 James B. Ames
James B. Ames- Department of Chemistry, University of California, Davis, Davis, CA, United States
Retinal membrane guanylate cyclases (RetGC1 and RetGC2) are expressed in photoreceptor rod and cone cells, where they promote the onset of visual recovery during phototransduction. The catalytic activity of RetGCs is regulated by their binding to regulatory proteins, guanylate cyclase activating proteins (GCAP1-5) and the retinal degeneration 3 protein (RD3). RetGC1 is activated by its binding to Ca2+-free/Mg2+-bound GCAP1 at low cytosolic Ca2+ levels in light-activated photoreceptors. By contrast, RetGC1 is inactivated by its binding to Ca2+-bound GCAP1 and/or RD3 at elevated Ca2+ levels in dark-adapted photoreceptors. The Ca2+ sensitive cyclase activation helps to replenish the cytosolic cGMP levels in photoreceptors during visual recovery. Mutations in RetGC1, GCAP1 or RD3 that disable the Ca2+-dependent regulation of cyclase activity are genetically linked to rod/cone dystrophies and other inherited forms of blindness. Here I review the structural interaction of RetGC1 with GCAP1 and RD3. I propose a two-state concerted model in which the dimeric RetGC1 allosterically switches between active and inactive conformational states with distinct quaternary structures that are oppositely stabilized by the binding of GCAP1 and RD3. The binding of Ca2+-free/Mg2+-bound GCAP1 is proposed to activate the cyclase by stabilizing RetGC1 in an active conformation (R-state), whereas Ca2+-bound GCAP1 and/or RD3 inhibit the cyclase by locking RetGC1 in an inactive conformation (T-state). Exposed hydrophobic residues in GCAP1 (residues H19, Y22, M26, F73, V77, W94) are essential for cyclase activation and could be targeted by rational drug design for the possible treatment of rod/cone dystrophies.
Introduction
Light activation of retinal rod and cone cells (called visual phototransduction) triggers a decrease in the cytosolic cGMP concentration that causes cyclic nucleotide gated (CNG) channels to close, which hyperpolarizes the plasma membrane to generate a neural signal (Stryer, 1991; Baylor, 1996; Figure 1). In dark-adapted photoreceptors, relatively high levels of cGMP keep CNG channels open, resulting in high cytosolic Ca2+ levels at or near 500 nM (Woodruff et al., 2002). By contrast, light-activation of the photoreceptor promotes the hydrolysis of cGMP and the light-induced lowering of cGMP causes CNG channels to close, resulting in a nearly 10-fold drop in the cytosolic Ca2+ level (Gray-Keller and Detwiler, 1994). The light-induced decrease in both cGMP and Ca2+ are important signals that promote the re-synthesis of cGMP by the enzyme retinal membrane guanylate cyclases (Dizhoor et al., 1994; Lowe et al., 1995). The re-synthesis of cGMP by the cyclase during visual recovery is highly regulated by the light-dependent cytosolic Ca2+ concentration (Koch and Stryer, 1988; Koutalos and Yau, 1996), and the Ca2+-dependent regulation of the cyclase is mediated by guanylate cyclase activating proteins discussed below. Retinal membrane guanylate cyclase is also regulated by the retinal degeneration 3 (RD3) protein (Friedman et al., 2006). The binding of RD3 to the cyclase has been shown to inhibit the cyclase enzymatic activity (Peshenko et al., 2011). In addition, RD3 binding promotes trafficking of RetGC1 from the endoplasmic reticulum (ER) to the rod outer segment disk membrane (Azadi et al., 2010; Zulliger et al., 2015).
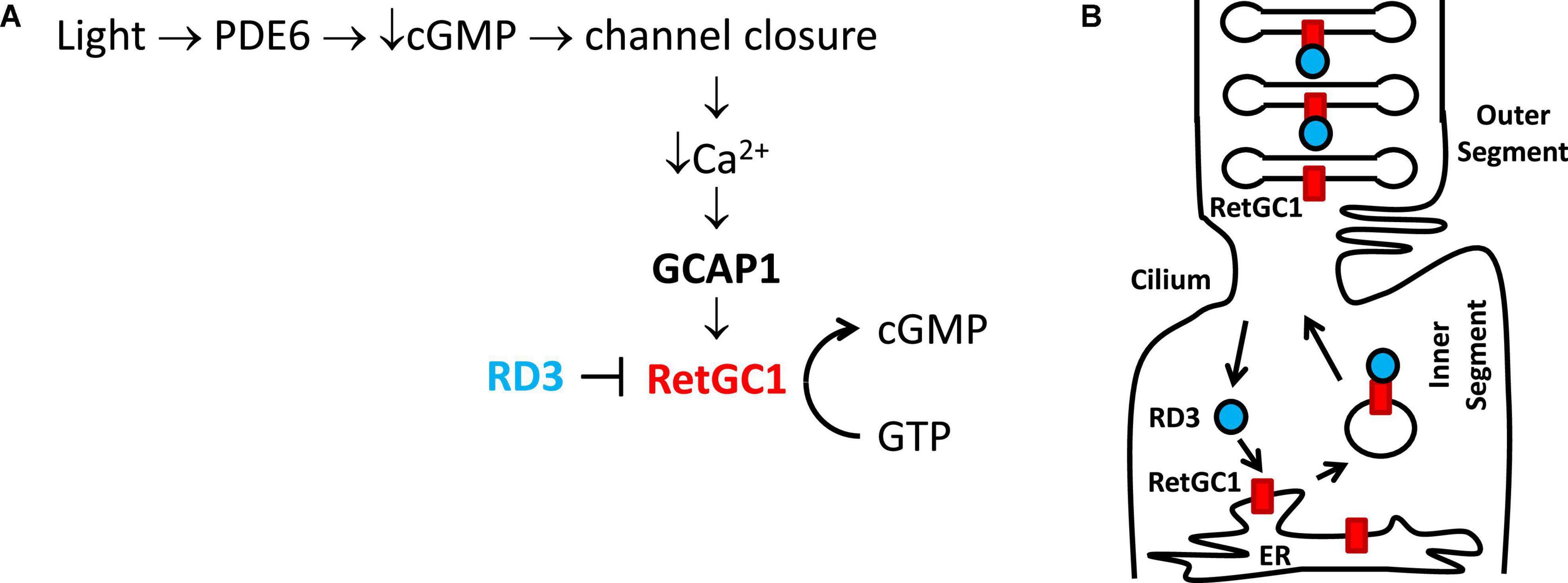
Figure 1. Physiological role of GCAP1 and RD3 in visual phototransduction. Adapted from Ames (2019). (A) Visual excitation pathway in retinal photoreceptor cells. Light-activated channel closure promotes a drop in cytosolic Ca2+ level that in turn causes Ca2+-free/Mg2+-bound GCAP1 to bind and activate RetGC1. RD3 binding to RetGC1 inhibits the cyclase activity. (B) RD3 (cyan) guides the trafficking of RetGC1 (red) to outer segment disk membranes.
