEN2, both of which are key components of the γ-secretase complex and therefore result in an increase in Aβ42 if improperly expressed (reviewed in Hernandez-Sapiens et al., 2022). PSEN1 is also responsible for regulating lysosomal calcium homeostasis and therefore the acidity status of lysosomes, making PSEN1 mutations detrimental to lysosomal pH levels (Lee J. H. et al., 2010; Lee et al., 2015). EOAD is a rare form of AD, and represents between 1 and 5% of AD patients (Reitz et al., 2012). Late-onset AD (LOAD) occurs in patients older than 65 years and, although it can be brought about by genetic mutations (Verghese et al., 2011), its causes are more often multifactorial, ranging from traumatic brain injury, diet and even environmental factors (Sarkar et al., 2014; Kell and Pretorius, 2015; Colacurcio and Nixon, 2017; An et al., 2018), with the primary cause being increased age (Alzheimer’s Association, 2019). Regardless of the classification that AD falls under, both share the presence of senile plaques brought about by the overexpression of Aβ42 as well as atrophy of the brain, resulting in the same clinical symptoms as the disease progresses.
In the past, the mechanism by which neurons release aggregated, insoluble Aβ into the extracellular space was poorly understood, however Bhattacharyya et al., 2021 have recently shed light on the role of mitochondrial associated ER membranes in their release (Bhattacharyya et al., 2021). The presence of co-localized Aβ42, p62 and ubiquitin within senile plaques implies that there an attempt at degrading these proteins through autophagy prior to their expulsion from the neuron (Komatsu et al., 2007; Nilsson et al., 2013). It may be the case that these proteins either aggregate to the point of becoming hydrophobic and are therefore unable to be degraded by the lysosome or that there are no sufficient acidified lysosomes that are readily available for their degradation (Lee J. H. et al., 2022; Lee S. E. et al., 2022). Once expelled from the neuron, Aβ42 aggregates and clumps together, resulting in the insoluble senile plaques that characterize the disease (Mucke and Selkoe, 2012). The extracellular presence of these plaques triggers an immune response from microglia and astrocytes, both of which begin producing ROS and reactive nitrogen species (RNS) against the unfamiliar, proteinaceous material (Heales et al., 2004; Kitazawa et al., 2004; Hickman et al., 2008). This RO/NS production appears to bring about several consequences. Indeed, studies have revealed that this increase produces a pro-inflammatory state within the extracellular region which contributes toward cytotoxicity, and anti-amyloid therapies may therefore be a poor choice in combating AD symptoms (Patel et al., 2005; García-Bueno et al., 2008; Jin et al., 2008; Huang et al., 2020). Additionally, according the amyloid cascade hypothesis, the manifestation of this pro-inflammatory state contributes toward the hyperphosphorylation of the microtubule-associated protein, tau, thereby leading to the appearance of NFTs (Hardy and Selkoe, 2002). However, other studies have indicated that it may be plausible that tauopathies arise prior to the formation of senile plaques (Braak et al., 2011; Theofilas et al., 2018). Indeed, tauopathies may also arise due to mutations in the gene that encodes for tau; MAPT and tauopathies that arise due to the initial appearance of Aβ42 aggregation are known as “secondary tauopathies” (Iovino et al., 2015; Leveille et al., 2021). Regardless of the order of molecular events unfolding, the appearance of both senile plaques and NFTs are commonplace in brains of AD patients (Näslund et al., 2000; Ando et al., 2014) and therefore deserve equal levels of attention. Since tau is responsible for microtubule assembly and stabilization (Binder et al., 1985; Page et al., 2010), its hyperphosphorylation will result in its dissociation from microtubules; causing the formation of oligomeric tau, the loss of microtubule stability and therefore a loss in microtubule-dependent transport (Iqbal et al., 2009) thereby impacting axonal transport (Page et al., 2010). Additionally, increased levels of oligomeric tau have been shown to disrupt the ETC by co-localizing with complex V, leading to a diminishment of ATP production (David et al., 2005). The overall loss of microtubule-dependent transport coupled with mitochondrial energy disruption has been shown to reduce synaptic activity as well as organelle transport throughout the neuron (Lasagna-Reeves et al., 2011). As a result, neurons undergo cell death and form NFTs, a key hallmark of tauopathies, a group of neurodegenerative disease that is often seen in AD patients (Braak and Braak, 1991; Jack et al., 2010), but has also been reported to develop alongside Parkinson’s disease or even independently (Dickson, 2010; Wills et al., 2010).
Evidence suggests that the presence of senile plaques and NFTs are in fact the end result of disrupted proteostasis. Senile plaques in particular have been suggested to represent an attempt made by the cell to expel harmful proteinaceous components to prevent their interactions with mitochondria and thereby decreasing further ROS generation within the cell (Nixon et al., 2005; Abramov et al., 2009; Serrano-Pozo et al., 2011) whereas NFTs represent a ‘left behind’ remnant after the oligomerization of hyperphosphorylated tau has induced neuronal cell death (Braak and Braak, 1991; Min et al., 2010; Page et al., 2010). Indeed, the soluble, intracellular Aβ42 and hyperphosphorylated tau oligomers, and not their aggregated, insoluble counterparts, appear to cause the decrease in synaptic transmission and the eventual death of neurons observed in AD, as these effects appear to occur prior to the formation of senile plaques and NFTs (Mucke et al., 2000; Manczak et al., 2006; Cirrito et al., 2008; Lasagna-Reeves et al., 2011; Polito et al., 2014).
Parkinson’s disease (PD) is histologically characterized by the presence of intracellular Lewy bodies; α-synuclein-containing protein inclusions that are particularly located within the cell bodies of dopaminergic neurons of the substantia nigra (Spillantini et al., 1998; Kouli et al., 2018; Kavuri et al., 2020). Motor symptoms associated with the disease, such as severe tremors, rigidity and slowed movement, are due to the dopamine toxicity (Sveinbjornsdottir, 2016) or failure of mitochondrial clearance through mitophagy (Chinta et al., 2010; Moskal et al., 2020). Additionally, Braak et al. (2004) have demonstrated the pattern of neuronal degeneration in other regions of the brain as PD progresses, such as the brain stem and hippocampus, thereby demonstrating additional characteristic symptoms of the disease, some of which overlap with AD symptoms such as memory loss and mood changes (Braak et al., 2004).
Similar to AD, PD manifests in either a genetic or sporadic manner, with genetic manifestations being due to either autosomal dominant or recessive mutations (Reviewed in Breydo et al., 2012), which distinguish themselves from one another at the molecular level despite leading to similar symptoms. The autosomal dominant form of PD is due to several possible mutations in the SNCA gene. As a result, α-synuclein; the protein expressed by SNCA transcription, is expressed with a misfolded conformation (Polymeropoulos et al., 1997; Le and Appel, 2004). Natively expressed α-synuclein plays an important role in synaptic vesicle transmission and recycling (Cheng et al., 2011). Additionally, α-synuclein regulates the synthesis of dopamine by interacting with and inhibiting tyrosine hydroxylase; a rate-limiting enzyme for the synthesis of dopamine (Perez et al., 2002; Di Maio et al., 2016). Since dopamine is highly reactive and causes ROS production levels must be properly maintained, especially in dopaminergic neurons (Kao et al., 2002). For this reason, dopamine is sequestered into vesicles by vesicle monoamine transporter 2 (VMAT2) such that its reactive potential is removed from the cytoplasmic environment (Kao et al., 2002; Lotharius and Brundin, 2002; Ulusoy et al., 2012).
However, misfolded, oligomeric α-synuclein, causes dysfunctional dopamine storage. Misfolded α-synuclein expression has been demonstrated to be accompanied by reduced levels of VMAT2 and increased cytoplasmic dopamine levels (Lotharius et al., 2002; Guo et al., 2008; Zhou et al., 2011). As a result, dopamine remains in the cytoplasm and reacts with oxygen to produce ROS. This ROS generation causes subsequent mitochondrial stress, thereby increasing ROS generation further and finally leading to the death of the dopaminergic cell (Whitworth et al., 2005; Ulusoy et al., 2012). Additionally, it has been shown that misfolded α-synuclein co-localizes with complex I of the mitochondrial ETC and thereby interferes with normal ATP generation, leading to the increased production of ROS as well as increasing the likelihood of inducing apoptosis (Chinta et al., 2010; Di Maio et al., 2016; Ding et al., 2018). Other factors contributing toward the genetic expression of PD have been reviewed and are discussed by Nagatsu et al. (2019). It is therefore clear that the role of α-synuclein in regulating dopamine production, especially within the dopaminergic neurons, is key in controlling and dampening its damaging potential toward the cell (Chu and Kordower, 2007). The degradation of wild-type α-synuclein appears to occur primarily through chaperone-mediated autophagy (CMA) which links cargo with LAMP2a for degradation by the lysosome (Mak et al., 2010). However, it has been reported that CMA as well as the UPS are both blocked in the brains of PD patients (Mcnaught et al., 2002; Massey et al., 2006; Ebrahimi-Fakhari et al., 2011). Indeed, Martinez-Vicente et al. (2008) have shown that the post-translational modification of wild-type α-synuclein by dopamine leads to a decreased clearance via lysosomes (Martinez-Vicente et al., 2008; Tang et al., 2015) thereby contributing toward their accumulation and the formation of Lewy bodies. Additionally, a study conducted by Ejlerskov et al. (2015) involved the deletion of an important component of the interferon. The result was the appearance of PD-associated symptoms, such as cognitive impairments and motor deficiencies in mice, as well as the appearance of Lewy bodies, all of which appeared to be accompanied with changes in autophagy as well as receptors p62 and NBR1, confirming the involvement of macroautophagy in PD pathology (Ejlerskov et al., 2015). Taken together, it becomes clear that oligomeric α-synuclein causes a dysfunction in autophagy flux as well as decreased ATP generation by mitochondria, thereby contributing toward cell death onset.
The misfolded structure of mutated α-synuclein makes it highly aggregate-prone and the risk of forming insoluble intracellular Lewy bodies is increased as the degradation capacity decreases (Spillantini et al., 1998; Zhou et al., 2011; Ejlerskov et al., 2015). Lewy bodies are composed primarily of α-synuclein but have also been shown to consist of other materials such as PINK1 and Parkin, as well as iron and many other constituents (Baba et al., 1998; Wakabayashi et al., 2013). The formation of Lewy bodies has been shown to lead to a decrease in tyrosine hydroxylase, therefore leading to a decrease in dopamine-dependent neurotoxicity in the substantia nigra (Mori et al., 2006) as well as minimizing the reactive activities of various components and therefore offers neuroprotective benefits to the neuron (Wakabayashi et al., 2006). However, Lewy bodies may act as neuronal obstructions that hinder axonal transport (Katsuse et al., 2003), thereby leading to decreased synapse firing and contributing toward the cognitive and motor impairment associated with the disease (Volpicelli-Daley et al., 2011; Games et al., 2013; Hassink et al., 2018). The autosomal recessive form of PD is a result of mutations in PARK2 and PARK6 genes (Kitada et al., 1998). PINK1, translated from PARK6 (Valente et al., 2004) and Parkin, translated from PARK2 work together to maintain the pool of healthy mitochondria within the neuron through mitophagy. Indeed, PINK1 has been shown to bear a mitochondrial targeting sequence and is thereby able to be recruited to mitochondria at basal levels. However due to the manner in which PINK1 is cleaved, it is later released into the cytosol where it is cleared by the UPS (Jin et al., 2010; Yamano and Youle, 2013). Should the mitochondria be depolarized or exposed to cellular stress, PINK1 will remain on the mitochondrial membrane in its uncleaved form where it acts as a receptor for Parkin; an E3 ligase, which aids in the degradation of depolarized mitochondria by acting as a receptor for autophagosomes (Narendra et al., 2008; Lazarou et al., 2012; Okatsu et al., 2013) (see section 6 and Figure 2). As such, the combined function of both PINK1 and Parkin are shown to enable successful mitophagy, thereby contributing toward the elimination of depolarized mitochondria and the ROS that would have been produced.
