 Bianka A. Holguin
Bianka A. Holguin Zacariah L. Hildenbrand
Zacariah L. Hildenbrand Ricardo A. Bernal
Ricardo A. Bernal- Department of Chemistry and Biochemistry, University of Texas at El Paso, El Paso, TX, United States
Small heat shock protein 27 is a critically important chaperone, that plays a key role in several essential and varied physiological processes. These include thermotolerance, apoptosis, cytoskeletal dynamics, cell differentiation, protein folding, among others. Despite its relatively small size and intrinsically disordered termini, it forms large and polydisperse oligomers that are in equilibrium with dimers. This equilibrium is driven by transient interactions between the N-terminal region, the α-crystallin domain, and the C-terminal region. The continuous redistribution of binding partners results in a conformationally dynamic protein that allows it to adapt to different functions where substrate capture is required. However, the intrinsic disorder of the amino and carboxy terminal regions and subsequent conformational variability has made structural investigations challenging. Because heat shock protein 27 is critical for so many key cellular functions, it is not surprising that it also has been linked to human disease. Charcot-Marie-Tooth and distal hereditary motor neuropathy are examples of neurodegenerative disorders that arise from single point mutations in heat shock protein 27. The development of possible treatments, however, depends on our understanding of its normal function at the molecular level so we might be able to understand how mutations manifest as disease. This review will summarize recent reports describing investigations into the structurally elusive regions of Hsp27. Recent insights begin to provide the required context to explain the relationship between a mutation and the resulting loss or gain of function that leads to Charcot-Marie Tooth disease and distal hereditary motor neuropathy.
Introduction
The proteome is a vast collection of proteins each of which contributes to the maintenance of cellular homeostasis (Kim et al., 2013; Bakthisaran et al., 2015; Webster et al., 2019). Diverse environmental and physiological stressors can lead to various deleterious effects including structural damage of proteins (Sun and MacRae, 2005; Kim et al., 2013; Webster et al., 2019). With so many threats overwhelming the integrity of the proteome it is imperative that the cell ensures a robust defense mechanism to keep its proteins structurally viable and functional (Eyles and Gierasch, 2010; Kim et al., 2013). Cellular stressors such as heat shock can often result in partial protein misfolding that can then lead to various disorders including neurodegeneration (Eyles and Gierasch, 2010; Webster et al., 2019). The integrity of protein structure and function is maintained by a class of proteins known as molecular chaperones (Sun and MacRae, 2005; Bhatt et al., 2018; Rodriguez et al., 2020). These molecular chaperones sustain protein homeostasis through assistance with protein folding or the stabilization of partially misfolded proteins (Sun and MacRae, 2005; Kim et al., 2013). A subclass of molecular chaperones, known as heat shock proteins, can assist in the folding of nascent polypeptides or aid in the stabilization and the refolding of non-native proteins (van Montfort et al., 2001; Fu, 2014; Enriquez et al., 2017; Bhatt et al., 2018). They are also involved in other pathways such as degradation of irreversibly damaged proteins and unfolding of other proteins for translocation across a membrane (Haslbeck et al., 1999; Shashidharamurthy et al., 2005; Richter et al., 2010).
Heat shock proteins are broadly conserved amongst six major classes. These include Hsp100, Hsp90, Hsp70, Hsp60, Hsp40, and the small heat shock proteins (Sun and MacRae, 2005; Richter et al., 2010; Fu, 2014; Bakthisaran et al., 2015; Enriquez et al., 2017; Rodriguez et al., 2020). Small heat shock proteins (sHsps) are molecular chaperones that do not require the hydrolysis of ATP (Bova et al., 2002; Sobott et al., 2002). Their primary role is to maintain a partially denatured protein in a folding-competent state to protect it from irreversible and detrimental aggregation (Borrelli et al., 2002; Bakthisaran et al., 2015). Small heat shock protein 27 is a ubiquitously expressed chaperone involved in many physiological and metabolic pathways (Parcellier et al., 2005). In addition to protein folding, it has been implicated in the regulation of redox states, stabilization of the cytoskeleton, anti-apoptotic activity, and in important interactions with transcription factors and mRNA processing proteins (D’Ydewalle et al., 2011; Chalova et al., 2014; Fu, 2014). Hsp27 (HspB1) belongs to the subgroup of small heat shock proteins that encompass 10 human sHsps. These small heat shock proteins are encoded by the genes HspB1–10 and have been categorized into two main classes (Bakthisaran et al., 2015; Muranova et al., 2015; Webster et al., 2019). Class I sHsp include HspB1 (Hsp27), HspB5 (α-B-crystallin), HspB6 (Hsp20), and HspB8 (Hsp22; Kappe et al., 2003; Bakthisaran et al., 2015). These sHsps are widely distributed with expression in nearly all cells and tissues where they perform various roles (Kappe et al., 2003; Mainz et al., 2015; Mymrikov et al., 2017). The second class of small heat shock proteins includes HspB2, HspB3, HspB4 (αA-crystallin), HspB7 (cvHsp), HspB9, and HspB10 (ODF1) and are restricted to certain types of tissues such as the eyes, heart, and testes (Kappe et al., 2003; Bakthisaran et al., 2015).
Small heat shock proteins are characterized by a conserved sequence of approximately 80–100 residues known as the α-crystallin domain (ACD; Fu, 2014; Bakthisaran et al., 2015; Muranova et al., 2015; Clouser et al., 2019). The ACD is flanked by a variable hydrophobic amino terminal region (NTR) and a short flexible c-terminal region (CTR; Garrido et al., 2012; Hochberg et al., 2014; Rajagopal et al., 2015; Figure 1). Small heat shock proteins form dimers via β-sheets formed by the ACD. These dimers serve as building blocks of class 1 sHsp that organize into large oligomeric complexes that are stabilized by their N-terminal and C-terminal regions (Lelj-Garolla and Mauk, 2012; McDonald et al., 2012; Aquilina et al., 2013; Rajagopal et al., 2015). The dimers are thought to be the smallest functional form of small heat shock proteins (Lambert et al., 1999; Hayes et al., 2009). These complexes engage in rapid dimer exchange creating a dynamic equilibrium between large multimers of various sizes and dimers (Bova et al., 2000, 2002; Sobott et al., 2002; Rajagopal et al., 2015; Figure 2). The stress pathway activates p38 mitogen-activated protein kinase, which in turn phosphorylates mitogen-activated protein kinase activated protein kinase (MAPKAPK2; Kostenko and Moens, 2009; Bakthisaran et al., 2015; Lang et al., 2021). Phosphorylation by MAPKAPK2 shifts the equilibrium to small oligomers and dimers whereas protein phosphatase 2A activity reverses this process and favors the formation of large multimers (Cairns et al., 1994; Kostenko and Moens, 2009; Katsogiannou et al., 2014; Bakthisaran et al., 2015; Figure 2). This system might be a way to store large amounts of Hsp27 to quickly react to stress without the need for de novo protein synthesis (Peschek et al., 2013).
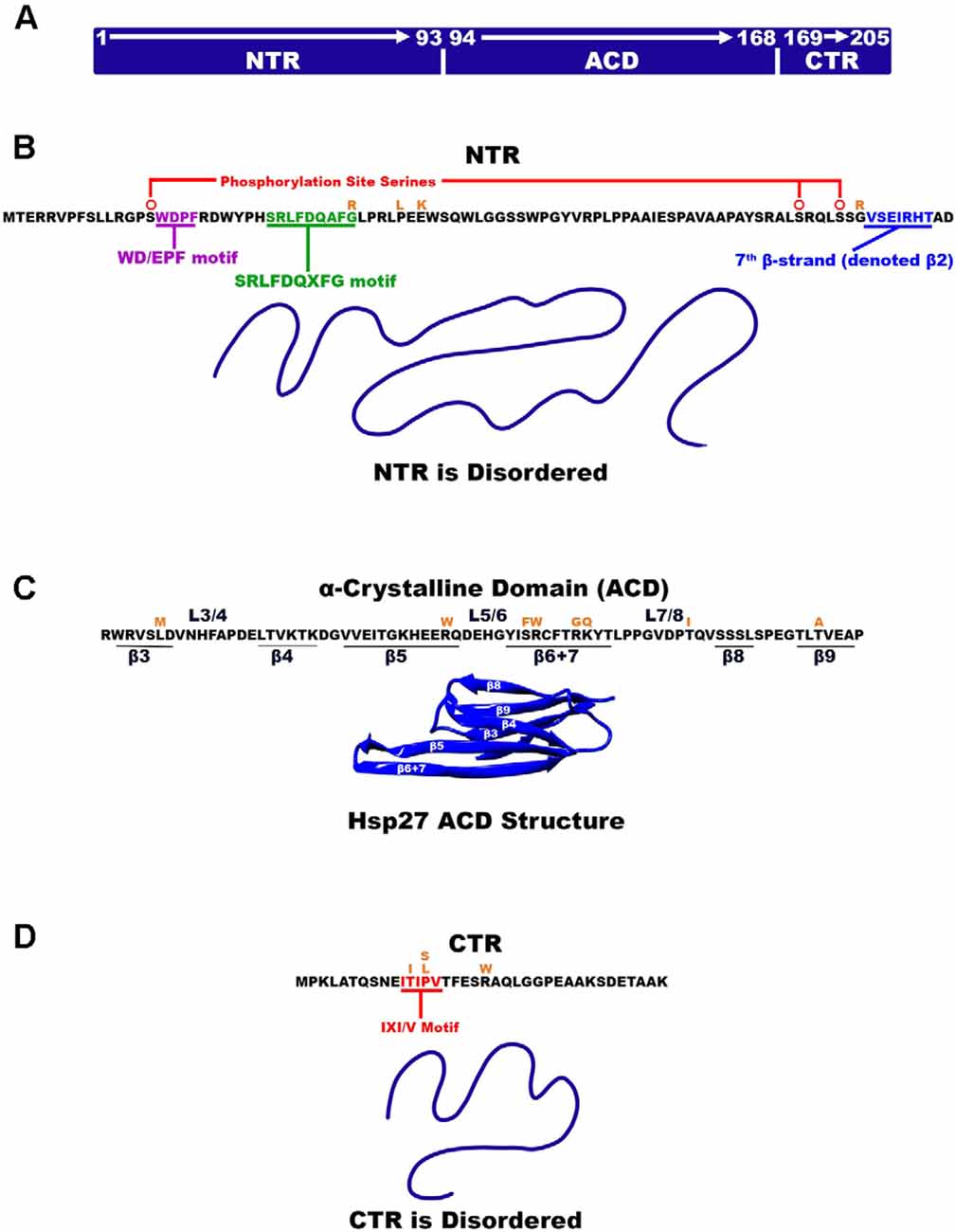
Figure 1. Hsp27 Structural organization. (A) Hsp27 is made up of three regions where each region is identified as NTR, ACD, and CTR. The residue numbers that make up each region are also denoted. (B) The amino acid sequence of the disordered NTR is listed and the two motifs WD/EPF and SRLFDQxFG are highlighted with red and green, respectively. β-strand β-2 is highlighted in cyan. (C) The ACD amino acid sequence is listed as well as the location of β-strands and their connecting loops. The ACD is the only part of Hsp27 that is ordered (PDB 2N3J). (D) Hsp27 disordered CTR highlighting the IXI/V motif in red. Amino acid mutations are illustrated in orange above the amino acid being mutated.