Retinal membrane guanylate cyclases (RetGC1 and RetGC2) (Dizhoor et al., 1994; Palczewski et al., 1994, 2004) are regulated by a family of guanylate cyclase activating proteins (GCAP1-5, see Figure 2). RetGC1 is known to interact with GCAP1 (Laura et al., 1996), whereas RetGC2 can interact with both GCAP1 and GCAP2 (Laura and Hurley, 1998). The functional differences between RetGC1 and RetGC2 are currently not well understood. RetGC1 knockout mice cause a loss of cone function but do not exhibit rod degeneration. By contrast, RetGC2 is important for rod function but the RetGC2 knockout has no effect on cones (Yang et al., 1999; Baehr et al., 2007; Boye et al., 2011, 2013). A related membrane guanylate cyclase (regulated by GCAP1) is also expressed in the olfactory bulb (Duda et al., 2001). RetGCs are regulated by up to 8 different vertebrate homologs of guanylate cyclase activating proteins (GCAP1-8) (Imanishi et al., 2004; Scholten and Koch, 2011). The binding of Ca2+-free GCAP1 to RetGC1 activates the cyclase enzymatic activity at low Ca2+ levels in light activated photoreceptors (Peshenko and Dizhoor, 2006; Lim et al., 2009). By contrast, the binding of Ca2+-bound GCAP1 to RetGC1 inhibits the cyclase activity at high Ca2+ levels in dark-adapted photoreceptors (Dizhoor and Hurley, 1996; Dizhoor et al., 1998). The Ca2+-bound GCAP1 can also activate the odorant surface receptor ONE-GC (Duda et al., 2012), which raises the question about how Ca2+-bound GCAP1 can oppositely regulate both RetGC1 and ONE-GC. The Ca2+-dependent regulation of RetGCs by GCAPs in the retina coordinates the recovery phase of visual phototransduction (Figure 1A). Light-activation of the photoreceptor causes a reduction in both cGMP and Ca2+ levels (Figure 1A), which triggers a need for RetGC1 to become activated by Ca2+-free GCAP1 when the cytosolic Ca2+ and cGMP levels are both low. This light-induced activation of RetGC1 is important for replenishing cGMP levels to recover the dark state during visual recovery (Figure 1A). The Ca2+-dependent cyclase regulation is abolished in GCAP1 knockout mice (Burns et al., 2002; Howes et al., 2002; Mendez and Chen, 2002; Pennesi et al., 2003). Particular GCAP1 mutants that weaken Ca2+ binding to GCAP1 (Dell’orco et al., 2010) cause constitutive activation of RetGC1 that directly leads to retinal degenerative diseases known as rod-cone dystrophies (Semple-Rowland et al., 1996; Sokal et al., 1998; Baehr and Palczewski, 2007; Behnen et al., 2010; Bondarenko et al., 2010; Jiang and Baehr, 2010; Dell’orco et al., 2014).
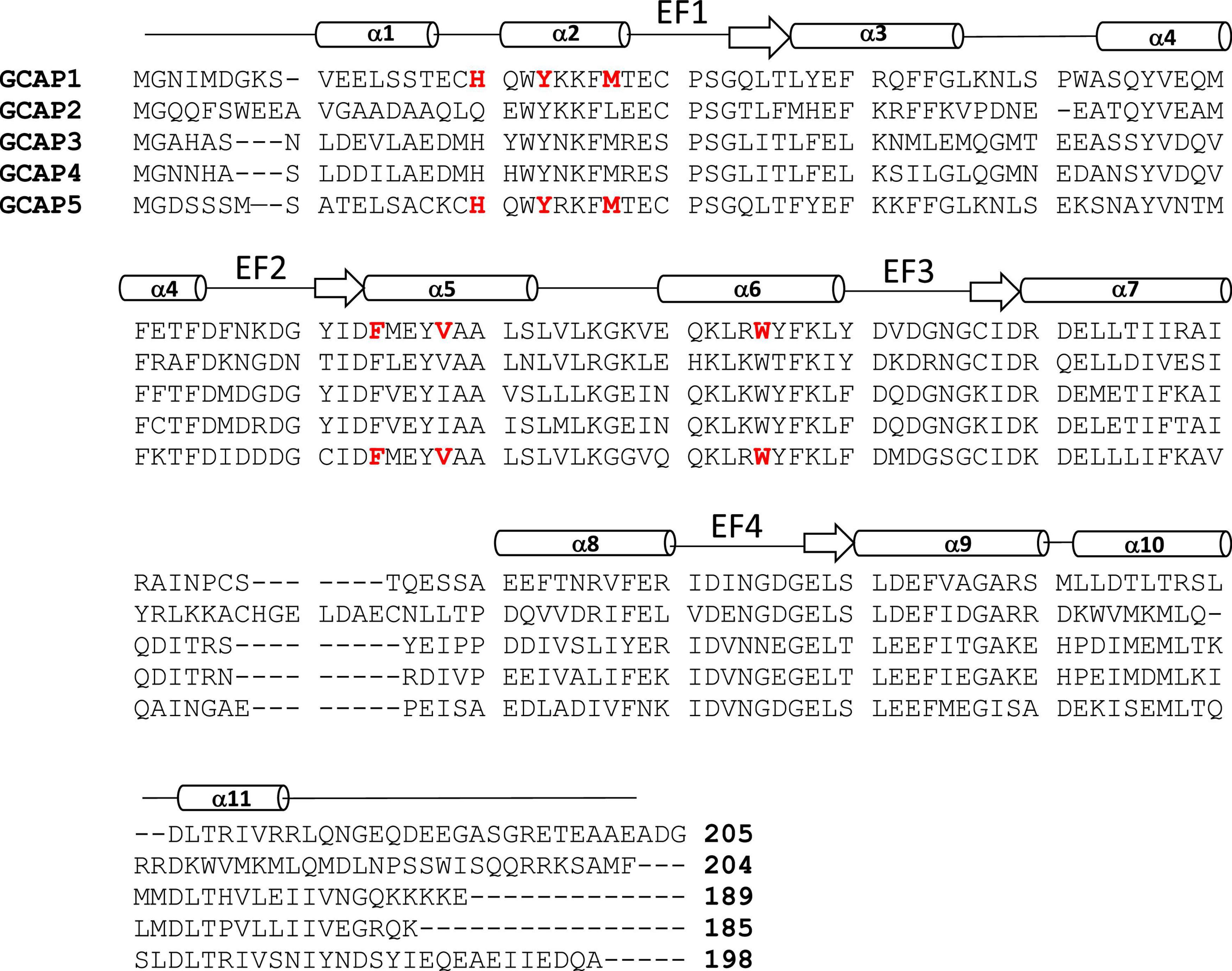
Figure 2. Amino acid sequence alignment of GCAP proteins. Swiss Protein Database accession numbers are P46065 (bovine GCAP1), P51177 (bovine GCAP2), O95843 (zebrafish GCAP3), Q6ZM98 (zebrafish GCAP4), and Q5MAC8 (zebrafish GCAP5). Exposed “hot spot” residues in GCAP1 and GCAP5 are highlighted red. Secondary structure (helices and strands) are shown above the sequence alignment depicted as cylinders and arrows.
RD3 is a 23-kDa retinal protein that is essential for proper photoreceptor function, and the deletion of RD3 causes retinal degeneration and blindness in human patients that possess recessive Leber Congenital Amaurosis 12 (LCA) (Friedman et al., 2006). The lack of RD3 expression in knockout mice leads to reduced levels of RetGC1 in the outer segment membrane of photoreceptors. This reduction of RetGC1 caused by a lack of RD3 is consistent with observations that RD3 promotes the trafficking of RetGC1 into the outer segment (Azadi et al., 2010; Zulliger et al., 2015; Figure 1B). The RetGC1 protein is first expressed inside the endoplasmic reticulum (ER), where it is processed and inserted into transport vesicles (Figure 1B). The RetGC1 containing transport vesicles in the inner segment then bind to RD3, which guides the trafficking of these vesicles into the outer segment. The binding of RD3 to RetGC1 facilitates the transfer of RetGC1 from transport vesicles into the outer segment disk membrane. RD3 binding to RetGC1 also inhibits the cyclase enzymatic activity, perhaps by obstructing the binding of GCAPs (Peshenko et al., 2011, 2016; Figure 1A). The RD3 binding site in RetGC1 involves multivalent contacts. Mutagenesis studies have suggested that RD3 contacts the RetGC1 dimerization domain (residues 800–851) that also interacts with GCAP1 (Peshenko et al., 2011, 2016). RD3 also interacts with a C-terminal region in RetGC1 that is downstream of the catalytic domain (Azadi et al., 2010). The RD3 binding to RetGC1 is important for keeping the cyclase enzymatic activity turned off during its vesicle trafficking to the disk membrane. The RetGC1 trafficking in the absence of RD3 causes elevated cGMP levels in the inner segment, which turns on apoptosis that leads to retinal degeneration (Dizhoor et al., 1998; Newbold et al., 2002). As a result, mutations in RD3 and/or RetGC1 that disable RD3 binding are believed to cause Lebering are belie amaurosis (Azadi et al., 2010; Zulliger et al., 2015) and various forms of retinal degeneration (Friedman et al., 2006; Azadi et al., 2010; Molday et al., 2013, 2014). The ability of RD3 to prevent aberrant activation of RetGC1 by GCAPs is essential for the survival of photoreceptors (Peshenko et al., 2016). Lastly, RD3 may also interact with guanylate kinase to promote the synthesis of GDP to control the recycling of nucleotides in the inner segment (Wimberg et al., 2018). However, more recent studies argue against any role for RD3 in GMP recycling (Dizhoor et al., 2021).