The genetic failure of the PINK1/Parkin relationship results in mitophagy dysfunction and the accumulation of depolarized mitochondria within neuronal cytoplasm. Patients with these recessive mutations have been shown to display symptoms that are clinically indistinguishable from sporadic PD aside from having an earlier onset (Yang and Tiffany-Castiglioni, 2005; Houlden et al., 2012; Chai and Lim, 2013). The exact reason why mitophagy dysfunction causes PD symptoms remains unclear, however due to the high mitochondrial actions required in the substantia nigra for the purpose of dopamine release, it stands to reason that although this dysfunction manifests across the entire brain, the substantia nigra is the first region to suffer from degeneration (Reviewed in Surmeier et al., 2017). In addition to AD, PD has been described primarily as a sporadic disorder (Gasser, 2009; Houlden et al., 2012). Sporadic PD has been suggested to be contributed toward and manifests as a result of exposure to environmental toxins, an increased aggregation of misfolded proteins or increased systemic oxidative stress resulting in the increased production of ROS (Schapira et al., 1990; Yang and Tiffany-Castiglioni, 2005; Parihar et al., 2008; Okatsu et al., 2013; Di Maio et al., 2016). Additionally, Studies using MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), the metabolites of which are known to inhibit mitochondrial complex I, have demonstrated the effects felt by dopaminergic neurons given increased oxidative damage. This appears to be similar to the pathological effects of misfolded α-synuclein, however the use of MPTP does not seem to induce the formation of Lewy bodies (Kowall et al., 2000; Shimoji et al., 2005). Indeed, Cleeter et al. (1992) have used MPTP in animal models and found that the resulting effects on dopaminergic neurons of the substantia nigra were similar to those observed in autosomal dominant PD. It has been shown that the induction of autophagy by rapamycin protected mice against MPTP induced dopaminergic neuron loss (Zhang et al., 2017; Pupyshev et al., 2019). Additionally, studies using MPTP have also shown that there is an increase in PARK1 activity. This increased PARK1 activity leads to increased expression of mutant α-synuclein and therefore an increase in cytoplasmic dopamine as described in the case of autosomal dominant PD (Vila et al., 2000) and provides crucial insight into the study of PD.
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by mutations in the huntingtin (HTT) gene and characterized by the initial loss of neurons along the striatum that slowly progresses into other brain regions including the cerebral cortex and hypothalamus (Vonsattel et al., 1985; DiFiglia et al., 1997; Tebbenkamp et al., 2011). As a result, the clinical symptoms of HD include dysfunctional motor capabilities (Dragatsis et al., 2000; Guo et al., 2012) followed by cognitive impairment (Bäckman et al., 1997; Montoya et al., 2006; Vinther-Jensen et al., 2014). Brains of affected patients have been shown to contain intranuclear inclusion bodies and dystrophic neurites (Davies et al., 1997; Iwata et al., 2009). These inclusions contain mutant huntingtin (mHtt) proteins with polyglutamine (polyQ) repeats at the NH2-terminal, demonstrating the detrimental role of this protein in the pathogenesis of the disease. In healthy cells, wild-type Htt appears to be localized in the cytoplasm and associates with organelles such as mitochondria, synaptic vesicles and cytoskeletal components (Li et al., 2001; Orr et al., 2008; Morfini et al., 2009; Shirendeb et al., 2011). As such, Htt has been suggested to play important roles in anti-apoptotic signaling, post-synaptic transmission and protein trafficking (Reviewed in Schulte and Littleton, 2011), however, the exact role remains mysterious.
Molecularly, HD has been shown to be as a result of a mutation that causes the expansion of polyQ repeats at the NH2-terminal of the Htt protein (MacDonald et al., 1993). The number of polyQ repeats vary according to the individual and appear to correlate with the rate at which the disease progresses (Arrasate et al., 2004). Studies have demonstrated that the NH2-terminal expansions of mHtt bear sites that are selectively cleaved by proteases such as caspases and calpains (Kim et al., 2001; Wellington et al., 2002; Tebbenkamp et al., 2011, 2012). The result is the production of mHtt fragments that have been shown to be aggregate-prone and therefore toxic to the cellular system. Indeed, it appears that it is these soluble, toxic proteins that are responsible for many of the cellular disruptions that arise in the case of HD such as dysfunctional autophagy cargo recognition, diminished microtubule-dependent transport of vesicles and inappropriate mitochondrial fission and therefore dysfunctional mitophagy (Martinez-Vicente et al., 2010; Shirendeb et al., 2011, 2012). The Increased production of mHtt suggests that there would be a downregulated production of natively-folded wild-type Htt and therefore a deficit in the regular necessary activities of Htt. The aggregate-prone nature of mHtt causes a disruption to the proteostatic balance of the cell unless levels are reduced. Iwata et al. (2005) have demonstrated that there is no clear autophagy activity visible within the nucleus. However, the nucleus has been found to be make use of the UPS to facilitate degradation, thereby implicating the nucleus as being wholly dependent upon the UPS for protein degradation (Iwata et al., 2005; Chow et al., 2012; Juenemann et al., 2013; Bhat et al., 2014). Consequently, highly aggregated mHtt has been found to inhibit the UPS. Despite its final nuclear destination, the fact that oligomeric mHtt is known to co-localize with key cellular structures, makes it clear that it does exist within the cytoplasm for some time. It stands to reason therefore that it is vital for autophagy and the UPS operating together to minimize oligomeric mHtt prior to its aggregation and co-localization with the nucleus. As is the case in AD and PD, mHtt monomers aggregate to the point where they are no longer able to be degraded by the proteasome and must therefore be degraded through autophagy. Indeed, the use of rapamycin and rilmenidine, respectively have been found to induce autophagy and lead to enhanced clearance of mHtt (Berger et al., 2006; Rose et al., 2010). However, it has been proposed that mHtt associates with p62 in such a way that there is improper cargo recognition by the autophagosome, causing a slower turnover of mHtt through autophagy (Stolz et al., 2014).
Furthermore, mHtt has been found to associate with the outer mitochondrial membrane, translocating to the inner membrane where it interferes with the ETC, thereby disrupting ATP generation and increasing ROS generation (Orr et al., 2008; Shirendeb et al., 2011). Additionally, aggregated mHtt has been shown to localize along the axon, leading to an inhibition of axonal transport (Li et al., 2001; Morfini et al., 2009). This leads to a decrease in mitochondrial trafficking and thus reduces the number of mitochondria present at pre-synaptic terminals. As a result, mitochondria are no longer able to reach ATP-demanding regions -such as pre-synaptic terminals- resulting in a reduction in neurotransmitter release (Orr et al., 2008; Shirendeb et al., 2012). Additionally, HD has been shown to exhibit physiological changes in glutamatergic signaling (Faideau et al., 2010; Miller et al., 2012) as well as NMDAR signaling (Beal et al., 1991; Lee et al., 2006; Dau et al., 2014), both of which contribute toward excitotoxicity expected in the brain.
Taken together, oligomeric mHtt causes disruptions in the proteostatic balance, leading to an accumulation of dysfunctional mitochondria, decreased autophagy and excitotoxicity. Aggregated mHtt appears to disrupt axonal transport systems, thereby inhibiting synaptic transmission. The relationship between various rates of autophagy and subsequent clearance of diffuse or possibly aggregated mHtt is, however, largely unknown, and deserves further study.
It becomes clear that the soluble oligomers are problematic and reducing their expression at a cellular level is key to restoring cellular homeostasis. The autophagy pathway relies heavily upon the availability of acidified lysosomes which in turn only achieve a sufficient acidification status through the ATP produced through oxidative phosphorylation. Mitochondria in turn depend on the substrates released from the autophagy pathway, i.e., glucogenic and ketogenic amino acids, following cargo degradation. Here we will collectively term this cross talk the “autophagy triad” (Figure 6).
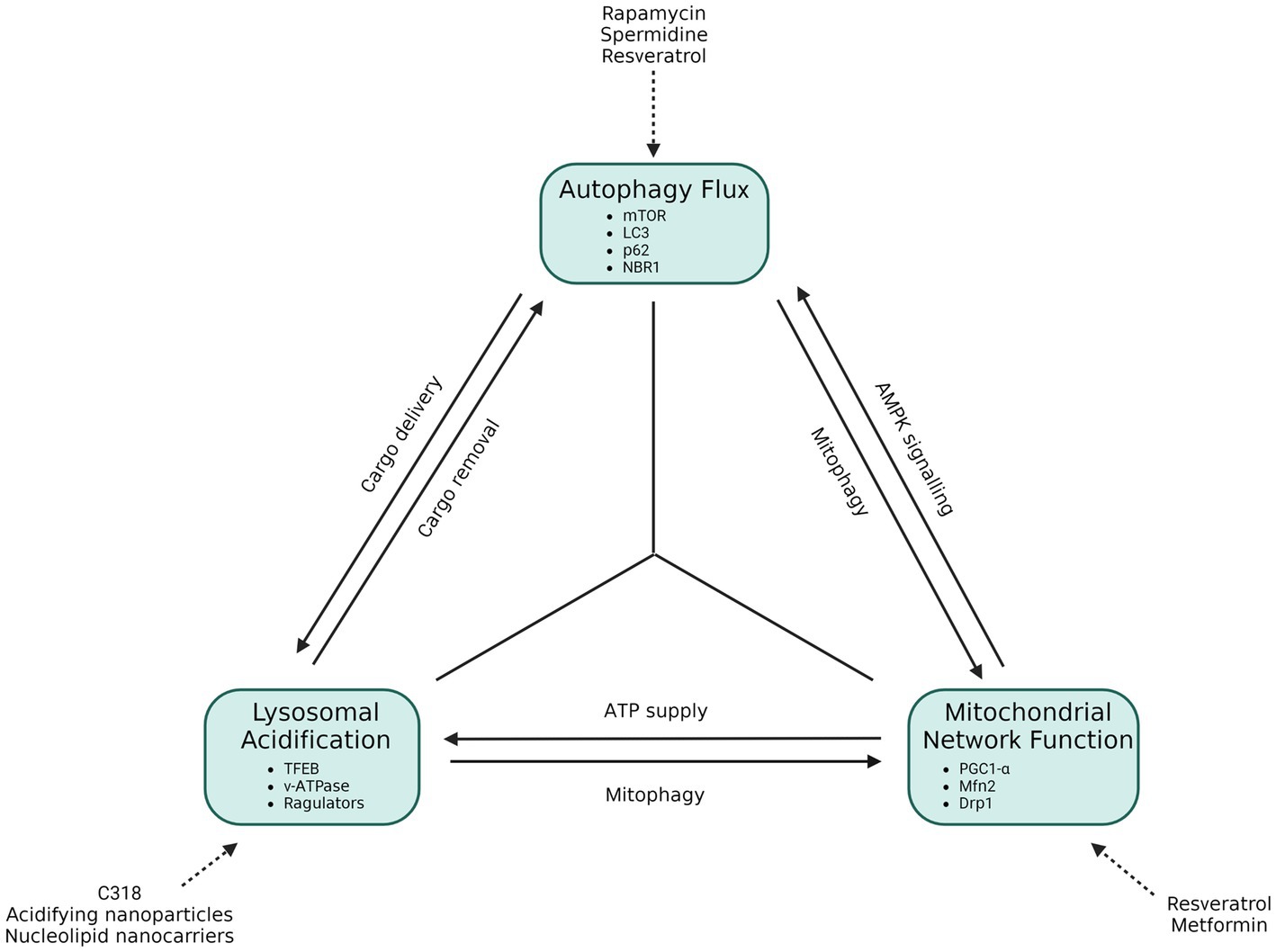
Figure 6. The functional autophagy triad. Autophagy flux requires the availability of acidified lysosomes, which are dependent on ATP output through oxidative phosphorylation. Mitochondria, in turn, are dependent on the amino acid by-products brought about from autophagy, but also require the autophagy process to eliminate dysfunctional mitochondria through mitophagy. Autophagy initiation is also dependent upon the health of mitochondria, as this determines autophagosome initiation activity through AMPK signaling. In this way an interdependent, triangular relationship between these components becomes apparent, which aids in the cellular stress response and cell survival. If one of these components is disrupted or dysfunctional, a direct or indirect knock-on effect on the next component emerges, and in so doing causes cellular stress that may contribute to neurodegenerative disease.
