
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 10 May 2022
Sec. Molecular Signalling and Pathways
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.861159
This article is part of the Research Topic Sub-molecular Mechanism of Genetic Epilepsy View all 13 articles
 Jing-Yang Wang1,2†
Jing-Yang Wang1,2† Jie Wang1,2†
Jie Wang1,2† Xin-Guo Lu3Wang Song1,2
Xin-Guo Lu3Wang Song1,2 Sheng Luo1,2
Sheng Luo1,2 Dong-Fang Zou3Li-Dong Hua4Qian Peng5Yang Tian6Liang-Di Gao1,2Wei-Ping Liao1,2
Dong-Fang Zou3Li-Dong Hua4Qian Peng5Yang Tian6Liang-Di Gao1,2Wei-Ping Liao1,2 Na He1,2*
Na He1,2*Objective: The PKD1 encodes polycystin-1, a large transmembrane protein that plays important roles in cell proliferation, apoptosis, and cation transport. Previous studies have identified PKD1 mutations in autosomal dominant polycystic kidney disease (ADPKD). However, the expression of PKD1 in the brain is much higher than that in the kidney. This study aimed to explore the association between PKD1 and epilepsy.
Methods: Trios-based whole-exome sequencing was performed in a cohort of 314 patients with febrile seizures or epilepsy with antecedent febrile seizures. The damaging effects of variants was predicted by protein modeling and multiple in silico tools. The genotype-phenotype association of PKD1 mutations was systematically reviewed and analyzed.
Results: Eight pairs of compound heterozygous missense variants in PKD1 were identified in eight unrelated patients. All patients suffered from febrile seizures or epilepsy with antecedent febrile seizures with favorable prognosis. All of the 16 heterozygous variants presented no or low allele frequencies in the gnomAD database, and presented statistically higher frequency in the case-cohort than that in controls. These missense variants were predicted to be damaging and/or affect hydrogen bonding or free energy stability of amino acids. Five patients showed generalized tonic-clonic seizures (GTCS), who all had one of the paired missense mutations located in the PKD repeat domain, suggesting that mutations in the PKD domains were possibly associated with GTCS. Further analysis demonstrated that monoallelic mutations with haploinsufficiency of PKD1 potentially caused kidney disease, compound heterozygotes with superimposed effects of two missense mutations were associated with epilepsy, whereas the homozygotes with complete loss of PKD1 would be embryonically lethal.
Conclusion: PKD1 gene was potentially a novel causative gene of epilepsy. The genotype-phenotype relationship of PKD1 mutations suggested a quantitative correlation between genetic impairment and phenotypic variation, which will facilitate the genetic diagnosis and management in patients with PKD1 mutations.
PKD1 (MIM 601313) is a gene with 46 exons located on chromosome 16p13. The encoded protein polycystin-1 (PC1) forms a complex with polycystin-2 (PC2), which regulates calcium permeable cation channels and intracellular calcium homeostasis (Semmo et al., 2014; Kim et al., 2016; Su et al., 2018). PC1 is distributed widely in multiple tissues, especially highly in the brain (Fagerberg et al., 2014). Its expression is also developmentally regulated with the highest level in fetal life and maintained throughout adulthood (Geng et al., 1997). Homozygous Pkd1 knockout mice have exhibited neural tube defects and embryonic or perinatal lethality (Lu et al., 2001; Lantinga-van Leeuwen et al., 2007). PC1 can also interact with homer 1/Vesl-1 and plays a role in synaptic plasticity in postnatal mouse hippocampus (Stokely et al., 2006). So far, PKD1 mutations were reported to be associated with autosomal dominant polycystic kidney disease (ADPKD, MIM 173900), which was featured by dilatation of the renal tubules leading to cyst formation and progressive renal failure. The relationship between PKD1 and diseases of the brain remains unknown.
Febrile seizures (FS) are the most common human convulsive event. Children with FS have a higher risk of developing spontaneous afebrile seizures (Baulac et al., 2004). Retrospective studies indicated that 10–15% of patients with epilepsy had antecedent FS (epilepsy with febrile seizures plus, EFS+) (Baulac et al., 2004). The gene associated with FS/EFS+ include ADGRV1, CPA6, DYRK1A, FEB2, FEB5, FEB6, FEB7, FEB9, FEB10, FGF13, GABRB3, GABRD, GABRG2, GEFSP4, GEFSP6, GEFSP8, HCN1, NPRL3, SCN1A, SCN1B, SCN2A, SCN9A, and STX1B (OMIM)1. In this study, we performed trios-based whole-exome sequencing (WES) in a cohort of patients of FS and epilepsy with antecedent FS. PKD1 compound heterozygous mutations were identified in eight unrelated cases, among which seven probands presented epilepsy with antecedent FS. This study suggested that PKD1 gene is potentially a candidate pathogenic gene of FS and epilepsy with antecedent FS.
A total of 314 cases with febrile seizures and epilepsy with antecedent FS were recruited for genetic screening during January 2018–July 2021 from four hospitals, including The Second Affiliated Hospital of Guangzhou Medical University, Shenzhen Children’s Hospital, Guangzhou Women and Children’s Medical Center of Guangzhou Medical University, and Dongguan City Maternal and Child Health Hospital. Clinical characteristics of the affected individuals were collected, including present age, gender, age at seizure onset, seizure course, family history, state of development, effective antiepileptic drugs (AEDs). Magnetic resonance imaging (MRI) scans were performed to detect any brain structure abnormalities. Long-term video-EEG examination was obtained that included open-close eyes test, intermittent photic stimulation, hyperventilation, and sleeping recording. The results were reviewed by two qualified electroencephalographers. Epileptic seizures and epilepsies were diagnosed according to the criteria of the Commission on Classification and Terminology of the ILAE (1981, 1989, 2001, 2010, and 2017). FS was diagnosed with the criteria: (1) a seizure occurring in childhood after age of 1 month to 6 years accompanied by a fever, (2) the febrile illness not caused by central nervous system infection, (3) not meeting criteria for other acute symptomatic seizures. The patients and their parents (trios), and other available family members were screened for genetic variants by the whole-exome sequencing.
For the controls, a cohort of 296 healthy Chinese volunteers was recruited as a normal control group as our previous report (Wang et al., 2021). Frequencies of the identified variants were also compared with that in the other control populations, including East Asian and general populations in the Genome Aggregation Database (gnomAD)2.
This study was approved by the Ethics Committee of The Second Affiliated Hospital of Guangzhou Medical University, and written informed consent was obtained from all patients and their parents.
Whole blood samples were collected from the probands, their parents, and other available family members. Qiagen Flexi Gene DNA kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from the whole blood samples. Trio-based whole-exome sequencing was performed on an Illumina HiSeq 2000 system by BGI-Shenzhen (Shenzhen, China) as previously described (Wang et al., 2021). The sequencing data were generated by massively parallel sequencing with >125 times average depth and >98% coverage in the capture region of the chip to obtain high-quality reads that were mapped to the Genome Reference Consortium Human Genome build 37 (GRCh37) by Burrows-Wheeler alignment. Single-nucleotide point variants and indels were called with the Genome Analysis Toolkit. To obtain the comprehensive list of candidate pathogenic variants in each trio, we adopted a case-by-case analytical pattern. We first prioritized the rare variants with a minor allele frequency <0.005 in the 1000 Genomes Projects, Exome Aggregation Consortium, and gnomAD, and then screened for possibly disease-causing variants in each case under the following five models: (1) epilepsy-associated gene (Wang et al., 2017); (2) de novo variant dominant; (3) autosomal recessive inheritance, including homozygous and compound heterozygous variants; (4) X-linked; and (5) co-segregation analysis. To identify novel epilepsy-associated genes, we put the known epilepsy-associated genes aside (Wang et al., 2017). Genes with repetitively identified de novo variants, bi-allelic variants, hemizygous variants, or variants with segregations, were selected for further studies to define the gene-disease association. PKD1 appeared as a candidate gene with recurrent compound heterozygous variants in this cohort. Sanger sequencing was used to validate the positive findings and the variant origination. All PKD1 variants identified in this study were annotated to reference transcript NM_001009944.2.
