
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 13 April 2022
Sec. Molecular Signalling and Pathways
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.860410
This article is part of the Research Topic Pathways and Processes Underpinning Axonal Biology and Pathobiology View all 11 articles
 Colin L. Crawford1
Colin L. Crawford1 Christina Antoniou2
Christina Antoniou2 Lina Komarek1,3
Lina Komarek1,3 Verena Schultz1
Verena Schultz1 Claire L. Donald4
Claire L. Donald4 Paul Montague1
Paul Montague1 Susan C. Barnett1Christopher Linington1
Susan C. Barnett1Christopher Linington1 Hugh J. Willison1
Hugh J. Willison1 Alain Kohl4
Alain Kohl4 Michael P. Coleman2*
Michael P. Coleman2* Julia M. Edgar1,3*
Julia M. Edgar1,3*Zika virus (ZIKV) is a neurotropic flavivirus recently linked to congenital ZIKV syndrome in children and encephalitis and Guillain-Barré syndrome in adults. Neurotropic viruses often use axons to traffic to neuronal or glial cell somas where they either remain latent or replicate and proceed to infect new cells. Consequently, it has been suggested that axon degeneration could represent an evolutionarily conserved mechanism to limit viral spread. Whilst it is not known if ZIKV transits in axons, we previously reported that ZIKV infection of glial cells in a murine spinal cord-derived cell culture model of the CNS is associated with a profound loss of neuronal cell processes. This, despite that postmitotic neurons are relatively refractory to infection and death. Here, we tested the hypothesis that ZIKV-associated degeneration of neuronal processes is dependent on activation of Sterile alpha and armadillo motif-containing protein 1 (SARM1), an NADase that acts as a central executioner in a conserved axon degeneration pathway. To test this, we infected wild type and Sarm1 homozygous or heterozygous null cell cultures with ZIKV and examined NAD+ levels as well as the survival of neurons and their processes. Unexpectedly, ZIKV infection led to a rapid SARM1-independent reduction in NAD+. Nonetheless, the subsequent profound loss of neuronal cell processes was SARM1-dependent and was preceded by early changes in the appearance of β-tubulin III staining. Together, these data identify a role for SARM1 in the pathogenesis of ZIKV infection, which may reflect SARM1's conserved prodegenerative function, independent of its NADase activity.
The emergence of mosquito-borne Zika virus (ZIKV; a positive-strand RNA virus of the Flaviviridae family) in Pacific Ocean islands and the Americas added this previously little-known pathogen to the list of neurotropic flaviviruses (Baud et al., 2017; Gubler et al., 2017; Pierson and Diamond, 2018; Pardy and Richer, 2019). The association of ZIKV with multiple neurological complications including congenital ZIKV syndrome, encephalitis and Guillain-Barré syndrome (Gulland, 2016) makes it particularly important to understand the underlying mechanism. We previously demonstrated that post-mitotic murine CNS neurons are relatively refractory to infection by ZIKV, which productively infects oligodendroglia and astroglia in murine models of the perinatal CNS (Cumberworth et al., 2017; Schultz et al., 2021). Surprisingly, despite the paucity of infected neuronal somas and insignificant death of neurons, ZIKV infection led to a profound loss of axons and dendrites (Cumberworth et al., 2017). Thus, we hypothesized this involves a mechanism related to Wallerian degeneration, where axons also die independently from the soma (Coleman and Höke, 2020), following insults that include physical injury, toxins, and genetic mutation (Osterloh et al., 2012; Conforti et al., 2014; Coleman and Höke, 2020).
SARM1 (Sterile alpha and armadillo motif-containing protein 1) has a well-established role in Wallerian degeneration and is regarded as a central executioner in this conserved axon degeneration pathway involving NAD-related metabolism. The SARM1 TIR domain has NAD+ consuming glycohydrolase (NADase) activity, considered critical for its prodegenerative capacity (Essuman et al., 2017; Horsefield et al., 2019), although it has additional NADP hydrolase and base exchange activities that could also play important roles (Angeletti et al., 2021). As axons are often used by neurotropic viruses to enter neuronal and glial somas where the cellular machinery they use to replicate is located (Richards et al., 2021) it has been suggested that the known response of SARM1 to bunyavirus or rabies virus infection (both negative strand RNA viruses) could represent a mechanism to limit the spread of neurotropic virus (Mukherjee et al., 2013; Sundaramoorthy et al., 2020).
Notably, SARM1 also has a role in innate immunity since it contains a highly conserved Toll/Il-1 receptor (TIR) domain-containing protein, classified as a member of the toll-like receptor (TLR) adaptor family (Carty and Bowie, 2019). However, unlike other TLR adapters, SARM1 is predominantly expressed in neurons (Lin et al., 2014) and was initially described as an inhibitor of TLR signaling (Carty et al., 2006). Subsequent studies have suggested positive contributions of SARM1 to innate immunity through activation of transcriptional programmes that regulate the production of cytokines and chemokines, including in the nervous system (Carty and Bowie, 2019; Hopkins et al., 2021).
The SARM1-dependent loss of axons and dendrites with absent or delayed neuronal cell death following infection by bunyavirus (Mukherjee et al., 2013) or rabies virus (Sundaramoorthy et al., 2020) could thus provide an intriguing link between innate immunity and axon degeneration mechanisms. To investigate any role SARM1 may play in ZIKV-induced death of neuronal processes, we infected CNS myelinating spinal cord cultures from wild type mice, or Sarm1 heterozygous or homozygous null mice (Kim et al., 2007) on a type I interferon receptor (Ifnar1) null background; the last to facilitate infection (Miner and Diamond, 2017). As previously (Cumberworth et al., 2017), Sarm1 wild type neuronal soma were relatively refractory to infection, but glial cell infection was accompanied by progressive degeneration of neuronal processes. In contrast, there was prolonged survival of neuronal processes in Sarm1 null cultures, accompanied by increased infection and death of neuronal somas. Surprisingly, this pro-degenerative role of SARM1 appeared to be independent of its NADase activity, although ZIKV infection itself caused NAD+ depletion in all three Sarm1 genotypes.
Type I interferon receptor deficient (Ifnar1−/−) mice, which recapitulate aspects of human ZIKV infection and disease (Miner and Diamond, 2017) (129S7/SvEvBrdBkl-Hprtb-m2 background; B&K Universal) and Sarm1−/− mice (maintained on a C57BL6/J background) (Kim et al., 2007) were bred to produce Ifnar1+/−::Sarm1+/− mice. These were further crossed to produce Ifnar1−/−::Sarm1+/− mice from which Ifnar1−/−::Sarm1+/+, +/− or −/− embryos were generated. Mice were maintained in Tecniplast 1284L Blue line IVC cages, in a 12-h light/dark cycle and provided ad libitum with sterile food and water. Mice were time-mated and pregnant females were killed by rising CO2 overdose and cervical dislocation on embryonic day (E) 13. All animal studies were approved by the Ethical Committee of the University of Glasgow and licensed by the UK Home Office (Project License number PPL P78DD6240).
Myelinating cell cultures were established using E13 mouse spinal cord as described in detail previously (Thomson et al., 2006; Bijland et al., 2019), with the exception that cultures were generated from single embryos rather than pooled embryos. A small piece of tail (~1 mm) was taken for retrospective genotyping.