In this review, I provide an overview of previous atomic-resolution structures of GCAP1 (Stephen et al., 2007; Lim et al., 2016), GCAP5 (Cudia et al., 2021) and RD3 (Peshenko et al., 2019), and propose a molecular mechanism for how RetGC1 might be regulated by GCAP1 and RD3.
Molecular structure and function of guanylate cyclase activating proteins
Guanylate cyclase activating proteins are a family of retinal Ca2+ sensors
The GCAP proteins (GCAP1-5, see Figure 2) are a family of Ca2+ binding proteins expressed exclusively in vertebrate photoreceptors (Polans et al., 1996; Ames, 2021). GCAP1 (Palczewski et al., 1994) and GCAP2 (Dizhoor et al., 1995) were originally discovered in mammalian photoreceptors, but up to seven GCAP homologs were later discovered in zebrafish and other vertebrate species (Imanishi et al., 2004; Scholten and Koch, 2011). The GCAP proteins consist of about 200 amino acid residues that contain N-terminal myristoylation, four EF-hand Ca2+ binding sites (EF1, EF2, EF3, and EF4 in Figure 2), and non-conserved residues in the N-terminal and C-terminal helices (α1 and α11 in Figure 2). The EF-hands in GCAP1 can bind to both Ca2+ and Mg2+ (Peshenko and Dizhoor, 2006, 2007). Mg2+ can bind to the second EF-hand (magenta sphere in Figure 3A) in GCAP1 (Peshenko and Dizhoor, 2004, 2007; Lim et al., 2009), which stabilizes the Ca2+-free/Mg2+-bound protein conformation (Figure 3A) that activates RetGC1 in light-activated photoreceptors (Dizhoor et al., 1994; Peshenko and Dizhoor, 2004; Marino et al., 2015). In dark-adapted photoreceptors, Ca2+ binds to GCAP1 (in place of Mg2+), which stabilizes the Ca2+-bound structure (Figure 3B) that is important for the inhibition of RetGC1 (Dizhoor et al., 1998). Ca2+ binds to GCAP1 at the second, third and fourth EF-hands (orange spheres in Figure 3B; Ames et al., 1999; Stephen et al., 2007) with an apparent dissociation constant of 100 nM (Lim et al., 2009; Dizhoor et al., 2010). Dark-adapted photoreceptors maintain relatively high cytosolic Ca2+ levels (500 nM) (Woodruff et al., 2002), which promotes the formation of Ca2+-bound GCAP1 (Dizhoor and Hurley, 1996; Dizhoor et al., 1998). Light-activation of the photoreceptor causes a 10-fold reduction in the cytosolic Ca2+ level (Gray-Keller and Detwiler, 1994; Woodruff et al., 2002) while the Mg2+ concentration remains fixed at ∼1 mM (Chen et al., 2003). Therefore, GCAP1 binds to Mg2+ instead of Ca2+ in light-activated photoreceptors, and the Ca2+-free/Mg2+-bound GCAP1 binds to RetGC1 to promote the synthesis of cGMP during visual recovery (Dizhoor et al., 1994, 1995; Gorczyca et al., 1995).
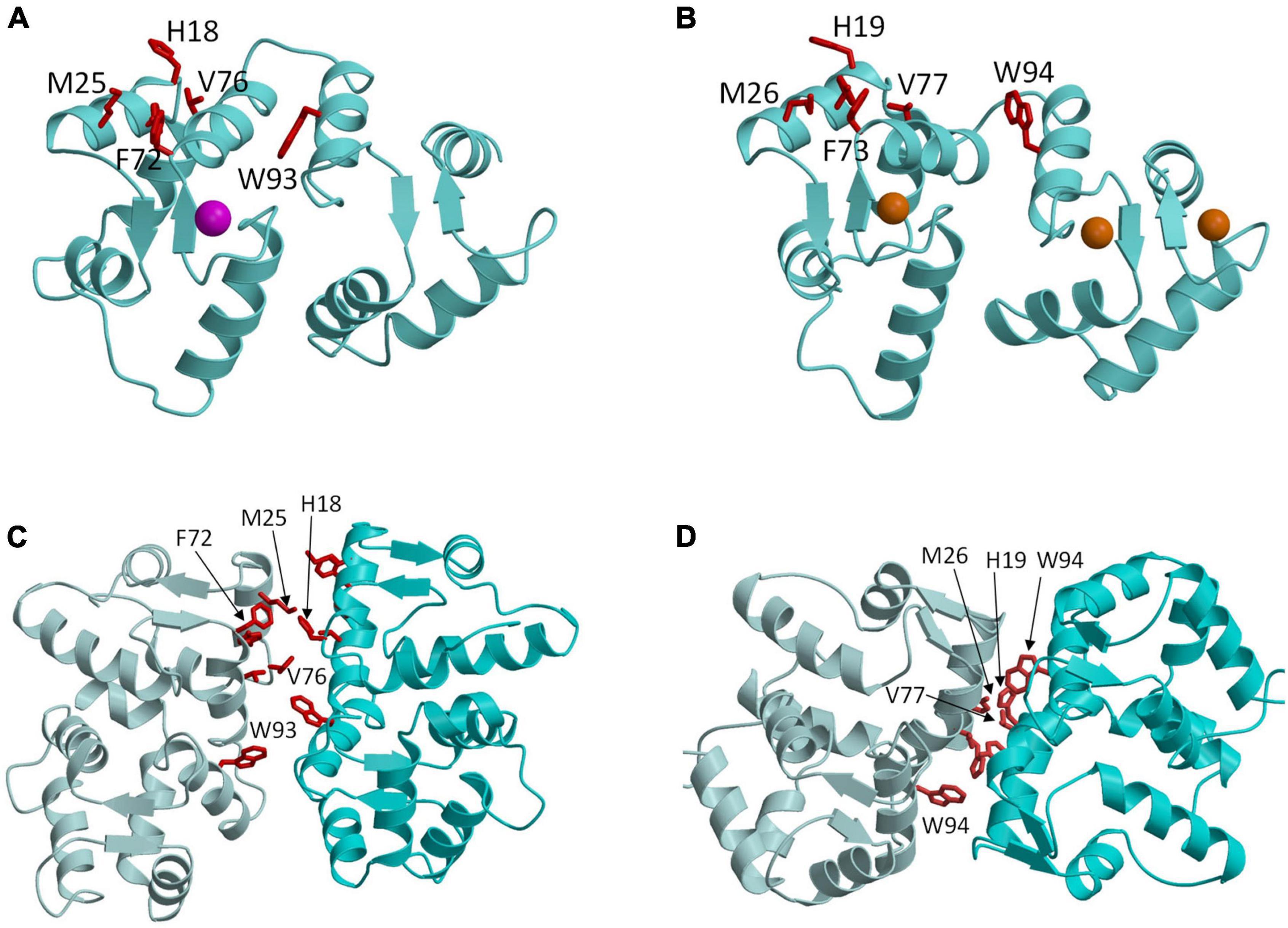
Figure 3. Molecular structures of GCAP1 and GCAP5. Main chain structures of Ca2+-free/Mg2+-bound GCAP5 (PDB ID: 7M2M) (A) and Ca2+-bound GCAP1 (PDB ID: 2R2I) (B). Bound Ca2+ and Mg2+ are orange and magenta. Dimeric structures of GCAP5 (C) and GCAP1 (D) determined by double electron electron resonance (DEER). Side chain atoms of exposed “hot spot” residues are shown as red sticks.