Autophagy activity is crucial in providing substrates for oxidative phosphorylation, and mitophagy is important for the maintenance of a healthy mitochondrial network by eliminating depolarized mitochondria (Burman et al., 2017; Rana et al., 2017). Furthermore, loss of autophagy cargo recognition has been shown to result in an increase in aggregate formation (Bhattacharjee et al., 2022) and contributes toward neurodegenerative disease manifestation. Indeed, a study investigating a p62 mutant in amyotrophic lateral sclerosis (ALS) showed that p62 behavior was changed in such a way that autophagy activity and mitochondrial respiration were both significantly decreased (Bartolome et al., 2017). Additionally, studies in which PINK1 and Parkin have been knocked out, respectively have demonstrated a decrease in mitochondrial activity, highlighting the key role of mitophagy in maintaining mitochondrial health (Song et al., 2017; Zhi et al., 2019).
Highlighted in section 6, mitochondria are dynamic organelles, undergoing constant fission and fusion events in response to the metabolic requirements of the cell. Fusion events occur through mitofusin-2 (Mfn2) and these events are shown to result in the increase of ATP, with cases of hyperfusion occurring due to a loss in ETC complex IV activity (Rolland et al., 2013). Fission events on the other hand, occur primarily through the dynamin-1-like protein (Drp1), which is responsible for separating the mitochondria in response to metabolic stress for the purpose of eliminating harmful, ROS producing mitochondria from the system (Rana et al., 2017; Fonseca et al., 2019). Indeed, Fang et al. (2019) have shown an accumulation of dysfunctional mitochondria in the neurons of AD patients, demonstrating mitophagy dysfunction early in disease progression (Fang et al., 2019). It becomes clear therefore that mitochondrial network health is vital for the maintenance of mitochondrial quality and for enabling acidification of lysosomes (Yagi et al., 2021; Huang et al., 2022).
Studies in which ATP6AP2, a key component of the lysosomal acidification machinery, had been knocked down in Drosophila and mouse models demonstrated an increase in autophagosomes along with diminishments in cognitive performance (Dubos et al., 2015; Hirose et al., 2019). Likewise, the loss of lysosomal acidity has been shown to be accompanied by an accumulation of autophagosomes as a result of a decline in autophagy flux (see section 3). The restoration of lysosomal acidification through novel nanoparticles and small molecules has been shown to decrease the numbers of autophagosomes present in the cell, demonstrating an increase in autophagy flux while also enhancing the presence of healthy mitochondria, likely through the induction of mitophagy (Vest et al., 2022; Brouillard et al., 2023; Zeng et al., 2023).
Dysfunction within any of these three entities may hence contribute toward the onset of neurodegenerative disease (Figure 6). Likewise, it is clear that therapeutic interventions that lead to the increased activity of one entity (as reviewed by Lumkwana et al., 2017; Lo and Zeng, 2023) will have subsequent knock-on effects that will enhance the functioning of the other.
AD, PD and HD constitute the most prevalent neurodegenerative diseases. Whilst all three have been shown to exhibit genetic inheritance, it is concerning to note that sporadic AD and PD are commonplace in individuals within aging populations and present without genetic factors (Greenamyre and Hastings, 2004; Alzheimer’s Association, 2022). As such, the incidence of AD and PD is of increasing concern as it reflects lifestyle as well as the environmental factors as contributing causes for neurodegenerative disorders (Winklhofer and Haass, 2010; Genuis and Kelln, 2015; Kell and Pretorius, 2015; Xu et al., 2016; Colacurcio and Nixon, 2017; An et al., 2018).
Arrasate et al. (2004) have demonstrated that a greater level of mHtt inclusion bodies is positively correlated with greater cell survival. In PD, Lewy bodies are shown to consist of iron as well as misfolded α-synuclein. By removing misfolded α-synuclein from the cytoplasm, it may be that there is less dopamine dysregulation, whereas removing iron prevents a reaction with dopamine that would result in the production of ROS (Castellani et al., 2000; Lotharius et al., 2002; Ulusoy et al., 2012). Aβ oligomers are released to the extracellular microenvironment, by a mechanism which is poorly described, but is likely to involve mitochondria-associated ER membrane, where they aggregate into senile plaques, thereby potentially eliminating the harmful oligomers from the cytoplasm (Ray et al., 2010; Nilsson et al., 2013; Bhattacharyya et al., 2021). However, prior to the formation of senile plaques, these proteins have been shown to trigger an immune response which leads to neuroinflammation in the extracellular milieu. Additionally, their presence in the extracellular millieu enables them to interact with NMDA receptors in such a way that induces excitotoxicity through excessive Ca2+ entry into the neuron (Kitazawa et al., 2004; Shankar et al., 2007; Goussakov et al., 2010; Ma et al., 2014). The immune response in particular may be exacerbated by increased hydrophobicity leading to the formation of senile plaques, thereby further increasing the neurotoxicity produced. Inclusion bodies are the hallmark characteristic used to histologically identify and distinguish between neurodegenerative diseases. They are composed of oligomeric forms of misfolded proteins which increase in hydrophobicity as they aggregate over time, forming the insoluble inclusion bodies that are deposited in regions of the brain that correspond with the symptoms of the disease at later stages of the disease. In addition to the characteristic oligomers, inclusion bodies have also been shown to consist of p62 and ubiquitin, demonstrating that autophagy plays a critical role in the formation of insoluble aggregations (Hara et al., 2006; Komatsu et al., 2007; Jackson et al., 2017). It is therefore plausible that inclusion bodies are not the root cause of neurotoxicity per se, but rather that they arise due to autophagy dysfunction brought about by the effects of the oligomeric constituents of the inclusion bodies that cause damage to intracellular organelles (Figure 6).
It is however of vital importance that their production be minimized in a timely manner so as to avoid disruptions of the mitochondrial ETC (Hansson Petersen et al., 2008; Orr et al., 2008; Parihar et al., 2008; Rao et al., 2014; Di Maio et al., 2016) or ER stress (Timmins et al., 2009; Gupta et al., 2010), that can lead to increased intracellular Ca2+ levels (Yu et al., 2009; Affaticati et al., 2011; Zündorf and Reiser, 2011; McBrayer and Nixon, 2013), both of which contribute toward a cytotoxic environment; therefore driving the likelihood of cell death onset. Additionally, inducing autophagy with mTOR inhibitors such as rapamycin has been shown to decrease the availability of these proteins (Caccamo et al., 2010; Malagelada et al., 2010; Spilman et al., 2010) and other inducers of autophagy such as spermidine, resveratrol and metformin, to name a few, have also been shown to result in reduced levels of oligomers and thus contributing toward the survival of the cell (Eisenberg et al., 2009; Shi et al., 2012; Ai et al., 2015; Pietrocola et al., 2015; Wang et al., 2018) although this should be performed with caution so as to avoid – albeit unlikely – autosis onset. It becomes clear therefore that the misfolded conformations of these key proteins are what drives cytotoxicity as their effects occur prior to the appearance of inclusion bodies, an important consideration when developing effective therapies against neurodegenerative disease in order to avoid consequential neuroinflammation.
The mild hydrophobicity of soluble oligomers creates a challenge to the autophagy system which increases autophagosome production as a means of increasing flux as a stress response to decrease the protein load (Nixon et al., 2005; Fujita et al., 2008a; Menzies et al., 2015; Bordi et al., 2016). Although basal autophagosome counts in neurons are generally low, treatment with lysosomal inhibitors such as bafilomycin A1 or chloroquine cause an accumulation of autophagosomes, lysosomes and autolysosomes demonstrating that neuronal autophagy flux is in fact very high (Boland et al., 2008; du Toit et al., 2018a). Regardless, patients still suffer from symptoms of neurodegeneration. This is likely because there is an overproduction and oligomerization of misfolded proteins, generated at a greater rate than the rate of their degradation through autophagy (Martinez-Vicente et al., 2010; Wu et al., 2015; Bordi et al., 2016). A possible reason for this may be due to the interaction between oligomers and mitochondria, resulting in decreased ATP output (Kim et al., 2001; Manczak et al., 2006; Hansson Petersen et al., 2008; Chinta et al., 2010; Di Maio et al., 2016). Since lysosomes require ATP to be reacidified after the degradation of cargo (Forgac, 2007; Zoncu et al., 2011), the decreased availability of ATP will lead to a decrease in protein degradation in spite of cargo sequestration by autophagosomes (Figure 6). Indeed, Lee J. H. et al. (2022) and Lee S. E. et al. (2022) have recently demonstrated the importance of acidified lysosome pool size in the context of AD and neuronal health (Colacurcio and Nixon, 2017; Lee J. H. et al., 2022). Additionally, this will result in dysfunctional mitophagy, leading to the accumulation of dysfunctional mitochondria and their continued production of ROS and pro-apoptotic signaling molecules (Narendra et al., 2008; Orr et al., 2008; Zhang and Ney, 2009). Taken together, it is plausible that the stress produced by mitochondria and the subsequent loss of lysosomal acidification lies at the heart of neurodegeneration as autophagy flux is dependent upon adequate mitochondrial output (Figure 6). It is important to note that these interactions occur intracellularly but create extracellular stressors that lead to knock-on effects to surrounding cells. Increasing the autophagy flux seems to be the gold standard to decreasing ND toxicity, however, as it is known that dysfunctional mitochondria do continue to accumulate during the pathogenesis of the disease, it is conceivable that increasing autophagy flux will work to a degree but reach a point where it is no longer effective as the lysosomal pool becomes increasingly deacidified. If our suggested “autophagy triad” is relevant (Figure 6), then it may be the case that inducing mitochondrial biogenesis or mitochondrial fusion, through treatments such as resveratrol or metformin, might be more beneficial than previously anticipated, as these will contribute toward lysosomal acidification and subsequently autophagy flux that effectively aids in the clearance of depolarized mitochondria as well as NDD proteins. This notion would strongly support a combined therapy that makes use of upregulated mitochondrial biogenesis paired with autophagy flux induction, therefore impacting lysosome function and subsequent mitophagy and proteinaceous cargo capacity. This deserves further study.
SdW conceptualized, wrote the manuscript, and developed the figures. BL conceptualized. BL and RT edited the manuscript. All authors contributed to the article and approved the submitted version.
