 Barbara Kaltschmidt
Barbara Kaltschmidt Laureen P. Helweg
Laureen P. Helweg Johannes F. W. Greiner
Johannes F. W. Greiner Christian Kaltschmidt2,3
Christian Kaltschmidt2,3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 02 August 2022
Sec. Molecular Signalling and Pathways
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.954541
This article is part of the Research Topic Transcription Regulation - Brain Development and Homeostasis - A Finely Tuned and Orchestrated Scenario in Physiology and Pathology, volume II View all 12 articles
The transcription factor NF-κB is commonly known to drive inflammation and cancer progression, but is also a crucial regulator of a broad range of cellular processes within the mammalian nervous system. In the present review, we provide an overview on the role of NF-κB in the nervous system particularly including its constitutive activity within cortical and hippocampal regions, neuroprotection as well as learning and memory. Our discussion further emphasizes the increasing role of human genetics in neurodegenerative disorders, namely, germline mutations leading to defects in NF-κB-signaling. In particular, we propose that loss of function mutations upstream of NF-κB such as ADAM17, SHARPIN, HOIL, or OTULIN affect NF-κB-activity in Alzheimer’s disease (AD) patients, in turn driving anatomical defects such as shrinkage of entorhinal cortex and the limbic system in early AD. Similarly, E3 type ubiquitin ligase PARKIN is positively involved in NF-κB signaling. PARKIN loss of function mutations are most frequently observed in Parkinson’s disease patients. In contrast to AD, relying on germline mutations of week alleles and a disease development over decades, somatic mutations affecting NF-κB activation are commonly observed in cells derived from glioblastoma multiforme (GBM), the most common malignant primary brain tumor. Here, our present review particularly sheds light on the mutual exclusion of either the deletion of NFKBIA or amplification of epidermal growth factor receptor (EGFR) in GBM, both resulting in constitutive NF-κB-activity driving tumorigenesis. We also discuss emerging roles of long non-coding RNAs such as HOTAIR in suppressing phosphorylation of IκBα in the context of GBM. In summary, the recent progress in the genetic analysis of patients, particularly those suffering from AD, harbors the potential to open up new vistas for research and therapy based on TNFα/NF-κB pathway and neuroprotection.
The transcription factor nuclear factor kappa light chain enhancer of activated B cells (NF-κB) is a key regulator of inflammation and cancer progression and vitally drives a broad range of cellular processes within the mammalian nervous system (Kaltschmidt and Kaltschmidt, 2009). In the present review, we will particularly shed light on this crucial role of NF-κB in the nervous system as well as in neuroprotection and associated diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), and glioblastoma multiforme (GBM). On the molecular level, the NF-κB family comprises five DNA-binding members REL (c-REL), RELA (p65), and RELB (RELB) as well as NFKB1 (p50) and NFKB2 (p52), with the latest lacking transactivation domains (Ghosh et al., 1995). NF-κB-signaling can be distinguished into the canonical, non-canonical and atypical pathway (Figure 1). Canonical and non-canonical NF-κB-signaling share a common regulatory element, the inhibitor of κB (IκBα) kinase (IKK). In non-canonical NF-κB-signaling, ligand binding to receptors like CD40 activates IKK1 via NF-κB-inducible kinase (NIK). Phosphorylation of IKK1 results in processing of p100 to p52 and subsequent nuclear translocation of p52/RELB (Kaltschmidt et al., 2018; Figure 1). Canonical NF-κB-signaling is mediated by phosphorylation of the NEMO/IKK1/IKK2-complex for instance upon binding of ligands to TNF receptor 1 (TNFR1) (Figure 1). Phosphorylated IKKs in turn phosphorylate IκBα, which keeps p65/p50 in an inactive cytoplasmic state (Zhang et al., 2017). IKK-dependent phosphorylation of IκBα results in its proteasomal degradation and enables nuclear translocation of p65/p50 and expression of target genes (Figure 1). In atypical NF-κB-signaling, exposure of cells to UV light leads to NF-κB activation via p38 MAP Kinase (MAPK) activation, in turn resulting in casein kinase 2 (CK2)-mediated phosphorylation and degradation of IκB. In contrast to canonical and non-canonical activation of NF-κB, CK2-dependent atypical phosphorylation of IκB is c-terminal (e.g., at Ser293) and independent to IKK, which phosphorylates IκB at n-terminal phosphorylation sites (such as Ser32 or Ser36) (McElhinny et al., 1996; Sayed et al., 2000; Kato et al., 2003; reviewed in Lin et al., 2010; Figure 1). Next to TNFα (Furukawa and Mattson, 1998; Kaltschmidt et al., 1999; Figure 1), NF-κB can be also activated by the neurotransmitter glutamate (Guerrini et al., 1995; Kaltschmidt et al., 1995) and its agonists like kainate (Kaltschmidt et al., 1995) and N-methyl-D-aspartate (NMDA) (Meffert et al., 2003) in the nervous system as discussed below.
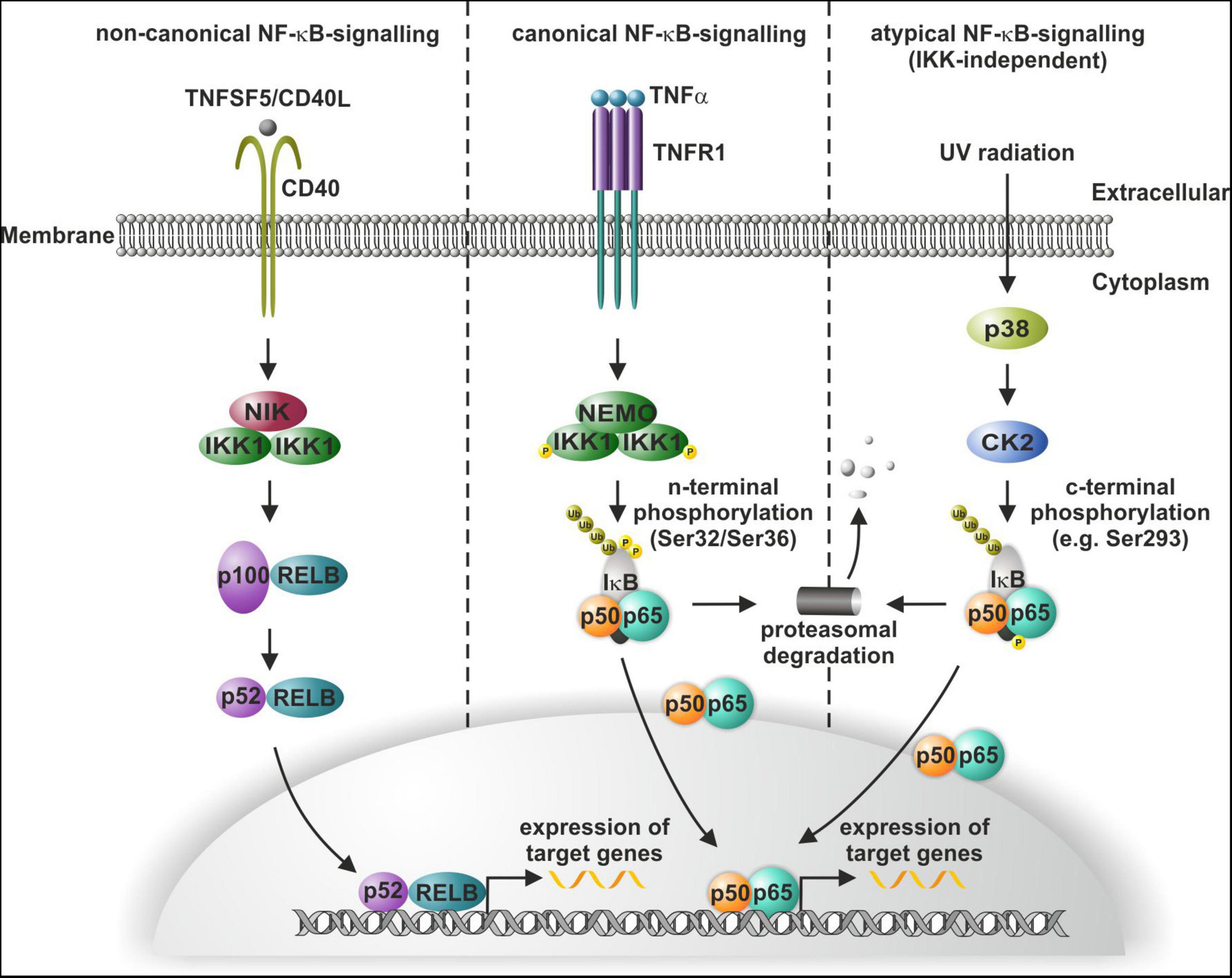
Figure 1. Non-canonical, canonical, and atypical NF-κB-signaling. Non-canonical signaling includes CD40-mediated activation of NF-κB-inducible kinase (NIK), which activates (IκBα) kinase (IKK) and leads to the processing of the p100/RELB and translocation of p52/RELB (left panel). In canonical NF-κB-signaling, TNFα-mediated activation of the IKK complex leads to phosphorylation of IκBα and allows the p50/p65 dimer to translocate to the nucleus (mid panel). UV light induces atypical activation of NF-κB-signaling through p38-MAPK activation and consequent IκBα phosphorylation independent to IKK, which again releases the p50/p65 dimer (right panel). Modified from Kaltschmidt et al. (2021).
