 Kelvin K. Hui1,2*†‡
Kelvin K. Hui1,2*†‡ Yukiko Goda
Yukiko Goda Motomasa Tanaka
Motomasa Tanaka
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 08 July 2022
Sec. Neuroplasticity and Development
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.893111
This article is part of the Research TopicThe Role of GABA-Shift in Neurodevelopment and Psychiatric DisordersView all 13 articles
Excitatory-inhibitory (E-I) imbalance has been shown to contribute to the pathogenesis of a wide range of neurodevelopmental disorders including autism spectrum disorders, epilepsy, and schizophrenia. GABA neurotransmission, the principal inhibitory signal in the mature brain, is critically coupled to proper regulation of chloride homeostasis. During brain maturation, changes in the transport of chloride ions across neuronal cell membranes act to gradually change the majority of GABA signaling from excitatory to inhibitory for neuronal activation, and dysregulation of this GABA-shift likely contributes to multiple neurodevelopmental abnormalities that are associated with circuit dysfunction. Whilst traditionally viewed as a phenomenon which occurs during brain development, recent evidence suggests that this GABA-shift may also be involved in neuropsychiatric disorders due to the “dematuration” of affected neurons. In this review, we will discuss the cell signaling and regulatory mechanisms underlying the GABA-shift phenomenon in the context of the latest findings in the field, in particular the role of chloride cotransporters NKCC1 and KCC2, and furthermore how these regulatory processes are altered in neurodevelopmental and neuropsychiatric disorders. We will also explore the interactions between GABAergic interneurons and other cell types in the developing brain that may influence the GABA-shift. Finally, with a greater understanding of how the GABA-shift is altered in pathological conditions, we will briefly outline recent progress on targeting NKCC1 and KCC2 as a therapeutic strategy against neurodevelopmental and neuropsychiatric disorders associated with improper chloride homeostasis and GABA-shift abnormalities.
The predominant form of neurotransmission in the brain occurs at chemical synapses where presynaptic neurons release neurotransmitters that bind to receptors on postsynaptic neurons. A major type of postsynaptic receptors are ion channels which allow the selective influx or efflux of monovalent (Na+, K+, and Cl–) or divalent (Ca2+) ions into and out of postsynaptic neurons. For any particular ion, the direction of flow is dictated by the electrochemical gradient, and the difference in concentrations of ions across the membrane results in a membrane potential. In the adult brain, the opening of ion channels permeable to cations such as Na+ and Ca2+ drives the membrane potential toward the threshold potential (depolarization) through their entry into the cell. Increased permeability for Cl– anions and their cell entry, in contrast, hyperpolarizes the cell to drive the membrane potential further away from the threshold potential, and reduces the chance of the cell firing an action potential.
The primary inhibitory neurotransmitter in the central nervous system (CNS), γ-aminobutyric acid (GABA), activates ionotropic GABAA receptors that are permeable to chloride, and the maintenance of intracellular and extracellular chloride concentrations ([Cl–]i and [Cl–]e, respectively) is crucial for effective GABAergic neurotransmission. The intracellular chloride concentration ([Cl–]i) relative to its extracellular concentration is generally lower in the mature brain than in the immature brain, and thus the opening of GABAA receptors results in the influx of chloride ions into the postsynaptic neuron and hyperpolarizes it (Fukuda et al., 1998; Kuner and Augustine, 2000). Since disruptions in the excitation-inhibition (E-I) balance has been strongly linked to a number of neurological disorders such as epilepsy and autism spectrum disorder (ASD) (Fritschy, 2008; Nelson and Valakh, 2015), and inhibition critically depends on chloride levels, there has been much research interest in recent years to determine whether and how impairments in chloride homeostasis contribute to these pathologies.
Neuronal chloride homeostasis is maintained primarily by K+-Cl– cotransporter-2 (KCC2) (Jarolimek et al., 1999; Kakazu et al., 1999; Rivera et al., 1999; Hübner et al., 2001b) and Na+-K+-2Cl– cotransporter-1 (NKCC1) (Kakazu et al., 1999; Lu et al., 1999), which act to export and import Cl– out of and into the cell, respectively. Because cells normally have a high [K+]i, KCC2 is able to transport the chloride ion (along with K+) against its concentration gradient in order to maintain a low [Cl–]i (Payne et al., 1996). Similarly, NKCC1 uses the electrochemical gradient of Na+ to move K+ and Cl– into the cell (Gamba et al., 1994; Figure 1A).
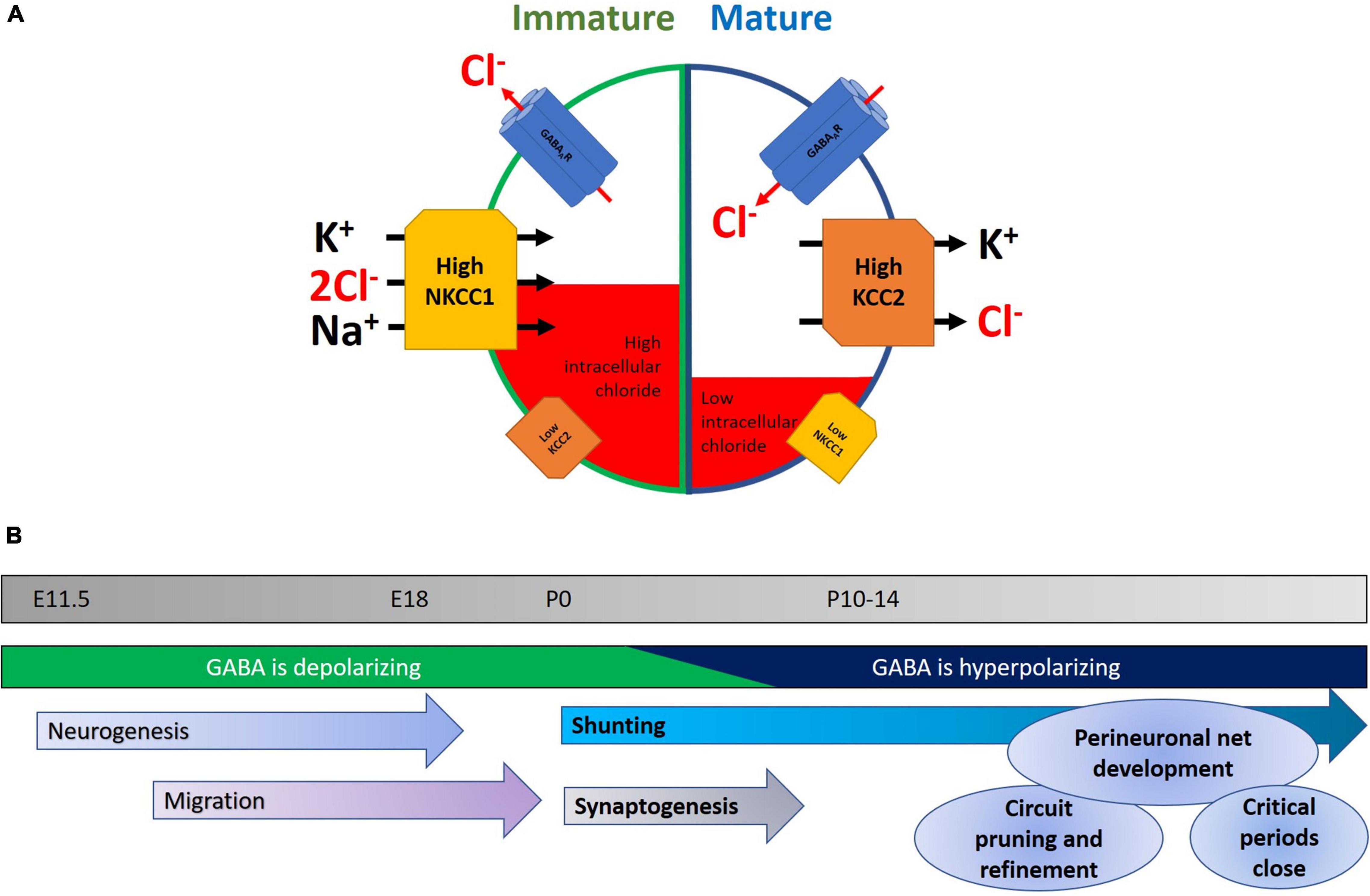
Figure 1. (A) Developmental changes in NKCC1 and KCC2 within neurons. In immature neurons, NKCC1 is expressed at high levels and is stabilized at neuronal plasma membranes, where it transports chloride into the cell, along with potassium and sodium. A concomitant low level of KCC2 expression keeps intracellular chloride levels high. The net result of this is that opening of GABAA receptors results in an efflux of chloride, leading to neuronal depolarization and a higher probability of neuronal activity. Later in development, the GABA-shift in neurons is primarily characterized by an upregulation of KCC2, potentially accompanied by a downregulation of NKCC1. This mature state is characterized by lower levels of intracellular chloride, and consequently chloride enters the cell upon GABAergic activity, and hyperpolarizes the neuron, reducing the likelihood of action potential generation. (B) Timeline highlighting some key events during early development. Normal development of the nervous system is characterized by a series of stages in which neurons are born, migrate to their final location, and then begin to make synapses. Early in the program, depolarizing GABA helps in aspects of neurogenesis, migration, and elevating activity in the nascent network. As the GABA-shift switches GABA to being hyperpolarizing, this coincides with the onset of an increase in glutamatergic synaptic drive. Later in development, interneuronal activity and structure continue to be refined with the formation of perineuronal nets and the closure of critical periods.
During the development and maturation of the nervous system, intracellular chloride levels in neurons change dramatically in concentration, from 37 mM at E18, to 12 mM at P16, however it should be noted that such measurements are not trivial (Owens et al., 1996; Arosio and Ratto, 2014). This initial higher [Cl–]i in immature neurons causes a chloride efflux when GABAA receptors are activated and ultimately results in membrane depolarization, thus effectively making GABA signaling excitatory in nature in the immature nervous system. While depolarizing responses are typically excitatory, there are counter-examples of GABA acting to depolarize cells leading to inhibition of network activity (for example see Kirmse et al., 2015). During early postnatal neurodevelopment, changes in expression of KCC2 and NKCC1 are believed to reduce the intracellular chloride concentration, thereby causing GABAergic neurotransmission to become inhibitory (Kakazu et al., 1999; Rivera et al., 1999; Yamada et al., 2004). In human neocortices, their expressions reach adult levels between postconceptional week (PCW) 40–50 (but see Sedmak et al., 2015 for an example of continuing postnatal increases), while in rodent hippocampus and cortex these changes primarily occur during the second week following birth (Rivera et al., 1999; Dzhala et al., 2005). The drop of [Cl–]i was largely attributed by earlier studies to the downregulation of NKCC1 in neurons as they mature (Plotkin et al., 1997; Hübner et al., 2001a; Yamada et al., 2004), although the exact expression pattern of NKCC1 still remains somewhat controversial (Virtanen et al., 2020). Subsequent studies have revealed that KCC2 upregulation, in particular the KCC2b isoform, likely plays a prominent role for the observed GABA-shift in the cortex during brain maturation (Rivera et al., 1999; Dzhala et al., 2005; Zhu et al., 2005; Uvarov et al., 2007). This is complicated, however, by observations of functionally inactive KCC2 in immature neuronal populations related to subcellular localization, oligomerization, and phosphorylation status (Balakrishnan et al., 2003; Khirug et al., 2005; Blaesse et al., 2006), thus suggesting that expression alone may not necessarily imply transporter function. Despite some controversies surrounding their specific contributions to the GABA-shift during brain development, KCC2 and NKCC1 both act to maintain a low intracellular chloride level in mature neurons, and thus ensure inward Cl– flow upon GABAA receptor activation.
Before discussing the GABA-shift in details, let us first describe the diverse roles of GABAergic signaling during normal neurodevelopment. The transition of highly plastic young brain into the relatively stable adult state involves multiple sets of overlapping processes (Figure 1B). These include the laying down of perineuronal nets, the buildup of extracellular matrix (Hensch, 2005), the refinement of excitatory and inhibitory neuronal networks, and the topic of this review: the change of GABA acting as a depolarizing, excitatory neurotransmitter to a hyperpolarizing inhibitory one. It is worth paying attention to the different roles demanded of the mature and immature nervous system, and how GABA acts on each in turn. In the developing nervous system, depolarizing GABA promotes calcium entry into cells (Leinekugel et al., 1995), which in turn triggers various signaling cascades leading to neurite outgrowth and synapse development (see below). Moreover, excitatory GABAergic synaptic activity combines with glutamatergic inputs to drive waves of activity called giant depolarizing potentials (GDPs) that can then shape synaptic connectivity (Ben-Ari et al., 1989). Notably, deficits in GDPs have been linked to neuropsychiatric disorders (Griguoli and Cherubini, 2017).
Interestingly, the GABA-shift is still retained in brain-derived organoids (Zafeiriou et al., 2020) and appears to be a widespread and robust phenomenon in developing neuronal systems across species. It has not been confirmed that the GABA-shift occurs in humans, however there is immunohistochemical evidence suggesting that human cortical GABAergic development follows a similar course (Pinto et al., 2010).
GABA signaling is active in the developing neuronal network prior to the formation of synapses (Andäng et al., 2008; Wang and Kriegstein, 2008). Following the first wave of synaptogenesis, synaptic GABA responses have been detected before birth in both hippocampus and cortex prior to the formation of excitatory inputs in both rat and mouse tissues (LoTurco et al., 1995; Owens et al., 1996, 1999; Demarque et al., 2002; Gozlan and Ben-Ari, 2003). At the time of birth, GABAergic inputs into CA1 neurons are formed before glutamatergic ones, and appear as the apical dendrite arborizes (Tyzio et al., 1999).
During these early stages, depolarizing GABA signals modulate multiple temporally-overlapping aspects of network development, including the migration of young neurons (Behar et al., 1996, 1998, 2000; Denter et al., 2010; see Barker et al., 1998; Luhmann et al., 2015 for review) to proliferation (both promoting or inhibiting proliferation dependent on the cell type/brain region, see Haydar et al., 2000 for examples of both). Once the components of the network are in place, then depolarizing GABA acts to drive neurite outgrowth, and subsequently promote synapse formation. An early study demonstrated that mouse neuroblastoma cultures treated with GABA had an increase in neurite length and branching (Eins et al., 1983) and this has been repeated across a diverse range of cell types (reviewed in Sernagor et al., 2010). The capacity for GABA to promote neurite development in the cortex (Cancedda et al., 2007) has been shown to depend on chloride homeostasis as inhibiting NKCC1 with the loop diuretic bumetanide reduces dendritic arbor complexity (Wang and Kriegstein, 2011) and furthermore knockdown of NKCC1 slows the development of dendritic arbors (Young et al., 2012). Similarly, GABA has been demonstrated to drive dendritic arborization in the hippocampus (Gascon et al., 2006; Ge et al., 2006; Duveau et al., 2011). Critically, the potentiating effects of GABA on neurite complexity are limited to depolarizing GABA and no longer observed after the GABA-shift (Maric et al., 2001), which suggest that the delayed or extended GABA-shift may result in overgrowth in neuronal arbors and a state of hyperconnectivity.
Earlier work on the depolarizing nature of GABA in young tissue was somewhat controversial (see Holmgren et al., 2010; Zilberter et al., 2010; Mukhtarov et al., 2011; Dzhala et al., 2012; reviewed in Bregestovski and Bernard, 2012; and rebutted in Ben-Ari et al., 2012) due to potential artifacts associated with in vitro slice preparations and the difficulty of making in vivo measurements of [Cl–]i. A recent optogenetic approach, where ChR2 was expressed in interneurons (Valeeva et al., 2016), strengthens the case that GABA can be directly depolarizing in the early network. In this study, in vitro stimulation of ChR2-expressing interneurons drove a marked increase in EPSC frequency, whereas the same stimulation in vivo in anesthetized animals led to a small decrease in EPSC frequency. Nonetheless, even in young neurons, GABA may still act to reduce postsynaptic activity by shunting excitatory currents (Khalilov et al., 1999; Wells et al., 2000), further complicating the picture.
Finally, the network undergoes a large burst in synapse formation during the final stages of depolarizing GABA, with GABA-dependent excitation acting to facilitate the generation of new synapses, both in the hippocampus and cortex (Ben-Ari et al., 1997; Griguoli and Cherubini, 2017), by helping relieve the Mg2+ block of NMDARs. Locally applied GABA alone is sufficient to generate new synapses during development (Oh et al., 2016). On the contrary, Salmon et al. (2020) found that blocking GABAA receptors immediately prior to the GABA-shift (in mouse organotypic hippocampal slice cultures) increases the number of glutamatergic synapses formed. However, the authors also assessed the effect of bumetanide inhibition of NKCC1 on excitatory synapse numbers; they found that even under these conditions, GABA inhibition still increased excitatory synaptic density.
