
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Mol. Neurosci., 16 September 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.974890
This article is part of the Research TopicMolecular Advances and Applications of Machine Learning in Understanding Autism and Comorbid Psychiatric DisordersView all 16 articles
 Rebecca A. DeGiosio1
Rebecca A. DeGiosio1 Melanie J. Grubisha1
Melanie J. Grubisha1 Matthew L. MacDonald1
Matthew L. MacDonald1 Brandon C. McKinney1
Brandon C. McKinney1 Carlos J. Camacho2
Carlos J. Camacho2 Robert A. Sweet1,3*
Robert A. Sweet1,3*Microtubule-associated protein 2 (MAP2) is the predominant cytoskeletal regulator within neuronal dendrites, abundant and specific enough to serve as a robust somatodendritic marker. It influences microtubule dynamics and microtubule/actin interactions to control neurite outgrowth and synaptic functions, similarly to the closely related MAP Tau. Though pathology of Tau has been well appreciated in the context of neurodegenerative disorders, the consequences of pathologically dysregulated MAP2 have been little explored, despite alterations in its immunoreactivity, expression, splicing and/or stability being observed in a variety of neurodegenerative and neuropsychiatric disorders including Huntington’s disease, prion disease, schizophrenia, autism, major depression and bipolar disorder. Here we review the understood structure and functions of MAP2, including in neurite outgrowth, synaptic plasticity, and regulation of protein folding/transport. We also describe known and potential mechanisms by which MAP2 can be regulated via post-translational modification. Then, we assess existing evidence of its dysregulation in various brain disorders, including from immunohistochemical and (phospho) proteomic data. We propose pathways by which MAP2 pathology could contribute to endophenotypes which characterize these disorders, giving rise to the concept of a “MAP2opathy”—a series of disorders characterized by alterations in MAP2 function.
The microtubule (MT) cytoskeleton is a fundamental coordinator of neuronal structure and function. MTs, comprised of heterodimers of α- and β-tubulin subunits, are present in all cell types and provide dynamic support to enable cell migration/division, modify and maintain cellular shape, and serve as tracks for intracellular trafficking (Goodson and Jonasson, 2018). In neurons, the MT network critically defines neurite morphology and mediates trafficking processes essential for synaptic transmission (Kapitein and Hoogenraad, 2015). This network is carefully regulated by a series of proteins which manipulate MT stability and/or arrangement.
Among these, the microtubule-associated proteins (MAPs) define a set of proteins which directly bind MTs, typically serving to stabilize them (Bodakuntla et al., 2019). MAPs additionally can have scaffolding functions, recruiting other cytoskeleton-modifying proteins or signaling pathway components to specific subcellular locations. Given the essential roles the MT cytoskeleton plays in neurons, it is unsurprising that pathology of both tubulin and various MAPs have been identified as direct precipitants of various brain disorders. The tubulinopathies, for instance, describe a set of cortical malformations caused by mutations in the several tubulin genes, while the tauopathies define a group of neurodegenerative diseases—most notably Alzheimer’s Disease—characterized by pathology of the axonal MAP Tau.
These conditions overlap in part with “opathies” generated by dysfunction of intrinsically disordered proteins (IDPs), which have several features that confer unique pathogenic potential (Box 1). The structurally fluid nature of IDPs, including Tau, makes them well suited to play roles in numerous, diverse signaling pathways (Dunker et al., 2005); thus, depending on the exact pathology, a range of pleiotropic effects can be observed in IDP pathogenesis. For instance, tauopathies are distinguishable by differences in the affected Tau isoform(s), affected cell type(s), and/or fibrillar Tau aggregate structure. A similar degree of heterogeneity exists in the synucleopathies, resulting from pathologies in the IDP alpha-synuclein. In such disorders, the IDP does not represent a specific locus of defined pathogenesis, but rather a shared hub of pathology which can lead to various effects depending on the exact nature of the pathology and what upstream factors precipitated it.
Box 1. Features of intrinsically disordered proteins and their pathology.
IDPs are proteins which lack a stable tertiary structure. They are present in all organisms, being particularly abundant in eukaryotes, where over 30% of all proteins are estimated to have disordered regions (Ward et al., 2004). Their ability to adopt a variety of transient conformations tailored to different interacting partners enables their engagement in both “one-to-many” and “many-to-one” signaling, as well as their ability to scaffold together proteins, e.g., components of a particular signaling pathway. IDPs are heavily modified by post-translational modification (Pejaver et al., 2014) and alternative splicing (Romero et al., 2006), and tend to have relatively low rates of synthesis and short half-lives (Gsponer et al., 2008), ensuring tight regulation of their expression and activity.
These features make IDPs efficient master regulators of diverse signaling pathways, but also leave them more prone to potential dysregulations which can give rise to disease. Genetic mutations or changes in expression, splicing, modifications, trafficking and/or degradation all can lead to aberrant complex formation, signaling activity, and/or folding. The “IDPopathies” encompass a large group of human disorders which include various cancers, neurodegenerative diseases, diabetes, and cardiovascular disease (Uversky, 2014). Frequently the misfolding of proteins including IDPs also leads to their aggregation, giving rise to the amyloidoses, in which more than 37 distinct proteins have been implicated (Chiti and Dobson, 2017).
MAP2 is an abundant dendritic MAP as well as an IDP which is closely related to Tau. It has been long appreciated for its roles in defining and maintaining dendritic structure; however, its neuropathological potential has been minimally explored. Evidence is beginning to suggest that MAP2 can be a similar pathogenic hub to other IDPs and MAPs, with various forms of MAP2 dysfunction having distinct causal factors and outcomes. For instance, ischemia leads to calpain-induced MAP2 cleavage which is thought to disrupt cytoskeletal integrity and may contribute to neuronal atrophy (Pettigrew et al., 1996). Similar MAP2 degradation occurs in prion disease (Guo et al., 2012). In contrast, in Huntington’s Disease, MAP2 splicing is altered, leading to an imbalance between high and low molecular weight MAP2 forms that is thought to contribute to the dendritic atrophy which characterizes the disorder (Cabrera and Lucas, 2017). Alternatively, our recent work in primary auditory cortex indicates that in schizophrenia, MAP2 is hyperphosphorylated, leading to changes in dendritic architecture (Grubisha et al., 2021).
The goal of this review is to summarize the current understanding of MAP2 function and regulation and review the existing evidence for MAP2 dysregulation in disorder, discussing potential forms and consequences of MAP2 pathology. From this, we contemplate a new conceptual framework for schizophrenia and other disorders as “MAP2opathies” in parallel to the tauopathies, bearing implications for their study and treatment.
MAP2 is comprised of five functionally distinct domains: the N-terminus, a projection domain, a proline-rich domain, the MT-binding domain, and the C-terminus (Figure 1A). The MT-binding domain in turn contains 3–4 MT binding imperfect repeats, though only the last of these seems to be necessary for the MT bundling activity of the protein (Ludin et al., 1996). Though this domain was originally identified as the critical domain for MT-binding functionality based on its affinity for bovine MTs (Lewis et al., 1988), subsequent data has suggested that it alone is not sufficient for full MT-binding of MAP2. Indeed, addition of flanking sequences from both the proline-rich and C-terminal domains is necessary to match full-length MT-binding activity in transfected HeLa cells (Ferralli et al., 1994). Though the precise mechanism of MAP2/MT interaction remains unknown, the “jaws” model proposed for MT binding by Tau (Gustke et al., 1994)—with which MAP2 shares 90% sequence homology (Figure 1B) —may provide a framework to visualize this interaction, wherein flanking domains are responsible for positioning MAP2/Tau on the MT while the MT-binding domain mediates tubulin polymerization.
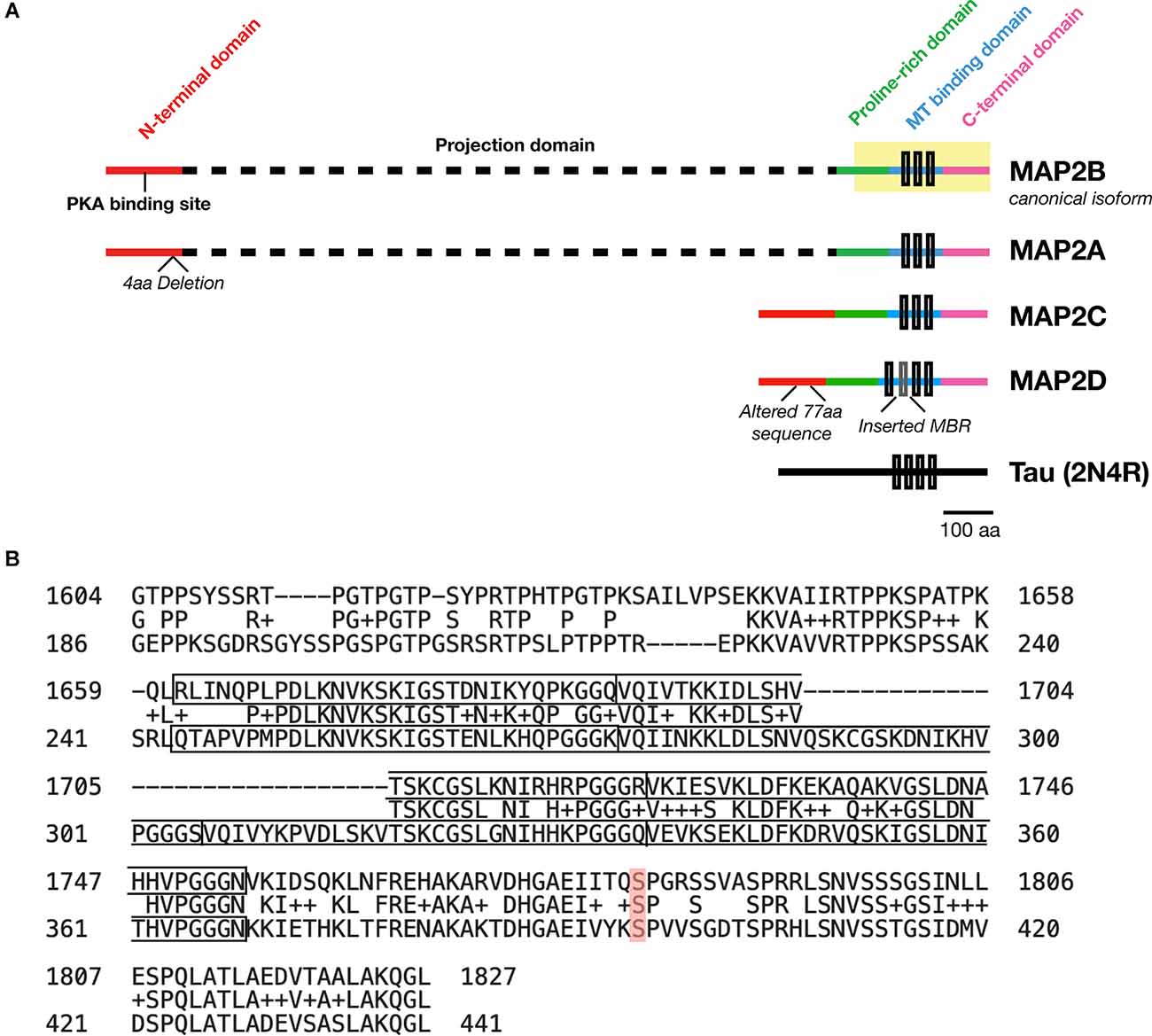
Figure 1. Domains of MAP2 and homology to MAP Tau. (A) Diagram depicting the domains of the four major isoforms of MAP2. Microtubule (MT)-binding repeats are denoted by black rectangles. Note that an additional MT binding repeat (MBR; gray box) is present in the low molecular weight (LMW) isoform MAP2D. Tau is also shown for size comparison. (B) Protein BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) alignment of the C-terminus (highlighted region of MAP2B in A) from MAP2B (Uniprot P11137-1; top) and 2N4R Tau (P10636-8; bottom). The MT-binding repeat sequences for each protein are indicated with solid boxes. The middle row indicates homologous or similar (+) residues. MAP2B S1782 (mentioned in the text) is highlighted in red. Figure generated in Microsoft Powerpoint.
In addition to facilitating MT binding, these flanking domains are likely to serve roles in mediating interactions with key binding partners. The closely homologous proline-rich domain of Tau, for instance, mediates interaction with SH3 domain-containing proteins, and MAP2 is known to interact with several such proteins such as the +TIP protein EB3 (Kapitein et al., 2011), Src and Grb2 (Lim and Halpain, 2000). The N-terminus of MAP2 also notably contains a binding site for the PKA regulatory subunit subtype II. As such, MAP2 is the primary dendritic A-kinase anchoring protein (AKAP; Zhong et al., 2009). In contrast, the projection domain appears to regulate MT spacing (Chen et al., 1992).