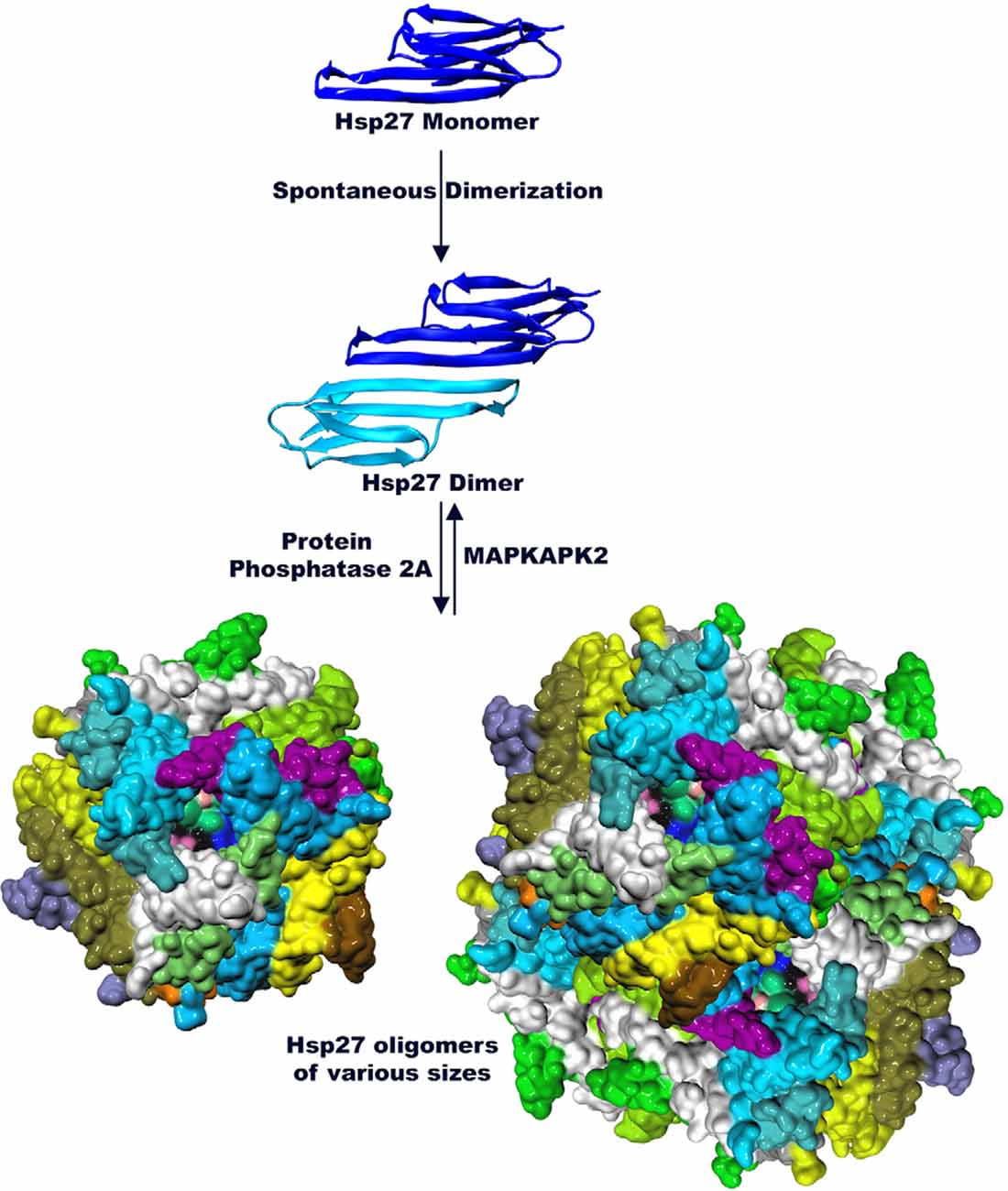
Figure 2. Hsp27 is organized into higher order structures (PDB 6DV5 and 2N3J). The monomer of Hsp27 assembles into dimers via the ACD. Dimers in turn interact with each other to form large highly dynamic multimers. Phosphorylation by MAPKAPK2 dissociates oligomers of Hsp27 and favors dimer formation while Protein Phosphatase 2A shifts the equilibrium towards multimer formation.
Small heat shock proteins can be perturbed by a myriad of inherited and sporadic mutations that result in the formation of neurological disorders. For example, various missense mutations in the small heat shock protein 27 (Hsp27) cause the neuropathies Charcot-Marie-Tooth Disease Axonal Type 2F (CMT2F) and Distal Hereditary Motor Neuropathy 2B (dHMN2B; Houlden et al., 2008; Tanabe et al., 2018). These neuropathies are commonly inherited disorders that exclusively affect the peripheral nervous system (Holmgren et al., 2013). They are closely related in their key features of disease progression and only differ by the point mutation that leads to the respective disease. CMT2F is a subset of the more broadly categorized forms of CMT, specifically characterized by axonal degeneration of the peripheral nerves (Ismailov et al., 2001; Schwartz, 2019). The characteristic symptom of CMT2F includes the slow progressive loss of motor function manifested as weakness and muscle atrophy of the distal limbs (Evgrafov et al., 2004; Gentile et al., 2020). Additionally, there can be decreased tendon reflexes of the ankle and legs, as well as physical deformities of the feet (Webster et al., 2019; Gentile et al., 2020). In cases of sensory neuron involvement, afflicted individuals lose pain and temperature sensation (Szigeti and Lupski, 2009). The disease, although progressive over many years, does not affect the life span (Schwartz, 2019). Individuals with CMT2F typically experience the onset of symptoms during adulthood and may require treatment through physical therapy, mobility aids, orthopedic shoes, and/or surgery (Evgrafov et al., 2004). More severe forms of CMT2F can result in hearing loss and vocal cord impairment, in addition to acute distal limb pain (Bakthisaran et al., 2015).
While CMT2F manifests as a motor and sensory neuropathy, Distal Hereditary Motor Neuropathy Type II (dHMNIIB) exclusively affects motor neurons (Geuens et al., 2017; Schwartz, 2019). dHMNIIB clinically presents similarly to CMT2F as they are related diseases caused by some of the same mutations (Tanabe et al., 2018). Individuals experience lower limb weakness due to muscle loss and deteriorating leg mobility (Dierick et al., 2007; Chung et al., 2008; Tanabe et al., 2018). The typical onset of the disease occurs in individuals between the ages of 15 and 45 and is a progressive disease that severely compromises the quality of life (Dierick et al., 2007; Chung et al., 2008; Schwartz, 2019). Both CMT2F and dHMNIIB have autosomal dominant patterns of inheritance indicating that only one copy of a mutated gene is sufficient to cause disease (Evgrafov et al., 2004; Schwartz, 2019). These neuropathies are described as being clinically and genetically heterogenous (Ackerley et al., 2006; Dierick et al., 2007; Echaniz-Laguna, 2015; Tanabe et al., 2018). This means that the same mutation can result in different clinical presentations and different mutations can manifest as the same disease, further complicating the understanding of the pathomechanisms. There are many mutations in Hsp27 that have been implicated in hereditary neuropathies (Muranova et al., 2019; Vendredy et al., 2020). A number of these mutations result in either CMT2F, dHMN2B, or both. There have been several reports linking mutated Hsp27 to disturbances in the cytoskeletal components of axons (Almeida-Souza et al., 2011; D’Ydewalle et al., 2011; Nefedova et al., 2017). Several substrates of Hsp27, such as Tau and α-synuclein, are involved in stabilizing cytoskeletal components and vesicle trafficking (Abisambra et al., 2010; Barbier et al., 2019; Baughman et al., 2020). These are processes that are required for normal motor neuron functioning (Abisambra et al., 2010; Barbier et al., 2019). These relationships have been implicated as indirect causes for many neurodegenerative diseases including CMT2F and dHMN2B (Webster et al., 2019; Muranova et al., 2020; Vendredy et al., 2020).
Hsp27 has been suspected in the progression of other neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease (Zhang et al., 2014; Hu et al., 2021). They are the top two most common neurological illnesses affecting roughly 12% of people over 60 years of age (Hu et al., 2021). Alzheimer’s disease (AD) is also a progressive neurodegenerative disorder localized to the brain, mainly affecting neurons of the cortex (Nussbaum and Ellis, 2003). The classical manifestations of Alzheimer’s are an accumulation of amyloid-β plaques and neurofibrillary tangles of hyperphosphorylated tau (Nussbaum and Ellis, 2003). Alzheimer’s is the number one cause of dementia and severely diminishes the quality of life of those affected. Parkinson’s disease (PD) results in the irreversible degeneration of dopaminergic neurons in the substantia nigra and includes the aggregation of α-synuclein contained within Lewy bodies and neuritis (Nussbaum and Ellis, 2003; Poewe et al., 2017). Parkinson’s disease, like Alzheimer’s, can also cause dementia in addition to the classical parkinsonian symptoms including tremors, rigidity, and bradykinesia (Williams and Litvan, 2013). Recently increased attention has targeted the involvement of Hsp27 in the progression of these neurological diseases from a therapeutic standpoint (Venugopal et al., 2019; Baughman et al., 2020; Vicente Miranda et al., 2020; Navarro-Zaragoza et al., 2021). The substrates of Hsp27 such as Tau and α-synuclein are major components in the pathological progression of both AD and PD. Although there is no evidence to suggest that mutations of Hsp27 are linked to AD and PD, the faulty chaperone may contribute to disease by ineffective regulation of these substrates.