The authors wish to thank the South African Medical Research Council (SAMRC) as well as the National Research Foundation (NRF) of South Africa for funding.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramov, E., Dolev, I., Fogel, H., Ciccotosto, G. D., Ruff, E., and Slutsky, I. (2009). Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 12, 1567–1576. doi: 10.1038/nn.2433
Affaticati, P., Mignen, O., Jambou, F., Potier, M. C., Klingel-Schmitt, I., Degrouard, J., et al. (2011). Sustained calcium signalling and caspase-3 activation involve NMDA receptors in thymocytes in contact with dendritic cells. Cell Death Differ. 18, 99–108. doi: 10.1038/cdd.2010.79
Ai, Z., Li, C., Li, L., and He, G. (2015). Resveratrol inhibits β-amyloid-induced neuronal apoptosis via regulation of p53 acetylation in PC12 cells. Mol. Med. Rep. 11, 2429–2434. doi: 10.3892/mmr.2014.3034
Alers, S., Loffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11. doi: 10.1128/mcb.06159-11
Alzheimer’s Association (2019). Alzheimer’s disease facts and figures. Alzheimer’s Dementia 15, 321–387. doi: 10.1016/j.jalz.2019.01.010
Alzheimer’s Association (2022). Alzheimer’s disease facts and figures. Alzheimer’s Dementia 18, 700–789. doi: 10.1002/alz.12638
Amaya, C., Fader, C. M., and Colombo, M. I. (2015). Autophagy and proteins involved in vesicular trafficking. FEBS Lett. 589, 3343–3353. doi: 10.1016/j.febslet.2015.09.021
An, Y., Varma, V. R., Varma, S., Casanova, R., Dammer, E., Pletnikova, O., et al. (2018). Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 14, 318–329. doi: 10.1016/j.jalz.2017.09.011
Andersen, J. K. (2003). Paraquat and iron exposure as possible synergistic environmental risk factors in Parkinson’s disease. Neurotox. Res. 5, 307–313. doi: 10.1007/BF03033150
Ando, K., Laborde, Q., Lazar, A., Godefroy, D., Youssef, I., Amar, M., et al. (2014). Inside Alzheimer brain with CLARITY: senile plaques, neurofibrillary tangles and axons in 3-D. Acta Neuropathol. 128, 457–459. doi: 10.1007/s00401-014-1322-y
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M. Y., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. J. Med. Res. 152, 879–884.
Bäckman, L., Robins-Wahlin, T. B., Lundin, A., Ginovart, N., and Farde, L. (1997). Cognitive deficits in Huntington’s disease are predicted by dopaminergic PET markers and brain volumes. Brain 120, 2207–2217. doi: 10.1093/brain/120.12.2207
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Bánréti, Á., Sass, M., and Graba, Y. (2013). The emerging role of acetylation in the regulation of autophagy. Autophagy 9, 819–829. doi: 10.4161/auto.23908
Bartolome, F., Esteras, N., Martin-Requero, A., Boutoleau-Bretonniere, C., Vercelletto, M., Gabelle, A., et al. (2017). Pathogenic p62/SQSTM1 mutations impair energy metabolism through limitation of mitochondrial substrates. Sci. Rep. 7, 1666–1614. doi: 10.1038/s41598-017-01678-4
Beal, M. F., Ferrante, J., Swartz, J., and Kowall, N. W. (1991). Chronic Quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 11, 1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. doi: 10.1126/science.292.5521.1552
Berger, Z., Ravikumar, B., Menzies, F. M., Oroz, L. G., Underwood, B. R., Pangalos, M. N., et al. (2006). Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 15, 433–442. doi: 10.1093/hmg/ddi458
Betz, C., and Hall, M. N. (2013). Where is mTOR and what is it doing there? J. Cell Biol. 203, 563–574. doi: 10.1083/jcb.201306041
Bhat, K. P., Yan, S., Wang, C. E., Li, S., and Li, X. J. (2014). Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc. Natl. Acad. Sci. U. S. A. 111, 5706–5711. doi: 10.1073/pnas.1402215111
Bhattacharjee, A., Ürmösi, A., Jipa, A., Kovács, L., Deák, P., Szabó, Á., et al. (2022). Loss of ubiquitinated protein autophagy is compensated by persistent cnc/NFE2L2/Nrf2 antioxidant responses. Autophagy 18, 2385–2396. doi: 10.1080/15548627.2022.2037852
Bhattacharyya, R., Black, S. E., Lotlikar, M. S., Fenn, R. H., Jorfi, M., Kovacs, D. M., et al. (2021). Axonal generation of amyloid-β from palmitoylated APP in mitochondria-associated endoplasmic reticulum membranes. Cell Rep. 35:109134. doi: 10.1016/j.celrep.2021.109134
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1985). The distribution of tau in the mammalian central nervous central nervous. J. Cell Biol. 101, 1371–1378. doi: 10.1083/jcb.101.4.1371
Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Øvervatn, A., et al. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614. doi: 10.1083/jcb.200507002
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Bordi, M., Berg, M. J., Mohan, P. S., Peterhoff, C. M., Alldred, M. J., Che, S., et al. (2016). Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12, 2467–2483. doi: 10.1080/15548627.2016.1239003
Boyle, K. B., and Randow, F. (2013). The role of “eat-me” signals and autophagy cargo receptors in innate immunity. Curr. Opin. Microbiol. 16, 339–348. doi: 10.1016/j.mib.2013.03.010
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Braak, H., Ghebremedhin, E., Rüb, U., Bratzke, H., and Del Tredici, K. (2004). Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318, 121–134. doi: 10.1007/s00441-004-0956-9
Braak, H., Thal, D.R., Ghebremedhin, E., and Tredici, K.Del. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969, doi: 10.1097/NEN.0b013e318232a379
Breydo, L., Wu, J. W., and Uversky, V. N. (2012). α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta Mol. basis Dis. 1822, 261–285. doi: 10.1016/j.bbadis.2011.10.002
Brouillard, M., Kinet, R., Joyeux, M., Dehay, B., Crauste-Manciet, S., and Desvergnes, V. (2023). Modulating lysosomal pH through innovative multimerized succinic acid-based nucleolipid derivatives. Bioconjug. Chem. 34, 572–580. doi: 10.1021/acs.bioconjchem.3c00041
Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261. doi: 10.1126/science.aam9080
Burman, J. L., Pickles, S., Wang, C., Sekine, S., Vargas, J. N. S., Zhang, Z., et al. (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 216, 3231–3247. doi: 10.1083/jcb.201612106
Caccamo, A., Ferreira, E., Branca, C., and Oddo, S. (2017). p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 22, 865–873. doi: 10.1038/mp.2016.139
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120. doi: 10.1074/jbc.M110.100420
Castellani, R. J., Siedlak, S. L., Perry, G., and Smith, M. A. (2000). Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 100, 111–114. doi: 10.1007/s004010050001
Cebollero, E., Van Der Vaart, A., Zhao, M., Rieter, E., Klionsky, D. J., Helms, J. B., et al. (2012). Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr. Biol. 22, 1545–1553. doi: 10.1016/j.cub.2012.06.029
Cha, M. Y., Han, S. H., Son, S. M., Hong, H. S., Choi, Y. J., Byun, J., et al. (2012). Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One 7, 1–11. doi: 10.1371/journal.pone.0034929
Chai, C., and Lim, K. (2013). Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genomics 14, 486–501. doi: 10.2174/1389202914666131210195808
Chen, S., Berthelier, V., Yang, W., and Wetzel, R. (2001). Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 311, 173–182. doi: 10.1006/jmbi.2001.4850
Cheng, F., Vivacqua, G., and Yu, S. (2011). The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 42, 242–248. doi: 10.1016/j.jchemneu.2010.12.001
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Chiti, F., and Dobson, C. M. (2006). Protein Misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Chiti, F., and Dobson, C. M. (2009). Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 5, 15–22. doi: 10.1038/nchembio.131
Chow, W. N. V., Luk, H. W., Chan, H. Y. E., and Lau, K. F. (2012). Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65. Biochem. J. 443, 681–689. doi: 10.1042/BJ20112175
Chu, Y., and Kordower, J. H. (2007). Age-associated increases of α-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol. Dis. 25, 134–149. doi: 10.1016/j.nbd.2006.08.021
Chung, S., Yao, H., Caito, S., Hwang, J.-W., Arunachalam, G., and Rahman, I. (2010). Regulation of SIRT1 in cellular functions: role of polyphenols. Arch. Biochem. Biophys. 501, 79–90. doi: 10.1016/j.abb.2010.05.003
Cirrito, J. R., Kang, J. E., Lee, J., Stewart, F. R., Verges, D. K., Silverio, L. M., et al. (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron 58, 42–51. doi: 10.1016/j.neuron.2008.02.003
Cleeter, M. W. J., Cooper, J. M., and Schapira, A. H. V. (1992). Irreversible inhibition of mitochondrial complex I by 1-Methyl-4-Phenylpyridinium: evidence for free radical involvement. J. Neurochem. 58, 786–789. doi: 10.1111/j.1471-4159.1992.tb09789.x
Colacurcio, D. J., and Nixon, R. A. (2017). Disorders of lysosomal acidification – the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 32, 75–88. doi: 10.1016/j.arr.2016.05.004.Disorders
D'Amelio, M., Cavallucci, V., Middei, S., Marchetti, C., Pacioni, S., Ferri, A., et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 14, 69–76. doi: 10.1038/nn.2709
Dau, A., Gladding, C. M., Sepers, M. D., and Raymond, L. A. (2014). Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol. Dis. 62, 533–542. doi: 10.1016/j.nbd.2013.11.013
David, D. C., Hauptmann, S., Scherping, I., Schuessel, K., Keil, U., Rizzu, P., et al. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. doi: 10.1074/jbc.M500356200
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal Intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cells 90, 537–548. doi: 10.1016/S0092-8674(00)80513-9
de Wet, S., Du Toit, A., and Loos, B. (2021). Spermidine and rapamycin reveal distinct autophagy flux response and cargo receptor clearance profile. Cells 10, 1–19. doi: 10.3390/cells10010095
Deosaran, E., Larsen, K. B., Hua, R., Sargent, G., Wang, Y., Kim, S., et al. (2013). NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 126, 939–952. doi: 10.1242/jcs.114819
di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8, 1–15. doi: 10.1126/scitranslmed.aaf3634
Dickson, D. W. (2010). Neuropathology of non-Alzheimer degenerative disorders. Int. J. Clin. Exp. Pathol. 3, 1–23.
