 Min Huang
Min Huang Qi Qi1,2†
Qi Qi1,2† Tao Xu
Tao Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 09 February 2023
Sec. Pain Mechanisms and Modulators
Volume 16 - 2023 | https://doi.org/10.3389/fnmol.2023.1128974
This article is part of the Research Topic Molecular Mechanism of Neuroimmune Modulation and Synaptic Plasticity in Acute and Chronic Pain View all 19 articles
Autism spectrum disorder (ASD) includes a group of multifactorial neurodevelopmental disorders characterized by impaired social communication, social interaction, and repetitive behaviors. Several studies have shown an association between cases of ASD and mutations in the genes of SH3 and multiple ankyrin repeat domain protein 3 (SHANK3). These genes encode many cell adhesion molecules, scaffold proteins, and proteins involved in synaptic transcription, protein synthesis, and degradation. They have a profound impact on all aspects of synaptic transmission and plasticity, including synapse formation and degeneration, suggesting that the pathogenesis of ASD may be partially attributable to synaptic dysfunction. In this review, we summarize the mechanism of synapses related to Shank3 in ASD. We also discuss the molecular, cellular, and functional studies of experimental models of ASD and current autism treatment methods targeting related proteins.
Autism spectrum disorder (ASD) is a term widely used to describe multiple multifactorial neurodevelopmental (Sharma et al., 2018) and grouping disorders (Becerra et al., 2014), including autism (Jiang and Ehlers, 2013), Asperger’s syndrome (Berkel et al., 2018), and no other general developmental disorders (Thapar et al., 2017). However, the updated diagnostic criteria for ASD mainly focus on two core areas: social communication disorder and alternative interests/repetitive behaviors (Kales et al., 2015; Kodak and Bergmann, 2020; Srivastava et al., 2021).
The prevalence of ASD has increased steadily (Kincaid et al., 2017; Hyman et al., 2020). Genetic factors, parental history of mental illness, premature birth, and oxygen exposure to psychotropic drugs or pesticides are associated with a higher risk of ASD (Kincaid et al., 2017; Masi et al., 2017; Crump et al., 2021). Various scales, such as the Childhood Autism Rating Scale (CARS; Moon et al., 2019), Childhood Autism Spectrum Disorder Observation (ASD-OC; Neal et al., 2012), and Developmental, Dimensional, and Diagnostic Interview (3Di; Randall et al., 2018), can be used to assess abnormal behaviors and symptoms in ASD efficiently. Nearly 75% of patients with ASD have coexisting mental illnesses or complications, including ADHD, epilepsy, anxiety, bipolar disorder, depression, Tourette’s syndrome, and inpatient diseases (Tuchman et al., 2010; Doshi-Velez et al., 2014).
Pharmacological and non-pharmacological interventions are used for ASD treatment (Sharma et al., 2018). Existing drug treatments, including psychostimulants, atypical antipsychotics, antidepressants, and α-2 adrenergic receptor agonists, can partially relieve the core symptoms of ASD and control comorbidities (Aman, 2004; Muskens et al., 2017). Moreover, non-pharmacological interventions, including music therapy and cognitive and social behavioral therapies, improve the social interaction and oral communication of patients with ASD (Sharma et al., 2018; Esposito et al., 2020). The combined use of vitamins, herbs, nutritional supplements, and behavioral therapy improves ASD symptoms; however, the specific efficacy needs further investigation (Guang et al., 2018; Sharma et al., 2018).
Shank (also known as ProSAP) protein has three main subtypes, Shank1, Shank2, and Shank3, with similar structural domains: N-terminal ankyrin repeats, an Src homology 3 (SH3) domain, a PSD-95/Discs large/ZO-1 (PDZ) domain, an extended proline-rich region, and a sterile alpha motif (SAM) domain (Saupe et al., 2011; Costales and Kolevzon, 2015; Varghese et al., 2017; Sgritta et al., 2019).
As the main scaffold protein of excitatory synaptic PSD, Shank protein interacts with more than 30 types of postsynaptic proteins through these domains. These domains are critical for synapse formation, glutamate receptor transport, and neuronal signal transmission (Monteiro and Feng, 2017; Orefice et al., 2019). Shank3 is encoded by the SHANK3 gene located on chromosome 22q13.3. The 22q13.3 deletion syndrome [also known as Phelan-McDermid syndrome (PMS)] is found and characterized by marked developmental deterioration (Phelan et al., 2001; Phelan and McDermid, 2012). More than 50% of patients with PMS have identified SHANK3 abnormalities, including complete deletions, insertions, splicing mutations, and point mutations (Durand et al., 2007; Gauthier et al., 2009; Boccuto et al., 2013). Mutations in SHANK3 are estimated to occur in 1–2% of people with autism and intellectual disabilities, while mutations in SHANK1 and SHANK2 are less common (Boccuto et al., 2013; Leblond et al., 2014; Roberts et al., 2014). Shank3 deficient neurons showed reduced overall expression levels of PSD protein, including GKAP, Homer1b/c, AMPAR subunit GluA1, and NMDAR subunit NR2A (Wang et al., 2011; Peça et al., 2011a). The disruption of the interaction and connection between Shank3, GKAP, and Homer1b/c may cause the redistribution and disruption of the activity-dependent GluA1 subunit. Consequently, posterior tonicity increases, and hippocampal LTP decreases (Wang et al., 2011). In patients with PMS, neurons induced by pluripotent stem cells (iPSCs) showed significantly impaired NMDAR and AMPAR-mediated synaptic transmission (Shcheglovitov et al., 2013; Speed et al., 2015; Qin et al., 2018; Tatavarty et al., 2020). Shank3-deficient mice showed repetitive self-harm and social interaction defects. In addition, mEPSC frequency and amplitude were significantly reduced, indicating a reduction in the number of functional synapses and a decrease in the postsynaptic responses of available synapses (Peça et al., 2011b; Lee et al., 2021). In contrast, overexpression of Shank3 resulted in a sharp increase in the amplitude of AMPAR-mediated NMDAR and NMDAR-mediated EPSC and a high frequency of AMPAR-mediated mEPSC (Arons et al., 2012).
The prevalence of ASD in boys is four to five times that in girls (Christensen et al., 2018). Patients with genetic and chromosomal diseases tend to show more symptoms of ASD (Ogata et al., 2014). About 10% of children with ASD also have Down syndrome or Fragile X syndrome (DiGuiseppi et al., 2010; Auerbach et al., 2021). The psychiatric history of biological parents, especially the history of schizophrenia and affective disorder, is associated with an increased incidence of ASD (Jokiranta et al., 2013). Fetal exposure to pesticides is associated with a decrease in infant weight and length, delay in psychomotor development, and high risk of autism (Landrigan, 2010). In addition, epidemiological studies have shown that exposure of pregnant mothers to viral or bacterial infections, especially in the first trimester or middle of pregnancy, promotes the mother’s immune activation (MIA) and increases the risks of children’s neuropsychiatric disease, including ASD (Estes and McAllister, 2016). MIA is related to an increase in neuroinflammatory cytokines, abnormal expression of synaptic proteins, and abnormal development of synaptic connections, all of which may contribute to the pathophysiology of ASD (Pendyala et al., 2017). Consuming psychotropic drugs during pregnancy is considered a risk factor for autism. Several studies have demonstrated that prescribing antidepressants to pregnant women increases the risk of autism moderately (Gidaya et al., 2014; Siu and Weksberg, 2017). Certain limitations, including failure to carefully adjust the mother’s psychiatric history (Hoover and Kaufman, 2018; Bai et al., 2020), genetic susceptibility to ASD (Pardo and Eberhart, 2007; Wei et al., 2021), and variable molecular and clinical effects of different antidepressants (McCarthy et al., 2016; Soler et al., 2018; Johannessen et al., 2019), may lead to differences in findings of current and repeated studies. However, a recent 12-year study (including full-term live births by mothers who received antidepressants during pregnancy) concluded that the period of antidepressant use did increase the risk of autism in children (Boukhris et al., 2016; Modabbernia et al., 2017). A previous pilot study evaluated medical screening results at the time of referral for children and adolescents with different mental disorders. They found newly developed somatic functions in 56% of the subjects (Muskens et al., 2015). These findings include a wide range of medical problems, including weight and height problems (Dhaliwal et al., 2019), high thyroid hormone levels (Needham et al., 2021), dyslipidemia (Panjwani et al., 2020), anemia (Wiegersma et al., 2019), vitamin D and B12 deficiency (Raghavan et al., 2018; Wang Z. et al., 2020), and malformations (Grafodatskaya et al., 2010). Some of these results require consultation with other medical experts. In contrast, other results directly impact daily medical practice, such as adjusting psycho-drug logic therapy and participating in overweight prevention plans (Vannucchi et al., 2014; Mansouri et al., 2020). These studies contribute to improving people’s understanding of the relationship between somatic and mental symptoms in developmental disorders. It points to common genetic pathways and other potential mechanisms that may be involved (Dovey et al., 2019; Frolli et al., 2021). In addition, the simultaneous assessment of medical and psychiatric disorders may be of great value. Clinicians can then relate somatic functions to different diagnostic considerations for medical and psychological intervention (Rodin et al., 2021).