Genomic DNA was extracted from ear (adult) or tail (embryo) biopsies using a protocol modified from Truett et al. (2000). Briefly, tissue was lysed at 95°C for 90 min in 50 mM NaOH. Following neutralization with 10% v/v 1 M Tris pH 5, the resultant solution was vortexed and diluted 1:10 in nuclease-free water. Ifnar1 genotyping was conducted as described previously (Cumberworth et al., 2017). For Sarm1 genotyping, 1 μl of the diluted genomic DNA lysate was amplified in a 12.5 μl PCR reaction in a 1x RedTaq Reaction mix (Sigma-Aldrich) comprising 0.4 mM dNTP, 0.06 U/μl RedTaq Polymerase, 3 mM MgCl2 and 0.2 μM Sarm1 primers. Cycling parameters were an initial denaturation at 95°C for 5 min, then 35 cycles (95°C 1 min; 60°C 1 min; 72°C 1 min) followed by a final elongation at 72°C for 10 min. The PCR products were resolved on standard 2% TAE agarose gels.
Sarm1 primers were described previously (Gilley et al., 2015). Primer pairs 5′-ACGCCTGGTTTCTTACTCTACG-3′ and 5′- CCTTACCTCTTGC GGGTGATGC-3′ amplify wild type Sarm1, producing a 508-bp product. Primers 5′-GGTAGCCGGATCAAGCGTATGC-3′ and 5′- CTCATCTCCGGGCCTTTCGACC-3′ amplifies the neomycin resistance cassette retained in the knockout allele (Kim et al., 2007), producing a 450-bp product.
The Brazilian strain of ZIKV, ZIKV/H. sapiens/Brazil/PE243/2015 (GenBank accession number KX197192; abbreviated ZIKV PE243; referred to subsequently as ZIKV) has been described previously (Donald et al., 2016). CNS cultures were infected with ZIKV at a multiplicity of infection (MOI) of 0.3 or 0.6 for 1 h at 37°C in 2% fetal bovine serum (FBS) in cell culture grade PBS (10010023, Gibco™) as described previously (Cumberworth et al., 2017). Controls (mock-infected) were treated in parallel with vehicle only (2% FBS in PBS). Following incubation, the inoculum was aspirated, and the cultures returned to serum-free differentiation medium (Bijland et al., 2019). Media was partially replenished every 2 days.
For immunocytochemistry, cultures were fixed at 24 h post infection (hpi) or 6 days post infection (dpi) in 8% paraformaldehyde for 1 h at room temperature and subsequently stored in PBS at 4°C before staining. For NAD+ assay, cells were collected at 24 hpi.
Samples were prepared for NAD+ quantification using the NAD/NADH-Glo™ assay (G9071, Promega) according to the manufacturer's instructions. Briefly, at 24 hpi, a cell scraper was used to remove cell monolayers from glass coverslips (2 × 35 mm dish per sample, 3 coverslips per dish) and these were transferred to a microcentrifuge tube. The cells were pelleted by centrifugation on a benchtop centrifuge at 13,000 RPM for 30 s. Supernatant was discarded and cells rinsed once in 1 ml PBS containing cOmplete™ EDTA-free protease inhibitor (11873580001, Sigma-Aldrich) before repeat centrifugation. The cells were lysed in 100 μl of bicarbonate-based lysis buffer (100 mM Na2CO3, 20 mM NaHCO3, 10 mM nicotinamide, 0.05% Triton-X 100, 1% dodecyltrimethylammonium bromide) by vortexing for ~1 min. Cell debris and insoluble material was then pelleted by centrifugation at 13,000 RPM for 10 min before the supernatant was transferred to a fresh 1.5 ml reaction tube. Pierce™ BCA Protein Assay Kit (23225, Thermo Fisher) was used according to the manufacturer's instructions to determine protein concentrations. Cell lysates were prepared to 0.5 mg/ml in lysis solution. 12.5 μg lysate was then incubated in 12.5 μl 0.4 M HCl at 60°C for 15 min, allowed to cool to room temperature, and neutralized with 12.5 μl 0.5 M Tris base. The samples were then snap-frozen and transported between institutions on dry ice. For determination of NAD+ levels, 10 μl of each neutralized reaction was mixed with 10 μl of the NAD-Glo detection reagent (prepared following manufacturer's instructions) on ice, in wells of a 384-well white polystyrene microplate (Corning). The plate and contents were incubated at room temperature for 50 min before luminescence was read using a GloMax® Explorer plate reader (Promega). Concentrations of NAD+ are expressed as nmol/mg of protein.
Post-fixation, cells were permeabilized in ethanol (−20°C; 10 min) and incubated in primary antibodies in blocking buffer (10% goat serum in PBS containing 2x NaCl [274 mM final] and 1% BSA) overnight at 4°C. Mouse anti-ZIKV (Aalto Bio, clone 0302156; 1 in 500) was used in combination with rabbit anti-NeuN (Sigma-Aldrich, ABN78; 1 in 500). Mouse IgG1 SMI31 anti-phosphorylated heavy and medium chain neurofilament (BioLegend, 801601; 1 in 1,500) and mouse IgG2a anti-β-Tubulin III (Sigma-Aldrich, T8578; 1 in 200) were applied simultaneously. After washing, secondary antibodies (goat anti-mouse IgG1 Alexa 488/568/647 and goat anti-rabbit IgG or goat anti-mouse IgG2a Alexa 568/647; 1 in 1,000; Invitrogen) were applied for 1 h at room temperature. Coverslips were mounted on glass slides in Mowiol® 4-88 (Sigma-Aldrich) mounting medium with DAPI (1 μg/ml; D1306, Invitrogen).
For quantification of cell density and neurofilament staining, fluorescence microscopy and image capture were performed using a Zeiss Axio Imager M2 fluorescent microscope with standard epifluorescence optics and Zen Blue software (Carl Zeiss AG, Germany). To avoid bias, fields of view (FoV) were selected in the blue (DAPI) channel and images were captured (10 images from across each coverslip) at x20 magnification in the red, far red, green and blue channels. Representative images for illustration were obtained using the same microscope and software or with a Zeiss LSM 880 Confocal Microscope using a Zeiss Plan-Apochromat 63 × /1.4 oil immersion objective and Zen Black software.
A rectangular area of interest (AoI) of 111,488 μm2 was placed on each image and immunostained cells or DAPI +ve nuclei, respectively, within and touching west and north borders were quantified. Only immunopositive cells with a DAPI +ve nucleus qualified. The average cell density per AOI was converted to cells/mm2 using the formula, cell density per AOI/area of AOI μm2 × 1,000,000. Pyknotic nuclei were distinguished from healthy nuclei on the basis of size and homogeneity and intensity of DAPI staining; pyknotic nuclei being condensed and intensely labeled (Cummings and Schnellmann, 2004). Experimenters were blinded to genotypes and three (of nine) sets of images were each quantified by two experimenters (average values were used) for quality control purposes.