To evaluate the pathogenicity of candidate variants, the structure of PC1 was modeled to predict the effect of missense mutations on protein structure by using the Iterative Threading ASSEmbly Refinement software (I-TASSER)3. The confidence of models was quantitatively measured by a C-score in the range of [–5,2]. Three models were predicted and utilized in this study with C-scores of −0.64, −1.14, and −3.02, respectively. PyMOL 2.3 software was used for three-dimensional protein structure visualization and analysis.
To evaluate the protein stability changes upon single nucleotide mutations, the free energy change value (ΔΔG, kcal/mol) was predicted by I-Mutant server4 (Capriotti et al., 2005). Variants were divided into three classes: large increase of protein stability (ΔΔG > 0.5 kcal/mol), large decrease of protein stability (ΔΔG < −0.5 kcal/mol), and neutral stability (−0.5 kcal/mol ≤ ΔΔG ≤ 0.5 kcal/mol). The consequences of all the missense variants were predicted by 21 in silico tools, including SIFT, PolyPhen2_HVAR, CADD, MutationTaster, Fathmm-MKL, fitCons, PhastCons, and GERP++.
Previously, PKD1 mutations were reported to be associated with ADPKD. To explore the genotype-phenotype association of PKD1 mutations, we systematically reviewed all PKD1 mutations in the HGMD database (version: HGMD Professional 2021.3)5 and the ADPKD Variant database (version 4.0)6. The registered variants were reviewed from the references indexed in the databases. The literature was also searched on the PubMed database using the following search items: “biallelic mutation PKD1,” “compound heterozygous mutation PKD1,” and “homozygous mutation PKD1.” Some biallelic PKD1 mutations were reviewed from the cited references of publications from the initial search using the above-mentioned terms. Monoallelic and biallelic PKD1 mutations identified in patients with ADPKD or sporadic PKD were systematically reviewed and classified.
R statistical software (version 4.0.3) was used for data processing. The frequencies of the PKD1 variants between the epilepsy cohort and the controls were compared by a two-sided Fisher exact test (Consortium, 2015). The burden of recessive variants was analyzed according to the method recommended recently (Martin et al., 2018). A p value of <0.05 was considered to be statistically significant.
The Clinical Validity Framework that was developed by Clinical Genome Resource (ClinGen) was performed to evaluate PKD1 as a novel candidate epilepsy gene (Strande et al., 2017).
Eight pairs of compound heterozygous missense variants, including c.3362G > A/p.S1121N and c.8680G > A/p.A2894T, c.5401C > T/p.P1801S and c.6878C > T/p.P2293L c.8744A > G/p.N2915S and c.11689C > T/p.L3897F, c.10315C > T/p.R3439W and c.12391_12392delinsTT/p.E4131L, c.3587C > T/p.T1196M and c.10733C > T/p.A3578V, c.5212C > T/p.L1738F and c.10102G > A/p.D3368N, c.6706T > C/p.F2236L and c.10760C > T/p.A3587V, and c.1966C > G/p.L656V and c.4817C > G/p.T1606S, were identified in eight unrelated cases with FS or EFS+ (Table 1 and Figure 1). The heterozygotes were inherited from their asymptomatic parents, indicating that PKD1 variants in FS and EFS+ followed an autosomal recessive mode of inheritance.
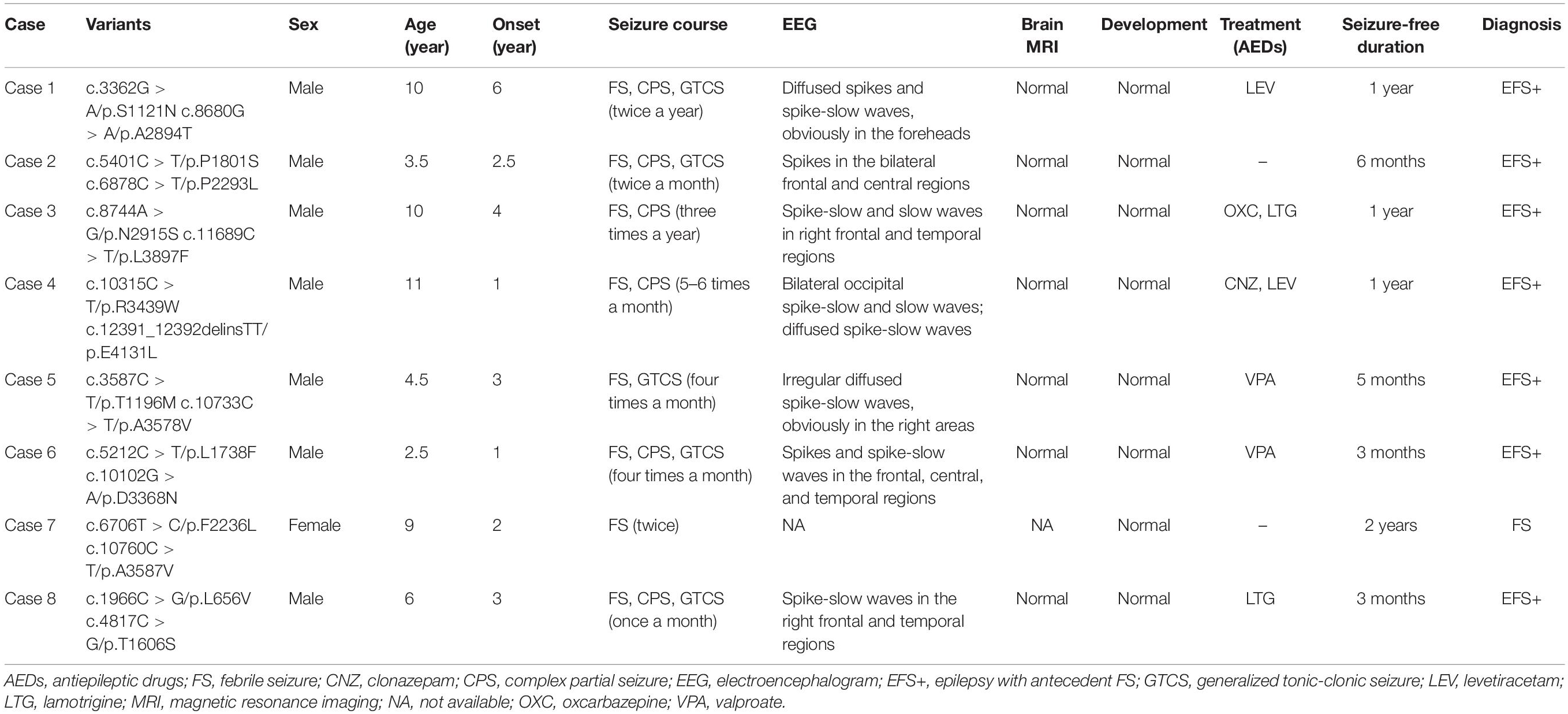
Table 1. Clinical feature of the individuals with PKD1 mutations.