When we started in 1992 to work on NF-κB in the nervous system not much was known on that topic. Our own review (Kaltschmidt B. et al., 1993) on this topic suggested an involvement of NF-κB based on the production of reactive oxygen intermediates in various neurological diseases, such as multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and others. Today, a Pubmed search with MeSH entries “nervous system disease” and “NF-kappa B” reveals 4,750 results (May 18, 2022), starting in 1993 with two of our own papers.
Our starting investigation was a biochemical study with fractionation of rodent brains in so called synaptosomes. Synaptosomes are prepared by shear stress with a Potter type tissue homogenizer followed by gradient fractionation from different brain regions (Whittaker et al., 1964). Why we used synaptosomes? This biochemical preparation provides the opportunity to fractionate the complex brain in simple subcellular compartments. In addition, synaptosomes are a way to enrich NF-κB from total brain lysates. Synaptosomes can be analyzed by standard DNA-binding techniques such as EMSA to identify transcription factors such as NF-κB.
Synaptosomes are composed of pre-synaptic elements stabilized by post-synaptic density elements. Within synapses derived from olfactory bulb, white and gray matter, cerebellum and brain stem we could detect inducible NF-κB activity, with highest activity in gray matter. DNA binding of NF-κB could be inhibited by recombinant IκBα (Kaltschmidt C. et al., 1993), previously called MAD-3. Later on, synaptic localization of NF-κB could be reproduced by several other groups using synaptosomes (e.g., Meberg et al., 1996; Suzuki et al., 1997; Meffert et al., 2003; Schmeisser et al., 2012). In this line, it was reported that RELA could exist in two pools, a soluble one in synaptosomes and a membrane bound in fractions containing post-synaptic density proteins. While the active RELA-pool in the synaptoplasm is initially activated during early memory consolidation, its subsequent translocation into the membranes of the synaptosomes seems important for consolidation of long-term memory in mice (Salles et al., 2015). Furthermore, localization in post-synaptic spines and regulation of synapse growth was shown by Meffert et al. (2003) (Boersma et al., 2011; Dresselhaus et al., 2018). The function of transcription factors in synaptosomes could be within “a dialogue between synapses and genes” as published by Kandel (2001). In this hypothesis a feedback of activated synapses back to the nucleus (so called retrograde transport, see below) can initiate changes in gene transcription. These might direct synaptic enhancement and regrowing of novel synaptic contacts. For this task inducible transcription factors are well suited. Collectively these gathered evidence suggest that NF-κB is a synaptic protein in different brain region such as cerebellum, cortex and hippocampus, olfactory bulb, and brain stem.
We could show that NF-κB could be retrogradely transported from the pre-synaptic site to the nucleus in hippocampal neurons (Wellmann et al., 2001). Retrograde transport could be studied by injection of radioactively labeled proteins in animals, like it was shown for transport of nerve growth factor (NGF) in sensory neurons (Stoeckel et al., 1975). Furthermore transport of green fluorescent protein (GFP) fusion proteins could be analyzed by life microscopy (Wellmann et al., 2001), which could be combined with the analysis of transport in photobleached regions (Meffert et al., 2003). Finally transport of single fusion proteins could be studied with blinking probes (Widera et al., 2016). Later on, we could extend this observation with super resolution microscopy (Mikenberg et al., 2007; Widera et al., 2016). In this line, it is important to introduce TNFα, which also can be retrogradely transported in inflammatory lesions of peripheral nerves (Shubayev and Myers, 2001). Accordingly, we demonstrated Hsc70 as a novel interactor of NF-κB potentially involved in retrograde transport in rodent brain (Klenke et al., 2013), while deoxyspergualin, a specific inhibitor of Hsc70, inhibited nuclear import of NF-κB. We have previously shown that NF-κB was activated in microglia of experimental allergic encephalomyelitis (EAE) rats (Kaltschmidt et al., 1994a). In this line, effective treatment of EAE with deoxyspergualin could be shown in two models of acute and chronic relapsing EAE (Schorlemmer and Seiler, 1991). Taken together these data provide evidence for a retrograde transport of NF-κB from activated synapses to the nucleus, one component of the transport complex is Hsc70, which could be inhibited by deoxyspergualin.
We discovered constitutive activity (always activated NF-κB in the nucleus) within cortical and hippocampal regions of rodent brains (Kaltschmidt et al., 1994b). Further analysis of NF-κB activity with reporter gene assays could reproduce this constitutive activity within the neurons of the cortical or the hippocampal region (Schmidt-Ullrich et al., 1996; Bhakar et al., 2002). In particular, Barker and coworkers showed that efficient blocking of endogenous NF-κB activity by recombinant adenovirus led to death of cortical neurons, whereas induction of NF-κB activity resulted in neuroprotection (Bhakar et al., 2002). Evidence for constitutive activity demonstrates that NF-κB is a sensor for neuronal activity of glutamatergic neurons. Anesthesia of cultured neurons with, e.g., NMDA receptor antagonists inhibits constitutive NF-κB activity.
Activation of NF-κB in neurons is possible by the neurotransmitter glutamate (Guerrini et al., 1995; Kaltschmidt et al., 1995) and agonists such as kainate (Kaltschmidt et al., 1995) and NMDA (Meffert et al., 2003), as well as by pro-inflammatory cytokines as TNFα (Furukawa and Mattson, 1998; Kaltschmidt et al., 1999; Figure 1). Taken together evidence indicated a major role of NF-κB in glutamatergic signaling as well as pro-inflammatory TNF mediated signaling.
In the beginning of the 21st century, several new transgenic mouse models with specific repression or activation of NF-κB in the nervous system were generated, allowing studies of behavior and learning. Mollie Meffert and coworkers discovered that p65-deficient mice have no synaptic NF-κB. These mice when crossed to TNFR1-deficient mice could survive from TNF-mediated hepatic failure and had a learning defect of spatial learning in a radial arm maze (Meffert et al., 2003). We discovered that expression of transdominant negative IκBα in glutamatergic neurons affected spatial memory formation and repressed expression of the novel NF-κB target gene protein kinase A (catalytic subunit α) (Kaltschmidt et al., 2006). Warner Greene and coworkers discovered that neuronal expression of super-repressor IκB in GABA-ergic interneurons led to enhanced spatial learning and memory (O’Mahony et al., 2006). Later on, it was shown that NF-κB-repression by transdominant negative IKK2 led to impaired learning and memory. The authors further identified insulin-like growth factor 2 (IGF2) as a novel IKK/NF-κB target gene. In this line, IGF2 was able to restore synapse density and promoted spine maturation in IKK/NF-κB signaling-deficient neurons within 24 h (Schmeisser et al., 2012). In contrast, expression of a constitutively active allele of IKK2 in forebrain neurons led to degeneration of microglia and astrocytes as well as spatial learning defects (Maqbool et al., 2013). Therefore evidence indicates NF-κB involvement in learning and memory and additionally in brain regeneration in vivo.
Alzheimer’s disease (AD) is a devastating neurodegenerative disease and was discovered by Alois Alzheimer in Munich when analyzing brain sections of his patient Auguste Deter, who died in a lunatic asylum in Frankfurt. Alois Alzheimer immediately discovered the presence of “miliary foci” (senile plaques) and very strange changes in neurofibrils, which clump into tangles that eventually replace dead neurons (Alzheimer, 1907). Later on, he extended this observation toward examples of neurofibril tangles and plaques (Alzheimer, 1911). Then, Konrad Beyreuther and coworkers purified plaque proteins and determined the amyloid plaque core protein sequence (Masters et al., 1985). Furthermore, the amyloid A4 beta precursor protein was cloned by the group of Müller-Hill (Kang et al., 1987). Notably, many AD-relevant genes such as BIN1, IκBα, APP, BASE1, COX-2, MnSOD, CuZnSOD, TNFR1, and others are target genes of NF-κB (Snow and Albensi, 2016; Figure 2).