Altogether, the downstream effects of GABA on aspects of brain organization and function differ across the GABA-shift. Let us next discuss how the beginning and end of this process is controlled.
When exactly does the GABA-shift happen? Interestingly, the exact timing of the switch from depolarizing to hyperpolarizing GABA varies substantially across neuronal type, species, brain region, and even sexes. The shift happens earlier in cortex than in hippocampus (Murata and Colonnese, 2020), earlier and longer-lasting in females than in males (Kyrozis et al., 2006; Nuñez and McCarthy, 2007; Galanopoulou, 2008; but reversed in cerebral cortex, see Roux et al., 2018), and earlier in GABAergic neurons compared to pyramidal cells (although this is also controversial, see Banke and McBain, 2006; Sauer and Bartos, 2010 for opposing examples). Presumably, the regulation of NKCC1 and KCC2 expression pattern that are unique to each experimental context being examined (cell type, brain region, animal sex, and species) contribute to variations in the shift of the actions of GABA.
Throughout the developing nervous system, spontaneous endogenous waves of activity play a profound role in shaping the network connectivity and function. For example, in rodents, eye opening happens over a brief period of one to two days during the second week of life, at a time when the visual network is already functional. Does the depolarizing nature of GABA in the young brain help this process? Interestingly, a recent study (Ge et al., 2021) demonstrated a role for retinal waves in guiding the formation of direction-selective visual network by simulating a naturalistic pattern of optic flow. This occurred at a similar developmental period (P8–11) to the GABA-shift during which rodent retina are either undergoing the GABA-shift (Vu et al., 2000) or have just finished it (Zhang et al., 2005). Previous work in retina has demonstrated also that the switch from depolarizing to hyperpolarizing GABA coincides with the end of the propagating retinal waves, with a role for the regulation of KCC2 (Vu et al., 2000; Sernagor et al., 2003). On the contrary, however, another study has found that intravitreous injection of a GABAA blocker (bicuculine) from birth to P15-18 did not prevent the development of direction-selective circuitry (Sun et al., 2011), raising questions about the necessity of depolarizing GABA for the proper visual network development.
As we have discussed above, given the highly variable timing of the GABA-shift, it is perhaps not surprising that the experimental evidence is mixed as to the identity of the signals that determine the start and end of the GABA-shift. Using rat hippocampal cultures, one study found that blockade of GABAA receptors prevented the increases in KCC2, and thus delayed the GABA-shift (Ganguly et al., 2001); whilst another study, using mouse cultured neurons and organotypic slices, found no contribution of GABAA signaling to the changes in KCC2 (Ludwig et al., 2003). These studies broadly agreed on some points, namely that blocking excitatory neurotransmission (using APV and CNQX) had no effect on the course of the GABA-shift, and that blockade of neuronal spiking with tetrodotoxin (TTX) did not inhibit the GABA-shift. Notably, Khirug et al. (2005) reported that cultured mouse hippocampal neurons showed a step-like change in KCC2 activity after two weeks in culture between DIV 13 and 14, whilst acute slices showed instead a gradual increase from P5 to 14.
In some preparations, neuronal activity does seem to be a critical part of the GABA-shift mechanism. In turtle retina, spontaneous activity begins well before hatching, and the end of spontaneous activity corresponds with KCC2 upregulation (Sernagor et al., 2003). Moreover, dark-rearing turtles delays the GABA-shift, with a correspondingly weaker KCC2 expression. Further work in turtle retina has demonstrated that at least in this model, GABAA activity is required for the switch (Leitch et al., 2005). Similar to the study by Ganguly et al. (2001), the chronic blockade of GABAA signaling in turtle retina prevented the switch. On the contrary, a study using mouse retina found that the GABA-shift normally could be detected between P7-9, which was complete by P9-11, and that mouse retinal explants cultured in blockers of GABAA/B/C signaling still underwent the GABA-shift (Barkis et al., 2010). In fact, the same study subsequently blocked nearly all activity using a combination of drugs including TTX and glutamatergic signaling and still observed the GABA-shift.
What could be the triggers of the GABA-shift? Barkis et al. (2010) found that retinal ganglion cells (RGCs) cultured alone failed to make the switch. Importantly, the impaired switch was rescued by co-culturing with cells from the superior colliculus, suggesting a diffusible factor originating from other cell types as a required component, while the possible contribution of brain-derived neurotrophic factor (BDNF) could be excluded. On the contrary, the BDNF precursor pro-BDNF has been shown to regulate KCC2 levels (Riffault et al., 2016), keeping it low during development. Intriguingly, BDNF itself, either exogenously applied (Ludwig et al., 2011) or using a BDNF-overexpressing mouse model (Aguado et al., 2003), increases KCC2 levels despite KCC2 levels being unaffected in Bdnf knockout (KO) mice (Puskarjov et al., 2015) (see below for further discussion on BDNF-dependent signaling that regulates KCC2 and NKCC1). Liu et al. (2006) have identified a role for cholinergic signaling in controlling the GABA switch in chick ciliary ganglion, where spontaneous nicotinic cholinergic activity modulates transporter levels, and thus the balance of Cl– across the membrane. Taken as a whole, the evidence largely points to the regulation of KCC2 as being the primary driver behind the onset of the GABA-shift, but the identities of the upstream signal and the basis for cell-type specificity remain to be fully elucidated.
Another example of a potential molecular player in triggering the GABA-shift is the oxytocin receptor, activation of which promotes cell surface expression of KCC2 and stabilizes it at the cell membrane (Leonzino et al., 2016). Neurons from oxytocin receptor KO mice show a delayed GABA-shift, which nevertheless fully completes a few days later than control animals. Additionally, in these cultures NKCC1 levels were not different in KO cells, again consistently with KCC2 being the key player (Leonzino et al., 2016). It is worth mentioning here that there is another, transient GABA-shift during childbirth, that is also triggered by oxytocin, and acts to protect the fetus against anoxic episodes (Tyzio et al., 2006). Interestingly, the target for this transient GABA-shift has been suggested to be in fact NKCC1 activity and not KCC2, thus demonstrating that under certain conditions NKCC1 can dramatically influence GABA-shift behavior. It is worth mentioning that other candidates have been proposed to explain the suppression of neuronal activity during birth (for example vasopressin, see Spoljaric et al., 2017)].
Finally, another aspect of the GABA-shift concerns the developmental change in the GABAA receptor activity itself. GABAA receptors are large pentameric channels composed of a mixture of α, β, and γ subunits, often accompanied by the accessory δ and ρ subunits (Hevers and Lüddens, 1998). An early study that characterized GABAA receptor subunit mRNA levels in rat brain tissue found a change in the α subunit at around P8, from α2/3 to α1 in several brain regions (Laurie et al., 1992). Similarly, immunohistochemical studies suggest that α1 expression ramps up following birth and is still increasing at around the time of the GABA-shift (Fritschy et al., 1994; Davis et al., 2000) with a concomitant decrease in α2 levels. Importantly, GABAA receptors containing α1 subunits are more sensitive to GABA than those containing α2/3 (Wafford et al., 1993; Ebert et al., 1994; Hevers and Lüddens, 1998) and as such, the switch in the receptor subunit composition may serve to exaggerate the GABA-shift.
A number of genetic and epigenetic mechanisms regulating KCC2 expression have been identified thus far (Figure 2A). KCC2 is encoded by the SLC12A5 gene to produce two isoforms (KCC2a and KCC2b), both of which are expressed only in neuronal cells (Payne et al., 1996; Uvarov et al., 2007). This is principally mediated by a 1.4 kb promoter fragment and two RE1-Silencing Transcription factor (REST)/Neuron-Restrictive Silencer Factor (NRSF) repressor elements identified in its 5′ promoter region and the first intron of the KCC2b isoform (Yeo et al., 2009). Notably, the 1.4 kb promoter fragment alone is sufficient to restrict KCC2 expression in neuronal cells (Uvarov et al., 2005), and the fragment contains binding sites for transcription factors such as Early growth response 4 (Egr4) (Uvarov et al., 2006) and Upstream stimulating factors 1 and 2 (USF-1/2) (Markkanen et al., 2008), suggesting these as key factors in controlling neuronal expression.

Figure 2. (A) Transcriptional and epigenetic mechanisms involved in regulating KCC2 and NKCC1 expression. Transcriptional regulatory elements including NRSE/RE-1, E-box, and Erg4 RE are involved in regulating the expression of KCC2 and NKCC1 by various stimuli such as oxytocin and BDNF-TrkB/p75NTR signaling. Additional regulatory mechanisms including DNA methylation and histone acetylation have also been demonstrated for KCC2 regulation. Given the association of chromatin regulators and other proteins that regulate gene expression with various neurodevelopmental disorders such as ASD, it will be of interest to determine how the expression of KCC2 and NKCC1, and ultimately the GABA-shift, may be affected in those pathologic conditions. A, acetylation; Me, DNA methylation. (B) Phospho-regulatory network by WNK/SPAK/OSR1 and other kinases to modulate KCC2 and NKCC1 activity. WNK kinases function both as a sensor and effector of intracellular chloride concentrations as their catalytic activities are influenced by the direct binding of Cl– ions. SPAK/OSR1 are phosphorylated by WNK kinases and in turn can phosphorylate KCC2 and NKCC1 proteins. Numerous phosphorylation sites have been identified on KCC2 and NKCC1, which ultimately modulate their activities directly or by influencing their surface expression or degradation. Other phosphorylation sites have been identified on KCC2 and NKCC1 (discussed in main text) but were found to have no effects on transporter functionality and/or surface expression. P, phosphorylation.
Given its role in mediating chloride homeostasis and consequently the direction of Cl–flow triggered by GABAA receptor signaling, it is not surprising that the regulation of KCC2 expression also depends on the maturation status of the neuron of interest. For instance, as mentioned above, BDNF is known to regulate KCC2 expression. In particular, BDNF enhances KCC2 expression in immature neurons but suppresses it in mature neurons (Rivera et al., 2002, 2004; Aguado et al., 2003; Wake et al., 2007; Shulga et al., 2008; Boulenguez et al., 2010; Ludwig et al., 2011). Such opposing effects appear to be mediated via distinct pathways as BDNF activates expression in immature neurons via the ERK1/2 pathway and Egr4 (Ludwig et al., 2011), whereas its inhibitory effects on KCC2 expression in mature neurons depends on a signaling cascade involving Src homology domain containing transforming protein/FGF receptor substrate 2 (Shc/FRS-2), phospholipase C γ (PLC-γ), and cAMP response element-binding protein CREB (Rivera et al., 2004). The regulation of KCC2 by BDNF is further complicated by the effects of its precursor form (pro-BDNF), which can also suppress KCC2 expression via binding to the p75 neurotrophin receptor (p75NTR) (Rivera et al., 2002; Riffault et al., 2016). Thus, the overall effects of BDNF and pro-BDNF on KCC2 expression likely depends on the combinatorial expression patterns of BDNF/pro-BDNF and their respective receptors in the developing brain. Notably, this is further supported by the observations that p75NTR is highly expressed during development (Yang et al., 2014; Menshanov et al., 2015; Winnubst et al., 2015) and that BDNF/pro-BDNF are involved in various activity-dependent changes in neurons (Gubellini et al., 2005; Langlois et al., 2013; Yang et al., 2014).
In contrast, much less is currently known about the regulation of NKCC1 expression (encoded by the SLC12A2 gene), but both BDNF and oxytocin have been observed to downregulate its expression (Ceanga et al., 2010; Eftekhari et al., 2014; Tyzio et al., 2014). The factors required to maintain high NKCC1 expression in immature neurons however remains to be identified.
In addition, although much less is known about epigenetic regulation of SLC12A2 and SLC12A5, several recent studies have also demonstrated that the expressions of NKCC1 and KCC2 may be modulated via epigenetic mechanisms. Exposure of cultured developing human and rat cortical neurons to Bisphenol A (BPA), a possible chemical toxicant used in the production of a wide range of consumer products linked to increased incidence of neurodevelopmental disorders (Nesan and Kurrasch, 2019), was found to delay the KCC2 expression and increase [Cl–]i during neuronal maturation (Yeo et al., 2013). BPA treatment was observed to alter both the methyl-CpG binding protein 2 (MECP2) binding to the KCC2 promoter and its histone H3K9 acetylation. Furthermore, DNA methylation of the KCC2 regulatory region was found to be increased by BPA treatment while the administration of a DNA methyltransferase inhibitor partially rescued the reduction of KCC2 expression caused by BPA. Consistent with other studies using chemical or genetic HDAC inhibition (Yeo et al., 2009; Hou et al., 2018; Ouyang et al., 2019), this study also found Hdac1/2 knockdown to increase SLC12A5 mRNA levels and rescue the BPA-induced KCC2 downregulation (Yeo et al., 2013). Conversely, DNA methylation of the NKCC1 promoter region increases significantly during postnatal neurodevelopment and in vitro DNA methylation of the NKCC1 promoter region reduces NKCC1 expression (Lee H. A. et al., 2010). Together, these studies point to possible epigenetic mechanisms regulating KCC2 and NKCC1 expression to mediate the GABA-shift during neurodevelopment.
Aside from the transcriptional mechanisms described above, KCC2 and NKCC1 are also regulated via post-translational modifications that modulate their surface expression and activity levels. While other kinases have been shown to regulate NKCC1 and KCC2, the with-no-lysine (K) (WNK)-serine/threonine protein kinase 39 (SPAK)/OSR1 oxidative stress-responsive 1 (OSR1) pathway is a principal regulator for both transporters to coordinate their activities based on intracellular chloride concentrations (Alessi et al., 2014; Shekarabi et al., 2017).
Of all four WNK isoforms (WNK1-4) in the human genome, WNK1 is believed to be the principal WNK kinase in the human CNS (Shekarabi et al., 2013). This is supported by the observation that WNK1 mutations result in an autosomal recessive disease characterized by congenital pain insensitivity (Shekarabi et al., 2008). WNK kinases effectively function as intracellular chloride sensors, through Cl– binding to their catalytic domains in an inhibitory manner that suppress their autophosphorylation and kinase activity (Piala et al., 2014). Active WNK kinases phosphorylate SPAK and OSR1 (Moriguchi et al., 2005), which in turn directly phosphorylate KCC2 and NKCC1 to suppress and enhance their activities, respectively (Pacheco-Alvarez et al., 2006; Vitari et al., 2006; de los Heros et al., 2014; Figure 2B). When [Cl–]i is low, chloride-unbound WNK kinases are active and consequently suppress KCC2 (to reduce Cl– export) and increase NKCC1 activation (to enhance Cl– import). In contrast, WNK kinases are inhibited by high [Cl–]i, thereby reversing these changes to enhance KCC2-mediated Cl– export while suppressing further import by NKCC1.
Specifically, activated SPAK/OSR1 targets threonine residues T906 and T1007 (all residue numbers refer to human sequences) located on the C-terminal intracellular domain of KCC2 (de los Heros et al., 2014), which significantly suppress its Cl– transport activity (Rinehart et al., 2009). Notably, while these sites were partially phosphorylated in neonatal mouse brains, there was no evidence of their phosphorylation from adult brains, thus providing fully active KCC2 to export chloride ions to maintain the inhibitory nature of GABAergic signaling. Moreover, a separate study directly implicated the WNK1-SPAK/OSR1 signaling pathway in modulating KCC2 function and GABA activity by enhancing its phosphorylation at these two sites in immature neurons (Friedel et al., 2015). Specifically, the authors observed that in utero knockdown of Wnk1 resulted in lower [Cl–]i and a hyperpolarizing shift for GABA specifically in immature neurons (P3-5), as there were no observable effects on mature neurons (P30). These findings thus support a role for WNK kinases in maintaining the high intracellular chloride concentration necessary for GABA to have depolarizing effects in the immature brain and may potentially be involved in regulating the GABA-shift. As described above, WNK kinases in theory should be inhibited by the high [Cl–]i present in immature neurons and in turn reduce KCC2 phosphorylation to enhance Cl– export. However, although Wnk1 knockdown was found to reduce [Cl–]i in immature neurons, Friedel et al. (2015) did observe higher T906 and T1007 phosphorylation on KCC2 in immature neurons compared to their mature counterparts; thus suggesting that either the WNK-SPAK/OSR1 pathway remained sufficiently active despite the high [Cl–]i in immature neurons or that additional kinases are at work during this stage. Further studies comparing the WNK-SPAK/OSR1 pathway between immature and mature neurons are necessarily to fully delineate the importance of this pathway to [Cl–]i regulation and the GABA-shift during neurodevelopment. More recently, two groups independently produced knock-in mice with phosphomimetic mutations in these two phosphorylation sites (i.e., T906E and T1007E) to evaluate their physiologic significance. The authors observed reduced postnatal survival of homozygous mutants due to respiratory arrest and epileptic seizures (Watanabe et al., 2019), while heterozygotes displayed abnormal ASD-like impairments in ultrasonic vocalization and social behavior (Pisella et al., 2019), thereby demonstrating further the physiological importance of these two phosphorylation sites. Pharmacologically, N-ethylmaleimide (NEM) was found to activate KCC2 activity by enhancing the phosphorylation and dephosphorylation of S940 (to be discussed below) and T1007, respectively, to stabilize its surface expression (Conway et al., 2017). It was revealed that NEM mediates its effects on KCC2 by reducing WNK-dependent SPAK phosphorylation (S373) and depends on the dephosphorylation of T1007 on KCC2 as the overexpression of the T1007E phosphomimic mutant abolished the effects of NEM on KCC2 activity. Conversely, experiments using knock-in mice with alanine replacement at these phosphorylation sites demonstrated that they are critical to the timing of the GABA-shift (Moore et al., 2019). Moreover, the S906A/T1007A mutations altered social behaviors and led to slight enhancements in specific cognitive functions (Moore et al., 2019), while also protecting against chemoconvulsant-induced seizures (Moore et al., 2018). In addition, the WNK-SPAK/OSR1 pathway is also known to phosphorylate the N-terminal T6 of KCC2a, which is missing in KCC2b due to alternative splicing of exon-1a (Uvarov et al., 2007; Markkanen et al., 2017). Similar to its C-terminal counterparts (T906 and T1007), phosphorylation of T6 by SPAK was found to reduce its transporter activity (Markkanen et al., 2017).