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Ding, H., Xiong, Y., Sun, J., Chen, C., Gao, J., and Xu, H. (2018). Asiatic acid prevents oxidative stress and apoptosis by inhibiting the translocation of α-Synuclein into mitochondria. Front. Neurosci. 12, 1–10. doi: 10.3389/fnins.2018.00431
Diot, A., Agnew, T., Sanderson, J., Liao, C., Carver, J., das Neves, R. P., et al. (2018). Validating the RedMIT/GFP-LC3 mouse model by studying Mitophagy in autosomal dominant optic atrophy due to the OPA1Q285STOP mutation. Front. Cell Develop. Biol. 6, 1–12. doi: 10.3389/fcell.2018.00103
Dragatsis, I., Levine, M. S., and Zeitlin, S. (2000). Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 26, 300–306. doi: 10.1038/81593
du Toit, A., de Wet, S., Hofmeyr, J.-H., Müller-Nedebock, K., and Loos, B. (2018b). The precision control of Autophagic flux and vesicle dynamics—A micropattern approach. Cells 7:94. doi: 10.3390/cells7080094
du Toit, A., Hofmeyr, J. H. S., Gniadek, T.J., and Loos, B. (2018a). Measuring autophagosome flux. Autophagy 14, 1060–1071. doi: 10.1080/15548627.2018.1469590
Dubos, A., Castells-Nobau, A., Meziane, H., Oortveld, M. A. W., Houbaert, X., Iacono, G., et al. (2015). Conditional depletion of intellectual disability and parkinsonism candidate gene ATP6AP2 in fly and mouse induces cognitive impairment and neurodegeneration. Hum. Mol. Genet. 24, 6736–6755. doi: 10.1093/hmg/ddv380
Ebrahimi-Fakhari, D., Cantuti-Castelvetri, I., Fan, Z., Rockenstein, E., Masliah, E., Hyman, B. T., et al. (2011). Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-Synuclein. J. Neurosci. 31, 14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011
Eisenberg, T., Knauer, H., Schauer, A., Büttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. doi: 10.1038/ncb1975
Ejlerskov, P., Hultberg, J. G., Wang, J., Carlsson, R., Ambjørn, M., Kuss, M., et al. (2015). Lack of neuronal IFN-β-IFNAR causes Lewy body- and Parkinson’s disease-like dementia. Cells 163, 324–339. doi: 10.1016/j.cell.2015.08.069
Eunjung, K., Goraksha-Hicks, P., Li, L., Neufeld, T. P., and Guan, K.-L. (2008). Regulation of TORC1 by rag GFPases in nutrient response. Nat. Cell Biol. 10, 935–945. doi: 10.1038/ncb1753
Evans, C. S., and Holzbaur, E. L. F. (2020). Quality control in neurons: Mitophagy and other selective autophagy mechanisms. J. Mol. Biol. 432, 240–260. doi: 10.1016/j.jmb.2019.06.031
Faideau, M., Kim, J., Cormier, K., Gilmore, R., Welch, M., Auregan, G., et al. (2010). In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum. Mol. Genet. 19, 3053–3067. doi: 10.1093/hmg/ddq212
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Fass, E., Shvets, E., Degani, I., Hirschberg, K., and Elazar, Z. (2006). Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 281, 36303–36316. doi: 10.1074/jbc.M607031200
Fonseca, T. B., Sánchez-Guerrero, Á., Milosevic, I., and Raimundo, N. (2019). Mitochondrial fission requires DRP1 but not dynamins. Nature 570, E34–E42. doi: 10.1038/s41586-019-1296-y
Forgac, M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929. doi: 10.1038/nrm2272
Frank, M., Duvezin-Caubet, S., Koob, S., Occhipinti, A., Jagasia, R., Petcherski, A., et al. (2012). Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta, Mol. Cell Res. 1823, 2297–2310. doi: 10.1016/j.bbamcr.2012.08.007
Fujita, N., Hayashi-Nishino, M., Fukumoto, H., Omori, H., Yamamoto, A., Noda, T., et al. (2008a). An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 19, 4651–4659. doi: 10.1091/mbc.E08
Fujita, N., Itoh, T., Omori, H., Fukuda, M., Noda, T., and Yoshimori, T. (2008b). The Atg16L complex specifies the site of LC3 Lipidation for membrane biogenesis in autophagy. Mole. Biol. Cell. 19, 2092–2100. doi: 10.1091/mbc.E07
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Games, D., Seubert, P., Rockenstein, E., Patrick, C., Trejo, M., Ubhi, K., et al. (2013). Axonopathy in an α-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of c-terminal-truncated α-synuclein. J. Med. Res. 182, 940–953. doi: 10.1016/j.ajpath.2012.11.018
García-Bueno, B., Caso, J. R., and Leza, J. C. (2008). Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci. Biobehav. Rev. 32, 1136–1151. doi: 10.1016/j.neubiorev.2008.04.001
Gasser, T. (2009). Mendelian forms of Parkinson’s disease. BBA Molec Basis Disease. 1792, 587–596. doi: 10.1016/j.bbadis.2008.12.007
Genuis, S. J., and Kelln, K. L. (2015). Toxicant exposure and bioaccumulation: a common and potentially reversible cause of cognitive dysfunction and dementia. Behav. Neurol. 2015, 1–10. doi: 10.1155/2015/620143
Goussakov, I., Miller, M. B., and Stutzmann, G. E. (2010). NMDA-mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J. Neurosci. 30, 12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010
Greenamyre, J. T., and Hastings, T. G. (2004). Parkinson’s- divergent causes, convergent mechanisms. Science 304, 1120–1122. doi: 10.1126/science.1098966
Guo, J. T., Chen, A. Q., Kong, Q., Zhu, H., Ma, C. M., and Qin, C. (2008). Inhibition of vesicular monoamine transporter-2 activity in α-synuclein stably transfected SH-SY5Y cells. Cell. Mol. Neurobiol. 28, 35–47. doi: 10.1007/s10571-007-9227-0
Guo, Z., Rudow, G., Pletnikova, O., Codispoti, K. E., Orr, B. A., Crain, B. J., et al. (2012). Striatal neuronal loss correlates with clinical motor impairment in Huntington’s disease. Mov. Disord. 27, 1379–1386. doi: 10.1002/mds.25159
Gupta, S., Deepti, A., Deegan, S., Lisbona, F., Hetz, C., and Samali, A. (2010). HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1 a -XBP1 signaling through a physical interaction. PLoS Biol. 8, 1–15. doi: 10.1371/journal.pbio.1000410
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Haigis, M. C., and Sinclair, D. A. (2010). Mammalian sirtuins: biological insights and disease relevance. Ann. Rev. Pathol. 5, 253–295. doi: 10.1146/annurev.pathol.4.110807.092250
Hamano, T., Gendron, T. F., Causevic, E., Yen, S.-H., Lin, W.-L., Isidoro, C., et al. (2008). Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 27, 1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U. S. A. 105, 13145–13150. doi: 10.1073/pnas.0806192105
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hassink, G. C., Raiss, C. C., Segers-Nolten, I. M. J., Van Wezel, R. J. A., Subramaniam, V., Le Feber, J., et al. (2018). Exogenous α-synuclein hinders synaptic communication in cultured cortical primary rat neurons. PLoS One 13, 1–21. doi: 10.1371/journal.pone.0193763
Heales, S. J. R., Lam, A. A. J., Duncan, A. J., and Land, J. M. (2004). Neurodegeneration or neuroprotection: the pivotal role of astrocytes. Neurochem. Res. 29, 513–519. doi: 10.1023/B:NERE.0000014822.69384.0f
Hernandez-Sapiens, M. A., Reza-Zaldívar, E. E., Márquez-Aguirre, A. L., Gómez-Pinedo, U., Matias-Guiu, J., Cevallos, R. R., et al. (2022). Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease. Neural Regen. Res. 17, 31–37. doi: 10.4103/1673-5374.313016
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hickman, S. E., Allison, E. K., and Khoury, J. E. (2008). Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. Neurobiol. Dis. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hirose, T., Cabrera-Socorro, A., Chitayat, D., Lemonnier, T., Féraud, O., Cifuentes-Diaz, C., et al. (2019). ATP6AP2 variant impairs CNS development and neuronal survival to cause fulminant neurodegeneration. J. Clin. Investig. 129, 2145–2162. doi: 10.1172/JCI79990
Houlden, H., Singleton, A. B., Lrrk, S. Á., and Park, Á. P. Á. (2012). The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 124, 325–338. doi: 10.1007/s00401-012-1013-5
Huang, L. K., Chao, S. P., and Hu, C. J. (2020). Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 27, 1–13. doi: 10.1186/s12929-019-0609-7
Huang, Y. C., Hsu, S. M., Shie, F. S., Shiao, Y. J., Chao, L. J., Chen, H. W., et al. (2022). Reduced mitochondria membrane potential and lysosomal acidification are associated with decreased oligomeric Aβ degradation induced by hyperglycemia: A study of mixed glia cultures. PLoS One 17, e0260966–e0260925. doi: 10.1371/journal.pone.0260966
Huse, J. T., Pijak, D. S., Leslie, G. J., Lee, V.-Y., and Doms, R. W. (2000). Maturation and endosomal targeting of β-site amyloid precursor protein-cleaving enzyme. J. Biol. Chem. 275, 33729–33737. doi: 10.1074/jbc.M004175200
Iovino, M., Agathou, S., González-Rueda, A., del Castillo Velasco-Herrera, M., Borroni, B., Alberici, A., et al. (2015). Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138, 3345–3359. doi: 10.1093/brain/awv222
Iqbal, K., Liu, F., Gong, C.-X., Alonso Alejandra, D. C., and Grundke-Iqbal, I. (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69. doi: 10.1007/s00401-009-0486-3.Mechanisms
Iwata, A., Christianson, J. C., Bucci, M., Ellerby, L. M., Nukina, N., Forno, L. S., et al. (2005). Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. PNAS 102, 13135–13140. doi: 10.1073/pnas.0505801102
Iwata, A., Nagashima, Y., Matsumoto, L., Suzuki, T., Yamanaka, T., Date, H., et al. (2009). Intranuclear degradation of polyglutamine aggregates by the ubiquitin-proteasome system. J. Biol. Chem. 284, 9796–9803. doi: 10.1074/jbc.M809739200
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13, 45–53.
Jack, C. R., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/S1474-4422(09)70299-6
Jackson, K. L., Lin, W., Miriyala, S., Dayton, R. D., Panchatcharam, M., McCarthy, K. J., et al. (2017). p62 pathology model in the rat substantia Nigra with filamentous inclusions and progressive neurodegeneration. PLoS One 12, e0169291–e0169217. doi: 10.1371/journal.pone.0169291
Jahreiss, L., Menzies, F. M., and Rubinsztein, D. C. (2008). The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 9, 574–587. doi: 10.1111/j.1600-0854.2008.00701.x
Jin, J., Kim, H., Maxwell, J. A., and Li, L. (2008). Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflammation 45, 1–10. doi: 10.1186/1742-2094-5-23
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. doi: 10.1083/jcb.143.7.1883
Juenemann, K., Schipper-Krom, S., Wiemhoefer, A., Kloss, A., Sanz, A. S., and Reits, E. A. J. (2013). Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 288, 27068–27084. doi: 10.1074/jbc.M113.486076
Kaizuka, T., Morishita, H., Hama, Y., Tsukamoto, S., Matsui, T., Toyota, Y., et al. (2016). An Autophagic flux probe that releases an internal control. Mol. Cell 64, 835–849. doi: 10.1016/j.molcel.2016.09.037
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Kao, S., Eye, M., Infirmary, E., and Lee, F. (2002). Dopamine-dependent neurotoxicity of α -synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 8, 600–606. doi: 10.1038/nm0602-600
Katsuse, O., Iseki, E., Marui, W., and Kosaka, K. (2003). Developmental stages of cortical Lewy bodies and their relation to axonal transport blockage in brains of patients with dementia with Lewy bodies. J. Neurol. Sci. 211, 29–35. doi: 10.1016/S0022-510X(03)00037-6
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Kavuri, S., Sivanesan, S., Howell, M. D., Vijayaraghavan, R., and Rajadas, J. (2020). Studies on Parkinson’s-disease-linked genes, brain urea levels and histopathology in rotenone induced Parkinson’s disease rat model. World J. Neurosci. 10, 216–234. doi: 10.4236/wjns.2020.104021
Kell, D. B., and Pretorius, E. (2015). On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr. Biol. 7, 1339–1377. doi: 10.1039/c5ib00158g
Kihara, A., Kabeya, Y., Ohsumi, Y., and Yoshimori, T. (2001). Beclin–phosphatidylinositol 3‐kinase complex functions at the trans‐Golgi network. EMBO Rep. 2, 330–335. doi: 10.1093/embo-reports/kve061
Kim, P. K., Hailey, D. W., Mullen, R. T., and Lippincott-Schwartz, J. (2008). Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. U. S. A. 105, 20567–20574. doi: 10.1073/pnas.0810611105
Kim, M. J., Hwang, G. Y., Cho, M. J., Chi, B. H., Park, S. I., Chang, I. H., et al. (2019). Depletion of NBR1 in urothelial carcinoma cells enhances rapamycin-induced apoptosis through impaired autophagy and mitochondrial dysfunction. J. Cell. Biochem. 120, 19186–19201. doi: 10.1002/jcb.29248
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, Y. J., Yi, Y., Sapp, E., Wang, Y., Cuiffo, B., Kegel, K. B., et al. (2001). Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain- dependent proteolysis. PNAS 98, 12784–12789. doi: 10.1073/pnas.221451398
Kirkin, V., Lamark, T., Sou, Y. S., Bjørkøy, G., Nunn, J. L., Bruun, J. A., et al. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516. doi: 10.1016/j.molcel.2009.01.020
Kishida, K. T., and Klann, E. (2007). Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 9, 233–244. doi: 10.1089/ars.2007.9.ft-8.Sources
Kitada, T., Aakawa, S., Hattori, N., Matsumine, H., Yokochi, M., Mizuno, Y., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Letters Nature 169, 166–169.