MAP2 has four major isoforms in vivo derived from alternative splicing: high molecular weight (HMW) MAP2A and MAP2B (the canonical isoform), and low molecular weight (LMW) MAP2C and MAP2D. These four MAP2 isoforms share four of the five functional domains, with the projection domain being selective to the HMW forms. In human, MAP2A possesses a 4-residue deletion relative to MAP2B, and MAP2D contains an additional MT-binding repeat and a modified 77-residue segment in the N-terminal domain relative to the other three isoforms (see Figure 1A). Whereas the subcellular localization of Tau is predominantly axonal (although some Tau has been observed in dendrites and dendritic spines), MAP2 are predominantly somatodendritic (Caceres et al., 1984), although MAP2C can be detected in axons (Meichsner et al., 1993), and MAP2D may be expressed in some glia (Doll et al., 1993). Additional splice variants—including protein coding transcripts—of MAP2 may also exist but remain uncharacterized (Howe et al., 2021).
Following transcription, MAP2 mRNA localizes to neuronal dendrites for local translation (Garner et al., 1988). In rat neurons, translocation of this mRNA is dependent upon the trans-acting protein MARTA2—which has high affinity for a cis-acting 3’ UTR dendritic targeting element (Blichenberg et al., 1999)—as well as the kinesin KIF5 (Rehbein et al., 2000; Zivraj et al., 2013). Evidence suggests that local MAP2 protein translation in dendrites can be activity-dependent; prolonged high-frequency stimulation of the middle molecular layer of rat dentate gyrus causes an increase in MAP2 immunoreactivity in surrounding lamina, which is diminished by inhibition of protein synthesis (Steward and Halpain, 1999). This may be mediated by mTOR signaling, as rapamycin blocks tetanus-induced dendritic synthesis of MAP2 protein in hippocampal slice (Gong et al., 2006).
An enhanced selectivity of the HMW forms for the dendritic compartment may be due to presence of the projection domain, which inhibits MAP2A/B access to axons (Kanai and Hirokawa, 1995), perhaps via interactions with axonal initial segment proteins such as AnkyrinG (Hedstrom et al., 2008). Differential MT organization in dendrites and axons is subsequently thought to result from MAP2 vs. Tau binding (Chen et al., 1992). In addition to the dendritic shaft, MAP2 has also been observed in dendritic spines as early as 1989 by immunogold electron microscopy (Morales and Fifkova, 1989), though it is found less abundantly in spines in the absence of synaptic stimulus (see Section “Synaptic plasticity”).
MAP2 is expressed shortly after the switch from neuronal precursor to neuron, and its isoforms are differentially expressed across neurodevelopment. MAP2B is expressed throughout development; MAP2A levels increase during development; MAP2C levels decrease during development, although it remains present in adulthood (Jalava et al., 2007). As a result, HMW MAP2 forms the vast majority of MAP2 in adulthood. In addition, MAP2C is expressed in the mature testis where it is involved in spermatogenesis (Sun and Handel, 2011).
It has been long accepted that the primary function of MAP2, like Tau, is to bind MTs to increase their polymerization and bundling (Herzog and Weber, 1978; Stearns and Brown, 1979; Lewis et al., 1989). MAP2 is also commonly called an MT stabilizer, though importantly it does not suppress MT dynamics; rather, it reduces the frequency of depolymerizing “catastrophe” events in assembling MTs to enable their growth (Itoh and Hotani, 1994). This mechanism of MAP2 (and Tau; Baas and Qiang, 2019) can be thought of as a “dynamics-preserving” stabilization, as opposed to that of the “genuine” stabilizer MAP6, knockdown of which increases MT dynamics in the axonal compartment (Tortosa et al., 2017).
In addition to MTs, MAP2 can also bind and bundle actin, like Tau (Correas et al., 1990; Moraga et al., 1993; Roger et al., 2004). Thus, both proteins act as cross-linkers between the MT and actin networks (Mohan and John, 2015). Such crosstalk is important for various aspects of fundamental cellular functions such as cell motility and division. However, both proteins may affect actin filaments differently; for instance, phosphatidylinositol disrupts MAP2C- but not Tau-induced actin bundles (Yamauchi and Purich, 1993).
The MT polymerizing and bundling effects of MAP2 facilitate process formation and maintain mature dendritic structure. Indeed, expression of MAP2 has repeatedly been shown to induce process formation in multiple heterologous cell lines (Edson et al., 1993; Leclerc et al., 1996). In neurons, MAP2 may act as the primary target of a number of endogenous agents which affect neurite structure via MT dynamics. For instance, pregnenolone—a neurosteroid with potential antidepressant and neuroprotective properties (Osuji et al., 2010; Marx et al., 2011, 2014)—enhances neurite growth by binding directly to MAP2 to simulate tubulin polymerization (Murakami et al., 2000; Fontaine-Lenoir et al., 2006). Melatonin increases neurite length, cellular MT content and MAP2 expression in a variety of contexts (Meléndez et al., 1996; Prieto-Gómez et al., 2008; Shu et al., 2016).
The neurite-forming and elongating effects of MAP2 can be disrupted by multiple interventions. The process appears to be negatively regulated by the projection domain, as MAP2B has a lower capacity for process formation than MAP2C in Sf9 cells (Bélanger et al., 2002). Expression of antisense oligonucleotides against MAP2 reduces the number of neurites and MTs within neurites in primary cortical cultures (Sharma et al., 1994). Similarly, in vivo knockout of MAP2 decreases dendritic length and MT density in hippocampal neurons (Harada et al., 2002). Expression of mutant MAP2 constructs can also be used to disrupt dendritic morphogenesis; overexpression of a phosphomimetic MAP2C construct which cannot bind to MTs (S319E/S350E/S382E; Ozer and Halpain, 2000) yields shortened neurites both in dissociated neuronal culture (Huang et al., 2013) and in Neuro-2a cells (Dehmelt et al., 2003) relative to wild-type MAP2C expression. Therefore, changes in MAP2 expression, splicing, or phosphorylation could drive pathological changes in dendritic morphology, namely in reduced dendritic outgrowth.
MAP2 disruption can also affect structural and functional plasticity of synapses. As shown in the work of Kim et al. (2020); HMW MAP2 is recruited to spines following chemical long-term potentiation (LTP) induction, whereas knockdown of HMW MAP2 impairs LTP induction, LTP-induced growth of dendritic spines, and AMPAR recruitment to spines. This function appears to be selective to the HMW forms of MAP2, as MAP2C-GFP fails to translocate into spines in an activity-dependent manner. Notably, knockdown of HMW MAP2 did not alter spine density in this study; however, we have recently established that at least one MAP2 phosphomimetic mutant (S1782E) is associated with a reduction in spine density in cortical pyramidal neurons of CRISPR knock-in mice (Grubisha et al., 2021). Thus, MAP2 can also contribute to spine formation and/or maintenance, however these functions may be compensated for by related cytoskeletal proteins (such as MAP1B; Teng et al., 2001) in the case of knockdown/knockout.
The role of MT dynamics in mediating the structural and functional plasticity of dendritic spines has become well-appreciated following the discovery that MTs transiently invade spines in an activity-dependent manner (Gu et al., 2008; Hu et al., 2008; Mitsuyama et al., 2008; Jaworski et al., 2009). Such invasions facilitate NMDAR-dependent spine head growth (Merriam et al., 2011), delivering cargo to the spine head in response to synaptic activity (McVicker et al., 2016). The link between regulation of MT dynamics and spine structure is further supported by findings that inhibition of MT polymerization leads to impaired LTP, regression of mature-appearing spines to immature spine shapes, and loss of spines (Jaworski et al., 2009). Thus, it is plausible that MAP2 mediates its effects on synaptic plasticity via MT dynamics. Indeed, the binding of MAP2 to MTs is itself an activity-dependent process (Vaillant et al., 2002), likely guided via phosphorylation (see below). Recent studies have also illuminated the importance of MT-actin crosstalk to spine morphogenesis, with actin remodeling at activated spines promoting MT entry (Merriam et al., 2013; Schätzle et al., 2018), although to date it is unknown if the MT-actin crosslinking function of MAP2 plays a role in this process.
In addition to its direct interaction with cytoskeletal networks, MAP2 also may mediate its effects on plasticity via its interactome. MAP2 creates local dendritic reservoirs of proteins critical to LTP/LTD, including PKA, EB3, RNA particles and polyribosomes (Lim and Halpain, 2000; Nielsen et al., 2002; Ostroff et al., 2002; Angenstein et al., 2005; Farah et al., 2005; Zhong et al., 2009; Kapitein et al., 2011; Sontag et al., 2012; Chirillo et al., 2019; Kim et al., 2020). MAP2 is known to mediate the relocalization of EB3 to the dendritic shaft following chemical long-term depression (cLTD) stimulus, concurrent with a reduction in MT invasion events (Kapitein et al., 2011). Conversely, translocation of HMW MAP2 to spine heads following chemical LTP (Kim et al., 2020) raises the possibility that MAP2 may contribute to activity-dependent protein trafficking into spines. Additionally, as an AKAP, MAP2 is thought to contribute to a spatial gradient of type II PKA formed between the dendritic shaft and spines following cAMP elevation, which in turn impacts synaptic strength and LTP induction (Zhong et al., 2009). Moreover, our own proteomic screen of the MAP2 interactome identified MAP2 interactions with RNA binding proteins regulating translation which served to inhibit protein synthesis (Grubisha et al., 2021), potentially impacting dendritic spine plasticity. Such proteins included FMR1, PCBP1–3, and hnRNPK, which has been previously shown to regulate spine density and LTP as well as mediate the effects of BDNF on dendritic mRNA metabolism and NMDAR function (Folci et al., 2014; Leal et al., 2017). MAP2 protein has previously been shown to interact directly with IMP1 via its KH domains, which facilitate nucleic acid binding in a number of RNA-binding proteins (Nielsen et al., 2002). Thus, at least some of the observed interactions with RNA-binding proteins may be direct as opposed to indirect associations via tubulin or associated motor proteins. However, these interactions remain to be verified.
The association of MAP2 with ribosomal proteins is not a novel discovery (Farah et al., 2005) but has remained a relatively underappreciated aspect of the protein. We have found that this association has functional consequence, as overexpressed MAP2C inhibits protein synthesis in HEK cells (Grubisha et al., 2021). The mechanism of such regulation is unknown, but may depend on its association with RNA-binding proteins (see above). Overexpression of MAP2 may ultimately sequester such proteins to disrupt typical RNA trafficking and subsequently, protein synthesis. In addition to regulating protein synthesis, MAP2 has also been shown to exhibit chaperone-like qualities, preventing protein aggregation and facilitating enzyme refolding (Sarkar et al., 2004; Mitra et al., 2015). Of particular interest is the association MAP2 shares with Tau; MAP2 prevents the arachidonic acid-induced aggregation of Tau protein (Mitra et al., 2015), although the mechanism thereof is still unclear. MAP2 also regulates protein transport, predominantly by steric inhibition of the MT motors kinesin and dynein (Lopez and Sheetz, 1993; Hagiwara et al., 1994; Seitz et al., 2002). This can also contribute to selective axonal transport; using a sensory neuron model, Gumy et al. (2017) demonstrated that MAP2B generates a pre-axonal filtering zone controlling the activity of kinesin-1 and -5. Thus, MAP2 can shape the expression, conformation and transport of other proteins. However, these capacities remain understudied, particularly in the context of synaptic plasticity, where it may facilitate protein translocation into spines (Kim et al., 2020).
Neuron loss, regardless of cause, will be accompanied by corresponding loss of MAP2. However, in the context of induced brain injury, loss of MAP2 has been observed at early time points which precede neuronal death (Deshpande et al., 1992), raising the question of whether MAP2 loss may be involved casually in such death, presumably via disruption of the cytoskeletal network. MAP2 expression is significantly and consistently reduced early after induction of ischemia in both rodents (Dawson and Hallenbeck, 1996; Mages et al., 2021) and humans (Kühn et al., 2005), a loss which is thought to occur through calpain-mediated proteolysis (Pettigrew et al., 1996). However, the loss is also transient (Huh et al., 2003), suggesting that it does not correspond strictly to neuronal death. Moreover, MAP2 loss can be observed in regions of minimal cell death in mild traumatic brain injury models (Folkerts et al., 1998). Thus, whether MAP2 loss can precipitate or exacerbate neuronal death remains an open question which warrants further investigation, e.g., through characterization of neuronal survival following MAP2 knockout/knockdown, and/or rescue experiments exogenously expressing MAP2 during or post-injury. Such studies can illuminate whether MAP2 itself may represent a viable target for novel neuroprotective agents and strategies.