Features of the Small Heat Shock Protein 27 Regions
Three major structural regions of Hsp27 can be defined by the structural and functional distinctions between them. The N-terminal region (NTR) of Hsp27 is composed of the first 93 residues and nearly 50% of the Hsp27 primary sequence (Clouser et al., 2019; Figures 1A,B). The NTR is inherently disordered and composed of predominantly hydrophobic and aromatic residues. It is thought to be primarily responsible for driving/stabilizing higher order multimers of Hsp27 (Bakthisaran et al., 2015). A less conserved region within the NTR is the WD/EPF motif made of residues 16-WDPF-19 that have been shown to correlate with protein chaperoning activity (Thériault et al., 2004; Lelj-Garolla and Mauk, 2012). Hsp27 is phosphorylated via the p38 MAPK pathway by MAPKAPK2, which occurs at serine residues 15, 78, 82 of the NTR (Lambert et al., 1999; Shashidharamurthy et al., 2005; Figure 1B). The phosphorylation site at serine-15 precedes the NTR WD/EPF domain while phosphorylation sites 78 and 82 reside near the end of the NTR (Lelj-Garolla and Mauk, 2012). In its unphosphorylated state Hsp27 exists as a large polydisperse oligomeric species with a molecular weight of up to ~650 kDa (Rogalla et al., 1999). Experimental evidence has demonstrated that tunability of Hsp27 oligomeric size can be achieved through stepwise phosphorylation of the three phosphorylation sites (Hayes et al., 2009; Jovcevski et al., 2015). Phosphorylation of all three sites yields smaller oligomers and dimers whereas phosphorylation of one or two sites results in intermediately sized oligomers (Hayes et al., 2009; Jovcevski et al., 2015). At residue positions, 26–34 and near the center of the NTR is the conserved SRLFDQxFG motif (Pasta et al., 2003). Truncation studies have confirmed that this motif plays a large part in the production and stability of higher order multimers (Pasta et al., 2003). The NTR appears to be a highly flexible region that can adopt several conformations to facilitate oligomerization.
The Hsp27 α crystalline domain (ACD) is comprised of residues 94–168 that are arranged into six β-strands that form two β-sheets (Baranova et al., 2011; Alderson et al., 2019; Nappi et al., 2020; Figure 1C). This immunoglobulin-like fold of the ACD is conserved amongst various organisms in both sequence and structure (Weeks et al., 2018; Clouser et al., 2019). The β-strands denoted as 4, 5, and extended strands 6 + 7 make up one β-sheet, whereas β-strands 3, 8, and 9 form another β-sheet (Hochberg et al., 2014; Klevit, 2020). Residues on the carboxy-terminal side of the NTR (84–91) periodically form a seventh β-strand (denoted β2) that integrates into the β3/β8/β9 β-sheet (Collier et al., 2019; Klevit, 2020; Figure 1B). The driving force for Hsp27 dimerization is hydrogen bonding and electrostatic interactions between ACDs. This occurs by pairwise alignment of the extended β6 + 7 strands in an antiparallel 2 register (AP2) creating the dimer interface and an extended β-sheet (Aquilina et al., 2013; Rajagopal et al., 2015; Clouser et al., 2019; Boelens, 2020; Figure 3A). The extended β-sheet is the sum of two β6 + 7 containing β-sheets forming one longer extended symmetry related β-sheet. The extended β-sheet and two β3/β8/β9 β-sheets from each monomer create a groove that is located right above the dimer interface and between the two β3/β8/β9 β-sheets, therein described as the dimer interface groove (Klevit, 2020; Figure 3B). In total, 18 residues with their side chains pointing inward, create the dimer interface groove. Eight of these residues are from each β6 + 7 strand and 10 are from loop regions that connect the β-strands 3 and 4 (L3/4) and β-strands 5 and 6 + 7 (L5/6; Klevit, 2020).
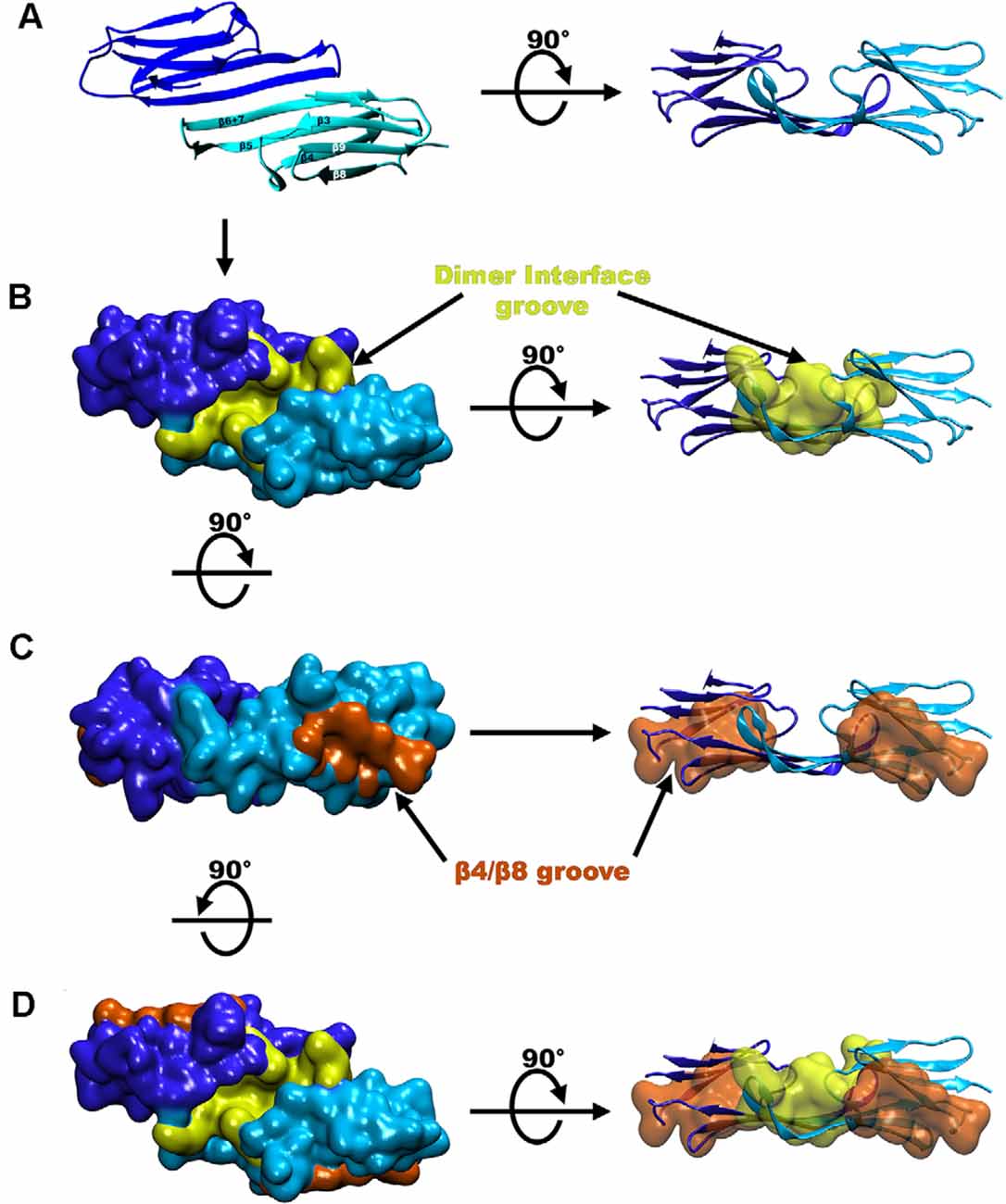
Figure 3. Groove locations in the crystal structure of Hsp27. (A) A ribbon diagram of the Hsp27 dimer (PDB 4MJH). (B) Hsp27 structure seen in panel (A) but in surface representation where the dimer interface groove is shaded yellow. (C) Hsp27 β4/β8 groove (colored in orange). (D) Representation of the three grooves on a dimeric unit colored the same as in panels (B,C).
Another structurally notable feature of the ACD is the β4/β8 groove which includes a hydrophobic pocket centered between β-strands 4 and 8 and is located on the outer edge of the ACD β-sandwich (Ehrnsperger et al., 1997; Klevit, 2020; Reid Alderson et al., 2021; Figure 3C). Therefore, within a dimer of Hsp27 there are two hydrophobic grooves located at the edges of each monomer and one shared dimer interface groove created by the linkage of two monomers (Clouser et al., 2019; Boelens, 2020; Figure 3D). These grooves provide regions for several client protein and oligomer binding opportunities and will be discussed later in this review.
The C-terminal region (CTR) and extension make up the remaining 37 residues of the Hsp27 primary sequence (Figure 1A). The CTR has an abundance of polar and charged residues and is partly responsible for oligomer formation and is thought to aid in the solubility of Hsp27 oligomers and or oligomer/substrate complexes (Pasta et al., 2004; Chalova et al., 2014; Carver et al., 2017; Boelens, 2020). The CTR contains a well-conserved region known as the IXI/V motif at residue positions 179–183 characterized by alternating isoleucine and valine residues with the central residue being a canonical proline (Lelj-Garolla and Mauk, 2012; Carver et al., 2017; Figure 1D). This motif contributes to the stabilization of oligomers by interlinking adjacent dimers. The C-terminal extension consists of the last 22 residues and does not contain any secondary structure (Alderson et al., 2017; Carver et al., 2017). The extension including the IXI/V motif is inherently disordered and highly mobile (Alderson et al., 2017). This is partly due to cis-trans isomerization about the 182 and 194 proline residues which causes changes in the adopted disordered states (Alderson et al., 2017). Interestingly, however, the residues near these pralines were shown to convert into a low residual β-strand structure which is thought to have implications in regulating oligomerization stability (Alderson et al., 2017).
Hsp27 NTR ACD and CTR Interact to Form Higher Order Complexes
Recent studies have revealed that the involvement and dynamics of the NTR in oligomer formation is far more complicated than originally thought (Clouser et al., 2019; Collier et al., 2019; Nappi et al., 2020). Various techniques including NMR, HX-MS, and modeling were employed to characterize the NTR of Hsp27 (Clouser et al., 2019). These revealed that various hydrophobic peptide regions of the NTR make multiple contacts with two grooves of the ACD (Clouser et al., 2019). Using six truncated variations of the NTR, a map of the binding combinations of NTR and ACD displayed four separate interactions (Clouser et al., 2019). First, the distal region of the NTR that encompasses the first 13 residues interacts with the β4/β8 groove of the ACD (Figure 4). This interaction is thought to be the result of alternating hydrophobic residues on the distal region of the NTR (6-VPFSLL-11) considered to be a motif that can bind the β4/β8 groove (Clouser et al., 2019). The next set of residues of the NTR, that contains serine-15 and the WD/EPF motif (12–27), was shown to bind a negatively charged area made of loops L3/4 and L5/6 (Figure 4; Clouser et al., 2019). Interestingly, serine residues 78 and 82, although distant in sequence, were also found near this NTR binding region (Clouser et al., 2019). It is suggested that this phosphorylated serine congregation facilitates the regulation of phosphorylation (Clouser et al., 2019). Additional negative charges, introduced by phosphorylation, disturb the region, and promote dissociation of the NTR (12–27) from the negatively charged loops (Hayes et al., 2009; Clouser et al., 2019).