In contrast, the activity of NKCC1 is enhanced by the phosphorylation of T203, T207, T212, T217, and T230 by the WNK-SPAK/OSR1 pathway (Darman and Forbush, 2002; Flemmer et al., 2002; Kahle et al., 2005; Vitari et al., 2006). Specifically, it was demonstrated that T217 phosphorylation is necessary for NKCC1 activity while the other sites serve modulatory roles (Darman and Forbush, 2002). Thus, through the coordinated phosphorylation of KCC2 and NKCC1, the WNK-SPAK/OSR1 pathway serves to monitor [Cl–]i and alter KCC2/NKCC1 activities to maintain an optimal intracellular chloride concentration. This control of KCC2/NKCC1 activity by the WNK-SPAK/OSR1 pathway is primarily achieved by influencing the surface expression of these two cotransporters on the cell membrane (Rinehart et al., 2009).
In addition to modulation by the WNK-SPAK/OSR1 pathway, the activities of KCC2 and NKCC1 are also regulated by other phosphorylation events. The phosphorylation of KCC2 at serine residue S940 is mediated by protein kinase C (PKC) and enhances its surface expression to increase overall transporter activity. The physiological importance of this phosphorylation site was revealed as knock-in mice incapable of S940 phosphorylation (i.e., S940A mutants) exhibit ASD-like deficits in social behaviors and cognitive functions (Moore et al., 2019), while also showing accelerated onset of status epilepticus and lethality from chemical-induced seizure activity (Silayeva et al., 2015). As for its regulatory signaling, both oxytocin and 5-hydroxytryptamine type 2A receptor activation are known to increase KCC2 activity by promoting S940 phosphorylation via PKC-dependent mechanisms (Lee et al., 2007; Bos et al., 2013; Leonzino et al., 2016). Notably, PKC can also indirectly influence KCC2 surface expression by enhancing complex formation between KCC2 and kainate receptors via the phosphorylation of subunit GluK2 (Mahadevan et al., 2014; Pressey et al., 2017; Garand et al., 2019). Conversely, S940 dephosphorylation can be triggered by calcium influx via NMDA receptors through the actions of protein phosphatase 1 (PP1) (Lee et al., 2011).
Aside from these serine and threonine residues, KCC2 tyrosine phosphorylation at Y903 and Y1087 have also been observed by as yet unidentified tyrosine kinases. These C-terminal phosphorylation sites are believed to enhance KCC2 internalization and lysosomal degradation (Lee H. H. C. et al., 2010), as the phosphorylation-incompetent Y903F/Y1087F mutant showed increases in both total and surface KCC2 expression (Lee H. H. C. et al., 2010). Notably, Strange et al. (2000) had originally drew similar conclusions about Y1087 phosphorylation as they observed the phosphomimic Y1087D mutation to completely abolish KCC2 activity in Xenopus oocytes without any changes in surface expression of the mutants. It remains unclear, however, how phosphorylated Y1087 could directly impact KCC2 activity. In addition, other groups have also found the Y1087D variant to disrupt KCC2 activity in subsequent studies (Akerman and Cline, 2006; Pellegrino et al., 2011). By contrast, an independent study using the same aspartate substitution at Y1087 but considered it as a non-phosphorylatable mutant concluded that Y1087 phosphorylation activates KCC2 activity by influencing its distribution on the cell membrane and regulating its clustering (Watanabe et al., 2009). Notably, the authors also used genistein or lavendustin A, and sodium vanadate as tyrosine kinase inhibitor and tyrosine phosphatase inhibitor, respectively, to manipulate Y1087 phosphorylation status to examine its effects; it must be taken into consideration that these compounds are non-specific and therefore KCC2 Y1087 phosphorylation-independent effects cannot be dismissed. Given this, and that aspartate (as well as glutamate) substitutions are typically considered as phosphomimics rather than a non-phosphorylatable state, the findings by Watanabe et al. (2009) may in fact be in agreement with the previous studies about the effects of Y1087 phosphorylation even though the precise molecular mechanism underlying its influence on KCC2 function remains largely unknown. Apart from Y903 and Y1087, additional phosphorylation sites at both the N- (S25, S26, S31, and T34) and C-termini (S932, S934, S937, T999, T1009, S1022, S1025, and S1026) have been identified to date (Weber et al., 2014; Cordshagen et al., 2018). Using alanine and aspartate mutants, the authors demonstrated S932D, S934D, S937D, and T1009A substitutions to enhance KCC2 functions, whereas mutations at the other sites had no effects. Importantly, the S932D, S934D, S937D, and T1009A mutations did not influence surface expression of KCC2 and thus suggested that the effects were directly on its transport activity.
As for NKCC1, AMPK has been shown to phosphorylate it at S77 and S242, which in turn suppresses its activity (Sid et al., 2010; Fraser et al., 2014). In both studies, AMPK was shown by both groups to directly phosphorylate N-terminal fragments of NKCC1 in vitro using recombinant proteins. While phosphorylation of NKCC1 at these sites appear to be AMPK-dependent, additional studies are necessary to determine whether they occur and are mediated directly by AMPK in vivo, especially in the context of neural cells, given the contrasting evidence from these and other groups (Sid et al., 2010; Miraucourt et al., 2016; King et al., 2019). Altogether, the above studies reveal the dynamic regulation of KCC2 and NKCC1 activity at multiple levels by phosphorylation.
KCC2 glycosylation has also been observed on at least six residues (N283, N291, N310, N328, N338, and N339) and are believed to regulate its membrane targeting (Agez et al., 2017). Notably, three KCC2 mutations associated with severe early-onset epileptic encephalopathy (L311H, L426P, and G551D) have been shown to reduce glycosylation and surface expression (Stödberg et al., 2015).
It is clear that the correct expression, surface targeting, and phosphorylation status of NKCC1 and KCC2 are critically important for healthy circuit development and function, but how does this impact the myriad of different cell types in the brain? We will discuss this topic next.
Changes in the balance of chloride across neuronal membranes are not expected to strongly affect postsynaptic neuronal activity unless there is concomitant release of GABA from presynaptic inhibitory neurons. The development of the inhibitory network has been widely studied across brain regions (review by Kubota et al., 2016; Lim et al., 2018; Ferrer and García, 2022), and mistimed development or inappropriate levels of inhibitory neuronal activity has been linked to multiple pathological states, with epilepsy being the primary example, but also schizophrenia and autism (reviewed by Marín, 2012; Chu and Anderson, 2015). The mammalian cortex contains a wide range of inhibitory neurons (Petilla Interneuron Nomenclature Group et al., 2008), and they change in number throughout development and past the end of the GABA-shift (for more details on interneuronal development we refer the reader to Gonchar et al., 2008; Bartolini et al., 2013; Ouellet and de Villers-Sidani, 2014). The activity and development of the interneuronal network is implicated in various aspects of circuit maturation, including ending critical periods (Hensch, 2005) and dynamically modulating axonal pruning and dendritic spine formation (Anderson et al., 1995).
Interneuronal development proceeds in stages, beginning at neurogenesis, followed by migration, and finally embedding into and modulating the circuit (Lim et al., 2018). This process is still ongoing throughout the period of the GABA shift, with some subpopulations of interneurons becoming mature earlier than others. Interestingly, the developmental period of the inhibitory neuronal network is temporally extended in larger animals such as humans, and this may contribute to the high levels of cognitive flexibility exhibited by these creatures, whilst also leaving them susceptible to many of the disease states that arise from imprecise GABAergic activity (Kim and Paredes, 2021).
Currently, one critical missing piece of the puzzle is the contribution of glial cell types to the shift in GABA’s effect. The presence of astrocytes has been shown to increase the speed of the GABA-shift in vitro (Li et al., 1998), however it is difficult to repeat this kind of experiment in vivo. Expression of NKCC1 and KCC2 can act to regulate neuronal intracellular [Cl–] levels, but functions of the network critically depend also on the ionic balance of the extracellular milieu for which astrocytes play a central role, particularly of potassium levels (see Hertz and Chen, 2016; Bellot-Saez et al., 2017 for review). In support of such a role, dysfunction in glial potassium homeostasis has pathological downstream effects on neuronal activity (Robel and Sontheimer, 2016). The KCC2 protein requires a low extracellular potassium concentration to carry chloride out of the cell. There is a substantial body of evidence indicating that astrocytes are largely responsible for the clearance of extracellular K+ following bouts of neuronal activity (Hertz, 1965; Ransom et al., 2000; Wang et al., 2012), either by local uptake or redistribution and buffering throughout the gap-junction connected astrocyte network. Astrocytes can also clear GABA from the extracellular milieu through GAT1-3 expression and activity (Boddum et al., 2016), and themselves express GABA receptors (MacVicar et al., 1989; Boisvert et al., 2018). Moreover, there is a wealth of data demonstrating that GABA signals can bidirectionally modulate astrocytic calcium levels (Doengi et al., 2009; Matos et al., 2018; Yu et al., 2018), which in turn controls the release of ATP and other gliotransmitters (Kang et al., 1998; Liu et al., 2004; Serrano et al., 2006). Neuronal activity, extracellular Cl– levels, and astrocytic Cl– have been demonstrated to be dynamically linked, and neuronal activity can trigger the release of Cl– from astrocytes into the extracellular space, where it contributes to subsequent GABAergic activity (Egawa et al., 2013). Astrocytes and GABAergic neurons are known to signal to one another (for examples of interneuron to astrocyte, see Mariotti et al., 2018; Mederos et al., 2021; for astrocyte to interneuron, see Shigetomi et al., 2012; reviewed in Mederos and Perea, 2019) in a manner that appears to be specific to particular classes of GABAergic interneurons (Mariotti et al., 2018; Matos et al., 2018), adding an extra layer of complexity to the system.
NKCC1 is expressed in murine astrocytes (Su et al., 2002) and astrocytic internal Cl– levels remains high throughout development, at around 30–50 mM in cultures (Bekar and Walz, 2002) and brain slices (Untiet et al., 2017). In vivo measurements have proven tricky, however (see Arosio and Ratto, 2014 for review). Although NKCC1 levels are largely stable in astrocytes, under various pathological conditions NKCC1 expression can be upregulated, undergoes phosphorylation, and is stabilized at the astrocytic plasma membrane (Jayakumar and Norenberg, 2010). Astrocytes from Slc12a2 KO mice are unable to regulate their volume in response to a hyperkalemic (75 mM K+) challenge (Su et al., 2002), and NKCC1 has been implicated in astrocytic swelling in pathological conditions such as ischemia and brain edema caused by liver dysfunction (Yan et al., 2001; Jayakumar et al., 2008; reviewed in Jayakumar and Norenberg, 2010). Astrocytic NKCC1 is upregulated in response to trauma (Jayakumar et al., 2011) and both inhibiting NKCC1 activity pharmacologically with bumetanide or genetically with an anti-NKCC1 siRNA significantly reduces trauma-induced increase in astrocytic volume. It is worth mentioning that the Slc12a2 KO mouse is viable (Delpire et al., 1999; Dixon et al., 1999; Flagella et al., 1999); however, the loss of the NKCC1 protein causes deficits in neuronal proliferation (Magalhães and Rivera, 2016) and exacerbates the severity of a mouse model of epilepsy (Hampel et al., 2021), amongst other issues. Whereas neuronal loss of NKCC1 appears to be compensated for at the network level (Sipila et al., 2009), the consequences on glia are much less clear. In adult tissue, NKCC1 has been implicated in astrocytic responses to neuronal potentiation (Henneberger et al., 2020), helping coordinate the withdrawal of fine astrocytic processes from synapses. All of the above indicates that deficits in NKCC1 astrocytic expression, function, or surface trafficking may all have pathological consequences. Nevertheless, experimental evidence using astrocytic-specific knockdown or knockout of NKCC1 are currently lacking. As discussed below, NKCC1 has joined the long-list of proteins whose mutation or absence in humans is linked to diseases (reviewed in Koumangoye et al., 2020), although it remains unclear how the loss of NKCC1 function in the affected individuals may have influenced the GABA-shift during their development. In fact, very little is known about the consequences of disrupted astrocytic NKCC1 expression or function on the GABA-shift, and this is a ripe field for study.
Conversely, how about the role of glia in the case of KCC2? Multiple studies have demonstrated a link between chronic inflammatory pain and KCC2 downregulation in the spinal cord (Coull et al., 2003, 2005; Okada-Ogawa et al., 2015; Tsuruga et al., 2016). Interestingly, the trigger for changes in neuronal KCC2 levels seems to be BDNF that is potentially released from nearby glia. Originally, microglia were thought to be the culprits; however, recent evidence suggests astrocytic signaling to be important for regulating both KCC2 and NKCC1. There is further evidence for a more direct role of astrocytes in modulating KCC2 levels. IL-6 released from astrocytes can elevate BDNF expression, and this in turn triggers downregulation of KCC2 in a model of neuropathic pain (Kitayama et al., 2016). It remains unclear if the source of this BDNF is neuronal or astrocytic, or potentially both.
Due to their critical role in maintaining chloride homeostasis and consequently the GABA-shift during brain development, it is not surprising that a number of studies have revealed changes in KCC2 and NKCC1 in patients with various neurodevelopmental and neuropsychiatric disorders.
Several recent studies have identified mutations in SLC12A5 from children affected by epilepsy in infancy with migrating focal seizures (EIMFS) (Table 1; Stödberg et al., 2015; Saitsu et al., 2016; Saito et al., 2017). The loss-of-function mutations (L311H, L426P, and G551D as mentioned above) identified by Stödberg et al. (2015), reduce KCC2 surface expression in neurons accompanied by depolarized chloride equilibrium potential (ECl), thereby potentially reducing GABAAR-mediated hyperpolarization compared to neurons carrying wild-type KCC2 protein. In a separate study (Saitsu et al., 2016), two of the mutations identified from EIMFS patients (E50_Q93del and M415V) significantly elevated ECl even though no changes in KCC2 cell surface expression were observed. Electrophysiological recordings demonstrated that the presence of these mutations suppressed the ability of the transporter to extrude chloride from the cell’s interior, and overexpression of E50_Q93del and M415V mutants of KCC2 resulted in elevated intracellular chloride concentrations. Furthermore, another study found a distinct mutation (R952H) in KCC2 from patients from an Australian family with febrile seizures that similarly disrupted chloride homeostasis by reducing KCC2 surface expression and function (Puskarjov et al., 2014). Another group also found the same mutation (R952H) along with R1049C to associate with idiopathic generalized epilepsy (IGE) (Kahle et al., 2014). Notably, the authors detected a reduction of S940 phosphorylation in the mutant proteins, previously demonstrated to enhance KCC2 surface expression by reducing its internalization (Lee et al., 2007). More recently, the R952H and R1049C mutations in SLC12A5 were also found in ASD (Merner et al., 2015). In addition, the same study identified the R1048W mutation in ASD patients, likely to cause similar disruption in KCC2 expression and function as R1049C due to their proximity. Notably, exome sequencing data suggest that ASD patients are more likely than healthy controls to have synonymous mutations predicted to create or disrupt CpG sites, thereby potentially affecting gene expression via alterations in DNA methylation patterns. Aside from these mutations affecting KCC2, recent studies have also identified multiple SLC12A2 mutations in patients affected by various neurodevelopmental disorders (Table 1; Delpire et al., 2016; Marchese et al., 2016; Anazi et al., 2017; Macnamara et al., 2019; McNeill et al., 2020; Stödberg et al., 2020). The pathogenic effects of these mutations on NKCC1 expression or functions, however, remain largely unknown.