Significantly, many MAP2 functions—such as its interactions with MTs and actin, as well as its chaperone-like properties—are regulated by phosphorylation. Phosphosites span the entirety of MAP2; however, phosphosites in the proline-rich, MT-binding and C-terminal domains share a higher degree of conservation across species and with Tau (Sánchez et al., 2000). Generally, phosphorylation tends to reduce the MT-binding affinity of MAP2 and therefore inhibit its MT bundling and polymerization-promoting functions, though the exact effects are site-specific. MAP2 phosphorylation is developmentally regulated (Riederer et al., 1995; Quinlan and Halpain, 1996; Sánchez et al., 2000), presumably mediating different phases of MT growth state and organization as neurons mature. Moreover, long-standing findings have established that MAP2 is also phosphorylated in response to synaptic activity that induces plasticity (Quinlan and Halpain, 1996; Li et al., 2016). This activity-dependent phosphorylation appears to be mediated at least in part by the Ras-MAPK pathway (Llansola et al., 2001; Kim et al., 2020), though to our knowledge, activity-dependent phosphosites of MAP2 have not yet been confirmed. Conversely, sensory deprivation appears to affect MAP2 phosphorylation as well; for instance, olfactory restriction sharply reduces immunoreactivity of AP18, a MAP2 antibody which recognizes pS136 (Philpot et al., 1997).
Other kinases known to phosphorylate MAP2 include PKA, CaMKII, PKC, GSK-3β, CDKs, and MARKs (Sánchez et al., 2000) as well as JNK1. JNK1 represents a notable exception to the general principle of MAP2 phosphorylation in that its phosphorylation of several proline-rich domain residues (T1619, T1622, and T1625) increases—rather than decreases—the protein’s binding to MTs, as well as enhances dendritic arborization (Komulainen et al., 2014). This diversity of upstream regulators allows highly precise regulation of MAP2 function under a variety of cellular conditions. Notably, while phosphosites in MAP2 are frequently conserved in Tau, the position of sites phosphorylated in each MAP by a single kinase can differ, even in regions of high homology. This is the case for PKA, which mainly phosphorylates MAP2C at S435, whereas its major phosphosite in Tau is S214 (Jansen et al., 2017). Thus, the two can be differentially regulated by shared upstream kinases.
Interestingly, upstream kinases of MAP2 overlap substantially with those implicated in neuropsychiatric and neurodegenerative disorders. For instance, MAPK3 (ERK1), a component of the Ras-MAPK pathway, has been identified as the probable causal gene underlying the association signal at its locus in the largest schizophrenia genome-wide association study conducted to date (Schizophrenia Working Group of the Psychiatric Genomics Consortium et al., 2020). MAPK3 is also present at the 16p.11.2 locus, deletion of which is associated with autism (Pucilowska et al., 2015). GSK-3β, another kinase of MAP2, is a major regulator of neuronal development and has been strongly implicated in Alzheimer’s disease (Maqbool et al., 2016). It is also thought to be a major effective target of various antipsychotics, antidepressants and mood stabilizers such as lithium (Beaulieu et al., 2009). PKA, a binding partner and kinase of MAP2, acts downstream of voltage-gated calcium channels (Davare et al., 1999), which are now clearly identified by unbiased genomic studies as contributing to risk in schizophrenia, bipolar disorder, depression, and autism spectrum disorders (Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011; Lu et al., 2012; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Rao et al., 2016).
In addition to phosphorylation, MAP2 is likely regulated through other PTMs. Glycosylation (Ding and Vandré, 1996), ADP-ribosylation (Scaife et al., 1992), and lysine acetylation (Hwang et al., 2016) of MAP2 have all been described, though their functions are unclear. One study found that phosphorylation of MAP2 by GSK3β and/or PKA inhibited its O-glycosylation by 50%-90% (Khatra et al., 2013), suggesting that these regulatory mechanisms share an inverse relationship. Similarly, Tau is significantly less O-GlcNAc glycosylated when hyperphosphorylated (Lefebvre et al., 2003; Robertson et al., 2004). Beyond PTMs, functions of MAP2 are also regulated by alterative splicing. Besides the differences between HMW and LMW MAP2 described here (see Section “Domains and isoforms of MAP2”), alternative splice variants (Howe et al., 2021) have yet to be confirmed and/or characterized, but could also have distinct functions. Finally, interacting partners, including proteins and other biomolecules, can affect the actions of MAP2. For instance, as previously mentioned, Tau and phosphatidylinositol influence the MT-binding and actin-bundling properties of MAP2, respectively. Phosphatidylinositol additionally inhibits its MT assembling ability (Yamauchi and Purich, 1987).
The first half of this review has highlighted the diverse roles MAP2 plays in the development and maintenance of neuronal function, including in neurite extension/outgrowth, synaptic plasticity, and protein folding/transport. We have also described how these functions can be modified by a series of mechanisms, including downregulation/knockdown, splicing variants, and PTMs. Below we review the evidence of dysregulated MAP2 present in neuropsychiatric and neurodegenerative disorders (summarized in Table 1), and further discuss how MAP2 may contribute to their characteristic endophenotypes based on the known functions of the protein.
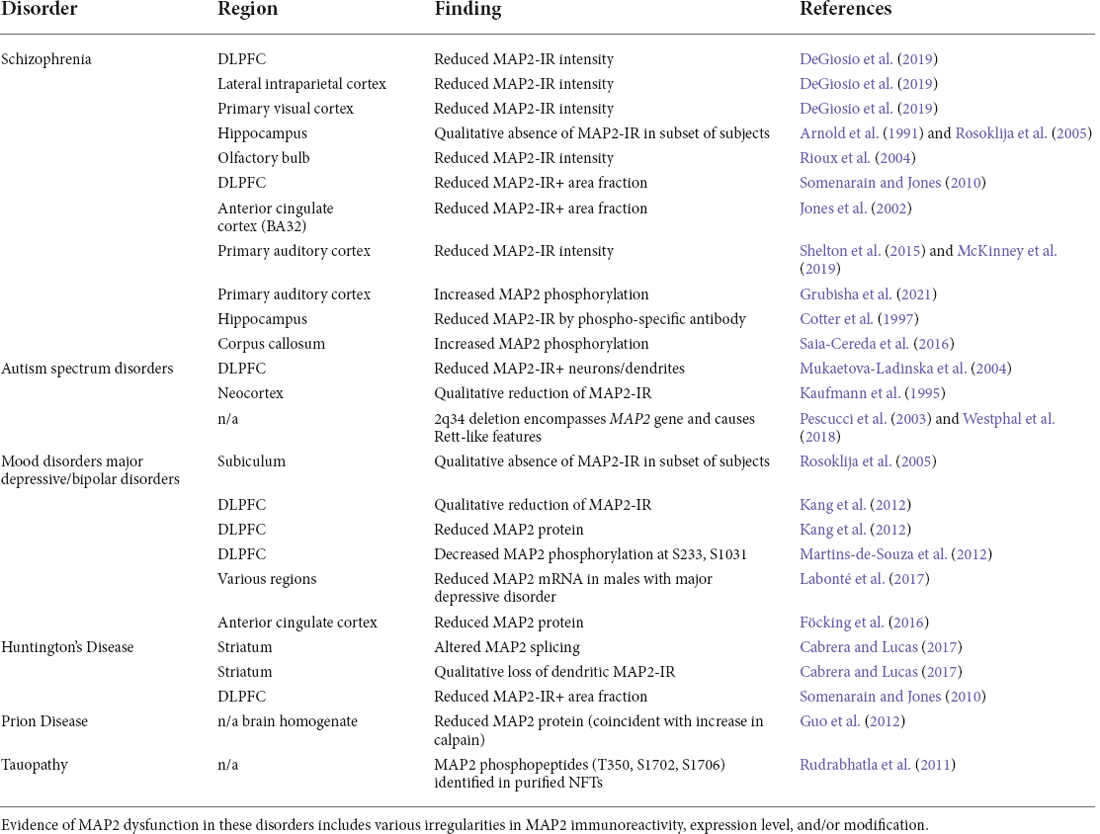
Table 1. Evidence of MAP2 dysfunction in neuropsychiatric and neurodegenerative disorder.
Schizophrenia is a psychiatric disorder characterized by psychotic symptoms as well as a variety of cognitive dysfunctions. Reduced MAP2 immunoreactivity (MAP2-IR) has been reported in diverse brain regions in schizophrenia and has been described as a “molecular hallmark” of the disorder (Marchisella et al., 2016). This observation has been made in the subiculum, the olfactory bulbs, entorhinal cortex, BA9, BA32 and, in our studies, dorsolateral prefrontal cortex (DLPFC), lateral intraparietal cortex, primary visual cortex, and primary auditory cortex (BA41; Arnold et al., 1991; Jones et al., 2002; Rioux et al., 2004; Rosoklija et al., 2005; Somenarain and Jones, 2010; Shelton et al., 2015; DeGiosio et al., 2019; McKinney et al., 2019). The reduced MAP2-IR does not appear to result from reductions in neuron number/density, as this remains unchanged in areas where MAP2-IR loss is profound (Jones et al., 2002; Dorph-Petersen et al., 2009; Somenarain and Jones, 2010). In our own studies we have also excluded potential effects of several clinical and technical confounds such as postmortem interval (the time from death until tissue fixation), duration of tissue freezer storage, use of psychotropic medications, or substance use status at time of death (Shelton et al., 2015; DeGiosio et al., 2019; McKinney et al., 2019). While lower tissue pH (a possible indicator of brain health prior to death) was correlated with lower MAP2-IR in one study, this effect did not account for reductions present across the cortex in schizophrenia (DeGiosio et al., 2019). As previously alluded to (see Section “Neuronal death—marker or effector?”), cerebral ischemia/hypoxia have previously been shown to reduce numbers of MAP2-IR-positive neurons in human neocortex and hippocampus; however, although causes of death associated with cerebral hypoxia or ischemia were more common in our schizophrenia subjects, they also did not explain the differences in MAP2-IR (DeGiosio et al., 2019). These analyses indicate that MAP2-IR deficit can represent a selective impairment in MAP2 that is not simply consequential to common factors affecting neuronal health.
With the additional power available from a combined cohort of 45 schizophrenia subjects paired with non-psychiatric comparison subjects, we were able to conduct formal mediation tests demonstrating that lower MAP2-IR mediated reductions in the density of dendritic spines in primary auditory cortex, suggesting that pathology of MAP2 is related, potentially in a causal manner, to pathogenic processes. Interestingly, in this study we determined that MAP2 protein levels were not reduced (McKinney et al., 2019). Moreover, MAP2 mRNA levels are unchanged in the hippocampal formation in schizophrenia subjects (Law et al., 2004). Therefore, MAP2-IR loss in schizophrenia does not appear to reflect reductions in mRNA/protein levels. Instead, MAP2 may undergo aberrant PTM to change its function. This could also preclude homeostatic compensation by other MAPs, previously proposed to occur in MAP2 knockout mice, which have shown that MAP2 expression is dispensable to mouse survival (Teng et al., 2001; Harada et al., 2002).
In support of this idea, several studies have suggested that the phosphorylation state of MAP2 is altered in schizophrenia. The immunohistochemical studies of Cotter et al. (1997) showed a trend toward decreased immunoreactivity by Ab305—a MAP2 antibody which recognizes pT1616/pT1619 in the proline-rich domain—in hippocampus of individuals with schizophrenia. The findings of Saia-Cereda et al. (2016) indicate that MAP2 phosphorylation is globally altered in the corpus callosum of schizophrenia subjects. In addition, using phosphoproteomics methods we have recently shown that MAP2 is differentially phosphorylated in primary auditory cortex, with phosphorylation events tending to be upregulated in disorder. Moreover, in this study we demonstrated that a subset of identified MAP2 phosphopeptides were significantly correlated with dendritic spine density and synaptic protein levels in BA41 (Grubisha et al., 2021).
This provided the first evidence indicating a potential pathogenic role for MAP2 phosphorylation in schizophrenia. As a proof of concept, we showed that a MAP2 mutant mimicking the most highly upregulated phosphorylation event identified, pS1782 (pS426 in MAP2C; Figure 1B), reduced the MT binding affinity of the protein, and that CRISPR mice harboring this mutation displayed reduced spine density and dendritic complexity in auditory cortex (Grubisha et al., 2021), paralleling findings from postmortem tissue (reviewed in Glausier and Lewis, 2013). This indicates that at least one schizophrenia-associated MAP2 phosphorylation event may be capable of causing dendritic pathology in vivo. Interestingly, this residue is homologous to S396 in Tau- a residue which is associated with AD and regulates Tau localization as well as LTD (Bramblett et al., 1993; Mondragón-Rodríguez et al., 2014; Regan et al., 2015; Xia et al., 2015; Wesseling et al., 2020). We further found that levels of the pS1782-bearing phosphopeptide were significantly lower in schizophrenia subjects on antipsychotic medication at time of death, suggesting that this phosphorylation event may represent a target of antipsychotic treatment, although the effect was not recapitulated in a monkey model of long-term antipsychotic exposure (Grubisha et al., 2021). In summary, aberrant MAP2 phosphorylation in schizophrenia may both underlie the profound reductions in MAP2-IR observed postmortem and have direct consequences for neuronal structure and function.