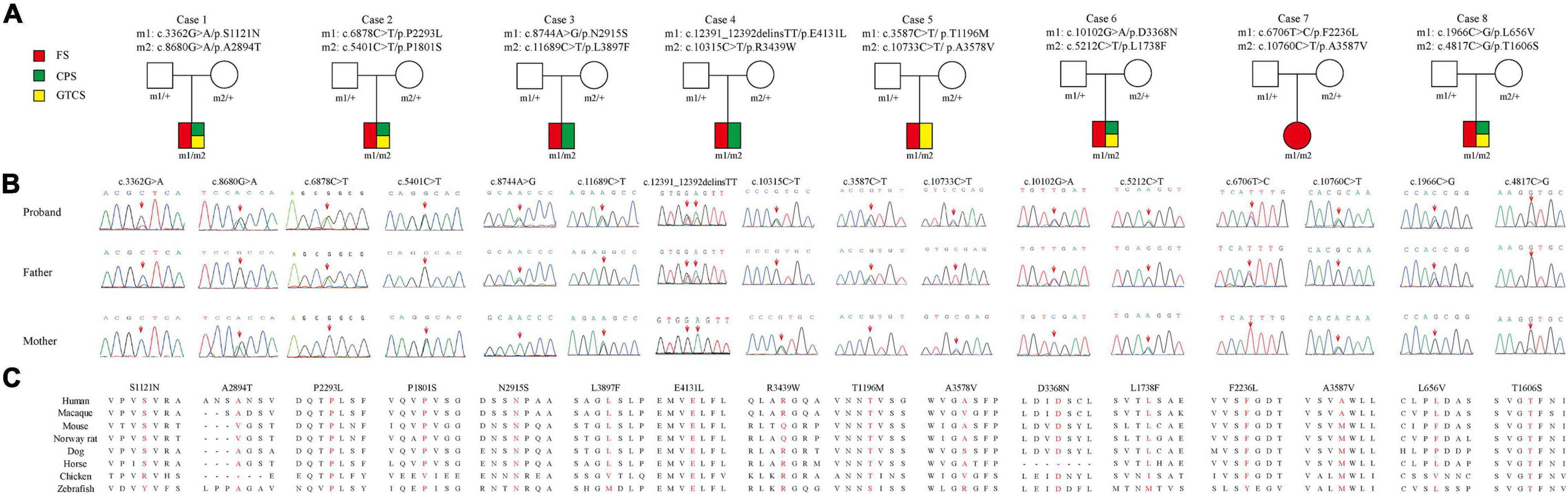
Figure 1. Genetic data of cases with PKD1 mutations. (A) Pedigrees of eight cases with compound heterozygous PKD1 missense mutations and their corresponding phenotypes. FS, febrile seizure; CPS, complex partial seizure; GTCS, generalized tonic-clonic seizure. (B) DNA sequencing chromatogram of PKD1 mutations. Arrows indicate the positions of the mutations. (C) Amino acid sequence alignment of the sixteen missense variants show that residues P2293L, N2915S, E4131L, D3368N, and T1606S are highly conserved in various species; S1121N, P1801S, L3897F, T1196M, L1738F, and F2236L were likely to be highly conserved across mammalian species; while L656V, A2894T, R3439W, A3578V, and A3587V are less conserved.
All heterozygous variants presented no or low allele frequencies in the gnomAD database (Table 2). Homozygotes of these variants were not listed in any public databases. None of the variants were observed in the 296 normal controls. When the burden of recessive variants was analyzed (Martin et al., 2018), the PKD1 variants in the present cohort were significantly more than the expected number by chance in the East Asian population [minor allele frequency (MAF) < 0.005, p = 0.0004208]. Furthermore, the aggregate frequency of the variants in this cohort was significantly higher than that in the five controls (Table 2), including the gnomAD-all population (16/628 vs. 248/221464, p = 2.2 × 10–16), the controls of gnomAD-all population (vs. 112/88270, p = 1.43 × 10–15), the gnomAD-East Asian population (vs. 162/16258, p = 0.001014), the controls of the gnomAD-East Asian population (vs. 76/7610, p = 0.002089), and the 296 normal controls (vs. 0/296, p = 3.06 × 10–5). None of the eight affected patients had pathogenic or likely pathogenic mutations in the 977 genes known to be associated with epileptic phenotypes (Wang et al., 2017).
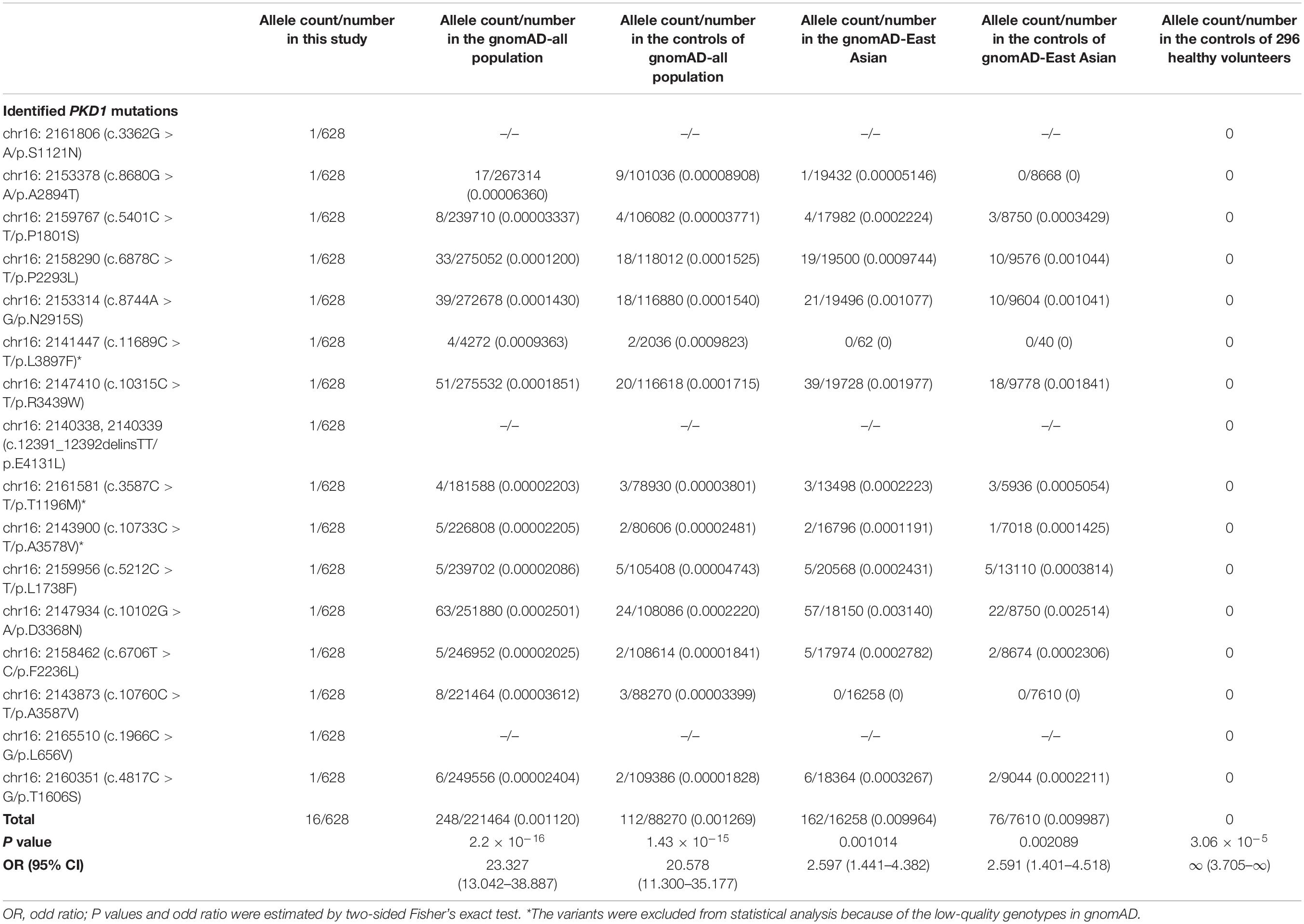
Table 2. A gene-based burden analysis for PKD1 mutations identified in this study.
The missense mutations P2293L, N2915S, E4131L, D3368N, and T1606S affected amino acid residues that are highly conserved in various species, and the residues S1121N, P1801S, L3897F, T1196M, L1738F, and F2236L were likely to be highly conserved across mammalian species, while the missense mutation L656V, A3578V, and A3587V were less conserved by sequence alignment, but were predicted to be conserved by in silico tools (Supplementary Table 1).
The 4303-residue PC1 comprises a long N-terminal extracellular region, eleven transmembrane helices, and a C-terminal coiled-coil domain7. The N-terminal extracellular portion contains leucine-rich repeats (LRR), a C-type lectin domain, immunoglobulin (Ig)-like PKD repeat domains, a single low-density lipoprotein (LDL) receptor motif, the receptor for egg jelly (REJ) domain, and G protein-coupled receptor proteolysis site (GPS; Hughes et al., 1995; Rondeau, 1995; Gallagher et al., 2010; Arac et al., 2012; Ha et al., 2020). The majority (10/16) of missense mutations were located in the N-terminal extracellular region, among which five variants were located in the PKD repeat domains, including S1121N, T1196M, T1606S, L1738F, and P1801S. This region of PC1 likely plays an important role in cell-cell and cell-matrix interactions (Babich et al., 2004). The other six variants were located in transmembrane helices and C-terminal coiled-coil domain, including R3439W and L3368F in TM3–TM4 linker, A3578V in TM4 domain, A3587V in TM5 domain, L3897F in S1–S2 linker and E4131L in C-terminal (Figure 2). No hotspot variant or region was observed.