Table 1. Mutations in SLC12A5 and SLC12A2 identified from patients with neurodevelopmental and neuropsychiatric disorders.
Recent studies have also observed altered SLC12A5 and SLC12A2 expression in patients. Consistent with their opposing roles in maintaining chloride homeostasis and GABA-shift, whereas NKCC1 protein levels were observed to be increased in patients with temporal lobe epilepsy (TLE) (Sen et al., 2007), SLC12A5 (KCC2) mRNA expression was found to be downregulated (Huberfeld et al., 2007). In addition, patients with tuberous sclerosis or type IIb cortical dysplasia, both of which are common causes of refractory epilepsy, also displayed changes in KCC2 and NKCC1 expression and levels (Talos et al., 2012; Ruffolo et al., 2016).
As described above, previous studies suggest that the expression of KCC2 and NKCC1 can be regulated via epigenetic mechanisms. Notably, a strong link between disrupted chromatin regulation and neurodevelopmental disorders has been implicated in recent years (Hui et al., 2018). Specifically, mutations in a number of chromatin regulators have been associated with neurodevelopmental disorders such as Rett syndrome (MECP2), intellectual disability (CHD1, CHD2, CHD7, CHD8, ATRX, and KDM5C), epilepsy (CHD1, CHD2, CHD7, CHD8) (Consortium et al., 2013), and ASD (MECP2, CHD1, CHD2, CHD3, CHD7, CHD8, MBD1, MACROD2, H2AFY, ARID1B, SMARCC1, SMARCC2, and JMJD1C) (reviewed in LaSalle, 2013). Two recent studies found KCC2 RNA expression to be significantly reduced in Rett syndrome patients (Gogliotti et al., 2018; Hinz et al., 2019). At the protein level, KCC2 levels were reduced in cerebrospinal fluid collected from patients with Rett syndrome without a change in NKCC1 levels, thereby disturbing the KCC2/NKCC1 ratio (Duarte et al., 2013). It will be of particular interest to examine how KCC2 and NKCC1 expression are altered in patients carrying mutations in the other chromatin regulators and determine how potential disruptions of the GABA-shift may contribute to those neurodevelopmental disorders.
Recent studies have also identified alterations of NKCC1 and/or KCC2 expression in patient-derived brain samples or animal models of neuropsychiatric disorders such as SZ and bipolar disorder (BD). For example, a significant reduction in KCC2 expression and thus an reducing trend for the KCC2/NKCC1 transcript ratio (as NKCC1 mRNA levels were not different between patients and controls) were observed in the hippocampi of SZ patients, suggesting the possibility of an immature GABA-shift (Hyde et al., 2011). Consistent with this, a separate group also observed reduced protein levels of KCC2 in the dorsal lateral prefrontal cortex of SZ patients (Sullivan et al., 2015). A follow-up study of alternative KCC2 transcripts further identified increased expression of transcript EXON6B (with a new exon 6) in SZ patients but the same transcript was reduced in patients with major depressive disorder (MDD), while other alternatively spliced transcripts examined including AK098371 (truncated KCC2 transcript), EXON2B (with a new exon 2), and ΔEXON6 (with exon 6 deletion) were not found to be different (Tao et al., 2012). In contrast, Morita et al. (2014) identified reduced expression of various alternatively spliced transcripts of NKCC1 (NKCC1b and 1-2a) in SZ patients compared to healthy controls. Although it is unclear how these alternative transcripts may affect overall KCC2 and NKCC1 functions, the studies highlight the potential involvement for differentially spliced KCC2 and NKCC1 transcripts in SZ and MDD pathophysiology.
SLC12A5 and SLC12A2 mutations have also been identified in SZ patients, including the R952H mutation in SLC12A5 described above (Merner et al., 2015). The same group later identified a Y199C mutation in the SLC12A2 gene specifically in SZ patients but not in ASD nor intellectual disability (ID) patients (Merner et al., 2016). Functional analysis of this NKCC1 mutant revealed an increase in its transporter activity, thus potentially altering chloride homeostasis and consequently GABAergic signaling in affected neurons. In addition, increased expression of OSR1 and WNK3 were detected in SZ patients compared to healthy controls (Arion and Lewis, 2010), thus potentially altering KCC2 and NKCC1 phospho-regulation and ultimately disturbing the GABA-shift. Similar to changes in KCC2 and NKCC1 expression discussed above, it remains to be determined how precisely these alterations are involved in pathophysiology, given that neuropsychiatric disorders do not typically manifest until late adolescence or early adulthood even though the mutations have been present during neurodevelopment.
To this end, it is of interest that several studies have recently identified transcriptomic signatures of “immaturity” in the dentate gyri and prefrontal cortices of adult patients with ASD, SZ, or BD (Gandal et al., 2012; Walton et al., 2012; Hagihara et al., 2014; Murano et al., 2019), suggestive of a reversal of the mature state (“dematuration”). Nevertheless, interpretation of the observations in dentate gyri are complicated by the fact that it is a major site of adult neurogenesis and thus the changes may reflect a difference in the process or the state of developmental maturation rather than signifying dematuration per se. Furthermore, multiple studies have also observed immature morphological and functional properties in neurons of SZ animal models or mutant animals exhibiting behavioral deficits relevant to neuropsychiatric disorders (Miyakawa et al., 2003; Yamasaki et al., 2008; Ohira et al., 2013; Shin et al., 2013; Takao et al., 2013; Kobayashi et al., 2014; Zhao et al., 2018; Sawada et al., 2020). Given the importance of the GABA-shift from excitatory to inhibitory during brain development and maturation, any disruptions in this process or even its reversal in the form of dematuration of neuronal phenotypes could potentially have profound effects on brain network activity and contribute to neurological disorders. Interestingly, a state of neuronal dematuration can be induced by different types of stimuli such as inflammation, hyperactivity, and drug treatment (Kobayashi et al., 2010, 2012; Takao et al., 2013). In parallel, various studies have shown that KCC2 and NKCC1 expression, phosphorylation status, or function could be affected by various types of stress (Hewitt et al., 2009; Sarkar et al., 2011; Miller and Maguire, 2014; Corradini et al., 2017; Furukawa et al., 2017; Hu et al., 2017) and may coincide with a disruption of neuronal maturation or the induction of a dematuration process. For example, maternal immune activation (MIA) by the injection of double stranded RNA poly(I:C) was found to alter the KCC2/NKCC1 ratio via a proinflammatory cytokine-dependent mechanism, and consequently delay the GABA-shift (Corradini et al., 2017). An independent study found that a similar MIA effectively abolished the GABA-shift and maintained GABA as an excitatory signal (Fernandez et al., 2018). Interestingly, a NKCC1-specific inhibitor was able to restore the hyperpolarizing actions of GABA in hippocampal slices from poly(I:C)-treated animals, thus highlighting the involvement of disrupted KCC2/NKCC1-mediated chloride homeostasis in the neurological deficits caused by MIA. More importantly, such findings indicate that pharmacologic intervention is possible and could have therapeutic benefits for certain patients with neurodevelopmental and neuropsychiatric disorders that have disturbed GABA-shifts.
In addition to the above study that demonstrated the beneficial effects of NKCC1-specific inhibitor bumetanide, commonly used clinically as a diuretic, it has been shown to be a useful treatment in mouse models of SZ (Kim et al., 2021), Down Syndrome (DS) (Deidda et al., 2015), Rett Syndrome (Banerjee et al., 2016), 22q11.2 deletion (DiGeorge) syndrome (Amin et al., 2017), neonatal epilepsy (Dzhala et al., 2005), and ID (Maset et al., 2021). More recent studies have further identified a novel therapeutic molecule known as ARN23746 (Savardi et al., 2020; Borgogno et al., 2021) and a series of KCC2 expression-enhancing compounds (KEECs) to be effective against the core symptoms exhibited by DS and ASD mouse models (Tang et al., 2019). Whereas ARN23746 targets NKCC1 selectively (thus limiting off-target diuretic side effects exhibited by bumetanide), the KEECs act via distinct cell signaling pathways [activation of the sirtuin 1 (SIRT1) or transient receptor potential cation channel subfamily V member 1 (TRPV1) pathways, or inhibition of fms-like tyrosine kinase 3 (FLT3) or glycogen synthase kinase 3β (GSK3β) pathways] to upregulate KCC2 expression at both mRNA and protein levels. In addition, anti-NKCC1 gene therapy or positive modulation of KCC2 overexpression by IGF1 and oxytocin have also been shown to be useful in treating cognitive deficits in a DS (Parrini et al., 2021) and Rett Syndrome mouse models (Tang et al., 2016; Bertoni et al., 2021), respectively.
Thus far, clinical trials have provided mixed results for the use of the NKCC1 blocker bumetanide to help reduce ASD symptoms in patients. While there have been reports hinting at successes in correcting for some symptoms in open-label pilot studies (Lemonnier and Ben-Ari, 2010; Hadjikhani et al., 2018; Fernell et al., 2021) and randomized double-blind clinical trials (NCT01078714 and NCT03156153) (Lemonnier et al., 2012, 2017; Zhang et al., 2020; Dai et al., 2021), other studies (NCT03715153, NCT03715166, and 2014-001560-35) have failed to show significant differences compared to placebo in their primary outcome measures (Sprengers et al., 2020; Crutel et al., 2021; Georgoula et al., 2022). Similarly, there are contrasting findings from small scale studies which attempt to treat patients with other types of neurodevelopmental and neuropsychiatric disorders such as epilepsy, fragile X syndrome, and schizophrenia using bumetanide (Eftekhari et al., 2013; Lemonnier et al., 2016; Rahmanzadeh et al., 2017; Soul et al., 2021), clearly highlighting the need for larger studies to definitively verify its effects. A particular issue with bumetanide potentially hindering its beneficial effects in these trials is its poor penetration into the CNS. Fortunately, the aforementioned ARN23746 appears to have a better in vivo pharmacokinetic profile in mice as compared to bumetanide (Savardi et al., 2020), thus offering a glimpse of hope that ARN23746 and its derivatives may have more potent effects in human patients. Furthermore, in dealing with complex disorders such as ASD and schizophrenia in which the precise pathogenic cause is typically unknown for each patient, pharmacologic strategies such as bumetanide may simply not be effective for every patient since it is likely that only a subpopulation are experiencing E-I imbalance due to a dysregulated GABA-shift. As such, brain organoids derived from patient-specific induced pluripotent stem cells (iPSCs) (Zafeiriou et al., 2020) could be used to screen for patients with pathologic alterations in the GABA-shift to determine whether they are likely going to benefit from NKCC1 inhibitors. Finally, another issue with understanding the potentially negative consequences of disrupted NKCC1 function and its contribution to GABA-shift is the widespread expression of the protein (as well as its relatives like NKCC2, for example), and the resulting broad spectrum of problems that may occur throughout the body when they are inhibited. Thus, continued efforts to identify new therapeutic molecules such as ARN23746 and KEECs which do not have peripheral effects like bumetanide may prove to be fruitful.
This review has attempted to lay out the changes that occur in GABAergic signaling during early development, with a particular focus on the GABA-shift, both in how it proceeds in healthy tissue, and how it is disrupted in various pathological states. Abnormalities in the progression of the GABA-shift have been linked to several neurodevelopmental and neuropsychiatric disorders. Potential therapeutic targets typically focus on a handful of proteins: the transporters that move Cl– across the neuronal membrane, and regulators of those proteins, such as WNK kinases. The balance of Cl– across the membrane is primarily regulated by KCC2 and NKCC1, and as such, they are prime targets for pharmacological modulation. However, the widespread expression of NKCC1 in other tissues of the body complicates the utility of the pharmacological approach for therapy. Furthermore, our understanding of disturbances in the normal GABA-shift is hampered by the high variability: it happens at different times, in different cell types, that may be intermingled within a particular brain region. Moreover, in human cases with a mutated or missing chloride transporter, it is not clear how much of a patient’s symptoms may be attributable to an abnormal GABA-shift, and how much is related to ongoing network dysfunction. Further research is required to understand the pathological consequences of a mis-timed or incomplete GABA-shift, and treatment options, whilst currently available, are limited.
A better grasp of the factors that control the GABA-shift during development may provide a greater understanding of what happens in the process of dematuration within the adult network. What are the exact triggers for the onset and successful completion of the GABA-shift? The impact and participation of different cell types in the GABA-shift, and the ongoing maintenance of GABAergic signaling, and both intra- and extracellular chloride regulation is an extremely important topic, and the extent of the contribution of glial cells remains a topic of much interest.
KH and TC conceptualized and wrote the manuscript. YG and MT provided intellectual input and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the RIKEN Center for Brain Science, Japan Agency for Medical Research and Development (AMED; JP21wm0525014h, JP15dm0207001, and 21gm1410009h to MT), Core-to-Core Program (JPJSCCA20220007 to YG) by Japan Society for the Promotion of Science (JSPS), Grants-in-Aid for Transformative Research Area (A) (21H05257 to MT) from Ministry of Education, Culture, Sports, Science and Technology (MEXT, Japan), and Japan Epilepsy Research Foundation (to MT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agez, M., Schultz, P., Medina, I., Baker, D. J., Burnham, M. P., Cardarelli, R. A., et al. (2017). Molecular architecture of potassium chloride co-transporter KCC2. Sci. Rep. 7:16452. doi: 10.1038/s41598-017-15739-1
Aguado, F., Carmona, M. A., Pozas, E., Aguiloì, A., Martiìnez-Guijarro, F. J., Alcantara, S., et al. (2003). BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development 130, 1267–1280. doi: 10.1242/dev.00351
Akerman, C. J., and Cline, H. T. (2006). Depolarizing GABAergic Conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26, 5117–5130. doi: 10.1523/jneurosci.0319-06.2006
Alessi, D. R., Zhang, J., Khanna, A., Hochdorfer, T., Shang, Y., and Kahle, K. T. (2014). The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci. Signal. 7:re3. doi: 10.1126/scisignal.2005365
Amin, H., Marinaro, F., Tonelli, D. D. P., and Berdondini, L. (2017). Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci. Rep. 7:15752. doi: 10.1038/s41598-017-15793-9
Anazi, S., Maddirevula, S., Salpietro, V., Asi, Y. T., Alsahli, S., Alhashem, A., et al. (2017). Expanding the genetic heterogeneity of intellectual disability. Hum. Genet. 136, 1419–1429. doi: 10.1007/s00439-017-1843-2
Andäng, M., Hjerling-Leffler, J., Moliner, A., Lundgren, T. K., Castelo-Branco, G., Nanou, E., et al. (2008). Histone H2AX-dependent GABA(A) receptor regulation of stem cell proliferation. Nature 451, 460–464. doi: 10.1038/nature06488
Anderson, S. A., Classey, J. D., Condé, F., Lund, J. S., and Lewis, D. A. (1995). Synchronous development of pyramidal neuron dendritic spines and parvalbumin-immunoreactive chandelier neuron axon terminals in layer III of monkey prefrontal cortex. Neuroscience 67, 7–22. doi: 10.1016/0306-4522(95)00051-j
Arion, D., and Lewis, D. A. (2010). Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch. Gen. Psychiatry 68, 21–31. doi: 10.1001/archgenpsychiatry.2010.114
Arosio, D., and Ratto, G. M. (2014). Twenty years of fluorescence imaging of intracellular chloride. Front. Cell. Neurosci. 8:258. doi: 10.3389/fncel.2014.00258
Balakrishnan, V., Becker, M., Löhrke, S., Nothwang, H. G., Güresir, E., and Friauf, E. (2003). Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory Brainstem. J. Neurosci. 23, 4134–4145. doi: 10.1523/jneurosci.23-10-04134.2003
Banerjee, A., Rikhye, R. V., Breton-Provencher, V., Tang, X., Li, C., Li, K., et al. (2016). Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, E7287–E7296. doi: 10.1073/pnas.1615330113
Banke, T. G., and McBain, C. J. (2006). GABAergic Input onto CA3 hippocampal interneurons remains shunting throughout development. J. Neurosci. 26, 11720–11725. doi: 10.1523/jneurosci.2887-06.2006
Barker, J. L., Behar, T., Li, Y. X., Liu, Q. Y., Ma, W., Maric, D., et al. (1998). GABAergic cells and signals in CNS development. Perspect. Dev. Neurobiol. 5, 305–322.