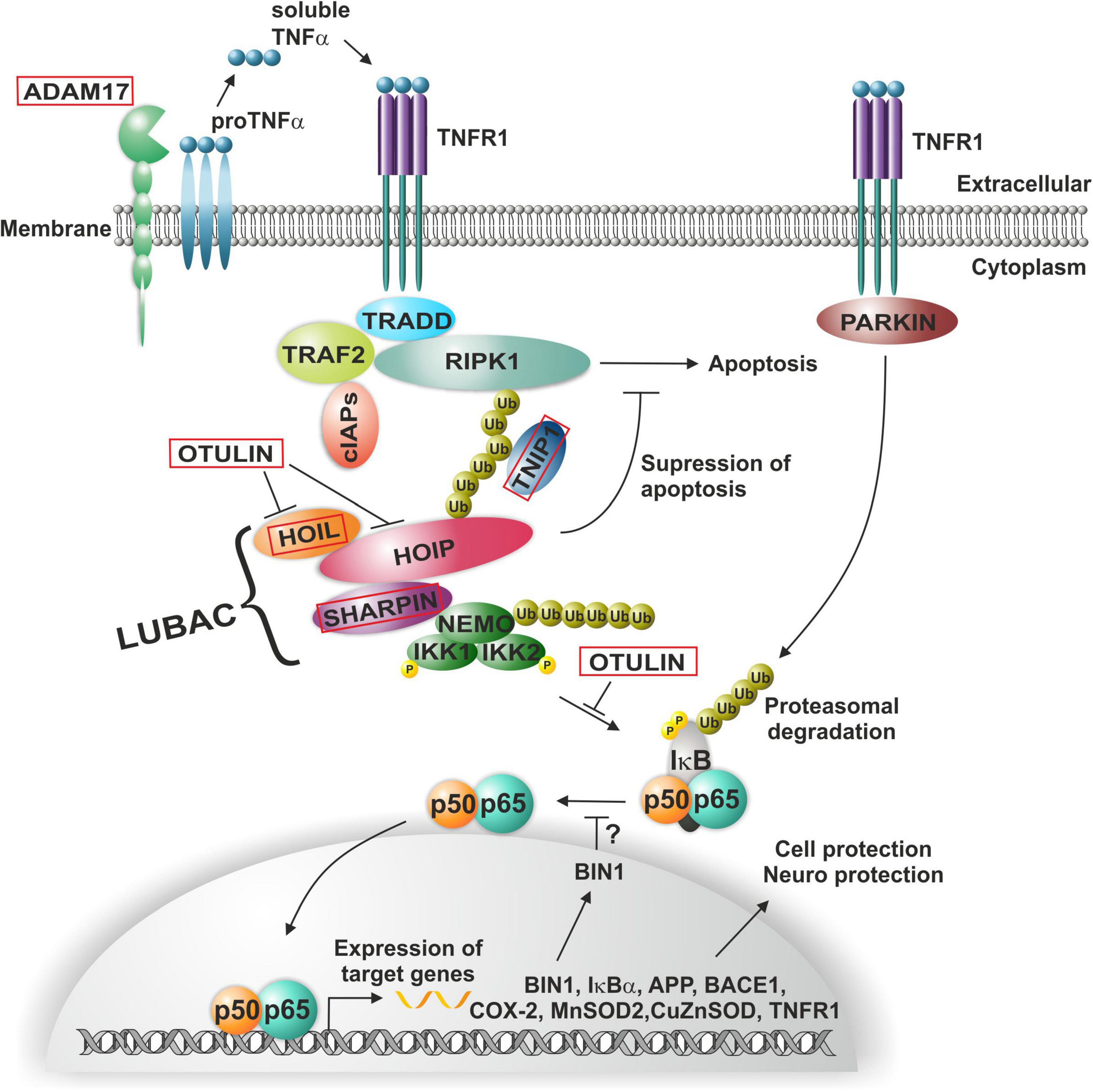
Figure 2. NF-κB-signaling in Alzheimer’s and Parkinson’s disease. ADAM17-induced soluble TNFα mediates the activation of the TNF receptor complex, where poly-ubiquitination leads to the recruitment of the LUBAC complex. Linear ubiquitination of NEMO mediates the activation and phosphorylation of the (IκBα) kinase (IKK) complex and consequent activation of NF-κB p50/p65, in turn driving expression of a range of AD-relevant genes such as BIN1, IκBα, APP, BASE1, COX-2, MnSOD, CuZnSOD, or TNFR1. In PD, activation of NF-κB is mediated by PARKIN in a TNFR1-dependent manner.
However, a recent genetic study using genome wide association of 111,326 clinically diagnosed AD cases and 677,663 controls challenges this one and only hypothesis of the A beta concept and provides additional evidence for AD-linked genes with higher significance. Notably, BIN1 (bridging integrator 1) alleles (p-value: 6 × 10^−118) were most significantly associated to AD, whereas APP (precursors of A beta) had a significance of correlation of only 1 × 10^−12 (Bellenguez et al., 2022). Most interestingly, BIN1 knockout in breast cancers was linked to increased nuclear NF-κB (Ghaneie et al., 2006), suggesting a dual role as inhibitor of NF-κB and as a target gene (Mao et al., 1999; Figure 2). BIN1 encodes several isoforms of a nucleocytoplasmic adaptor protein, initially described with tumor suppressor functions. BIN1 may be involved in the regulation of MYC activity and the control of cell proliferation (Sakamuro et al., 1996). Three isoforms of BIN1 were detected in neurons and astrocytes (isoforms 1, 2, and 3) and four isoforms in microglia (isoforms 6, 9, 10, and 12) of human brain (Taga et al., 2020) showing a strong association of BIN1 isoforms expressed by neurons/astrocytes and tangles that might contribute to cognitive decline in AD. Its role as a tumor suppressor gene suggests a function of wt BIN1 allele as an inhibitor of NF-κB activation (Elliott et al., 2000). Additionally, BIN1 transcript levels were increased in AD brains and BIN1 mediates AD risk by modulating Tau pathology (Chapuis et al., 2013). In particular, Chapuis et al. (2013) identified a novel 3 bp insertion allele upstream of BIN1, which is directly linked to increased BIN1 expression and risk for developing AD. In addition, this novel insertion was associated with Tau accumulation but not with amyloid loads in AD brains. On mechanistic level, knockdown of the BIN1 ortholog Amph suppressed Tau-toxicity in an amyloid-independent manner in Drosophila. Coimmunoprecipitation studies further substantiated a direct interaction between Tau and BIN1 in human cells and mouse brains. Although the particular pathogenic mechanism linking BIN1 and Tau in AD progression remains unclear, BIN1 may modulate Tau aggregation/oligomer formation in earlier disease stages (Chapuis et al., 2013). In human induced neurons, BIN1-knockout reduced early endosome signaling and overexpression of the AD-relevant isoform of BIN1 (isoform 1) increased the size of early endosomes and led to neurodegeneration (Lambert et al., 2022). The TNFα pathway with linear ubiquitination was also included in the new genetically associated processes within AD patients (Bellenguez et al., 2022). Interestingly, ADAM17, an extracellular protease leading to soluble TNFα, the TNF receptor interactor TNIP1, and components of the linear ubiquitination machinery linear ubiquitination chain assembly complex (LUBAC): HOIL, SHARPIN, and the negative regulator OTULIN are all affected by mutations in AD patients (Figure 2, red boxes). In this context, it might be interesting to note that Mattson and coworkers discovered a neuroprotective effect of TNFα (Cheng et al., 1994). Furthermore, they discovered that TNFα protected against A beta/glutamate toxicity (Barger et al., 1995; Albensi and Mattson, 2000). Activated NF-κB might direct the expression of antioxidant enzymes such as MnSOD and Calcium-binding proteins such as calbindin. Calcium buffering and reduction of reactive oxygen intermediates by antioxidant enzymes can actively protect against glutamate and Ab mediated induction of apoptosis (see for further discussion: Neuroprotective signal transduction edt. M.P. Mattson Springer Science Press 1998). We could reproduce this observation in cerebellar granule cells and additionally could show that treatment with a low dose of A beta (100 nM) activates p65 specifically and fortifies neurons against a neurotoxic high dose of A beta (Kaltschmidt et al., 1999). In addition also TNFα activated NF-κB at a very sharp peak of 2 ng/ml leading to neuroprotection whereas higher doses of TNF had adversive effects. Furthermore, we could demonstrate that activated NF-κB was restricted to cells in the close vicinity of early plaques in AD patient brains (Kaltschmidt et al., 1997; Ferrer et al., 1998). Interestingly, the overall NF-κB immunoreactivity was quite low in AD brains in comparison to age-matched controls. Recent genetic evidence of potential loss-of-function mutants involved in NF-κB signaling of AD patients might provide a genetic reason for this early correlation (Bellenguez et al., 2022). In this line, human SHARPIN mutants were shown to have less capability of NF-κB activation (Park et al., 2021). Furthermore, SHARPIN mutants (Park et al., 2021) of the LUBAC complex are highly significantly correlated with a reduction in the thickness of the entorhinal cortex, one of the earliest signs of AD (Kobro-Flatmoen et al., 2021; Figures 3A,B). Soheili-Nezhad et al. (2020) identified a SHARPIN mutant in AD associated with degeneration of the limbic system and its interconnecting white-matter. Since SHARPIN is a post-synaptic density protein and other components of the TNFα pathway (see also Figure 2) are also localized in neurons, we conclude that some mutations present in AD led to a reduced TNFα/NF-κB-driven neuroprotection. Accordingly, a transgenic mouse model with NF-κB ablation in basal forebrain neurons led to a severe degeneration of the dentate gyrus (Figures 3C,D), which could be rescued on the cellular level by reactivation of NF-κB (Imielski et al., 2012). In addition, behavioral deficits were completely repaired. In summary, our discussion emphasizes a potential neuroprotective, regenerative role of NF-κB in neurons of AD. Based on the evidences reported above, we suggest a testable hypothesis where loss of function mutations upstream of NF-κB such as ADAM17, SHARPIN, HOIL, or OTULIN (Figure 2) lead to defects in NF-κB signaling including retrograde transport of the signaling endosome and neuroprotection. Finally, these defects might account for the anatomical defects seen in the entorhinal cortex (early AD, Figure 3) followed up by the limbic system and finally shrinkage of the cortex (late AD). We hypothesize based on the evidence presented above that the development of the disease is observed over decades of continuous cognitive decline because the defects in AD patients all rely on weak alleles in contrast to knockouts in somatic cells as in GBM (see below). In summary these genetic data demonstrate the involvement of TNF mediated NF-κB activation in neurons of AD patients. Especially loss of function mutations of the NF-κB activator Sharpin alone or together with additional mutations suggest a loss of neuroprotection enhancing the development of AD.