The SHANK gene was first found to be related to neurodevelopmental disorders in the study of PMS (Costales and Kolevzon, 2015; Frank, 2021). PMS is a neurodevelopmental disorder caused by the deletion of 22q13.3, characterized by autistic behavior, hypotonia, and continuous or even nonexistent speech (Burdeus-Olavarrieta et al., 2021). Genome rearrangements in patients with PMS include deletions, chromosomal, mesenchymal deletions, and unbalanced translocations (Bonaglia et al., 2011; Leblond et al., 2014; Tammimies, 2019). In almost all reported cases (Wilson et al., 2003; Wilson et al., 2008), the loss of Shank3 was observed. These cases support the theory that PMS symptoms are caused by the loss of the SHANK3 haplotype (Phelan and McDermid, 2012) or complete SHANK3 gene on chromosome 22 (Grabrucker et al., 2011a,b). In addition to the symptoms of autism, genetic screening of patients with ASD who have not yet been diagnosed with PMS also revealed many SHANK3 mutations (Durand et al., 2007; Moessner et al., 2007; Gauthier et al., 2009; Boccuto et al., 2013; Leblond et al., 2014). These mutations include small deletions, nonsense mutations, breakpoints, and missense mutations. A meta-analysis study found that mutations or disruptions in the SHANK gene family accounted for about 1–2% of all patients with ASD (Leblond et al., 2014; Zhou et al., 2016). The degree of related mutations and cognitive impairment between SHANK1-3 also differs: patients with SHANK3 mutations suffer more cognitive impairment than those with SHANK1 or SHANK2 mutations (Qin et al., 2022). In addition, patients with SHANK3 mutations have severe cognitive deficits (Chevallier et al., 2012; Zhou et al., 2016). These findings together indicate that the common neurobiological effects shared by all members of the SHANK gene family may be related to the pathophysiology of ASD. Furthermore, the degree of cognitive impairment in ASD may be due to mutations in the SHANK family members, the most significant being SHANK3 (Chevallier et al., 2012; Baum et al., 2015; Monteiro and Feng, 2017; Banker et al., 2021). The difference in the severity of symptoms can be determined by the mutation of the specific SHANK gene (Jiang and Ehlers, 2013; Gong and Wang, 2015; Li and Pozzo-Miller, 2020). The results of the study suggest that mutations in the SHANK3 gene are the primary cause of ASD and that the expression of the other two remaining SHANK subtypes can (or cannot) compensate for its loss (Mashayekhi et al., 2021; Salomaa et al., 2021). Family mutations are comprehensive and should be screened in clinical practice. Many human SHANK3 mutations map to exon 21 and are associated with moderate to severe intellectual disability (D'Antoni et al., 2014; Mossa et al., 2021; Purushotham et al., 2022). Mutations in the pro-domain region of exon 21 were not associated with altered pathophysiology (Shi et al., 2017; Moutin et al., 2021). One possible explanation is that exon 21 is present in most SHANK3 isotype surrogates (Moutin et al., 2021). Therefore, mutations in this exon may have more severe effects (Speed et al., 2019). Given the nature of these genes in ASD, it becomes imperative to understand their usual role in synapses and how mutations disrupt them (Figure 1).
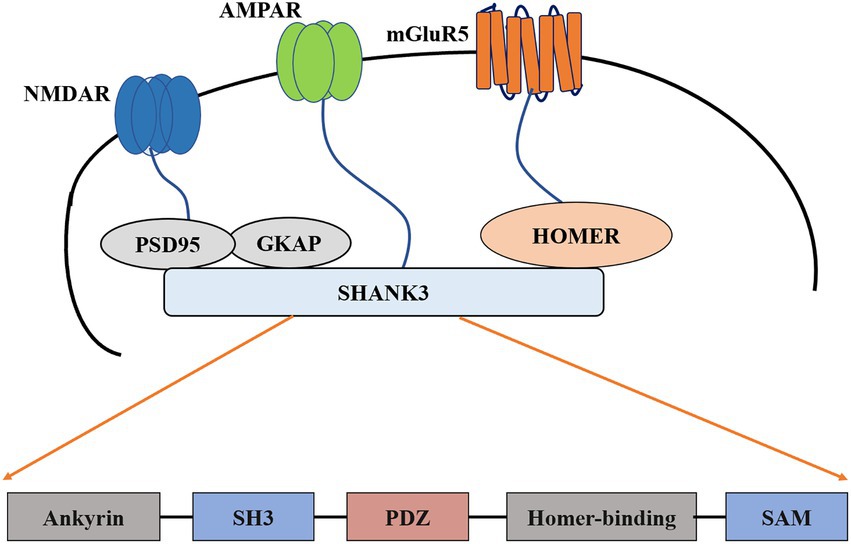
Figure 1. Schematic diagram of postsynaptic density and construction of Shank3. PSD-95, Postsynaptic density-95; SH3, Src homology 3; PDZ, PSD95/DlgA/Zo-1; SAM, Sterile alpha motif; mGluR5, metabotropic glutamate receptor 5; AMPAR, The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; NMDAR, The N-methyl-D-aspartate receptor; GKAP, guanylate kinase-associated protein.
The expression of different Shank3 subtypes varies with different brain regions and ages (Wang et al., 2014b; Vyas et al., 2021), complicating the analysis and establishment of animal models. Studies on various mouse models of Shank3 heterozygous deletion have recorded changes in the transport of glutamate transmitters or synapses (Yi et al., 2016; Lutz et al., 2020). For example, the Shank3 (Shank3α) model, by deleting exons 4–9, shows reduced immunoreactivity of the glutamate receptor 1 in hippocampal CA1 (Bozdagi et al., 2010; Epstein et al., 2014). In Shank3B−/− mice, gene deletion of two consecutive Shank3 isoforms (Shank3α and β) increases the length and complexity of dendrites and decreases the synaptic density of spinal neurons in the striatum and PSD length and thickness (Peça et al., 2011a; Balaan et al., 2019; Rendall et al., 2019). Purkinje cells of Shank3+/ΔC mice have Shank3 C-terminal deletions, although the density does not change, and they have a robust dendritic complexity in the body and reduced synaptic density (Kloth et al., 2015; Zhu et al., 2018). The expression of NMDA receptors in the PFC of these mice was reduced; however, the density of synapses did not change (Duffney et al., 2015). The dendrites on the CA1 neurons of Shank3ΔC/ΔC mice reduced LTP and NMDA/AMPA ratios in the hippocampus; however, their complexity or synapse density did not change (Bangash et al., 2011; Kouser et al., 2013). In a KO mouse model (Shank3Δe4–22−/− mice) that eliminated all Shank3 subtypes, the PSD length and thickness of the striatum were reduced (Pagliardini et al., 2005; Bey et al., 2018). Restoring Shank3 expression in adult Shank3-deficient mice can reverse the destruction of dendritic spines in the striatum and stimulate synaptic function (Peça et al., 2011a; Mei et al., 2016; Hsueh, 2019). The above findings confirmed the role of Shank3 in mobilizing multiple glutamate receptor assemblies at the PSD and synaptic signal substitution and dynamics (Jeong et al., 2021). Although the loss of heterozygosity in Shank3 is more representative of the defects observed in human PMS, the researchers still focused on the homozygous Shank3 KO (Han et al., 2016; Yi et al., 2016; Jacot-Descombes et al., 2020).
Based on the molecular and physiological abnormalities in SHANK3-deficient animals (Delling and Boeckers, 2021), various pharmaceutical compounds have been proposed as possible therapeutic options for SHANK3-related neuropsychiatric disorders (Wang X. et al., 2016; Vicidomini et al., 2017). For example, modulation of glutamate receptors may be beneficial, as AMPAR, NMDAR, or GRM5 hypofunction may lead to excitation/inhibition imbalance, contributing to ASD-like phenotypes in SHANK3-mutant mice (Guo et al., 2019; Rhine et al., 2019; Nuzzo et al., 2020).
Hypersensitivity, hyposensitivity, or abnormal interest in sensory stimuli are common features of patients with ASD (Lord et al., 2020) and PMS, who usually show increased pain tolerance (Zencica et al., 2010; Soorya et al., 2013). Abnormal sensory function in SHANK3-deficient rodents can be analyzed in various ways. The most used paradigm focuses on the perception of injury or somatic sensory function. Examples of the methods used include the tail-flick, Frey, and hot plate tests. However, other methods have also been investigated, including auditory, olfactory, visual, vestibular function, and sensorimotor gating (Bruno et al., 2021). In rat models, exons 4–22 (Han et al., 2013; Drapeau et al., 2018), exon 11 (Vicidomini et al., 2017), exon 21 (Kouser et al., 2013), and exons 11–21 (Song et al., 2019) describe general somatic dysfunction, especially low pain sensitivity. Targeted destruction of SHANK3 in tail embryo cells, somatosensory neurons [exon 13–16 (Orefice et al., 2019)], and cells expressing SCN10A [commonly known as Nav1.8, exon 4–22 (Han et al., 2013)] can induce somatosensory dysfunction. Two studies examining the constitutive SHANK3 defect model exon 13–16 (Chen et al., 2020; Kabitzke et al., 2020) described somatosensory dysfunction related to light touch stimulation. In contrast, in other studies, the somatosensory function examined by heat/injury perception was not affected in these mice (Dhamne et al., 2017; Schroeder et al., 2017). Additionally, there is mixed evidence regarding the mouse model of exons 4–9 (Yang et al., 2012; Orefice et al., 2019), while the somatic sensory function of mice with exon 8 appears unaffected (Yoo et al., 2019).
These previous studies suggest that ASD subjects express their pain perception through various behaviors, including typical reactions similar to those seen in the general population. Crying, shouting, protecting the painful parts, or seeking comfort are examples. However, the behaviors are more specific and less easily recognized as pain-related behaviors (making specific sounds, playing posture, etc.; Dubois et al., 2017). Despite these findings, the version of the Diagnostic and Statistical Manual of Mental Disorders (DSM 5) still mentions sensitivity to pain as a diagnostic standard and uses the term “obvious pain indication.” Pain sensitivity is a critical issue for patients with autism because they experience pain more frequently than others (Gillberg and Coleman, 1996; Lee et al., 2008). The sensitivity of patients with autism to pain may affect their medical pain management. This may lead to an insufficient treatment or a lack of evaluation. Therefore, this assumption needs to be tested.
Although some adults with ASD may also experience reduced pain sensitivity, lack of pain perception may not be considered a classic characteristic of ASD populations. To understand the pain in ASD and explain its lack of reactivity, it is necessary to focus on multiple components of pain rather than just the mechanism of pain and consider pain as part of a broader conceptual framework (Hadjistavropoulos and Craig, 2002; Dubois et al., 2010; Rattaz et al., 2013; Moore, 2015). Conceptualizing pain expression from the perspective of multiple factors requires individual pain management strategies and evaluation according to the clinical characteristics of patients with autism.