Digital images of representative fields of view obtained from cell cultures immunostained with antibody SMI31 that recognizes phosphorylated heavy (H) and medium (M) chain neurofilament, were used to quantify the densities of neuronal processes. A Fiji (Schindelin et al., 2012) macro was recorded to analyse all images identically. To specifically select linear structures, a threshold between 12 and 30 (depending on staining) was used and debris was filtered out using the “analyze particles” function with the following parameters: size = 250.00-infinity, circularity = 0.00–0.30. The “histogram” function was then used to obtain the number of positive pixels. Macro to quantify axon densities in Fiji:
setOption(“ScaleConversions”, true);
run(“8-bit”);
setAutoThreshold(“Default dark”);
//run(“Threshold...”);
setThreshold(12, 255);
run(“Analyze Particles...”, “size=250.00-Infinity circularity=0.00-0.30 show=Masks display in_situ”);
getHistogram(values, counts, 2);
Array.print(counts);
Analyses were performed using GraphPad Prism 9 software (GraphPad Software Inc., San Diego, CA). Significance is indicated as <0.05 (*), <0.001 (***) and <0.0001 (****). A one-way ANOVA was used to compare cell densities or neurofilament stained area across three Sarm1 genotypes, using independent cultures each generated from a single embryo from a minimum of 3–4 independent litters per analysis. A two-way ANOVA was used to compare NAD+ concentrations between mock-infected and infected cultures, across all three genotypes, using independent cultures each generated from a single embryo from 3 independent dams.
Embryonic murine spinal cord-derived myelinating cell cultures contain all major neural cell types including choline acetyltransferase positive motor neurons that project to the body's periphery in vivo, oligodendrocyte progenitors (OPCs), myelinating oligodendrocytes, astrocytes and microglia. To determine if SARM1 depletion (Sarm1−/−) or haploinsufficiency (Sarm1+/−) conserves axons, we infected cultures generated from Sarm1+/+ (control), Sarm1+/− or Sarm1−/− mice with MOI 0.3 or 0.6 ZIKV (Table 1) and stained them with antibody to phosphorylated H- and M-neurofilament or β-tubulin III, at 6 dpi (Figure 1). Quantification of neurofilament staining demonstrated statistically significant preservation of neuronal cell processes in infected Sarm1−/− cultures compared to Sarm1+/+ controls. SARM1 haploinsufficiency conferred no benefit at this time point (Figure 1).

Table 1. MOI ZIKV used in experiments quantifying neurofilament and β-tubulin III staining.
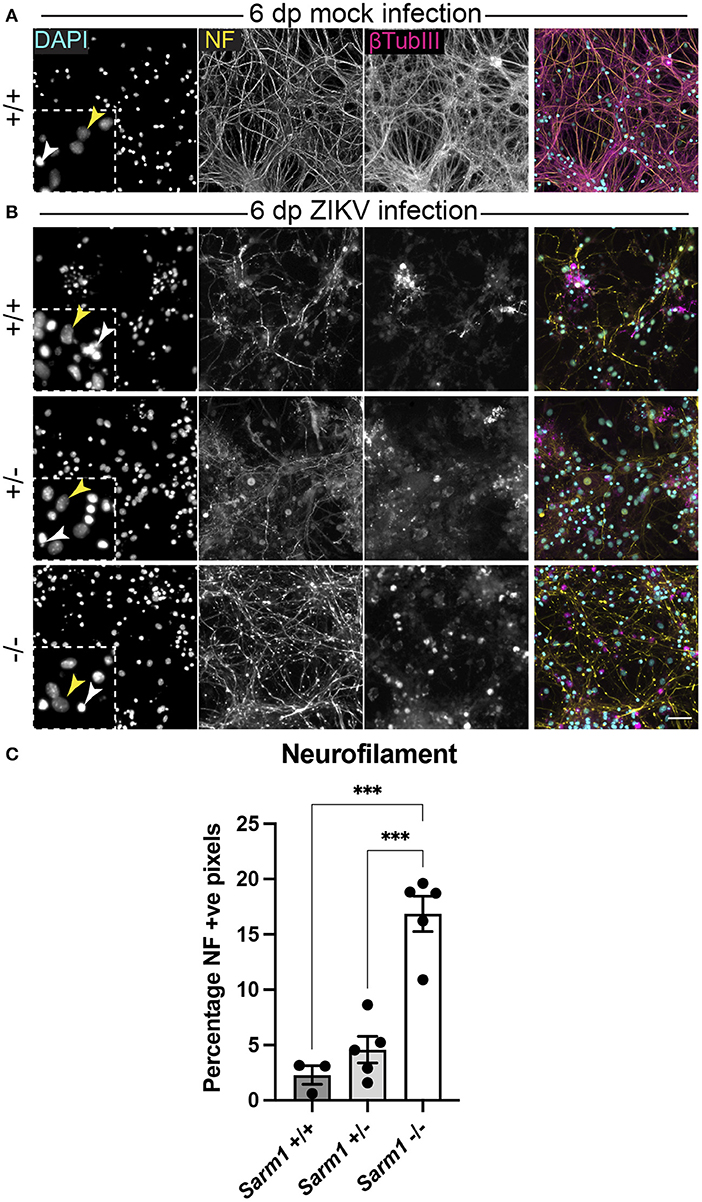
Figure 1. SARM1 depletion delays degeneration of neuronal processes. (A) Mock infected Sarm1+/+ cultures appeared healthy at 6 dpi, with numerous pale DAPI +ve nuclei (yellow arrowhead in inset), relatively few pyknotic nuclei (white arrowhead) and dense neurofilament (NF) and β-tubulin III (β-TubIII) positive cell processes. (B) ZIKV infected cultures appeared altered at the same time point, with several pyknotic nuclei (white arrowheads in insets) and fragmented neurofilament and β-tubulin III-stained neuronal processes. This was most obvious in Sarm1+/+ and Sarm1+/− cultures whereas neurofilament appeared relatively preserved in Sarm1−/− cultures. Bar: 50 μm. (C) Quantification of neurofilament positive pixels as a proportion of all pixels demonstrated a significant preservation of neuronal processes in Sarm1−/− cultures compared to Sarm1+/+ or +/−. Bars represent mean ± S.E.M. ***p < 0.001.
To confirm that the observed decrease in degeneration of neuronal processes in the absence of SARM1 does not simply reflect reduced infection in Sarm1−/− cultures, we quantified the proportion of DAPI +ve cell nuclei (both healthy-appearing and pyknotic) that were ZIKV +ve, in each of the three genotypes. We ruled out a decrease in the proportion of productively infected cells at 6 dpi, instead, observing a trend toward an increase in the proportion of infected cells in the Sarm1−/− cultures (Figures 2A,B). We ruled out the possibility this was due to differences in cell densities between genotypes (Figures 2A,C). Thus, protection of neuronal processes does not result from there being reduced numbers of productively infected cells in the absence of SARM1.
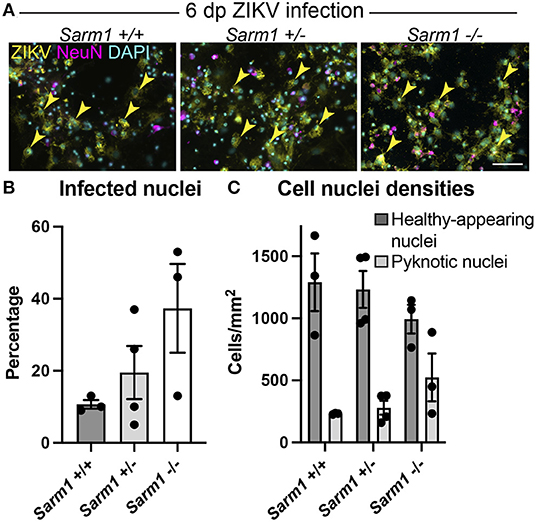
Figure 2. SARM1 does not enhance susceptibility to ZIKV infection. (A) Fluorescence micrographs of ZIKV infected cells at 6 dpi in Sarm1 +/+, +/− and −/− cultures. Across all three genotypes, many cells are ZIKV +ve (yellow arrowheads). Few of these are NeuN +ve, indicating that most infected cells are glia. Bar: 50 μm. (B) Graph of percentage DAPI +ve nuclei that are ZIKV +ve, demonstrates that SARM1 status does not result in significant difference in proportions of infected cells, although there is a trend toward an increased proportion of infected cells in the absence of SARM1. (C) Graphs of DAPI +ve cell densities demonstrates there is no significant difference in cell densities across the three Sarm1 genotypes. However, there appears to be a tendency for increased cell death (pyknotic nuclei) in the absence of SARM1. Bars represent mean ± S.E.M.