Kitazawa, M., Yamasaki, T. R., and LaFerla, F. M. (2004). Microglia as a potential bridge between the amyloid β-peptide and tau. Ann. N. Y. Acad. Sci. 1035, 85–103. doi: 10.1196/annals.1332.006
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Arozena, A. A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Kloda, A., Martinac, B., and Adams, D. H. (2007). Polymodal regulation of NMDA receptor channels. Channels 1, 334–343. doi: 10.4161/chan.5044
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cells 131, 1149–1163. doi: 10.1016/j.cell.2007.10.035
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. doi: 10.1016/S0962-8924(00)01852-3
Kouli, A., Torsney, K. M., and Kuan, W.-L. (2018). Parkinson’s disease: etiology, neuropathology, and pathogenesis Parkinson’s disease: etiology, neuropathology, and pathogenesis. In: Parkinson’s Disease: Pathogenesis and Clinical Aspects. Brisbane, Australia: Codon Publications. 3–26.
Kowall, N. W., Hantraye, C. A. P., Brouillet, E., Beal, M. F., McKee, A. C., and Ferrante, R. J. (2000). MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 11, 211–213. doi: 10.1097/00001756-200001170-00041
Koyama-Honda, I., Itakura, E., Fujiwara, T. K., Koyama-honda, I., Itakura, E., Fujiwara, T. K., et al. (2013). Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 9, 1491–1499. doi: 10.4161/auto.25529
Kraft, C., Peter, M., and Hofmann, K. (2010). Selective autophagy: ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 12, 836–841. doi: 10.1038/ncb0910-836
Kriel, J., and Loos, B. (2019). The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 26, 640–652. doi: 10.1038/s41418-018-0267-4
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Clos, A. L., Jackson, G. R., and Kayed, R. (2011). Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6, 1–14. doi: 10.1186/1750-1326-6-39
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Le, W., and Appel, S. H. (2004). Mutant genes responsible for Parkinson’s disease. Curr. Opin. Pharmacol. 4, 79–84. doi: 10.1016/j.coph.2003.09.005
Lee, S., Chu, K., Park, J., Kang, L., Ko, S., Jung, K., et al. (2006). Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 1118, 199–207. doi: 10.1016/j.brainres.2006.08.035
Lee, I. H., and Finkel, T. (2009). Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 284, 6322–6328. doi: 10.1074/jbc.M807135200
Lee, S. E., Kwon, D., Shin, N., Kong, D., Kim, N. G., Kim, H. Y., et al. (2022). Accumulation of APP-CTF induces mitophagy dysfunction in the iNSCs model of Alzheimer’s disease. Cell Death Discovery 8, 1–10. doi: 10.1038/s41420-021-00796-3
Lee, J. H., McBrayer, M. K., Wolfe, D. M., Haslett, L. J., Kumar, A., Sato, Y., et al. (2015). Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444. doi: 10.1016/j.celrep.2015.07.050
Lee, K. J., Moussa, C. E. H., Lee, Y., and Sung, Y. (2010). Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 169, 344–356. doi: 10.1016/j.neuroscience.2010.04.078
Lee, S., Sato, Y., and Nixon, R. A. (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 31, 7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011
Lee, J. H., Yang, D. S., Goulbourne, C. N., Im, E., Stavrides, P., Pensalfini, A., et al. (2022). Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 25, 688–701. doi: 10.1038/s41593-022-01084-8
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cells 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Leveille, E., Ross, O. A., and Gan-Or, Z. (2021). Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Related Disord. 90, 142–154. doi: 10.1016/j.parkreldis.2021.09.008
Li, M., Khambu, B., Zhang, H., Kang, J. H., Chen, X., Chen, D., et al. (2013). Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 288, 35769–35780. doi: 10.1074/jbc.M113.511212
Li, H., Li, S. H., Yu, Z. X., Shelbourne, P., and Li, X. J. (2001). Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. Rapid Commun. 21, 8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001
Li, R., Lindholm, K., Yang, L. B., Yue, X., Citron, M., Yan, R., et al. (2004). Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. U. S. A. 101, 3632–3637. doi: 10.1073/pnas.0205689101
Lim, J., Lachenmayer, M. L., Wu, S., Liu, W., Kundu, M., Wang, R., et al. (2015). Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective Autophagic clearance of protein aggregates. PLoS Genet. 11, e1004987–e1004928. doi: 10.1371/journal.pgen.1004987
Lo, C. H., and Zeng, J. (2023). Defective lysosomal acidification: a new prognostic marker and therapeutic target for neurodegenerative diseases. Transl. Neurodegen. 12:29. doi: 10.1186/s40035-023-00362-0
Loos, B., du Toit, A., and Hofmeyr, J. H. S. (2014). Defining and measuring autophagosome flux - concept and reality. Autophagy 10, 2087–2096. doi: 10.4161/15548627.2014.973338
Loos, B., Engelbrecht, A. M., Lockshin, R. A., Klionsky, D. J., and Zakeri, Z. (2013). The variability of autophagy and cell death susceptibility: unanswered questions. Autophagy 9, 1270–1285. doi: 10.4161/auto.25560
Lotharius, J., Barg, S., Wiekop, P., Lundberg, C., Raymon, H. K., and Brundin, P. (2002). Effect of mutant α-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem. 277, 38884–38894. doi: 10.1074/jbc.M205518200
Lotharius, J., and Brundin, P. (2002). Pathogenesis of Parkinson’s disease: dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 3, 932–942. doi: 10.1038/nrn983
Lumkwana, D., du Toit, A., Kinnear, C., and Loos, B. (2017). Autophagic flux control in neurodegeneration: progress and precision targeting—where do we stand? Prog. Neurobiol. 153, 64–85. doi: 10.1016/j.pneurobio.2017.03.006
Ma, J., Choi, B. R., Chung, C., Min, S. S., Jeon, W. K., and Han, J. S. (2014). Chronic brain inflammation causes a reduction in GluN2A and GluN2B subunits of NMDA receptors and an increase in the phosphorylation of mitogen-activated protein kinases in the hippocampus. Mol. Brain 7, 1–10. doi: 10.1186/1756-6606-7-33
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cells 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Mak, S. K., McCormack, A. L., Manning-Bog, A. B., Cuervo, A. M., and Di Monte, D. A. (2010). Lysosomal degradation of α-synuclein in vivo. J. Biol. Chem. 285, 13621–13629. doi: 10.1074/jbc.M109.074617
Malagelada, C., Jin, Z. H., Jackson-lewis, V., Przedborski, S., and Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Marceau, K., Ruttle, P. L., Shirtcliff, E. A., Essex, M. J., Susman, E. J., Studies, A., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 57, 742–768. doi: 10.1038/nature14893.The
Mardakheh, F. K., Auciello, G., Dafforn, T. R., Rappoport, J. Z., and Heath, J. K. (2010). Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol. Cell. Biol. 30, 5672–5685. doi: 10.1128/mcb.00878-10
Marsh, T., Kenific, C. M., Suresh, D., Gonzalez, H., Shamir, E. R., Mei, W., et al. (2020). Autophagic degradation of NBR1 restricts metastatic outgrowth during mammary tumor progression. Dev. Cell 52, 591–604.e6. doi: 10.1016/j.devcel.2020.01.025
Martina, J. A., Chen, Y., Gucek, M., and Puertollano, R. (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914. doi: 10.4161/auto.19653
Martínez, G., Duran-Aniotz, C., Cabral-Miranda, F., Vivar, J. P., and Hetz, C. (2017). Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 16, 615–623. doi: 10.1111/acel.12599
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806.somal
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 13, 567–576. doi: 10.1038/nn.2528
Martín-Maestro, P., Gargini, R., A Sproul, A., García, E., Antón, L. C., Noggle, S., et al. (2017). Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer’s disease-associated presenilin 1 mutation. Front. Mol. Neurosci. 10, 1–15. doi: 10.3389/fnmol.2017.00291
Massey, A. C., Kaushik, S., Sovak, G., Kiffin, R., and Cuervo, A. M. (2006). Consequences of the selective blockage of chaperone-mediated autophagy. PNAS 103, 5805–5810. doi: 10.1073/pnas.0507436103
McBrayer, M., and Nixon, R. A. (2013). Lysosome and calcium dysregulation in Alzheimer’s disease: partners in crime. Biochem. Soc. Trans. 41, 1495–1502. doi: 10.1042/BST20130201
McNaught, K. S. P., Bj??rklund, L. M., Belizaire, R., Isacson, O., Jenner, P., Olanow, C. W., et al. (2002). Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport 13, 1437–1441. doi: 10.1097/00001756-200208070-00018
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Meredith, G. E., Totterdell, S., Petroske, E., Santa Cruz, K., Callison, R. C., and Lau, Y. S. (2002). Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson’s disease. Brain Res. 956, 156–165. doi: 10.1016/S0006-8993(02)03514-X
Miller, B. R., Dorner, J. L., Bunner, K. D., Gaither, T. W., Klein, E. L., Barton, S. J., et al. (2012). Up-regulation of GLT1 reverses the deficit in cotcally evoked striatal ascorbate efflux in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 121, 629–638. doi: 10.1111/j.1471-4159.2012.07691.x
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. doi: 10.1101/gad.1599207.eralize
Mizushima, N., Kuma, A., Kobayashi, Y., Yamamoto, A., Matsubae, M., Takao, T., et al. (2003). Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116, 1679–1688. doi: 10.1242/jcs.00381
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Mizushima, N., Noda, T., and Ohsumi, Y. (1999). Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 18, 3888–3896. doi: 10.1093/emboj/18.14.3888
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., and Ohsumi, Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111. doi: 10.1091/mbc.E03
Montoya, A., Pelletier, M., Menear, M., Duplessis, E., Richer, F., and Lepage, M. (2006). Episodic memory impairment in Huntington’s disease: A meta-analysis. Neuropsychologia 44, 1984–1994. doi: 10.1016/j.neuropsychologia.2006.01.015