Current treatment options for ASD include pharmacological and non-pharmacological interventions. Pharmacological interventions include psychostimulants, atypical antipsychotics, antidepressants, alpha-2 adrenergic receptor agonists, NMDA receptor antagonists, and antiepileptic mood stabilizers (Aman et al., 2008; Costales and Kolevzon, 2015; Bicker et al., 2021). This section focuses on the safety and tolerability profiles of the main strategies used to treat children and adults with ASD (Table 1).
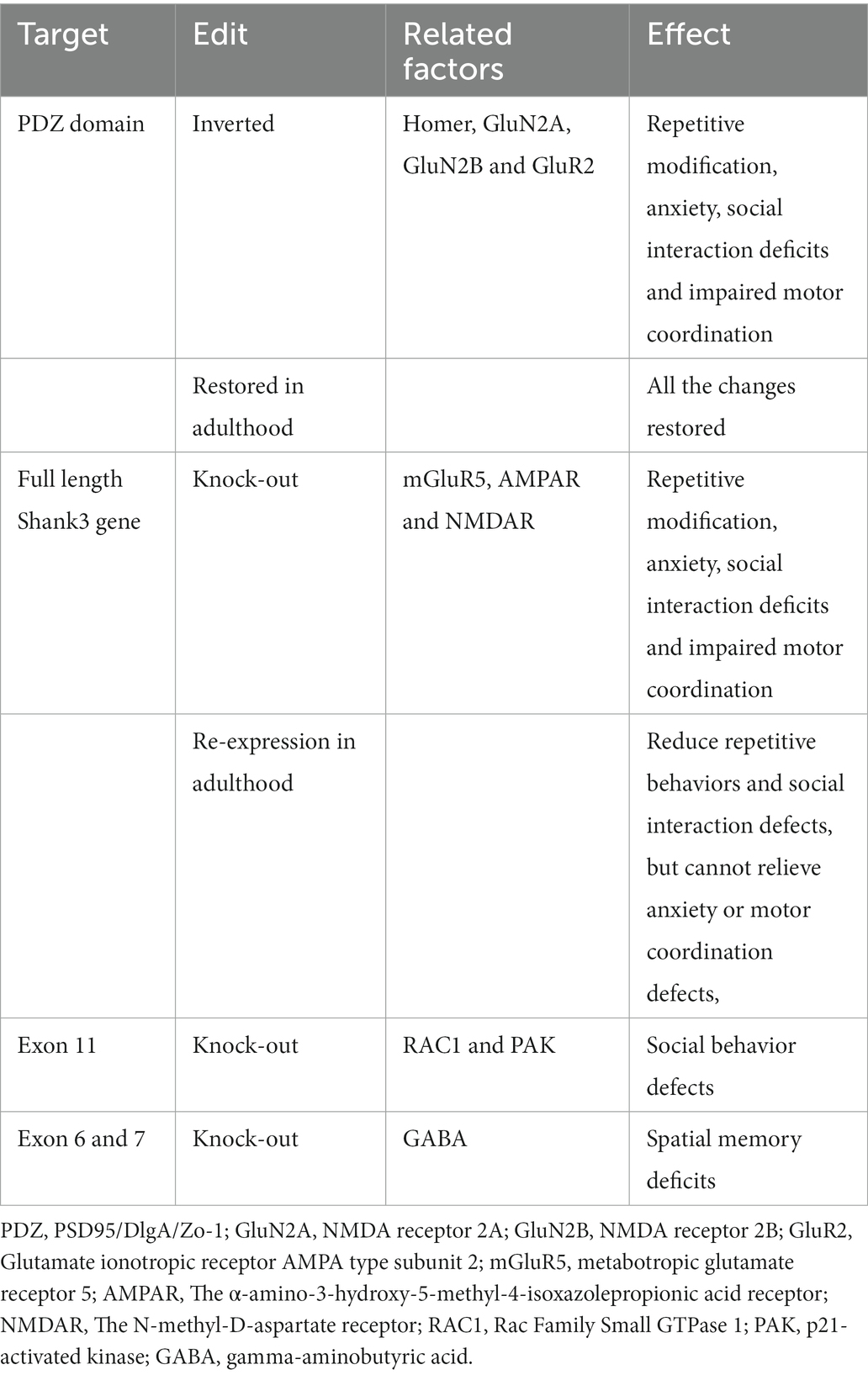
Table 1. Targets in Shank3 and treatment effects.
Because the genome of human mutations associated with ASD is very complex, animal models are the basis for studying specific mutations and establishing beneficial genotype-cell phenotypes (De Rubeis et al., 2018; Zhou et al., 2019; Reed et al., 2020). New approaches to preclinical animal studies should take into account evidence that certain therapeutic windows affect specific circuits and associated behavioral phenotypes, but also potentially reopen those critical plasticity periods to enhance therapeutic effectiveness. In particular, the advantage of the SHANK3 gene is that different mouse models can be designed, including different Shank3 gene deletions or failures (Engineer et al., 2018; Poleg et al., 2021). In addition, the SHANK3 protein has a precise location in the glutamatergic synapse. The study of the Shank3 gene is less complicated than that of other autism-related genes (Uchino and Waga, 2015; Tao-Cheng et al., 2016; Wang L. et al., 2020; Golden et al., 2021). Since autism is a neurodevelopmental disorder (symptoms appearing before age three; Lord et al., 2000; Bonaglia et al., 2001), one of the critical questions in autism research is whether the symptoms are reversible in adulthood. Recently, in a study, mice with Shank3-KO (with the deletion of the PDZ domain) had an inverted PDZ domain so that they could be repositioned at any point in the growth phase to re-express the Shank3 gene (Jaramillo et al., 2016, 2017; Jacot-Descombes et al., 2020). This gene design is crucial because it keeps the Shank3 gene under the control of its endogenous genome and avoids the expression of the SHANK3 gene at a non-physiological level, which may cause potential confusion (Saré et al., 2020). These Shank3-KO mice had defects in neurotransmission in the striatum. As a result, the striatum’s synapse density and the levels of essential PSD proteins (SAPAP3, Homer, GluN2A, GluN2B, and GluR2) were reduced. Behaviorally, these Shank3-KO mice showed repetitive self-harm modification, anxiety, social interaction deficits (decreased frequency and duration of social interaction), and impaired motor coordination. By restoring the expression of the Shank3 gene, all these changes could be restored in adulthood (Mei et al., 2016; Guo et al., 2019; Lee et al., 2021).
Re-expression of Shank3 in adulthood can reduce repetitive self-harm behaviors and social interaction deficits but not anxiety or motor coordination defects. Accordingly, this re-expression can only save a portion of the behavioral manifestations of autism (Grabrucker et al., 2011a; Peça et al., 2011a; Tai et al., 2020). A similar study showed that early postpartum intervention could improve irreversible behavioral defects in adulthood (Jin et al., 2018; Bukatova et al., 2021). Therefore, this phenomenon emphasizes the unique performance of SHANK3 expression at specific developmental stages and throughout life. Relying on the emergence of novel gene editing methods (such as CRISPR; Liu et al., 2018; Tu et al., 2019; Chiola et al., 2021), repairing the SHANK3 gene in adulthood can alleviate some synaptic and behavioral disorders related to SHANK3 mutations. Although there are still technical limitations to the genetic manipulation of mature neurons in the fully adult brain, recent research has promoted the application of CRISPR to adult brain repair (Swiech et al., 2015; Lee et al., 2017). More importantly, this work shows the possibility of treating patients with SHANK3 mutations or deletions during adulthood (either pharmacologically or through future genetic modification methods).
Some studies have shown that the re-expression of the Shank3 gene in the brain leads to a complete reversal of the expression of the SHANK3 protein. Previous studies have verified this result (Speed et al., 2015). However, we cannot conclude that this biochemical rescue leads to the rescue of some behaviors or synaptic phenotypes in mutants. Transgene seems to have complex effects on wild-type mouse synaptic transmission (Han et al., 2013; Lin R. et al., 2021). Nevertheless, some scholars have successfully replicated the previous behavioral and electrophysiological findings in Shank3 mutant mouse models (Kouser et al., 2013; Speed et al., 2015).
In summary, these studies indicate that restoring Shank3 levels or downstream signals in adults may be one of the therapeutic ways to alleviate certain synaptic and behavioral disorders associated with Shank3 mutations (Bariselli et al., 2016; Jaramillo et al., 2020; Reichova et al., 2020). As downstream mediators and proteins related to the Shank3 network are regulated, two groups have recently studied mGluR5 and Homer as potential therapeutic targets in ASD (Kouser et al., 2013; Wang X. et al., 2016; Vicidomini et al., 2017; Huang et al., 2021). Using complete Shank3-KO mice (Wang X. et al., 2016), they demonstrated that inhibition of mGluR5 activity could reduce excessive licking, while positive agonists of mGluR5 aggravated self-licking. In another study (Vicidomini et al., 2017), the pharmacological enhancement of mGluR5 activity improved repetitive behavior and rescued other behavioral defects in Shank3-KO mice (Verpelli et al., 2011; Lin et al., 2014). Although these studies may seem contradictory, the mGluR5 positive agonist CDPPB exacerbated self-modification in these studies. These results are based on different Shank3-mutant transgenic mice, which can lead to different results. However, the findings from both studies are consistent in other respects. According to another study (Vicidomini et al., 2017), pharmacological activation of mGluR5 activity alleviated functional (NMDA-induced synaptic membrane depolarization; Arvanov and Wang, 1997; Pisani et al., 2001) and behavioral defects (social interaction and Morris water maze; Wang Y. et al., 2016) in mice with exon 11 deletion. Another study reported that the cortical actin filaments of mice lacking exon 11 were significantly reduced (Duffney et al., 2015). This was attributed to the reduced activity of RAC1 and PAK, as well as the enhanced activity of cofilin (the main factor involved in actin depolymerization; Al-Ayadhi and Halepoto, 2013; Sarowar and Grabrucker, 2016; Yi et al., 2016). This suggests that actin modulators may be another potential molecular target for treating ASD. The increase of RAC1 activity in the PFC of these mice improved their social behavior defects and NMDAR function (Park et al., 2003; Duffney et al., 2013, 2015). In contrast, inhibition of PAK or RAC1 function resulted in social behavior defects and dysregulation of NMDAR function in wild-type mice (Bennett and Lagopoulos, 2014; Lin Y. et al., 2021). The use of drugs in Shank3 mutant mice to reverse their symptoms highlights the potential target pathway. NMDA hypofunction is a potential mechanism of ASD behavior (Won et al., 2012; Lee et al., 2015). Social interaction could be improved by treating mice with CDPPB (probably due to the enhancement of NMDAR function through mGluR5 activation; Won et al., 2012). In recent years, many studies have investigated NMDA-dependent inhibitors, demonstrating their therapeutic efficacy in ASD (King et al., 2001; Minshawi et al., 2016; Wink et al., 2017). The mechanism of the NMDAR antagonist, ketamine, acting on the nervous system, significantly overlaps with the pathophysiological theory of ASD, including destroying synaptic connections and neuronal networks (Wang et al., 2014a,b; Krüttner et al., 2022). However, despite the broad interest throughout psychopharmacology research, ketamine has not been explored in clinical trials of ASD. Furthermore, mice lacking exons 6 and 7 have impaired GABAergic neurotransmission (Orefice et al., 2016; Lim et al., 2017). Collectively, these studies suggest that NMDAR hypofunction leads to specific ASD-like phenotypes in Shank-mutant mice, and other related molecular targets may be used to regulate NMDAR activity. Direct gene targeting in humans appears to be a future treatment option for some types of ASDs, as the recent success of many techniques related to detecting and treating genetic disorders may provide the necessary tools.