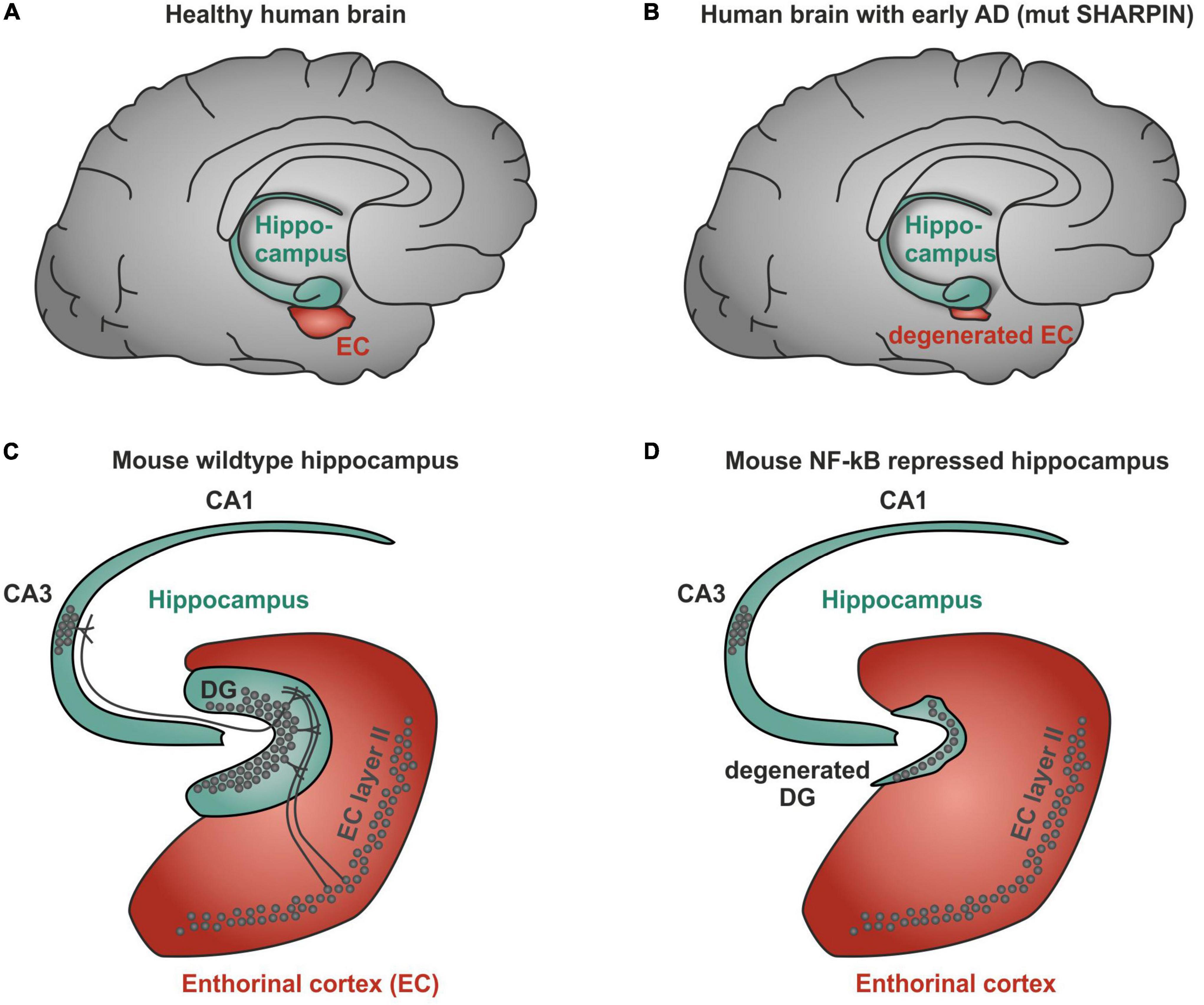
Figure 3. NF-κB-mediated degeneration of human and mouse brain. (A) Scheme of healthy human brain showing a normal sized entorhinal cortex. (B) Human SHARPIN mutated Alzheimer’s disease (AD) brain showing a degenerated entorhinal cortex. (C) Scheme of healthy NF-κB wildtype mouse brain with normal dentate gyrus. (D) NF-κB repression in mice brains leads to a degenerated dentate gyrus.
Parkinson’s disease is another important neurodegenerative disease affecting more than approximately 1% of individuals older than 60 years. It is suggested that oxidative stress induces apoptosis of dopaminergic neurons in the substantia nigra, resulting in neurodegenerative disorders observed in patients diagnosed with PD. PD was first described by James Parkinson in 1817 (Parkinson, 2002), as a disease separated from other neurological disorders and characterized by typical involuntary tremulous motions of body parts, even when not in action. Much later Arvid Carlson discovered the presence of dopamine in the brain (Carlsson et al., 1958). He used a fluorimetric method in combination with ion exchange chromatography. Furthermore he discovered that reserpine isolated from the plant Rauwolfia led to a Parkinson like phenotype in rabbits. Reserpine lead to complete depletion of dopamine which could be restored by injection of L-Dopa. This discovery was rewarded in 2000 with the Nobel Price. The biochemical discovery of replenishing dopamine by L-Dopa was translated to a clinical L-Dopa therapy for PD patients (Ehringer and Hornykiewicz, 1960). Neuropathological analysis revealed neuronal lesions in regions of the motor system rich in dopamine such as the substantia nigra but also within the limbic system and the brain stem. The cellular basis of dopamine release are dopaminergic neurons which release dopamine to other neurons without any direct synaptic contacts (volume transmission) and are relatively few (about 4,00,000) in the human brain (reviewed in Schultz, 2007). Lewy (1912) discovered a hallmark of PD pathology the so called Lewy bodies as neuronal eosinophilic cytoplasmic inclusion bodies in brain stems of PD patients. Neurons dying during the progression of PD are characterized by the presence of Lewy bodies in their perikarya and Lewy neurites in their neuronal processes. Lewy bodies can be immunostained with antibodies raised against α-synuclein. This type of immunostaining reveals many kinds of Lewy bodies ranging from dot- or thread-like forms to very large types (Braak and Braak, 2000). Similar to AD in PD changes in the neuronal cytoskeleton develop but in only a few susceptible types of nerve cells. α-synuclein is a small protein involved in synaptic dopamine release and a major component which is aggregated in a prion-like fashion in Lewy bodies (Steiner et al., 2018). Braak et al. (2003a) developed a new staging system for PD severity using anti- alpha synuclein histochemistry. Based on these data, they proposed a new hypothesis on the pathology of PD: alpha synuclein pathology typical for PD might spread from gut to brain presumable along the vagus nerve (Braak et al., 2003b). Later on indeed gut brain transmission of pathology with concomitant loss of dopaminergic neurons could be observed in mice after injection of prion-like alpha synuclein fibrils into gut muscles (Kim et al., 2019). While exogenous alpha synuclein could lead to mitochondrial disfunction, overexpression of parkin could rescue the mitochondrial disfunction (Wilkaniec et al., 2021).
