 Meaghan Navarrete
Meaghan Navarrete Yi Zhou
Yi Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 14 March 2022
Sec. Brain Disease Mechanisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.857495
This article is part of the Research Topic Biology of Brain Disorders – Cellular substrates for disrupted synaptic function and experience-dependent plasticity View all 14 articles
Schizophrenia is a debilitating mental disorder that affects approximately 1% of the world population, yet the disorder is not very well understood. The genetics of schizophrenia is very heterogenous, making it hard to pinpoint specific alterations that may cause the disorder. However, there is growing evidence from human studies suggesting a link between alterations in the 14-3-3 family and schizophrenia. The 14-3-3 proteins are abundantly expressed in the brain and are involved in many important cellular processes. Knockout of 14-3-3 proteins in mice has been shown to cause molecular, structural, and behavioral alterations associated with schizophrenia. Thus, 14-3-3 animal models allow for further exploration of the relationship between 14-3-3 and schizophrenia as well as the study of schizophrenia pathology. This review considers evidence from both human and animal model studies that implicate the 14-3-3 family in schizophrenia. In addition, possible mechanisms by which alterations in 14-3-3 proteins may contribute to schizophrenia-like phenotypes such as dopaminergic, glutamatergic, and cytoskeletal dysregulations are discussed.
Schizophrenia is a psychiatric disorder that affects both cognition and behavior. The symptoms of schizophrenia are generally grouped into positive, negative, and cognitive symptoms including hallucinations, delusions, anhedonia, and reductions in attention and memory. The onset of schizophrenia typically occurs in late adolescence to early adulthood (Jablensky, 2000). However, schizophrenia symptoms can manifest differently for each individual and can vary in severity over that individual’s lifetime (Freedman, 2003). Certain genetic variations, stressful life circumstances, and altered brain structure or functions have been linked to a higher incidence of schizophrenia. Nevertheless, both the etiology and the pathology of schizophrenia remain elusive. No single gene is responsible for the development of the disorder and there are no biomarkers to aid in diagnosing patients. These limitations hinder our ability to both diagnose and properly treat schizophrenia. Antipsychotic drugs are typically prescribed to treat the symptoms of schizophrenia and have been a useful therapeutic. These drugs can help alleviate the positive symptoms, and to a lesser extent the negative symptoms of the disorder, but are ineffective in treating cognitive symptoms (Freedman, 2003). In addition, many of these drugs come with undesired side effects like movement disorders (Mentzel et al., 2017) and weight gain (Volavka et al., 2002); which can lead to inconsistent usage and reduced treatment effectiveness (Kane et al., 2013). Therefore, further investigation of the etiology and pathophysiology of schizophrenia can facilitate our understanding of this disorder and is critical for the development of more effective treatments. Several genes which show promise in helping us decipher the developmental risks and mechanisms behind schizophrenia belong to the 14-3-3 family.
The 14-3-3 family of proteins has been identified in all eukaryotic organisms (van Hemert et al., 2001). There are seven known mammalian 14-3-3 genes, each of which expresses a distinct protein isoform; beta (β), gamma (γ), epsilon (ε), zeta (ζ), eta (η), theta (θ), and sigma (σ) (Berg et al., 2003). The highest concentration of 14-3-3 proteins is found in the brain, where they encompass around 1% of the total soluble proteins (Boston et al., 1982). The crystalline structure of 14-3-3 shows that two L shaped monomers come together in a dimerized pair to form a cup-shaped structure (Liu et al., 1995). All 14-3-3 isoforms can form either hetero or homodimers (Takahashi, 2003). This allows for the binding of two regions of the same interacting protein (Berg et al., 2003) or the binding of two different ligands (Fu et al., 2000). Thus, 14-3-3 proteins have a diverse range of hundreds of binding partners. Inside the concave face of the 14-3-3 dimer, polar-charged and hydrophobic amino acids create an amphipathic groove that interacts with phosphoserine and phosphothreonine containing motifs located on its binding partners (Yaffe et al., 1997; Wang et al., 1998). Consequently, 14-3-3 can modulate the function or subcellular location of its binding partners through phosphorylation-dependent protein–protein interactions. Many cellular processes and pathways have been linked to 14-3-3 function and expression. In the nervous system, published studies have indicated the involvement of 14-3-3 proteins in intracellular signaling, cell division and differentiation, apoptosis, and ion channel function (Berg et al., 2003). In addition, both human and animal studies have implicated 14-3-3 in several neurodegenerative and psychiatric diseases, including schizophrenia (Foote and Zhou, 2012).
Several human genome-wide association studies (GWAS) have revealed a genetic link between the 14-3-3 family and schizophrenia. In addition, 14-3-3 knockout animal models have shown schizophrenia-like phenotypes, providing further evidence for this connection. This review will discuss the results of some of the genetic and animal model studies that provide evidence for the link between 14-3-3 and schizophrenia.
Genetic linkage and proteomic studies have sought to identify gene or protein expression that may be altered in individuals who are affected by schizophrenia. However, the genetics of the disorder can be heterogeneous; no single gene nor mutation has been identified as the sole cause of schizophrenia. Nevertheless, the 14-3-3 family has been implicated in several studies of schizophrenia patients (Table 1). These results suggest that 14-3-3 alterations may contribute to the development of the disorder; therefore, further investigation of this relationship is warranted.

Table 1. 14-3-3 Human Studies.