Although Shank3 mutation is heterozygous in humans, the analysis and identification of Shank3 homozygous mutant mice are imperative for understanding the physiological role of Shank3 and its functional consequences. In addition, the mutation has destructive effects. In terms of the treatment window, the earlier the treatment is administered, the better the outcome. However, interventions in adulthood may still be useful for reducing some of the symptoms associated with SHANK3 mutations. It is necessary to carefully analyze the specific phenotype of a genotype before trying a drug alone. Exploring the SHANK3 gene may help uncover some of the neurobiological aspects of autism.
MH and TX performed the conceptualization. MH and QQ searched the literature and prepared the draft. All authors contributed to the article and approved the submitted version.
This work was supported by grant from the Natural Science Foundation of China (82171486), Natural Science Foundation of Shanghai to TX (21ZR1448400), the Interdisciplinary Program of Shanghai Jiao Tong University to TX (YG2021ZD23), and General Science Foundation of Shanghai Sixth People’s Hospital to TX (YNMS202114).
We thank the lab members for their help on this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ZQ declared a shared parent affiliation with the authors to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Ayadhi, L., and Halepoto, D. M. (2013). Role of proteomics in the discovery of autism biomarkers. J. Coll. Physicians Surg. Pak. 23, 137–143.
Aman, M. G. (2004). Management of hyperactivity and other acting-out problems in patients with autism spectrum disorder. Semin. Pediatr. Neurol. 11, 225–228. doi: 10.1016/j.spen.2004.07.006
Aman, M. G., Farmer, C. A., Hollway, J., and Arnold, L. E. (2008). Treatment of inattention, overactivity, and impulsiveness in autism spectrum disorders. Child Adolesc. Psychiatr. Clin. N. Am. 17, 713–738. doi: 10.1016/j.chc.2008.06.009
Arons, M. H., Thynne, C. J., Grabrucker, A. M., Li, D., Schoen, M., Cheyne, J. E., et al. (2012). Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 32, 14966–14978. doi: 10.1523/JNEUROSCI.2215-12.2012
Arvanov, V. L., and Wang, R. Y. (1997). NMDA-induced response in pyramidal neurons of the rat medial prefrontal cortex slices consists of NMDA and non-NMDA components. Brain Res. 768, 361–364. doi: 10.1016/S0006-8993(97)00842-1
Auerbach, B. D., Manohar, S., Radziwon, K., and Salvi, R. (2021). Auditory hypersensitivity and processing deficits in a rat model of fragile X syndrome. Neurobiol. Dis. 161:105541. doi: 10.1016/j.nbd.2021.105541
Bai, D., Marrus, N., Yip, B. H. K., Reichenberg, A., Constantino, J. N., and Sandin, S. (2020). Inherited risk for autism through maternal and paternal lineage. Biol. Psychiatry 88, 480–487. doi: 10.1016/j.biopsych.2020.03.013
Balaan, C., Corley, M. J., Eulalio, T., Leite-Ahyo, K., Pang, A. P. S., Fang, R., et al. (2019). Juvenile Shank3b deficient mice present with behavioral phenotype relevant to autism spectrum disorder. Behav. Brain Res. 356, 137–147. doi: 10.1016/j.bbr.2018.08.005
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cells 145, 758–772. doi: 10.1016/j.cell.2011.03.052
Banker, S. M., Gu, X., Schiller, D., and Foss-Feig, J. H. (2021). Hippocampal contributions to social and cognitive deficits in autism spectrum disorder. Trends Neurosci. 44, 793–807. doi: 10.1016/j.tins.2021.08.005
Bariselli, S., Tzanoulinou, S., Glangetas, C., Prévost-Solié, C., Pucci, L., Viguié, J., et al. (2016). SHANK3 controls maturation of social reward circuits in the VTA. Nat. Neurosci. 19, 926–934. doi: 10.1038/nn.4319
Baum, S. H., Stevenson, R. A., and Wallace, M. T. (2015). Behavioral, perceptual, and neural alterations in sensory and multisensory function in autism spectrum disorder. Prog. Neurobiol. 134, 140–160. doi: 10.1016/j.pneurobio.2015.09.007
Becerra, T. A., Von Ehrenstein, O. S., Heck, J. E., Olsen, J., Arah, O. A., Jeste, S. S., et al. (2014). Autism spectrum disorders and race, ethnicity, and nativity: a population-based study. Pediatrics 134, e63–e71. doi: 10.1542/peds.2013-3928
Bennett, M. R., and Lagopoulos, J. (2014). Stress and trauma: BDNF control of dendritic-spine formation and regression. Prog. Neurobiol. 112, 80–99. doi: 10.1016/j.pneurobio.2013.10.005
Berkel, S., Eltokhi, A., Fröhlich, H., Porras-Gonzalez, D., Rafiullah, R., Sprengel, R., et al. (2018). Sex hormones regulate SHANK expression. Front. Mol. Neurosci. 11:337. doi: 10.3389/fnmol.2018.00337
Bey, A. L., Wang, X., Yan, H., Kim, N., Passman, R. L., Yang, Y., et al. (2018). Brain region-specific disruption of Shank3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl. Psychiatry 8:94. doi: 10.1038/s41398-018-0142-6
Bicker, F., Nardi, L., Maier, J., Vasic, V., and Schmeisser, M. J. (2021). Criss-crossing autism spectrum disorder and adult neurogenesis. J. Neurochem. 159, 452–478. doi: 10.1111/jnc.15501
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Bonaglia, M. C., Giorda, R., Beri, S., De Agostini, C., Novara, F., Fichera, M., et al. (2011). Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet. 7:e1002173. doi: 10.1371/journal.pgen.1002173
Bonaglia, M. C., Giorda, R., Borgatti, R., Felisari, G., Gagliardi, C., Selicorni, A., et al. (2001). Disruption of the ProSAP2 gene in a t(12,22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 69, 261–268. doi: 10.1086/321293
Boukhris, T., Sheehy, O., Mottron, L., and Bérard, A. (2016). Antidepressant use during pregnancy and the risk of autism spectrum disorder in children. JAMA Pediatr. 170, 117–124. doi: 10.1001/jamapediatrics.2015.3356
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism. 1:15. doi: 10.1186/2040-2392-1-15
Bruno, L. P., Doddato, G., Valentino, F., Baldassarri, M., Tita, R., Fallerini, C., et al. (2021). New candidates for autism/intellectual disability identified by whole-exome sequencing. Int. J. Mol. Sci. 22:22. doi: 10.3390/ijms222413439
Bukatova, S., Renczes, E., Reichova, A., Filo, J., Sadlonova, A., Mravec, B., et al. (2021). Shank3 deficiency is associated with altered profile of neurotransmission markers in pups and adult mice. Neurochem. Res. 46, 3342–3355. doi: 10.1007/s11064-021-03435-6
Burdeus-Olavarrieta, M., San José-Cáceres, A., García-Alcón, A., González-Peñas, J., Hernández-Jusdado, P., and Parellada-Redondo, M. (2021). Characterisation of the clinical phenotype in Phelan-McDermid syndrome. J. Neurodev. Disord. 13:26. doi: 10.1186/s11689-021-09370-5
Chen, Q., Deister, C. A., Gao, X., Guo, B., Lynn-Jones, T., Chen, N., et al. (2020). Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a Shank3 mouse model of ASD. Nat. Neurosci. 23, 520–532. doi: 10.1038/s41593-020-0598-6
Chevallier, C., Kohls, G., Troiani, V., Brodkin, E. S., and Schultz, R. T. (2012). The social motivation theory of autism. Trends Cogn. Sci. 16, 231–239. doi: 10.1016/j.tics.2012.02.007
Chiola, S., Napan, K. L., Wang, Y., Lazarenko, R. M., Armstrong, C. J., Cui, J., et al. (2021). Defective AMPA-mediated synaptic transmission and morphology in human neurons with hemizygous SHANK3 deletion engrafted in mouse prefrontal cortex. Mol. Psychiatry 26, 4670–4686. doi: 10.1038/s41380-021-01023-2
Christensen, D. L., Braun, K. V. N., Baio, J., Bilder, D., Charles, J., Constantino, J. N., et al. (2018). Prevalence and characteristics of autism Spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2012. MMWR Surveill. Summ. 65, 1–23. doi: 10.15585/mmwr.ss6513a1
Costales, J. L., and Kolevzon, A. (2015). Phelan-McDermid syndrome and SHANK3: implications for treatment. Neurotherapeutics 12, 620–630. doi: 10.1007/s13311-015-0352-z
Crump, C., Sundquist, J., and Sundquist, K. (2021). Preterm or early term birth and risk of autism. Pediatrics 148:e2020032300. doi: 10.1542/peds.2020-032300
D'antoni, S., Spatuzza, M., Bonaccorso, C. M., Musumeci, S. A., Ciranna, L., Nicoletti, F., et al. (2014). Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with intellectual disability and autism. Neurosci. Biobehav. Rev. 46, 228–241. doi: 10.1016/j.neubiorev.2014.02.003
De Rubeis, S., Siper, P. M., Durkin, A., Weissman, J., Muratet, F., Halpern, D., et al. (2018). Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism. 9:31. doi: 10.1186/s13229-018-0205-9