Morfini, G. A., You, Y.-M., Pollema, S. L., Kaminska, A., Liu, K., Yoshioka, K., et al. (2009). Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 12, 864–871. doi: 10.1038/nn.2346
Mori, F., Nishie, M., Kakita, A., Yoshimoto, M., Takahashi, H., and Wakabayashi, K. (2006). Relationship among α-synuclein accumulation, dopamine synthesis, and neurodegeneration in Parkinson disease substantia nigra. J. Neuropathol. Exp. Neurol. 65, 808–815. doi: 10.1097/01.jnen.0000230520.47768.1a
Morselli, E., Mariño, G., Bennetzen, M. V., Eisenberg, T., Megalou, E., Schroeder, S., et al. (2011). Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 192, 615–629. doi: 10.1083/jcb.201008167
Moscat, J., and Diaz-Meco, M. T. (2009). p62 at the crossroads of autophagy, apoptosis, and cancer. Cells 137, 1001–1004. doi: 10.1016/j.cell.2009.05.023
Moskal, N., Riccio, V., Bashkurov, M., Taddese, R., Datti, A., Lewis, P. N., et al. (2020). ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 11, 1–14. doi: 10.1038/s41467-019-13781-3
Mucke, L., Masliah, E., Yu, G., Mallory, M., Rockenstein, E., Tatsuno, G., et al. (2000). High-level neuronal expression of Aβ1–42in Wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, 1–18. doi: 10.1101/cshperspect.a006338
Nagatsu, T., Nakashima, A., Ichinose, H., and Kobayashi, K. (2019). Human tyrosine hydroxylase in Parkinson’s disease and in related disorders. J. Neu. Tran. 126, 397–409. doi: 10.1007/s00702-018-1903-3
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Näslund, J., Haroutunian, V., Mohs, R., Davis, K. L., Davies, P., Greengard, P., et al. (2000). Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. J. Am. Med. Assoc. 283, 1571–1577. doi: 10.1001/jama.283.12.1571
Nilsson, P., Loganathan, K., Sekiguchi, M., Matsuba, Y., Hui, K., Tsubuki, S., et al. (2013). Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69. doi: 10.1016/j.celrep.2013.08.042
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091. doi: 10.1242/jcs.019265
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613.Amyloid
Oh, E. S., Savonenko, A. V., King, J. F., Fangmark, S. M., Rudow, G. L., Xu, G., et al. (2009). Amyloid precursor protein increases cortical neuron size in transgenic mice. Neurobiol. Aging 30, 1238–1244. doi: 10.1016/j.neurobiolaging.2007.12.024
Okatsu, K., Uno, M., Koyano, F., Go, E., Kimura, M., Oka, T., et al. (2013). A dimeric pink1-containing complex on depolarized mitochondria stimulates parkin recruitment. J. Biol. Chem. 288, 36372–36384. doi: 10.1074/jbc.M113.509653
Orr, A. L., Li, S., Wang, C., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. Rapid Commun. 28, 2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008
Padman, B. S., Nguyen, T. N., Uoselis, L., Skulsuppaisarn, M., Nguyen, L. K., and Lazarou, M. (2019). LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 10, 1–13. doi: 10.1038/s41467-019-08335-6
Page, A., Shepherd, D., and Mudher, A. (2010). Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 120, 593–604. doi: 10.1007/s00401-010-0716-8
Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 860–864. doi: 10.1038/nature05853
Pankiv, S., Alemu, E. A., Brech, A., Bruun, J. A., Lamark, T., Øvervatn, A., et al. (2010). FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end - directed vesicle transport. J. Cell Biol. 188, 253–269. doi: 10.1083/jcb.200907015
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
Parone, P. A., Da, S., Tondera, D., Mattenberger, Y., James, D. I., and Martinou, J.-C. (2008). Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 3, 1–9. doi: 10.1371/journal.pone.0003257
Patel, N. S., Paris, D., Mathura, V., Quadros, A. N., Crawford, F. C., and Mullan, M. J. (2005). Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflammation 2, 1–10. doi: 10.1186/1742-2094-2-9
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cells 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pechmann, S., Levy, E. D., Tartaglia, G.-G., and Vendruscolo, M. (2009). Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc. Natl. Acad. Sci. 106, 10159–10164. doi: 10.1073/pnas.0812414106
Perciavalle, R. M., Stewart, D. P., Koss, B., Lynch, J., Milasta, S., Bathina, M., et al. (2012). Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 14, 575–583. doi: 10.1038/ncb2488
Perez, R. G., Waymire, J. C., Lin, E., Liu, J. J., Guo, F., and Zigmond, M. J. (2002). A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002
Pietrocola, F., Lachkar, S., Enot, D. P., Niso-Santano, M., Bravo-San Pedro, J. M., Sica, V., et al. (2015). Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 22, 509–516. doi: 10.1038/cdd.2014.215
Pineda-Ramírez, N., Alquisiras-burgos, I., Ortiz-plata, A., Ruiz-Tachiquin, M.-E., Espinoza-Rojo, M., and Aguilera, P. (2020). Resveratrol activates neuronal autophagy through AMPK in the ischemic brain. Mol. Neurobiol. 57, 1055–1069. doi: 10.1007/s12035-019-01803-6
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., and Rizzuto, R. (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418. doi: 10.1038/onc.2008.308
Polito, V. A., Li, H., Martini-Stoica, H., Wang, B., Yang, L., Xu, Y., et al. (2014). Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 6, 1142–1160. doi: 10.15252/emmm.201303671
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pupyshev, A. B., Tikhonova, M. A., Akopyan, A. A., Tenditnik, M. V., Dubrovina, N. I., and Korolenko, T. A. (2019). Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 177, 1–11. doi: 10.1016/j.pbb.2018.12.005
Quintanilla, R. A., Matthews-Roberson, T. A., Dolan, P. J., and Johnsion, G. V. W. (2009). Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J. Biol. Chem. 284, 18754–18766. doi: 10.1074/jbc.M808908200
Rabinovitch, R. C., Samborska, B., Faubert, B., Ma, E. H., Gravel, S. P., Andrzejewski, S., et al. (2017). AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. CellReports. 21, 1–9. doi: 10.1016/j.celrep.2017.09.026
Rabinowitz, J. D., and White, E. (2010). Autophagy and metabolism. Science 330, 1344–1348. doi: 10.1126/science.1193497
Rana, A., Oliveira, M. P., Khamoui, A. V., Aparicio, R., Rera, M., Rossiter, H. B., et al. (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8, 1–14. doi: 10.1038/s41467-017-00525-4
Rao, V. K., Carlson, E. A., and Yan, S. S. (2014). Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta Mol. basis Dis. 1842, 1267–1272. doi: 10.1016/j.bbadis.2013.09.003
Ravikumar, B., Acevedo-Arozena, A., Imarisio, S., Berger, Z., Vacher, C., O’Kane, C. J., et al. (2005). Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 37, 771–776. doi: 10.1038/ng1591
Ravikumar, B., Sarkar, S., Davies, J. E., Futter, M., Garcia-Arencibia, M., Green-Thompson, Z. W., et al. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435. doi: 10.1152/physrev.00030.2009
Ray, B., Banerjee, P. K., Greig, N. H., and Lahiri, D. K. (2010). Memantine treatment decreases levels of secreted Alzheimer’s amyloid precursor protein (APP) and amyloid beta (Aβ) peptide in the human neuroblastoma cells. Neurosci. Lett. 470, 1–5. doi: 10.1016/j.neulet.2009.11.016
Reitz, C., Brayne, C., and Mayeux, R. (2012). Epidemiology of Alzheimer disease. Nat Rev Neurol 7, 137–152. doi: 10.1038/nrneurol.2011.2.Epidemiology
Rideout, H. J., Lang-Rollin, I., and Stefanis, L. (2004). Involvement of macroautophagy in the dissolution of neuronal inclusions. Int. J. Biochem. Cell Biol. 36, 2551–2562. doi: 10.1016/j.biocel.2004.05.008
Rolland, S. G., Motori, E., Memar, N., Hench, J., Frank, S., Winklhofer, K. F., et al. (2013). Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. PNAS 110. doi: 10.1073/pnas.1303872110
Rose, C., Menzies, F. M., Renna, M., Acevedo-Arozena, A., Corrochano, S., Sadiq, O., et al. (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 19, 2144–2153. doi: 10.1093/hmg/ddq093
Russell, R. C., Tian, Y., Yuan, H., Park, H. W., Chang, Y. Y., Kim, J., et al. (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15, 741–750. doi: 10.1038/ncb2757
Sahani, M. H., Itakura, E., and Mizushima, N. (2014). Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10, 431–441. doi: 10.4161/auto.27344
Salminen, A., Kaarniranta, K., Haapasalo, A., Hiltunen, M., Soininen, H., and Alafuzoff, I. (2012). Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 96, 87–95. doi: 10.1016/j.pneurobio.2011.11.005
Sancak, Y., Bar-Peled, L., Zoncu, R., Markhard, A. L., Nada, S., and Sabatini, D. M. (2010). Regulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cells 141, 290–303. doi: 10.1016/j.cell.2010.02.024
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-peled, L., et al. (2008). The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. doi: 10.1126/science.1157535
Sardiello, M., Palmieri, M., di Ronza, A., Medina, D. L., Valenza, M., Gennarino, V. A., et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. doi: 10.1126/science.1174447
Sarkar, S. (2013). Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 41, 1103–1130. doi: 10.1042/BST20130134
Sarkar, S., Floto, R. A., Berger, Z., Imarisio, S., Cordenier, A., Pasco, M., et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111. doi: 10.1083/jcb.200504035
Sarkar, C., Zhao, Z., Aungst, S., Sabirzhanov, B., Faden, A. I., and Lipinski, M. M. (2014). Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 10, 2208–2222. doi: 10.4161/15548627.2014.981787
Schapira, A. H. V., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x
Scherz-Shouval, R., and Elazar, Z. (2007). ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427. doi: 10.1016/j.tcb.2007.07.009
Schulte, J., and Littleton, J. T. (2011). The biological function of the huntingtin protein and its relevance to Huntington’s disease pathology. Curr. Trends Neurol. 5, 65–78.