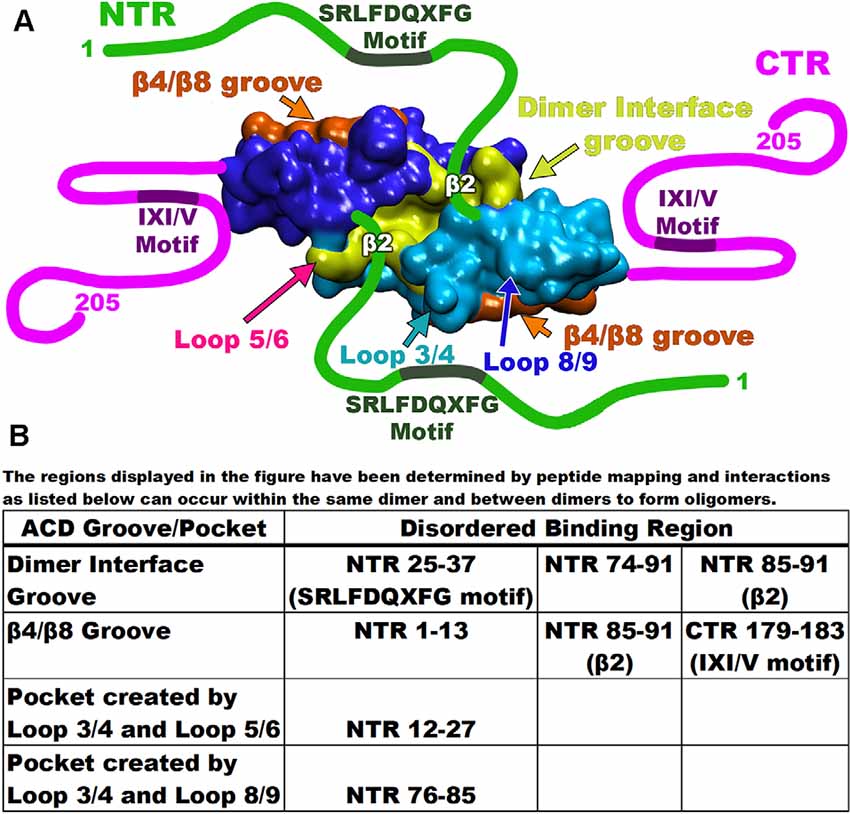
Figure 4. Map of Hsp27 interacting regions. (A) Each of the monomers is depicted as either blue or cyan with the dimer interface groove in yellow and the β4/β8 groove for each monomer in orange. Disordered regions are drawn as either green or magenta lines with the binding motifs as labeled. Important interaction loops are indicated by arrows. (B) The table at the bottom lists each of the disordered binding regions and the groove/pocket that bind as described by peptide mapping.
The remaining segments of the NTR (residues 25–37 and 74–91) are found to bind the groove created by the dimer interface (Figure 4). For example, the NTR (25–37) contains the conserved SRLFDQxFG motif (Pasta et al., 2003; Clouser et al., 2019). This motif binds at the dimer interface groove much like the NTR (74–91) that contains the phosphorylation site serine residues (Klevit, 2020). This suggests that at any given time, several regions of the NTR may be bound to different grooves within the ACD. Another study proposed an additional NTR-ACD binding combination (Collier et al., 2019). Using a previous structure and co-crystallization strategy, an ACD construct was built that included NTR phosphorylation site serines 78 and 82 (Collier et al., 2019). The β2 strand of Hsp27 (residues 84–93) transiently binds the adjacent β3 strand in an antiparallel orientation (Clouser et al., 2019; Collier et al., 2019; Klevit, 2020). The presence of the β2 strand, therefore, alters the space allotted within the dimer interface groove (Collier et al., 2019; Klevit, 2020). Various peptide mimics of the NTR crystallized with an ACD construct (84–171) confirm a hydrogen bonding network between the β2 and β3 strands (Collier et al., 2019). Additionally, a third Hsp27 binding combination has revealed that the β2 strand becomes extended and transiently binds the β4/β8 groove of an adjacent dimer (Figure 4; Collier et al., 2019). As mentioned previously, the NTR (12–27) containing serine 15, binds a space between negatively charged loops L3/4 and L5/6. This was found to perturb the phosphorylation site residues S78 and S82 indicating that the three phosphorylation sites are close (Clouser et al., 2019). This is confirmed by the crystal structure (6GJH) revealing that a peptide segment containing the phosphorylation site serines 78 and 82 binds a pocket created by loops L3/4 and L8/9 within the ACD (Figure 4; Collier et al., 2019). Therefore, the binding pockets for the NTR regions share the center L3/4 loop indicating that the phosphorylation site serines 15, 78, and 82 are close in space. Recently, an x-ray structure defined a spherical Hsp27 oligomer complex composed of 12 dimers (Nappi et al., 2020). The structure revealed that serine residues 78 and 82 from each monomer share the same area. Collectively, these studies reveal that between adjacent dimers, six phosphorylation sites are arranged near each other. This supports the claim that charge repulsion is the driving force of oligomer dissociation that results in a molecule with increased flexibility (Collier et al., 2019).
The CTR of the small heat shock proteins forms transient interactions with regions on the ACD (Pasta et al., 2004). As seen with many sHsp the IXI/V motif within the CTR fits into the hydrophobic β4/β8 groove in an antiparallel orientation (Figure 5; Clouser et al., 2019; Baughman et al., 2020). This interaction is like the NTR distal motif and the NTR β2 strand binding within the groove (Figure 4). The outer residues of the IXI/V motif fit into the holes of the hydrophobic groove due to the kink created by the center proline-182 residue (Freilich et al., 2018; Klevit, 2020). This is described as an extended formation of the CTR in the trans-conformation as the canonical proline can undergo cis-trans isomerization about the I-P peptide bond (Alderson et al., 2017). The interaction is found to occur within the same dimeric subunit and between adjacent dimers as well (Reid Alderson et al., 2021). The lowly populated residual β-strand structure may further encourage binding between the ACD and CTR and other binding partners (Alderson et al., 2017). The IXI/V motif is considered a short linear motif (SLiM) that modulates interactions between proteins (Reid Alderson et al., 2021). In the case of Hsp27, those interactions are between the CTR, the ACD, and possibly client proteins (Reid Alderson et al., 2021). A crystal structure of the truncated Hsp27 ACD co-crystallized with a peptide mimicking the C-terminal IXI/V motif has been solveds (Figure 5; Hochberg et al., 2014). This assembly shows the C-terminal SLiM in an antiparallel alignment to the β8 strand. The binding of the CTR IXI/V motif within the hydrophobic groove of the ACD acts to further stabilize high molecular weight homo-oligomers of Hsp27. It accomplishes this by transiently linking neighboring dimers indicating that the population of bound and unbound IXI/V motifs is mixed (Boelens, 2020). The binding combinations for these regions occur within a dimer and between adjacent dimers. The transient nature of these interacting regions creates a flexible protein that is unexpectedly stabilized. Hsp27 is a dynamic protein that is continuously exchanging its binding regions allowing for substrate capture.
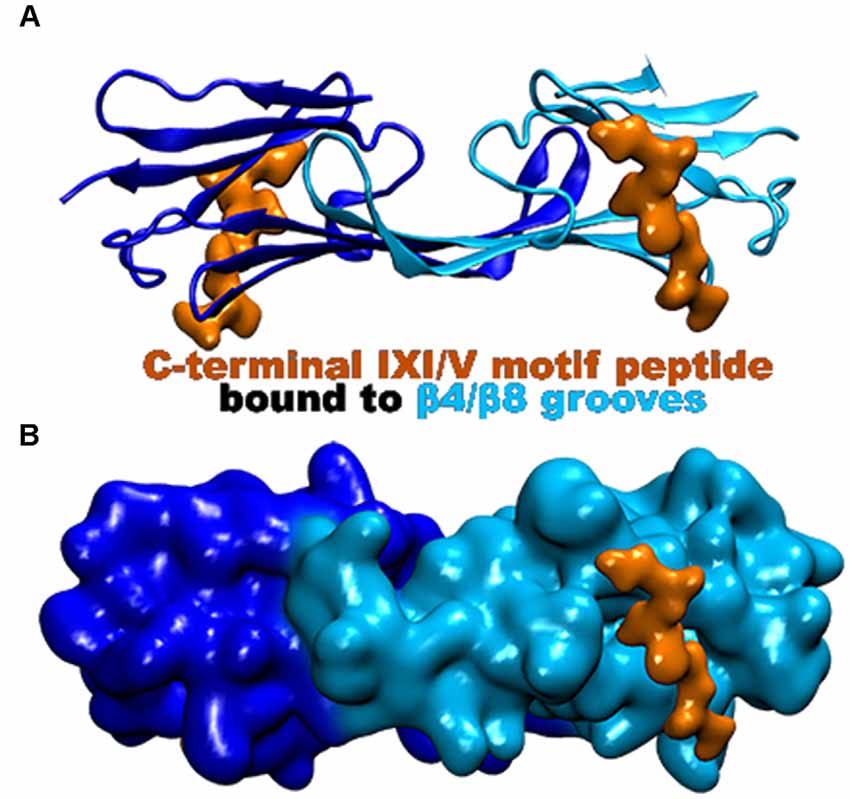
Figure 5. Hsp27 crystal structure (PDB 4MJH) co-crystallized with peptide mimic of the IXI/V motif. The peptide (colored in orange) was found bound to the β4/β8 groove as indicated. (A) Ribbon representation of the dimer illustrating the location of the two bound peptides. (B) Surface representation of the dimer that better illustrates the β4/β8 groove with the bound peptide.