GCAP1 and GCAP5 form dimers in the absence of retinal membrane guanylate cyclases 1
GCAP1 and GCAP5 both form homodimers in solution in the absence of RetGC1 (Figures 3C,D). The dimerization of GCAP1 and GCAP5 are both Ca2+-independent and have a dimerization dissociation constant in the micromolar range (Lim et al., 2018; Boni et al., 2020; Cudia et al., 2021). Structural models of GCAP1 and GCAP5 dimers were both determined previously by measuring intermolecular DEER distances that served as restraints for molecular docking (Lim et al., 2018; Cudia et al., 2021; Figures 3C,D). The overall structures of both dimers are fairly similar (RMSD = 2.4 Å) and residues at the dimer interface are highly conserved (see red residues in Figure 3). An important structural difference is that the GCAP5 dimer forms an intermolecular salt bridge between R22 and D71 that is not seen in the GCAP1 dimer, and the GCAP5 mutation R22A abolishes dimerization (Cudia et al., 2021). The dimer structures of GCAP1 and GCAP5 are both stabilized by hydrophobic contacts at the dimer interface (Figures 3C,D). The most prominent intermolecular contacts involve conserved hydrophobic residues, H19, Y22, M26, F73, V77, and W94 in GCAP1 (Figure 3). In particular, the methyl side-chain atoms of V77 (V76 in GCAP5) each contact one another at the dimer interface and therefore explain why the V77E (or V76E in GCAP5) mutation significantly weakens protein dimerization for both GCAP1 and GCAP5 (Lim et al., 2016; Cudia et al., 2021). Individual point mutations at the dimer interface in GCAP1 (H19A, Y22A, F73A, V77E, and W94A) or GCAP5 (H18A, Y21A, F72A, V76E, and W93E) each weaken the dimerization dissociation constant and completely abolish the activation of RetGC1 by GCAP1 (Lim et al., 2018) or GCAP5 (Cudia et al., 2021). Thus, the exposed hydrophobic residues at the dimer interface are essential for both GCAP dimerization and activation of RetGC1.
Mutational hotspot residues in GCAP1 and GCAP5
The structures of GCAP1 (Figure 3B; Stephen et al., 2007) and GCAP5 (Figure 3A; Cudia et al., 2021) reveal exposed hydrophobic residues (H19, Y22, M26, F73, V77, and W94 in GCAP1 or H18, Y21, F72, V76, and W93 in GCAP5) that I propose could be targeted by rational drug design (see exposed residues highlighted red in Figure 3). The exposed hydrophobic residues are located at the dimer interface for both GCAP1 and GCAP5 (Figures 3C,D). Mutating the exposed residues in GCAP1 (H19A, Y22A, M26A, F73A, V77E, and W94E) each weaken dimerization and abolish cyclase activation (Lim et al., 2018). The corresponding mutations in GCAP5 (H18E, Y21E, M25E, F72E, V76E, and W93E) also weaken dimerization and abolish cyclase activation (Cudia et al., 2021). These results initially suggested that the dimeric structures of GCAP1 (and GCAP5) might be essential for cyclase activation. However, the GCAP5 mutant (R22A) was recently shown to abolish GCAP5 dimerization but still caused a threefold activation of the cyclase, which suggests that formation of a GCAP5 dimer (as seen in Figure 3C) may not be required to activate the cyclase. The enhanced cyclase activation caused by the monomeric R22A mutant is consistent with a previous suggestion that the exposed hotspot residues in GCAP1 may interact directly with RetGC1 (Peshenko et al., 2014) rather than mediate GCAP dimerization. If the exposed hotspot were to bind to RetGC1 with higher affinity than it binds to itself, then the apparent dimerization of GCAP1 and GCAP5 that occurs in the absence of RetGC1 might be an artifact of not having the cyclase present. Future studies are needed to test whether RetGC1 binding to GCAP1 prevents GCAP1 dimerization to distinguish whether the GCAP1 hotspot binds to RetGC1. Regardless of whether the exposed hotspot facilitates GCAP1/GCAP5 dimerization (Figure 3) or binds to RetGC1 (Figure 4), this hotspot (highlighted red in Figure 4) could serve as a binding site for an inhibitor whose binding would increase the apparent Km value of the cyclase activity. The binding of small molecules or peptides that selectively target the hotspot should prevent GCAP1 from activating RetGC1. In particular, small molecule inhibitors that selectively target the hot spot of constitutively active GCAP1 mutants [Y99C (Payne et al., 1998), D100G (Nong et al., 2014), E111V (Marino et al., 2018), and E155G (Wilkie et al., 2001)] should block the constitutive activation of RetGC1. Future studies are needed to screen for small molecule inhibitors that specifically target the exposed hotspot in GCAP1 mutants and test whether these drugs might slow the progression of cone-rod dystrophies.
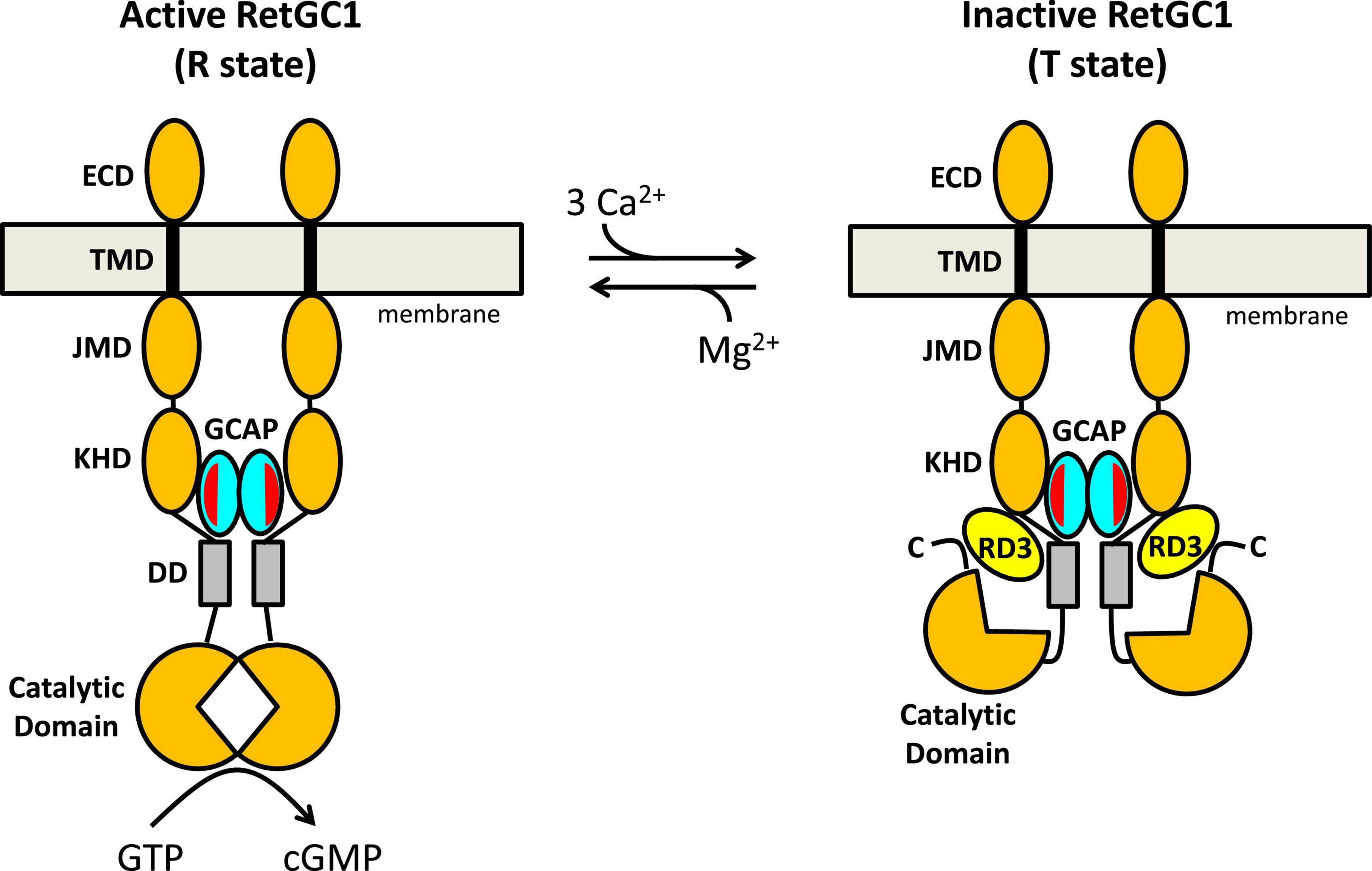
Figure 4. Schematic model of RetGC1 (orange) regulation by GCAP1 (cyan) and RD3 (yellow). Abbreviations of RetGC1 domains: ECD, extracellular domain; TMD, transmembrane domain; JMD, juxtamembrane domain; KHD, kinase homology domain; DD, dimerization domain (gray). The exposed hydrophobic patch in GCAP1 is red.