A proteomic pathway analysis revealed that changes in the hippocampus of schizophrenia patients prominently implicate 14-3-3 signaling (Schubert et al., 2015). Further, several studies have revealed that there are 14-3-3 isoform specific changes associated with schizophrenia. In an expression analysis of peripheral leukocytes of drug-naïve first-episode schizophrenia patients, there were four down and one upregulated 14-3-3 mRNA isoforms, and five downregulated protein isoforms (Qing et al., 2016). There was a positive correlation between these isoform specific expression changes and schizophrenia. In addition, there was a negative correlation between the expression of the ε, θ and ζ isoforms and the positive symptoms of schizophrenia. While in a more recent study of peripheral blood expression levels, five of the seven 14-3-3 family members showed significantly higher baseline expression and significant changes in expression in schizophrenia patients who converted to psychosis compared to those that did not convert (Demars et al., 2020). Interestingly, there also is evidence that the 14-3-3 isoform expression levels respond differentially to antipsychotic treatment (Middleton et al., 2005; Rivero et al., 2015). Together these findings suggest that each of the 14-3-3 isoforms may be involved in schizophrenia in different capacities and are worthy of individual investigation. In fact, the genetic link between schizophrenia and individual isoforms has been further studied, particularly the ε, η, and ζ isoforms.
The 14-3-3ε isoform is encoded by the YWHAE gene, which has been proposed to be a schizophrenia susceptibility gene. Gene-based analyses have shown that common variants in the YWHAE gene contribute to schizophrenia (Torrico et al., 2020). In the study of one Japanese population, the rs28365859 single nucleotide polymorphism (SNP) of the YWHAE gene showed a significant difference between schizophrenia patients and controls (Ikeda et al., 2008). The minor allele was more frequent in controls and corresponded to higher protein expression, indicating that a major allele may be a risk factor for schizophrenia. In subsequent MRI studies, the same SNP as well as several others were shown to be related to changes in the orbitofrontal sulcogyral pattern and changes in the volume of the insula, putamen, and hippocampus of schizophrenia patients, possible developmental abnormalities that could contribute to the disorder (Kido et al., 2014; Takahashi et al., 2014).
Among other chromosomal loci, susceptibility for schizophrenia has been identified at the 8p and 22q locations (Badner and Gershon, 2002). 14-3-3ζ is genetically encoded by the YWHAZ gene at the 8p23 location. In addition to being located on a susceptible locus, genetic studies have releveled associations between SNPs and ultra-rare variants of the YWHAZ gene and schizophrenia (Jia et al., 2004; Wong et al., 2005; Torrico et al., 2020). The YWHAH gene which encodes 14-3-3η is located at 22q12.3, another susceptibility locus. Interestingly, a deletion in 22q11.2 leads to 22q11.2 Deletion Syndrome and a phenotype that often includes schizophrenia (Bassett and Chow, 2008). An estimated 1% of schizophrenia patients have also been diagnosed with 22q11.2 Deletion Syndrome. The proximity of 22q11.2 to the YWHAH gene and the overlapping association with schizophrenia seems to indicate a strong genetic link between 14-3-3η and schizophrenia. In further support of this link, both SNPs and variable number tandem repeats (VNTRs) in YWHAH have been associated with schizophrenia in genetic microarray studies (Toyooka et al., 1999; Wong et al., 2003; Grover et al., 2009). In addition, several studies have shown that the 14-3-3η protein is differentially expressed in schizophrenia (Vawter et al., 2001; Altar et al., 2009; Wu et al., 2012). Thus, both YWHAZ and YWHAH genes have been considered as schizophrenia risk genes because they are located chromosomally close to loci that have been genetically associated with schizophrenia (English et al., 2011).
Despite the evidence discussed above for the genetic link between 14-3-3 and schizophrenia, there are some incongruencies in the literature. One study found no association between YWHAE SNPs and schizophrenia (Liu et al., 2011). While several studies have failed to find significant associations between YWHAH and schizophrenia (Hayakawa et al., 1998; Bell et al., 2000; Duan et al., 2005; Wang et al., 2005). In recognition of these discrepancies, Rivero et al. (2015) performed a western blot analysis of schizophrenia subjects and controls. This study indicated that the outcomes of genetic and proteomic studies in schizophrenia are likely influenced by gender, postmortem delay, age, and pharmacological differences in the subjects being tested. When all schizophrenia subjects were grouped together, no differences in 14-3-3 immunoreactivity were found in comparison to controls. However, when the subjects were more appropriately grouped, the results did show genetic linkages (Rivero et al., 2015). While the symptoms, demographics, and environmental factors of schizophrenia are widely varied, it is also important to consider the genetic heterogeneity of the disorder. Therefore, studying the genetic link between just one family of proteins with schizophrenia is further complicated by the fact that over 200 genetic loci have been identified in association with schizophrenia (Legge et al., 2021), and that interaction between two or more of these loci may contribute to the development of the disorder. These data suggest that the heterogeneity of schizophrenia may contribute to mixed results in the literature, especially when there is not proper grouping or when controls are not careful case matched. Thus, it is important to consider these limitations when interpreting genetic linkage studies and their discrepancies. Taking these considerations into account in future work will help further clarify the genetic link between the 14-3-3 family and schizophrenia.