Barkis, W. B., Ford, K. J., and Feller, M. B. (2010). Non–cell-autonomous factor induces the transition from excitatory to inhibitory GABA signaling in retina independent of activity. Proc. Natl. Acad. Sci. U.S.A. 107, 22302–22307. doi: 10.1073/pnas.1008775108
Bartolini, G., Ciceri, G., and Marín, O. (2013). Integration of gabaergic interneurons into cortical cell assemblies: lessons from embryos and adults. Neuron 79, 849–864. doi: 10.1016/j.neuron.2013.08.014
Behar, T., Li, Y., Tran, H., Ma, W., Dunlap, V., Scott, C., et al. (1996). GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J. Neurosci. 16, 1808–1818. doi: 10.1523/jneurosci.16-05-01808.1996
Behar, T. N., Schaffner, A. E., Scott, C. A., Greene, C. L., and Barker, J. L. (2000). GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb. Cortex 10, 899–909. doi: 10.1093/cercor/10.9.899
Behar, T. N., Schaffner, A. E., Scott, C. A., O’Connell, C., and Barker, J. L. (1998). Differential response of cortical plate and ventricular zone cells to GABA as a Migration Stimulus. J. Neurosci. 18, 6378–6387. doi: 10.1523/jneurosci.18-16-06378.1998
Bekar, L. K., and Walz, W. (2002). Intracellular chloride modulates A-type potassium currents in astrocytes. Glia 39, 207–216. doi: 10.1002/glia.10096
Bellot-Saez, A., Kékesi, O., Morley, J. W., and Buskila, Y. (2017). Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci. Biobehav. Rev. 77, 87–97. doi: 10.1016/j.neubiorev.2017.03.002
Ben-Ari, Y., Khazipov, R., Leinekugel, X., Caillard, O., and Gaiarsa, J.-L. (1997). GABAA, NMDA and AMPA receptors: a developmentally regulated ‘ménage à trois’. Trends Neurosci. 20, 523–529. doi: 10.1016/s0166-2236(97)01147-8
Ben-Ari, Y., Woodin, M. A., Sernagor, E., Cancedda, L., Vinay, L., Rivera, C., et al. (2012). Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever! Front. Cell. Neurosci. 6:35. doi: 10.3389/fncel.2012.00035
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325. doi: 10.1113/jphysiol.1989.sp017762
Bertoni, A., Schaller, F., Tyzio, R., Gaillard, S., Santini, F., Xolin, M., et al. (2021). Oxytocin administration in neonates shapes hippocampal circuitry and restores social behavior in a mouse model of autism. Mol. Psychiatry 26, 7582–7595. doi: 10.1038/s41380-021-01227-6
Blaesse, P., Guillemin, I., Schindler, J., Schweizer, M., Delpire, E., Khiroug, L., et al. (2006). Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J. Neurosci. 26, 10407–10419. doi: 10.1523/jneurosci.3257-06.2006
Boddum, K., Jensen, T. P., Magloire, V., Kristiansen, U., Rusakov, D. A., Pavlov, I., et al. (2016). Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 7:13572. doi: 10.1038/ncomms13572
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Borgogno, M., Savardi, A., Manigrasso, J., Turci, A., Portioli, C., Ottonello, G., et al. (2021). Design, synthesis, in vitro and in vivo characterization of selective NKCC1 Inhibitors for the Treatment of Core Symptoms in Down Syndrome. J. Med. Chem. 64, 10203–10229. doi: 10.1021/acs.jmedchem.1c00603
Bos, R., Sadlaoud, K., Boulenguez, P., Buttigieg, D., Liabeuf, S., Brocard, C., et al. (2013). Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 110, 348–353. doi: 10.1073/pnas.1213680110
Boulenguez, P., Liabeuf, S., Bos, R., Bras, H., Jean-Xavier, C., Brocard, C., et al. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307. doi: 10.1038/nm.2107
Bregestovski, P., and Bernard, C. (2012). Excitatory GABA: how a correct observation may turn out to be an experimental artifact. Front. Pharmacol. 3:65. doi: 10.3389/fphar.2012.00065
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M.-M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. doi: 10.1523/jneurosci.5169-06.2007
Ceanga, M., Spataru, A., and Zagrean, A.-M. (2010). Oxytocin is neuroprotective against oxygen–glucose deprivation and reoxygenation in immature hippocampal cultures. Neurosci. Lett. 477, 15–18. doi: 10.1016/j.neulet.2010.04.024
Chu, J., and Anderson, S. A. (2015). Development of Cortical Interneurons. Neuropsychopharmacology 40, 16–23. doi: 10.1038/npp.2014.171
Consortium, E., Project, E. P., Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Conway, L. C., Cardarelli, R. A., Moore, Y. E., Jones, K., McWilliams, L. J., Baker, D. J., et al. (2017). N-Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J. Biol. Chem. 292, 21253–21263. doi: 10.1074/jbc.m117.817841
Cordshagen, A., Busch, W., Winklhofer, M., Nothwang, H. G., and Hartmann, A.-M. (2018). Phosphoregulation of the intracellular termini of K+-Cl− cotransporter 2 (KCC2) enables flexible control of its activity. J. Biol. Chem. 293, 16984–16993. doi: 10.1074/jbc.ra118.004349
Corradini, I., Focchi, E., Rasile, M., Morini, R., Desiato, G., Tomasoni, R., et al. (2017). Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691. doi: 10.1016/j.biopsych.2017.09.030
Coull, J. A. M., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Coull, J. A. M., Boudreau, D., Bachand, K., Prescott, S. A., Nault, F., Sík, A., et al. (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. doi: 10.1038/nature01868
Crutel, V., Lambert, E., Penelaud, P.-F., Severo, C. A., Fuentes, J., Rosier, A., et al. (2021). Bumetanide oral liquid formulation for the treatment of children and adolescents with autism spectrum disorder: design of two phase III Studies (SIGN Trials). J. Autism Dev. Disord. 51, 2959–2972. doi: 10.1007/s10803-020-04709-8
Dai, Y., Zhang, L., Yu, J., Zhou, X., He, H., Ji, Y., et al. (2021). Improved symptoms following bumetanide treatment in children aged 3-6 years with autism spectrum disorder: a randomized, double-blind, placebo-controlled trial. Sci. Bull. 66, 1591–1598. doi: 10.1016/j.scib.2021.01.008
Darman, R. B., and Forbush, B. (2002). A regulatory locus of phosphorylation in the N Terminus of the Na-K-Cl Cotransporter, NKCC1. J. Biol. Chem. 277, 37542–37550. doi: 10.1074/jbc.m206293200
Davis, A. M., Penschuck, S., Fritschy, J. M., and McCarthy, M. M. (2000). Developmental switch in the expression of GABA(A) receptor subunits alpha(1) and alpha(2) in the hypothalamus and limbic system of the rat. Brain Res. Dev. Brain Res. 119, 127–138. doi: 10.1016/s0165-3806(99)00150-9
de los Heros, P., Alessi, D. R., Gourlay, R., Campbell, D. G., Deak, M., Macartney, T. J., et al. (2014). The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+–Cl− co-transporters. Biochem. J. 458, 559–573. doi: 10.1042/bj20131478
Deidda, G., Parrini, M., Naskar, S., Bozarth, I. F., Contestabile, A., and Cancedda, L. (2015). Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 21, 318–326. doi: 10.1038/nm.3827
Delpire, E., Lu, J., England, R., Dull, C., and Thorne, T. (1999). Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 22, 192–195. doi: 10.1038/9713
Delpire, E., Wolfe, L., Flores, B., Koumangoye, R., Schornak, C. C., Omer, S., et al. (2016). A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb. Mol. Case Stud. 2:a001289. doi: 10.1101/mcs.a001289
Demarque, M., Represa, A., Becq, H., Khalilov, I., Ben-Ari, Y., and Aniksztejn, L. (2002). Paracrine intercellular communication by a Ca2+- and SNARE-independent release of GABA and glutamate prior to synapse formation. Neuron 36, 1051–1061. doi: 10.1016/s0896-6273(02)01053-x
Denter, D. G., Heck, N., Riedemann, T., White, R., Kilb, W., and Luhmann, H. J. (2010). GABAC receptors are functionally expressed in the intermediate zone and regulate radial migration in the embryonic mouse neocortex. Neuroscience 167, 124–134. doi: 10.1016/j.neuroscience.2010.01.049
Dixon, M. J., Gazzard, J., Chaudhry, S. S., Sampson, N., Schulte, B. A., and Steel, K. P. (1999). Mutation of the Na-K-Cl Co-Transporter Gene Slc12a2 Results in Deafness in Mice. Hum. Mol. Genet. 8, 1579–1584. doi: 10.1093/hmg/8.8.1579
Doengi, M., Hirnet, D., Coulon, P., Pape, H.-C., Deitmer, J. W., and Lohr, C. (2009). GABA uptake-dependent Ca(2+) signaling in developing olfactory bulb astrocytes. Proc. Natl. Acad. Sci. U.S.A. 106, 17570–17575. doi: 10.1073/pnas.0809513106
Duarte, S. T., Armstrong, J., Roche, A., Ortez, C., Pérez, A., O’Callaghan, M., et al. (2013). Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with rett syndrome. PLoS One 8:e68851. doi: 10.1371/journal.pone.0068851
Duveau, V., Laustela, S., Barth, L., Gianolini, F., Vogt, K. E., Keist, R., et al. (2011). Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur. J. Neurosci. 34, 362–373. doi: 10.1111/j.1460-9568.2011.07782.x
Dzhala, V., Valeeva, G., Glykys, J., Khazipov, R., and Staley, K. (2012). Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J. Neurosci. 32, 4017–4031. doi: 10.1523/jneurosci.5139-11.2012
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
Ebert, B., Wafford, K. A., Whiting, P. J., Krogsgaard-Larsen, P., and Kemp, J. A. (1994). Molecular pharmacology of gamma-aminobutyric acid type A receptor agonists and partial agonists in oocytes injected with different alpha, beta, and gamma receptor subunit combinations. Mol. Pharmacol. 46, 957–963.
Eftekhari, S., Habibabadi, J. M., Ziarani, M. N., Fesharaki, S. S. H., Gharakhani, M., Mostafavi, H., et al. (2013). Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia 54, e9–e12. doi: 10.1111/j.1528-1167.2012.03654.x
Eftekhari, S., Mehrabi, S., Soleimani, M., Hassanzadeh, G., Shahrokhi, A., Mostafavi, H., et al. (2014). BDNF modifies hippocampal KCC2 and NKCC1 expression in a temporal lobe epilepsy model. Acta Neurobiol. Exp. 74, 276–287.
Egawa, K., Yamada, J., Furukawa, T., Yanagawa, Y., and Fukuda, A. (2013). Cl− homeodynamics in gap junction-coupled astrocytic networks on activation of GABAergic synapses. J. Physiol. 591, 3901–3917. doi: 10.1113/jphysiol.2013.257162
Eins, S., Spoerri, P. E., and Heyder, E. (1983). GABA or sodium-bromide-induced plasticity of neurites of mouse neuroblastoma cells in culture. Cell Tissue Res. 229, 457–460. doi: 10.1007/bf00214987
Fernandez, A., Dumon, C., Guimond, D., Tyzio, R., Bonifazi, P., Lozovaya, N., et al. (2018). The GABA developmental shift is abolished by maternal immune activation already at birth. Cereb. Cortex 29, 3982–3992. doi: 10.1093/cercor/bhy279
Fernell, E., Gustafsson, P., and Gillberg, C. (2021). Bumetanide for autism: open-label trial in six children. Acta Paediatr. 110, 1548–1553. doi: 10.1111/apa.15723
Ferrer, C., and García, N. V. D. M. (2022). The role of inhibitory interneurons in circuit assembly and refinement across sensory cortices. Front. Neural Circuits 16:866999. doi: 10.3389/fncir.2022.866999
Flagella, M., Clarke, L. L., Miller, M. L., Erway, L. C., Giannella, R. A., Andringa, A., et al. (1999). Mice lacking the Basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 274, 26946–26955. doi: 10.1074/jbc.274.38.26946
Flemmer, A. W., Giménez, I., Dowd, B. F. X., Darman, R. B., and Forbush, B. (2002). Activation of the Na-K-Cl Cotransporter NKCC1 detected with a phospho-specific antibody. J. Biol. Chem. 277, 37551–37558. doi: 10.1074/jbc.m206294200
Fraser, S. A., Davies, M., Katerelos, M., Gleich, K., Choy, S.-W., Steel, R., et al. (2014). Activation of AMPK reduces the co-transporter activity of NKCC1. Mol. Membr. Biol. 31, 95–102. doi: 10.3109/09687688.2014.902128
Friedel, P., Kahle, K. T., Zhang, J., Hertz, N., Pisella, L. I., Buhler, E., et al. (2015). WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 8:ra65. doi: 10.1126/scisignal.aaa0354
Fritschy, J.-M. (2008). Epilepsy, E/I balance and GABAA receptor plasticity. Front. Mol. Neurosci. 1:5. doi: 10.3389/neuro.02.005.2008
Fritschy, J. M., Paysan, J., Enna, A., and Mohler, H. (1994). Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J. Neurosci. 14, 5302–5324. doi: 10.1523/JNEUROSCI.14-09-05302.1994
Fukuda, A., Tanaka, M., Yamada, Y., Muramatsu, K., Shimano, Y., and Nishino, H. (1998). Simultaneous optical imaging of intracellular Cl− in neurons in different layers of rat neocortical slices: advantages and limitations. Neurosci. Res. 32, 363–371. doi: 10.1016/s0168-0102(98)00099-6
Furukawa, M., Tsukahara, T., Tomita, K., Iwai, H., Sonomura, T., Miyawaki, S., et al. (2017). Neonatal maternal separation delays the GABA excitatory-to-inhibitory functional switch by inhibiting KCC2 expression. Biochem. Biophy. Res. Commun. 493, 1243–1249. doi: 10.1016/j.bbrc.2017.09.143
Galanopoulou, A. S. (2008). Dissociated gender-specific effects of recurrent seizures on GABA Signaling in CA1 Pyramidal Neurons: role of GABAA Receptors. J. Neurosci. 28, 1557–1567. doi: 10.1523/jneurosci.5180-07.2008
Gamba, G., Miyanoshita, A., Lombardi, M., Lytton, J., Lee, W. S., Hediger, M. A., et al. (1994). Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 269, 17713–17722. doi: 10.1016/s0021-9258(17)32499-7
Gandal, M. J., Nesbitt, A. M., McCurdy, R. M., and Alter, M. D. (2012). Measuring the maturity of the fast-spiking interneuron transcriptional program in autism, schizophrenia, and bipolar disorder. PLoS One 7:e41215. doi: 10.1371/journal.pone.0041215
Ganguly, K., Schinder, A. F., Wong, S. T., and Poo, M. (2001). GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105, 521–532. doi: 10.1016/s0092-8674(01)00341-5
Garand, D., Mahadevan, V., and Woodin, M. A. (2019). Ionotropic and metabotropic kainate receptor signalling regulates Cl− homeostasis and GABAergic inhibition. J. Physiol. 597, 1677–1690. doi: 10.1113/jp276901
Gascon, E., Dayer, A. G., Sauvain, M.-O., Potter, G., Jenny, B., Roo, M. D., et al. (2006). GABA regulates dendritic growth by stabilizing lamellipodia in newly generated interneurons of the olfactory bulb. J. Neurosci. 26, 12956–12966. doi: 10.1523/jneurosci.4508-06.2006
Ge, S., Goh, E. L. K., Sailor, K. A., Kitabatake, Y., Ming, G., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593. doi: 10.1038/nature04404
Ge, X., Zhang, K., Gribizis, A., Hamodi, A. S., Sabino, A. M., and Crair, M. C. (2021). Retinal waves prime visual motion detection by simulating future optic flow. Science 373:eabd0830. doi: 10.1126/science.abd0830