A long established genetic linkage is the loss of function of Parkin E3 ubiquitin ligase (Panicker et al., 2021). In this line, recent in vitro analysis showed that overexpression of PARKIN strongly enhances TNFα-mediated NF-κB activation in HEK-293 cells, suggesting an involvement of this E3 ubiquitin ligase in TNFα-mediated NF-κB signaling by stabilizing LUBAC (Meschede et al., 2020; Figure 2).
Furthermore reactive oxygen-mediated neurodegeneration was shown to regulate mRNA expression and survival in neurons derived from neural crest derived stem cells in a sex-specific manner (Ruiz-Perera et al., 2018). In particular we observed that female neurons are more susceptible to oxidative stress-mediated cell death. However, activation of NF-κB (RELA) by TNFα let to significant neuroprotection against oxidative stress-induced cell death in both sexes. While female neurons upregulated the NF-κB target genes SOD2 and IGF2 to induce neuroprotection, male neurons showed elevated expression of the neuroprotective NF-κB target gene PKA cat alpha (Ruiz-Perera et al., 2018). But when cRel, a crucial regulator of neuronal development from neural crest is inhibited, neural crest derived stem cells shift their fate from the neuronal to the oligodendrocytic lineage (Ruiz-Perera et al., 2020; reviewed in Greiner et al., 2019). We therefore conclude that NF-κB seems to be a key player in neuroprotection of both sexes, although the protective gene expression program beneath is sexually dimorphic.
Glioblastoma multiforme (GBM) is the most common malignant primary brain and CNS tumor with a very poor prognosis. GBM rapidly developing de novo are classified as primary GBM, while GBM developing from low-grade astrocytoma are classified as secondary GBM and show IDH1 mutations (Ohgaki and Kleihues, 2013). NF-κB signaling has been shown to play a central role in glioblastoma growth (reviewed in Smith et al., 2008). Consistently, mesenchymal differentiation mediated by NF-κB has been shown to promote radiation resistance in GBM (Bhat et al., 2013) and inhibition of NF-κB attenuates mesenchymal characteristics and cell proliferation (Wang et al., 2018). Several studies revealed constitutive activation of NF-κB in GBM cells, which promoted invasiveness, angiogenesis and stem cell features such as self-renewal (Raychaudhuri et al., 2007; Xie et al., 2010; Rinkenbaugh et al., 2016; reviewed in Kaltschmidt et al., 2022). Accordingly, we previously reported the GO term “NF-κB binding” to be enriched in a global transcriptome analysis of three primary glioblastoma stem cell populations (Witte et al., 2021).
The oncogenic long non-coding RNA (lncRNA) HOTAIR induces altered histone H3 lysine 27 methylation and thus has been shown to promote the enrichment of the TNFα/NF-κB signaling protein complex, the IκB kinase complex, and the IKK1-IKK2 complex in glioma cells (Wang et al., 2021). Additionally, HOTAIR suppresses the expression of the NF-κB upstream inhibitor UBXN1 via histone modification, leading to enhanced IκBα phosphorylation and consequent NF-κB activation, which in turn induces the expression of PD-L1 and enhances T cell killing resistance and immune escape (Chernorudskiy and Gainullin, 2013; Wang et al., 2021; Figure 4). In addition to enhanced phosphorylation, a heterozygous deletion of NFKBIA (IκBα) has been reported by Bredel and coworkers in 28% of GBM and 22% of cancer stem-like cells. Analyzing 790 human GBMs revealed a mutual exclusion of either NFKBIA deletion or EGFR amplification with similar clinical outcomes (Bredel et al., 2011; Figure 4). Of note, viral-mediated expression of NFKBIA in established GBM cell lines with NFKBIA deletions or EGFR amplification reduced cell viability, but it did not affect NFKBIA/EGFR wildtype GBM cells. EGFR mutations and consequent overexpression are frequent in GBM, with a majority of GBM containing the constitutively active mutant EGFR, which shows a genetic deletion of exon 2–7 and is named EGFRvIII (Heimberger et al., 2005; Figure 4). Other EGFR mutations in GBM include a N-terminal deletion (EGFRvI), deletion of exons 14–15 (EGFRvII), deletion of exons 25–27 (EGFRvIV), and deletion of exons 25–28 (EGFRvV) (An et al., 2018). EGFRvIII overexpression in GBM patients with EGFR amplification was significantly correlated to poor overall survival (Shinojima et al., 2003). On cellular level, several studies linked expression of EGFRvIII in GBM with tumor invasion as well as angiogenesis and inhibition of EGFRvIII inhibited these processes (Zheng et al., 2014; Camorani et al., 2015; Eskilsson et al., 2016; reviewed in Keller and Schmidt, 2017; Figure 4). In EGFRvIII overexpressing glioma cells, angiogenesis and tumor growth were promoted by EGFRvIII-mediated activation of NF-κB (Bonavia et al., 2012). EGFRvIII is also linked to self-renewal of GBM cells and connected to GBM stem-like cell populations (Emlet et al., 2014; Kim et al., 2021). Consistently, EGFRvIII and EGFR are downregulated upon differentiation of GBM neurospheres as well as inhibited EGFR signaling induced differentiation with decreased tumorigenic and stem-like cell potential (Stockhausen et al., 2014).
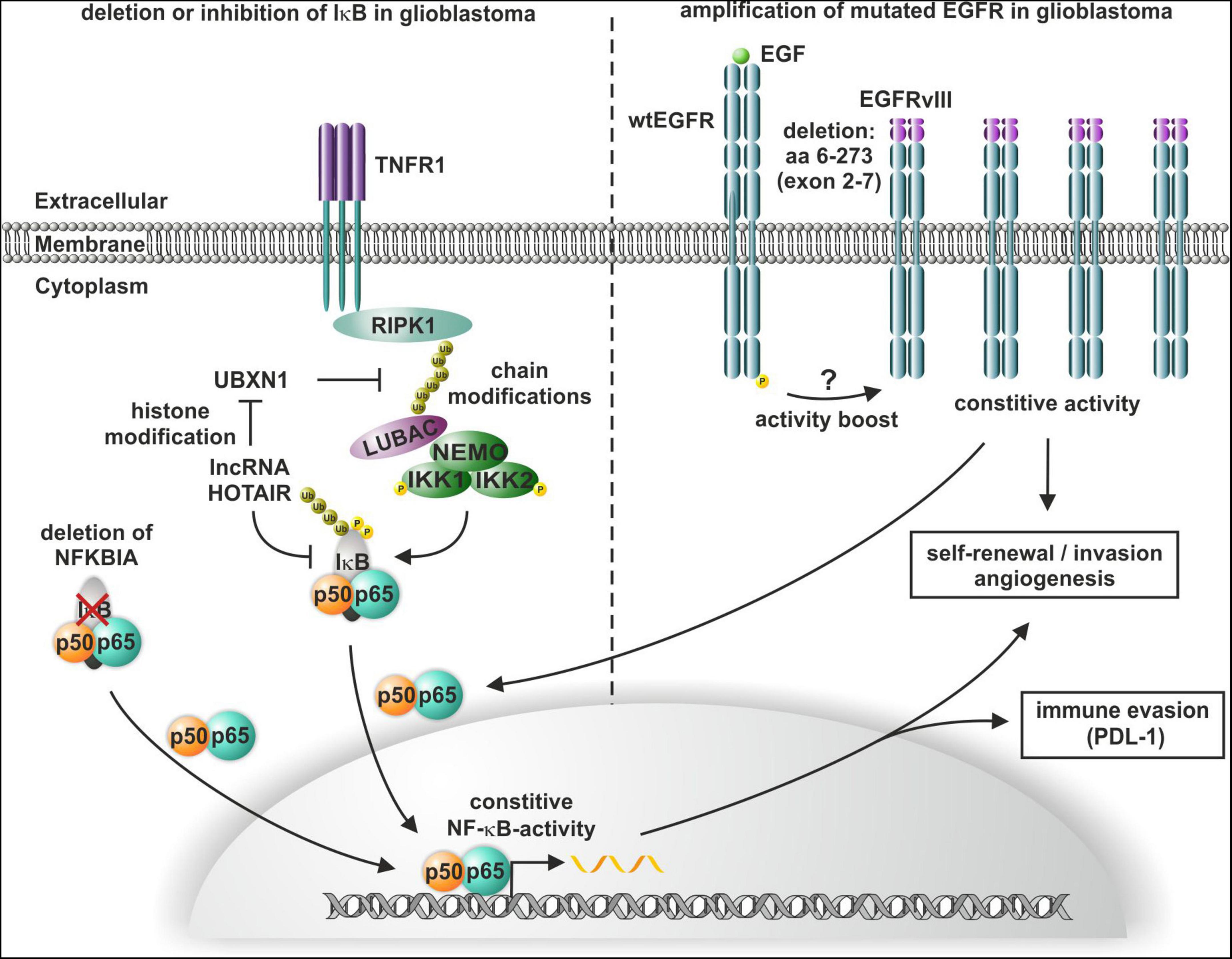
Figure 4. Constitutive activation of NF-κB in glioblastoma multiforme (GBM). Heterozygous deletion of NFKBIA or HOTAIR-mediated phosphorylation of IκB leads to the constitutive activation of NF-κB, which in turn induces self-renewal, angiogenesis, and immune escape. NF-κB is also activated by constitutive activity of mutated EGFRvIII, which also confers self-renewal and invasion in GBM.