SARM1 is an NADase that depletes axons of NAD+ following a variety of insults. Furthermore, the NS3 domain of ZIKV itself has been shown to deplete NAD+ by a PARP1-dependent mechanism in HeLa cells (Xu et al., 2019). To determine if there is depletion of NAD+ upon ZIKV infection of CNS cells, we first determined a time point to assay NAD+ concentrations, prior to overt cellular injury. At 24 hpi, staining of DAPI +ve cell nuclei revealed little difference between infected cultures of either of the three Sarm1 genotypes and their mock-infected controls (Figures 3A,B and Supplementary Figure 1), suggesting the absence of overt ZIKV infection-related cell death at this time point, as quantified previously (Cumberworth et al., 2017). Nonetheless, despite that staining for phosphorylated H- and M-neurofilament (antibody clone SMI31) demonstrated similar positive pixel densities across all the three genotypes (Figure 3C), subtle changes in morphology of some neuronal processes in ZIKV-infected Sarm1+/+ cultures could be seen at this time point (arrows; Figure 3B). This was confirmed by β-tubulin III staining which provided evidence for fragmentation of the microtubule network in ZIKV-infected Sarm1+/+ and Sarm1+/−, but not Sarm1−/− cultures or mock-infected controls (Figures 3A,B and Supplementary Figure 1). Quantification of NAD+ levels at 24 hpi demonstrated a similar significant decrease in NAD+ levels in ZIKV-infected cultures of each of the three Sarm1 genotypes, in comparison to their matched mock-infected controls (Figure 3D). The degree of NAD+ reduction was most consistent in Sarm1−/− cultures and more variable in wild type and heterozygous controls, most particularly when NAD+ levels were below 2 nmol/mg−1 protein in mock-infected control cultures. On average, NAD+ levels were reduced to ~60% of mock-infected control levels, across all three genotypes (Figure 3E). Together, these data demonstrate that whilst degeneration of neuronal processes is expediated by SARM1, NAD+ depletion at 24 hpi is independent of SARM1. NAD+ levels were not assessed at later time points due to confounding consequences of leakage of NAD+ from dying cells and injured axons.
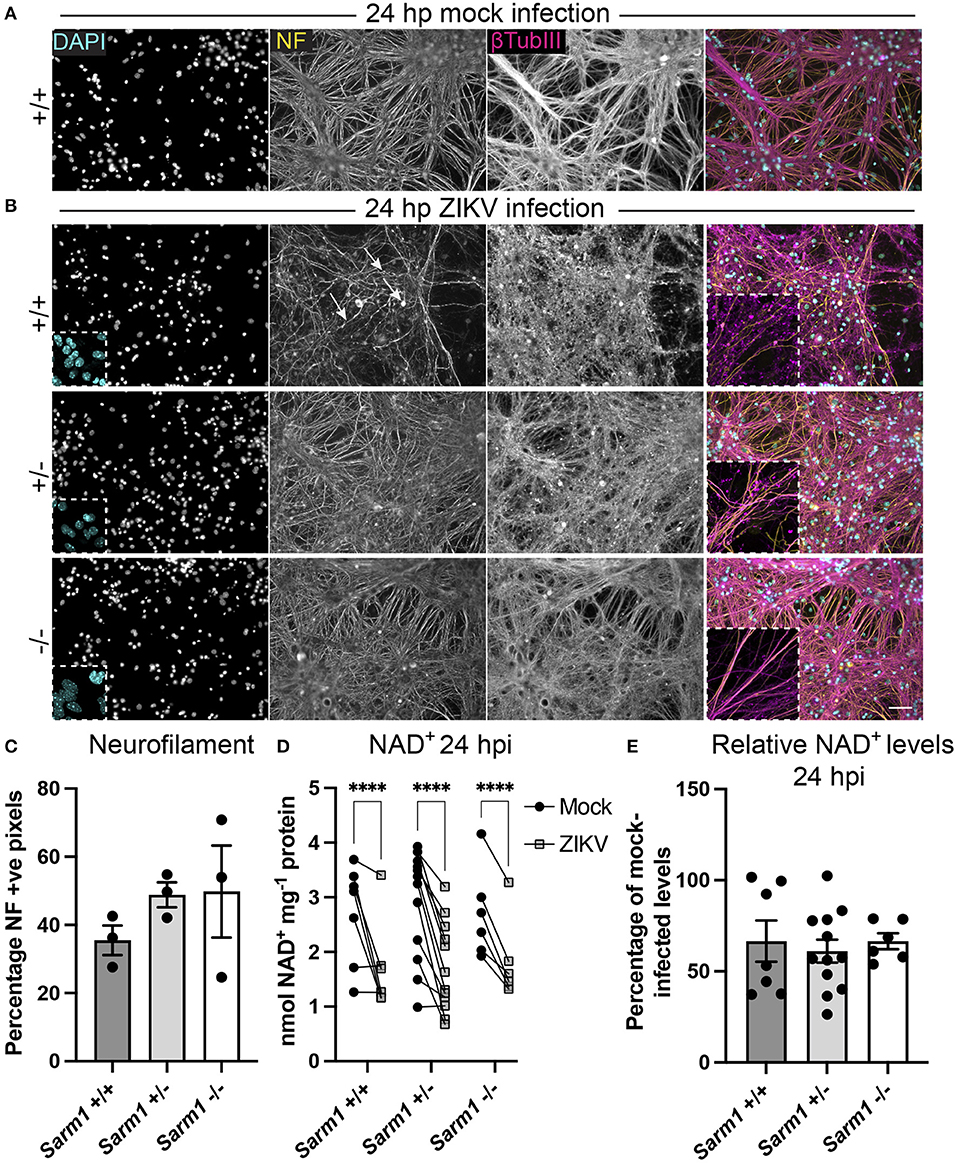
Figure 3. ZIKV infection depletes NAD+ levels independent of SARM1 at 24 hpi. (A) Mock infected Sarm1+/+ cultures appear healthy at 24 hpi. DAPI +ve cell nuclei are abundant and neurofilament (NF) and β-tubulin III-stained neuronal processes appear dense and smooth. Bar: 50 μm. (B) At 24 hpi with ZIKV, DAPI +ve cell nuclei appear similar in terms of density and appearance across all three Sarm1 genotypes, although there is already evidence of subtle injury to neuronal cell processes in Sarm1+/+ (arrows indicate irregularly stained neurofilament +ve structures) and Sarm1+/− cultures, most obvious with β-tubulin III staining. In contrast, neuronal processes in Sarm1−/− cultures appear dense and smooth, as in the mock-infected controls (see also Supplementary Figure 1). (C) Neurofilament positive pixels as a percentage of all pixels per AOI is similar across all three genotypes suggesting that neuronal processes remain viable at this timepoint. (D) Compared to levels in matched mock-infected controls (black circles), NAD+ levels are significantly reduced in ZIKV-infected cultures (gray squares) at 24 hpi. (E) The relative reduction in NAD+ in ZIKV infected cultures in comparison to their mock-infected controls is similar across all three genotypes. Bars represent mean ± S.E.M. ****p < 0.0001.