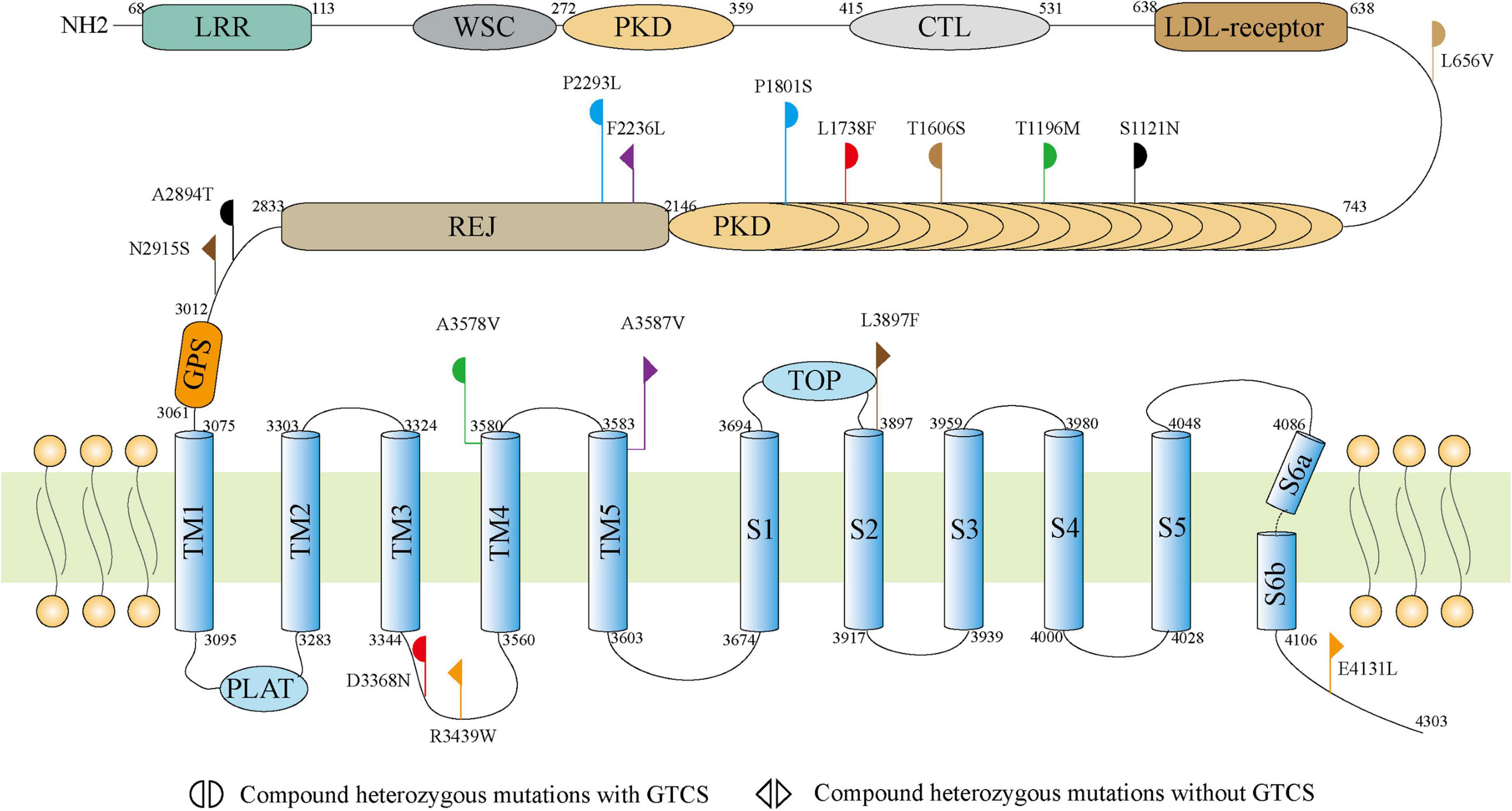
Figure 2. Schematic illustration of the PC1 and locations of the PKD1 mutations identified in this study.
The molecular effect of the missense variants was further analyzed by protein modeling using I-TASSER (Figure 3). Two pairs of compound heterozygous variants (c.8744A > G/p.N2915S and c.11689C > T/p.L3897F, c.12391_12392delinsTT/p.E4131L and c.10315C > T/p.R3439W) had hydrogen bonding changes in both biallelic variants. Another three pairs (c.3362G > A/p.S1121N and c.8680G > A/p.A2894T, c.10102G > A/p.D3368N and c.5212C > T/p.L1738F, c.1966C > G/p.L656V and c.4817C > G/p.T1606S) had hydrogen bonding changes in one of the paired variants. The other three pairs (c.5401C > T/p.P1801S and c.6878C > T/p.P2293L, c.3587C > T/p.T1196M and c.10733C > T/p.A3578V, c.6706T > C/p.F2236L and c.10760C > T/p.A3587V) showed no hydrogen bond changes, but P1801S and F2236L were predicted to decrease the protein stability measured by ΔΔG.
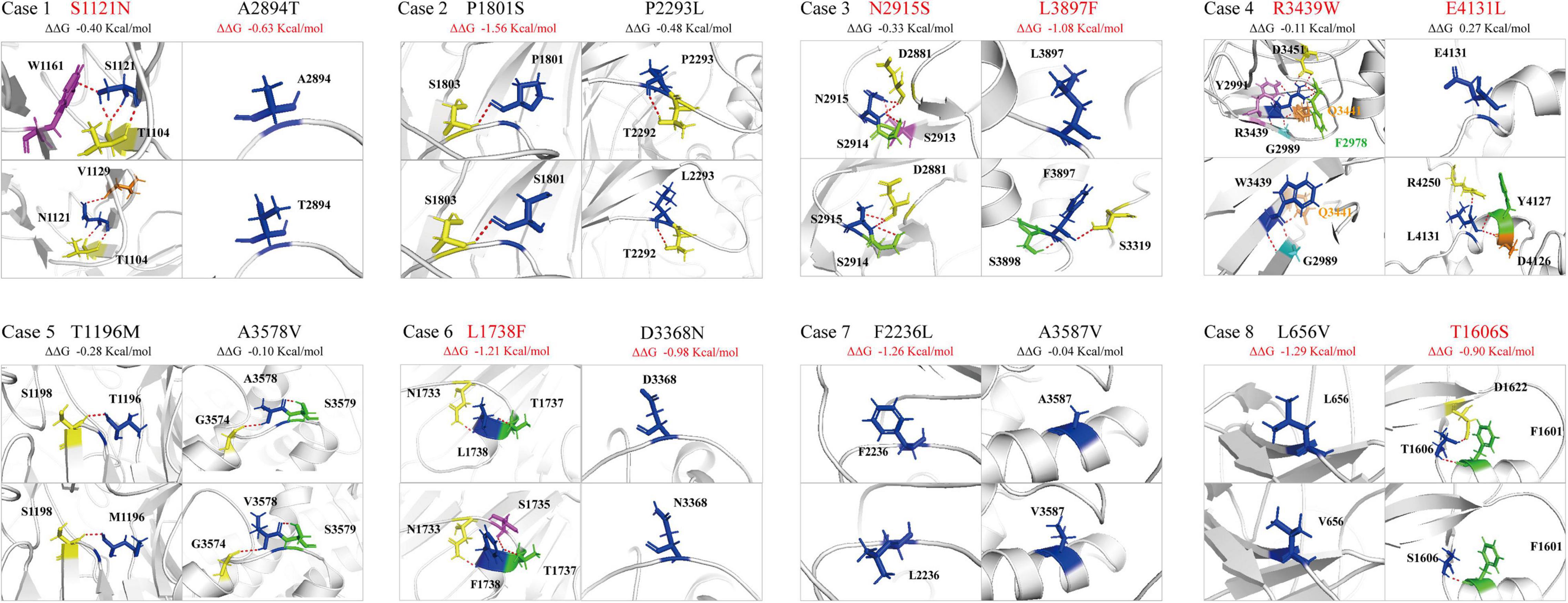
Figure 3. Hydrogen bond changes of the PKD1 mutants. Variants with changes on hydrogen bonding or free energy stability are highlighted in red.