Considering the evidence that 14-3-3 protein expressions are changed in the brain of schizophrenia patients, investigation of 14-3-3 levels in the cerebrospinal fluid (CSF) may serve as a promising new direction to take in the diagnosis of schizophrenia. Although the CSF is an indirect representation of neurochemistry, changes in mRNA and protein levels of various 14-3-3 isoforms could potentially be reflected in the CSF of schizophrenia patients. The 14-3-3 family has been implicated in several other neurodegenerative, neurodevelopmental, and neuropsychiatric disorders (Foote and Zhou, 2012). In fact, CSF levels of 14-3-3 have been used as a biomarker for several of these neurological diseases (Van Everbroeck et al., 2005; Antonell et al., 2020; Figgie and Appleby, 2021; Nilsson et al., 2021) as well as several other diseases (Neal and Yu, 2010; Zeng and Tan, 2018; Morales et al., 2012). Early diagnosis of schizophrenia is difficult, yet early treatment can often make a difference in the prognosis of disease progression and severity. The history of 14-3-3 being used as a biomarker suggests that it could potentially serve as an indicator of schizophrenia, thus further study into this diagnostic possibility is warranted.
Although human studies have provided valuable insight into the link between the 14-3-3 family and schizophrenia, they do not provide sufficient information about the role of 14-3-3 in the pathogenesis of schizophrenia. In order to better understand the pathology of schizophrenia and how 14-3-3 proteins may be involved; animal models are needed. Several animal models have been used to study the 14-3-3 family in the context of schizophrenia and have provided further support for the link suggested by human studies (Table 2).
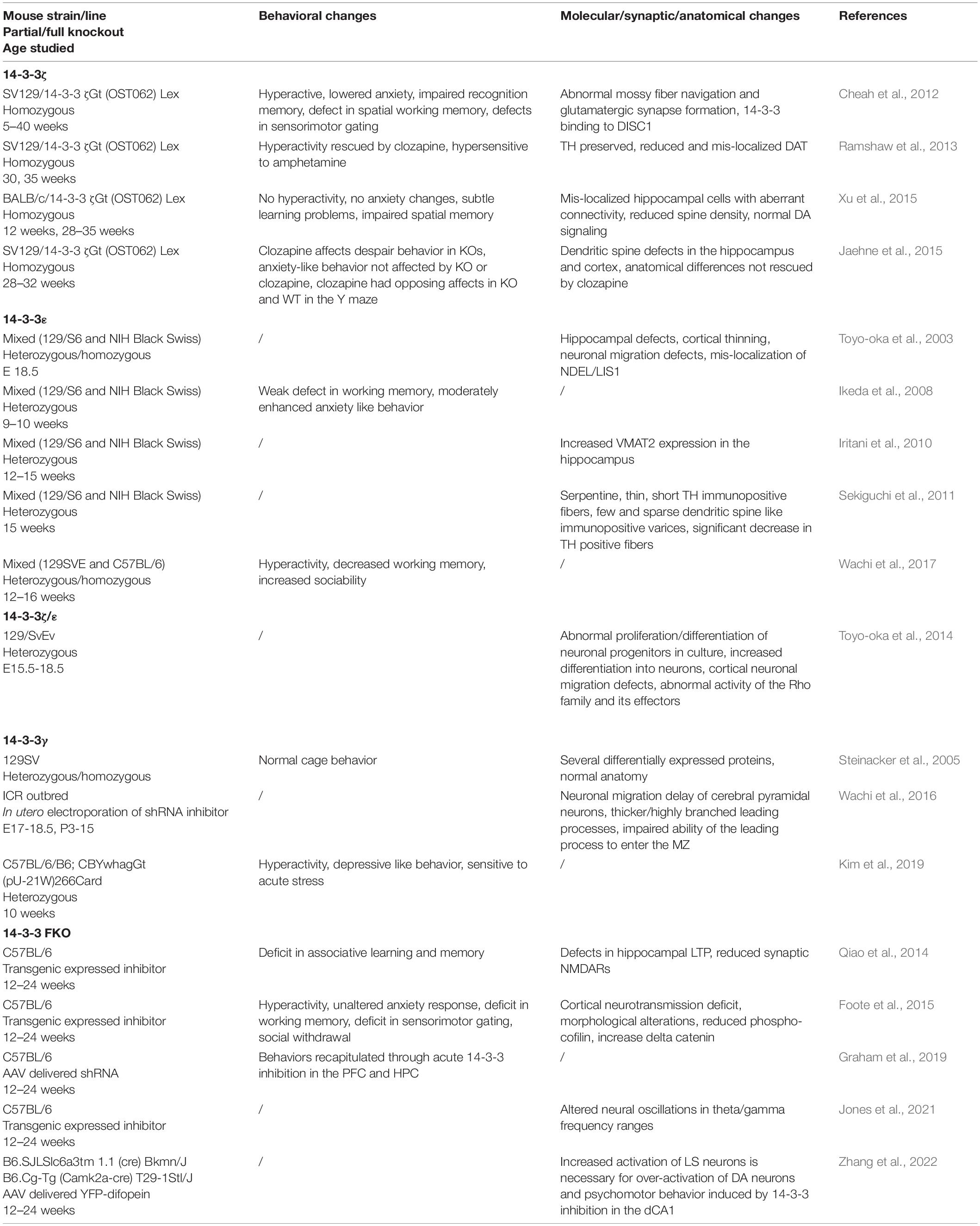
Table 2. 14-3-3 Animal Models.