As some neurotropic viruses use microtubule-dependent axon transport to reach the neuronal soma, the changes observed in microtubule staining at 24 hpi led us to ask whether SARM1-dependent mechanisms might limit infection of neuronal somas. To address this, we examined infected NeuN +ve somas across the three Sarm1 genotypes (Figure 4A). Compared to Sarm1+/+, we observed a significant increase in the proportion of infected neuronal somas in Sarm1−/− cultures and a similar trend in Sarm1+/− cultures (Figure 4B), that did not reflect differences in the total number of NeuN positive cells of any genotype (Figure 4C). Correspondingly, we observed a significant increase in the proportion of pyknotic neurons in Sarm1−/− cultures compared to Sarm1+/+ cultures. Sarm1+/− cultures had, on average, an intermediate proportion of pyknotic neurons, likely reflecting SARM1 haploinsufficiency (Figure 4D). Together, these data suggest that SARM1-dependent degeneration of neuronal processes limits infection and death of neuronal somas.
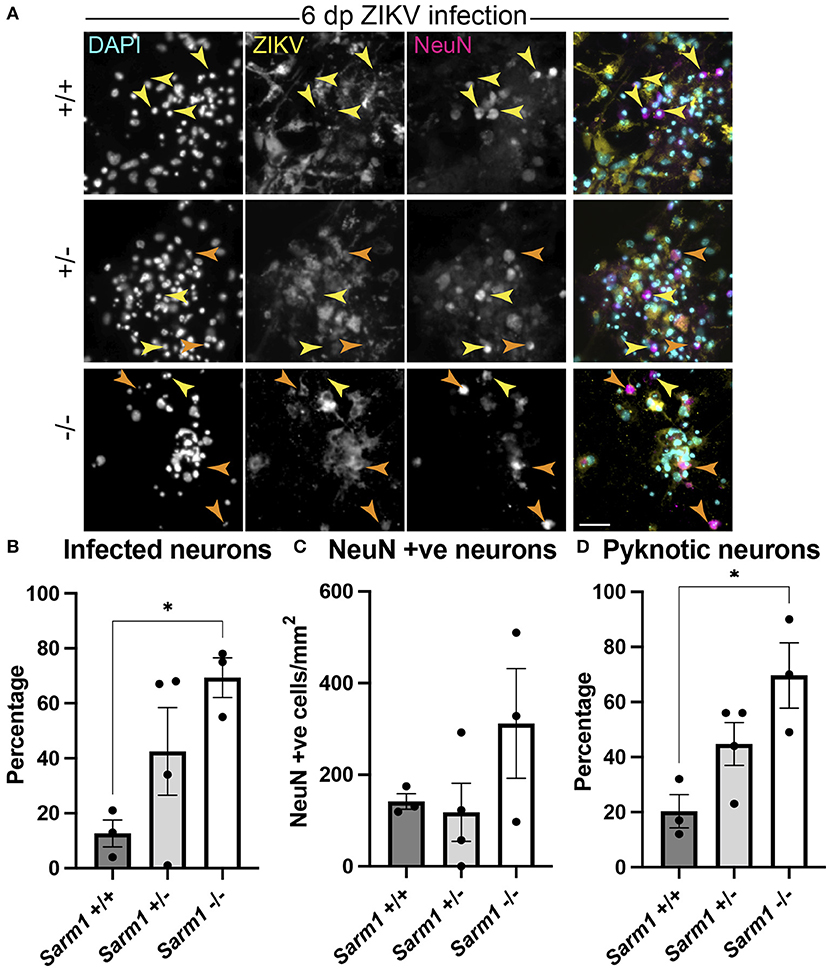
Figure 4. SARM1-dependent degeneration of neuronal processes limits infection and death of neurons. (A) Fluorescence micrographs of ZIKV infected Sarm1+/+, Sarm1+/− and Sarm1−/− cultures at 6 dpi. In wild type cultures (Sarm1+/+), most NeuN +ve neurons are not ZIKV +ve (yellow arrowheads) whereas in Sarm1−/− cultures, many NeuN +ve neurons are ZIKV +ve (orange arrowheads). In Sarm1+/− cultures, both ZIKV +ve (orange arrowheads) and -ve (yellow arrowheads) NeuN +ve cells are evident. Bar: 50 μm. (B) Graph of proportions of NeuN +ve infected cells, demonstrates a significantly greater proportion of NeuN +ve neurons are ZIKV +ve in Sarm1−/− cultures compared to Sarm1+/+ control. (C) This does not reflect a significant difference in NeuN +ve cell densities in Sarm1−/− cultures compared to Sarm1+/+ controls. (D) There is a significant increase in the proportion of pyknotic NeuN +ve nuclei in Sarm1−/− cultures compared to Sarm1+/+ controls. Bars represent mean ± S.E.M. *p < 0.05 (defined in M&M).
Our data provide novel evidence of a SARM1-dependent mechanism of degeneration of neuronal processes following ZIKV infection. Further, this involves neither an increase in ZIKV infected cells nor increased susceptibility of neuronal somas to cell death. On the contrary, at least at the time point tested, the proportions of infected and/or dying neurons were significantly enhanced in the absence of SARM1. Furthermore, we provide evidence in primary CNS cell cultures for the physiological relevance of a previous report of ZIKV-induced NAD+ depletion in HeLa cells (Xu et al., 2019). Surprisingly, although NAD+ depletion has been suggested to make SARM1 activation more likely (Figley et al., 2021) we found no clear evidence of any additional depletion of NAD+ under conditions (ZIKV-infection) where SARM1 contributes to axon loss. Indeed, since most infected cells in our mixed neural cell cultures are glia (Cumberworth et al., 2017), it may be that depletion of glial NAD+ makes the largest contribution to the fall we observed. Irrespective of the cellular localization of NAD+ depletion, it appears that the mechanism by which SARM1 contributes to degeneration of neuronal processes following ZIKV infection is independent of its NADase activity.
One possible explanation for these findings is that degeneration of neuronal processes in this context could result from one of the additional enzyme activities of SARM1, which include NADP hydrolysis and cyclisation, and base exchange of nicotinamide of NAD(P) with other endogenous pyridine bases such as nicotinic acid (Zhao et al., 2019; Angeletti et al., 2021; Figley et al., 2021). These may lead to reduced ROS buffering capability due to lowering of NADPH, or to calcium mobilization due to generation of NaADP, the most potent known calcium mobilizing signal (Galione et al., 2000; Angeletti et al., 2021). Indeed, a circa 50% decline in NAD+, as observed here at 24 hpi, is not sufficient to kill axons or cells since doses of NAMPT inhibitor FK866 that depletes NAD+ to or beyond this level, are not lethal (Di Stefano et al., 2015). Transected Sarm1−/− sciatic nerve axons also survive more than a week after the fall in NAD+ passes 50% of basal levels (Gilley et al., 2015). Thus, it will be important to investigate these other potential mediators of SARM1 activity, or indeed any novel activities that are found for this multifunctional enzyme, in future work.