Georgoula, C., Ferrin, M., Pietraszczyk-Kedziora, B., Hervas, A., Marret, S., Oliveira, G., et al. (2022). A Phase III Study of Bumetanide Oral Liquid Formulation for the Treatment of Children and Adolescents Aged Between 7 and 17 Years with Autism Spectrum Disorder (SIGN 1 Trial): participant Baseline Characteristics. Child Psychiatry Hum. Dev. [Epub ahead of print]. doi: 10.1007/s10578-022-01328-5
Gogliotti, R., Fisher, N., Stansley, B., Jones, C., Lindsley, C., Conn, J., et al. (2018). Total RNA-sequencing of Rett syndrome autopsy samples identifies the M4 muscarinic receptor as a novel therapeutic target. J. Pharmacol. Exp. Ther. 365, 291–300 doi: 10.1124/jpet.117.246991
Gonchar, Y., Wang, Q., and Burkhalter, A. (2008). Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front. Neuroanat. 1:3. doi: 10.3389/neuro.05.003.2007
Gozlan, H., and Ben-Ari, Y. (2003). Interneurons are the Source and the Targets of the First Synapses Formed in the Rat Developing Hippocampal Circuit. Cereb. Cortex 13, 684–692. doi: 10.1093/cercor/13.6.684
Griguoli, M., and Cherubini, E. (2017). Early correlated network activity in the hippocampus: its putative role in shaping neuronal circuits. Front. Cell. Neurosci. 11:255. doi: 10.3389/fncel.2017.00255
Gubellini, P., Ben-Ari, Y., and Gaïarsa, J.-L. (2005). Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J. Neurosci. 25, 5796–5802. doi: 10.1523/jneurosci.0824-05.2005
Hadjikhani, N., Johnels, J. Å, Lassalle, A., Zürcher, N. R., Hippolyte, L., Gillberg, C., et al. (2018). Bumetanide for autism: more eye contact, less amygdala activation. Sci. Rep. 8:3602. doi: 10.1038/s41598-018-21958-x
Hagihara, H., Ohira, K., Takao, K., and Miyakawa, T. (2014). Transcriptomic evidence for immaturity of the prefrontal cortex in patients with schizophrenia. Mol. Brain 7:41. doi: 10.1186/1756-6606-7-41
Hampel, P., Johne, M., Gailus, B., Vogel, A., Schidlitzki, A., Gericke, B., et al. (2021). Deletion of the Na-K-2Cl cotransporter NKCC1 results in a more severe epileptic phenotype in the intrahippocampal kainate mouse model of temporal lobe epilepsy. Neurobiol. Dis. 152:105297. doi: 10.1016/j.nbd.2021.105297
Haydar, T. F., Wang, F., Schwartz, M. L., and Rakic, P. (2000). Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J. Neurosci. 20, 5764–5774. doi: 10.1523/jneurosci.20-15-05764.2000
Henneberger, C., Bard, L., Panatier, A., Reynolds, J. P., Kopach, O., Medvedev, N. I., et al. (2020). ltp induction boosts glutamate spillover by driving withdrawal of Perisynaptic Astroglia. Neuron 108, 919–936.e11. doi: 10.1016/j.neuron.2020.08.030
Hensch, T. K. (2005). Critical period mechanisms in developing visual cortex. Curr. Top. Dev. Biol. 69, 215–237. doi: 10.1016/s0070-2153(05)69008-4
Hertz, L. (1965). Possible role of neuroglia: a potassium-Mediated Neuronal – Neuroglial – Neuronal Impulse Transmission System. Nature 206, 1091–1094. doi: 10.1038/2061091a0
Hertz, L., and Chen, Y. (2016). Importance of astrocytes for potassium ion (K+) homeostasis in brain and glial effects of K+ and its transporters on learning. Neurosci. Biobehav. Rev. 71, 484–505. doi: 10.1016/j.neubiorev.2016.09.018
Hevers, W., and Lüddens, H. (1998). The diversity of GABAA receptors: pharmacological and electrophysiological properties of GABAA channel subtypes. Mol. Neurobiol. 18, 35–86. doi: 10.1007/bf02741459
Hewitt, S. A., Wamsteeker, J. I., Kurz, E. U., and Bains, J. S. (2009). Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 12, 438–443. doi: 10.1038/nn.2274
Hinz, L., Barrufet, J. T., and Heine, V. M. (2019). KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol. Commun. 7:196. doi: 10.1186/s40478-019-0852-x
Holmgren, C. D., Mukhtarov, M., Malkov, A. E., Popova, I. Y., Bregestovski, P., and Zilberter, Y. (2010). Energy substrate availability as a determinant of neuronal resting potential, GABA signaling and spontaneous network activity in the neonatal cortex in vitro. J. Neurochem. 112, 900–912. doi: 10.1111/j.1471-4159.2009.06506.x
Hou, X., Weng, Y., Wang, T., Ouyang, B., Li, Y., Song, Z., et al. (2018). Suppression of HDAC2 in spinal cord alleviates mechanical hyperalgesia and restores KCC2 expression in a rat model of bone cancer pain. Neuroscience 377, 138–149. doi: 10.1016/j.neuroscience.2018.02.026
Hu, D., Yu, Z.-L., Zhang, Y., Han, Y., Zhang, W., Lu, L., et al. (2017). Bumetanide treatment during early development rescues maternal separation-induced susceptibility to stress. Sci. Rep. 7:11878. doi: 10.1038/s41598-017-12183-z
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., et al. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873. doi: 10.1523/jneurosci.2761-07.2007
Hübner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001b). Disruption of KCC2 reveals an essential role of K-Cl Cotransport Already in Early Synaptic Inhibition. Neuron 30, 515–524. doi: 10.1016/s0896-6273(01)00297-5
Hübner, C. A., Lorke, D. E., and Hermans-Borgmeyer, I. (2001a). Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech. Dev. 102, 267–269. doi: 10.1016/s0925-4773(01)00309-4
Hui, K., Katayama, Y., Nakayama, K. I., Nomura, J., and Sakurai, T. (2018). Characterizing vulnerable brain areas and circuits in mouse models of autism: towards understanding pathogenesis and new therapeutic approaches. Neurosci. Biobehav. Rev. 110, 77–91. doi: 10.1016/j.neubiorev.2018.08.001
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095. doi: 10.1523/jneurosci.1234-11.2011
Jarolimek, W., Lewen, A., and Misgeld, U. (1999). A Furosemide-Sensitive K+–Cl-cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. J. Neurosci. 19, 4695–4704. doi: 10.1523/jneurosci.19-12-04695.1999
Jayakumar, A. R., Liu, M., Moriyama, M., Ramakrishnan, R., Forbush, B., Reddy, P. V. B., et al. (2008). Na-K-Cl cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J. Biol. Chem. 283, 33874–33882. doi: 10.1074/jbc.m804016200
Jayakumar, A. R., and Norenberg, M. D. (2010). The Na–K–Cl Co-transporter in astrocyte swelling. Metab. Brain Dis. 25, 31–38. doi: 10.1007/s11011-010-9180-3
Jayakumar, A. R., Panickar, K. S., Curtis, K. M., Tong, X. Y., Moriyama, M., and Norenberg, M. D. (2011). Na-K-Cl cotransporter-1 in the mechanism of cell swelling in cultured astrocytes after fluid percussion injury. J. Neurochem. 117, 437–448. doi: 10.1111/j.1471-4159.2011.07211.x
Kahle, K. T., Merner, N. D., Friedel, P., Silayeva, L., Liang, B., Khanna, A., et al. (2014). Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–774. doi: 10.15252/embr.201438840
Kahle, K. T., Rinehart, J., de los Heros, P., Louvi, A., Meade, P., Vazquez, N., et al. (2005). WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788. doi: 10.1073/pnas.0508307102
Kakazu, Y., Akaike, N., Komiyama, S., and Nabekura, J. (1999). Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J. Neurosci. 19, 2843–2851. doi: 10.1523/jneurosci.19-08-02843.1999
Kang, J., Jiang, L., Goldman, S. A., and Nedergaard, M. (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692. doi: 10.1038/3684
Khalilov, I., Dzhala, V., Ben-Ari, Y., and Khazipov, R. (1999). Dual Role of GABA in the neonatal rat hippocampus. Dev. Neurosci. 21, 310–319. doi: 10.1159/000017380
Khirug, S., Huttu, K., Ludwig, A., Smirnov, S., Voipio, J., Rivera, C., et al. (2005). Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur. J. Neurosci. 21, 899–904. doi: 10.1111/j.1460-9568.2005.03886.x
Kim, H. R., Rajagopal, L., Meltzer, H. Y., and Martina, M. (2021). Depolarizing GABAA current in the prefrontal cortex is linked with cognitive impairment in a mouse model relevant for schizophrenia. Sci. Adv. 7:eaba5032. doi: 10.1126/sciadv.aba5032
Kim, J.-Y., and Paredes, M. F. (2021). Implications of extended inhibitory neuron development. Int. J. Mol. Sci. 22:5113. doi: 10.3390/ijms22105113
King, S. J., Bunz, M., Chappell, A., Scharl, M., Docherty, M., Jung, B., et al. (2019). AMPK mediates inhibition of electrolyte transport and NKCC1 activity by reactive oxygen species. Am. J. Physiol. Gastrointest. Liver Physiol. 317, G171–G181. doi: 10.1152/ajpgi.00317.2018
Kirmse, K., Kummer, M., Kovalchuk, Y., Witte, O. W., Garaschuk, O., and Holthoff, K. (2015). GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 6:7750. doi: 10.1038/ncomms8750
Kitayama, T., Morita, K., Motoyama, N., and Dohi, T. (2016). Down-regulation of zinc transporter-1 in astrocytes induces neuropathic pain via the brain-derived neurotrophic factor - K+-Cl− co-transporter-2 signaling pathway in the mouse spinal cord. Neurochem. Int. 101, 120–131. doi: 10.1016/j.neuint.2016.11.001
Kobayashi, K., Haneda, E., Higuchi, M., Suhara, T., and Suzuki, H. (2012). Chronic fluoxetine selectively upregulates dopamine D1-like receptors in the hippocampus. Neuropsychopharmacology 37, 1500–1508. doi: 10.1038/npp.2011.335
Kobayashi, K., Ikeda, Y., Sakai, A., Yamasaki, N., Haneda, E., Miyakawa, T., et al. (2010). Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proc. Natl. Acad. Sci. U.S.A. 107, 8434–8439. doi: 10.1073/pnas.0912690107
Kobayashi, M., Nakatani, T., Koda, T., Matsumoto, K., Ozaki, R., Mochida, N., et al. (2014). Absence of BRINP1 in mice causes increase of hippocampal neurogenesis and behavioral alterations relevant to human psychiatric disorders. Mol. Brain 7:12. doi: 10.1186/1756-6606-7-12
Koumangoye, R., Bastarache, L., and Delpire, E. (2020). NKCC1: newly found as a human disease-causing ion transporter. Function 2:zqaa028. doi: 10.1093/function/zqaa028
Kubota, Y., Karube, F., Nomura, M., and Kawaguchi, Y. (2016). The diversity of cortical inhibitory synapses. Front. Neural Circuits 10:27. doi: 10.3389/fncir.2016.00027
Kuner, T., and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459. doi: 10.1016/s0896-6273(00)00056-8
Kyrozis, A., Chudomel, O., Moshé, S. L., and Galanopoulou, A. S. (2006). Sex-dependent maturation of GABAA receptor-mediated synaptic events in rat substantia nigra reticulata. Neurosci. Lett. 398, 1–5. doi: 10.1016/j.neulet.2005.12.018
Langlois, A., Diabira, D., Ferrand, N., Porcher, C., and Gaiarsa, J.-L. (2013). NMDA-Dependent Switch of proBDNF Actions on Developing GABAergic Synapses. Cereb. Cortex 23, 1085–1096. doi: 10.1093/cercor/bhs071
LaSalle, J. (2013). Autism genes keep turning up chromatin. OA Autism 1:14. doi: 10.13172/2052-7810-1-2-610
Laurie, D., Wisden, W., and Seeburg, P. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172.
Lee, H. H. C., Deeb, T. Z., Walker, J. A., Davies, P. A., and Moss, S. J. (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor–mediated currents. Nat. Neurosci. 14, 736–743. doi: 10.1038/nn.2806
Lee, H. A., Hong, S., Kim, J., and Jang, I. (2010). Possible involvement of DNA methylation in NKCC1 gene expression during postnatal development and in response to ischemia. J. Neurochem. 114, 520–529. doi: 10.1111/j.1471-4159.2010.06772.x
Lee, H. H. C., Jurd, R., and Moss, S. J. (2010). Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179. doi: 10.1016/j.mcn.2010.06.008
Lee, H. H. C., Walker, J. A., Williams, J. R., Goodier, R. J., Payne, J. A., and Moss, S. J. (2007). Direct Protein Kinase C-dependent Phosphorylation Regulates the Cell Surface Stability and Activity of the Potassium Chloride Cotransporter KCC2. J. Biol. Chem. 282, 29777–29784. doi: 10.1074/jbc.m705053200
Leinekugel, X., Tseeb, V., Ben-Ari, Y., and Bregestovski, P. (1995). Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J. Physiol. 487, 319–329. doi: 10.1113/jphysiol.1995.sp020882
Leitch, E., Coaker, J., Young, C., Mehta, V., and Sernagor, E. (2005). GABA type-A activity controls its own developmental polarity switch in the maturing retina. J. Neurosci. 25, 4801–4805. doi: 10.1523/jneurosci.0172-05.2005
Lemonnier, E., and Ben-Ari, Y. (2010). The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 99, 1885–1888. doi: 10.1111/j.1651-2227.2010.01933.x
Lemonnier, E., Degrez, C., Phelep, M., Tyzio, R., Josse, F., Grandgeorge, M., et al. (2012). A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2:e202. doi: 10.1038/tp.2012.124
Lemonnier, E., Lazartigues, A., and Ben-Ari, Y. (2016). Treating Schizophrenia with the diuretic bumetanide: a case report. Clin. Neuropharmacol. 39, 115–117. doi: 10.1097/wnf.0000000000000136
Lemonnier, E., Villeneuve, N., Sonie, S., Serret, S., Rosier, A., Roue, M., et al. (2017). Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7:e1056. doi: 10.1038/tp.2017.10
Leonzino, M., Busnelli, M., Antonucci, F., Verderio, C., Mazzanti, M., and Chini, B. (2016). The Timing of the Excitatory-to-Inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 15, 96–103. doi: 10.1016/j.celrep.2016.03.013
Li, Y., Schaffner, A. E., Walton, M. K., and Barker, J. L. (1998). Astrocytes regulate developmental changes in the chloride ion gradient of embryonic rat ventral spinal cord neurons in culture. J. Physiol. 509, 847–858. doi: 10.1111/j.1469-7793.1998.847bm.x
Lim, L., Mi, D., Llorca, A., and Marín, O. (2018). Development and functional diversification of cortical interneurons. Neuron 100, 294–313. doi: 10.1016/j.neuron.2018.10.009
Liu, Q., Xu, Q., Arcuino, G., Kang, J., and Nedergaard, M. (2004). Astrocyte-mediated activation of neuronal kainate receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 3172–3177. doi: 10.1073/pnas.0306731101
Liu, Z., Neff, R. A., and Berg, D. K. (2006). Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science 314, 1610–1613. doi: 10.1126/science.1134246
LoTurco, J. J., Owens, D. F., Heath, M. J. S., Davis, M. B. E., and Kriegstein, A. R. (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298. doi: 10.1016/0896-6273(95)90008-x
Lu, J., Karadsheh, M., and Delpire, E. (1999). Developmental regulation of the neuronal-specific isoform of K-CL cotransporter KCC2 in postnatal rat brains. J. Neurobiol. 39, 558–568.