Delling, J. P., and Boeckers, T. M. (2021). Comparison of SHANK3 deficiency in animal models: phenotypes, treatment strategies, and translational implications. J. Neurodev. Disord. 13:55. doi: 10.1186/s11689-021-09397-8
Dhaliwal, K. K., Orsso, C. E., Richard, C., Haqq, A. M., and Zwaigenbaum, L. (2019). Risk factors for unhealthy weight gain and obesity among children with autism Spectrum disorder. Int. J. Mol. Sci. 20:3285. doi: 10.3390/ijms20133285
Dhamne, S. C., Silverman, J. L., Super, C. E., Lammers, S. H. T., Hameed, M. Q., Modi, M. E., et al. (2017). Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism. 8:26. doi: 10.1186/s13229-017-0142-z
Diguiseppi, C., Hepburn, S., Davis, J. M., Fidler, D. J., Hartway, S., Lee, N. R., et al. (2010). Screening for autism spectrum disorders in children with down syndrome: population prevalence and screening test characteristics. J. Dev. Behav. Pediatr. 31, 181–191. doi: 10.1097/DBP.0b013e3181d5aa6d
Doshi-Velez, F., Ge, Y., and Kohane, I. (2014). Comorbidity clusters in autism spectrum disorders: an electronic health record time-series analysis. Pediatrics 133, e54–e63. doi: 10.1542/peds.2013-0819
Dovey, T. M., Kumari, V., and Blissett, J. (2019). Eating behaviour, behavioural problems and sensory profiles of children with avoidant/restrictive food intake disorder (ARFID), autistic spectrum disorders or picky eating: same or different? Eur. Psychiatry 61, 56–62. doi: 10.1016/j.eurpsy.2019.06.008
Drapeau, E., Riad, M., Kajiwara, Y., and Buxbaum, J. D. (2018). Behavioral phenotyping of an improved mouse model of Phelan-McDermid syndrome with a complete deletion of the Shank3 gene. eNeuro 5:ENEURO.0046-18.2018. doi: 10.1523/ENEURO.0046-18.2018
Dubois, A., Michelon, C., Rattaz, C., Zabalia, M., and Baghdadli, A. (2017). Daily living pain assessment in children with autism: exploratory study. Res. Dev. Disabil. 62, 238–246. doi: 10.1016/j.ridd.2017.01.003
Dubois, A., Rattaz, C., Pry, R., and Baghdadli, A. (2010). Autism and pain - a literature review. Pain Res. Manag. 15, 245–253. doi: 10.1155/2010/749275
Duffney, L. J., Wei, J., Cheng, J., Liu, W., Smith, K. R., Kittler, J. T., et al. (2013). Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. J. Neurosci. 33, 15767–15778. doi: 10.1523/JNEUROSCI.1175-13.2013
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Engineer, C. T., Rahebi, K. C., Borland, M. S., Buell, E. P., Im, K. W., Wilson, L. G., et al. (2018). Shank3-deficient rats exhibit degraded cortical responses to sound. Autism Res. 11, 59–68. doi: 10.1002/aur.1883
Epstein, I., Tushev, G., Will, T. J., Vlatkovic, I., Cajigas, I. J., and Schuman, E. M. (2014). Alternative polyadenylation and differential expression of Shank mRNAs in the synaptic neuropil. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 369:20130137. doi: 10.1098/rstb.2013.0137
Esposito, D., Belli, A., Ferri, R., and Bruni, O. (2020). Sleeping without prescription: Management of Sleep Disorders in children with autism with non-pharmacological interventions and over-the-counter treatments. Brain Sci. 10:441. doi: 10.3390/brainsci10070441
Estes, M. L., and Mcallister, A. K. (2016). Maternal immune activation: implications for neuropsychiatric disorders. Science 353, 772–777. doi: 10.1126/science.aag3194
Frank, Y. (2021). The neurological manifestations of Phelan-McDermid syndrome. Pediatr. Neurol. 122, 59–64. doi: 10.1016/j.pediatrneurol.2021.06.002
Frolli, A., Bosco, A., Di Carmine, F., Cavallaro, A., Lombardi, A., Sergi, L., et al. (2021). Parent training and therapy in children with autism. Pediatr. Rep. 13, 216–226. doi: 10.3390/pediatric13020030
Gauthier, J., Spiegelman, D., Piton, A., Lafrenière, R. G., Laurent, S., St-Onge, J., et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150b, 421–424. doi: 10.1002/ajmg.b.30822
Gidaya, N. B., Lee, B. K., Burstyn, I., Yudell, M., Mortensen, E. L., and Newschaffer, C. J. (2014). In utero exposure to selective serotonin reuptake inhibitors and risk for autism spectrum disorder. J. Autism Dev. Disord. 44, 2558–2567. doi: 10.1007/s10803-014-2128-4
Gillberg, C., and Coleman, M. (1996). Autism and medical disorders: a review of the literature. Dev. Med. Child Neurol. 38, 191–202. doi: 10.1111/j.1469-8749.1996.tb15081.x
Golden, C. E. M., Wang, V. X., Harony-Nicolas, H., Hof, P. R., and Buxbaum, J. D. (2021). Reduced brain volume and white matter alterations in Shank3-deficient rats. Autism Res. 14, 1837–1842. doi: 10.1002/aur.2568
Gong, X., and Wang, H. (2015). SHANK1 and autism spectrum disorders. Sci. China Life Sci. 58, 985–990. doi: 10.1007/s11427-015-4892-6
Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M., et al. (2011a). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581. doi: 10.1038/emboj.2010.336
Grabrucker, A. M., Schmeisser, M. J., Schoen, M., and Boeckers, T. M. (2011b). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603. doi: 10.1016/j.tcb.2011.07.003
Grafodatskaya, D., Chung, B., Szatmari, P., and Weksberg, R. (2010). Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry 49, 794–809. doi: 10.1016/j.jaac.2010.05.005
Guang, S., Pang, N., Deng, X., Yang, L., He, F., Wu, L., et al. (2018). Synaptopathology involved in autism spectrum disorder. Front. Cell. Neurosci. 12:470. doi: 10.3389/fncel.2018.00470
Guo, B., Chen, J., Chen, Q., Ren, K., Feng, D., Mao, H., et al. (2019). Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat. Neurosci. 22, 1223–1234. doi: 10.1038/s41593-019-0445-9
Hadjistavropoulos, T., and Craig, K. D. (2002). A theoretical framework for understanding self-report and observational measures of pain: a communications model. Behav. Res. Ther. 40, 551–570. doi: 10.1016/s0005-7967(01)00072-9
Han, K., Holder, J. L. Jr., Schaaf, C. P., Lu, H., Chen, H., Kang, H., et al. (2013). SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503, 72–77. doi: 10.1038/nature12630
Han, Q., Kim, Y. H., Wang, X., Liu, D., Zhang, Z. J., Bey, A. L., et al. (2016). SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron 92, 1279–1293. doi: 10.1016/j.neuron.2016.11.007
Hoover, D. W., and Kaufman, J. (2018). Adverse childhood experiences in children with autism spectrum disorder. Curr. Opin. Psychiatry 31, 128–132. doi: 10.1097/YCO.0000000000000390
Hsueh, Y. P. (2019). Synaptic formation, neural circuits and neurodevelopmental disorders controlled by signaling, translation, and epigenetic regulation. Dev. Neurobiol. 79, 2–7. doi: 10.1002/dneu.22655
Huang, M., Pu, S., Jiang, W., Worley, P. F., and Xu, T. (2021). Deficiency of SHANK3 isoforms impairs thermal hyperalgesia and dysregulates the expression of postsynaptic proteins in the spinal cord. Neurosci. Res. 163, 26–33. doi: 10.1016/j.neures.2020.03.001
Hyman, S. L., Levy, S. E., and Myers, S. M. (2020). Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics 145:e20193447. doi: 10.1542/peds.2019-3447
Jacot-Descombes, S., Keshav, N. U., Dickstein, D. L., Wicinski, B., Janssen, W. G. M., Hiester, L. L., et al. (2020). Altered synaptic ultrastructure in the prefrontal cortex of Shank3-deficient rats. Mol. Autism. 11:89. doi: 10.1186/s13229-020-00393-8
Jaramillo, T. C., Speed, H. E., Xuan, Z., Reimers, J. M., Escamilla, C. O., Weaver, T. P., et al. (2017). Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res. 10, 42–65. doi: 10.1002/aur.1664
Jaramillo, T. C., Speed, H. E., Xuan, Z., Reimers, J. M., Liu, S., and Powell, C. M. (2016). Altered striatal synaptic function and abnormal behaviour in Shank3 Exon4-9 deletion mouse model of autism. Autism Res. 9, 350–375. doi: 10.1002/aur.1529
Jaramillo, T. C., Xuan, Z., Reimers, J. M., Escamilla, C. O., Liu, S., and Powell, C. M. (2020). Early restoration of Shank3 expression in Shank3 Knock-out mice prevents Core ASD-like behavioral phenotypes. eNeuro 7:ENEURO.0332-19.2020. doi: 10.1523/ENEURO.0332-19.2020
Jeong, J., Li, Y., and Roche, K. W. (2021). CaMKII phosphorylation regulates synaptic enrichment of Shank3. eNeuro 8, ENEURO.0481–ENEU20.2021. doi: 10.1523/ENEURO.0481-20.2021
Jiang, Y. H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Jin, C., Kang, H., Ryu, J. R., Kim, S., Zhang, Y., Lee, Y., et al. (2018). Integrative brain transcriptome analysis reveals region-specific and broad molecular changes in Shank3-overexpressing mice. Front. Mol. Neurosci. 11:250. doi: 10.3389/fnmol.2018.00250