Structural basis of rd3 binding to retinal membrane guanylate cyclases 1
Retinal degeneration 3 forms an elongated four-helix bundle
The NMR structure of RD3 (Peshenko et al., 2019) reveals an elongated overall structure (70 Å long by 30 Å wide) that adopts a four-helix bundle (helix α1: P21–V51; α2: P75–K87; α3: P90–Q107; α4: V111–T139) (Figure 5A). The four helices in RD3 are bundled together in an antiparallel fashion with interhelical contacts formed by hydrophobic residues on the inner surface of helices α1 (residues L29 and L33), α3 (F100), and α4 (V114, F118, L122) whose side chain atoms point inward toward the hydrophobic core. The N-terminal and C-terminal ends of helices α1 and α4 are solvent exposed and the ends of these helices are rigidified by a series of salt bridge interactions (see blue and red side chain atoms in Figure 5A), which generates a long end-to-end distance in the elongated structure. A map of the electrostatic surface potential of RD3 reveals a negatively charged protein surface on one side of the protein (Figure 5B), in which many negatively charged glutamate side chain atoms (from E106, E108, E110, E113, E127, E132, E134) are clustered together on the protein surface and are suggested to make electrostatic contacts with RetGC1. A separate set of exposed residues in RD3 (see H89, C93, P95, I97, R99, R101, Q102 in Figure 5C) are clustered on the opposite side of the protein surface located near the center of the structure (highlighted red in Figure 5C). Site-specific mutations of these solvent exposed residues (see H89, C93, P95, I97, R99, R101, Q102, S120 in Figure 5C) have been shown to abolish RetGC1 regulation (Peshenko et al., 2016), suggesting that these residues might make direct contact with RetGC1.
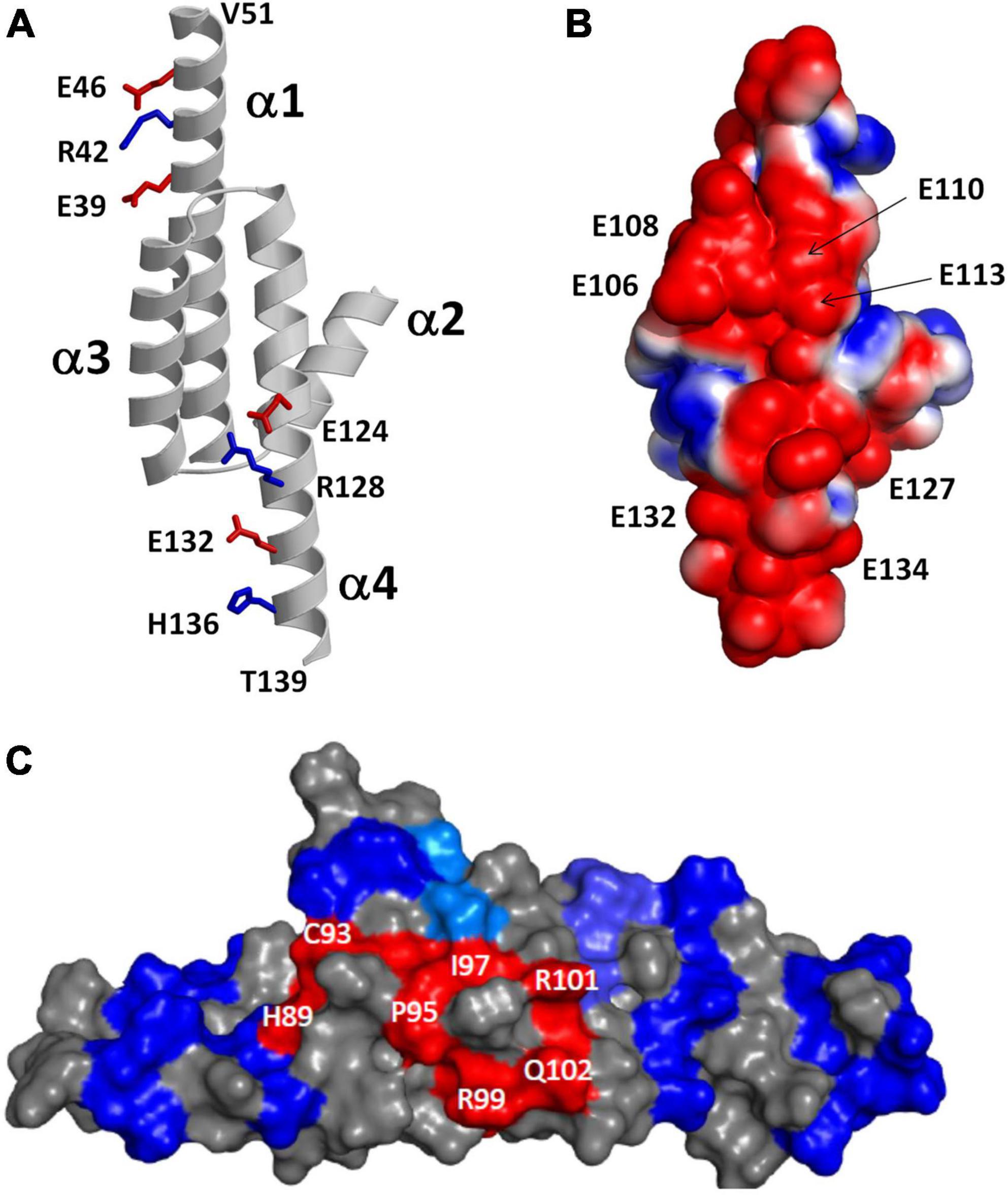
Figure 5. Structural analysis of RD3. Adapted from Peshenko et al. (2019). (A) Main chain structure of RD3. Salt bridges near the N-terminus (E39, R42, and E46) and C-terminus (E124, R128, E132, and H136) rigidify α1 and α4 to form an elongated 4-helix bundle. (B) Map of surface electrostatic potential of RD3 illustrates negatively charged protein surface (red). (C) Space-filling representation of RD3 shows residues on the protein surface (highlighted in red) that contact RetGC1.
Retinal degeneration 3 binding to retinal membrane guanylate cyclases 1
The three-dimensional structure of RD3 (Figure 5) provides clues about how RD3 might bind to and regulate RetGC1. Previous studies suggested that RD3 and GCAP1 may competitively bind to RetGC1 (Peshenko et al., 2011, 2016). The RD3 binding site on RetGC1 was therefore initially suggested to overlap at least partially with the GCAP1 binding site in RetGC1. GCAP1 and GCAP2 have been shown to interact primarily with the kinase homology and dimerization domains within RetGC1 (Peshenko et al., 2015a,b). The W708R mutation within the kinase homology domain of RetGC1 was recently shown to abolish RetGC1 binding with both RD3 and GCAP1, which is consistent with a partial overlapping of the GCAP1 and RD3 binding sites (Peshenko et al., 2015b). However, RD3 binding to RetGC1 was also disrupted by the removal of a C-terminal fragment downstream of the cyclase catalytic domain (Azadi et al., 2010). This same C-terminal deletion does not affect RetGC1 binding to GCAP1 (Peshenko et al., 2015b). Furthermore, particular point mutations in the RetGC1 dimerization domain (R822A and M823A) that block RetGC1 binding to GCAP1 have no effect on RetGC1 binding to RD3 (Peshenko et al., 2015a). These different binding properties suggest that RD3 and GCAP1 may have non-overlapping binding regions or perhaps a non-localized and multivalent binding site in each case. Future studies are needed to map the entire RD3 binding site in RetGC1.