In summary, therapy development for neurological diseases might be successful for PD with stem cell therapy (Piao et al., 2021), perhaps with a combined approach where growth promoting factors are used to enhance the performance of midbrain dopaminergic neuron grafts (Bjorklund and Parmar, 2021). However, more than 50 years of AD research failed to deliver new therapeutic approaches (Yiannopoulou et al., 2019). Despite the complexity of AD, a recent progress in the genetic analysis of AD patients reviewed here might open up new vistas for research and therapy based on TNFα/NF-κB pathway and neuroprotection. We conclude that TNFα-mediated NF-κB activation essential for neuroprotection is hampered by germ line mutations in many AD patients. This evidence provides a testable hypothesis for reduced NF-κB activation in neurons of AD patients. Mutations may act alone as shown for Sharpin in combinations.
BK and CK: conceptualization and funding acquisition. BK, LH, JG, and CK: writing–original draft preparation and writing–review and editing. LH and JG: visualization. All authors read and agreed to the published version of the manuscript.
This research was funded by the University of Bielefeld. LH was funded by an internal grant of the Bethel Foundation, Bielefeld, Germany. We acknowledged support for the publication costs by the Deutsche Forschungsgemeinschaft and the Open Access Publication Fund of Bielefeld University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Albensi, B. C., and Mattson, M. P. (2000). Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse 35, 151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8<3.0.CO;2-P
Alzheimer, A. (1907). Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Z. Psychatrie Psychisch Gerichtliche Medizin 64, 146–148.
Alzheimer, A. (1911). über eigenartige Krankheitsfälle des späteren Alters. Z. Die Gesamte Neurologie Psychiatrie 4:356. doi: 10.1007/BF02866241
An, Z., Aksoy, O., Zheng, T., Fan, Q. W., and Weiss, W. A. (2018). Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 37, 1561–1575. doi: 10.1038/s41388-017-0045-7
Barger, S. W., Horster, D., Furukawa, K., Goodman, Y., Krieglstein, J., and Mattson, M. P. (1995). Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc. Natl. Acad. Sci. U.S.A. 92, 9328–9332. doi: 10.1073/pnas.92.20.9328
Bellenguez, C., Kucukali, F., Jansen, I. E., Kleineidam, L., Moreno-Grau, S., Amin, N., et al. (2022). New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436. doi: 10.1038/s41588-022-01024-z
Bhakar, A. L., Tannis, L. L., Zeindler, C., Russo, M. P., Jobin, C., Park, D. S., et al. (2002). Constitutive nuclear factor-kappa B activity is required for central neuron survival. J. Neurosci. 22, 8466–8475.
Bhat, K. P. L., Balasubramaniyan, V., Vaillant, B., Ezhilarasan, R., Hummelink, K., Hollingsworth, F., et al. (2013). Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346. doi: 10.1016/j.ccr.2013.08.001
Bjorklund, A., and Parmar, M. (2021). Dopamine cell therapy: from cell replacement to circuitry repair. J. Parkinsons Dis. 11, S159–S165. doi: 10.3233/JPD-212609
Boersma, M. C., Dresselhaus, E. C., De Biase, L. M., Mihalas, A. B., Bergles, D. E., and Meffert, M. K. (2011). A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J. Neurosci. 31, 5414–5425. doi: 10.1523/jneurosci.2456-10.2011
Bonavia, R., Inda, M. M., Vandenberg, S., Cheng, S. Y., Nagane, M., Hadwiger, P., et al. (2012). EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene 31, 4054–4066. doi: 10.1038/onc.2011.563
Braak, H., and Braak, E. (2000). Pathoanatomy of Parkinson’s disease. J. Neurol. 247, II3–II10. doi: 10.1007/PL00007758
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003a). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Braak, H., Rüb, U., Gai, W. P., and Del Tredici, K. (2003b). Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Trans. 110, 517–536. doi: 10.1007/s00702-002-0808-2
Bredel, M., Scholtens, D. M., Yadav, A. K., Alvarez, A. A., Renfrow, J. J., Chandler, J. P., et al. (2011). NFKBIA deletion in glioblastomas. N. Engl. J. Med. 364, 627–637. doi: 10.1056/NEJMoa1006312
Camorani, S., Crescenzi, E., Colecchia, D., Carpentieri, A., Amoresano, A., Fedele, M., et al. (2015). Aptamer targeting EGFRvIII mutant hampers its constitutive autophosphorylation and affects migration, invasion and proliferation of glioblastoma cells. Oncotarget 6, 37570–37587. doi: 10.18632/oncotarget.6066
Carlsson, A., Lindqvist, M., Magnusson, T., and Waldeck, B. (1958). On the presence of 3-hydroxytyramine in brain. Science 127:471. doi: 10.1126/science.127.3296.471
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Cheng, B., Christakos, S., and Mattson, M. P. (1994). Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron 12, 139–153. doi: 10.1016/0896-6273(94)90159-7
Chernorudskiy, A. L., and Gainullin, M. R. (2013). Ubiquitin system: direct effects join the signaling. Sci. Signal. 6:e22. doi: 10.1126/scisignal.2004251
Dresselhaus, E. C., Boersma, M. C. H., and Meffert, M. K. (2018). Targeting of NF-kappaB to dendritic spines is required for synaptic signaling and spine development. J. Neurosci. 38, 4093–4103. doi: 10.1523/JNEUROSCI.2663-16.2018
Ehringer, H., and Hornykiewicz, O. (1960). [Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system]. Klin. Wochenschr. 38, 1236–1239. doi: 10.1007/BF01485901
Elliott, K., Ge, K., Du, W., and Prendergast, G. C. (2000). The c-Myc-interacting adaptor protein Bin1 activates a caspase-independent cell death program. Oncogene 19, 4669–4684. doi: 10.1038/sj.onc.1203681
Emlet, D. R., Gupta, P., Holgado-Madruga, M., Del Vecchio, C. A., Mitra, S. S., Han, S. Y., et al. (2014). Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 74, 1238–1249. doi: 10.1158/0008-5472.CAN-13-1407
Eskilsson, E., Rosland, G. V., Talasila, K. M., Knappskog, S., Keunen, O., Sottoriva, A., et al. (2016). EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol. 18, 1644–1655. doi: 10.1093/neuonc/now113
Ferrer, I., Marti, E., Lopez, E., and Tortosa, A. (1998). NF-kB immunoreactivity is observed in association with beta A4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 24, 271–277. doi: 10.1046/j.1365-2990.1998.00116.x
Furukawa, K., and Mattson, M. P. (1998). The transcription factor NF-kappaB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor-alpha in hippocampal neurons. J. Neurochem. 70, 1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x
Ghaneie, A., Duhadaway, J. B., Zemba-Palko, V., and Prendergast, G. C. (2006). Bin1 expression is lost in breast cancer. J. Clin. Oncol. 24(Suppl. 18), 10772–10772. doi: 10.1200/jco.2006.24.18_suppl.10772
Ghosh, G., van Duyne, G., Ghosh, S., and Sigler, P. B. (1995). Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature 373, 303–310. doi: 10.1038/373303a0
Greiner, J. F. W., Merten, M., Kaltschmidt, C., and Kaltschmidt, B. (2019). Sexual dimorphisms in adult human neural, mesoderm-derived, and neural crest-derived stem cells. FEBS Lett. 593, 3338–3352. doi: 10.1002/1873-3468.13606
Guerrini, L., Blasi, F., and Denis-Donini, S. (1995). Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc. Natl. Acad. Sci. U.S.A. 92, 9077–9081. doi: 10.1073/pnas.92.20.9077
Heimberger, A. B., Suki, D., Yang, D., Shi, W., and Aldape, K. (2005). The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 3:38. doi: 10.1186/1479-5876-3-38