Johannessen, M., Haugen, I. B., Bakken, T. L., and Braaten, Ø. (2019). A 22q13.33 duplication harbouring the SHANK3 gene: does it cause neuropsychiatric disorders? BMJ Case Rep. 12:e228258. doi: 10.1136/bcr-2018-228258
Jokiranta, E., Brown, A. S., Heinimaa, M., Cheslack-Postava, K., Suominen, A., and Sourander, A. (2013). Parental psychiatric disorders and autism spectrum disorders. Psychiatry Res. 207, 203–211. doi: 10.1016/j.psychres.2013.01.005
Kabitzke, P., Morales, D., He, D., Cox, K., Sutphen, J., Thiede, L., et al. (2020). Mouse model systems of autism spectrum disorder: replicability and informatics signature. Genes Brain Behav. 19:e12676. doi: 10.1111/gbb.12676
Kales, H. C., Gitlin, L. N., and Lyketsos, C. G. (2015). Assessment and management of behavioral and psychological symptoms of dementia. BMJ 350:h369. doi: 10.1136/bmj.h369
Kincaid, D. L., Doris, M., Shannon, C., and Mulholland, C. (2017). What is the prevalence of autism spectrum disorder and ASD traits in psychosis? A systematic review. Psychiatry Res. 250, 99–105. doi: 10.1016/j.psychres.2017.01.017
King, B. H., Wright, D. M., Handen, B. L., Sikich, L., Zimmerman, A. W., Mcmahon, W., et al. (2001). Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 40, 658–665. doi: 10.1097/00004583-200106000-00010
Kloth, A. D., Badura, A., Li, A., Cherskov, A., Connolly, S. G., Giovannucci, A., et al. (2015). Cerebellar associative sensory learning defects in five mouse autism models. elife 4:e06085. doi: 10.7554/eLife.06085
Kodak, T., and Bergmann, S. (2020). Autism Spectrum disorder: characteristics, associated behaviors, and early intervention. Pediatr. Clin. N. Am. 67, 525–535. doi: 10.1016/j.pcl.2020.02.007
Kouser, M., Speed, H. E., Dewey, C. M., Reimers, J. M., Widman, A. J., Gupta, N., et al. (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 33, 18448–18468. doi: 10.1523/JNEUROSCI.3017-13.2013
Krüttner, S., Falasconi, A., Valbuena, S., Galimberti, I., Bouwmeester, T., Arber, S., et al. (2022). Absence of familiarity triggers hallmarks of autism in mouse model through aberrant tail-of-striatum and prelimbic cortex signaling. Neuron 110, 1468–1482.e5. doi: 10.1016/j.neuron.2022.02.001
Landrigan, P. J. (2010). What causes autism? Exploring the environmental contribution. Curr. Opin. Pediatr. 22, 219–225. doi: 10.1097/MOP.0b013e328336eb9a
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. doi: 10.1371/journal.pgen.1004580
Lee, J., Chung, C., Ha, S., Lee, D., Kim, D. Y., Kim, H., et al. (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell. Neurosci. 9:94. doi: 10.3389/fncel.2015.00094
Lee, L. C., Harrington, R. A., Chang, J. J., and Connors, S. L. (2008). Increased risk of injury in children with developmental disabilities. Res. Dev. Disabil. 29, 247–255. doi: 10.1016/j.ridd.2007.05.002
Lee, D. K., Li, S. W., Bounni, F., Friedman, G., Jamali, M., Strahs, L., et al. (2021). Reduced sociability and social agency encoding in adult Shank3-mutant mice are restored through gene re-expression in real time. Nat. Neurosci. 24, 1243–1255. doi: 10.1038/s41593-021-00888-4
Lee, B., Zhang, Y., Kim, Y., Kim, S., Lee, Y., and Han, K. (2017). Age-dependent decrease of GAD65/67 mRNAs but normal densities of GABAergic interneurons in the brain regions of Shank3-overexpressing manic mouse model. Neurosci. Lett. 649, 48–54. doi: 10.1016/j.neulet.2017.04.016
Li, W., and Pozzo-Miller, L. (2020). Dysfunction of the corticostriatal pathway in autism spectrum disorders. J. Neurosci. Res. 98, 2130–2147. doi: 10.1002/jnr.24560
Lim, C. S., Kim, H., Yu, N. K., Kang, S. J., Kim, T., Ko, H. G., et al. (2017). Enhancing inhibitory synaptic function reverses spatial memory deficits in Shank2 mutant mice. Neuropharmacology 112, 104–112. doi: 10.1016/j.neuropharm.2016.08.016
Lin, C. W., Chen, C. Y., Cheng, S. J., Hu, H. T., and Hsueh, Y. P. (2014). Sarm1 deficiency impairs synaptic function and leads to behavioral deficits, which can be ameliorated by an mGluR allosteric modulator. Front. Cell. Neurosci. 8:87. doi: 10.3389/fncel.2014.00087
Lin, R., Learman, L. N., Bangash, M. A., Melnikova, T., Leyder, E., Reddy, S. C., et al. (2021). Homer1a regulates Shank3 expression and underlies behavioral vulnerability to stress in a model of Phelan-McDermid syndrome. Cell Rep. 37:110014. doi: 10.1016/j.celrep.2021.110014
Lin, Y., Li, H., Peng, J., Li, C., Zhu, C., Zhou, Y., et al. (2021). Decrease of morphine-CPP by sinomenine via mediation of tyrosine hydroxylase, NMDA receptor subunit 2B and opioid receptor in the zebrafish brain. Pak. J. Pharm. Sci. 34, 1659–1665.
Liu, C. X., Li, C. Y., Hu, C. C., Wang, Y., Lin, J., Jiang, Y. H., et al. (2018). CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism. 9:23. doi: 10.1186/s13229-018-0204-x
Lord, C., Brugha, T. S., Charman, T., Cusack, J., Dumas, G., Frazier, T., et al. (2020). Autism spectrum disorder. Nat. Rev. Dis. Primers 6:5. doi: 10.1038/s41572-019-0138-4
Lord, C., Cook, E. H., Leventhal, B. L., and Amaral, D. G. (2000). Autism spectrum disorders. Neuron 28, 355–363. doi: 10.1016/S0896-6273(00)00115-X
Lutz, A. K., Pfaender, S., Incearap, B., Ioannidis, V., Ottonelli, I., Föhr, K. J., et al. (2020). Autism-associated SHANK3 mutations impair maturation of neuromuscular junctions and striated muscles. Sci. Transl. Med. 12:eaaz3267. doi: 10.1126/scitranslmed.aaz3267
Mansouri, M., Pouretemad, H., Roghani, M., Wegener, G., and Ardalan, M. (2020). Autistic-like behaviours and associated brain structural plasticity are modulated by oxytocin in maternally separated rats. Behav. Brain Res. 393:112756. doi: 10.1016/j.bbr.2020.112756
Mashayekhi, F., Mizban, N., Bidabadi, E., and Salehi, Z. (2021). The association of SHANK3 gene polymorphism and autism. Minerva Pediatr. 73, 251–255. doi: 10.23736/S2724-5276.16.04539-4
Masi, A., Demayo, M. M., Glozier, N., and Guastella, A. J. (2017). An overview of autism Spectrum disorder, heterogeneity and treatment options. Neurosci. Bull. 33, 183–193. doi: 10.1007/s12264-017-0100-y
Mccarthy, A., Wafford, K., Shanks, E., Ligocki, M., Edgar, D. M., and Dijk, D. J. (2016). REM sleep homeostasis in the absence of REM sleep: effects of antidepressants. Neuropharmacology 108, 415–425. doi: 10.1016/j.neuropharm.2016.04.047
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Minshawi, N. F., Wink, L. K., Shaffer, R., Plawecki, M. H., Posey, D. J., Liu, H., et al. (2016). A randomized, placebo-controlled trial of D-cycloserine for the enhancement of social skills training in autism spectrum disorders. Mol. Autism. 7:2. doi: 10.1186/s13229-015-0062-8
Modabbernia, A., Velthorst, E., and Reichenberg, A. (2017). Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol. Autism. 8:13. doi: 10.1186/s13229-017-0121-4
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297. doi: 10.1086/522590
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157. doi: 10.1038/nrn.2016.183
Moon, S. J., Hwang, J. S., Shin, A. L., Kim, J. Y., Bae, S. M., Sheehy-Knight, J., et al. (2019). Accuracy of the childhood autism rating scale: a systematic review and meta-analysis. Dev. Med. Child Neurol. 61, 1030–1038. doi: 10.1111/dmcn.14246
Moore, D. J. (2015). Acute pain experience in individuals with autism spectrum disorders: a review. Autism 19, 387–399. doi: 10.1177/1362361314527839
Mossa, A., Pagano, J., Ponzoni, L., Tozzi, A., Vezzoli, E., Sciaccaluga, M., et al. (2021). Developmental impaired Akt signaling in the Shank1 and Shank3 double knock-out mice. Mol. Psychiatry 26, 1928–1944. doi: 10.1038/s41380-020-00979-x
Moutin, E., Sakkaki, S., Compan, V., Bouquier, N., Giona, F., Areias, J., et al. (2021). Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 26, 7596–7609. doi: 10.1038/s41380-021-01230-x
Muskens, J. B., Velders, F. P., and Staal, W. G. (2017). Medical comorbidities in children and adolescents with autism spectrum disorders and attention deficit hyperactivity disorders: a systematic review. Eur. Child Adolesc. Psychiatry 26, 1093–1103. doi: 10.1007/s00787-017-1020-0
Muskens, J. B., Vermeulen, K., Van Deurzen, P. A., Tomesen, E. M., Van Der Gaag, R. J., Buitelaar, J. K., et al. (2015). Somatic screening in child and adolescent psychiatry: a descriptive pilot study. Tijdschr. Psychiatr. 57, 710–718.