The detailed clinical characteristics of the eight cases with compound heterozygous PKD1 variants were summarized in Table 1. All patients began with FS, which started ranged from 1 to 6 years old, with a median onset age of 2.7 years. Seven patients developed afebrile seizures and were diagnosed as epilepsy with antecedent FS, and one patient (case 7) was affected by only FS. Six of the patients had complex partial seizures. Five patients showed generalized tonic-clonic seizures (GTCS), whom all had one of paired variants that was located in the PKD domains. All patients suffered infrequent seizures that ranged from several episodes to monthly seizures and became seizure-free finally, including two without AEDs treatment (case 2 and 7), four after monotherapy of AEDs (case 1, 5, 6, and 8), and two with combination of two AEDs (case 3 and 4). The EEGs in the seven patients with epilepsy showed focal discharges or diffused discharges with predominance in focal regions (Table 1 and Figure 4). Brain MRI was normal in all cases. All the patients had normal development and showed no enlarged kidneys or kidney cysts.
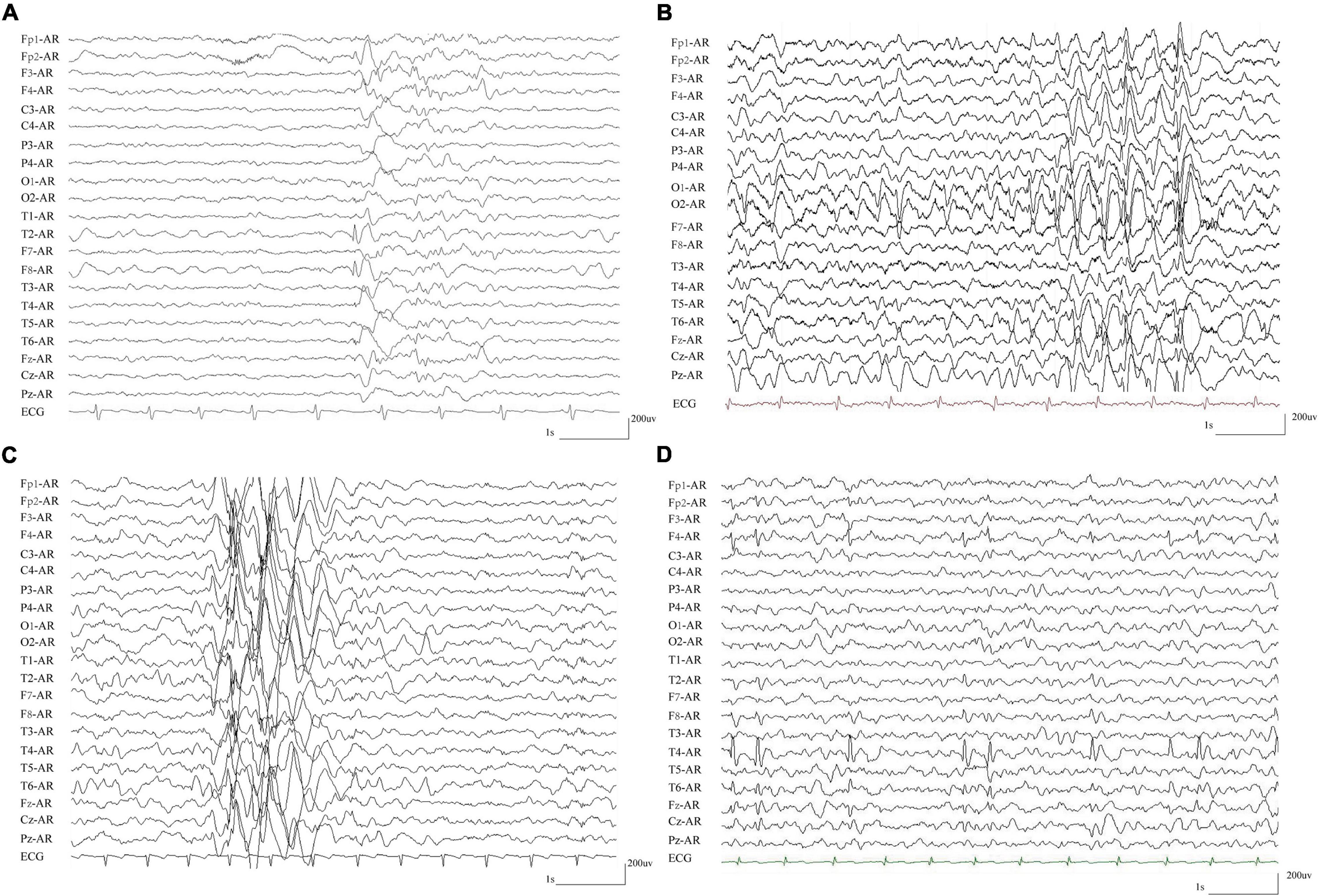
Figure 4. Representative EEG recordings from patients with compound heterozygous PKD1 mutations. (A) Interictal EEG in case 3 showed spike-slow and slow waves in the right anterior frontal and temporal regions. (B) Interictal EEG in case 4 showed spike-slow and slow waves in the bilateral occipital lobes and diffused spike-slow waves. (C) Interictal EEG in case 5 showed irregular diffused spike-slow waves with predominance in the right areas. (D) Interictal EEG in case 8 showed spike-slow waves in the right frontal and temporal regions.
In summary, the patients with compound heterozygous PKD1 variants showed several common features: all began with febrile seizures; suffered infrequent seizures and became seizure-free; focal discharges in EEGs; and normal neurodevelopment.
The expression of PC1 is ubiquitously distributed and developmentally regulated, predominantly during embryonic, infant, and adult stage. Tissue-specific expression is the basis of gene function and subsequently the clinical phenotype. We thus compared the expression of PKD1 in the human brain and the kidney. In UniGene database, PKD1 in the brain is expressed 1.79 times as much as that in the kidney (Figure 5A)8. Furthermore, data from the GTEx database showed that PKD1 was highly and widely expressed in all sub-regions of the brain, including the cortex, hypothalamus and hippocampus. Expression of PKD1 in these sub-regions of the brain was higher than that in the kidney, for example, the expression in the brain cortex was 6.10 times higher than that in the kidney cortex (Figure 5B)9.