Autism spectrum disorders are neurodevelopmental disorders characterized by alterations in social communication and repetitive behaviors, as well as varying levels of intellectual disability. In several case reports of intellectual disability and autism, reduced numbers of MAP2-IR+ neurons and dendrites has been noted, without a change in total neuron number (Kaufmann et al., 1995; Mukaetova-Ladinska et al., 2004). Interestingly, a significant change in MAP2 expression was noted in a recent proteomic analysis of autism (though this did not survive correction for false discovery rate; Abraham et al., 2019). Moreover, case study reports of rare 2q34 deletions—which encompass the MAP2 gene—have described autism-like outcomes (Pescucci et al., 2003; Westphal et al., 2018). Though inconclusive, these data raise the possibility that MAP2 levels are reduced in at least some cases of autism. Such change, however, is likely to originate post-transcriptionally, as mRNA levels appear to be unaltered (Gandal et al., 2018).
A depletion of MAP2, even partial, could feasibly contribute to endophenotypes which characterize autism spectrum disorders, particularly with respect to synaptic plasticity. As described earlier, knockdown of MAP2 demonstrably impairs functional and structural plasticity of dendritic spines (Kim et al., 2020), which can be expected to alter learning and memory. Indeed, deficits in verbal learning/memory and working memory are among the most consistently impaired cognitive domains in adults with autism (Velikonja et al., 2019). Additionally, in postmortem brain tissue from individuals with autism, dendritic spine morphology shifts towards more immature phenotypes (Martínez-Cerdeño, 2017), which could result from a lack of MAP2-mediated activity-dependent spine growth. This may warrant further investigation of MAP2 abundance and function in autism.
MAP2-IR reduction has additionally been reported in the DLPFC of individuals with major depressive disorder (Kang et al., 2012) and in the hippocampus of major depression and bipolar disorder subjects (Rosoklija et al., 2005). The origin of this reduction is unclear. In the work of Kang et al. (2012), reduced DLPFC MAP2 levels were observed by western blot; however, it is unclear from these data if this is solely the result of reduced neuron density, which has also been noted in the region (Rajkowska et al., 1999). In contrast, microarray data indicates no change in MAP2 mRNA expression in depression (Seney et al., 2018). However, a separate study utilizing RNA sequencing to study sex-specific transcriptomic changes in depression identified a male-specific reduction in MAP2 mRNA level in multiple cortical regions (Labonté et al., 2017). Decreased MAP2 was also recently observed in bipolar disorder via proteomic analysis of the postsynaptic density (Föcking et al., 2016)—though not, to date, at the whole homogenate level—while it fails to exhibit change at the mRNA level by RNA sequencing (Gandal et al., 2018).
Two MAP2 phosphorylation events in the projection domain (pS233 and pS1031) have been reported to be decreased (case:control ratio = 0.82–0.83) in DLPFC of individuals with major depression, though these changes did not remain significant after controlling for false discovery rate (Martins-de-Souza et al., 2012). Interestingly, however, chronic antidepressant treatment has been observed to affect MAP2 expression and/or phosphorylation and subsequently, tubulin polymerization kinetics. Subchronic treatment of naïve rats with imipramine increases MAP2-IR in hippocampus (Iwata et al., 2006), while treatments with fluvoxamine, desipramine, maprotiline, and citalopram increase MAP2 phosphorylation (as assessed using phosphospecific antibodies or 32P incorporation), in turn suppressing tubulin polymerization (Miyamoto et al., 1995, 1997; Perez et al., 1995). It is unknown if and how such modification of MAP2 might contribute to antidepressant effects; however, previous data indicates that transient phosphorylation of MAP2 could support the dendritic outgrowth observed in response to antidepressant treatment (Seo et al., 2014). Indeed, 32P incorporation in MAP2 has previously been shown to correlate with dendritic arborization across time in cultured hippocampal neurons (Díez-Guerra and Avila, 1993). This observation was soon after substantiated by data indicating that treatment with protein kinase inhibitors reduces dendritic branching in these cells, while protein phosphatase inhibitors increase branching (Audesirk et al., 1997). However, our work on MAP2 S1782E—in which pseudo-phosphorylation of MAP2 reduced dendritic length and branching (Grubisha et al., 2021)—suggests that effects of MAP2 phosphorylation on dendritic arborization are complex and could depend on site, brain area and/or duration of signal. These data warrant further investigation of MAP2 phosphorylation state in depression, and of the specific interactions between MAP2 and antidepressants to better understand their mechanism of action.
Huntington’s disease is a heritable neurodegenerative disorder caused by mutation in huntingtin, a microtubule-associated protein involved in axonal transport. The work of Cabrera and Lucas (2017) established that MAP2 splicing is altered in striatum of Huntington’s subjects, favoring the LMW forms and reducing overall MAP2 levels. The authors proposed that this aberrant splicing of MAP2 results from altered activity of splicing factor SRSF6, previously implicated in the disease. The loss of MAP2 protein was accompanied by a substantial loss of MAP2-IR in the region, complimenting prior work showing reduced MAP2-IR in Brodmann area 9 of Huntington’s subjects (Somenarain and Jones, 2010).
As described above (see Section “Domains and isoforms of MAP2”), the HMW and LMW forms of the protein differ by inclusion or exclusion of the projection domain. This yields differential subcellular localization patterns and MT spacing characteristics. Additionally, they appear to differ in their ability to support activity-dependent spine plasticity, with HMW forms selectively translocating into spines following chemical LTP stimulus (Kim et al., 2020). In Huntington’s, such MAP2-mediated plasticity may be diminished, with consequences for learning and memory. Additionally, LMW forms appear to have greater affinity for neurite formation than HMW forms (Bélanger et al., 2002). Thus, the shift towards LMW MAP2 could be expected to yield more dendrites, and indeed this has been reported in prefrontal cortex of a small cohort of Huntington’s subjects (Sotrel et al., 1993). Therefore, aberrant MAP2 splicing may represent a crucial precipitant to the structural and functional abnormalities seen in neurons affected by Huntington’s disease.
Prion diseases are a group of transmissible neurodegenerative diseases caused by prions—misfolded proteins which can transmit their misfolded shape onto normal variants of the same protein, causing aggregation and altered protein function. Work by Guo et al. (2012) described significant reductions of MAP2 protein in scrapies-infected hamsters, attributable to a concomitant increase in the protease calpain. Interestingly, this parallels findings from brain injury models, such as ischemia and traumatic spinal cord injury, which also describe calpain-mediated MAP2 degradation (Pettigrew et al., 1996; Springer et al., 1997). Aβ oligomers can similarly induce such degradation (Fifre et al., 2006). Further, one recent study has demonstrated the downregulation of MAP2 protein in SMN-deficient motor neuron-like cells modeling spinal muscular atrophy, which the authors speculate may also be due to calpain hyperactivity (Özer et al., 2022). Thus, MAP2 appears to be a frequent target of calpain to mediate MT destabilization and potentially cell death under a variety of circumstances. Further study can clarify what therapeutic value MAP2 may have in preventing neuron loss (see Section “Neuronal death—marker or effector?”).
The tauopathies are a group of neurodegenerative disorders characterized by the hyperphosphorylation of MAP Tau and its subsequent assembly into neurofibrillary tangles (NFTs). MAP2 has generally not been considered as a potential pathogenic agent in the tauopathies, as its presence in NFTs has been a subject of some debate (Xie et al., 2014) and it forms no analogous aggregates. However, recent works have called into question whether NFTs themselves drive neuropathology, or if they are by products of pathology which is driven by soluble tau forms (Kopeikina et al., 2012). Indeed, the work of Xie et al. (2014) showed that while MAP2 and Tau have different aggregation properties, pan-neuronal expression of either can elicit severe neurotoxic effects. In this revised framework, interactions between MAP2 and soluble Tau may be important in shaping tauopathy. MAP2 and Tau have been known to cross-regulate each other; for instance, hyperphosphorylated Tau can sequester HMW MAP2 and thereby inhibit MT assembly (Alonso et al., 1997). Conversely, MAP2 prevents the arachidonic acid-induced aggregation of Tau, though MAP2 phosphorylation impairs this chaperoning ability (Mitra et al., 2015). Thus, MAP2 and Tau seem to share a reciprocal relationship whereby dysregulation of one can lead to that of the other. Mass spectrometry methods have revealed the presence of MAP2 phosphopeptides in purified Alzheimer’s NFTs (Rudrabhatla et al., 2011), indicating that MAP2 may be hyperphosphorylated in the disorder. Thus, MAP2 remains an active element to consider in the pathogenesis of tauopathies. Better understanding the origin of its aberrant phosphorylation in tauopathies and the nature of its interactions with Tau could lead to new therapeutic targets for the prevention of Tau aggregation.
MAP2 is often viewed solely as an endpoint marker of neuronal health. However, it is also a critical cytoskeletal regulator, dysfunction of which drastically affects neuronal structure and function. We have here described the known roles of MAP2 in neurite outgrowth, synaptic plasticity and protein folding/transport, as well as mechanisms of its regulation, including most prominently alternative splicing and phosphorylation. As such, MAP2 is poised to become dysregulated in various ways, potentially leading to distinct, pleiotropic effects. We have reviewed the current evidence for alterations in MAP2 function present in various neuropsychiatric and neurodegenerative disorders such as schizophrenia and Huntington’s disease. MAP2 pathology appears to originate at different levels across these disorders; for instance, Huntington’s is marked by aberrant RNA splicing, autism may be associated with lowered MAP2 protein abundance, while schizophrenia is distinguished by aberrant post-translational modification of MAP2.
This parallels the multiple distinct mechanisms by which tau can become dysregulated in tauopathy, which is not limited to mutations in MAPT genes, but also includes protein modification (particularly phosphorylation), cis/trans isomerization, and more. Thus, a “MAP2opathy” may similarly exist, characterizing varied neurodevelopmental and neurodegenerative disorders; akin to Tau, MAP2 could represent a molecular bottleneck situated downstream of genetic and environmental risk factors and upstream of neuronal pathology (Figure 2). This would position MAP2 as a flexible therapeutic target, able to ameliorate pathology while remaining effective in a large proportion of affected individuals regardless of their unique set of risk factors. However, classification of MAP2opathy as a genuine proteinopathy is still premature and awaits further research. Table 2 outlines various forms of evidence in favor of classification of a proteinopathy, comparing the existing evidence in the case of either tau or MAP2. An overall lack of genetic evidence implicating MAP2 mutation in disorder distinguishes it from tau and requires consideration. This could be due to the role of LMW MAP2 in spermatogenesis (Sun and Handel, 2011), which may make the gene mutation-intolerant [loss of function observed/expected upper bound fraction (LOEUF) = 0.1 (Karczewski et al., 2020)] and limit the ability of organisms to transmit MAP2 mutations that could later cause neuropathology. Instead, pathogenic MAP2 is more likely to arise from integrated upstream processes including kinase/phosphatase function, splicing machinery and proteolytic processes, analogous to those at play in tauopathy.
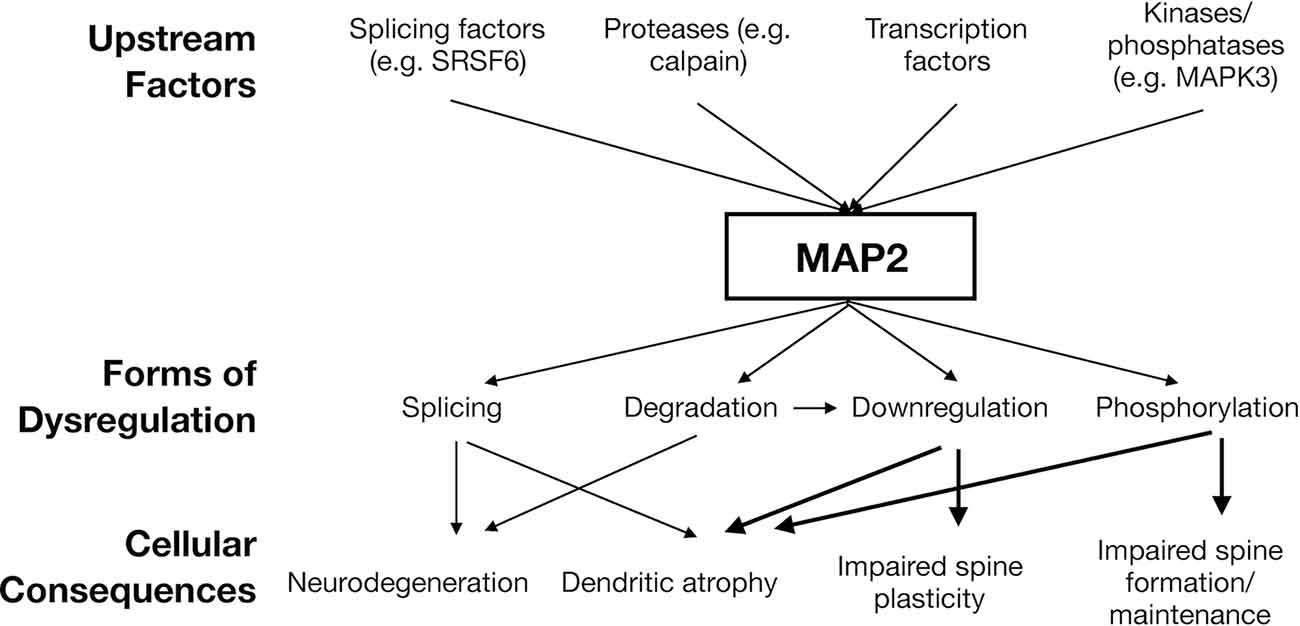
Figure 2. Hypothesized model of “MAP2opathy”. Genetically- or environmentally-precipitated upstream risk factors are expected to affect MAP2 in a variety of ways, leading to diverse, overlapping cellular consequences. Thick arrows indicate causal relationships established in in vivo models (Harada et al., 2002; Kim et al., 2020; Grubisha et al., 2021). Thin arrows indicate noted correlational relationships (Pettigrew et al., 1996; Guo et al., 2012; Cabrera and Lucas, 2017). Figure generated in Microsoft Powerpoint.