One isoform of particular interest in regards to animal models of schizophrenia is 14-3-3ζ. Several 14-3-3ζ knockout models exhibit schizophrenia-like phenotypes. Homozygous 14-3-3ζ knockout mice of the Sv/129 background display behavioral abnormalities including hyperactivity, impaired recognition memory, reduced anxiety, dysfunction in hippocampal-dependent memory, and altered sensorimotor gating (Cheah et al., 2012). It is important to note that 14-3-3ζ knockout in this model is prominent in the hippocampus and dentate gyrus, pointing to the role of these regions in schizophrenia-like behavior. Defects in the hippocampus that may underlie these behavioral changes were apparent before the region was fully developed. The defects included neuronal migration defects, abnormal mossy fiber navigation, and altered glutamatergic synapse formation (Cheah et al., 2012). While the glutamate system may be altered, there is also evidence that 14-3-3ζ knockout affects the dopamine system within this model. The hyperactivity of 14-3-3ζ knockout mice is rescued by clozapine administration and knockout animals are more sensitive to amphetamine administration when compared to wildtype controls (Ramshaw et al., 2013). The mechanisms of action of these drugs and their effects on 14-3-3ζ knockout animals suggest that the dopamine system may underlie some of the behavioral abnormalities in this model. In fact, 14-3-3ζ was shown to have a physical association with the dopamine transporter (DAT). Further, decreased DAT levels found as a result of 14-3-3ζ knockout also led to increased striatal dopamine (Ramshaw et al., 2013). However, clozapine administration was not able to rescue altered anxiety behavior, the structural abnormalities in spine formation, or neuronal mis-localization in 14-3-3ζ knockout mice (Jaehne et al., 2015). This result suggests that the loss of 14-3-3ζ causes changes beyond the dopamine system as well.
Although these studies provide evidence that alterations in 14-3-3ζ can lead to schizophrenia-like phenotypes, these abnormalities have not been fully recapitulated in 14-3-3ζ knockout mice from other genetic backgrounds. When backcrossed into a BALB/c background, homozygous 14-3-3ζ knockout mice can live to adulthood. However, these mice show only weak learning disability and do not differ from controls in many of the same behavioral tests that Sv/129 14-3-3ζ knockout animals display schizophrenia-like behavior (Xu et al., 2015). In addition, the dopamine system appears intact in this model, as dopamine signaling and DAT expression are unaltered in the 14-3-3ζ knockout mice. Despite the discrepancies in behavior and dopamine function, BALB/c homozygous 14-3-3ζ knockout mice do display many of the same structural abnormalities in the brain as 14-3-3ζ knockout mice from the Sv/129 background. These abnormalities included mis-localized hippocampal neurons, reduced CA3 spine density, and abhorrent mossy fiber tracts (Xu et al., 2015).
Interestingly, overexpression of 14-3-3ζ can lead to increased spine density in primary hippocampal neuron culture (Angrand et al., 2006). This is consistent with the reduction in spine density seen in 14-3-3ζ knockout mice, indicating that 14-3-3ζ positively regulates spine density. Overall there is evidence that 14-3-3ζ knockout does induce schizophrenia-like defects in mice, but these defects seem to depend on the genetic background of the animal models. Thus, the models discussed above provide further support for the importance of 14-3-3ζ to many structural processes in the brain that may underlie schizophrenia-like phenotypes when altered.
Several human studies have found a link between 14-3-3ε and schizophrenia, prompting further studies in 14-3-3ε knockout animals. Homozygous knockout of 14-3-3ε is prenatally lethal in in-bred genetic backgrounds. Thus, Toyo-oka et al. (2003) examined the brains of homozygous and heterozygous 14-3-3ε knockout mice at embryonic day 18.5, prior to homozygous lethality. Both genotypes had hippocampal defects and cortical thinning, structural issues which were underscored by shortened neuronal migration and mis-localization of key proteins involved in migration processes. The lethality of homozygous knockout mice points to the increased severity of these defects with complete loss of 14-3-3ε. Additional studies of heterozygous 14-3-3ε knockout mice revealed further molecular, structural, and behavioral alterations. One such alteration seen in the hippocampal formation was significantly increased levels of VMAT2, a protein involved in the transport of monoamine neurotransmitters into neuronal vesicles (Iritani et al., 2010). Another study found that 14-3-3ε knockout mice have decreased numbers of tyrosine hydroxylase positive fibers that also exhibit altered functional structure (Sekiguchi et al., 2011). Behavioral testing of this heterozygous 14-3-3ε knockout model revealed weak deficits in working memory and moderately enhanced anxiety like behavior (Ikeda et al., 2008). However, a mixed genetic background model of 14-3-3ε knockout showed different behavioral results, including weaker motor activity, hyperactivity, visual/spatial memory defects, and unaltered anxiety-like behavior (Wachi et al., 2017). Thus, the above evidence suggests that loss of 14-3-3ε causes both structural and behavioral abnormalities in mice that resemble those seen in schizophrenia patient populations. However, these results may be affected by the genetic background of the mouse models in use. Nonetheless, 14-3-3ε knockout models may be a valuable tool in studying the molecular, structural, and behavioral aspects of schizophrenia.