Imielski, Y., Schwamborn, J. C., Luningschror, P., Heimann, P., Holzberg, M., Werner, H., et al. (2012). Regrowing the adult brain: NF-kappaB controls functional circuit formation and tissue homeostasis in the dentate gyrus. PLoS One 7:e30838. doi: 10.1371/journal.pone.0030838
Kaltschmidt, B., and Kaltschmidt, C. (2009). NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 1:a001271. doi: 10.1101/cshperspect.a001271
Kaltschmidt, B., Baeuerle, P. A., and Kaltschmidt, C. (1993). Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol. Aspects Med. 14, 171–190. doi: 10.1016/0098-2997(93)90004-w
Kaltschmidt, B., Greiner, J. F. W., Kadhim, H. M., and Kaltschmidt, C. (2018). Subunit-specific role of NF-kappaB in cancer. Biomedicines 6:44. doi: 10.3390/biomedicines6020044
Kaltschmidt, B., Ndiaye, D., Korte, M., Pothion, S., Arbibe, L., Prullage, M., et al. (2006). NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol. Cell Biol. 26, 2936–2946. doi: 10.1128/MCB.26.8.2936-2946.2006
Kaltschmidt, B., Uherek, M., Volk, B., Baeuerle, P. A., and Kaltschmidt, C. (1997). Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 2642–2647. doi: 10.1073/pnas.94.6.2642
Kaltschmidt, B., Uherek, M., Wellmann, H., Volk, B., and Kaltschmidt, C. (1999). Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc. Natl. Acad. Sci. U.S.A. 96, 9409–9414. doi: 10.1073/pnas.96.16.9409
Kaltschmidt, B., Witte, K. E., Greiner, J. F. W., Weissinger, F., and Kaltschmidt, C. (2022). Targeting NF-kappaB signaling in cancer stem cells: a narrative review. Biomedicines 10:261. doi: 10.3390/biomedicines10020261
Kaltschmidt, C., Greiner, J. F. W., and Kaltschmidt, B. (2021). The Transcription factor NF-kappaB in stem cells and development. Cells 10:2042. doi: 10.3390/cells10082042
Kaltschmidt, C., Kaltschmidt, B., and Baeuerle, P. A. (1993). Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech. Dev. 43, 135–147. doi: 10.1016/0925-4773(93)90031-r
Kaltschmidt, C., Kaltschmidt, B., and Baeuerle, P. A. (1995). Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc. Natl. Acad. Sci. U.S.A. 92, 9618–9622. doi: 10.1073/pnas.92.21.9618
Kaltschmidt, C., Kaltschmidt, B., Lannes-Vieira, J., Kreutzberg, G. W., Wekerle, H., Baeuerle, P. A., et al. (1994a). Transcription factor NF-kappa B is activated in microglia during experimental autoimmune encephalomyelitis. J. Neuroimmunol. 55, 99–106. doi: 10.1016/0165-5728(94)90151-1
Kaltschmidt, C., Kaltschmidt, B., Neumann, H., Wekerle, H., and Baeuerle, P. A. (1994b). Constitutive NF-kappa B activity in neurons. Mol. Cell Biol. 14, 3981–3992. doi: 10.1128/mcb.14.6.3981-3992.1994
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Kato, T. Jr., Delhase, M., Hoffmann, A., and Karin, M. (2003). CK2 is a C-terminal IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol. Cell 12, 829–839. doi: 10.1016/s1097-2765(03)00358-7
Keller, S., and Schmidt, M. H. H. (2017). EGFR and EGFRvIII promote angiogenesis and cell invasion in glioblastoma: combination therapies for an effective treatment. Int. J. Mol. Sci. 18:1295. doi: 10.3390/ijms18061295
Kim, H. J., Kim, J. Y., Jung, C. W., Lee, Y. S., An, J. Y., Kim, E. H., et al. (2021). ANO1 regulates the maintenance of stemness in glioblastoma stem cells by stabilizing EGFRvIII. Oncogene 40, 1490–1502. doi: 10.1038/s41388-020-01612-5
Kim, S., Kwon, S. H., Kam, T. I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627–641.e7. doi: 10.1016/j.neuron.2019.05.035
Klenke, C., Widera, D., Engelen, T., Muller, J., Noll, T., Niehaus, K., et al. (2013). Hsc70 is a novel interactor of NF-kappaB p65 in living hippocampal neurons. PLoS One 8:e65280. doi: 10.1371/journal.pone.0065280
Kobro-Flatmoen, A., Lagartos-Donate, M. J., Aman, Y., Edison, P., Witter, M. P., and Fang, E. F. (2021). Re-emphasizing early Alzheimer’s disease pathology starting in select entorhinal neurons, with a special focus on mitophagy. Ageing Res. Rev. 67:101307. doi: 10.1016/j.arr.2021.101307
Lambert, E., Saha, O., Soares Landeira, B., Melo de Farias, A. R., Hermant, X., Carrier, A., et al. (2022). The Alzheimer susceptibility gene BIN1 induces isoform-dependent neurotoxicity through early endosome defects. Acta Neuropathol. Commun. 10:4. doi: 10.1186/s40478-021-01285-5
Lewy, F. (1912). “Paralysis agitans. I. Pathologische anatomie,” in Handbuch der Neurologie, ed. M. Lewandowsky (Berlin: Springer).
Lin, Y., Bai, L., Chen, W., and Xu, S. (2010). The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 14, 45–55. doi: 10.1517/14728220903431069
Mao, N. C., Steingrimsson, E., DuHadaway, J., Wasserman, W., Ruiz, J. C., Copeland, N. G., et al. (1999). The murine Bin1 gene functions early in myogenesis and defines a new region of synteny between mouse chromosome 18 and human chromosome 2. Genomics 56, 51–58. doi: 10.1006/geno.1998.5709
Maqbool, A., Lattke, M., Wirth, T., and Baumann, B. (2013). Sustained, neuron-specific IKK/NF-kappaB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegener. 8:40. doi: 10.1186/1750-1326-8-40
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
McElhinny, J. A., Trushin, S. A., Bren, G. D., Chester, N., and Paya, C. V. (1996). Casein kinase II phosphorylates I kappa B alpha at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol. Cell Biol. 16, 899–906. doi: 10.1128/MCB.16.3.899
Meberg, P. J., Kinney, W. R., Valcourt, E. G., and Routtenberg, A. (1996). Gene expression of the transcription factor NF-kappa B in hippocampus: regulation by synaptic activity. Brain Res. Mol. Brain Res. 38, 179–190. doi: 10.1016/0169-328x(95)00229-l
Meffert, M. K., Chang, J. M., Wiltgen, B. J., Fanselow, M. S., and Baltimore, D. (2003). NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 6, 1072–1078. doi: 10.1038/nn1110
Meschede, J., Sadic, M., Furthmann, N., Miedema, T., Sehr, D. A., Muller-Rischart, A. K., et al. (2020). The parkin-coregulated gene product PACRG promotes TNF signaling by stabilizing LUBAC. Sci. Signal. 13:eaav1256. doi: 10.1126/scisignal.aav1256
Mikenberg, I., Widera, D., Kaus, A., Kaltschmidt, B., and Kaltschmidt, C. (2007). Transcription factor NF-kappaB is transported to the nucleus via cytoplasmic dynein/dynactin motor complex in hippocampal neurons. PLoS One 2:e589. doi: 10.1371/journal.pone.0000589
Ohgaki, H., and Kleihues, P. (2013). The definition of primary and secondary glioblastoma. Clin. Cancer Res. 19, 764–772. doi: 10.1158/1078-0432.Ccr-12-3002
O’Mahony, A., Raber, J., Montano, M., Foehr, E., Han, V., Lu, S. M., et al. (2006). NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol. Cell Biol. 26, 7283–7298. doi: 10.1128/MCB.00510-06
Panicker, N., Ge, P., Dawson, V. L., and Dawson, T. M. (2021). The cell biology of Parkinson’s disease. J. Cell Biol. 220:e202012095. doi: 10.1083/jcb.202012095
Park, J. Y., Lee, D., Lee, J. J., Gim, J., Gunasekaran, T. I., Choi, K. Y., et al. (2021). A missense variant in SHARPIN mediates Alzheimer’s disease-specific brain damages. Transl. Psychiatry 11:590. doi: 10.1038/s41398-021-01680-5
Parkinson, J. (2002). An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14, 223–236; discussion 222. doi: 10.1176/jnp.14.2.223
Piao, J., Zabierowski, S., Dubose, B. N., Hill, E. J., Navare, M., Claros, N., et al. (2021). Preclinical efficacy and safety of a human embryonic stem cell-derived midbrain dopamine progenitor product, MSK-DA01. Cell Stem Cell 28, 217–229.e7. doi: 10.1016/j.stem.2021.01.004
Raychaudhuri, B., Han, Y., Lu, T., and Vogelbaum, M. A. (2007). Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J. Neurooncol. 85, 39–47. doi: 10.1007/s11060-007-9390-7