Allosteric mechanism of retinal membrane guanylate cyclases 1 regulation by guanylate cyclase activating proteins 1 and retinal degeneration 3
A schematic model for the allosteric regulation of RetGC1 by GCAP1 and RD3 is presented in Figure 4. RetGC1 is known to form a dimer in the disk membrane (Liu et al., 1997; Olshevskaya et al., 1999; Yu et al., 1999). The dimeric RetGC1 is proposed to adopt two distinct quaternary structures: The R-state conformation places the two catalytic domains in close proximity to assemble the cyclase active site at the dimer interface in order to provide maximal enzymatic activity (Figure 4, left panel). The T-state conformation causes the catalytic domains to disassociate and disassemble the cyclase catalytic site, which should abolish the cyclase activity (Figure 4, right panel). The dimeric RetGC1 is believed to bind two molecules of GCAP1 to form a 2:2 complex (Peshenko et al., 2010). Previous studies have observed that GCAP1 may adopt a pre-formed dimer before binding to the RetGC1 dimer (Lim et al., 2018; Boni et al., 2020). However, more recent studies suggest that GCAP5 dimerization is not required for cyclase binding and activation (Cudia et al., 2021). Therefore, the exposed hydrophobic patch in GCAP1 (red residues in Figure 3) that facilitates GCAP1 dimerization in the absence of RetGC1 (Figures 3C,D) is proposed here to interact directly with RetGC1 as suggested previously (Peshenko et al., 2014) (see red patch in Figure 4). RetGC1 binding to Ca2+-free/Mg2+-bound GCAP1 at low Ca2+ levels (in light-activated photoreceptors) is proposed to stabilize the RetGC1 dimer in the active conformation (R), which shifts the equilibrium in favor of the active R state and turns on cyclase activity. The binding of Ca2+-bound GCAP1 to RetGC1 at high Ca2+ levels (in dark-adapted photoreceptors) is proposed to stabilize the RetGC1 dimer in the inactive conformation (T). In essence, GCAP1 is suggested here to serve as both a positive and negative allosteric effector for RetGC1: Ca2+-free GCAP1 shifts the allosteric equilibrium in favor of R, in contrast to Ca2+-bound GCAP1 that shifts the equilibrium in favor of T. In the absence of GCAP1, the RetGC1 dimer can occupy both conformational states (T and R). However, the conformational equilibrium in the absence of GCAP1 is shifted toward the inactive T-state, because the basal cyclase activity (in the absence of GCAP1) is more than 100-fold lower than it is in the presence of GCAP1.
The structural model of the RetGC1 dimer also explains how RD3 (yellow in Figure 4) might inactivate the cyclase activity (Figure 4, right panel). RD3 is proposed to bind to the RetGC1 dimer by contacting the KHD/dimerization domains (Peshenko et al., 2011, 2016) and the C-terminal region downstream of the catalytic domain (Azadi et al., 2010). This multivalent interaction is proposed to lock the RetGC1 dimer in the inactive T-state conformation. The binding of RD3 to RetGC1 (without the binding of GCAP1) must be sufficient to stabilize the inactive T-state, because RD3 alone can bind to and inactivate RetGC1 in the inner segment, which lacks GCAP1. Therefore, the binding of RD3 to inactive T-state is suggest here to be independent of the binding of Ca2 + -bound GCAP1, which would imply that the binding of either RD3 or Ca2+-bound GCAP1 are each sufficient to stabilize the inactive T-state. This hypothesis could be further tested by overexpressing RD3 (Peshenko et al., 2021) in the GCAP-/- double knockout strain (Pennesi et al., 2003) to verify whether the RD3 overexpression will cause lower basal RetGC1 activity and reduced dark current. Lastly, at low cytosolic Ca2+ levels, the binding of Ca2+-free/Mg2+-bound GCAP1 to the RetGC1 R-state (Figure 4 left panel) should prevent RD3 from binding to RetGC1, because the structural model in Figure 4 predicts that RD3 would not be able to form multivalent contacts with the active R-state. Therefore, an important prediction of this model is that Ca2+-free/Mg2+-bound GCAP1 should weaken RD3 binding to RetGC1 at low Ca2+ levels, in contrast to Ca2+-bound GCAP1 that can bind simultaneously with RD3 to the inactive T-state of RetGC1. Future studies are needed to test the predictions of the model and to determine atomic-resolution structures of RetGC1 bound to GCAP1 and RD3.
Conclusion
Retinal membrane guanylate cyclases (RetGC1 and RetGC2) are expressed in photoreceptor rod and cone cells where they promote the visual recovery phase of phototransduction. The cyclase enzymatic activity is oppositely regulated by GCAPs and RD3. Mutations in RetGC1, GCAP1 and RD3 that disable the Ca2+-dependent cyclase regulation are genetically linked to retinal degenerative diseases and inherited forms of blindness. A molecular model (Figure 4) was presented that explains how a RetGC1 dimer is allosterically regulated by the binding of GCAP1 and RD3. The binding of Ca2+-free/Mg2+-bound GCAP1 is proposed to stimulate cyclase activity by stabilizing RetGC1 in an active conformational state (R), whereas RD3 binding is proposed to decrease cyclase activity by locking RetGC1 in the inactive conformational state (T). The model predicts that Ca2+-free GCAP1 should inhibit RD3 binding to RetGC1, in contrast to Ca2+-bound GCAP1 that should enhance RetGC1 binding to RD3. These predictions could be tested by overexpressing RD3 in a GCAP-/- double knockout photoreceptor and measuring its effect on dark current. Exposed hydrophobic residues in GCAP1 (residues H19, Y22, M26, F73, V77, W94) are essential for cyclase activation and could be targeted by rational drug design for the possible treatment of rod/cone dystrophies.
Author contributions
JA wrote and conceived the entire manuscript.
Funding
This work was supported by the NIH (EY012347) to JA.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ames, J. B. (2019). Retinal Degeneration-3 (RD3) Protein is a Molecular Linchpin for Retinal Disease. EC Ophthalmol. 10, 10–12.
Ames, J. B. (2021). Structural Insights into Retinal Guanylate Cyclase Activator Proteins (GCAPs). Int. J. Mol. Sci. 22:8731.
Ames, J. B., Dizhoor, A. M., Ikura, M., Palczewski, K., and Stryer, L. (1999). Three-dimensional structure of guanylyl cyclase activating protein-2, a calcium-sensitive modulator of photoreceptor guanylyl cyclases. J. Biol. Chem. 274, 19329–19337. doi: 10.1074/jbc.274.27.19329
Azadi, S., Molday, L. L., and Molday, R. S. (2010). RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163. doi: 10.1073/pnas.1010460107
Baehr, W., Karan, S., Maeda, T., Luo, D. G., Li, S., Bronson, J. D., et al. (2007). The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 282, 8837–8847.
Baehr, W., and Palczewski, K. (2007). Guanylate cyclase-activating proteins and retina disease. Subcell. Biochem. 45, 71–91.
Behnen, P., Dell’ Orco, D., and Koch, K. W. (2010). Involvement of the calcium sensor GCAP1 in hereditary cone dystrophies. Biol. Chem. 391, 631–637.
Bondarenko, V. A., Hayashi, F., Usukura, J., and Yamazaki, A. (2010). Involvement of rhodopsin and ATP in the activation of membranous guanylate cyclase in retinal photoreceptor outer segments (ROS-GC) by GC-activating proteins (GCAPs): A new model for ROS-GC activation and its link to retinal diseases. Mol. Cell Biochem. 334, 125–139. doi: 10.1007/s11010-009-0323-y
Boni, F., Marino, V., Bidoia, C., Mastrangelo, E., Barbiroli, A., Dell’orco, D., et al. (2020). Modulation of Guanylate Cyclase Activating Protein 1 (GCAP1) Dimeric Assembly by Ca(2+) or Mg(2+): Hints to Understand Protein Activity. Biomolecules 10:1408. doi: 10.3390/biom10101408
Boye, S. L., Conlon, T., Erger, K., Ryals, R., Neeley, A., Cossette, T., et al. (2011). Long-term preservation of cone photoreceptors and restoration of cone function by gene therapy in the guanylate cyclase-1 knockout (GC1KO) mouse. Invest. Ophthalmol. Vis. Sci. 52, 7098–7108. doi: 10.1167/iovs.11-7867
Boye, S. L., Peshenko, I. V., Huang, W. C., Min, S. H., Mcdoom, I., Kay, C. N., et al. (2013). AAV-mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum. Gene Ther. 24, 189–202. doi: 10.1089/hum.2012.193
Burns, M. E., Mendez, A., Chen, J., and Baylor, D. A. (2002). Dynamics of Cyclic GMP Synthesis in Retinal Rods. Neuron 36, 81–91.
Chen, C., Nakatani, K., and Koutalos, Y. (2003). Free magnesium concentration in salamander photoreceptor outer segments. J. Physiol. 553, 125–135. doi: 10.1113/jphysiol.2003.053280
Cudia, D., Roseman, G. P., Assafa, T. E., Shahu, M. K., Scholten, A., Menke-Sell, S. K., et al. (2021). NMR and EPR-DEER Structure of a Dimeric Guanylate Cyclase Activator Protein-5 from Zebrafish Photoreceptors. Biochemistry 60, 3058–3070. doi: 10.1021/acs.biochem.1c00612
Dell’orco, D., Behnen, P., Linse, S., and Koch, K. W. (2010). Calcium binding, structural stability and guanylate cyclase activation in GCAP1 variants associated with human cone dystrophy. Cell Mol. Life Sci. 67, 973–984.