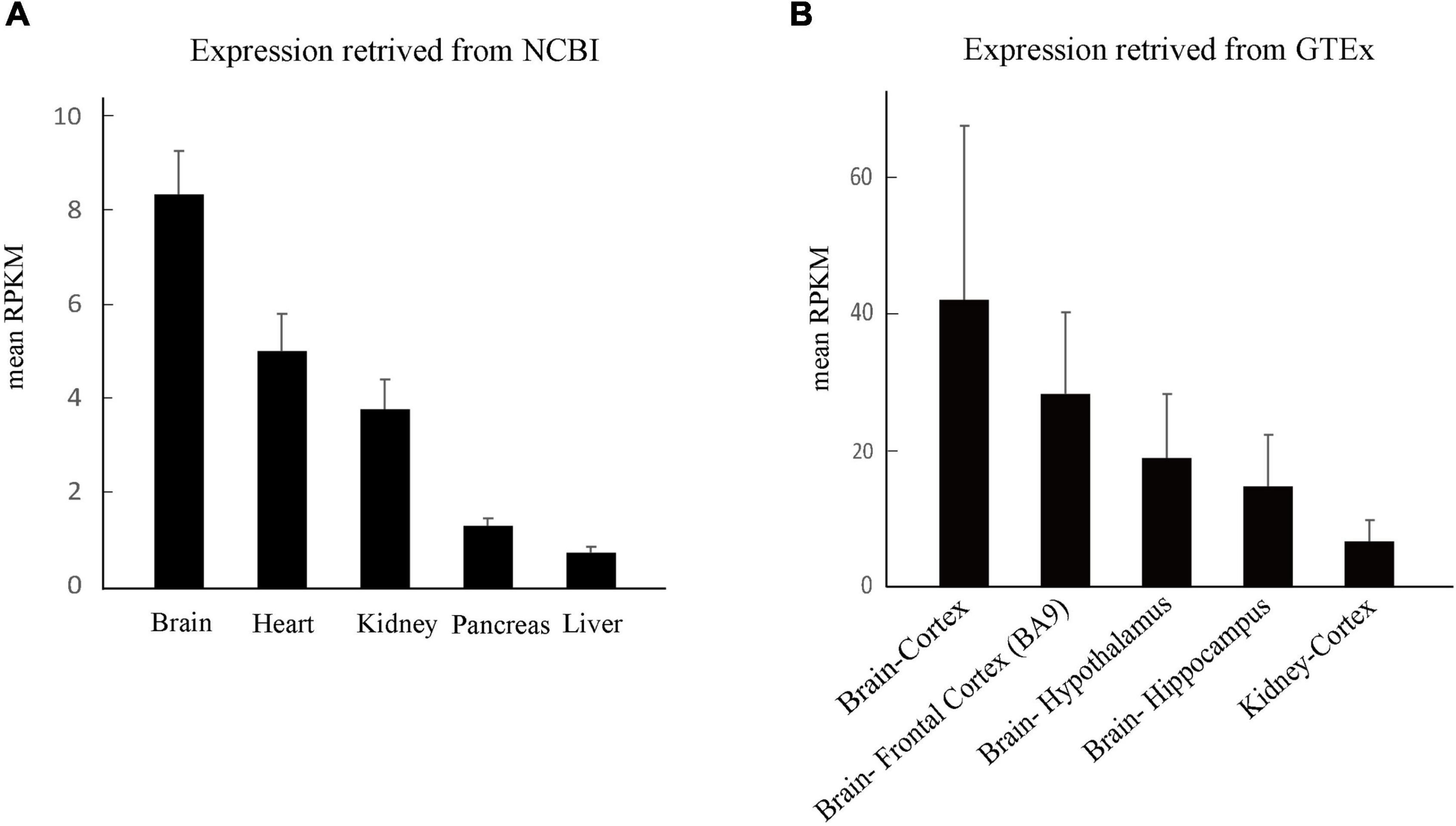
Figure 5. Tissue expression of PKD1. (A) The overall expression of PKD1 in different tissues retrieved from NCBI. (B) Comparison of PKD1 expression in the sub-regions of human brain and kidney retrieved from GTEx.
To explore the mechanism underlying phenotypic variations, we systematically reviewed PKD1 mutations and analyzed correlations between genotypes and phenotypes. To date, a total of 2599 mutations have been registered to be associated with ADPKD or sporadic PKD in the HGMD database and ADPKD Mutation Database. ADPKD/PKD-associated PKD1 mutations were mainly monoallelic mutations (90.9%, 2360/2599), among which majority (72.3%, 1706/2360) were destructive mutations, including nonsense, splicing defects, frameshifting, in frame deletion/insertions, and large rearrangements. A small portion (9.1%) of the 2599 mutations were reported as biallelic mutations in ADPKD/PKD, including 114 pairs of compound heterozygotes and nine pairs of homozygotes. It was noticed that the nine pairs of PKD1 homozygotes were associated with a severe phenotype in 14 individuals, and 21.4% of the patients with homozygotes (3/14) were premature neonate died and others (71.4%, 10/14) exhibited in utero onset ADPKD (Cornec-Le Gall et al., 2018; Al-Hamed et al., 2019; Garel et al., 2019; Durkie et al., 2021).
In contrast, in the present study, FS/EFS+ associated PKD1 mutations were all compound heterozygous missense mutations (100%, 16/16).
We evaluated the PKD1-epilepsy correlation by using ClinGen Clinical-Validity Framework. The total allowable points from clinical-genetic aspect were 7.5 points and that from experimental aspect were 6 points. The results of clinical validity summary matrix were 13.5 points that was categorized as “Strong,” supporting the association between PKD1 variants and FS/EFS+ (Table 3).
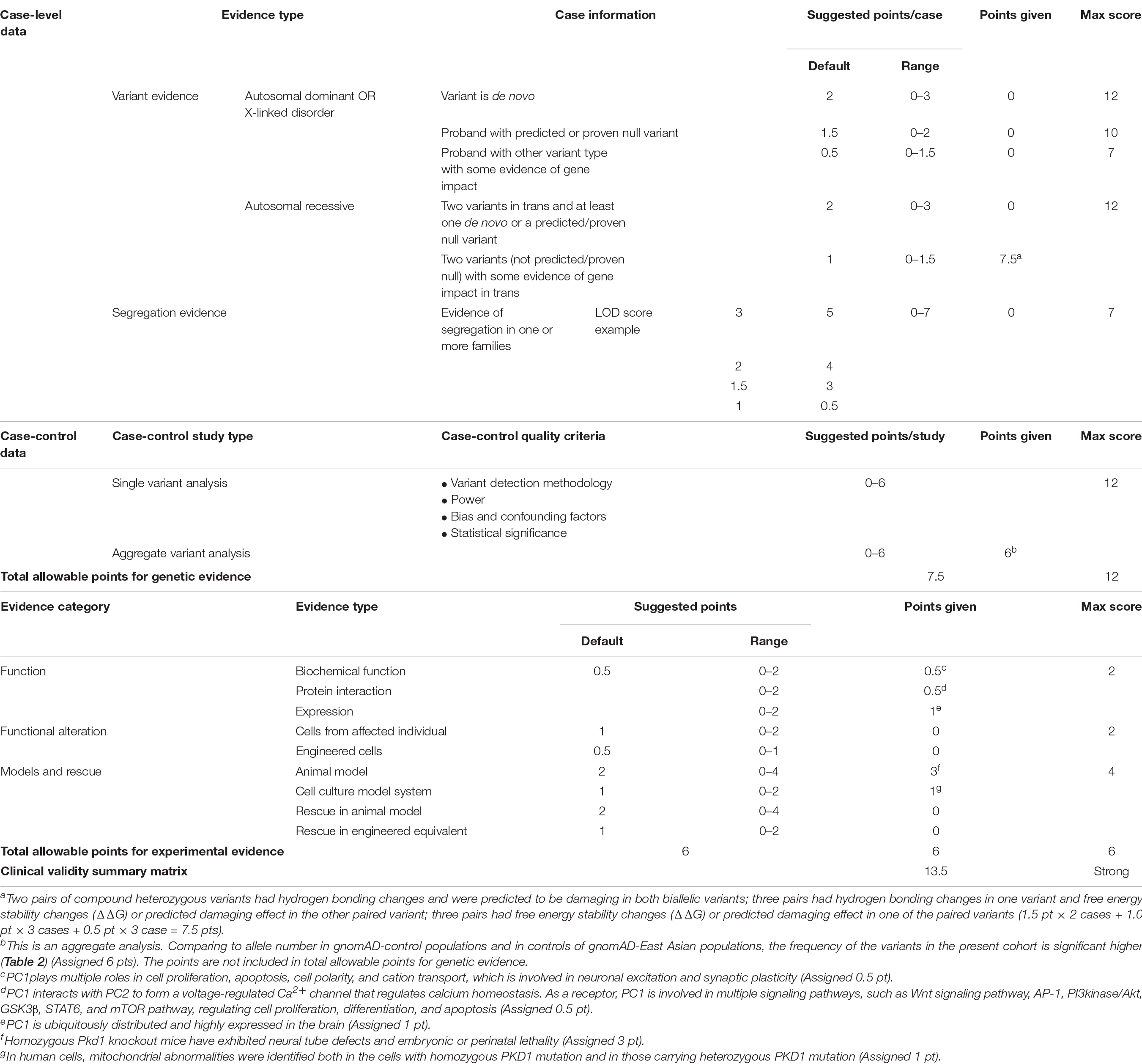
Table 3. Evaluating the clinical validity of PKD1-epilepsy associations based on the framework developed by the clinical genome resource.