Neal, D., Matson, J. L., and Hattier, M. A. (2012). A comparison of diagnostic criteria on the autism Spectrum disorder observation for children (ASD-OC). Dev. Neurorehabil. 15, 329–335. doi: 10.3109/17518423.2012.697492
Needham, B. D., Adame, M. D., Serena, G., Rose, D. R., Preston, G. M., Conrad, M. C., et al. (2021). Plasma and fecal metabolite profiles in autism spectrum disorder. Biol. Psychiatry 89, 451–462. doi: 10.1016/j.biopsych.2020.09.025
Nuzzo, T., Sekine, M., Punzo, D., Miroballo, M., Katane, M., Saitoh, Y., et al. (2020). Dysfunctional d-aspartate metabolism in BTBR mouse model of idiopathic autism. Biochim. Biophys. Acta, Proteins Proteomics 1868:140531. doi: 10.1016/j.bbapap.2020.140531
Ogata, H., Ihara, H., Murakami, N., Gito, M., Kido, Y., and Nagai, T. (2014). Autism spectrum disorders and hyperactive/impulsive behaviors in Japanese patients with Prader-Willi syndrome: a comparison between maternal uniparental disomy and deletion cases. Am. J. Med. Genet. A 164a, 2180–2186. doi: 10.1002/ajmg.a.36615
Orefice, L. L., Mosko, J. R., Morency, D. T., Wells, M. F., Tasnim, A., Mozeika, S. M., et al. (2019). Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cells 178, 867–886.e824. doi: 10.1016/j.cell.2019.07.024
Orefice, L. L., Zimmerman, A. L., Chirila, A. M., Sleboda, S. J., Head, J. P., and Ginty, D. D. (2016). Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cells 166, 299–313. doi: 10.1016/j.cell.2016.05.033
Pagliardini, S., Ren, J., Wevrick, R., and Greer, J. J. (2005). Developmental abnormalities of neuronal structure and function in prenatal mice lacking the prader-willi syndrome gene necdin. Am. J. Pathol. 167, 175–191. doi: 10.1016/S0002-9440(10)62964-1
Panjwani, A. A., Ji, Y., Fahey, J. W., Palmer, A., Wang, G., Hong, X., et al. (2020). Maternal dyslipidemia, plasma branched-chain amino acids, and the risk of child autism spectrum disorder: evidence of sex difference. J. Autism Dev. Disord. 50, 540–550. doi: 10.1007/s10803-019-04264-x
Pardo, C. A., and Eberhart, C. G. (2007). The neurobiology of autism. Brain Pathol. 17, 434–447. doi: 10.1016/j.cell.2019.12.036
Park, E., Na, M., Choi, J., Kim, S., Lee, J. R., Yoon, J., et al. (2003). The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J. Biol. Chem. 278, 19220–19229. doi: 10.1074/jbc.M301052200
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011a). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peça, J., Ting, J., and Feng, G. (2011b). SnapShot: autism and the synapse. Cells 147:706.e701. doi: 10.1016/j.cell.2011.10.015
Pendyala, G., Chou, S., Jung, Y., Coiro, P., Spartz, E., Padmashri, R., et al. (2017). Maternal immune activation causes behavioral impairments and altered cerebellar cytokine and synaptic protein expression. Neuropsychopharmacology 42, 1435–1446. doi: 10.1038/npp.2017.7
Phelan, K., and Mcdermid, H. E. (2012). The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2, 186–201. doi: 10.1159/000334260
Phelan, M. C., Rogers, R. C., Saul, R. A., Stapleton, G. A., Sweet, K., Mcdermid, H., et al. (2001). 22q13 deletion syndrome. Am. J. Med. Genet. 101, 91–99. doi: 10.1177/000992280404300106
Pisani, A., Gubellini, P., Bonsi, P., Conquet, F., Picconi, B., Centonze, D., et al. (2001). Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience 106, 579–587. doi: 10.1016/s0306-4522(01)00297-4
Poleg, S., Kourieh, E., Ruban, A., Shapira, G., Shomron, N., Barak, B., et al. (2021). Behavioral aspects and neurobiological properties underlying medical cannabis treatment in Shank3 mouse model of autism spectrum disorder. Transl. Psychiatry 11:524. doi: 10.1038/s41398-021-01612-3
Purushotham, S. S., Reddy, N. M. N., D'souza, M. N., Choudhury, N. R., Ganguly, A., Gopalakrishna, N., et al. (2022). A perspective on molecular signalling dysfunction, its clinical relevance and therapeutics in autism spectrum disorder. Exp. Brain Res. 240, 2525–2567. doi: 10.1007/s00221-022-06448-x
Qin, Y., Du, Y., Chen, L., Liu, Y., Xu, W., Liu, Y., et al. (2022). A recurrent SHANK1 mutation implicated in autism spectrum disorder causes autistic-like core behaviors in mice via downregulation of mGluR1-IP3R1-calcium signaling. Mol. Psychiatry 27, 2985–2998. doi: 10.1038/s41380-022-01539-1
Qin, L., Ma, K., Wang, Z. J., Hu, Z., Matas, E., Wei, J., et al. (2018). Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 21, 564–575. doi: 10.1038/s41593-018-0110-8
Raghavan, R., Riley, A. W., Volk, H., Caruso, D., Hironaka, L., Sices, L., et al. (2018). Maternal multivitamin intake, plasma folate and vitamin B (12) levels and autism Spectrum disorder risk in offspring. Paediatr. Perinat. Epidemiol. 32, 100–111. doi: 10.1111/ppe.12414
Randall, M., Egberts, K. J., Samtani, A., Scholten, R. J., Hooft, L., Livingstone, N., et al. (2018). Diagnostic tests for autism spectrum disorder (ASD) in preschool children. Cochrane Database Syst. Rev. 7:Cd009044. doi: 10.1002/14651858.CD009044
Rattaz, C., Dubois, A., Michelon, C., Viellard, M., Poinso, F., and Baghdadli, A. (2013). How do children with autism spectrum disorders express pain? A comparison with developmentally delayed and typically developing children. Pain 154, 2007–2013. doi: 10.1016/j.pain.2013.06.011
Reed, M. D., Yim, Y. S., Wimmer, R. D., Kim, H., Ryu, C., Welch, G. M., et al. (2020). IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature 577, 249–253. doi: 10.1038/s41586-019-1843-6
Reichova, A., Bacova, Z., Bukatova, S., Kokavcova, M., Meliskova, V., Frimmel, K., et al. (2020). Abnormal neuronal morphology and altered synaptic proteins are restored by oxytocin in autism-related SHANK3 deficient model. Mol. Cell. Endocrinol. 518:110924. doi: 10.1016/j.mce.2020.110924
Rendall, A. R., Perrino, P. A., Buscarello, A. N., and Fitch, R. H. (2019). Shank3B mutant mice display pitch discrimination enhancements and learning deficits. Int. J. Dev. Neurosci. 72, 13–21. doi: 10.1016/j.ijdevneu.2018.10.003
Rhine, M. A., Parrott, J. M., Schultz, M. N., Kazdoba, T. M., and Crawley, J. N. (2019). Hypothesis-driven investigations of diverse pharmacological targets in two mouse models of autism. Autism Res. 12, 401–421. doi: 10.1002/aur.2066
Roberts, J. L., Hovanes, K., Dasouki, M., Manzardo, A. M., and Butler, M. G. (2014). Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 535, 70–78. doi: 10.1016/j.gene.2013.10.020
Rodin, R. E., Dou, Y., Kwon, M., Sherman, M. A., Dgama, A. M., Doan, R. N., et al. (2021). The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat. Neurosci. 24, 176–185. doi: 10.1038/s41593-020-00765-6
Salomaa, S. I., Miihkinen, M., Kremneva, E., Paatero, I., Lilja, J., Jacquemet, G., et al. (2021). SHANK3 conformation regulates direct actin binding and crosstalk with Rap1 signaling. Curr. Biol. 31, 4956–4970.e4959. doi: 10.1016/j.cub.2021.09.022
Saré, R. M., Lemons, A., Song, A., and Smith, C. B. (2020). Sleep duration in mouse models of neurodevelopmental disorders. Brain Sci. 11:31. doi: 10.3390/brainsci11010031
Sarowar, T., and Grabrucker, A. M. (2016). Actin-dependent alterations of dendritic spine morphology in Shankopathies. Neural Plast. 2016:8051861. doi: 10.1155/2016/8051861
Saupe, J., Roske, Y., Schillinger, C., Kamdem, N., Radetzki, S., Diehl, A., et al. (2011). Discovery, structure-activity relationship studies, and crystal structure of nonpeptide inhibitors bound to the Shank3 PDZ domain. ChemMedChem 6, 1411–1422. doi: 10.1002/cmdc.201100094
Schroeder, J. C., Reim, D., Boeckers, T. M., and Schmeisser, M. J. (2017). Genetic animal models for autism spectrum disorder. Curr. Top. Behav. Neurosci. 30, 311–324. doi: 10.1007/7854_2015_407
Sgritta, M., Dooling, S. W., Buffington, S. A., Momin, E. N., Francis, M. B., Britton, R. A., et al. (2019). Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron 101, 246–259.e246. doi: 10.1016/j.neuron.2018.11.018
Sharma, S. R., Gonda, X., and Tarazi, F. I. (2018). Autism spectrum disorder: classification, diagnosis and therapy. Pharmacol. Ther. 190, 91–104. doi: 10.1016/j.pharmthera.2018.05.007
Shcheglovitov, A., Shcheglovitova, O., Yazawa, M., Portmann, T., Shu, R., Sebastiano, V., et al. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. doi: 10.1038/nature12618
Shi, R., Redman, P., Ghose, D., Hwang, H., Liu, Y., Ren, X., et al. (2017). Shank proteins differentially regulate synaptic transmission. eNeuro 4:ENEURO.0163-15.2017. doi: 10.1523/ENEURO.0163-15.2017
Siu, M. T., and Weksberg, R. (2017). Epigenetics of autism spectrum disorder. Adv. Exp. Med. Biol. 978, 63–90. doi: 10.1007/978-3-319-53889-1_4