Both 14-3-3ζ and 14-3-3ε are critical proteins when it comes to proper brain development, and there is evidence that loss of either can cause defects similar to those seen in schizophrenia patients. The underpinnings of these changes have been further elucidated through the study of a double knockout mouse model in which mice were heterozygous knockout for one isoform and homozygous knockout for the other 14-3-3 isoform (Ywhae+/flox; YwhazKO/KO and Ywhaeflox/flox; Ywhaz+/KO) (Toyo-oka et al., 2014). Double knockout mice displayed neuronal differentiation and migration defects as well as seizures. The same phenotypes are seen in single knockout models for these proteins but are more pronounced in double knockout animals. These results point to the critical involvement of 14-3-3ζ and 14-3-3ε proteins in the developing brain, as well as the functional redundancy between isoforms. A critical pathway through which these 14-3-3 proteins can regulate neuronal differentiation is the catenin/Rho GTPase/Limk1/cofilin signaling pathway, where 14-3-3 proteins directly interact with phosphorylated delta-catenin to promote F-actin formation. 14-3-3 double knockout mice were shown to have increased levels of delta-catenin, as well as decreased levels of beta-catenin and alphaN-catenin (Toyo-oka et al., 2014). Deletion of delta-catenin did not rescue neuronal migration abnormalities in double knockout mice; but mutants of the Ndel1 protein were able to do so, indicating that 14-3-3 proteins are also involved in a separate pathway that controls neuronal migration (Toyo-oka et al., 2014). Thus, 14-3-3 proteins are important regulators of several different pathways and the loss of one or more isoforms can detrimentally impact neural development and result in behavioral abnormality.
14-3-3γ is particularly enriched in the brain and is typically expressed in the developing cortex. Reduction in the γ isoform of 14-3-3 has yielded mixed outcomes when it comes to behavioral and morphological changes. One study found no obvious behavioral alterations or histological differences in the cortex of either heterozygous or homozygous 14-3-3γ knockout mice (Steinacker et al., 2005). While in another study, depletion of 14-3-3γ through in utero electroporation of a specific small hairpin RNA (shRNA) resulted in a delay of neural migration and morphological abnormalities in the cortex (Wachi et al., 2016). In a study of behavior, heterozygous 14-3-3γ knockout mice were hyperactive and more sensitive to acute stress when compared to wildtype littermates, while homozygous 14-3-3γ knockout mice died before birth (Kim et al., 2019). Although there are some conflicting results, there is evidence that loss of 14-3-3γ can cause abnormalities that resemble those found in psychiatric disorders like schizophrenia.
With the many roles of 14-3-3 proteins in neuronal processes and the genetic evidence linking the proteins to schizophrenia, our lab sought to create a mammalian model to study the synaptic and cognitive functions of the 14-3-3 protein family. Transgenic 14-3-3 functional knock-out (FKO) mice were generated through the expression of yellow fluorescent protein (YFP) fused difopein (dimeric 14-3-3 peptide inhibitor), which inhibits all isoforms of 14-3-3 from interacting with endogenous binding partners (Qiao et al., 2014). An important consideration is that 14-3-3 inhibition during embryonic development can be lethal. To avoid prenatal lethality, the transgenic expression of YFP-difopein was driven by the neuronal specific Thy-1 promotor, which is normally expressed in the perinatal period. The Thy-1 promotor created several founder mice in which the expression pattern of YFP-difopein varied but was preserved within the founder line. One of these founder lines had transgene expression that was relatively higher in the hippocampus (HPC) and the pre-frontal cortex (PFC). This line was found to display several behavioral, electrophysiological, and molecular abnormalities. During the contextual fear conditioning and passive avoidance tests, these 14-3-3FKO mice displayed significantly reduced freezing behavior and reduced latency to dark chamber, indicating impairments in associative learning and memory. Electrophysiological investigation of these mice reveled that they also exhibit defects in long-term synaptic plasticity of the hippocampus. Consistently, evidence for NMDAR dysfunction in the 14-3-3FKO line was observed, including significant reductions in the NMDAR/AMPAR ratio and in NMDAR mediated currents, as well as lowered levels of the GluN1 and GluN2a NMDA receptor subunits.
Further investigation of the 14-3-3FKO line revealed additional behavioral and synaptic defects that can be considered schizophrenia-like phenotypes. Increased activity in the open field test (OFT), decreased alteration in the Y maze test, decreased pre-pulse inhibition percentage, and decreased social interaction in the three-chamber test were observed in FKO mice vs. their wildtype (WT) littermates (Foote et al., 2015). These behavioral outcomes reveal deficits in psychomotor behavior, working memory, sensorimotor gating control, and social behavior respectively; all of which can be likened to schizophrenia-related phenotypes. In addition to these behavioral changes, whole-cell voltage-clamp recording of YFP-difopein infected cells in cortical neurons of the FKO mice show significant reductions in the frequencies of spontaneous excitatory and inhibitory post synaptic potentials, as well as alterations in miniature excitatory and inhibitory post synaptic potentials. Further, the cortical layer-5 and hippocampal CA1 pyramidal neurons of FKO mice had decreased distal apical dendrite complexity and decreased spine density when compared to cells from WT littermates. Potential molecular mechanisms of these changes may come from reduced levels of phospho-cofilin and increased levels of delta-catenin in FKO brain tissue.