Senechal, Y., Kelly, P. H., and Dev, K. K. (2008). Amyloid precursor protein knockout mice show age-dependent deficits in passive avoidance learning. Bahav. Brain Res. 186, 126–132. doi: 10.1016/j.bbr.2007.08.003
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, 1–24. doi: 10.1101/cshperspect.a006189
Settembre, C., di Malta, C., Polito, V. A., Arencibia, M. G., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA- type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Sharoar, M. G., Hu, X., Ma, X. M., Zhu, X., and Yan, R. (2019). Sequential formation of different layers of dystrophic neurites in Alzheimer’s brains. Mol. Psychiatry 24, 1369–1382. doi: 10.1038/s41380-019-0396-2
Shen, H. M., and Mizushima, N. (2014). At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 39, 61–71. doi: 10.1016/j.tibs.2013.12.001
Shi, W. Y., Xiao, D., Wang, L., Dong, L. H., Yan, Z. X., Shen, Z. X., et al. (2012). Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Disease. 3, e275–e279. doi: 10.1038/cddis.2012.13
Shi, R. Y., Zhu, S. H., Li, V., Gibson, S. B., Xu, X. S., and Kong, J. M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Shimoji, M., Zhang, L., Mandir, A. S., Dawson, V. L., and Dawson, T. M. (2005). Absence of inclusion body formation in the MPTP mouse model of Parkinson’s disease. Mol. Brain Res. 134, 103–108. doi: 10.1016/j.molbrainres.2005.01.012
Shirendeb, U. P., Calkins, M. J., Manczak, M., Anekonda, V., Dufour, B., McBride, J. L., et al. (2012). Mutant Huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 21, 406–420. doi: 10.1093/hmg/ddr475
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Song, L., McMackin, M., Nguyen, A., and Cortopassi, G. (2017). Parkin deficiency accelerates consequences of mitochondrial DNA deletions and parkinsonism. Neurobiol. Dis. 100, 30–38. doi: 10.1016/j.nbd.2016.12.024
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One 5, 1–8. doi: 10.1371/journal.pone.0009979
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. doi: 10.1038/ncb2979
Sumpter, R., and Levine, B. (2010). Autophagy and innate immunity: triggering, targeting and tuning. Semin. Cell Dev. Biol. 21, 699–711. doi: 10.1016/j.semcdb.2010.04.003
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson’s disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1016/bs.pbr.2020.02.005
Sveinbjornsdottir, S. (2016). The clinical symptoms of Parkinson’s disease. J. Neurochem. 139, 318–324. doi: 10.1111/jnc.13691
Swart, C., Haylett, W., Kinnear, C., Johnson, G., Bardien, S., and Loos, B. (2014). Neurodegenerative disorders: dysregulation of a carefully maintained balance? Exp. Gerontol. 58, 279–291. doi: 10.1016/j.exger.2014.09.003
Sweeney, P., Park, H., Baumann, M., Dunlop, J., Frydman, J., Kopito, R., et al. (2017). Protein misfolding in neurodegenerative diseases: implications and strategies. Transl. Neurodegen. 6, 6–13. doi: 10.1186/s40035-017-0077-5
Takuma, K., Yan, S.S.Du, Stern, D.M., and Yamada, K. (2005). Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J. Pharmacol. Sci. 97, 312–316. doi: 10.1254/jphs.CPJ04006X
Tanaka, K., and Matsuda, N. (2014). Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta, Mol. Cell Res. 1843, 197–204. doi: 10.1016/j.bbamcr.2013.03.012
Tang, F. L., Erion, J. R., Tian, Y., Liu, W., Yin, D. M., Ye, J., et al. (2015). VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for -Synuclein degradation and prevention of pathogenesis of Parkinson's disease. J. Neurosci. 35, 10613–10628. doi: 10.1523/JNEUROSCI.0042-15.2015
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518. doi: 10.1016/j.biocel.2004.05.009
Tebbenkamp, A. T. N., Crosby, K. W., Siemienski, Z. B., Brown, H. H., Golde, T. E., Borchelt, D. R., et al. (2012). Analysis of proteolytic processes and enzymatic activities in the generation of huntingtin N-terminal fragments in an HEK293 cell model. PLoS One 7, 1–10. doi: 10.1371/journal.pone.0050750
Tebbenkamp, A. T. N., Green, C., Xu, G., Denovan-Wright, E. M., Rising, A. C., Fromholt, S. E., et al. (2011). Transgenic mice expressing caspase-6-derived N-terminal fragments of mutant huntingtin develop neurologic abnormalities with predominant cytoplasmic inclusion pathology composed largely of a smaller proteolytic derivative. Hum. Mol. Genet. 20, 2770–2782. doi: 10.1093/hmg/ddr176
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C., and Blenis, J. (2003). Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268. doi: 10.1016/S0960-9822(03)00506-2
Theofilas, P., Ehrenberg, A. J., Nguy, A., Thackrey, J. M., Dunlop, S., Mejia, M. B., et al. (2018). Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol. Aging 61, 1–12. doi: 10.1016/j.neurobiolaging.2017.09.007
Timmins, J. M., Ozcan, L., Seimon, T. A., Li, G., Malagelada, C., Backs, J., et al. (2009). Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Invest. 119, 2925–2941. doi: 10.1172/JCI38857
Twig, G., Elorza, A., Molina, A. J. A., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Ulusoy, A., Björklund, T., Buck, K., and Kirik, D. (2012). Dysregulated dopamine storage increases the vulnerability to α-synuclein in nigral neurons. Neurobiol. Dis. 47, 367–377. doi: 10.1016/j.nbd.2012.05.012
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson's disease caused by mutations inPINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Verghese, P. B., Castellano, J. M., and Holtzman, D. M. (2011). Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 10, 241–252. doi: 10.1016/S1474-4422(10)70325-2
Vest, R. T., Chou, C. C., Zhang, H., Haney, M. S., Li, L., Laqtom, N. N., et al. (2022). Small molecule C381 targets the lysosome to reduce inflammation and ameliorate disease in models of neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 119, 1–12. doi: 10.1073/pnas.2121609119
Vicencio, J. M., Ortiz, C., Criollo, A., Jones, A. W. E., Kepp, O., Galluzzi, L., et al. (2009). The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 16, 1006–1017. doi: 10.1038/cdd.2009.34
Vila, M., Vukosavic, S., Jackson-lewis, V., Neystat, M., Jakowec, M., and Przedborski, S. (2000). α-Synuclein up-regulation in substantia Nigra dopaminergic neurons following Administration of the Parkinsonian Toxin MPTP. J. Neurochem. 74, 721–729. doi: 10.1046/j.1471-4159.2000.740721.x
Villegas, R., Martinez, N. W., Lillo, J., Pihan, P., Hernandez, D., Twiss, J. L., et al. (2014). Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 34, 7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014
Vinther-Jensen, T., Larsen, I. U., Hjermind, L. E., Budtz-Jørgensen, E., Nielsen, T. T., Nørremølle, A., et al. (2014). A clinical classification acknowledging neuropsychiatric and cognitive impairment in Huntington’s disease. Orphanet J. Rare Dis. 9, 1–9. doi: 10.1186/s13023-014-0114-8
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-Synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577. doi: 10.1097/00005072-198511000-00003
Wakabayashi, K., Mori, F., and Takahashi, H. (2006). Progression patterns of neuronal loss and Lewy body pathology in the substantia nigra in Parkinson’ s disease. Parkinson Related Disord 12, 92–98. doi: 10.1016/j.parkreldis.2006.05.028
Wakabayashi, K., Tanji, K., Odagiri, S., Miki, Y., Mori, F., and Takahashi, H. (2013). The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 47, 495–508. doi: 10.1007/s12035-012-8280-y
Wang, P., Jiang, L., Zhou, N., Zhou, H., Liu, H., Zhao, W., et al. (2018). Resveratrol ameliorates autophagic flux to promote functional recovery in rats after spinal cord injury. Oncotarget 9, 8427–8440. doi: 10.18632/oncotarget.23877
Wang, N., Ma, Q., Peng, P., Yu, Y., Xu, S., Wang, G., et al. (2020). Autophagy and ubiquitin-proteasome system coordinate to regulate the protein quality control of neurodegenerative disease-associated DCTN1. Neurotox. Res. 37, 48–57. doi: 10.1007/s12640-019-00113-y
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wellington, C. L., Ellerby, L. M., Gutekunst, C., Rogers, D., Warby, S., Graham, R. K., et al. (2002). Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci. 22, 7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002
Whitworth, A. J., Theodore, D. A., Greene, J. C., Beneš, H., Wes, P. D., and Pallanck, L. J. (2005). Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 102, 8024–8029. doi: 10.1073/pnas.0501078102
Wills, J., Jones, J., Haggerty, T., Duka, V., Joyce, J. N., and Sidhu, A. (2010). Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp. Neurol. 225, 210–218. doi: 10.1016/j.expneurol.2010.06.017
Winklhofer, K. F., and Haass, C. (2010). Mitochondrial dysfunction in Parkinson’s disease. Molec Basis Disease 1802, 216–231. doi: 10.1111/jnc.13731
Wu, F., Xu, H. D., Guan, J. J., Hou, Y. S., Gu, J. H., Zhen, X. C., et al. (2015). Rotenone impairs autophagic flux and lysosomal functions in Parkinson’s disease. Neuroscience 284, 900–911. doi: 10.1016/j.neuroscience.2014.11.004
Wu, S., Zhou, F., Zhang, Z., and Xing, D. (2011). Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission – fusion proteins. FEBS J. 278, 941–954. doi: 10.1111/j.1742-4658.2011.08010.x
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cells 124, 471–484. doi: 10.1016/j.cell.2006.01.016
Xiao, Y., Ma, B., McElheny, D., Parthasarathy, S., Long, F., Hoshi, M., et al. (2015). Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 22, 499–505. doi: 10.1038/nsmb.2991
Xie, R., Nguyen, S., McKeehan, W. L., and Liu, L. (2010). Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 11:89. doi: 10.1186/1471-2121-11-89
Xu, L., Nguyen, J. V., Lehar, M., Menon, A., Rha, E., Arena, J., et al. (2016). Repetitive mild traumatic brain injury with impact acceleration in the mouse: multifocal axonopathy, neuroinflammation, and neurodegeneration in the visual system repetitive mild traumatic brain injury with impact acceleration in the mouse: multifocal. Exp. Neurol. 275, 436–449. doi: 10.1016/j.expneurol.2014.11.004
Yagi, M., Toshima, T., Amamoto, R., Do, Y., Hirai, H., Setoyama, D., et al. (2021). Mitochondrial translation deficiency impairs NAD+ −mediated lysosomal acidification. EMBO J. 40, 1–17. doi: 10.15252/embj.2020105268
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Yang, W., and Tiffany-Castiglioni, E. (2005). The bipyridyl herbicide paraquat produces oxidative stress-mediated toxicity in human neuroblastoma SH-SY5Y cells: relevance to the dopaminergic pathogenesis. J. Toxicol. Environ. Health 68, 1939–1961. doi: 10.1080/15287390500226987
Yu, J., Chang, R. C., and Tan, L. (2009). Calcium dysregulation in Alzheimer’s disease: from mechanisms to therapeutic opportunities. Prog. Neurobiol. 89, 240–255. doi: 10.1016/j.pneurobio.2009.07.009
Yu, Z. X., Li, S. H., Evans, J., Pillarisetti, A., Li, H., and Li, X. J. (2003). Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington’s disease. J. Neurosci. 23, 2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003
Yu, L., McPhee, C. K., Zheng, L., Mardones, G. A., Rong, Y., Peng, J., et al. (2010). Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946. doi: 10.1038/nature09076
Zeng, J., Acin-Perez, R., Assali, E. A., Martin, A., Brownstein, A. J., Petcherski, A., et al. (2023). Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease. Communications 14, 1–17. doi: 10.1038/s41467-023-38165-6
Zhang, Y., He, X., Wu, X., Lei, M., Wei, Z., Zhang, X., et al. (2017). Rapamycin upregulates glutamate transporter and IL-6 expression in astrocytes in a mouse model of Parkinson’s disease. Cell Death Dis. 8, 1–13. doi: 10.1038/cddis.2016.491
Zhang, J., and Ney, P. A. (2009). Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 16, 939–946. doi: 10.1038/cdd.2009.16
Zhi, L., Qin, Q., Muqeem, T., Seifert, E. L., Liu, W., Zheng, S., et al. (2019). Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol. Aging 75, 1–10. doi: 10.1016/j.neurobiolaging.2018.10.025
Zhou, Z., Kim, J., Insolera, R., Peng, X., Fink, D. J., and Mata, M. (2011). Rho GTPase regulation of α-synuclein and VMAT2: implications for pathogenesis of Parkinson’s disease. Mol. Cell. Neurosci. 48, 29–37. doi: 10.1016/j.mcn.2011.06.002
Zoncu, R., Bar-Peled, L., Efeyan, A., Wang, S., Sancak, Y., and Sabatini, D. M. (2011). mTORC1 Senses Lysosomal Amino Acids. Science 334, 678–683. doi: 10.1109/IVS.2006.1689629
Keywords: autophagy, mitochondrial function, lysosomes, mitophagy, neurodegenerative diseases, proteostasis
Citation: de Wet S, Theart R and Loos B (2023) Cogs in the autophagic machine—equipped to combat dementia-prone neurodegenerative diseases. Front. Mol. Neurosci. 16:1225227. doi: 10.3389/fnmol.2023.1225227
Edited by:
Ramesh Kandimalla, Indian Institute of Chemical Technology (CSIR), IndiaReviewed by:
Chih Hung Lo, Brigham and Women's Hospital and Harvard Medical School, United StatesCopyright © 2023 de Wet, Theart and Loos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Loos, Ymxvb3NAc3VuLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.