Hsp27 Regions Involved in Substrate Capture
The three regions of Hsp27 have distinct roles in substrate capture (Jaya et al., 2009; Hochberg et al., 2014; Mainz et al., 2015). The ACD is thought to bind proteins that aggregate via the amyloid fibril aggregation pathway (Bakthisaran et al., 2015; Mainz et al., 2015; Freilich et al., 2018; Santhanagopalan et al., 2018; Webster et al., 2019). Client proteins that aggregate via this mechanism tend to form β-sheet conformations that resemble β-amyloid fibrils so the attraction between the ACD and the amyloid fibril-like substrate is robust (Mainz et al., 2015; Liu et al., 2020). The neuronal protein α-synuclein is an established substrate of Hsp27 that aggregates via the amyloid fibrillar pathway. It was found that full-length Hsp27 and truncated ACD transiently bind monomers of α-synuclein preventing their formation into fibrils (Cox et al., 2016, 2018; Jia et al., 2019). The truncated ACD displayed a lower binding affinity for α-synuclein monomers than the WT indicating that the α-synuclein monomer binding is in large part through the NTR (Jia et al., 2019). Additionally, the isolated ACD of Hsp27 cannot bind to α-synuclein fibrils that are already aggregated (Cox et al., 2018; Selig et al., 2020). Co-sedimentation experiments of Hsp27 lacking the CTR and in the presence of α-synuclein fibrils confirmed that the CTR is required to bind the fibrils (Selig et al., 2020). Therefore, involvement of the NTR and CTR is required when binding fibrillar aggregates of α-synuclein to prevent their elongation (Cox et al., 2018; Selig et al., 2020). These experiments suggest that the ACD alone is not sufficient to protect against amyloidogenic aggregation and that the involvement of all three regions is required.
Further evidence suggests that there is a strict interplay between the three Hsp27 regions to regulate chaperone activity (Baughman et al., 2018, 2020; Freilich et al., 2018). It has been observed that Hsp27 binds Tau, another protein that aggregates via the amyloidogenic pathway (Abisambra et al., 2010; Baughman et al., 2018, 2020; Freilich et al., 2018). This is mediated by an interaction between an IXI/V motif of Tau and the ACD hydrophobic groove suggesting that the groove provides protective benefits against fibril formation (Freilich et al., 2018). However, this study also revealed that using truncated forms of Hsp27 lacking the NTR completely abolished this protective activity, further complicating the mechanism by which Hsp27 binds Tau (Freilich et al., 2018). One study suggested that the chaperone activity of Hsp27 extends beyond the ACD hydrophobic groove and is actually dependent on interactions of NTR to client protein (Baughman et al., 2020). It was subsequently determined that a terminal peptide representing the first 13 residues of the Hsp27 NTR binds the ACD hydrophobic groove that binds Tau 50 residues downstream of the highly disordered NTR region (Clouser et al., 2019). These experiments demonstrate a possibility that a binding competition for the ACD groove exists beyond IXI/V-containing proteins such as Hsp27 CTR, Tau, PCBP1, and others. Mutation of the B4/B8 hydrophobic groove to discourage binding between the groove and any IXI/V motif showed an unexpected increase in chaperone activity towards Tau (Freilich et al., 2018). This suggests that the ACD region is not the actual site for chaperoning activity (Baughman et al., 2020). Instead, there may be a competition between the NTR, CTD, and other IXI/V containing entities for the hydrophobic groove of Hsp27. Therefore, the role of the ACD hydrophobic pocket may be to act as a regulatory limiting factor that controls the extent to which Hsp27 binds and chaperones proteins (Chalova et al., 2014; Freilich et al., 2018; Santhanagopalan et al., 2018). In summary, Hsp27 can protect a large array of structurally diverse client proteins by utilizing its dynamic conformational variability. Mutations of Hsp27 can easily disturb this tightly regulated balance and lead to neurodegenerative disorders.
Missense Mutations Associated With CMT2F and dHMN2B
Mutations within all three regions of Hsp27 are implicated in the neurodegenerative disorders CMT2F and dHMN2B (Arbach et al., 2017; Muranova et al., 2019, 2020; Vendredy et al., 2020). As previously mentioned, there is an abundance of clinical and genetic overlap between the two neuropathies further complicating the elucidation of disease progression. The NTR mutants (Table 1) are associated with dHMN and cause overstabilization of oligomers accompanied by a slight increase in size (Muranova et al., 2015). These mutants have decreased chaperoning activity and are resistant to phosphorylation (Nefedova et al., 2013b; Muranova et al., 2015). The NTR is a dynamic region that is constantly undergoing conformational modifications to accommodate oligomerization and substrate binding. Amino acid changes within this region directly affect the interaction of the NTR with the ACD. The effects are either enhanced or diminished binding to the ACD, both of which have negative consequences that can have a direct impact on the oligomerization, phosphorylation, and chaperoning activity since the three are interdependent. Furthermore, mutations of the NTR could potentially result in diminished chaperone activity toward Tau and α-synuclein. Experimental evidence suggests that the NTR is critical in the effective chaperoning of Tau and α-synuclein (Baughman et al., 2020; Selig et al., 2020). Perhaps disease mutations of the NTR could result in diminished chaperone activity of these proteins aggravating the progression of AD and PD. Several models demonstrate that Hsp27 is critical in preventing the formation of α-synuclein inclusions further supporting the notion that ineffective chaperoning will exacerbate disease progression (Cox et al., 2016, 2018; Cox and Ecroyd, 2017).
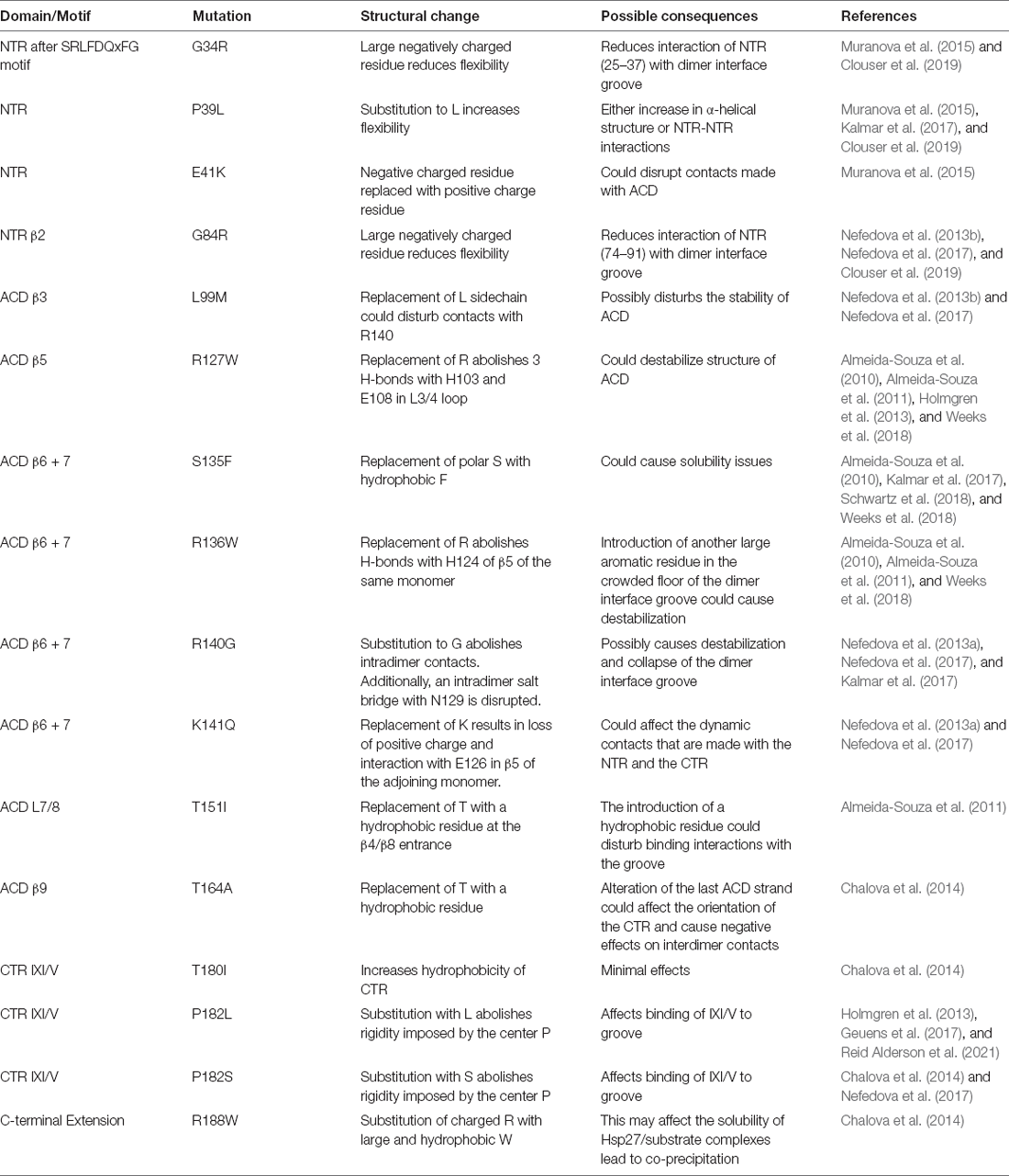
Table 1. Hsp27 mutations associated with CMT2F and dHMN2B.
The ACD is considered a hotspot for mutations that result in CMT2F and dHMN2B (Kalmar et al., 2017). Mutants R127W, S135F, and R136W destabilize the dimer interface groove resulting in increased monomerization that correlates with increased chaperoning activity (Almeida-Souza et al., 2010; Weeks et al., 2018; Alderson et al., 2019). These mutants were shown to be hyperactive and displayed elevated binding to α and β tubulin (Almeida-Souza et al., 2010, 2011; Weeks et al., 2018). This increased binding was correlated with an increased stability of the microtubule network and is an unfavorable consequence since the dynamic polymerizing nature of microtubules is essential to its function (Almeida-Souza et al., 2011; Kevenaar and Hoogenraad, 2015). Hsp27 mutations R140G and K141Q were found to decrease the thermal stability of the oligomers (Nefedova et al., 2013a). This causes changes in the oligomerization and chaperoning ability of Hsp27 as the two are connected. The T151I mutant occurs at the end of the β4/β8 groove within an α-helix and surprisingly did not affect chaperone activity and did not have increased binding to tubulin/microtubules (Almeida-Souza et al., 2010; Klevit, 2020). The T164A mutant was shown to cause dissociation of oligomers accompanied by a decrease in thermal stability (Chalova et al., 2014). The ACD mutations lead to destabilization of the secondary structure at the dimer interface. This may lead to aberrations within the dimer interface groove that disrupt contacts made by the NTR. If the NTR is not regulated properly by the ACD, changes to the quaternary structure and chaperoning ability arise. It was demonstrated that mutations of the ACD to enhance the release of the NTR increased Tau substrate capture (Baughman et al., 2020). Disease mutations of the ACD could strengthen the interaction between the NTR and the ACD. The point mutations of the ACD occur at the dimer interface groove and one near the β4/β8 groove, both regions known to transiently bind stretches of the NTR. A mutation from a charged residue to a hydrophobic residue could amplify the interaction between these ACD binding regions and the hydrophobic NTR resulting in a protein that can no longer effectively chaperone Tau.