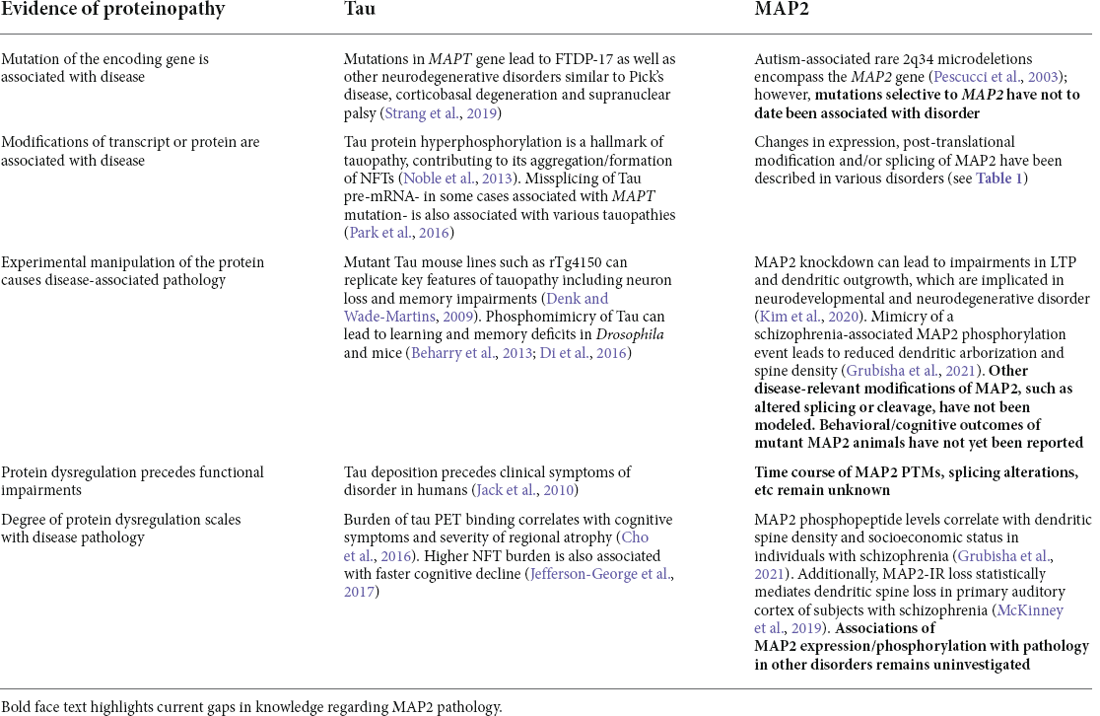
Table 2. Evidences of proteinopathy for tau vs. MAP2.
Existing data regarding MAP2 pathology is largely limited to correlational study, leaving open the possibility that changes to MAP2 immunoreactivity, expression or modification do not contribute to pathogenesis, but instead are merely incidental changes incurred by upstream processes. Despite clear evidence that disruptions to MAP2 expression and/or modification can lead to changes in dendritic and synaptic structure and function (Sharma et al., 1994; Ozer and Halpain, 2000; Harada et al., 2002; Akulinin and Dahlstrom, 2003; Dehmelt et al., 2003; Huang et al., 2013; Kim et al., 2020; Grubisha et al., 2021), this largely remains to be demonstrated in disease-relevant contexts. For example, our modeling of a schizophrenia-associated MAP2 phosphorylation event demonstrates a potential causal role of dysregulated MAP2 in the altered dendritic outgrowth which characterizes this disorder (Grubisha et al., 2021). Similarly, the altered splicing of MAP2 observed in Huntington’s might be modeled experimentally by manipulation of SRSF6 splicing factor, and rescue experiments can be performed by exogenous expression of HMW MAP2 to observe consequential effects on dendritic growth. Better understanding patterns of calpain-mediated cleavage of MAP2 could enable modeling of its degradation using truncation-inducing mutations or overexpression of truncated MAP2 constructs to explore roles of MAP2 degradation in neuronal atrophy of prion diseases or brain injury. MAP2 phosphorylation events observed in NFTs also have yet to be modeled, which could be used to illuminate what roles—if any—MAP2 phosphorylation plays in Tau aggregation/pathology.
Additionally, recent works revealing new or understudied functions of MAP2, such as that in LTP induction and AMPAR translocation (Kim et al., 2020), neuronal polarization via axonal filtering (Gumy et al., 2017), and our own work on phosphomimetic MAP2 (Grubisha et al., 2021) make clear that there is still much to learn about the basic biology of MAP2 protein. Firstly, thorough characterization of MAP2 PTM state on a residue-specific level in both healthy and diseased contexts will prove informative and can be readily achieved through modern mass spectrometry methods. Roles of such modifications in cytoskeletal filament organization, neurite outgrowth, synaptic plasticity and neuronal viability remain to be examined. Such studies will likely benefit from the extant literature regarding Tau, as these proteins structurally and functionally parallel one another—for example, through conserved phosphosites such as S396Tau/S1782MAP2 (Grubisha et al., 2021). Further, a bias exists in the literature favoring the in vitro study of immature MAP2c relative to mature, HMW forms of the protein, likely due to its ease of production and purification. However, these forms have differential structure and functions that remain to be fully understood. Such distinctions will provide insights to the developmental regulation of MAP2 functions. In conclusion, future studies of MAP2 will not only enhance our understanding of basic neurobiology but can also shed light on pathogenesis of a potential therapeutic target in neuropsychiatric and neurodegenerative disorders.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
RD and RS conceived the idea for this manuscript. RD performed the literature search and drafted the work with assistance from RS. MG, MM, BM, and CC critically revised the work. All authors contributed to the article and approved the submitted version.
This work was supported by National Institute of Mental Health (grant numbers: MH116046, MH071533, MH118497, and MH125235), National Institute on Aging (grant number: AG027224), and National Institutes of Health (grant number: GM97082).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abraham, J. R., Szoko, N., Barnard, J., Rubin, R. A., Schlatzer, D., Lundberg, K., et al. (2019). Proteomic investigations of autism brain identify known and novel pathogenetic processes. Sci. Rep. 9:13118. doi: 10.1038/s41598-019-49533-y
Akulinin, V. A., and Dahlstrom, A. (2003). Quantitative analysis of MAP2 immunoreactivity in human neocortex of three patients surviving after brain ischemia. Neurochem. Res. 28, 373–378. doi: 10.1023/a:1022401922669
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U S A 94, 298–303. doi: 10.1073/pnas.94.1.298
Angenstein, F., Evans, A. M., Ling, S.-C., Settlage, R. E., Ficarro, S., Carrero-Martinez, F. A., et al. (2005). Proteomic characterization of messenger ribonucleoprotein complexes bound to nontranslated or translated poly(A) mRNAs in the rat cerebral cortex. J. Biol. Chem. 280, 6496–6503. doi: 10.1074/jbc.M412742200
Arnold, S. E., Lee, V. M., Gur, R. E., and Trojanowski, J. Q. (1991). Abnormal expression of two microtubule-associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc. Natl. Acad. Sci. U S A 88, 10850–10854. doi: 10.1073/pnas.88.23.10850
Audesirk, G., Cabell, L., and Kern, M. (1997). Modulation of neurite branching by protein phosphorylation in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 102, 247–260. doi: 10.1016/s0165-3806(97)00100-4
Baas, P. W., and Qiang, L. (2019). Tau: it’s not what you think. Trends Cell Biol. 29, 452–461. doi: 10.1016/j.tcb.2019.02.007
Beaulieu, J.-M., Gainetdinov, R. R., and Caron, M. G. (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Ann. Rev. Pharmacol. Toxicol. 49, 327–347. doi: 10.1146/annurev.pharmtox.011008.145634
Beharry, C., Alaniz, M. E., and Alonso, A. D. C. (2013). Expression of Alzheimer-like pathological human tau induces a behavioral motor and olfactory learning deficit in Drosophila melanogaster. J. Alzheimers Dis. 37, 539–550. doi: 10.3233/JAD-130617
Bélanger, D., Farah, C. A., Nguyen, M. D., Lauzon, M., Cornibert, S., and Leclerc, N. (2002). The projection domain of MAP2b regulates microtubule protrusion and process formation in Sf9 cells. J. Cell Sci. 115, 1523–1539. doi: 10.1242/jcs.115.7.1523
Blichenberg, A., Schwanke, B., Rehbein, M., Garner, C. C., Richter, D., and Kindler, S. (1999). Identification of a cis-acting dendritic targeting element in MAP2 mRNAs. J. Neurosci. 19, 8818–8829. doi: 10.1523/JNEUROSCI.19-20-08818.1999
Bodakuntla, S., Jijumon, A., Villablanca, C., Gonzalez-Billault, C., and Janke, C. (2019). Microtubule-associated proteins: structuring the cytoskeleton. Trends Cell Biol. 29, 804–819. doi: 10.1016/j.tcb.2019.07.004
Bramblett, G. T., Goedert, M., Jakes, R., Merrick, S. E., Trojanowski, J. Q., and Lee, V. M. Y. (1993). Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 10, 1089–1099. doi: 10.1016/0896-6273(93)90057-x
Cabrera, J. R., and Lucas, J. J. (2017). MAP2 splicing is altered in Huntington’s disease. Brain Pathol. 27, 181–189. doi: 10.1111/bpa.12387
Caceres, A., Binder, L., Payne, M., Bender, P., Rebhun, L., and Steward, O. (1984). Differential subcellular localization of tubulin and the microtubule-associated protein MAP2 in brain tissue as revealed by immunocytochemistry with monoclonal hybridoma antibodies. J. Neurosci. 4, 394–410. doi: 10.1523/JNEUROSCI.04-02-00394.1984
Chen, J., Kanai, Y., Cowan, N., and Hirokawa, N. (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677. doi: 10.1038/360674a0
Chirillo, M. A., Waters, M. S., Lindsey, L. F., Bourne, J. N., and Harris, K. M. (2019). Local resources of polyribosomes and SER promote synapse enlargement and spine clustering after long-term potentiation in adult rat hippocampus. Sci. Rep. 9:3861. doi: 10.1038/s41598-019-40520-x
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi: 10.1146/annurev-biochem-061516-045115
Cho, H., Choi, J. Y., Hwang, M. S., Lee, J. H., Kim, Y. J., Lee, H. M., et al. (2016). Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 87, 375–383. doi: 10.1212/WNL.0000000000002892
Correas, I., Padilla, R., and Avila, J. (1990). The tubulin-binding sequence of brain microtubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem. J. 269, 61–64. doi: 10.1042/bj2690061
Cotter, D., Kerwin, R., Doshi, B., Martin, C. S., and Everall, I. P. (1997). Alterations in hippocampal non-phosphorylated MAP2 protein expression in schizophrenia. Brain Res. 765, 238–246. doi: 10.1016/s0006-8993(97)00575-1
Díez-Guerra, F. J., and Avila, J. (1993). MAP2 phosphorylation parallels dendrite arborization in hippocampal neurones in culture. Neuroreport 4, 419–422. doi: 10.1097/00001756-199304000-00020
Davare, M. A., Dong, F., Rubin, C. S., and Hell, J. W. (1999). The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J. Biol. Chem. 274, 30280–30287. doi: 10.1074/jbc.274.42.30280
Dawson, D. A., and Hallenbeck, J. M. (1996). Acute focal ischemia-induced alterations in MAP2 immunostaining: description of temporal changes and utilization as a marker for volumetric assessment of acute brain injury. J. Cereb. Blood Flow Metab. 16, 170–174. doi: 10.1097/00004647-199601000-00020
DeGiosio, R., Kelly, R. M., DeDionisio, A. M., Newman, J. T., Fish, K. N., Sampson, A. R., et al. (2019). MAP2 immunoreactivity deficit is conserved across the cerebral cortex within individuals with schizophrenia. NPJ Schizophr. 5:13. doi: 10.1038/s41537-019-0081-0
Dehmelt, L., Smart, F. M., Ozer, R. S., and Halpain, S. (2003). The role of microtubule-associated protein 2c in the reorganization of microtubules and lamellipodia during neurite initiation. J. Neurosci. 23, 9479–9490. doi: 10.1523/JNEUROSCI.23-29-09479.2003
Denk, F., and Wade-Martins, R. (2009). Knock-out and transgenic mouse models of tauopathies. Neurobiol. Aging 30, 1–13. doi: 10.1016/j.neurobiolaging.2007.05.010
Deshpande, J., Bergstedt, K., Lindén, T., Kalimo, H., and Wieloch, T. (1992). Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp. Brain Res. 88, 91–105. doi: 10.1007/BF02259131
Di, J., Cohen, L., Corbo, C., Phillips, G. R., El Idrissi, A., and Alonso, A. D. (2016). Abnormal tau induces cognitive impairment through two different mechanisms: synaptic dysfunction and neuronal loss. Sci. Rep. 6:20833. doi: 10.1038/srep20833
Ding, M., and Vandré, D. D. (1996). High molecular weight microtubule-associated proteins contain O-linked-N-acetylglucosamine. J. Biol. Chem. 271, 12555–12561. doi: 10.1074/jbc.271.21.12555
Doll, T., Meichsner, M., Riederer, B., Honegger, P., and Matus, A. (1993). An isoform of microtubule-associated protein 2 (MAP2) containing four repeats of the tubulin-binding motif. J. Cell Sci. 106, 633–639. doi: 10.1242/jcs.106.2.633
Dorph-Petersen, K.-A., Delevich, K. M., Marcsisin, M. J., Zhang, W., Sampson, A. R., Gundersen, H. J. G., et al. (2009). Pyramidal neuron number in layer 3 of primary auditory cortex of subjects with schizophrenia. Brain Res. 1285, 42–57. doi: 10.1016/j.brainres.2009.06.019
Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005). Flexible nets: the roles of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x
Edson, K., Weisshaar, B., and Matus, A. (1993). Actin depolymerisation induces process formation on MAP2-transfected non-neuronal cells. Development 117, 689–700. doi: 10.1242/dev.117.2.689
Farah, C. A., Liazoghli, D., Perreault, S., Desjardins, M., Guimont, A., Anton, A., et al. (2005). Interaction of Microtubule-associated Protein-2 and p63: a new link between microtubules and rough endoplasmic reticulum membranes in neurons. J. Biol. Chem. 280, 9439–9449. doi: 10.1074/jbc.M412304200
Ferralli, J., Doll, T., and Matus, A. (1994). Sequence analysis of MAP2 function in living cells. J. Cell Sci. 107, 3115–3125. doi: 10.1242/jcs.107.11.3115
Fifre, A., Sponne, I., Koziel, V., Kriem, B., Potin, F. T. Y., Bihain, B. E., et al. (2006). Microtubule-associated protein MAP1A, MAP1B and MAP2 proteolysis during soluble amyloid β-peptide-induced Neuronal apoptosis: synergistic involvement of calpain and caspase-3. J. Biol. Chem. 281, 229–240. doi: 10.1074/jbc.M507378200
Föcking, M., Dicker, P., Lopez, L. M., Hryniewiecka, M., Wynne, K., English, J. A., et al. (2016). Proteomic analysis of the postsynaptic density implicates synaptic function and energy pathways in bipolar disorder. Transl. Psychiatry 6:e959. doi: 10.1038/tp.2016.224
Folci, A., Mapelli, L., Sassone, J., Prestori, F., D’Angelo, E., Bassani, S., et al. (2014). Loss of hnRNP K impairs synaptic plasticity in hippocampal neurons. J. Neurosci. 34, 9088–9095. doi: 10.1523/JNEUROSCI.0303-14.2014
Folkerts, M. M., Berman, R. F., Muizelaar, J. P., and Rafols, J. A. (1998). Disruption of MAP-2 immunostaining in rat hippocampus after traumatic brain injury. J. Neurotrauma 15, 349–363. doi: 10.1089/neu.1998.15.349
Fontaine-Lenoir, V., Chambraud, B., Fellous, A., David, S., Duchossoy, Y., Baulieu, E.-E., et al. (2006). Microtubule-associated protein 2 (MAP2) is a neurosteroid receptor. Proc. Natl. Acad. Sci. U S A 103, 4711–4716. doi: 10.1073/pnas.0600113103
Gandal, M. J., Zhang, P., Hadjimichael, E., Walker, R. L., Chen, C., Liu, S., et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia and bipolar disorder. Science 362:eaat8127. doi: 10.1126/science.aat8127
Garner, C. C., Tucker, R. P., and Matus, A. (1988). Selective localization of messenger RNA for cytoskeletal protein MAP2 in dendrites. Nature 336, 674–677. doi: 10.1038/336674a0
Glausier, J. R., and Lewis, D. A. (2013). Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107. doi: 10.1016/j.neuroscience.2012.04.044
Gong, R., Park, C. S., Abbassi, N. R., and Tang, S.-J. (2006). Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J. Biol. Chem. 281, 18802–18815. doi: 10.1074/jbc.M512524200
Goodson, H. V., and Jonasson, E. M. (2018). Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol. 10:a022608. doi: 10.1101/cshperspect.a022608
Grubisha, M., Sun, X., MacDonald, M., Garver, M., Sun, Z., Paris, K., et al. (2021). MAP2 is differentially phosphorylated in schizophrenia, altering its function. Mol. Psychiatry 26, 5371–5388. doi: 10.1038/s41380-021-01034-z
Gsponer, J., Futschik, M. E., Teichmann, S. A., and Babu, M. M. (2008). Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368. doi: 10.1126/science.1163581
Gu, J., Firestein, B. L., and Zheng, J. Q. (2008). Microtubules in dendritic spine development. J. Neurosci. 28, 12120–12124. doi: 10.1523/JNEUROSCI.2509-08.2008
Gumy, L. F., Katrukha, E. A., Grigoriev, I., Jaarsma, D., Kapitein, L. C., Akhmanova, A., et al. (2017). MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron 94, 347–362.e7. doi: 10.1016/j.neuron.2017.03.046
Guo, Y., Gong, H.-S., Zhang, J., Xie, W.-L., Tian, C., Chen, C., et al. (2012). Remarkable reduction of MAP2 in the brains of scrapie-infected rodents and human prion disease possibly correlated with the increase of calpain. PLoS One 7:e30163. doi: 10.1371/journal.pone.0030163
Gustke, N., Trinczek, B., Biernat, J., Mandelkow, E.-M., and Mandelkow, E. (1994). Domains of tau protein and interactions with microtubules. Biochemistry 33, 9511–9522. doi: 10.1021/bi00198a017
Hagiwara, H., Yorifuji, H., Sato-Yoshitake, R., and Hirokawa, N. (1994). Competition between motor molecules (kinesin and cytoplasmic dynein) and fibrous microtubule-associated proteins in binding to microtubules. J. Biol. Chem. 269, 3581–3589. doi: 10.1016/S0021-9258(17)41903-X
Harada, A., Teng, J., Takei, Y., Oguchi, K., and Hirokawa, N. (2002). MAP2 is required for dendrite elongation, PKA anchoring in dendrites and proper PKA signal transduction. J. Cell Biol. 158, 541–549. doi: 10.1083/jcb.200110134
Hedstrom, K. L., Ogawa, Y., and Rasband, M. N. (2008). AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J. Cell. Biol. 183, 635–640. doi: 10.1083/jcb.200806112
Herzog, W., and Weber, K. (1978). Fractionation of brain microtubule-associated proteins: isolation of two different proteins which stimulate tubulin polymerization in vitro. Eur. J. Biochem. 92, 1–8. doi: 10.1111/j.1432-1033.1978.tb12716.x
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2021). Ensembl 2021. Nucleic Acids Res. 49, D884–D891. doi: 10.1093/nar/gkaa942
Hu, X., Viesselmann, C., Nam, S., Merriam, E., and Dent, E. W. (2008). Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 28, 13094–13105. doi: 10.1523/JNEUROSCI.3074-08.2008
Huang, Y.-A., Kao, J.-W., Tseng, D. T.-H., Chen, W.-S., Chiang, M.-H., and Hwang, E. (2013). Microtubule-associated type II protein kinase A is important for neurite elongation. PLoS One 8:e73890. doi: 10.1371/journal.pone.0073890
Huh, J., Raghupathi, R., Laurer, H. L., Helfaer, M. A., and Saatman, K. E. (2003). Transient loss of microtubule-associated protein 2 immunoreactivity after moderate brain injury in mice. J. Neurotrauma 20, 975–984. doi: 10.1089/089771503770195821
Hwang, A. W., Trzeciakiewicz, H., Friedmann, D., Yuan, C.-X., Marmorstein, R., Lee, V. M. Y., et al. (2016). Conserved lysine acetylation within the microtubule-binding domain regulates MAP2/tau family members. PLoS One 11:e0168913. doi: 10.1371/journal.pone.0168913
Itoh, T. J., and Hotani, H. (1994). Microtubule-stabilizing activity of microtubule-associated proteins (MAPs) is due to increase in frequency of rescue in dynamic instability: shortening length decreases with binding of MAPs onto microtubules. Cell Struct. Funct. 19, 279–290. doi: 10.1247/csf.19.279
Iwata, M., Shirayama, Y., Ishida, H., and Kawahara, R. (2006). Hippocampal synapsin I, growth-associated protein-43 and microtubule-associated protein-2 immunoreactivity in learned helplessness rats and antidepressant-treated rats. Neuroscience 141, 1301–1313. doi: 10.1016/j.neuroscience.2006.04.060
Jack, C. R., Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/S1474-4422(09)70299-6
Jalava, N. S., Lopez-Picon, F. R., Kukko-Lukjanov, T.-K., and Holopainen, I. E. (2007). Changes in microtubule-associated protein-2 (MAP2) expression during development and after status epilepticus in the immature rat hippocampus. Int. J. Dev. Neurosci. 25, 121–131. doi: 10.1016/j.ijdevneu.2006.12.001
Jansen, S., Melková, K., Trošanová, Z., Hanáková, K., Zachrdla, M., Nováček, J., et al. (2017). Quantitative mapping of microtubule-associated protein 2c (MAP2c) phosphorylation and regulatory protein 14-3-3ζ-binding sites reveals key differences between MAP2c and its homolog Tau. J. Biol. Chem. 292:10316. doi: 10.1074/jbc.A116.771097
Jaworski, J., Kapitein, L. C., Gouveia, S. M., Dortland, B. R., Wulf, P. S., Grigoriev, I., et al. (2009). Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100. doi: 10.1016/j.neuron.2008.11.013
Jefferson-George, K. S., Wolk, D. A., Lee, E. B., and McMillan, C. T. (2017). Cognitive decline associated with pathological burden in primary age-related tauopathy. Alzheimers Dement. 13, 1048–1053. doi: 10.1016/j.jalz.2017.01.028
Jones, L. B., Johnson, N., and Byne, W. (2002). Alterations in MAP2 immunocytochemistry in areas 9 and 32 of schizophrenic prefrontal cortex. Psychiatry Res. 114, 137–148. doi: 10.1016/s0925-4927(02)00022-7
Kanai, Y., and Hirokawa, N. (1995). Sorting mechanisms of tau and MAP2 in neurons: suppressed axonal transit of MAP2 and locally regulated microtubule binding. Neuron 14, 421–432. doi: 10.1016/0896-6273(95)90298-8
Kang, H. J., Voleti, B., Hajszan, T., Rajkowska, G., Stockmeier, C. A., Licznerski, P., et al. (2012). Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413–1417. doi: 10.1038/nm.2886
Kapitein, L. C., and Hoogenraad, C. C. (2015). Building the neuronal microtubule cytoskeleton. Neuron 87, 492–506. doi: 10.1016/j.neuron.2015.05.046
Kapitein, L. C., Yau, K. W., Gouveia, S. M., van der Zwan, W. A., Wulf, P. S., Keijzer, N., et al. (2011). NMDA receptor activation suppresses microtubule growth and spine entry. J. Neurosci. 31, 8194–8209. doi: 10.1523/JNEUROSCI.6215-10.2011
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kaufmann, W. E., Naidu, S., and Budden, S. (1995). Abnormal expression of microtubule-associated protein 2 (MAP-2) in neocortex in rett syndrome. Neuropediatrics 26, 109–113. doi: 10.1055/s-2007-979738
Khatra, B. S., Juarez, M., Johnson, D., and DeTure, M. (2013). Phosphorylation of tau-4R and microtubule binding region of MAP-2C by PKA and GSK-3β inhibits their O-glycosylation by OGT. FASEB J. 27:1038.8. doi: 10.1096/fasebj.27.1_supplement.1038.8
Kim, Y., Jang, Y.-N., Kim, J.-Y., Kim, N., Noh, S., Kim, H., et al. (2020). Microtubule-associated protein 2 mediates induction of long-term potentiation in hippocampal neurons. FASEB J. 34, 6965–6983. doi: 10.1096/fj.201902122RR
Komulainen, E., Zdrojewska, J., Freemantle, E., Mohammad, H., Kulesskaya, N., Deshpande, P., et al. (2014). JNK1 controls dendritic field size in L2/3 and L5 of the motor cortex, constrains soma size and influences fine motor coordination. Front. Cell Neurosci. 8:272. doi: 10.3389/fncel.2014.00272
Kopeikina, K., Hyman, B., and Spires-Jones, T. (2012). Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 3, 223–233. doi: 10.2478/s13380-012-0032-y
Kühn, J., Meissner, C., and Oehmichen, M. (2005). Microtubule-associated protein 2 (MAP2)—a promising approach to diagnosis of forensic types of hypoxia-ischemia. Acta Neuropathol. 110, 579–586. doi: 10.3174/ajnr.A7606
Labonté, B., Engmann, O., Purushothaman, I., Menard, C., Wang, J., Tan, C., et al. (2017). Sex-specific transcriptional signatures in human depression. Nat. Med. 23, 1102–1111. doi: 10.1038/nm.4386
Law, A. J., Weickert, C. S., Hyde, T. M., Kleinman, J. E., and Harrison, P. J. (2004). Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am. J. Psychiatry 161, 1848–1855. doi: 10.1176/ajp.161.10.1848