Ludwig, A., Li, H., Saarma, M., Kaila, K., and Rivera, C. (2003). Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur. J. Neurosci. 18, 3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x
Ludwig, A., Uvarov, P., Soni, S., Thomas-Crusells, J., Airaksinen, M. S., and Rivera, C. (2011). Early Growth Response 4 Mediates BDNF Induction of Potassium Chloride Cotransporter 2 Transcription. J. Neurosci. 31, 644–649. doi: 10.1523/jneurosci.2006-10.2011
Luhmann, H. J., Fukuda, A., and Kilb, W. (2015). Control of cortical neuronal migration by glutamate and GABA. Front. Cell. Neurosci. 9:4. doi: 10.3389/fncel.2015.00004
Macnamara, E. F., Koehler, A. E., D’Souza, P., Estwick, T., Lee, P., Vezina, G., et al. (2019). Kilquist syndrome: a novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum. Mutat. 40, 532–538. doi: 10.1002/humu.23722
MacVicar, B., Tse, F., Crichton, S., and Kettenmann, H. (1989). GABA-activated Cl− channels in astrocytes of hippocampal slices. J. Neurosci. 9, 3577–3583. doi: 10.1523/jneurosci.09-10-03577.1989
Magalhães, A. C., and Rivera, C. (2016). NKCC1-deficiency results in abnormal proliferation of neural progenitor cells of the lateral ganglionic eminence. Front. Cell. Neurosci. 10:200. doi: 10.3389/fncel.2016.00200
Mahadevan, V., Pressey, J. C., Acton, B. A., Uvarov, P., Huang, M. Y., Chevrier, J., et al. (2014). Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 7, 1762–1770. doi: 10.1016/j.celrep.2014.05.022
Marchese, M., Valvo, G., Moro, F., Sicca, F., and Santorelli, F. M. (2016). Targeted Gene Resequencing (Astrochip) to explore the tripartite synapse in autism–epilepsy phenotype with macrocephaly. Neuromol. Med. 18, 69–80. doi: 10.1007/s12017-015-8378-2
Maric, D., Liu, Q. Y., Maric, I., Chaudry, S., Chang, Y. H., Smith, S. V., et al. (2001). GABA expression dominates neuronal lineage progression in the embryonic rat neocortex and facilitates neurite outgrowth via GABA(A) autoreceptor/Cl− channels. J. Neurosci. 21, 2343–2360. doi: 10.1523/JNEUROSCI.21-07-02343.2001
Marín, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Mariotti, L., Losi, G., Lia, A., Melone, M., Chiavegato, A., Gómez-Gonzalo, M., et al. (2018). Interneuron-specific signaling evokes distinctive somatostatin-mediated responses in adult cortical astrocytes. Nat. Commun. 9:82. doi: 10.1038/s41467-017-02642-6
Markkanen, M., Ludwig, A., Khirug, S., Pryazhnikov, E., Soni, S., Khiroug, L., et al. (2017). Implications of the N-terminal heterogeneity for the neuronal K-Cl cotransporter KCC2 function. Brain Res. 1675, 87–101. doi: 10.1016/j.brainres.2017.08.034
Markkanen, M., Uvarov, P., and Airaksinen, M. S. (2008). Role of upstream stimulating factors in the transcriptional regulation of the neuron-specific K–Cl cotransporter KCC2. Brain Res. 1236, 8–15. doi: 10.1016/j.brainres.2008.08.007
Maset, A., Galla, L., Francia, S., Cozzolino, O., Capasso, P., Goisis, R. C., et al. (2021). Altered Cl− homeostasis hinders forebrain GABAergic interneuron migration in a mouse model of intellectual disability. Proc. Natl. Acad. Sci. U.S.A. 118:e2016034118. doi: 10.1073/pnas.2016034118
Matos, M., Bosson, A., Riebe, I., Reynell, C., Vallée, J., Laplante, I., et al. (2018). Astrocytes detect and upregulate transmission at inhibitory synapses of somatostatin interneurons onto pyramidal cells. Nat. Commun. 9:4254. doi: 10.1038/s41467-018-06731-y
McNeill, A., Iovino, E., Mansard, L., Vache, C., Baux, D., Bedoukian, E., et al. (2020). SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain 143, 2380–2387. doi: 10.1093/brain/awaa176
Mederos, S., and Perea, G. (2019). Monitoring interneuron-astrocyte signaling and its consequences on synaptic transmission. Methods Mol. Biol. 1938, 117–129. doi: 10.1007/978-1-4939-9068-9_9
Mederos, S., Sánchez-Puelles, C., Esparza, J., Valero, M., Ponomarenko, A., and Perea, G. (2021). GABAergic signaling to astrocytes in the prefrontal cortex sustains goal-directed behaviors. Nat. Neurosci. 24, 82–92. doi: 10.1038/s41593-020-00752-x
Menshanov, P. N., Lanshakov, D. A., and Dygalo, N. N. (2015). proBDNF is a Major product of bdnf Gene Expressed in the Perinatal Rat Cortex. Physiol. Res. 64, 925–934. doi: 10.33549/physiolres.932996
Merner, N. D., Chandler, M. R., Bourassa, C., Liang, B., Khanna, A. R., Dion, P., et al. (2015). Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci. 9:386. doi: 10.3389/fncel.2015.00386
Merner, N. D., Mercado, A., Khanna, A. R., Hodgkinson, A., Bruat, V., Awadalla, P., et al. (2016). Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J. Psychiatr. Res. 77, 22–26. doi: 10.1016/j.jpsychires.2016.02.016
Miller, S., and Maguire, J. (2014). Deficits in KCC2 and activation of the HPA axis lead to depressionlike behavior following social defeat. Horm. Stud. 2, 1–10. doi: 10.7243/2052-8000-2-2
Miraucourt, L. S., Tsui, J., Gobert, D., Desjardins, J.-F., Schohl, A., Sild, M., et al. (2016). Endocannabinoid signaling enhances visual responses through modulation of intracellular chloride levels in retinal ganglion cells. eLife 5:e15932. doi: 10.7554/elife.15932
Miyakawa, T., Leiter, L. M., Gerber, D. J., Gainetdinov, R. R., Sotnikova, T. D., Zeng, H., et al. (2003). Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 100, 8987–8992. doi: 10.1073/pnas.1432926100
Moore, Y. E., Conway, L. C., Wobst, H. J., Brandon, N. J., Deeb, T. Z., and Moss, S. J. (2019). Developmental Regulation of KCC2 phosphorylation has long-term impacts on cognitive function. Front. Mol. Neurosci. 12:173. doi: 10.3389/fnmol.2019.00173
Moore, Y. E., Deeb, T. Z., Chadchankar, H., Brandon, N. J., and Moss, S. J. (2018). Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. U.S.A. 115, 10166–10171. doi: 10.1073/pnas.1810134115
Moriguchi, T., Urushiyama, S., Hisamoto, N., Iemura, S., Uchida, S., Natsume, T., et al. (2005). WNK1 Regulates Phosphorylation of Cation-Chloride-coupled Cotransporters via the STE20-related Kinases, SPAK and OSR1. J. Biol. Chem. 280, 42685–42693. doi: 10.1074/jbc.m510042200
Morita, Y., Callicott, J. H., Testa, L. R., Mighdoll, M. I., Dickinson, D., Chen, Q., et al. (2014). Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and schizophrenia. J. Neurosci. 34, 4929–4940. doi: 10.1523/jneurosci.1423-13.2014
Mukhtarov, M., Ivanov, A., Zilberter, Y., and Bregestovski, P. (2011). Inhibition of spontaneous network activity in neonatal hippocampal slices by energy substrates is not correlated with intracellular acidification. J. Neurochem. 116, 316–321. doi: 10.1111/j.1471-4159.2010.07111.x
Murano, T., Hagihara, H., Tajinda, K., Matsumoto, M., and Miyakawa, T. (2019). Transcriptomic immaturity inducible by neural hyperexcitation is shared by multiple neuropsychiatric disorders. Commun. Biol. 2:32. doi: 10.1038/s42003-018-0277-2
Murata, Y., and Colonnese, M. T. (2020). GABAergic interneurons excite neonatal hippocampus in vivo. Sci. Adv. 6:eaba1430. doi: 10.1126/sciadv.aba1430
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Nesan, D., and Kurrasch, D. M. (2019). Gestational exposure to common endocrine disrupting chemicals and their impact on neurodevelopment and behavior. Annu. Rev. Physiol. 82, 177–202. doi: 10.1146/annurev-physiol-021119-034555
Nuñez, J. L., and McCarthy, M. M. (2007). Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus. Dev. Neurobiol. 67, 1879–1890. doi: 10.1002/dneu.20567
Oh, W. C., Lutzu, S., Castillo, P. E., and Kwon, H.-B. (2016). De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353, 1037–1040. doi: 10.1126/science.aaf5206
Ohira, K., Kobayashi, K., Toyama, K., Nakamura, H. K., Shoji, H., Takao, K., et al. (2013). Synaptosomal-associated protein 25 mutation induces immaturity of the dentate granule cells of adult mice. Mol. Brain 6:12. doi: 10.1186/1756-6606-6-12
Okada-Ogawa, A., Nakaya, Y., Imamura, Y., Kobayashi, M., Shinoda, M., Kita, K., et al. (2015). Involvement of medullary GABAergic system in extraterritorial neuropathic pain mechanisms associated with inferior alveolar nerve transection. Exp. Neurol. 267, 42–52. doi: 10.1016/j.expneurol.2015.02.030
Ouellet, L., and de Villers-Sidani, E. (2014). Trajectory of the main GABAergic interneuron populations from early development to old age in the rat primary auditory cortex. Front. Neuroanat. 8:40. doi: 10.3389/fnana.2014.00040
Ouyang, B., Chen, D., Hou, X., Wang, T., Wang, J., Zou, W., et al. (2019). Normalizing HDAC2 Levels in the Spinal Cord Alleviates Thermal and Mechanical Hyperalgesia After Peripheral Nerve Injury and Promotes GAD65 and KCC2 Expression. Front. Neurosci. 13:346. doi: 10.3389/fnins.2019.00346
Owens, D. F., Boyce, L. H., Davis, M. B. E., and Kriegstein, A. R. (1996). Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 16, 6414–6423. doi: 10.1523/jneurosci.16-20-06414.1996
Owens, D. F., Liu, X., and Kriegstein, A. R. (1999). Changing Properties of GABA A Receptor–mediated signaling during early neocortical development. J. Neurophysiol. 82, 570–583. doi: 10.1152/jn.1999.82.2.570
Pacheco-Alvarez, D., Cristóbal, P. S., Meade, P., Moreno, E., Vazquez, N., Muñoz, E., et al. (2006). The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 281, 28755–28763. doi: 10.1074/jbc.m603773200
Parrini, M., Naskar, S., Alberti, M., Colombi, I., Morelli, G., Rocchi, A., et al. (2021). Restoring neuronal chloride homeostasis with anti-NKCC1 gene therapy rescues cognitive deficits in a mouse model of Down syndrome. Mol. Ther. 29, 3072–3092. doi: 10.1016/j.ymthe.2021.05.023
Payne, J. A., Stevenson, T. J., and Donaldson, L. F. (1996). Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 271, 16245–16252. doi: 10.1074/jbc.271.27.16245
Pellegrino, C., Gubkina, O., Schaefer, M., Becq, H., Ludwig, A., Mukhtarov, M., et al. (2011). Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J. Physiol. 589, 2475–2496. doi: 10.1113/jphysiol.2010.203703
Petilla Interneuron Nomenclature Group, Ascoli, G. A., Alonso-Nanclares, L., Anderson, S. A., Barrionuevo, G., Benavides-Piccione, R., et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568. doi: 10.1038/nrn2402
Piala, A. T., Moon, T. M., Akella, R., He, H., Cobb, M. H., and Goldsmith, E. J. (2014). Chloride Sensing by WNK1 involves inhibition of autophosphorylation. Sci. Signal. 7:ra41. doi: 10.1126/scisignal.2005050
Pinto, J. G. A., Hornby, K. R., Jones, D. G., and Murphy, K. M. (2010). Developmental Changes in GABAergic mechanisms in human visual cortex across the lifespan. Front. Cell. Neurosci. 4:16. doi: 10.3389/fncel.2010.00016
Pisella, L. I., Gaiarsa, J.-L., Diabira, D., Zhang, J., Khalilov, I., Duan, J., et al. (2019). Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 12:eaay0300. doi: 10.1126/scisignal.aay0300
Plotkin, M. D., Snyder, E. Y., Hebert, S. C., and Delpire, E. (1997). Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J. Neurobiol. 33, 781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5
Pressey, J. C., Mahadevan, V., Khademullah, C. S., Dargaei, Z., Chevrier, J., Ye, W., et al. (2017). A kainate receptor subunit promotes the recycling of the neuron-specific K+-Cl− co-transporter KCC2 in hippocampal neurons. J. Biol. Chem. 292, 6190–6201. doi: 10.1074/jbc.m116.767236
Puskarjov, M., Ahmad, F., Khirug, S., Sivakumaran, S., Kaila, K., and Blaesse, P. (2015). BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology 88, 103–109. doi: 10.1016/j.neuropharm.2014.09.005
Puskarjov, M., Seja, P., Heron, S. E., Williams, T. C., Ahmad, F., Iona, X., et al. (2014). A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep. 15, 723–729. doi: 10.1002/embr.201438749
Rahmanzadeh, R., Shahbazi, A., Ardakani, M. K., Mehrabi, S., Rahmanzade, R., and Joghataei, M. T. (2017). Lack of the effect of bumetanide, a selective NKCC1 inhibitor, in patients with schizophrenia: a double-blind randomized trial. Psychiatry Clin. Neurosci. 71, 72–73. doi: 10.1111/pcn.12475
Ransom, C. B., Ransom, B. R., and Sontheimer, H. (2000). Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J. Physiol. 522, 427–442. doi: 10.1111/j.1469-7793.2000.00427.x
Riffault, B., Kourdougli, N., Dumon, C., Ferrand, N., Buhler, E., Schaller, F., et al. (2016). Pro-Brain-Derived Neurotrophic Factor (proBDNF)-Mediated p75NTR Activation Promotes Depolarizing Actions of GABA and Increases Susceptibility to Epileptic Seizures. Cereb. Cortex 28, 510–527. doi: 10.1093/cercor/bhw385
Rinehart, J., Maksimova, Y. D., Tanis, J. E., Stone, K. L., Hodson, C. A., Zhang, J., et al. (2009). Sites of Regulated Phosphorylation that Control K-Cl Cotransporter Activity. Cell 138, 525–536. doi: 10.1016/j.cell.2009.05.031
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., et al. (2002). BDNF-induced TrkB activation down-regulates the K+–Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 159, 747–752. doi: 10.1083/jcb.200209011
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipilä, S., et al. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl Cotransporter KCC2. J. Neurosci. 24, 4683–4691. doi: 10.1523/jneurosci.5265-03.2004
Robel, S., and Sontheimer, H. (2016). Glia as drivers of abnormal neuronal activity. Nat. Neurosci. 19, 28–33. doi: 10.1038/nn.4184
Roux, S., Lohof, A., Ben-Ari, Y., Poulain, B., and Bossu, J.-L. (2018). Maturation of GABAergic transmission in cerebellar purkinje cells is sex dependent and altered in the valproate model of autism. Front. Cell. Neurosci. 12:232. doi: 10.3389/fncel.2018.00232
Ruffolo, G., Iyer, A., Cifelli, P., Roseti, C., Mühlebner, A., van Scheppingen, J., et al. (2016). Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions. Neurobiol. Dis. 95, 93–101. doi: 10.1016/j.nbd.2016.07.014
Saito, T., Ishii, A., Sugai, K., Sasaki, M., and Hirose, S. (2017). A de novo missense mutation in SLC12A5 found in a compound heterozygote patient with epilepsy of infancy with migrating focal seizures. Clin. Genet. 92, 654–658. doi: 10.1111/cge.13049
Saitsu, H., Watanabe, M., Akita, T., Ohba, C., Sugai, K., Ong, W. P., et al. (2016). Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci. Rep. 6:30072. doi: 10.1038/srep30072
Salmon, C. K., Pribiag, H., Gizowski, C., Farmer, W. T., Cameron, S., Jones, E. V., et al. (2020). Depolarizing GABA transmission restrains activity-dependent glutamatergic synapse formation in the developing hippocampal circuit. Front. Cell. Neurosci. 14:36. doi: 10.3389/fncel.2020.00036
Sarkar, J., Wakefield, S., MacKenzie, G., Moss, S. J., and Maguire, J. (2011). Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J. Neurosci. 31, 18198–18210. doi: 10.1523/jneurosci.2560-11.2011
Sauer, J.-F., and Bartos, M. (2010). Recruitment of early postnatal parvalbumin-positive hippocampal interneurons by GABAergic Excitation. J. Neurosci. 30, 110–115. doi: 10.1523/jneurosci.4125-09.2010
Savardi, A., Borgogno, M., Narducci, R., Sala, G. L., Ortega, J. A., Summa, M., et al. (2020). Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem 6, 2073–2096. doi: 10.1016/j.chempr.2020.06.017
Sawada, T., Chater, T. E., Sasagawa, Y., Yoshimura, M., Fujimori-Tonou, N., Tanaka, K., et al. (2020). Developmental excitation-inhibition imbalance underlying psychoses revealed by single-cell analyses of discordant twins-derived cerebral organoids. Mol. Psychiatry 25, 2695–2711. doi: 10.1038/s41380-020-0844-z
Sedmak, G., Jovanov-Milošević, N., Puskarjov, M., Ulamec, M., Krušlin, B., Kaila, K., et al. (2015). Developmental expression patterns of KCC2 and functionally associated molecules in the human brain. Cereb. Cortex 26, 4574–4589. doi: 10.1093/cercor/bhv218