Lastly, mutations in the CTR and extension are causative for dHMN. The T180I mutant significantly improves the stability of Hsp27 oligomers and does not alter chaperoning activity (Chalova et al., 2014). However, this mutation could impact the interconversion rate between cis and trans states about the I180-P182 bond (Alderson et al., 2017). Disturbances near this region could explain the increased oligomer stability that is not conducive to the mobile nature of Hsp27. The proline at position 182 is the central residue of the IXI/V motif and is susceptible to two different mutations, P182L and P182S. These mutations, as listed in Table 1, result in an increase in oligomerization resulting in aggregation and diminished chaperone activity (Chalova et al., 2014). The P182L mutation similarly results in very large oligomers that have diminished chaperone activity. This was confirmed both in vitro and in vivo with dHMN patient-derived stem cells (Reid Alderson et al., 2021). It was found that the P182L mutant decreases the affinity of the IXI/V for the β4/β8 groove possibly due to the disruption of the trans configuration of the proline residue which is evidenced to facilitate binding to the ACD (Alderson et al., 2017; Reid Alderson et al., 2021). This liberates the interaction between the CTR and ACD and results in increased binding of the NTR to the ACD groove (Reid Alderson et al., 2021). This explains why the P182L and possibly the P182S result in large oligomers that are unable to chaperone client proteins. The NTR regions are erroneously bound to the ACD limiting their availability to chaperone, resulting in large oligomeric complexes (Freilich et al., 2018; Reid Alderson et al., 2021). This study further revealed that other IXI/V-containing proteins have the potential to bind the Hsp27 hydrophobic groove. One such protein is poly(rC)-binding protein 1 (PCBP1; Geuens et al., 2017; Reid Alderson et al., 2021). The single-stranded nucleic acid binding protein has several neuronal transcript targets suggesting that disruption of PCBP1 may have deleterious effects on downstream proteins that are vital for neuronal activity (Geuens et al., 2017). P182L bound with higher affinity to PCBP1 compared to the WT when using a mutant cell line (Geuens et al., 2017). The increased interaction between the Hsp27, P182L, and PCBP1 caused an overall reduction in translational repression of PCBP1, resulting in elevated protein expression levels (Geuens et al., 2017). The R188W mutation does not alter oligomeric size or stability but does have a significant effect on chaperoning activity (Chalova et al., 2014). The substitution of a charged amino acid with a large hydrophobic residue may affect the solubility of Hsp27/substrate complexes (Chalova et al., 2014). In addition, it is suspected that the R188 mutation could interfere with the formation of the residual β-strand structure formed by residues 193–199 which is hypothesized to regulate oligomer and substrate dynamics (Alderson et al., 2017).
Concluding Remarks
Hsp27 plays a central role in several physiological processes, including maintaining cytoskeletal dynamics, apoptosis, signal transduction, activation of the proteasome, and protein folding (Parcellier et al., 2005; Garrido et al., 2012). It is comprised of several structurally distinct regions, including the NTR, the ACD, and the CTR. The NTR region is primarily responsible for substrate capture with sequences that are equipped to bind substrates. The NTR is generally bound to the ACD, which regulates the NTR, so it is not continuously and arbitrarily fishing for the substrate. The CTR regulates this interaction between the NTR and the ACD by competing with binding sites on the ACD that then render the NTR available for substrate capture. This delicate interplay is disturbed by a myriad of point mutations that cause neurodegenerative diseases. These mutations affect the three key regions of Hsp27 in both divergent and overlapping fashions, thus further complicating our current understanding of disease progression of CMT2F and dHMN2B. Their role in pathological progression may lie in the consequential inability to protect vital neuronal proteins. Similarly, the inability to properly chaperone these client proteins may worsen diseases such as Alzheimer’s and Parkinson’s disease. Although there have been many breakthroughs during the investigation of these relationships, further work is required to better elucidate the link between disrupted Hsp27 structure and the progression of these debilitating neuropathies. Furthermore, subsequent efforts are required to better understand how the conformational variability of Hsp27 regulates oligomerization, phosphorylation, and substrate capture with respect to Hsp27’s encompassing role in human physiology.
Author Contributions
BH wrote and prepared the manuscript. RB, ZH, and BH contributed to the editing of the manuscript at all points of the preparation process. All authors contributed to the article and approved the submitted version.
Funding
This work was made possible by the Welch Foundation award (AH-1649) awarded to RB. This work was supported by Grant 5U54MD007592 from the National Institute on Minority Health and Health Disparities (NIMHD), a component of the National Institutes of Health (NIH). We would like to thank the BBRC for partial funding of the work presented here.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the UTEP Department of Chemistry and Biochemistry for the use of core facilities.
References
Abisambra, J. F., Blair, L. J., Hill, S. E., Jones, J. R., Kraft, C., Rogers, J., et al. (2010). Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J. Neurosci. 30, 15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010
Ackerley, S., James, P. A., Kalli, A., French, S., Davies, K. E., and Talbot, K. (2006). A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 15, 347–354. doi: 10.1093/hmg/ddi452
Alderson, T. R., Benesch, J. L. P., and Baldwin, A. J. (2017). Proline isomerization in the C-terminal region of HSP27. Cell Stress Chaperones 22, 639–651. doi: 10.1007/s12192-017-0791-z
Alderson, T. R., Roche, J., Gastall, H. Y., Dias, D. M., Pritisanac, I., Ying, J., et al. (2019). Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun. 10:1068. doi: 10.1038/s41467-019-08557-8
Almeida-Souza, L., Asselbergh, B., D’Ydewalle, C., Moonens, K., Goethals, S., De Winter, V., et al. (2011). Small heat-shock protein HSPB1 mutants stabilize microtubules in charcot-marie-tooth neuropathy. J. Neurosci. 31, 15320–15328. doi: 10.1523/JNEUROSCI.3266-11.2011
Almeida-Souza, L., Goethals, S., de Winter, V., Dierick, I., Gallardo, R., Durme Van, J., et al. (2010). Increased monomerization of mutant HSPB1 leads to protein hyperactivity in Charcot-Marie-Tooth neuropathy. J. Biol. Chem. 285, 12778–12786. doi: 10.1074/jbc.M109.082644
Aquilina, J. A., Shrestha, S., Morris, A. M., and Ecroyd, H. (2013). Structural and functional aspects of hetero-oligomers formed by the small heat shock proteins αB-crystallin and HSP27. J. Biol. Chem. 288, 13602–13609. doi: 10.1074/jbc.M112.443812
Arbach, H., Butler, C., and McMenimen, K. A. (2017). Chaperone activity of human small heat shock protein-GST fusion proteins. Cell Stress Chaperones 22, 503–515. doi: 10.1007/s12192-017-0764-2
Bakthisaran, R., Tangirala, R., and Rao Ch, M. (2015). Small heat shock proteins: role in cellular functions and pathology. Biochim. Biophys. Acta 1854, 291–319. doi: 10.1016/j.bbapap.2014.12.019
Baranova, E. V., Weeks, S. D., Beelen, S., Bukach, O. V., N. Gusev, B., and Strelkov, S. V. (2011). Three-dimensional structure of α-crystallin domain dimers of human small heat shock proteins HSPB1 and HSPB6. J. Mol. Biol. 411, 110–122. doi: 10.1016/j.jmb.2011.05.024
Barbier, P., Zejneli, O., Martinho, M., Lasorsa, A., Belle, V., Smet-Nocca, C., et al. (2019). Role of tau as a microtubule-associated protein: structural and functional aspects. Front. Aging Neurosci. 11:204. doi: 10.3389/fnagi.2019.00204
Baughman, H. E. R., Clouser, A. F., R. Klevit, E., and Nath, A. (2018). HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 293, 2687–2700. doi: 10.1074/jbc.M117.803411
Baughman, H. E. R., Pham, T.-H. T., Adams, C. S., Nath, A., and Klevit, R. E. (2020). Release of a disordered domain enhances HspB1 chaperone activity toward tau. Proc. Nat. Acad. Sci. U S A 117, 2923–2929. doi: 10.1073/pnas.1915099117
Bhatt, J. M., Enriquez, A. S., Wang, J., Rojo, H. M., Molugu, S. K., Hildenbrand, Z. L., et al. (2018). Single-ring intermediates are essential for some chaperonins. Front. Mol. Biosci. 5:42. doi: 10.3389/fmolb.2018.00042
Boelens, W. C. (2020). Structural aspects of the human small heat shock proteins related to their functional activities. Cell Stress Chaperones 25, 581–591. doi: 10.1007/s12192-020-01093-1
Borrelli, M. J., Bernock, L. J., Landry, J., Spitz, D. R., Weber, L. A., Hickey, E., et al. (2002). Stress protection by a fluorescent Hsp27 chimera that is independent of nuclear translocation or multimeric dissociation. Cell Stress Chaperones 7, 281–296. doi: 10.1002/prot.26319
Bova, M. P., Huang, Q., Ding, L., and Horwitz, J. (2002). Subunit exchange, conformational stability and chaperone-like function of the small heat shock protein 16.5 from Methanococcus jannaschii. J. Biol. Chem. 277, 38468–38475. doi: 10.1074/jbc.M205594200
Bova, M. P., McHaourab, H. S., Han, Y., and Fung, B. K. (2000). Subunit exchange of small heat shock proteins. Analysis of oligomer formation of αA-crystallin and Hsp27 by fluorescence resonance energy transfer and site-directed truncations. J. Biol. Chem. 275, 1035–1042. doi: 10.1074/jbc.275.2.1035
Cairns, J., Qin, S., Philp, R., Y. Tan, H., and Guy, G. R. (1994). Dephosphorylation of the small heat shock protein Hsp27 in vivo by protein phosphatase 2A. J. Biol. Chem. 269, 9176–9183.