Rinkenbaugh, A. L., Cogswell, P. C., Calamini, B., Dunn, D. E., Persson, A. I., Weiss, W. A., et al. (2016). IKK/NF-kappaB signaling contributes to glioblastoma stem cell maintenance. Oncotarget 7, 69173–69187. doi: 10.18632/oncotarget.12507
Ruiz-Perera, L. M., Greiner, J. F. W., Kaltschmidt, C., and Kaltschmidt, B. (2020). A Matter of choice: inhibition of c-Rel shifts neuronal to oligodendroglial fate in human stem cells. Cells 9:1037. doi: 10.3390/cells9041037
Ruiz-Perera, L. M., Schneider, L., Windmoller, B. A., Muller, J., Greiner, J. F. W., Kaltschmidt, C., et al. (2018). NF-kappaB p65 directs sex-specific neuroprotection in human neurons. Sci. Rep. 8:16012. doi: 10.1038/s41598-018-34394-8
Sakamuro, D., Elliott, K. J., Wechsler-Reya, R., and Prendergast, G. C. (1996). BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat. Genet. 14, 69–77. doi: 10.1038/ng0996-69
Salles, A., Boccia, M., Blake, M., Corbi, N., Passananti, C., Baratti, C. M., et al. (2015). Hippocampal dynamics of synaptic NF-kappa B during inhibitory avoidance long-term memory consolidation in mice. Neuroscience 291, 70–80. doi: 10.1016/j.neuroscience.2015.01.063
Sayed, M., Kim, S. O., Salh, B. S., Issinger, O. G., and Pelech, S. L. (2000). Stress-induced activation of protein kinase CK2 by direct interaction with p38 mitogen-activated protein kinase. J. Biol. Chem. 275, 16569–16573. doi: 10.1074/jbc.M000312200
Schmeisser, M. J., Baumann, B., Johannsen, S., Vindedal, G. F., Jensen, V., Hvalby, O. C., et al. (2012). IkappaB kinase/nuclear factor kappaB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J. Neurosci. 32, 5688–5703. doi: 10.1523/JNEUROSCI.0111-12.2012
Schmidt-Ullrich, R., Memet, S., Lilienbaum, A., Feuillard, J., Raphael, M., and Israel, A. (1996). NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122, 2117–2128. doi: 10.1242/dev.122.7.2117
Schorlemmer, H. U., and Seiler, F. R. (1991). 15-Deoxyspergualin (15-DOS) for therapy in an animal model of multiple sclerosis (MS): disease modifying activity on acute and chronic relapsing experimental allergic encephalomyelitis (EAE). Agents Actions 34, 156–160. doi: 10.1007/BF01993265
Schultz, W. (2007). Multiple dopamine functions at different time courses. Annu. Rev. Neurosci. 30, 259–288. doi: 10.1146/annurev.neuro.28.061604.135722
Shinojima, N., Tada, K., Shiraishi, S., Kamiryo, T., Kochi, M., Nakamura, H., et al. (2003). Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 63, 6962–6970.
Shubayev, V. I., and Myers, R. R. (2001). Axonal transport of TNF-alpha in painful neuropathy: distribution of ligand tracer and TNF receptors. J. Neuroimmunol. 114, 48–56. doi: 10.1016/s0165-5728(00)00453-7
Smith, D., Shimamura, T., Barbera, S., and Bejcek, B. E. (2008). NF-kappaB controls growth of glioblastomas/astrocytomas. Mol. Cell Biochem. 307, 141–147. doi: 10.1007/s11010-007-9593-4
Snow, W. M., and Albensi, B. C. (2016). Neuronal gene targets of NF-kappaB and their dysregulation in Alzheimer’s disease. Front. Mol. Neurosci. 9:118. doi: 10.3389/fnmol.2016.00118
Soheili-Nezhad, S., Jahanshad, N., Guelfi, S., Khosrowabadi, R., Saykin, A. J., Thompson, P. M., et al. (2020). Imaging genomics discovery of a new risk variant for Alzheimer’s disease in the postsynaptic SHARPIN gene. Hum. Brain Mapp. 41, 3737–3748. doi: 10.1002/hbm.25083
Steiner, J. A., Quansah, E., and Brundin, P. (2018). The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res. 373, 161–173. doi: 10.1007/s00441-018-2814-1
Stockhausen, M. T., Kristoffersen, K., Stobbe, L., and Poulsen, H. S. (2014). Differentiation of glioblastoma multiforme stem-like cells leads to downregulation of EGFR and EGFRvIII and decreased tumorigenic and stem-like cell potential. Cancer Biol. Ther. 15, 216–224. doi: 10.4161/cbt.26736
Stoeckel, K., Schwab, M., and Thoenen, H. (1975). Specificity of retrograde transport of nerve growth factor (NGF) in sensory neurons: a biochemical and morphological study. Brain Res. 89, 1–14. doi: 10.1016/0006-8993(75)90129-8
Suzuki, T., Mitake, S., Okumura-Noji, K., Yang, J. P., Fujii, T., and Okamoto, T. (1997). Presence of NF-kappaB-like and IkappaB-like immunoreactivities in postsynaptic densities. Neuroreport 8, 2931–2935. doi: 10.1097/00001756-199709080-00025
Taga, M., Petyuk, V. A., White, C., Marsh, G., Ma, Y., Klein, H. U., et al. (2020). BIN1 protein isoforms are differentially expressed in astrocytes, neurons, and microglia: neuronal and astrocyte BIN1 are implicated in tau pathology. Mol. Neurodegener. 15:44. doi: 10.1186/s13024-020-00387-3
Wang, H., Li, L., and Yin, L. (2018). Silencing LncRNA LOXL1-AS1 attenuates mesenchymal characteristics of glioblastoma via NF-kappaB pathway. Biochem. Biophys. Res. Commun. 500, 518–524. doi: 10.1016/j.bbrc.2018.04.133
Wang, Y., Yi, K., Liu, X., Tan, Y., Jin, W., Li, Y., et al. (2021). HOTAIR up-regulation activates NF-kappaB to induce immunoescape in gliomas. Front. Immunol. 12:785463. doi: 10.3389/fimmu.2021.785463
Wellmann, H., Kaltschmidt, B., and Kaltschmidt, C. (2001). Retrograde transport of transcription factor NF-kappa B in living neurons. J. Biol. Chem. 276, 11821–11829. doi: 10.1074/jbc.M009253200
Whittaker, V. P., Michaelson, I. A., and Kirkland, R. J. (1964). The separation of synaptic vesicles from nerve-ending particles (‘synaptosomes’). Biochem. J. 90, 293–303. doi: 10.1042/bj0900293
Widera, D., Klenke, C., Nair, D., Heidbreder, M., Malkusch, S., Sibarita, J. B., et al. (2016). Single-particle tracking uncovers dynamics of glutamate-induced retrograde transport of NF-kappaB p65 in living neurons. Neurophotonics 3:041804. doi: 10.1117/1.NPh.3.4.041804
Wilkaniec, A., Lenkiewicz, A. M., Babiec, L., Murawska, E., Jȩśko, H. M., Cieślik, M., et al. (2021). Exogenous alpha-synuclein evoked parkin downregulation promotes mitochondrial dysfunction in neuronal cells. Implications for Parkinson’s disease pathology. Front. Aging Neurosci. 13:591475. doi: 10.3389/fnagi.2021.591475
Witte, K. E., Hertel, O., Windmöller, B. A., Helweg, L. P., Höving, A. L., Knabbe, C., et al. (2021). Nanopore sequencing reveals global transcriptome signatures of mitochondrial and ribosomal gene expressions in various human cancer stem-like cell populations. Cancers (Basel) 13:1136. doi: 10.3390/cancers13051136
Xie, T. X., Xia, Z., Zhang, N., Gong, W., and Huang, S. (2010). Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 23, 725–732.
Yiannopoulou, K. G., Anastasiou, A. I., Zachariou, V., and Pelidou, S. H. (2019). Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 7:97. doi: 10.3390/biomedicines7040097
Zhang, Q., Lenardo, M. J., and Baltimore, D. (2017). 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168, 37–57. doi: 10.1016/j.cell.2016.12.012
Keywords: NF-κB–nuclear factor kappa B, nervous system, Alzheimer’s disease, Parkinson’s disease, glioblastoma multiforme (GBM), SHARPIN, PARKIN, HOTAIR
Citation: Kaltschmidt B, Helweg LP, Greiner JFW and Kaltschmidt C (2022) NF-κB in neurodegenerative diseases: Recent evidence from human genetics. Front. Mol. Neurosci. 15:954541. doi: 10.3389/fnmol.2022.954541
Received: 27 May 2022; Accepted: 11 July 2022;
Published: 02 August 2022.
Edited by:
Jun Yan, The University of Queensland, AustraliaReviewed by:
Efthimios M. C. Skoulakis, Alexander Fleming Biomedical Sciences Research Center, GreeceCopyright © 2022 Kaltschmidt, Helweg, Greiner and Kaltschmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Kaltschmidt, YmFyYmFyYS5rYWx0c2NobWlkdEB1bmktYmllbGVmZWxkLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.