Soler, J., Fañanás, L., Parellada, M., Krebs, M. O., Rouleau, G. A., and Fatjó-Vilas, M. (2018). Genetic variability in scaffolding proteins and risk for schizophrenia and autism-spectrum disorders: a systematic review. J. Psychiatry Neurosci. 43, 223–244. doi: 10.1503/jpn.170066
Song, T. J., Lan, X. Y., Wei, M. P., Zhai, F. J., Boeckers, T. M., Wang, J. N., et al. (2019). Altered behaviors and impaired synaptic function in a novel rat model with a complete Shank3 deletion. Front. Cell. Neurosci. 13:111. doi: 10.3389/fncel.2019.00111
Soorya, L., Kolevzon, A., Zweifach, J., Lim, T., Dobry, Y., Schwartz, L., et al. (2013). Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism. 4:18. doi: 10.1186/2040-2392-4-18
Speed, H. E., Kouser, M., Xuan, Z., Liu, S., Duong, A., and Powell, C. M. (2019). Apparent genetic rescue of adult Shank3 exon 21 insertion mutation mice tempered by appropriate control experiments. eNeuro 6:ENEURO.0317-19.2019. doi: 10.1523/ENEURO.0317-19.2019
Speed, H. E., Kouser, M., Xuan, Z., Reimers, J. M., Ochoa, C. F., Gupta, N., et al. (2015). Autism-associated insertion mutation (InsG) of Shank3 exon 21 causes impaired synaptic transmission and behavioral deficits. J. Neurosci. 35, 9648–9665. doi: 10.1523/JNEUROSCI.3125-14.2015
Srivastava, S., Condy, E., Carmody, E., Filip-Dhima, R., Kapur, K., Bernstein, J. A., et al. (2021). Parent-reported measure of repetitive behavior in Phelan-McDermid syndrome. J. Neurodev. Disord. 13:53. doi: 10.1186/s11689-021-09398-7
Swiech, L., Heidenreich, M., Banerjee, A., Habib, N., Li, Y., Trombetta, J., et al. (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 102–106. doi: 10.1038/nbt.3055
Tai, C., Chang, C. W., Yu, G. Q., Lopez, I., Yu, X., Wang, X., et al. (2020). Tau reduction prevents key features of autism in mouse models. Neuron 106, 421–437.e411. doi: 10.1016/j.neuron.2020.01.038
Tammimies, K. (2019). Genetic mechanisms of regression in autism spectrum disorder. Neurosci. Biobehav. Rev. 102, 208–220. doi: 10.1016/j.neubiorev.2019.04.022
Tao-Cheng, J. H., Toy, D., Winters, C. A., Reese, T. S., and Dosemeci, A. (2016). Zinc stabilizes Shank3 at the postsynaptic density of hippocampal synapses. PLoS One 11:e0153979. doi: 10.1371/journal.pone.0153979
Tatavarty, V., Torrado Pacheco, A., Groves Kuhnle, C., Lin, H., Koundinya, P., Miska, N. J., et al. (2020). Autism-associated Shank3 is essential for homeostatic compensation in rodent V1. Neuron 106, 769–777.e764. doi: 10.1016/j.neuron.2020.02.033
Thapar, A., Cooper, M., and Rutter, M. (2017). Neurodevelopmental disorders. Lancet. Psychiatry 4, 339–346. doi: 10.1016/S2215-0366(16)30376-5
Tu, Z., Zhao, H., Li, B., Yan, S., Wang, L., Tang, Y., et al. (2019). CRISPR/Cas9-mediated disruption of SHANK3 in monkey leads to drug-treatable autism-like symptoms. Hum. Mol. Genet. 28, 561–571. doi: 10.1093/hmg/ddy367
Tuchman, R., Cuccaro, M., and Alessandri, M. (2010). Autism and epilepsy: historical perspective. Brain Dev. 32, 709–718. doi: 10.1016/j.braindev.2010.04.008
Uchino, S., and Waga, C. (2015). Novel therapeutic approach for autism spectrum disorder: focus on SHANK3. Curr. Neuropharmacol. 13, 786–792. doi: 10.2174/1570159x13666151029105547
Vannucchi, G., Masi, G., Toni, C., Dell'osso, L., Erfurth, A., and Perugi, G. (2014). Bipolar disorder in adults with Asperger′s syndrome: a systematic review. J. Affect. Disord. 168, 151–160. doi: 10.1016/j.jad.2014.06.042
Varghese, M., Keshav, N., Jacot-Descombes, S., Warda, T., Wicinski, B., Dickstein, D. L., et al. (2017). Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 134, 537–566. doi: 10.1007/s00401-017-1736-4
Verpelli, C., Dvoretskova, E., Vicidomini, C., Rossi, F., Chiappalone, M., Schoen, M., et al. (2011). Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 286, 34839–34850. doi: 10.1074/jbc.M111.258384
Vicidomini, C., Ponzoni, L., Lim, D., Schmeisser, M. J., Reim, D., Morello, N., et al. (2017). Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22, 689–702. doi: 10.1038/mp.2016.30
Vyas, Y., Cheyne, J. E., Lee, K., Jung, Y., Cheung, P. Y., and Montgomery, J. M. (2021). Shankopathies in the developing brain in autism Spectrum disorders. Front. Neurosci. 15:775431. doi: 10.3389/fnins.2021.775431
Wang, L., Adamski, C. J., Bondar, V. V., Craigen, E., Collette, J. R., Pang, K., et al. (2020). A kinome-wide RNAi screen identifies ERK2 as a druggable regulator of Shank3 stability. Mol. Psychiatry 25, 2504–2516. doi: 10.1038/s41380-018-0325-9
Wang, X., Bey, A. L., Chung, L., Krystal, A. D., and Jiang, Y. H. (2014a). Therapeutic approaches for shankopathies. Dev. Neurobiol. 74, 123–135. doi: 10.1002/dneu.22084
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7:11459. doi: 10.1038/ncomms11459
Wang, Z., Ding, R., and Wang, J. (2020). The association between vitamin D status and autism Spectrum disorder (ASD): a systematic review and meta-analysis. Nutrients 13:86. doi: 10.3390/nu13010086
Wang, Y., Ma, Y., Hu, J., Zhang, X., Cheng, W., Jiang, H., et al. (2016). Sex-specific effects of prenatal chronic mild stress on adult spatial learning capacity and regional glutamate receptor expression profiles. Exp. Neurol. 281, 66–80. doi: 10.1016/j.expneurol.2016.04.016
Wang, X., Mccoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wang, X., Xu, Q., Bey, A. L., Lee, Y., and Jiang, Y. H. (2014b). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism. 5:30. doi: 10.1186/2040-2392-5-30
Wei, H., Zhu, Y., Wang, T., Zhang, X., Zhang, K., and Zhang, Z. (2021). Genetic risk factors for autism-spectrum disorders: a systematic review based on systematic reviews and meta-analysis. J. Neural Transm. (Vienna) 128, 717–734. doi: 10.1007/s00702-021-02360-w
Wiegersma, A. M., Dalman, C., Lee, B. K., Karlsson, H., and Gardner, R. M. (2019). Association of Prenatal Maternal Anemia with Neurodevelopmental Disorders. JAMA Psychiat. 76, 1294–1304. doi: 10.1001/jamapsychiatry.2019.2309
Wilson, H. L., Crolla, J. A., Walker, D., Artifoni, L., Dallapiccola, B., Takano, T., et al. (2008). Interstitial 22q13 deletions: genes other than SHANK3 have major effects on cognitive and language development. Eur. J. Hum. Genet. 16, 1301–1310. doi: 10.1038/ejhg.2008.107
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C., et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40, 575–584. doi: 10.1136/jmg.40.8.575
Wink, L. K., Minshawi, N. F., Shaffer, R. C., Plawecki, M. H., Posey, D. J., Horn, P. S., et al. (2017). D-Cycloserine enhances durability of social skills training in autism spectrum disorder. Mol. Autism. 8:2. doi: 10.1186/s13229-017-0116-1
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Yang, M., Bozdagi, O., Scattoni, M. L., Wöhr, M., Roullet, F. I., Katz, A. M., et al. (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 32, 6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. doi: 10.1126/science.aaf2669
Yoo, Y. E., Yoo, T., Lee, S., Lee, J., Kim, D., Han, H. M., et al. (2019). Shank3 mice carrying the human Q321R mutation display enhanced self-grooming, abnormal electroencephalogram patterns, and suppressed neuronal excitability and seizure susceptibility. Front. Mol. Neurosci. 12:155. doi: 10.3389/fnmol.2019.00155
Zencica, P., Chaloupka, R., Hladíková, J., and Krbec, M. (2010). Adjacent segment degeneration after lumbosacral fusion in spondylolisthesis: a retrospective radiological and clinical analysis. Acta Chir. Orthop. Traumatol. Cechoslov. 77, 124–130.
Zhou, Y., Kaiser, T., Monteiro, P., Zhang, X., Van Der Goes, M. S., Wang, D., et al. (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162. doi: 10.1016/j.neuron.2015.11.023
Zhou, Y., Sharma, J., Ke, Q., Landman, R., Yuan, J., Chen, H., et al. (2019). Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 570, 326–331. doi: 10.1038/s41586-019-1278-0
Keywords: autism spectrum disorder, ASD, synapse, Shank3, treatment
Citation: Huang M, Qi Q and Xu T (2023) Targeting Shank3 deficiency and paresthesia in autism spectrum disorder: A brief review. Front. Mol. Neurosci. 16:1128974. doi: 10.3389/fnmol.2023.1128974
Edited by:
Linlin Zhang, Tianjin Medical University General Hospital, ChinaReviewed by:
Shunmei Lu, Wuxi People’s Hospital Affiliated to Nanjing Medical University, ChinaCopyright © 2023 Huang, Qi and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Xu,  YmFsb3JAc2p0dS5lZHUuY24=
YmFsb3JAc2p0dS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.