Carver, J. A., Grosas, A. B., Ecroyd, H., and Quinlan, R. A. (2017). The functional roles of the unstructured N- and C-terminal regions in αB-crystallin and other mammalian small heat-shock proteins. Cell Stress Chaperones 22, 627–638. doi: 10.1007/s12192-017-0789-6
Chalova, A. S., Sudnitsyna, M. V., S. Strelkov, V., and Gusev, N. B. (2014). Characterization of human small heat shock protein HspB1 that carries C-terminal domain mutations associated with hereditary motor neuron diseases. Biochim. Biophys. Acta 1844, 2116–2126. doi: 10.1016/j.bbapap.2014.09.005
Chung, K. W., Kim, S.-B., Cho, S. Y., Hwang, S. J., Park, S. W., Kang, S. H., et al. (2008). Distal hereditary motor neuropathy in Korean patients with a small heat shock protein 27 mutation. Exp. Mol. Med. 40:304. doi: 10.3858/emm.2008.40.3.304
Clouser, A. F., Baughman, H. E., Basanta, B., Guttman, M., Nath, A., and Klevit, R. E. (2019). Interplay of disordered and ordered regions of a human small heat shock protein yields an ensemble of “quasi-ordered” states. eLife 8:e50259. doi: 10.7554/eLife.50259
Collier, M. P., Alderson, T. R., de Villiers, C. P., Nicholls, D., Gastall, H. Y., Allison, T. M., et al. (2019). HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci. Adv. 5:eaav8421. doi: 10.1126/sciadv.aav8421
Cox, D., and Ecroyd, H. (2017). The small heat shock proteins αB-crystallin (HSPB5) and Hsp27 (HSPB1) inhibit the intracellular aggregation of α-synuclein. Cell Stress Chaperones 22, 589–600. doi: 10.1007/s12192-017-0785-x
Cox, D., Selig, E., Griffin, M. D. W., Carver, J. A., and Ecroyd, H. (2016). Small heat-shock proteins prevent α-synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation. J. Biol. Chem. 291, 22618–22629. doi: 10.1074/jbc.M116.739250
Cox, D., Whiten, D. R., Brown, J. W. P., Horrocks, M. H., Gil San, R., Dobson, C. M., et al. (2018). The small heat shock protein Hsp27 binds alpha-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 293, 4486–4497. doi: 10.1074/jbc.M117.813865
Dierick, I., Baets, J., Irobi, J., Jacobs, A., Vriendt De, E., Deconinck, T., et al. (2007). Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study. Brain 131, 1217–1227. doi: 10.1093/brain/awn029
D’Ydewalle, C., Krishnan, J., Chiheb, D. M., Damme Van, P., Irobi, J., Kozikowski, A. P., et al. (2011). HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 17, 968–974. doi: 10.1038/nm.2396
Echaniz-Laguna, A. (2015). The shifting paradigm of Charcot-Marie-Tooth disease. Rev. Neurol. (Paris) 171, 498–504. doi: 10.1016/j.neurol.2014.12.003
Ehrnsperger, M., Graber, S., Gaestel, M., and Buchner, J. (1997). Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 16, 221–229. doi: 10.1093/emboj/16.2.221
Enriquez, A. S., Rojo, H. M., Bhatt, J. M., Molugu, S. K., Z. Hildenbrand, L., and Bernal, R. A. (2017). The human mitochondrial Hsp60 in the APO conformation forms a stable tetradecameric complex. Cell Cycle 16, 1309–1319. doi: 10.1080/15384101.2017.1321180
Evgrafov, O. V., Mersiyanova, I., Irobi, J., Van Den Bosch, L., Dierick, I., Leung, C. L., et al. (2004). Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606. doi: 10.1038/ng1354
Eyles, S. J., and Gierasch, L. M. (2010). Nature’s molecular sponges: small heat shock proteins grow into their chaperone roles. Proc. Natl. Acad. Sci. U S A 107, 2727–2728. doi: 10.1073/pnas.0915160107
Freilich, R., Betegon, M., Tse, E., Mok, S. A., Julien, O., Agard, D. A., et al. (2018). Competing protein-protein interactions regulate binding of Hsp27 to its client protein tau. Nat. Commun. 9:4563. doi: 10.1038/s41467-018-07012-4
Fu, X. (2014). Chaperone function and mechanism of small heat-shock proteins. Acta Biochim. Biophys. Sin. (Shanghai) 46, 347–356. doi: 10.1093/abbs/gmt152
Garrido, C., Paul, C., Seigneuric, R., and Kampinga, H. H. (2012). The small heat shock proteins family: the long forgotten chaperones. Int. J. Biochem. Cell Biol. 44, 1588–1592. doi: 10.1016/j.biocel.2012.02.022
Gentile, L., Russo, M., Fabrizi, G. M., Taioli, F., Ferrarini, M., Testi, S., et al. (2020). Charcot-Marie-Tooth disease: experience from a large Italian tertiary neuromuscular center. Neurol. Sci. 41, 1239–1243. doi: 10.1007/s10072-019-04219-1
Geuens, T., Winter De, V., Rajan, N., Achsel, T., Mateiu, L., Almeida-Souza, L., et al. (2017). Mutant HSPB1 causes loss of translational repression by binding to PCBP1, an RNA binding protein with a possible role in neurodegenerative disease. Acta Neuropathol. Commun. 5:5. doi: 10.1186/s40478-016-0407-3
Haslbeck, M., Walke, S., Stromer, T., Ehrnsperger, M., White, H. E., Chen, S., et al. (1999). Hsp26: a temperature-regulated chaperone. EMBO J. 18, 6744–6751. doi: 10.1093/emboj/18.23.6744
Hayes, D., Napoli, V., Mazurkie, A., W. Stafford, F., and Graceffa, P. (2009). Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 284, 18801–18807. doi: 10.1074/jbc.M109.011353
Hochberg, G. K., Ecroyd, H., Liu, C., Cox, D., Cascio, D., Sawaya, M. R., et al. (2014). The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U S A 111, E1562–1570. doi: 10.1073/pnas.1322673111
Holmgren, A., Bouhy, D., Winter De, V., Asselbergh, B., J.-Timmermans, P., Irobi, J., et al. (2013). Charcot-Marie-Tooth causing HSPB1 mutations increase Cdk5-mediated phosphorylation of neurofilaments. Acta Neuropathol. 126, 93–108. doi: 10.1007/s00401-013-1133-6
Houlden, H., Laura, M., Wavrant-De Vrieze, F., Blake, J., Wood, N., and Reilly, M. M. (2008). Mutations in the HSP27 (HSPB1) gene cause dominant, recessive and sporadic distal HMN/CMT type 2. Neurology 71, 1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67
Hu, S., Tan, J., Qin, L., Lv, L., Yan, W., Zhang, H., et al. (2021). Molecular chaperones and Parkinson’s disease. Neurobiol. Dis. 160:105527. doi: 10.1016/j.nbd.2021.105527
Ismailov, S., Fedotov, V., Dadali, E., Polyakov, A., Broeckhoven Van, C., Ivanov, V., et al. (2001). A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2F) maps to chromosome 7q11–q21. Eur. J. Hum. Genet. 9, 646–650. doi: 10.1038/sj.ejhg.5200686
Jaya, N., Garcia, V., and Vierling, E. (2009). Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc. Natl. Acad. Sci. U S A 106, 15604–15609. doi: 10.1073/pnas.0902177106
Jia, C., Ma, X., Liu, Z., Gu, J., Zhang, X., Li, D., et al. (2019). Different heat shock proteins bind α-synuclein with distinct mechanisms and synergistically prevent its amyloid aggregation. Front. Neurosci. 13:1124. doi: 10.3389/fnins.2019.01124
Jovcevski, B., Kelly, M. A., Rote, A. P., Berg, T., Gastall, H. Y., Benesch, J. L., et al. (2015). Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol. 22, 186–195. doi: 10.1016/j.chembiol.2015.01.001
Kalmar, B., Innes, A., Wanisch, K., Kolaszynska, A. K., Pandraud, A., Kelly, G., et al. (2017). Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced charcot marie tooth disease. Hum. Mol. Genet. 26, 3313–3326. doi: 10.1093/hmg/ddx216
Kappe, G., Franck, E., Verschuure, P., Boelens, W. C., Leunissen, J. A., and de Jong, W. W. (2003). The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1–10. Cell Stress Chaperones 8, 53–61. doi: 10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2
Katsogiannou, M., Andrieu, C., and Rocchi, P. (2014). Heat shock protein 27 phosphorylation state is associated with cancer progression. Front. Genet. 5:346. doi: 10.3389/fgene.2014.00346
Kevenaar, J. T., and Hoogenraad, C. C. (2015). The axonal cytoskeleton: from organization to function. Front. Mol. Neurosci. 8:44. doi: 10.3389/fnmol.2015.00044
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Hartl, F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. doi: 10.1146/annurev-biochem-060208-092442
Klevit, R. E. (2020). Peeking from behind the veil of enigma: emerging insights on small heat shock protein structure and function. Cell Stress Chaperones 25, 573–580. doi: 10.1007/s12192-020-01092-2
Kostenko, S., and Moens, U. (2009). Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell. Mol. Life Sci. 66, 3289–3307. doi: 10.1007/s00018-009-0086-3
Lambert, H., Charette, S. J., Bernier, A. F., Guimond, A., and Landry, J. (1999). HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem. 274, 9378–9385. doi: 10.1074/jbc.274.14.9378
Lang, B. J., Guerrero, M. E., Prince, T. L., Okusha, Y., Bonorino, C., and Calderwood, S. K. (2021). The functions and regulation of heat shock proteins; key orchestrators of proteostasis and the heat shock response. Arch. Toxicol. 95, 1943–1970. doi: 10.1007/s00204-021-03070-8
Lelj-Garolla, B., and Mauk, A. G. (2012). Roles of the N- and C-terminal sequences in Hsp27 self-association and chaperone activity. Protein Sci. 21, 122–133. doi: 10.1002/pro.761
Liu, Z., Zhang, S., Gu, J., Tong, Y., Li, Y., Gui, X., et al. (2020). Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nat. Struct. Mol. Biol. 27, 363–372. doi: 10.1038/s41594-020-0399-3
Mainz, A., Peschek, J., Stavropoulou, M., Back, K. C., Bardiaux, B., Asami, S., et al. (2015). The chaperone alphaB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 22, 898–905. doi: 10.1038/nsmb.3108
McDonald, E. T., Bortolus, M., H. Koteiche, A., and McHaourab, H. S. (2012). Sequence, structure and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry 51, 1257–1268. doi: 10.1021/bi2017624
Muranova, L. K., Ryzhavskaya, A. S., Sudnitsyna, M. V., V. Shatov, M., and Gusev, N. B. (2019). Small heat shock proteins and human neurodegenerative diseases. Biochemistry (Moscow) 84, 1256–1267. doi: 10.1134/S000629791911004X
Muranova, L. K., Sudnitsyna, M. V., S. Strelkov, V., and Gusev, N. B. (2020). Mutations in HspB1 and hereditary neuropathies. Cell Stress Chaperones 25, 655–665. doi: 10.1007/s12192-020-01099-9
Muranova, L. K., Weeks, S. D., S. Strelkov, V., and Gusev, N. B. (2015). Characterization of mutants of human small heat shock protein HspB1 carrying replacements in the N-Terminal domain and associated with hereditary motor neuron diseases. PLoS One 10:e0126248. doi: 10.1371/journal.pone.0126248
Mymrikov, E. V., Daake, M., Richter, B., Haslbeck, M., and Buchner, J. (2017). The chaperone activity and substrate spectrum of human small heat shock proteins. J. Biol. Chem. 292, 672–684. doi: 10.1074/jbc.M116.760413
Nappi, L. Aguda, A. H., Nakouzi, N. A., Lelj-Garolla, B., Beraldi, E., Lallous, N., et al. (2020). Ivermectin inhibits HSP27 and potentiates efficacy of oncogene targeting in tumor models. J. Clin. Invest. 130, 699–714. doi: 10.1172/JCI130819
Navarro-Zaragoza, J., Cuenca-Bermejo, L., Almela, P., Laorden, M.-L., and Herrero, M.-T. (2021). Could small heat shock protein HSP27 be a first-line target for preventing protein aggregation in Parkinson’s disease. Int. J. Mol. Sci. 22:3038. doi: 10.3390/ijms22063038
Nefedova, V. V., Datskevich, P. N., Sudnitsyna, M. V., Strelkov, S. V., and Gusev, N. B. (2013a). Physico-chemical properties of R140G and K141Q mutants of human small heat shock protein HspB1 associated with hereditary peripheral neuropathies. Biochimie 95, 1582–1592. doi: 10.1016/j.biochi.2013.04.014
Nefedova, V. V., Sudnitsyna, M. V., S. Strelkov, V., and Gusev, N. B. (2013b). Structure and properties of G84R and L99M mutants of human small heat shock protein HspB1 correlating with motor neuropathy. Arch. Biochem. Biophys. 538, 16–24. doi: 10.1016/j.abb.2013.07.028
Nefedova, V. V., Sudnitsyna, M. V., and Gusev, N. B. (2017). Interaction of small heat shock proteins with light component of neurofilaments (NFL). Cell Stress Chaperones 22, 467–479.