PKD1 encodes PC1, a large transmembrane protein that plays multiple roles in cell proliferation, apoptosis, cell polarity, and cation transport (Paul and Vanden Heuvel, 2014). Previous studies have demonstrated that PKD1 is the major causative gene of ADPKD, of which mutations are responsible for 85% of ADPKD cases (Paul and Vanden Heuvel, 2014; Kim and Park, 2016). However, the expression of PC1 in the brain is much higher than that in the kidney (Figures 5A,B), suggesting a role in the function of the brain. In the present study, compound heterozygous PKD1 mutations were identified in eight unrelated cases with FS and epilepsy with antecedent FS. The frequency of the PKD1 variants in the cohort of epilepsy was significantly higher than that in control populations in gnomAD. These findings suggested that PKD1 was potentially associated with epilepsy. The evaluation from ClinGen Clinical-Validity Framework also supports a strong association between PKD1 mutations and epilepsy.
The transmembrane protein PC1 comprises a large extracellular N terminus, eleven membrane-spanning domains, and a short cytoplasmic C terminus. It functions as a channel subunit, cell surface receptor, and G-protein coupled receptor (Hu and Harris, 2020). PC1 may form ion channel pore by itself or contribute to channel pore via formation of heteromultimeric channels with PC2 (Babich et al., 2004). Generally, PC1 interacts with PC2 through the C-terminal tail to form a voltage-regulated Ca2+ channel, which regulates intracellular calcium influx. The simultaneous presence of both PC1 and PC2 amplify the inositol trisphosphate-induced Ca2+ release (Mekahli et al., 2012). Expression of PC1 can also regulate channel activity independent of the channel activity of PC2 (Babich et al., 2004). As a constitutive activator of G-proteins, PC1 activates endogenous voltage-activated Ca2+ channels and G protein-activated inward rectifying K+ channels in sympathetic neurons (Delmas et al., 2002). Deletion of PC1 caused complete loss of ionic currents (Babich et al., 2004). The calcium homeostasis is critical for neuronal stability and excitation, and the intracellular Ca2+ is essential for many basic cellular processes of neurons. Besides, PC1 is extensively expressed in the brain, especially in the cortex and hippocampus. These findings provide an electrophysiological and an anatomical basis for epileptogenesis.
As a receptor, PC1 is involved in multiple signaling pathways, such as Wnt signaling pathway, AP-1, PI3kinase/Akt, GSK3β, STAT6, and the mammalian target of rapamycin (mTOR) pathway (Boca et al., 2007; Boletta, 2009; Holditch et al., 2019). PC1 inhibits mTOR activity by stabilizing the tuberous sclerosis 1-tuberous sclerosis 2 (TSC1–TSC2) complex, which is known as a negative regulator of the mTOR complex (Boletta, 2009; Distefano et al., 2009). Through mediating signaling, PC1 regulates cell proliferation, differentiation, and apoptosis (Paul and Vanden Heuvel, 2014). Homozygous Pkd1 knockout mice have exhibited neural tube defects and embryonic lethality (Lu et al., 2001; Lantinga-van Leeuwen et al., 2007), indicating that PKD1 is important for the development of the brain. In the present study, the seven patients with EFS+ and PKD1 mutations had complex partial seizures and/or focal discharges in EEGs, suggesting potential neurodevelopmental abnormalities. However, whether the PKD1 variants were associated with brain malformation warrants further studies with large cohorts and advanced neuroimaging techniques.
It was noticed that five patients with GTCS had one of the paired missense variants that was located in the Ig-like PKD repeat domains of the N terminus. The PKD repeat domains play an important role in the PC1-dependent channel activity (Babich et al., 2004), possibly through regulating homophilic cell-cell/cell-matrix interactions (Ibraghimov-Beskrovnaya et al., 2000). Extracellular application of antibodies against the Ig-like PKD domains disrupted cell-cell adhesion, and reduces PC1-dependent ionic currents (Ibraghimov-Beskrovnaya et al., 2000; Babich et al., 2004). These data suggested that mutations in the PKD domains were possibly associated with GTCS, which warrants further validation by large cohorts and functional studies.
Previously reported PKD1 mutations were mainly associated with ADPKD. From the genotype aspect, ADPKD-associated PKD1 mutations were mainly monoallelic, and majority of the mutations were destructive (Sandford, 2009; Cornec-Le Gall et al., 2014; Lanktree et al., 2021), implying that monoallelic mutations with haploinsufficiency of PKD1 potentially caused kidney disease. Less commonly, truncating PKD1 mutations were identified in families ADPKD (Lanktree et al., 2021). Homozygotes of PKD1 mutations caused severe phenotype with in utero onset ADPKD or premature neonate death (Cornec-Le Gall et al., 2018; Al-Hamed et al., 2019; Garel et al., 2019; Durkie et al., 2021), consistent with the embryonic lethality in homozygous Pkd1 knockout mice (Lu et al., 2001; Lantinga-van Leeuwen et al., 2007). A quantitative correlation between genetic impairment and phenotypic severity was suggested. In the present study, the PKD1 mutations identified in our patients with FS/EFS+ were all compound heterozygous missense mutations. All patients showed infrequent seizures and became seizure-free without AEDs treatment or after treatments of one or two AEDs. All the children showed no enlarged kidney or kidney cysts at present. Whether they will develop kidney abnormalities in later adulthood needs further follow-up. On the other hand, attention should be paid to whether the PKD patients with compound heterozygous variants had self-limited FS or mild seizures in their early life.
This study has several limitations. The direct functional effects of the mutations were not examined. The whole spectrum of phenotype of PKD1 mutations warrants further determination with large cohorts.
This study identified eight pairs of compound heterozygous missense mutations in patients with FS/EFS+. These mutations presented significantly higher frequency in case cohort than that in the control populations. Taken together the data from gene expression profile, gene functions, and PKD1 deficiency animal model, it is suggested that PKD1 was potentially a novel cause of epilepsy. Further analysis revealed that monoallelic mutations with haploinsufficiency of PKD1 were associated with PKD, homozygotes with complete loss of PC1 would be embryonically lethal, whereas compound heterozygotes with superimposed effects of two missense mutations were potentially associated with epilepsy with good prognosis. The genotype-phenotype correlation helps explaining phenotypical variations.
The original contributions presented in the study are publicly available. This data can be found here: NCBI, OM969823, and OM969870.
The studies involving human participants were reviewed and approved by the Ethics Committee of The Second Affiliated Hospital of Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
NH, J-YW, and JW contributed to the conception of the study, interpretation of clinical data, and drafting of the manuscript. X-GL, WS, SL, D-FZ, L-DH, QP, YT, L-DG, and W-PL examined the patients and participated in drafting of the manuscript. W-PL provided critical review and substantially revised the manuscript. All authors read and approved the manuscript before sending the manuscript to the journal for publication.