One limitation of our study is that the NAD+ assay is based on cell culture lysates and therefore it cannot discriminate cell or cell compartment-specific effects. Regarding this, it is worth noting that ZIKV protein NS3 itself potently activates a nuclear poly (ADP-ribose) polymerase, PARP1, in HeLa and glioma cells that results in NAD+ depletion and cell death at 48 h post-treatment in the former (Xu et al., 2019). As PARP1 is also one of the main NAD+ consuming enzymes in neural cells (Klimova and Kristian, 2019), it seems reasonable to speculate that it is responsible for the circa 50% NAD+ depletion at 24 hpi in our ZIKV infected neural cell cultures. Consequently, we cannot exclude the possibility that a SARM1-dependent depletion of NAD+ specifically in neuronal processes is masked in these mixed neural cell type cultures. We also cannot rule out a SARM1-dependent fall in NAD+ at later timepoints, rapidly followed by degeneration of neuronal processes, since if this were to happen it would be confounded by cell death and extremely difficult to capture the optimal time point to detect it.
Degeneration of neuronal processes following ZIKV infection occurs independently of overt infection or death of neuronal somas in Sarm1 wild type cultures [the current study and Cumberworth et al. (2017)]. This raises the intriguing possibility that it represents a neuroprotective mechanism designed to limit viral spread, as proposed previously (Mukherjee et al., 2013; Sundaramoorthy et al., 2020). Neuronal processes, especially axons, are highly reliant on microtubule-dependent transport for function and survival (De Vos et al., 2008; Magiera et al., 2018; Guedes-Dias and Holzbaur, 2019; Kelliher et al., 2019; Koppers and Farías, 2021), and several neurotropic viruses including Theiler's virus, herpes simplex virus, poliovirus, rabies virus, and West Nile virus (WNV), transit in axons, likely relying on microtubule-dependent transport to reach their targets (Salinas et al., 2009 and reviewed in Richards et al., 2021). Thus, the significant increase in infection and death of neuronal somas in SARM1-depleted cultures (with delayed degeneration of neuronal processes) suggests that ZIKV can be transported in this way too and that SARM1-dependent degeneration of neuronal processes limits its spread to neuronal somas. Nonetheless, the possibility that the SARM1-dependent intrinsic immune capacity of neurons (Wang et al., 2018) protects these cells in Sarm1+/+ cultures, should not be discounted.
It is noteworthy that widespread changes in neuronal-specific β-tubulin III immunostaining were observed as early as 24 h post-ZIKV infection. This raises the intriguing possibility that, more generally, activation of evolutionarily conserved SARM1 hampers the retrograde transportation of some viruses from the site of entry at the body's periphery, even prior to axon destruction. In support of this suggestion is the fact that SARM1 participates in microtubule posttranslational modification (Chen et al., 2011), suggesting that activation of SARM1 could have a rapid impact on motor protein binding and cargo (including virus) transport.
The initial trigger for SARM1-dependent degeneration of neuronal processes following ZIKV infection could act through gain or loss of function, for example, through cytotoxicity, failure of trophic support from neighboring glia, or a combination of both. If a soluble cytotoxic factor is involved, neuronal processes seem particularly susceptible since all cell types are, in principle, equally exposed in cell culture where they are bathed in media. Nitric oxide is one potential cytotoxic molecule that causes Wallerian degeneration changes in electrically active axons at low micromolar concentrations (Smith et al., 2001). Failure of support from oligodendrocytes and astrocytes (Stassart et al., 2018; Duncan et al., 2021; Veloz Castillo et al., 2021) infected with ZIKV is another possibility. These cells provide energy metabolites to neurons (Pellerin et al., 1998; Fünfschilling et al., 2012; Lee et al., 2012), although energy insufficiency due to failed energy substrate provision is unlikely in this context because neuronal processes are suffused in a glucose-rich media in vitro. Nonetheless, oligodendrocytes and astrocytes confer axon support through multiple mechanisms that could be relevant in the context of ZIKV infection, such as regulating ion and glutamate homeostasis (Verkhratsky and Nedergaard, 2018; Hart and Karimi-Abdolrezaee, 2021), performing anti-oxidant functions (Baxter and Hardingham, 2016; Mukherjee et al., 2020), and enhancing axonal energy metabolism by deacetylating mitochondrial proteins (Chamberlain et al., 2021).
CNS complications of ZIKV infection in adults are rare, but include encephalitis, meningitis, myelitis, meningoencephalitis, transverse myelitis and neuropsychiatric symptoms (Carteaux et al., 2016; Galliez et al., 2016; Mécharles et al., 2016; da Silva et al., 2017; Zucker et al., 2017; Brito Ferreira et al., 2020). However, ZIKV infection has been linked to the peripheral neuropathy, Guillain-Barré syndrome (Oehler et al., 2014; Pinto-Díaz et al., 2017). Roles for SARM1 in other viral neuropathies have been reported. Mice lacking SARM1 are more susceptible to WNV infection, with reduced survival and enhanced neuronal death in the brainstem (Szretter et al., 2009), and this has been confirmed in a new CRISPR/Cas9-generated SARM1 null mouse that excludes any contribution from passenger mutations co-inherited with SARM1 from the original 129 strain ES cells (Uccellini et al., 2020). Similarly, a recent study demonstrated that different field strains of lyssavirus (“rabies”) cause SARM1-dependent axon degeneration in CNS and PNS mouse neurons, further supporting a role of SARM1 in neurotropic viral infection (Sundaramoorthy et al., 2020). The authors showed that the SARM1-mediated axon loss prevents the spread of virus along interconnected neurons, suggesting a possible host-defense mechanism restricting virus infection. The subsequent loss of neuronal processes in this and our study, possibly as a result of such a neuronal defense response, would be consistent with our observation that ZIKV-induced neuronal soma death increases in the absence of SARM1, and could thus represent a mechanism underlying neurological complications in rabies and ZIKV infection. It may even help to explain the evolutionary advantage of a mechanism for rapid axon degeneration, which otherwise appears paradoxical. It will be of great interest to further understand the effects of these and other viruses on SARM1-dependent axon loss, on NAD+ levels, and the roles of these mechanisms in viral neuropathies.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Ethics Review Committee of the University of Glasgow.
JE and MC: study design and supervision. CC, CA, LK, and VS: experimentation. CD: viral propagation. PM: genotyping. CC, LK, and JE: data analysis. JE, MC, CA, and CC: manuscript draft. JE, CC, and LK: figure composition. All authors: manuscript editing and approval. HW, JE, AK, MC, SB, and CL: funding. All authors contributed to the article and approved the submitted version.