Sen, A., Martinian, L., Nikolic, M., Walker, M. C., Thom, M., and Sisodiya, S. M. (2007). Increased NKCC1 expression in refractory human epilepsy. Epilepsy Res. 74, 220–227. doi: 10.1016/j.eplepsyres.2007.01.004
Sernagor, E., Chabrol, F., Bony, G., and Cancedda, L. (2010). GABAergic control of neurite outgrowth and remodeling during development and adult neurogenesis: general rules and differences in diverse systems. Front. Cell. Neurosci. 4:11. doi: 10.3389/fncel.2010.00011
Sernagor, E., Young, C., and Eglen, S. J. (2003). Developmental modulation of retinal wave dynamics: shedding light on the GABA saga. J. Neurosci. 23, 7621–7629. doi: 10.1523/jneurosci.23-20-07621.2003
Serrano, A., Haddjeri, N., Lacaille, J.-C., and Robitaille, R. (2006). GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J. Neurosci. 26, 5370–5382. doi: 10.1523/jneurosci.5255-05.2006
Shekarabi, M., Girard, N., Rivière, J.-B., Dion, P., Houle, M., Toulouse, A., et al. (2008). Mutations in the nervous system–specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J. Clin. Invest. 118, 2496–2505. doi: 10.1172/jci34088
Shekarabi, M., Lafrenière, R. G., Gaudet, R., Laganière, J., Marcinkiewicz, M. M., Dion, P. A., et al. (2013). Comparative analysis of the expression profile of Wnk1 and Wnk1/Hsn2 Splice variants in developing and adult mouse tissues. PLoS One 8:e57807. doi: 10.1371/journal.pone.0057807
Shekarabi, M., Zhang, J., Khanna, A. R., Ellison, D. H., Delpire, E., and Kahle, K. T. (2017). WNK kinase signaling in ion homeostasis and human disease. Cell Metab. 25, 285–299. doi: 10.1016/j.cmet.2017.01.007
Shigetomi, E., Tong, X., Kwan, K. Y., Corey, D. P., and Khakh, B. S. (2012). TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 15, 70–80. doi: 10.1038/nn.3000
Shin, R., Kobayashi, K., Hagihara, H., Kogan, J. H., Miyake, S., Tajinda, K., et al. (2013). The immature dentate gyrus represents a shared phenotype of mouse models of epilepsy and psychiatric disease. Bipolar Disord. 15, 405–421. doi: 10.1111/bdi.12064
Shulga, A., Thomas-Crusells, J., Sigl, T., Blaesse, A., Mestres, P., Meyer, M., et al. (2008). Posttraumatic GABAA-Mediated [Ca2+]i Increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci. 28, 6996–7005. doi: 10.1523/jneurosci.5268-07.2008
Sid, B., Miranda, L., Vertommen, D., Viollet, B., and Rider, M. H. (2010). Stimulation of human and mouse erythrocyte Na+–K+–2Cl− cotransport by osmotic shrinkage does not involve AMP-activated protein kinase, but is associated with STE20/SPS1-related proline/alanine-rich kinase activation. J. Physiol. 588, 2315–2328. doi: 10.1113/jphysiol.2009.185900
Silayeva, L., Deeb, T. Z., Hines, R. M., Kelley, M. R., Munoz, M. B., Lee, H. H. C., et al. (2015). KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U.S.A. 112, 3523–3528. doi: 10.1073/pnas.1415126112
Sipila, S. T., Huttu, K., Yamada, J., Afzalov, R., Voipio, J., Blaesse, P., et al. (2009). Compensatory Enhancement of Intrinsic Spiking upon NKCC1 Disruption in Neonatal Hippocampus. J. Neurosci. 29, 6982–6988. doi: 10.1523/jneurosci.0443-09.2009
Soul, J. S., Bergin, A. M., Stopp, C., Hayes, B., Singh, A., Fortuno, C. R., et al. (2021). A pilot randomized, controlled, double-blind trial of bumetanide to treat neonatal seizures. Ann. Neurol. 89, 327–340. doi: 10.1002/ana.25959
Spoljaric, A., Seja, P., Spoljaric, I., Virtanen, M. A., Lindfors, J., Uvarov, P., et al. (2017). Vasopressin excites interneurons to suppress hippocampal network activity across a broad span of brain maturity at birth. Proc. Natl. Acad. Sci. U.S.A. 114, E10819–E10828. doi: 10.1073/pnas.1717337114
Sprengers, J. J., van Andel, D. M., Zuithoff, N. P. A., Keijzer-Veen, M. G., Schulp, A. J. A., Scheepers, F. E., et al. (2020). Bumetanide for Core Symptoms of Autism Spectrum Disorder (BAMBI): a single center, double-blinded, participant-randomized, placebo-controlled, phase-2 superiority trial. J. Am. Acad. Child Adolesc. Psychiatry 60, 865–876. doi: 10.1016/j.jaac.2020.07.888
Stödberg, T., Magnusson, M., Lesko, N., Wredenberg, A., Munoz, D. M., Stranneheim, H., et al. (2020). SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol. Genet. 6:e478. doi: 10.1212/nxg.0000000000000478
Stödberg, T., McTague, A., Ruiz, A. J., Hirata, H., Zhen, J., Long, P., et al. (2015). Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 6:8038. doi: 10.1038/ncomms9038
Strange, K., Singer, T. D., Morrison, R., and Delpire, E. (2000). Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am. J. Physiol. Cell Physiol. 279, C860–C867. doi: 10.1152/ajpcell.2000.279.3.c860
Su, G., Kintner, D. B., Flagella, M., Shull, G. E., and Sun, D. (2002). Astrocytes from Na+-K+-Cl-cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol. 282, C1147–C1160. doi: 10.1152/ajpcell.00538.2001
Sullivan, C. R., Funk, A. J., Shan, D., Haroutunian, V., and McCullumsmith, R. E. (2015). Decreased chloride channel expression in the dorsolateral prefrontal cortex in schizophrenia. PLoS One 10:e0123158. doi: 10.1371/journal.pone.0123158
Sun, L., Han, X., and He, S. (2011). Direction-selective circuitry in rat retina develops independently of GABAergic, cholinergic and action potential activity. PLoS One 6:e19477. doi: 10.1371/journal.pone.0019477
Takao, K., Kobayashi, K., Hagihara, H., Ohira, K., Shoji, H., Hattori, S., et al. (2013). Deficiency of Schnurri-2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology 38, 1409–1425. doi: 10.1038/npp.2013.38
Talos, D. M., Sun, H., Kosaras, B., Joseph, A., Folkerth, R. D., Poduri, A., et al. (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. doi: 10.1002/ana.22696
Tang, X., Drotar, J., Li, K., Clairmont, C. D., Brumm, A. S., Sullins, A. J., et al. (2019). Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 11:eaau0164. doi: 10.1126/scitranslmed.aau0164
Tang, X., Kim, J., Zhou, L., Wengert, E., Zhang, L., Wu, Z., et al. (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 751–756. doi: 10.1073/pnas.1524013113
Tao, R., Li, C., Newburn, E. N., Ye, T., Lipska, B. K., Herman, M. M., et al. (2012). Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J. Neurosci. 32, 5216–5222. doi: 10.1523/jneurosci.4626-11.2012
Till, Á, Szalai, R., Hegyi, M., Kövesdi, E., Büki, G., Hadzsiev, K., et al. (2019). [A rare form of ion channel gene mutation identified as underlying cause of generalized epilepsy]. Orv. Hetil. 160, 835–838. doi: 10.1556/650.2019.31404
Tsuruga, K., Hashimoto, T., Kato, R., Kato, R., Uchida, Y., Hase, T., et al. (2016). Plantar injection of formalin in rats reduces the expression of a potassium chroride cotransporter KCC2 in the spinal cord and a kinase inhibitor suppresses this reduction. Biomed. Res. 37, 243–249. doi: 10.2220/biomedres.37.243
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hübner, C. A., Represa, A., et al. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792. doi: 10.1126/science.1133212
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Aniksztejn, L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19, 10372–10382. doi: 10.1523/JNEUROSCI.19-23-10372.1999
Untiet, V., Kovermann, P., Gerkau, N. J., Gensch, T., Rose, C. R., and Fahlke, C. (2017). Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia 65, 388–400. doi: 10.1002/glia.23098
Uvarov, P., Ludwig, A., Markkanen, M., Pruunsild, P., Kaila, K., Delpire, E., et al. (2007). A Novel N-terminal Isoform of the Neuron-specific K-Cl Cotransporter KCC2. J. Biol. Chem. 282, 30570–30576. doi: 10.1074/jbc.m705095200
Uvarov, P., Ludwig, A., Markkanen, M., Rivera, C., and Airaksinen, M. S. (2006). Upregulation of the Neuron-Specific K+/Cl− Cotransporter Expression by Transcription Factor Early Growth Response 4. J. Neurosci. 26, 13463–13473. doi: 10.1523/jneurosci.4731-06.2006
Uvarov, P., Pruunsild, P., Timmusk, T., and Airaksinen, M. S. (2005). Neuronal K+/Cl− co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J. Neurochem. 95, 1144–1155. doi: 10.1111/j.1471-4159.2005.03434.x
Valeeva, G., Tressard, T., Mukhtarov, M., Baude, A., and Khazipov, R. (2016). An Optogenetic Approach for Investigation of Excitatory and Inhibitory Network GABA Actions in Mice Expressing Channelrhodopsin-2 in GABAergic Neurons. J. Neurosci. 36, 5961–5973. doi: 10.1523/jneurosci.3482-15.2016
Virtanen, M. A., Uvarov, P., Hübner, C. A., and Kaila, K. (2020). NKCC1, an elusive molecular target in brain development: making sense of the existing data. Cells 9:2607. doi: 10.3390/cells9122607
Vitari, A. C., Thastrup, J., Rafiqi, F. H., Deak, M., Morrice, N. A., Karlsson, H. K. R., et al. (2006). Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem. J. 397, 223–231. doi: 10.1042/bj20060220
Vu, T. Q., Payne, J. A., and Copenhagen, D. R. (2000). Localization and developmental expression patterns of the neuronal K-Cl cotransporter (KCC2) in the rat retina. J. Neurosci. 20, 1414–1423. doi: 10.1523/JNEUROSCI.20-04-01414.2000
Wafford, K. A., Bain, C. J., Whiting, P. J., and Kemp, J. A. (1993). Functional comparison of the role of gamma subunits in recombinant human gamma-aminobutyric acidA/benzodiazepine receptors. Mol. Pharmacol. 44, 437–442.
Wake, H., Watanabe, M., Moorhouse, A. J., Kanematsu, T., Horibe, S., Matsukawa, N., et al. (2007). Early Changes in KCC2 Phosphorylation in Response to Neuronal Stress Result in Functional Downregulation. J. Neurosci. 27, 1642–1650. doi: 10.1523/jneurosci.3104-06.2007
Walton, N. M., Zhou, Y., Kogan, J. H., Shin, R., Webster, M., Gross, A. K., et al. (2012). Detection of an immature dentate gyrus feature in human schizophrenia/bipolar patients. Transl. Psychiatry 2:e135. doi: 10.1038/tp.2012.56
Wang, D. D., and Kriegstein, A. R. (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28, 5547–5558. doi: 10.1523/jneurosci.5599-07.2008
Wang, D. D., and Kriegstein, A. R. (2011). Blocking Early GABA Depolarization with Bumetanide Results in Permanent Alterations in Cortical Circuits and Sensorimotor Gating Deficits. Cereb. Cortex 21, 574–587. doi: 10.1093/cercor/bhq124
Wang, F., Smith, N. A., Xu, Q., Fujita, T., Baba, A., Matsuda, T., et al. (2012). Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci. Signal. 5:ra26. doi: 10.1126/scisignal.2002334
Watanabe, M., Wake, H., Moorhouse, A. J., and Nabekura, J. (2009). Clustering of Neuronal K+-Cl− Cotransporters in Lipid Rafts by Tyrosine Phosphorylation. J. Biol. Chem. 284, 27980–27988. doi: 10.1074/jbc.m109.043620
Watanabe, M., Zhang, J., Mansuri, M. S., Duan, J., Karimy, J. K., Delpire, E., et al. (2019). Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 12:eaaw9315. doi: 10.1126/scisignal.aaw9315
Weber, M., Hartmann, A.-M., Beyer, T., Ripperger, A., and Nothwang, H. G. (2014). A Novel Regulatory Locus of Phosphorylation in the C Terminus of the Potassium Chloride Cotransporter KCC2 That Interferes with N-Ethylmaleimide or Staurosporine-mediated Activation. J. Biol. Chem. 289, 18668–18679. doi: 10.1074/jbc.m114.567834
Wells, J. E., Porter, J. T., and Agmon, A. (2000). GABAergic inhibition suppresses paroxysmal network activity in the neonatal rodent hippocampus and neocortex. J. Neurosci. 20, 8822–8830. doi: 10.1523/jneurosci.20-23-08822.2000
Winnubst, J., Cheyne, J. E., Niculescu, D., and Lohmann, C. (2015). Spontaneous activity drives local synaptic plasticity in vivo. Neuron 87, 399–410. doi: 10.1016/j.neuron.2015.06.029
Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J., and Fukuda, A. (2004). Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 557, 829–841. doi: 10.1113/jphysiol.2004.062471
Yamasaki, N., Maekawa, M., Kobayashi, K., Kajii, Y., Maeda, J., Soma, M., et al. (2008). Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol. Brain 1:6. doi: 10.1186/1756-6606-1-6
Yan, Y., Dempsey, R. J., and Sun, D. (2001). Na+-K+-Cl− Cotransporter in Rat Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 21, 711–721. doi: 10.1097/00004647-200106000-00009
Yang, J., Harte-Hargrove, L. C., Siao, C.-J., Marinic, T., Clarke, R., Ma, Q., et al. (2014). proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 7, 796–806. doi: 10.1016/j.celrep.2014.03.040
Yeo, M., Berglund, K., Augustine, G., and Liedtke, W. (2009). Novel Repression of Kcc2 Transcription by REST–RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 29, 14652–14662. doi: 10.1523/jneurosci.2934-09.2009
Yeo, M., Berglund, K., Hanna, M., Guo, J. U., Kittur, J., Torres, M. D., et al. (2013). Bisphenol A delays the perinatal chloride shift in cortical neurons by epigenetic effects on the Kcc2 promoter. Proc. Natl. Acad. Sci. U.S.A. 110, 4315–4320. doi: 10.1073/pnas.1300959110
Young, S. Z., Taylor, M. M., Wu, S., Ikeda-Matsuo, Y., Kubera, C., and Bordey, A. (2012). NKCC1 knockdown decreases neuron production through GABAA-regulated neural progenitor proliferation and delays dendrite development. J. Neurosci. 32, 13630–13638. doi: 10.1523/jneurosci.2864-12.2012
Yu, X., Taylor, A. M. W., Nagai, J., Golshani, P., Evans, C. J., Coppola, G., et al. (2018). Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187.e9. doi: 10.1016/j.neuron.2018.08.015
Zafeiriou, M.-P., Bao, G., Hudson, J., Halder, R., Blenkle, A., Schreiber, M.-K., et al. (2020). Developmental GABA polarity switch and neuronal plasticity in Bioengineered Neuronal Organoids. Nat. Commun. 11:3791. doi: 10.1038/s41467-020-17521-w
Zhang, L., Huang, C.-C., Dai, Y., Luo, Q., Ji, Y., Wang, K., et al. (2020). Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 10:9. doi: 10.1038/s41398-020-0692-2
Zhang, L.-L., Pathak, H. R., Coulter, D. A., Freed, M. A., and Vardi, N. (2005). Shift of intracellular chloride concentration in ganglion and amacrine cells of developing mouse retina. J. Neurophysiol. 95, 2404–2416. doi: 10.1152/jn.00578.2005
Zhao, X.-F., Kohen, R., Parent, R., Duan, Y., Fisher, G. L., Korn, M. J., et al. (2018). PlexinA2 Forward Signaling through Rap1 GTPases regulates dentate gyrus development and schizophrenia-like behaviors. Cell Rep. 22, 456–470. doi: 10.1016/j.celrep.2017.12.044
Zhu, L., Lovinger, D., and Delpire, E. (2005). Cortical Neurons Lacking KCC2 expression show impaired regulation of intracellular chloride. J. Neurophysiol. 93, 1557–1568. doi: 10.1152/jn.00616.2004
Keywords: potassium chloride cotransporter-2 (KCC2), Na+-K+-2Cl– cotransporter-1 (NKCC1), neuropsychiatric disorders (NPD), neurodevelopmental disorders (NDD), chloride homeostasis, GABA-shift
Citation: Hui KK, Chater TE, Goda Y and Tanaka M (2022) How Staying Negative Is Good for the (Adult) Brain: Maintaining Chloride Homeostasis and the GABA-Shift in Neurological Disorders. Front. Mol. Neurosci. 15:893111. doi: 10.3389/fnmol.2022.893111
Received: 10 March 2022; Accepted: 10 June 2022;
Published: 08 July 2022.
Edited by:
Werner Kilb, Johannes Gutenberg University Mainz, GermanyReviewed by:
Kiyoshi Egawa, Hokkaido University, JapanCopyright © 2022 Hui, Chater, Goda and Tanaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelvin K. Hui, a2VsdmluLmh1aUBhbHVtLnV0b3JvbnRvLmNh; Thomas E. Chater, dGhvbWFzLmNoYXRlckByaWtlbi5qcA==
†ORCID: Kelvin K. Hui, orcid.org/0000-0003-2699-4536; Thomas E. Chater, orcid.org/0000-0001-6133-0001; Yukiko Goda, orcid.org/0000-0003-0352-9498; Motomasa Tanaka, orcid.org/0000-0002-2994-7703
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.