The behavioral, electrophysiological, and molecular results discussed above indicate that 14-3-3 inhibition in the PFC and HPC can lead to a variety of schizophrenia-like phenotypes. However, the individual roles for each of these brain regions was not distinguishable. In order to determine if inhibition in either the PFC and/or the HPC is necessary and sufficient to induce schizophrenia-like phenotypes, 14-3-3 function was regionally restored through an adeno-associated virus (AAV) delivered shRNA that knocks down YFP-difopein (Graham et al., 2019). Delivery of the shRNA to both the PFC and the HPC lead to significant reductions in OFT locomotor activity, while delivery to one region alone did not. Interestingly, shRNA injection to the HPC alone significantly increased the GluN1 levels in 14-3-3FKO animals, but not to the level of WT littermates. This was likely due to the fact that shRNA was not able to fully inhibit the YFP-difopein transgene. Due to this incomplete inhibition and the fact that the difopein expression in FKO mice is not strictly limited to the PFC and HPC, our lab sought to investigate the effect of region specific difopein expression. A virus using the CamKIIa promotor to drive YFP-difopein expression in excitatory neurons was created to determine if 14-3-3 inhibition in the PFC and/or the HPC is sufficient to induce schizophrenia related phenotypes (Graham et al., 2019). Behavioral testing revealed that WT mice with YFP-difopein injections to the HPC alone, but not the PFC alone, exhibit significantly less freezing behavior in the contextual fear conditioning test and decreased pre-pulse inhibition. In addition, injection of YFP-difopein to both the PFC and HPC, as well as the HPC alone, lead to increased locomotor activity in the OFT, while injection to the PFC alone did not. These results indicate that 14-3-3 inhibition in the HPC is sufficient to induce schizophrenia-like behaviors. In further support of this claim is the finding that YFP-difopein injection to the HPC has a direct effect on NMDA receptor regulation, resulting in decreased GluN1, GluN2A, and PSD95 levels.
To build upon the behavioral and molecular abnormalities found in our models of 14-3-3 inhibition, our lab investigated the role of 14-3-3 proteins in neural oscillations, which are dysfunctional in schizophrenia patients (Jones et al., 2021). FKO animals exhibited a range of changes in power, coherence, and phase-amplitude coupling in both resting and task-related theta and gamma oscillations. WT animals with 14-3-3 inhibited in the HPC alone showed similar yet distinct changes in these same measures. However, 14-3-3 inhibition to the PFC alone leads to few changes in neural oscillations. Overall, our in vivo electrophysiological results indicated that acute 14-3-3 inhibition in the HPC largely disrupts theta oscillations and is sufficient to cause neural oscillation defects in the HPC as well as the PFC.
Interestingly, FKO mice exhibit hyperactive dopamine signaling in the ventral tegmental area (VTA) and some of their altered behavior can be attenuated with antipsychotic administration (Foote et al., 2015). Yet there is no YFP-difopein expression detected in the VTA, indicating that 14-3-3 inhibition in other brain areas has an influence on VTA dopamine signaling. Acute 14-3-3 inhibition in the dorsal HPC (dHPC) alone causes c-Fos expression in the dHPC and increased locomotor activity in the OFT that is responsive to antipsychotics (Zhang et al., 2022). This overexcitation in the dHPC is accompanied by robust c-Fos expression in the VTA, indicating a connection between 14-3-3 inhibition induced dHPC overexcitation, hyperactive dopamine signaling, and hyperlocomotion. However, the neural circuitry underlying this connection is not clear, as the dHPC does not directly communicate with the VTA. Through neural tracing techniques, we found that the lateral septum (LS), which also shows increased c-Fos activity after OFT in dHPC injected mice, has mono-synaptic connections with the dHPC (Zhang et al., 2022). Previous studies have shown that the LS also communicates with the VTA, where its connections act on GABAergic interneurons to disinhibit VTA DA neurons (Vega-Quiroga et al., 2018). We confirmed that VTA projecting LS neurons are activated following OFT in difopein injected mice. Thus, the LS appears to be both activated during 14-3-3 inhibition induced hyperlocomotion and anatomically connected with the dHPC and VTA. To further confirm the role of the LS in this neural circuitry we used chemogenetic Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) to manipulate the activity of the LS in mice. Our results demonstrated that chemogenetic inhibition of the LS attenuates difopein induced hyperlocomotion as well as upregulated DA activity, while chemogenetic activation of LS projecting dHPC neurons elicits hyperlocomotion in WT mice. These results indicate that increased activity in the LS is both necessary and sufficient to induce changes in psycholocomotor behavior. Overall, our results provide evidence for a polysynaptic pathway from the dHPC to the LS to the VTA in which 14-3-3 inhibition causes an imbalance in the ratio of excitatory to inhibitory signaling that results in psychomotor behavior (Zhang et al., 2022).
Through a series of studies, our lab has created a mouse model of 14-3-3 inhibition that can be useful in the study schizophrenia related phenotypes at the behavioral, molecular, and circuitry levels. The family of 14-3-3 proteins is involved in several critical neuronal processes and has been associated with schizophrenia through genetic linkage studies. We found that transgenic expression of a 14-3-3 inhibitor in key forebrain areas results in a mouse line that displays several behavioral, electrophysiological, and molecular phenotypes which resembles those seen in schizophrenia patients. Through acute viral delivery of our inhibitor, we saw that the functional loss of 14-3-3 proteins in the hippocampus is sufficient to induce these disease-related outcomes. Using our 14-3-3 inhibited mice, we delineated a previously unknown polysynaptic circuit that connects 14-3-3 inhibition induced imbalances in neuronal signaling to changes in psychomotor behavior. Together, our work provides further evidence for the role of 14-3-3 dysfunction in schizophrenia and a tool which we can use to further explore the mechanisms of pathology in schizophrenia.
Human genetic and proteomic linkage studies have provided several lines of evidence that the 14-3-3 protein family and their associated genes may be altered in schizophrenia. Because the 14-3-3 proteins mediate such a wide range of cellular and molecular processes, any mutation to or change in expression of these proteins may contribute to abnormalities in these processes and potentially to disease states such as schizophrenia.