This project was partially funded through the European Union's Horizon 2020 research and innovation programme under ZikaPLAN Grant Agreement No. 734584 (HW, JE, SB, and CL); the UK Medical Research Council (MC_UU_12014/8 and MR/N017552/1) (AK); CSO and University of Glasgow SPRINT PhD programme studentship to CLC (JE); an MRC DTP Award and Gates Scholarship (CA); the John and Lucille van Geest Foundation (MC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.860410/full#supplementary-material
Angeletti, C., Amici, A., Gilley, J., Loreto, A., Trapanotto, A. G., Antoniou, C., et al. (2021). SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated by multiple physiologically-relevant NAD metabolites. iScience. 25:103812. doi: 10.1016/j.isci.2022.103812
Baud, D., Musso, D., Vouga, M., Alves, M. P., and Vulliemoz, N. (2017). Zika virus: a new threat to human reproduction. Am. J. Reprod. Immunol. 77. doi: 10.1111/aji.12614
Baxter, P. S., and Hardingham, G. E. (2016). Adaptive regulation of the brain's antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 100, 147–152. doi: 10.1016/j.freeradbiomed.2016.06.027
Bijland, S., Thomson, G., Euston, M., Michail, K., Thümmler, K., Mücklisch, S., et al. (2019). An in vitro model for studying CNS white matter: functional properties and experimental approaches. [version 1; peer review: 2 approved]. F1000Research 8, 117. doi: 10.12688/f1000research.16802.1
Brito Ferreira, M. L., Militão de Albuquerque, M., de, F. P., de Brito, C. A. A., de Oliveira França, R. F., Porto Moreira, Á. J., de Morais Machado, et al. (2020). Neurological disease in adults with Zika and chikungunya virus infection in Northeast Brazil: a prospective observational study. Lancet Neurol. 19, 826–839. doi: 10.1016/S1474-4422(20)30232-5
Carteaux, G., Maquart, M., Bedet, A., Contou, D., Brugières, P., Fourati, S., et al. (2016). Zika Virus associated with meningoencephalitis. N. Engl. J. Med. 374, 1595–1596. doi: 10.1056/NEJMc1602964
Carty, M., and Bowie, A. G. (2019). SARM: from immune regulator to cell executioner. Biochem. Pharmacol. 161, 52–62. doi: 10.1016/j.bcp.2019.01.005
Carty, M., Goodbody, R., Schröder, M., Stack, J., Moynagh, P. N., and Bowie, A. G. (2006). The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 7, 1074–1081. doi: 10.1038/ni1382
Chamberlain, K. A., Huang, N., Xie, Y., LiCausi, F., Li, S., Li, Y., et al. (2021). Oligodendrocytes enhance axonal energy metabolism by deacetylation of mitochondrial proteins through transcellular delivery of SIRT2. Neuron 109, 3456–3472.e8. doi: 10.1016/j.neuron.2021.08.011
Chen, C.-Y., Lin, C.-W., Chang, C.-Y., Jiang, S.-T., and Hsueh, Y.-P. (2011). Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J. Cell Biol. 193, 769–784. doi: 10.1083/jcb.201008050
Coleman, M. P., and Höke, A. (2020). Programmed axon degeneration: from mouse to mechanism to medicine. Nat. Rev. Neurosci. 21, 183–196. doi: 10.1038/s41583-020-0269-3
Conforti, L., Gilley, J., and Coleman, M. P. (2014). Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409. doi: 10.1038/nrn3680
Cumberworth, S. L., Barrie, J. A., Cunningham, M. E., de Figueiredo, D. P. G., Schultz, V., Wilder-Smith, A. J., et al. (2017). Zika virus tropism and interactions in myelinating neural cell cultures: CNS cells and myelin are preferentially affected. Acta Neuropathol. Commun. 5, 50. doi: 10.1186/s40478-017-0450-8
Cummings, B. S., and Schnellmann, R. G. (2004). Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. Chapter 12, Unit 12.18. doi: 10.1002/0471141755.ph1208s25
da Silva, I. R. F., Frontera, J. A., Bispo de Filippis, A. M., and Nascimento, O. J. M. do, RIO-GBS-ZIKV Research Group (2017). Neurologic complications associated with the zika virus in brazilian adults. JAMA Neurol. 74, 1190–1198. doi: 10.1001/jamaneurol.2017.1703
De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C. J. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151–173. doi: 10.1146/annurev.neuro.31.061307.090711
Di Stefano, M., Nascimento-Ferreira, I., Orsomando, G., Mori, V., Gilley, J., Brown, R., et al. (2015). A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 22, 731–742. doi: 10.1038/cdd.2014.164
Donald, C. L., Brennan, B., Cumberworth, S. L., Rezelj, V. V., Clark, J. J., Cordeiro, M. T., et al. (2016). Full genome sequence and sfRNA interferon antagonist activity of zika virus from recife, Brazil. PLoS Negl. Trop. Dis. 10, e0005048. doi: 10.1371/journal.pntd.0005048
Duncan, G. J., Simkins, T. J., and Emery, B. (2021). Neuron-oligodendrocyte interactions in the structure and integrity of axons. Front. Cell Dev. Biol. 9, 653101. doi: 10.3389/fcell.2021.653101
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 toll/interleukin-1 receptor domain possesses intrinsic nad+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022
Figley, M. D., Gu, W., Nanson, J. D., Shi, Y., Sasaki, Y., Cunnea, K., et al. (2021). SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 109, 1118–1136.e11. doi: 10.1016/j.neuron.2021.02.009
Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. doi: 10.1038/nature11007
Galione, A., Patel, S., and Churchill, G. C. (2000). NAADP-induced calcium release in sea urchin eggs. Biol. Cell 92, 197–204. doi: 10.1016/S0248-4900(00)01070-4
Galliez, R. M., Spitz, M., Rafful, P. P., Cagy, M., Escosteguy, C., Germano, C. S. B., et al. (2016). Zika virus causing encephalomyelitis associated with immunoactivation. Open Forum Infect. Dis. 3, ofw203. doi: 10.1093/ofid/ofw203
Gilley, J., Orsomando, G., Nascimento-Ferreira, I., and Coleman, M. P. (2015). Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 10, 1974–1981. doi: 10.1016/j.celrep.2015.02.060
Gubler, D. J., Vasilakis, N., and Musso, D. (2017). History and emergence of zika virus. J. Infect. Dis. 216, S860–S867. doi: 10.1093/infdis/jix451
Guedes-Dias, P., and Holzbaur, E. L. F. (2019). Axonal transport: driving synaptic function. Science 366, eaaw9997. doi: 10.1126/science.aaw9997
Gulland, A.. (2016). Zika virus is a global public health emergency, declares WHO. BMJ 352, i657. doi: 10.1136/bmj.i657
Hart, C. G., and Karimi-Abdolrezaee, S. (2021). Recent insights on astrocyte mechanisms in CNS homeostasis, pathology, and repair. J. Neurosci. Res. 99, 2427–2462. doi: 10.1002/jnr.24922
Hopkins, E. L., Gu, W., Kobe, B., and Coleman, M. P. (2021). A novel NAD signaling mechanism in axon degeneration and its relationship to innate immunity. Front. Mol. Biosci. 8, 703532. doi: 10.3389/fmolb.2021.703532
Horsefield, S., Burdett, H., Zhang, X., Manik, M. K., Shi, Y., Chen, J., et al. (2019). NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 365, 793–799. doi: 10.1126/science.aax1911
Kelliher, M. T., Saunders, H. A., and Wildonger, J. (2019). Microtubule control of functional architecture in neurons. Curr. Opin. Neurobiol. 57, 39–45. doi: 10.1016/j.conb.2019.01.003