Dell’orco, D., Sulmann, S., Zagel, P., Marino, V., and Koch, K. W. (2014). Impact of cone dystrophy-related mutations in GCAP1 on a kinetic model of phototransduction. Cell Mol. Life Sci. 71, 3829–3840. doi: 10.1007/s00018-014-1593-4
Dizhoor, A. M., Boikov, S. G., and Olshevskaya, E. V. (1998). Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1. Possible role in causing human autosomal dominant cone degeneration. J. Biol. Chem. 273, 17311–17314. doi: 10.1074/jbc.273.28.17311
Dizhoor, A. M., and Hurley, J. B. (1996). Inactivation of EF-hands makes GCAP-2 (p24) a constitutive activator of photoreceptor guanylyl cyclase by preventing a Ca2+-induced “activator-to-inhibitor” transition. J. Biol. Chem. 271, 19346–19350. doi: 10.1074/jbc.271.32.19346
Dizhoor, A. M., Lowe, D. G., Olsevskaya, E. V., Laura, R. P., and Hurley, J. B. (1994). The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352. doi: 10.1016/0896-6273(94)90449-9
Dizhoor, A. M., Olshevskaya, E. V., Henzel, W. J., Wong, S. C., Stults, J. T., Ankoudinova, I., et al. (1995). Cloning, sequencing and expression of a 24-kDa Ca2+-binding protein activating photoreceptor guanylyl cyclase. J. Biol. Chem. 270, 25200–25206. doi: 10.1074/jbc.270.42.25200
Dizhoor, A. M., Olshevskaya, E. V., and Peshenko, I. V. (2010). Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): Role in regulation of photoreceptor guanylyl cyclase. Mol. Cell Biochem. 334, 117–124. doi: 10.1007/s11010-009-0328-6
Dizhoor, A. M., Olshevskaya, E. V., and Peshenko, I. V. (2021). Retinal degeneration-3 protein promotes photoreceptor survival by suppressing activation of guanylyl cyclase rather than accelerating GMP recycling. J. Biol. Chem. 296:100362. doi: 10.1016/j.jbc.2021.100362
Duda, T., Jankowska, A., Venkataraman, V., Nagele, R. G., and Sharma, R. K. (2001). A novel calcium-regulated membrane guanylate cyclase transduction system in the olfactory neuroepithelium. Biochemistry 40, 12067–12077. doi: 10.1021/bi0108406
Duda, T., Pertzev, A., Koch, K. W., and Sharma, R. K. (2012). Antithetical modes of and the Ca(2+) sensors targeting in ANF-RGC and ROS-GC1 membrane guanylate cyclases. Front. Mol. Neurosci. 5:44. doi: 10.3389/fnmol.2012.00044
Friedman, J. S., Chang, B., Kannabiran, C., Chakarova, C., Singh, H. P., Jalali, S., et al. (2006). Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070. doi: 10.1086/510021
Gorczyca, W., Polans, A., Surgucheva, I., Subbaraya, I., Baehr, W., and Palczewski, K. (1995). Guanylyl cyclase activating protein. A calcium-sensitive regulator of phototransduction. J. Biol. Chem. 270, 22029–22036. doi: 10.1074/jbc.270.37.22029
Gray-Keller, M. P., and Detwiler, P. B. (1994). The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron 13, 849–861.
Howes, K. A., Pennesi, M. E., Sokal, I., Church-Kopish, J., Schmidt, B., Margolis, D., et al. (2002). GCAP1 rescues rod photoreceptor response in GCAP1/GCAP2 knockout mice. EMBO J. 21, 1545–1554.
Imanishi, Y., Yang, L., Sokal, I., Filipek, S., Palczewski, K., and Baehr, W. (2004). Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish: Characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1-8) in pufferfish (Fugu rubripes). J. Mol. Evol. 59, 204–217. doi: 10.1007/s00239-004-2614-y
Jiang, L., and Baehr, W. (2010). GCAP1 Mutations Associated with Autosomal Dominant Cone Dystrophy. Adv. Exp. Med. Biol. 664, 273–282.
Koch, K. W., and Stryer, L. (1988). Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature 334, 64–66. doi: 10.1038/334064a0
Koutalos, Y., and Yau, K. W. (1996). Regulation of sensitivity in vertebrate rod photoreceptors by calcium. Trends Neurosci. 19, 73–81.
Laura, R. P., Dizhoor, A. M., and Hurley, J. B. (1996). The membrane guanylyl cyclase, retinal guanylyl cyclase-1, is activated through its intracellular domain. J. Biol. Chem. 271, 11646–11651.
Laura, R. P., and Hurley, J. B. (1998). The kinase homology domain of retinal guanylyl cyclases 1 and 2 specifies the affinity and cooperativity of interaction with guanylyl cyclase activating protein-2. Biochemistry 37, 11264–11271. doi: 10.1021/bi9809674
Lim, S., Peshenko, I. V., Dizhoor, A. M., and Ames, J. B. (2009). Effects of Ca2+, Mg2+, and myristoylation on guanylyl cyclase activating protein 1 structure and stability. Biochemistry 48, 850–862. doi: 10.1021/bi801897p
Lim, S., Peshenko, I. V., Olshevskaya, E. V., Dizhoor, A. M., and Ames, J. B. (2016). Structure of Guanylyl Cyclase Activator Protein 1 (GCAP1) Mutant V77E in a Ca2+-free/Mg2+-bound Activator State. J. Biol. Chem. 291, 4429–4441. doi: 10.1074/jbc.M115.696161
Lim, S., Roseman, G., Peshenko, I., Manchala, G., Cudia, D., Dizhoor, A., et al. (2018). Retinal Guanylyl Cyclase Activating Protein 1 Forms a Functional Dimer. PLoS One 13:e0193947. doi: 10.1371/journal.pone.0193947
Liu, Y., Ruoho, A. E., Rao, V. D., and Hurley, J. B. (1997). Catalytic mechanism of the adenylyl and guanylyl cyclases: Modeling and mutational analysis. Proc. Natl. Acad. Sci. U.S A. 94, 13414–13419. doi: 10.1073/pnas.94.25.13414
Lowe, D. G., Dizhoor, A. M., Liu, K., Gu, Q., Spencer, M., Laura, R., et al. (1995). Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 6, 5535–5539. doi: 10.1073/pnas.92.12.5535
Marino, V., Dal Cortivo, G., Oppici, E., Maltese, P. E., D’esposito, F., Manara, E., et al. (2018). A novel p.(Glu111Val) missense mutation in GUCA1A associated with cone-rod dystrophy leads to impaired calcium sensing and perturbed second messenger homeostasis in photoreceptors. Hum. Mol. Genet. 27, 4204–4217. doi: 10.1093/hmg/ddy311
Marino, V., Sulmann, S., Koch, K. W., and Dell’orco, D. (2015). Structural effects of Mg(2)(+) on the regulatory states of three neuronal calcium sensors operating in vertebrate phototransduction. Biochim. Biophys. Acta. 1853, 2055–2065. doi: 10.1016/j.bbamcr.2014.10.026
Mendez, A., and Chen, J. (2002). Mouse models to study GCAP functions in intact photoreceptors. Adv. Exp. Med. Biol. 514, 361–388.
Molday, L. L., Djajadi, H., Yan, P., Szczygiel, L., Boye, S. L., Chiodo, V. A., et al. (2013). RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum. Mol. Genet. 22, 3894–3905. doi: 10.1093/hmg/ddt244
Molday, L. L., Jefferies, T., and Molday, R. S. (2014). Insights into the role of RD3 in guanylate cyclase trafficking, photoreceptor degeneration, and Leber congenital amaurosis. Front. Mol. Neurosci. 7:44. doi: 10.3389/fnmol.2014.00044
Newbold, R. J., Deery, E. C., Payne, A. M., Wilkie, S. E., Hunt, D. M., and Warren, M. J. (2002). Guanylate cyclase activating proteins, guanylate cyclase and disease. Adv. Exp. Med. Biol. 514, 411–438.