Several neurotransmitter systems appear to be altered in schizophrenia, including the dopamine, glutamate, and GABA systems. For many years the dopamine hypothesis and the glutamate hypothesis were separate lenses in which researchers and physicians studied and tried to treat the neuropathology of schizophrenia. More recently, it has been proposed that dysfunction in the dopamine system may be a downstream consequence of dysregulated glutamate neurotransmission in the forebrain. As the major excitatory neurotransmitter, glutamate has influence all over the brain, including in the dopamine system. However, it is unknown how the two systems could be interacting in the disease state, making it difficult to merge the two hypotheses for schizophrenia. The 14-3-3 protein family may serve as a potential link between dopamine and glutamate dysfunction in schizophrenia, as there is evidence that changes in 14-3-3 have effects in both neurotransmitter systems. Alterations in NMDA receptor (NMDAR) activity is reported in several 14-3-3 knockout models, and decreased NMDAR activity can alter the excitation and inhibition balance in neural networks and circuits (Moghaddam and Javitt, 2012). Surface expression of NMDA receptors is regulated and promoted by 14-3-3 proteins, particularly the ζ and ε isoforms, through their interactions with particular NMDAR subunits (Chen and Roche, 2009; Lee et al., 2021). This relationship is further highlighted by the finding that knockdown of the NMDAR subunit NR1 leads to synaptic reduction of 14-3-3ε (Ramsey et al., 2011; Ferris et al., 2014). Further, a recent study from our lab has elucidated a polysynaptic pathway in which 14-3-3 dysfunction in the dorsal hippocampus underlies altered psychomotor behavior mediated by dopamine in the VTA (Zhang et al., 2022). These results offer evidence on how the glutamate and dopamine systems may be linked in schizophrenia, as well as a potential role for 14-3-3 dysfunction in the mechanism of the pathology.
The 14-3-3 proteins have many binding partners and are involved in several critical molecular and cellular pathways; thus, the disruption of these proteins can have many potential impacts. One pathway affected by 14-3-3 disruption is the catenin/Rho GTPase/Limk1/cofilin signaling pathway, which plays a significant role in regulating actin cytoskeleton dynamics during brain development. Moreover, the Ndel1/Lis1/14-3-3ε complex has been proposed to be critical for neuronal migration (Foote and Zhou, 2012). Interestingly, the localization of the Ndel1/Lis1/14-3-3ε complex to axons is regulated by the schizophrenia related protein DISC1 (Taya et al., 2007). 14-3-3ε binds to the DISC1 binding region of Ndel1, maintaining its phosphorylation (Toyo-oka et al., 2003; Johnson et al., 2010), and deficiency of 14-3-3ε leads to mis-localization of Ndel1 and Lis1 (Toyo-oka et al., 2003). Additionally, 14-3-3 proteins have been shown to interact with many other cytoskeleton and dendritic spine related proteins. For example, 14-3-3ζ interacts with microtubule-associated protein/microtubule affinity-regulating kinase 3 (MARK3) (Angrand et al., 2006). The proper regulation of microtubules is necessary for neuronal migration and spine formation. In fact, knockout of microtubule associated protein 6 (MAP6) in mice results in many similar schizophrenia-like phenotypes as 14-3-3 knockout models. These include deficits in synaptic plasticity, abnormal glutamatergic signaling, and locomotor hyperactivity (Cuveillier et al., 2021). The similarities between MAP6 and 14-3-3 knockout models along with the physical interaction between 14-3-3 proteins and microtubule related proteins suggest that 14-3-3 are key players in appropriate cytoskeletal regulation. The association between 14-3-3 proteins and these specific binding partners and pathways may underlie the neuronal migration and synaptic defects observed in the 14-3-3 knockout animal models discussed above, as well as provide potential mechanistic insight into the pathogenesis of schizophrenia.
Several of these processes are critical to neurodevelopment, suggesting that any changes in 14-3-3 proteins during critical periods could potentially lead to abnormal structural and functional connections in the brain. In fact, the viability of knockout animals and the severity of their abnormalities in the brain are influenced by the timing of the 14-3-3 knockout. Similarly, the timing of the progression of schizophrenia in humans also points to the importance of these critical developmental periods. The molecular changes that underlie schizophrenia require further study to create a more holistic understanding of the disease and how 14-3-3 dependent regulatory pathways may be involved. The diverse and important roles for 14-3-3 proteins serve as promising points from which to study the mechanisms underlying the disorder.
Schizophrenia is a complicated mental disorder that greatly affects those who are diagnosed with it. The heterogeneity of the disorder makes it difficult to decipher the neurobiological basis of schizophrenia. One interesting and promising route of study in schizophrenia is the role of 14-3-3 proteins. The genetic link between 14-3-3 and schizophrenia suggests that studying schizophrenia through the 14-3-3 family can offer valuable insight to the disease. The results from 14-3-3 knockout models validate this idea and have allowed for a better understanding of the pathology of schizophrenia and how the 14-3-3 family may contribute to its progression. The 14-3-3 protein family has a wide variety of binding partners and functions, the exact mechanisms behind how alterations in 14-3-3 proteins can lead to schizophrenia-like phenotypes is not fully understood. However, some potential mechanisms include abnormal neural development and neuronal signaling following altered neurotransmitter receptor levels, mis-localized protein complexes, and interrupted cellular pathways. Future study will be needed to fully elucidate the causes of these observed phenotypes but will undoubtedly provide further understanding of the complex pathology of schizophrenia.
MN was responsible for drafting, writing, reviewing, and editing this manuscript. YZ was responsible for writing, reviewing, and editing the manuscript with suggestive ideas on formatting and outlining this manuscript. Both authors contributed to the article and approved the submitted version.