Leal, G., Comprido, D., de Luca, P., Morais, E., Rodrigues, L., Mele, M., et al. (2017). The RNA-binding protein hnRNP K mediates the effect of BDNF on dendritic mRNA metabolism and regulates synaptic NMDA receptors in hippocampal neurons. eNeuro 4, ENEURO.0268–17.2017. doi: 10.1523/ENEURO.0268-17.2017
Leclerc, N., Baas, P. W., Garner, C. C., and Kosik, K. S. (1996). Juvenile and mature MAP2 isoforms induce distinct patterns of process outgrowth. Mol. Biol. Cell 7, 443–455. doi: 10.1091/mbc.7.3.443
Lefebvre, T., Ferreira, S., Dupont-Wallois, L., Bussière, T., Dupire, M.-J., Delacourte, A., et al. (2003). Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins—a role in nuclear localization. Biochim. Biophys. Acta 1619, 167–176. doi: 10.1016/s0304-4165(02)00477-4
Lewis, S. A., Ivanov, I. E., Lee, G. H., and Cowan, N. J. (1989). Organization of microtubules in dendrites and axons is determined by a short hydrophobic zipper in microtubule-associated proteins MAP2 and tau. Nature 342, 498–505. doi: 10.1038/342498a0
Lewis, S. A., Wang, D. H., and Cowan, N. J. (1988). Microtubule-associated protein MAP2 shares a microtubule binding motif with tau protein. Science 242, 936–939. doi: 10.1126/science.3142041
Li, J., Wilkinson, B., Clementel, V. A., Hou, J., O’Dell, T. J., and Coba, M. P. (2016). Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci. Signal. 9:rs8. doi: 10.1126/scisignal.aaf6716
Lim, R. W. L., and Halpain, S. (2000). Regulated association of microtubule-associated protein 2 (MAP2) with Src and Grb2: evidence for MAP2 as a scaffolding protein. J. Biol. Chem. 275, 20578–20587. doi: 10.1074/jbc.M001887200
Llansola, M., Sáez, R., and Felipo, V. (2001). NMDA-induced phosphorylation of the microtubule-associated protein MAP-2 is mediated by activation of nitric oxide synthase and MAP kinase. Eur. J. Neurosci. 13, 1283–1291. doi: 10.1046/j.0953-816x.2001.01497.x
Lopez, L. A., and Sheetz, M. P. (1993). Steric inhibition of cytoplasmic dynein and kinesin motility by MAP2. Cell Motil. Cytoskeleton 24, 1–16. doi: 10.1002/cm.970240102
Lu, A. T.-H., Dai, X., Martinez-Agosto, J. A., and Cantor, R. M. (2012). Support for calcium channel gene defects in autism spectrum disorders. Mol. Autism. 3:18. doi: 10.1186/2040-2392-3-18
Ludin, B., Ashbridge, K., Funfschilling, U., and Matus, A. (1996). Functional analysis of the MAP2 repeat domain. J. Cell Sci. 109, 91–99. doi: 10.1242/jcs.109.1.91
Mages, B., Fuhs, T., Aleithe, S., Blietz, A., Hobusch, C., Härtig, W., et al. (2021). The cytoskeletal elements MAP2 and NF-L show substantial alterations in different stroke models while elevated serum levels highlight especially MAP2 as a sensitive biomarker in stroke patients. Mol. Neurobiol. 58, 4051–4069. doi: 10.1007/s12035-021-02372-3
Maqbool, M., Mobashir, M., and Hoda, N. (2016). Pivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 107, 63–81. doi: 10.1016/j.ejmech.2015.10.018
Marchisella, F., Coffey, E. T., and Hollos, P. (2016). Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton (Hoboken) 73, 596–611. doi: 10.1002/cm.21300
Martínez-Cerdeño, V. (2017). Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 77, 393–404. doi: 10.1002/dneu.22417
Martins-de-Souza, D., Guest, P. C., Vanattou-Saifoudine, N., Rahmoune, H., and Bahn, S. (2012). Phosphoproteomic differences in major depressive disorder postmortem brains indicate effects on synaptic function. Eur. Arch. Psychiatry Clin. Neurosci. 262, 657–666. doi: 10.1007/s00406-012-0301-3
Marx, C. E., Bradford, D. W., Hamer, R. M., Naylor, J. C., Allen, T. B., Lieberman, J. A., et al. (2011). Pregnenolone as a novel therapeutic candidate in schizophrenia: emerging preclinical and clinical evidence. Neuroscience 191, 78–90. doi: 10.1016/j.neuroscience.2011.06.076
Marx, C. E., Lee, J., Subramaniam, M., Rapisarda, A., Bautista, D. C. T., Chan, E., et al. (2014). Proof-of-concept randomized controlled trial of pregnenolone in schizophrenia. Psychopharmacology (Berl) 231, 3647–3662. doi: 10.1007/s00213-014-3673-4
McKinney, B. C., MacDonald, M. L., Newman, J. T., Shelton, M. A., DeGiosio, R. A., Kelly, R. M., et al. (2019). Density of small dendritic spines and microtubule-associated-protein-2 immunoreactivity in the primary auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 44, 1055–1061. doi: 10.1038/s41386-019-0350-7
McVicker, D. P., Awe, A. M., Richters, K. E., Wilson, R. L., Cowdrey, D. A., Hu, X., et al. (2016). Transport of a kinesin-cargo pair along microtubules into dendritic spines undergoing synaptic plasticity. Nat. Commun. 7:12741. doi: 10.1038/ncomms12741
Meichsner, M., Doll, T., Reddy, D., Weisshaar, B., and Matus, A. (1993). The low molecular weight form of microtubule-associated protein 2 is transported into both axons and dendrites. Neuroscience 54, 873–880. doi: 10.1016/0306-4522(93)90581-y
Meléndez, J., Maldonado, V., and Ortega, A. (1996). Effect of melatonin on β-tubulin and MAP2 expression in NIE-115 cells. Neurochem. Res. 21, 653–658. doi: 10.1007/BF02527721
Merriam, E. B., Lumbard, D. C., Viesselmann, C., Ballweg, J., Stevenson, M., Pietila, L., et al. (2011). Dynamic microtubules promote synaptic NMDA receptor-dependent spine enlargement. PLoS One 6:e27688. doi: 10.1371/journal.pone.0027688
Merriam, E. B., Millette, M., Lumbard, D. C., Saengsawang, W., Fothergill, T., Hu, X., et al. (2013). Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin and drebrin. J. Neurosci. 33, 16471–16482. doi: 10.1523/JNEUROSCI.0661-13.2013
Mitra, G., Gupta, S., Poddar, A., and Bhattacharyya, B. (2015). MAP2c prevents arachidonic acid-induced fibril formation of tau: role of chaperone activity and phosphorylation. Biophys. Chem. 205, 16–23. doi: 10.1016/j.bpc.2015.06.003
Mitsuyama, F., Niimi, G., Kato, K., Hirosawa, K., Mikoshiba, K., Okuya, M., et al. (2008). Redistribution of microtubules in dendrites of hippocampal CA1 neurons after tetanic stimulation during long-term potentiation. Ital. J. Anat. Embryol. 113, 17–27.
Miyamoto, S., Asakura, M., Sasuga, Y., Imafuku, J., Gamo, Y., and Osada, K. (1995). Chronic antidepressant administration inhibits microtubule assembly in rat cerebral cortex. Eur. Neuropsychopharmacol. 5:280.
Miyamoto, S., Asakura, M., Sasuga, Y., Osada, K., Bodaiji, N., Imafuku, J., et al. (1997). Effects of long-term treatment with desipramine on microtubule proteins in rat cerebral cortex. Eur. J. Pharmacol. 333, 279–287. doi: 10.1016/s0014-2999(97)01140-0
Mohan, R., and John, A. (2015). Microtubule-associated proteins as direct crosslinkers of actin filaments and microtubules. IUBMB Life 67, 395–403. doi: 10.1002/iub.1384
Mondragón-Rodríguez, S., Perry, G., Luna-Muñoz, J., Acevedo-Aquino, M. C., and Williams, S. (2014). Phosphorylation of tau protein at sites Ser396–404 is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 40, 121–135. doi: 10.1111/nan.12084
Moraga, D. M., Nuñez, P., Garrido, J., and Maccioni, R. B. (1993). A τ fragment containing a repetitive sequence induces bundling of actin filaments. J. Neurochem. 61, 979–986. doi: 10.1111/j.1471-4159.1993.tb03611.x
Morales, M., and Fifkova, E. (1989). Distribution of MAP 2 in dendritic spines and its colocalization with actin. Cell Tissue Res. 256, 447–456. doi: 10.1007/BF00225592
Mukaetova-Ladinska, E. B., Arnold, H., Jaros, E., Perry, R., and Perry, E. (2004). Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol. Appl. Neurobiol. 30, 615–623. doi: 10.1111/j.1365-2990.2004.00574.x
Murakami, K., Fellous, A., Baulieu, E.-E., and Robel, P. (2000). Pregnenolone binds to microtubule-associated protein 2 and stimulates microtubule assembly. Proc. Natl. Acad. Sci. U S A 97, 3579–3584. doi: 10.1073/pnas.97.7.3579
Nielsen, F. C., Nielsen, J., Kristensen, M. A., Koch, G., and Christiansen, J. (2002). Cytoplasmic trafficking of IGF-II mRNA-binding protein by conserved KH domains. J. Cell Sci. 115, 2087–2097. doi: 10.1242/jcs.115.10.2087
Noble, W., Hanger, D. P., Miller, C. C., and Lovestone, S. (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 4:83. doi: 10.3389/fneur.2013.00083
Ostroff, L. E., Fiala, J. C., Allwardt, B., and Harris, K. M. (2002). Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35, 535–545. doi: 10.1016/s0896-6273(02)00785-7
Osuji, I. J., Vera-Bolaños, E., Carmody, T. J., and Brown, E. S. (2010). Pregnenolone for cognition and mood in dual diagnosis patients. Psychiatry Res. 178, 309–312. doi: 10.1016/j.psychres.2009.09.006
Ozer, R. S., and Halpain, S. (2000). Phosphorylation-dependent localization of microtubule-associated protein MAP2c to the actin cytoskeleton. Mol. Biol. Cell 11, 3573–3587. doi: 10.1091/mbc.11.10.3573
Özer, P. Z., Koyunoğlu, D., Son, Ç. D., Yurter, H. E., and Bora, G. (2022). SMN loss dysregulates microtubule-associated proteins in spinal muscular atrophy model. Mol. Cell. Neurosci. 120:103725. doi: 10.1016/j.mcn.2022.103725
Park, S. A., Ahn, S. I., and Gallo, J.-M. (2016). Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 49, 405–413. doi: 10.5483/bmbrep.2016.49.8.084
Pejaver, V., Hsu, W. L., Xin, F., Dunker, A. K., Uversky, V. N., and Radivojac, P. (2014). The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 23, 1077–1093. doi: 10.1002/pro.2494
Perez, J., Mori, S., Caivano, M., Popoli, M., Zanardi, R., Smeraldi, E., et al. (1995). Effects of fluvoxamine on the protein phosphorylation system associated with rat neuronal microtubules. Eur. Neuropsychopharmacol. 5, 65–69. doi: 10.1016/0924-977x(95)00024-j
Pescucci, C., Meloni, I., Bruttini, M., Ariani, F., Longo, I., Mari, F., et al. (2003). Chromosome 2 deletion encompassing the MAP2 gene in a patient with autism and Rett-like features. Clin. Genet. 64, 497–501. doi: 10.1046/j.1399-0004.2003.00176.x
Pettigrew, L. C., Holtz, M. L., Craddock, S. D., Minger, S. L., Hall, N., and Geddes, J. W. (1996). Microtubular proteolysis in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 16, 1189–1202. doi: 10.1097/00004647-199611000-00013
Philpot, B. D., Lim, J. H., Halpain, S., and Brunjes, P. C. (1997). Experience-dependent modifications in MAP2 phosphorylation in rat olfactory bulb. J. Neurosci. 17, 9596–9604. doi: 10.1523/JNEUROSCI.17-24-09596.1997
Prieto-Gómez, B., Velázquez-Paniagua, M., Cisneros, L. O., Reyes-Vázquez, C., Jiménez-Trejo, F., Reyes, M. E., et al. (2008). Melatonin attenuates the decrement of dendritic protein MAP-2 immuno-staining in the hippocampal CA1 and CA3 fields of the aging male rat. Neurosci. Lett. 448, 56–61. doi: 10.1093/brain/awac191
Psychiatric GWAS Consortium Bipolar Disorder Working Group (2011). Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983. doi: 10.1038/ng.943
Pucilowska, J., Vithayathil, J., Tavares, E. J., Kelly, C., Karlo, J. C., and Landreth, G. E. (2015). The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 35, 3190–3200. doi: 10.1523/JNEUROSCI.4864-13.2015