Nong, E., Lee, W., Merriam, J. E., Allikmets, R., and Tsang, S. H. (2014). Disease progression in autosomal dominant cone-rod dystrophy caused by a novel mutation (D100G) in the GUCA1A gene. Doc. Ophthalmol. 128, 59–67. doi: 10.1007/s10633-013-9420-z
Olshevskaya, E. V., Ermilov, A. N., and And Dizhoor, A. M. (1999). Dimerization of guanylyl cyclase-activating protein. J. Biol. Chem. 274, 25583–25587.
Palczewski, K., Sokal, I., and Baehr, W. (2004). Guanylate cyclase-activating proteins: Structure, function, and diversity. Biochem. Biophys. Res. Commun. 322, 1123–1130.
Palczewski, K., Subbaraya, I., Gorczyca, W. A., Helekar, B. S., Ruiz, C. C., Ohguro, H., et al. (1994). Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron 13, 395–404.
Payne, A. M., Downes, S. M., Bessant, D. A., Taylor, R., Holder, G. E., Warren, M. J., et al. (1998). A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum. Mol. Genet. 7, 273–277. doi: 10.1093/hmg/7.2.273
Pennesi, M. E., Howes, K. A., Baehr, W., and Wu, S. M. (2003). Guanylate cyclase-activating protein (GCAP) 1 rescues cone recovery kinetics in GCAP1/GCAP2 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 100, 6783–6788. doi: 10.1073/pnas.1130102100
Peshenko, I. V., and Dizhoor, A. M. (2004). Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: Implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J. Biol. Chem. 279, 16903–16906. doi: 10.1074/jbc.C400065200
Peshenko, I. V., and Dizhoor, A. M. (2006). Ca2+ and Mg2+ binding properties of GCAP-1. Evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J. Biol. Chem. 281, 23830–23841. doi: 10.1074/jbc.M600257200
Peshenko, I. V., and Dizhoor, A. M. (2007). Activation and inhibition of photoreceptor guanylyl cyclase by guanylyl cyclase activating protein 1 (GCAP-1): The functional role of Mg2+/Ca2+ exchange in EF-hand domains. J. Biol. Chem. 282, 21645–21652. doi: 10.1074/jbc.M702368200
Peshenko, I. V., Olshevskaya, E. V., Azadi, S., Molday, L. L., Molday, R. S., and Dizhoor, A. M. (2011). Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519.
Peshenko, I. V., Olshevskaya, E. V., and Dizhoor, A. M. (2015b). Evaluating the Role of Retinal Guanylyl Cyclase 1 (RetGC1) Domains In Binding Guanylyl Cyclase Activating Proteins (GCAP). J. Biol. Chem. 290, 6913–6924.
Peshenko, I. V., Olshevskaya, E. V., and Dizhoor, A. M. (2015a). Dimerization Domain of Retinal Membrane Guanylyl Cyclase 1 (RetGC1) Is an Essential Part of Guanylyl Cyclase-activating Protein (GCAP) Binding Interface. J. Biol. Chem. 290, 19584–19596. doi: 10.1074/jbc.M115.661371
Peshenko, I. V., Olshevskaya, E. V., and Dizhoor, A. M. (2016). Functional Study and Mapping Sites for Interaction with the Target Enzyme in Retinal Degeneration 3 (RD3) Protein. J. Biol. Chem. 291, 19713–19723. doi: 10.1074/jbc.M116.742288
Peshenko, I. V., Olshevskaya, E. V., and Dizhoor, A. M. (2021). Retinal degeneration-3 protein attenuates photoreceptor degeneration in transgenic mice expressing dominant mutation of human retinal guanylyl cyclase. J. Biol. Chem. 297:101201. doi: 10.1016/j.jbc.2021.101201
Peshenko, I. V., Olshevskaya, E. V., Lim, S., Ames, J. B., and Dizhoor, A. M. (2014). Identification of target binding site in photoreceptor guanylyl cyclase-activating protein 1 (GCAP1). J. Biol. Chem. 289, 10140–10154. doi: 10.1074/jbc.M113.540716
Peshenko, I. V., Olshevskaya, E. V., Yao, S., Ezzeldin, H. H., Pittler, S. J., and Dizhoor, A. M. (2010). Activation of retinal guanylyl cyclase RetGC1 by GCAP1: Stoichiometry of binding and effect of new LCA-related mutations. Biochemistry 49, 709–717. doi: 10.1021/bi901495y
Peshenko, I. V., Yu, Q., Lim, S., Cudia, D., Dizhoor, A. M., and Ames, J. B. (2019). Retinal degeneration 3 (RD3) protein, a retinal guanylyl cyclase regulator, forms a monomeric and elongated four-helix bundle. J. Biol. Chem. 294, 2318–2328. doi: 10.1074/jbc.RA118.006106
Polans, A., Baehr, W., and Palczewski, K. (1996). Turned on by Ca2+! The physiology and pathology of Ca(2+)-binding proteins in the retina. Trends Neurosci. 19, 547–554. doi: 10.1016/s0166-2236(96)10059-x
Scholten, A., and Koch, K. W. (2011). Differential calcium signaling by cone specific guanylate cyclase-activating proteins from the zebrafish retina. PLoS One 6:e23117. doi: 10.1371/journal.pone.0023117
Semple-Rowland, S. L., Gorczyca, W. A., Buczylko, J., Helekar, B. S., Ruiz, C. C., Subbaraya, I., et al. (1996). Expression of GCAP1 and GCAP2 in the retinal degeneration (rd) mutant chicken retina. FEBS Lett. 385, 47–52. doi: 10.1016/0014-5793(96)00345-6
Sokal, I., Li, N., Surgucheva, I., Warren, M. J., Payne, A. M., Bhattacharya, S. S., et al. (1998). GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol. Cell 2, 129–133.
Stephen, R., Bereta, G., Golczak, M., Palczewski, K., and Sousa, M. C. (2007). Stabilizing function for myristoyl group revealed by the crystal structure of a neuronal calcium sensor, guanylate cyclase-activating protein 1. Structure 15, 1392–1402. doi: 10.1016/j.str.2007.09.013
Wilkie, S. E., Li, Y., Deery, E. C., Newbold, R. J., Garibaldi, D., Bateman, J. B., et al. (2001). Identification and functional consequences of a new mutation (E155G) in the gene for GCAP1 that causes autosomal dominant cone dystrophy. Am. J. Hum. Genet. 69, 471–480. doi: 10.1086/323265
Wimberg, H., Janssen-Bienhold, U., and Koch, K. W. (2018). Control of the Nucleotide Cycle in Photoreceptor Cell Extracts by Retinal Degeneration Protein 3. Front. Mol. Neurosci. 11:52. doi: 10.3389/fnmol.2018.00052
Woodruff, M. L., Sampath, A. P., Mathews, H. R., Krasnoperova, N. V., Lem, J., and Fain, G. L. (2002). Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542, 843–854. doi: 10.1113/jphysiol.2001.013987
Yang, R.-B., Robinson, S. W., Xiong, W.-H., Yau, K. W., Birch, D. G., and Garbers, D. L. (1999). Disruption of a Retinal Guanylyl Cyclase Gene Leads to Cone-Specific Dystrophy and Paradoxical Rod Behavior. J. Neurosci. 19, 5889–5897. doi: 10.1523/JNEUROSCI.19-14-05889.1999
Yu, H., Olshevskaya, E., Duda, T., Seno, K., Hayashi, F., Sharma, R. K., et al. (1999). Activation of retinal guanylyl cyclase-1 by Ca2+-binding proteins involves its dimerization. J. Biol. Chem. 274, 15547–15555.
Keywords: calcium, GCAP1, GCAP5, RD3 protein, guanylate cyclase (guanylyl cyclase), NMR
Citation: Ames JB (2022) Structural basis of retinal membrane guanylate cyclase regulation by GCAP1 and RD3. Front. Mol. Neurosci. 15:988142. doi: 10.3389/fnmol.2022.988142
Received: 07 July 2022; Accepted: 22 August 2022;
Published: 08 September 2022.
Edited by:
Clint L. Makino, Boston University, United StatesReviewed by:
Ching-Kang Jason Chen, Baylor College of Medicine, United StatesTiansen Li, National Institutes of Health (NIH), United States
Copyright © 2022 Ames. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James B. Ames, amJhbWVzQHVjZGF2aXMuZWR1