This work was funded by the National Natural Science Foundation of China (Grant No. 81971216 to NH), Science and Technology Project of Guangdong Province (Grant No. 2017B030314159 to W-PL), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2020A1515011048 to NH), Science and Technology Project of Guangzhou (Grant Nos. 201904010292 to NH and 201904020028 to W-PL), and UCB Pharma Ltd., Joint Science Research Foundation of China Association Against Epilepsy (Grant No. 2020006B to NH). The funders had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The help of patients and clinicians participating in this work are greatly appreciated.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.861159/full#supplementary-material
Al-Hamed, M. H., Alsahan, N., Rice, S. J., Edwards, N., Nooreddeen, E., Alotaibi, M., et al. (2019). Bialleleic PKD1 mutations underlie early-onset autosomal dominant polycystic kidney disease in Saudi Arabian families. Pediatr. Nephrol. 34, 1615–1623. doi: 10.1007/s00467-019-04267-x
Arac, D., Boucard, A. A., Bolliger, M. F., Nguyen, J., Soltis, S. M., Sudhof, T. C., et al. (2012). A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31, 1364–1378. doi: 10.1038/emboj.2012.26
Babich, V., Zeng, W. Z., Yeh, B. I., Ibraghimov-Beskrovnaya, O., Cai, Y., Somlo, S., et al. (2004). The N-terminal extracellular domain is required for polycystin-1-dependent channel activity. J. Biol. Chem. 279, 25582–25589. doi: 10.1074/jbc.M402829200
Baulac, S., Gourfinkel-An, I., Nabbout, R., Huberfeld, G., Serratosa, J., Leguern, E., et al. (2004). Fever, genes, and epilepsy. Lancet Neurol. 3, 421–430. doi: 10.1016/S1474-4422(04)00808-7
Boca, M., D’Amato, L., Distefano, G., Polishchuk, R. S., Germino, G. G., and Boletta, A. (2007). Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol. Biol. Cell 18, 4050–4061. doi: 10.1091/mbc.e07-02-0142
Boletta, A. (2009). Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics 2:6. doi: 10.1186/1755-8417-2-6
Capriotti, E., Fariselli, P., and Casadio, R. (2005). I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310. doi: 10.1093/nar/gki375
Consortium, C. (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. doi: 10.1038/nature14659
Cornec-Le Gall, E., Audrezet, M. P., Le Meur, Y., Chen, J. M., and Ferec, C. (2014). Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum. Mutat. 35, 1393–1406. doi: 10.1002/humu.22708
Cornec-Le Gall, E., Torres, V. E., and Harris, P. C. (2018). Genetic complexity of autosomal dominant polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 29, 13–23. doi: 10.1681/ASN.2017050483
Delmas, P., Nomura, H., Li, X., Lakkis, M., Luo, Y., Segal, Y., et al. (2002). Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J. Biol. Chem. 277, 11276–11283. doi: 10.1074/jbc.M110483200
Distefano, G., Boca, M., Rowe, I., Wodarczyk, C., Ma, L., Piontek, K. B., et al. (2009). Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell Biol. 29, 2359–2371. doi: 10.1128/MCB.01259-08
Durkie, M., Chong, J., Valluru, M. K., Harris, P. C., and Ong, A. C. M. (2021). Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genet. Med. 23, 689–697. doi: 10.1038/s41436-020-01026-4
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Gallagher, A. R., Germino, G. G., and Somlo, S. (2010). Molecular advances in autosomal dominant polycystic kidney disease. Adv. Chronic Kidney Dis. 17, 118–130. doi: 10.1053/j.ackd.2010.01.002
Garel, J., Lefebvre, M., Cassart, M., Della Valle, V., Guilbaud, L., Jouannic, J. M., et al. (2019). Prenatal ultrasonography of autosomal dominant polycystic kidney disease mimicking recessive type: case series. Pediatr. Radiol. 49, 906–912. doi: 10.1007/s00247-018-4325-3
Geng, L., Segal, Y., Pavlova, A., Barros, E. J., Lohning, C., Lu, W., et al. (1997). Distribution and developmentally regulated expression of murine polycystin. Am. J. Physiol. 272, F451–F459. doi: 10.1152/ajprenal.1997.272.4.F451
Ha, K., Nobuhara, M., Wang, Q., Walker, R. V., Qian, F., Schartner, C., et al. (2020). The heteromeric PC-1/PC-2 polycystin complex is activated by the PC-1 N-terminus. eLife 9:e60684. doi: 10.7554/eLife.60684
Holditch, S. J., Brown, C. N., Atwood, D. J., Lombardi, A. M., Nguyen, K. N., Toll, H. W., et al. (2019). A study of sirolimus and mTOR kinase inhibitor in a hypomorphic Pkd1 mouse model of autosomal dominant polycystic kidney disease. Am. J. Physiol. Renal Physiol. 317, F187–F196. doi: 10.1152/ajprenal.00051.2019
Hu, J., and Harris, P. C. (2020). Regulation of polycystin expression, maturation and trafficking. Cell Signal 72:109630. doi: 10.1016/j.cellsig.2020.109630
Hughes, J., Ward, C. J., Peral, B., Aspinwall, R., Clark, K., San Millan, J. L., et al. (1995). The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10, 151–160. doi: 10.1038/ng0695-151
Ibraghimov-Beskrovnaya, O., Bukanov, N. O., Donohue, L. C., Dackowski, W. R., Klinger, K. W., and Landes, G. M. (2000). Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene. PKD1. Hum. Mol. Genet. 9, 1641–1649. doi: 10.1093/hmg/9.11.1641
Kim, D. Y., and Park, J. H. (2016). Genetic Mechanisms of ADPKD. Adv. Exp. Med. Biol. 933, 13–22. doi: 10.1007/978-981-10-2041-4_2
Kim, S., Nie, H., Nesin, V., Tran, U., Outeda, P., Bai, C. X., et al. (2016). The polycystin complex mediates Wnt/Ca(2+) signalling. Nat. Cell Biol. 18, 752–764. doi: 10.1038/ncb3363
Lanktree, M. B., Guiard, E., Akbari, P., Pourafkari, M., Iliuta, I. A., Ahmed, S., et al. (2021). Patients with Protein-Truncating PKD1 Mutations and Mild ADPKD. Clin. J. Am. Soc. Nephrol. 16, 374–383. doi: 10.2215/CJN.11100720
Lantinga-van Leeuwen, I. S., Leonhard, W. N., van der Wal, A., Breuning, M. H., de Heer, E., and Peters, D. J. (2007). Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum. Mol. Genet. 16, 3188–3196. doi: 10.1093/hmg/ddm299
Lu, W., Shen, X., Pavlova, A., Lakkis, M., Ward, C. J., Pritchard, L., et al. (2001). Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet. 10, 2385–2396. doi: 10.1093/hmg/10.21.2385
Martin, H. C., Jones, W. D., McIntyre, R., Sanchez-Andrade, G., Sanderson, M., Stephenson, J. D., et al. (2018). Quantifying the contribution of recessive coding variation to developmental disorders. Science 362, 1161–1164. doi: 10.1126/science.aar6731
Mekahli, D., Sammels, E., Luyten, T., Welkenhuyzen, K., van den Heuvel, L. P., Levtchenko, E. N., et al. (2012). Polycystin-1 and polycystin-2 are both required to amplify inositol-trisphosphate-induced Ca2+ release. Cell Calcium 51, 452–458. doi: 10.1016/j.ceca.2012.03.002
Paul, B. M., and Vanden Heuvel, G. B. (2014). Kidney: polycystic kidney disease. Wiley Interdiscip. Rev. Dev. Biol. 3, 465–487. doi: 10.1002/wdev.152
Rondeau, E. (1995). Polycystic kidney: complete structure of the PKD1 gene and its protein. Nephrologie 16, 338–339.
Sandford, R. N. (2009). The diversity of PKD1 alleles: implications for disease pathogenesis and genetic counseling. Kidney Int. 75, 765–767. doi: 10.1038/ki.2009.17
Semmo, M., Kottgen, M., and Hofherr, A. (2014). The TRPP subfamily and polycystin-1 proteins. Handb. Exp. Pharmacol. 222, 675–711. doi: 10.1007/978-3-642-54215-2_27
Stokely, M. E., Hwang, S. Y., Hwang, J. Y., Fan, B., King, M. A., Inokuchi, K., et al. (2006). Polycystin-1 can interact with homer 1/Vesl-1 in postnatal hippocampal neurons. J. Neurosci. Res. 84, 1727–1737. doi: 10.1002/jnr.21076
Strande, N. T., Riggs, E. R., Buchanan, A. H., Ceyhan-Birsoy, O., DiStefano, M., Dwight, S. S., et al. (2017). Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 100, 895–906. doi: 10.1016/j.ajhg.2017.04.015
Su, Q., Hu, F., Ge, X., Lei, J., Yu, S., Wang, T., et al. (2018). Structure of the human PKD1-PKD2 complex. Science 361:eaat9819. doi: 10.1126/science.aat9819
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Keywords: PKD1 gene, epilepsy with antecedent febrile seizures, genotype-phenotype association, monoallelic mutation, compound heterozygous mutations
Citation: Wang J-Y, Wang J, Lu X-G, Song W, Luo S, Zou D-F, Hua L-D, Peng Q, Tian Y, Gao L-D, Liao W-P and He N (2022) Recessive PKD1 Mutations Are Associated With Febrile Seizures and Epilepsy With Antecedent Febrile Seizures and the Genotype-Phenotype Correlation. Front. Mol. Neurosci. 15:861159. doi: 10.3389/fnmol.2022.861159
Received: 24 January 2022; Accepted: 05 April 2022;
Published: 10 May 2022.
Edited by:
Yuwu Jiang, Peking University First Hospital, ChinaReviewed by:
Saima Siddiqi, Institute of Biomedical and Genetic Engineering (IBGE), PakistanCopyright © 2022 Wang, Wang, Lu, Song, Luo, Zou, Hua, Peng, Tian, Gao, Liao and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na He, aGVuYWNoaWxsaUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.