Kim, Y., Zhou, P., Qian, L., Chuang, J.-Z., Lee, J., Li, C., et al. (2007). MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J. Exp. Med. 204, 2063–2074. doi: 10.1084/jem.20070868
Klimova, N., and Kristian, T. (2019). Multi-targeted effect of nicotinamide mononucleotide on brain bioenergetic metabolism. Neurochem. Res. 44, 2280–2287. doi: 10.1007/s11064-019-02729-0
Koppers, M., and Farías, G. G. (2021). Organelle distribution in neurons: logistics behind polarized transport. Curr. Opin. Cell Biol. 71, 46–54. doi: 10.1016/j.ceb.2021.02.004
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Lin, C.-W., Liu, H.-Y., Chen, C.-Y., and Hsueh, Y.-P. (2014). Neuronally-expressed Sarm1 regulates expression of inflammatory and antiviral cytokines in brains. Innate Immun. 20, 161–172. doi: 10.1177/1753425913485877
Magiera, M. M., Bodakuntla, S., Žiak, J., Lacomme, S., Marques Sousa, P., Leboucher, S., et al. (2018). Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 37, e100440. doi: 10.15252/embj.2018100440
Mécharles, S., Herrmann, C., Poullain, P., Tran, T.-H., Deschamps, N., Mathon, G., et al. (2016). Acute myelitis due to Zika virus infection. Lancet 387, 1481. doi: 10.1016/S0140-6736(16)00644-9
Miner, J. J., and Diamond, M. S. (2017). Zika virus pathogenesis and tissue tropism. Cell Host Microbe 21, 134–142. doi: 10.1016/j.chom.2017.01.004
Mukherjee, C., Kling, T., Russo, B., Miebach, K., Kess, E., Schifferer, M., et al. (2020). Oligodendrocytes provide antioxidant defense function for neurons by secreting ferritin heavy chain. Cell Metab. 32, 259–272.e10. doi: 10.1016/j.cmet.2020.05.019
Mukherjee, P., Woods, T. A., Moore, R. A., and Peterson, K. E. (2013). Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity 38, 705–716. doi: 10.1016/j.immuni.2013.02.013
Oehler, E., Watrin, L., Larre, P., Leparc-Goffart, I., Lastère, S., Valour, F., et al. (2014). Zika virus infection complicated by Guillain-Barré syndrome – case report, French Polynesia, December 2013. Eurosurveillance 19, 20720. doi: 10.2807/1560-7917.ES2014.19.9.20720
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Pardy, R. D., and Richer, M. J. (2019). Zika virus pathogenesis: from early case reports to epidemics. Viruses 11, 886. doi: 10.3390/v11100886
Pellerin, L., Pellegri, G., Bittar, P. G., Charnay, Y., Bouras, C., Martin, J. L., et al. (1998). Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 20, 291–299. doi: 10.1159/000017324
Pierson, T. C., and Diamond, M. S. (2018). The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581. doi: 10.1038/s41586-018-0446-y
Pinto-Díaz, C. A., Rodríguez, Y., Monsalve, D. M., Acosta-Ampudia, Y., Molano-González, N., Anaya, J.-M., et al. (2017). Autoimmunity in Guillain-Barré syndrome associated with Zika virus infection and beyond. Autoimmun. Rev. 16, 327–334. doi: 10.1016/j.autrev.2017.02.002
Richards, A., Berth, S. H., Brady, S., and Morfini, G. (2021). Engagement of neurotropic viruses in fast axonal transport: mechanisms, potential role of host kinases and implications for neuronal dysfunction. Front. Cell. Neurosci. 15, 684762. doi: 10.3389/fncel.2021.684762
Salinas, S., Bilsland, L. G., Henaff, D., Weston, A. E., Keriel, A., Schiavo, G., et al. (2009). CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog. 5, e1000442. doi: 10.1371/journal.ppat.1000442
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schultz, V., Cumberworth, S. L., Gu, Q., Johnson, N., Donald, C. L., McCanney, G. A., et al. (2021). Zika virus infection leads to demyelination and axonal injury in mature CNS cultures. Viruses 13. doi: 10.3390/v13010091
Smith, K. J., Kapoor, R., Hall, S. M., and Davies, M. (2001). Electrically active axons degenerate when exposed to nitric oxide. Ann. Neurol. 49, 470–476 doi: 10.1002/ana.96
Stassart, R. M., Möbius, W., Nave, K.-A., and Edgar, J. M. (2018). The axon-myelin unit in development and degenerative disease. Front. Neurosci. 12, 467. doi: 10.3389/fnins.2018.00467
Sundaramoorthy, V., Green, D., Locke, K., O'Brien, C. M., Dearnley, M., and Bingham, J. (2020). Novel role of SARM1 mediated axonal degeneration in the pathogenesis of rabies. PLoS Pathog. 16, e1008343. doi: 10.1371/journal.ppat.1008343
Szretter, K. J., Samuel, M. A., Gilfillan, S., Fuchs, A., Colonna, M., and Diamond, M. S. (2009). The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J. Virol. 83, 9329–9338. doi: 10.1128/JVI.00836-09
Thomson, C. E., Hunter, A. M., Griffiths, I. R., Edgar, J. M., and McCulloch, M. C. (2006). Murine spinal cord explants: a model for evaluating axonal growth and myelination in vitro. J. Neurosci. Res. 84, 1703–1715. doi: 10.1002/jnr.21084
Truett, G. E., Heeger, P., Mynatt, R. L., Truett, A. A., Walker, J. A., and Warman, M. L. (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). BioTechniques 29, 52–54. doi: 10.2144/00291bm09
Uccellini, M. B., Bardina, S. V., Sánchez-Aparicio, M. T., White, K. M., Hou, Y.-J., Lim, J. K., et al. (2020). Passenger mutations confound phenotypes of SARM1-deficient mice. Cell Rep. 31, 107498. doi: 10.1016/j.celrep.2020.03.062
Veloz Castillo, M. F., Magistretti, P. J., and Calì, C. (2021). l-lactate: food for thoughts, memory and behavior. Metabolites 11, 548. doi: 10.3390/metabo11080548
Verkhratsky, A., and Nedergaard, M. (2018). Physiology of astroglia. Physiol. Rev. 98, 239–389. doi: 10.1152/physrev.00042.2016
Wang, Q., Zhang, S., Liu, T., Wang, H., Liu, K., Wang, Q., et al. (2018). Sarm1/Myd88-5 regulates neuronal intrinsic immune response to traumatic axonal injuries. Cell Rep. 23, 716–724. doi: 10.1016/j.celrep.2018.03.071
Xu, G., Li, S., Liu, X., Gao, P., Chen, X., Wang, H., et al. (2019). PARP-1 mediated cell death is directly activated by ZIKV infection. Virology 537, 254–262. doi: 10.1016/j.virol.2019.08.024
Zhao, Z. Y., Xie, X. J., Li, W. H., Liu, J., Chen, Z., Zhang, B., et al. (2019). A cell-permeant mimetic of NMN activates SARM1 to produce cyclic ADP-ribose and induce non-apoptotic cell death. iScience 15, 452–466. doi: 10.1016/j.isci.2019.05.001
Keywords: Zika virus, neurofilament, tubulin (microtubules), glia, nicotinamide adenine dinucleotide
Citation: Crawford CL, Antoniou C, Komarek L, Schultz V, Donald CL, Montague P, Barnett SC, Linington C, Willison HJ, Kohl A, Coleman MP and Edgar JM (2022) SARM1 Depletion Slows Axon Degeneration in a CNS Model of Neurotropic Viral Infection. Front. Mol. Neurosci. 15:860410. doi: 10.3389/fnmol.2022.860410
Received: 22 January 2022; Accepted: 18 February 2022;
Published: 13 April 2022.
Edited by:
Pabitra Sahoo, University of South Carolina, United StatesReviewed by:
Yongcheol Cho, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South KoreaCopyright © 2022 Crawford, Antoniou, Komarek, Schultz, Donald, Montague, Barnett, Linington, Willison, Kohl, Coleman and Edgar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia M. Edgar, anVsaWEuZWRnYXJAZ2xhc2dvdy5hYy51aw==; Michael P. Coleman, bWM0NjlAY2FtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.