Nussbaum, R. L., and Ellis, C. E. (2003). Alzheimer’s disease and Parkinson’s disease. N Engl. J. Med. 348, 1356–1364. doi: 10.1056/NEJM2003ra020003
Parcellier, A., Schmitt, E., Brunet, M., Hammann, A., Solary, E., and Garrido, C. (2005). Small heat shock proteins HSP27 and αB-crystallin: cytoprotective and oncogenic functions. Antioxid. Redox Signal 7, 404–413. doi: 10.1089/ars.2005.7.404
Pasta, S. Y., Raman, B., Ramakrishna, T., and Rao, C. M. (2003). Role of the conserved SRLFDQFFG region of α-Crystallin, a small heat shock protein. J. Biol. Chem. 278, 51159–51166. doi: 10.1074/jbc.M307523200
Pasta, S. Y., Raman, B., Ramakrishna, T., and Rao Ch, M. (2004). The IXI/V motif in the C-terminal extension of alpha-crystallins: alternative interactions and oligomeric assemblies. Mol. Vis. 10, 655–662.
Peschek, J., Braun, N., Rohrberg, J., Back, K. C., Kriehuber, T., Kastenmuller, A., et al. (2013). Regulated structural transitions unleash the chaperone activity of αB-crystallin. Proc. Natl. Acad. Sci. U S A 110, E3780–3789. doi: 10.1073/pnas.1308898110
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Rajagopal, P., Liu, Y., Shi, L., A. Clouser, F., and Klevit, R. E. (2015). Structure of the alpha-crystallin domain from the redox-sensitive chaperone, HSPB1. J. Biomol. NMR 63, 223–228. doi: 10.1007/s10858-015-9973-0
Reid Alderson, T., Adriaenssens, E., Asselbergh, B., Pritišanac, I., Lent Van, J., Gastall, H. Y., et al. (2021). A weakened interface in the P182L variant of HSP27 associated with severe Charcot-Marie-Tooth neuropathy causes aberrant binding to interacting proteins. EMBO J. 40:e103811. doi: 10.15252/embj.2019103811
Richter, K., Haslbeck, M., and Buchner, J. (2010). The heat shock response: life on the verge of death. Mol. Cell 40, 253–266. doi: 10.1016/j.molcel.2010.10.006
Rodriguez, A., Salzen Von, D., B. Holguin, A., and Bernal, R. A. (2020). Complex destabilization in the mitochondrial chaperonin Hsp60 leads to disease. Front. Mol. Biosci. 7:159. doi: 10.3389/fmolb.2020.00159
Rogalla, T., Ehrnsperger, M., Preville, X., Kotlyarov, A., Lutsch, G., Ducasse, C., et al. (1999). Regulation of Hsp27 oligomerization, chaperone function and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J. Biol. Chem. 274, 18947–18956. doi: 10.1074/jbc.274.27.18947
Santhanagopalan, I., Degiacomi, M. T., Shepherd, D. A., Hochberg, G. K. A., Benesch, J. L. P., and Vierling, E. (2018). It takes a dimer to tango: Oligomeric small heat shock proteins dissociate to capture substrate. J. Biol. Chem. 293, 19511–19521. doi: 10.1074/jbc.RA118.005421
Schwartz, N. U. (2019). Charcot-marie-tooth 2F (Hsp27 mutations): a review. Neurobiol. Dis. 130:104505. doi: 10.1016/j.nbd.2019.104505
Schwartz, N. U., Linzer, R. W., Truman, J. P., Gurevich, M., Hannun, Y. A., C. Senkal, E., et al. (2018). Decreased ceramide underlies mitochondrial dysfunction in Charcot-Marie-Tooth 2F. FASEB J. 32, 1716–1728. doi: 10.1096/fj.201701067R
Selig, E. E., Zlatic, C. O., Cox, D., Mok, Y.-F., Gooley, P. R., Ecroyd, H., et al. (2020). N- and C-terminal regions of αB-crystallin and Hsp27 mediate inhibition of amyloid nucleation, fibril binding and fibril disaggregation. J. Biol. Chem. 295, 9838–9854. doi: 10.1074/jbc.RA120.012748
Shashidharamurthy, R., Koteiche, H. A., Dong, J., and McHaourab, H. S. (2005). Mechanism of chaperone function in small heat shock proteins: dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. J. Biol. Chem. 280, 5281–5289. doi: 10.1074/jbc.M407236200
Sobott, F., Benesch, J. L., Vierling, E., and Robinson, C. V. (2002). Subunit exchange of multimeric protein Real-time, complexes. monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J. Biol. Chem. 277, 38921–38929. doi: 10.1074/jbc.M206060200
Sun, Y., and MacRae, T. H. (2005). Small heat shock proteins: molecular structure and chaperone function. Cell Mol. Life Sci. 62, 2460–2476. doi: 10.1007/s00018-005-5190-4
Szigeti, K., and Lupski, J. R. (2009). Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 17, 703–710. doi: 10.1038/ejhg.2009.31
Tanabe, H., Higuchi, Y., J.-Yuan, H., Hashiguchi, A., Yoshimura, A., Ishihara, S., et al. (2018). Clinical and genetic features of Charcot-Marie-Tooth disease 2F and hereditary motor neuropathy 2B in Japan. J. Peripher. Nerv. Syst. 23, 40–48. doi: 10.1111/jns.12252
Thériault, J. R., Lambert, H., Chávez-Zobel, A. T., Charest, G., Lavigne, P., and Landry, J. (2004). Essential role of the NH2-terminal WD/EPF motif in the phosphorylation-activated protective function of mammalian Hsp27. J. Biol. Chem. 279, 23463–23471. doi: 10.1074/jbc.M402325200
van Montfort, R. L., Basha, E., Friedrich, K. L., Slingsby, C., and Vierling, E. (2001). Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8, 1025–1030. doi: 10.1038/nsb722
Vendredy, L., Adriaenssens, E., and Timmerman, V. (2020). Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones 25, 679–699. doi: 10.1007/s12192-020-01101-4
Venugopal, A., Sundaramoorthy, K., and Vellingiri, B. (2019). Therapeutic potential of Hsp27 in neurological diseases. Egypt. J. Med. Hum. Genet. 20:21. doi: 10.1186/s43042-019-0023-4
Vicente Miranda, H., Chegão, A., Oliveira, M. S., Gomes Fernandes, B., Enguita, F. J., and Outeiro, T. F. (2020). Hsp27 reduces glycation-induced toxicity and aggregation of alpha-synuclein. FASEB J. 34, 6718–6728. doi: 10.1096/fj.201902936R
Webster, J. M., Darling, A. L., Uversky, V. N., and Blair, L. J. (2019). Small heat shock proteins, big impact on protein aggregation in neurodegenerative disease. Front. Pharmacol. 10:1047. doi: 10.3389/fphar.2019.01047
Weeks, S. D., Muranova, L. K., Heirbaut, M., Beelen, S., Strelkov, S. V., and Gusev, N. B. (2018). Characterization of human small heat shock protein HSPB1 alpha-crystallin domain localized mutants associated with hereditary motor neuron diseases. Sci. Rep. 8:688. doi: 10.1038/s41598-017-18874-x
Williams, D. R., and Litvan, I. (2013). Parkinsonian syndromes. Continuum (Minneap Minn) 19, 1189–1212. doi: 10.1212/01.CON.0000436152.24038.e0
Keywords: small heat shock protein 27 (Hsp27), Charcot-Marie-Tooth disease (CMT), distal hereditary motor neuropathy (dHMN), α-crystallin domain (ACD), heat shock protein
Citation: Holguin BA, Hildenbrand ZL and Bernal RA (2022) Insights Into the Role of Heat Shock Protein 27 in the Development of Neurodegeneration. Front. Mol. Neurosci. 15:868089. doi: 10.3389/fnmol.2022.868089
Received: 02 February 2022; Accepted: 09 March 2022;
Published: 30 March 2022.
Edited by:
Beatriz Alvarez, Complutense University of Madrid, SpainReviewed by:
Soumya De, Indian Institute of Technology Kharagpur, IndiaMaxim Sokolov, West Virginia University, United States
Copyright © 2022 Holguin, Hildenbrand and Bernal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ricardo A. Bernal, cmJlcm5hbEB1dGVwLmVkdQ==