Quinlan, E. M., and Halpain, S. (1996). Emergence of activity-dependent, bidirectional control of microtubule-associated protein MAP2 phosphorylation during postnatal development. J. Neurosci. 16, 7627–7637. doi: 10.1523/JNEUROSCI.16-23-07627.1996
Rajkowska, G., Miguel-Hidalgo, J. J., Wei, J., Dilley, G., Pittman, S. D., Meltzer, H. Y., et al. (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45, 1085–1098. doi: 10.1016/s0006-3223(99)00041-4
Rao, S., Yao, Y., Zheng, C., Ryan, J., Mao, C., Zhang, F., et al. (2016). Common variants in CACNA1C and MDD susceptibility: a comprehensive meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 171, 896–903. doi: 10.1002/ajmg.b.32466
Regan, P., Piers, T., Yi, J.-H., Kim, D.-H., Huh, S., Park, S. J., et al. (2015). Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 35, 4804–4812. doi: 10.1523/JNEUROSCI.2842-14.2015
Rehbein, M., Kindler, S., Horke, S., and Richter, D. (2000). Two trans-acting rat-brain proteins, MARTA1 and MARTA2, interact specifically with the dendritic targeting element in MAP2 mRNAs. Mol. Brain Res. 79, 192–201. doi: 10.1016/s0169-328x(00)00114-5
Riederer, B. M., Draberova, E., Viklicky, V., and Draber, P. (1995). Changes of MAP2 phosphorylation during brain development. J. Histochem. Cytochem. 43, 1269–1284. doi: 10.1177/43.12.8537643
Rioux, L., Ruscheinsky, D., and Arnold, S. E. (2004). Microtubule-associated protein MAP2 expression in olfactory bulb in schizophrenia. Psychiatry Res. 128, 1–7. doi: 10.1016/j.psychres.2004.05.022
Robertson, L. A., Moya, K. L., and Breen, K. C. (2004). The potential role of tau protein O-glycosylation in Alzheimer’s disease. J. Alzheimers Dis. 6, 489–495. doi: 10.3233/jad-2004-6505
Roger, B., Al-Bassam, J., Dehmelt, L., Milligan, R. A., and Halpain, S. (2004). MAP2c, but not tau, binds and bundles F-actin via its microtubule binding domain. Curr. Biol. 14, 363–371. doi: 10.1016/j.cub.2004.01.058
Romero, P. R., Zaidi, S., Fang, Y. Y., Uversky, V. N., Radivojac, P., Oldfield, C. J., et al. (2006). Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. U S A 103, 8390–8395. doi: 10.1073/pnas.0507916103
Rosoklija, G., Keilp, J. G., Toomayan, G., Mancevski, B., Haroutunian, V., Liu, D., et al. (2005). Altered subicular MAP2 immunoreactivity in schizophrenia. Prilozi 26, 13–34. doi: 10.1097/00005072-199505000-00212
Rudrabhatla, P., Jaffe, H., and Pant, H. C. (2011). Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimers NFTs. FASEB J. 25, 3896–3905. doi: 10.1096/fj.11-181297
Sánchez, C., Díaz-Nido, J., and Avila, J. (2000). Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog. Neurobiol. 61, 133–168. doi: 10.1016/s0301-0082(99)00046-5
Saia-Cereda, V. M., Cassoli, J. S., Schmitt, A., Falkai, P., and Martins-de-Souza, D. (2016). Differential proteome and phosphoproteome may impact cell signaling in the corpus callosum of schizophrenia patients. Schizophr. Res. 177, 70–77. doi: 10.1016/j.schres.2016.03.022
Sarkar, T., Mitra, G., Gupta, S., Manna, T., Poddar, A., Panda, D., et al. (2004). MAP2 prevents protein aggregation and facilitates reactivation of unfolded enzymes. Eur. J. Biochem. 271, 1488–1496. doi: 10.1111/j.1432-1033.2004.04053.x
Scaife, R. M., Wilson, L., and Purich, D. L. (1992). Microtubule protein ADP-ribosylation in vitro leads to assembly inhibition and rapid depolymerization. Biochemistry 31, 310–316. doi: 10.1021/bi00116a042
Schätzle, P., Esteves da Silva, M., Tas, R. P., Katrukha, E. A., Hu, H. Y., Wierenga, C. J., et al. (2018). Activity-dependent actin remodeling at the base of dendritic spines promotes microtubule entry. Curr. Biol. 28, 2081–2093. doi: 10.1016/j.cub.2018.05.004
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke, S., Walters, J. T. R., and O’Donovan, M. C. (2020). Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv [Preprint]. doi: 10.1101/2020.09.12.20192922
Seitz, A., Kojima, H., Oiwa, K., Mandelkow, E.-M., Song, Y.-H., and Mandelkow, E. (2002). Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 21, 4896–4905. doi: 10.1093/emboj/cdf503
Seney, M. L., Huo, Z., Cahill, K., French, L., Puralewski, R., Zhang, J., et al. (2018). Opposite molecular signatures of depression in men and women. Biol. Psychiatry 84, 18–27. doi: 10.1016/j.biopsych.2018.01.017
Seo, M. K., Lee, C. H., Cho, H. Y., Lee, J. G., Lee, B. J., Kim, J. E., et al. (2014). Effects of antidepressant drugs on synaptic protein levels and dendritic outgrowth in hippocampal neuronal cultures. Neuropharmacology 79, 222–233. doi: 10.1016/j.neuropharm.2013.11.019
Sharma, N., Kress, Y., and Shafit-Zagardo, B. (1994). Antisense MAP-2 oligonucleotides induce changes in microtubule assembly and neuritic elongation in pre-existing neurites of rat cortical neurons. Cell Motil. Cytoskeleton 27, 234–247. doi: 10.1002/cm.970270305
Shelton, M. A., Newman, J. T., Gu, H., Sampson, A. R., Fish, K. N., MacDonald, M. L., et al. (2015). Loss of microtubule-associated protein 2 immunoreactivity linked to dendritic spine loss in schizophrenia. Biol. Psychiatry 78, 374–385. doi: 10.1016/j.biopsych.2014.12.029
Shu, T., Wu, T., Pang, M., Liu, C., Wang, X., Wang, J., et al. (2016). Effects and mechanisms of melatonin on neural differentiation of induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 474, 566–571. doi: 10.1016/j.bbrc.2016.04.108
Somenarain, L., and Jones, L. B. (2010). A comparative study of MAP2 immunostaining in areas 9 and 17 in schizophrenia and Huntington chorea. J. Psychiatr. Res. 44, 694–699. doi: 10.1016/j.jpsychires.2009.12.006
Sontag, J.-M., Nunbhakdi-Craig, V., White, C. L., III, Halpain, S., and Sontag, E. (2012). The protein phosphatase PP2A/Bα binds to the microtubule-associated proteins tau and MAP2 at a motif also recognized by the kinase Fyn. J. Biol. Chem. 287, 14984–14993. doi: 10.1074/jbc.M111.338681
Sotrel, A., Williams, R. S., Kaufmann, W. E., and Myers, R. H. (1993). Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington’s disease: A quantitative Golgi study. Neurology 43:2088. doi: 10.1212/wnl.43.10.2088
Springer, J. E., Azbill, R. D., Kennedy, S. E., George, J., and Geddes, J. W. (1997). Rapid calpain I activation and cytoskeletal protein degradation following traumatic spinal cord injury: attenuation with riluzole pretreatment. J. Neurochem. 69, 1592–1600. doi: 10.1046/j.1471-4159.1997.69041592.x
Stearns, M. E., and Brown, D. L. (1979). Purification of a microtubule-associated protein based on its preferential association with tubulin during microtubule initiation. FEBS Lett. 101, 15–20.
Steward, O., and Halpain, S. (1999). Lamina-specific synaptic activation causes domain-specific alterations in dendritic immunostaining for MAP2 and CAM kinase II. J. Neurosci. 19, 7834–7845. doi: 10.1523/JNEUROSCI.19-18-07834.1999
Strang, K. H., Golde, T. E., and Giasson, B. I. (2019). MAPT mutations, tauopathy and mechanisms of neurodegeneration. Lab. Investig. 99, 912–928. doi: 10.1038/s41374-019-0197-x
Sun, F., and Handel, M. A. (2011). A mutation in Mtap2 is associated with arrest of mammalian spermatocytes before the first meiotic division. Genes (Basel) 2, 21–35. doi: 10.3390/genes2010021
Teng, J., Takei, Y., Harada, A., Nakata, T., Chen, J., and Hirokawa, N. (2001). Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth and microtubule organization. J. Cell Biol. 155, 65–76. doi: 10.1083/jcb.200106025
Tortosa, E., Adolfs, Y., Fukata, M., Pasterkamp, R. J., Kapitein, L. C., and Hoogenraad, C. C. (2017). Dynamic palmitoylation targets MAP6 to the axon to promote microtubule stabilization during neuronal polarization. Neuron 94, 809–825. doi: 10.1016/j.neuron.2017.04.042
Uversky, V. N. (2014). Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators Front. Front. Mol. Biosci. 1:6. doi: 10.3389/fmolb.2014.00006
Vaillant, A. R., Zanassi, P., Walsh, G. S., Aumont, A., Alonso, A., and Miller, F. D. (2002). Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron 34, 985–998. doi: 10.1016/s0896-6273(02)00717-1
Velikonja, T., Fett, A.-K., and Velthorst, E. (2019). Patterns of nonsocial and social cognitive functioning in adults with autism spectrum disorder: a systematic review and meta-analysis. JAMA Psychiatry 76, 135–151. doi: 10.1001/jamapsychiatry.2018.3645
Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F., and Jones, D. T. (2004). Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645. doi: 10.1016/j.jmb.2004.02.002
Wesseling, H., Mair, W., Kumar, M., Schlaffner, C. N., Tang, S., Beerepoot, P., et al. (2020). Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 183, 1699–1713.e13. doi: 10.1016/j.cell.2020.10.029
Westphal, D. S., Andres, S., Makowski, C., Meitinger, T., and Hoefele, J. (2018). MAP2 - a candidate gene for epilepsy, developmental delay and behavioral abnormalities in a patient with microdeletion 2q34 [case report]. Front. Genet. 9, 927–936. doi: 10.3389/fgene.2018.00099
Xia, D., Li, C., and Götz, J. (2015). Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim. Biophys. Acta 1852, 913–924. doi: 10.1016/j.bbadis.2014.12.017
Xie, C., Miyasaka, T., Yoshimura, S., Hatsuta, H., Yoshina, S., Kage-Nakadai, E., et al. (2014). The homologous carboxyl-terminal domains of microtubule-associated protein 2 and TAU induce neuronal dysfunction and have differential fates in the evolution of neurofibrillary tangles. PLoS One 9:e89796. doi: 10.1371/journal.pone.0089796
Yamauchi, P. S., and Purich, D. L. (1987). Modulation of microtubule assembly and stability by phosphatidylinositol action on microtubule-associated protein-2. J. Biol. Chem. 262, 3369–3375.
Yamauchi, P. S., and Purich, D. L. (1993). Microtubule-associated protein interactions with actin filaments: evidence for differential behavior of neuronal MAP-2 and tau in the presence of phosphatidylinositol. Biochem. Biophys. Res. Commun. 190, 710–715. doi: 10.1006/bbrc.1993.1107
Zhong, H., Sia, G.-M., Sato, T. R., Gray, N. W., Mao, T., Khuchua, Z., et al. (2009). Subcellular dynamics of type II PKA in neurons. Neuron 62, 363–374. doi: 10.1016/j.neuron.2009.03.013
Keywords: MAP2, cytoskeleton, psychiatric disorder, neurodevelopment, neurodegeneration
Citation: DeGiosio RA, Grubisha MJ, MacDonald ML, McKinney BC, Camacho CJ and Sweet RA (2022) More than a marker: potential pathogenic functions of MAP2. Front. Mol. Neurosci. 15:974890. doi: 10.3389/fnmol.2022.974890
Received: 21 June 2022; Accepted: 29 July 2022;
Published: 16 September 2022
Edited by:
Michael E. Cahill, University of Wisconsin-Madison, United StatesReviewed by:
Sara Lagalwar, Skidmore College, United StatesCopyright © 2022 DeGiosio, Grubisha, MacDonald, McKinney, Camacho and Sweet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert A. Sweet, c3dlZXRyYUB1cG1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.