This work was supported by the National Institutes of Health (award no. R01 MH115188 to YZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Altar, C. A., Vawter, M. P., and Ginsberg, S. D. (2009). Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology 34, 18–54. doi: 10.1038/npp.2008.172
Angrand, P. O., Segura, I., Volkel, P., Ghidelli, S., Terry, R., Brajenovic, M., et al. (2006). Transgenic mouse proteomics identifies new 14-3-3-associated proteins involved in cytoskeletal rearrangements and cell signaling. Mol. Cell Proteomics 5, 2211–2227. doi: 10.1074/mcp.M600147-MCP200
Antonell, A., Tort-Merino, A., Rios, J., Balasa, M., Borrego-Ecija, S., Auge, J. M., et al. (2020). Synaptic, axonal damage and inflammatory cerebrospinal fluid biomarkers in neurodegenerative dementias. Alzheimers Dement. 16, 262–272. doi: 10.1016/j.jalz.2019.09.001
Badner, J. A., and Gershon, E. S. (2002). Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol. Psychiatry 7, 405–411. doi: 10.1038/sj.mp.4001012
Bassett, A. S., and Chow, E. W. (2008). Schizophrenia and 22q11.2 deletion syndrome. Curr. Psychiatry Rep. 10, 148–157. doi: 10.1007/s11920-008-0026-1
Bell, R., Munro, J., Russ, C., Powell, J. F., Bruinvels, A., Kerwin, R. W., et al. (2000). Systematic screening of the 14-3-3 eta (eta) chain gene for polymorphic variants and case-control analysis in schizophrenia. Am. J. Med. Genet. 96, 736–743.
Berg, D., Holzmann, C., and Riess, O. (2003). 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 4, 752–762. doi: 10.1038/nrn1197
Boston, P. F., Jackson, P., and Thompson, R. J. (1982). Human 14-3-3 protein: radioimmunoassay, tissue distribution, and cerebrospinal fluid levels in patients with neurological disorders. J. Neurochem. 38, 1475–1482. doi: 10.1111/j.1471-4159.1982.tb07928.x
Cheah, P. S., Ramshaw, H. S., Thomas, P. Q., Toyo-Oka, K., Xu, X., Martin, S., et al. (2012). Neurodevelopmental and neuropsychiatric behaviour defects arise from 14-3-3zeta deficiency. Mol. Psychiatry 17, 451–466. doi: 10.1038/mp.2011.158
Chen, B. S., and Roche, K. W. (2009). Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron 62, 471–478. doi: 10.1016/j.neuron.2009.04.015
Cuveillier, C., Boulan, B., Ravanello, C., Denarier, E., Deloulme, J. C., Gory-Faure, S., et al. (2021). Beyond neuronal microtubule stabilization: MAP6 and CRMPS. two converging stories. Front. Mol. Neurosci. 14:665693. doi: 10.3389/fnmol.2021.665693
Demars, F., Kebir, O., Marzo, A., Iftimovici, A., Schramm, C., Icaar Study Group, et al. (2020). Dysregulation of peripheral expression of the YWHA genes during conversion to psychosis. Sci. Rep. 10:9863. doi: 10.1038/s41598-020-66901-1
Duan, S., Gao, R., Xing, Q., Du, J., Liu, Z., Chen, Q., et al. (2005). A family-based association study of schizophrenia with polymorphisms at three candidate genes. Neurosci. Lett. 379, 32–36. doi: 10.1016/j.neulet.2004.12.040
English, J. A., Pennington, K., Dunn, M. J., and Cotter, D. R. (2011). The neuroproteomics of schizophrenia. Biol. Psychiatry 69, 163–172. doi: 10.1016/j.biopsych.2010.06.031
Ferris, M. J., Milenkovic, M., Liu, S., Mielnik, C. A., Beerepoot, P., John, C. E., et al. (2014). Sustained N-methyl-d-aspartate receptor hypofunction remodels the dopamine system and impairs phasic signaling. Eur. J. Neurosci. 40, 2255–2263. doi: 10.1111/ejn.12594
Figgie, M. P. Jr., and Appleby, B. S. (2021). Clinical use of improved diagnostic testing for detection of prion disease. Viruses 13:789. doi: 10.3390/v13050789
Foote, M., Qiao, H., Graham, K., Wu, Y., and Zhou, Y. (2015). Inhibition of 14-3-3 proteins leads to schizophrenia-related behavioral phenotypes and synaptic defects in mice. Biol. Psychiatry 78, 386–395. doi: 10.1016/j.biopsych.2015.02.015
Foote, M., and Zhou, Y. (2012). 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Biol. 3, 152–164.
Fu, H., Subramanian, R. R., and Masters, S. C. (2000). 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647. doi: 10.1146/annurev.pharmtox.40.1.617
Graham, K., Zhang, J., Qiao, H., Wu, Y., and Zhou, Y. (2019). Region-specific inhibition of 14-3-3 proteins induces psychomotor behaviors in mice. NPJ Schizophr. 5:1. doi: 10.1038/s41537-018-0069-1
Grover, D., Verma, R., Goes, F. S., Mahon, P. L., Gershon, E. S., McMahon, F. J., et al. (2009). Family-based association of YWHAH in psychotic bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 977–983. doi: 10.1002/ajmg.b.30927
Hayakawa, T., Ishiguro, H., Toru, M., Hamaguchi, H., and Arinami, T. (1998). Systematic search for mutations in the 14-3-3 eta chain gene on chromosome 22 in schizophrenics. Psychiatr. Genet. 8, 33–36. doi: 10.1097/00041444-199800810-00006
Ikeda, M., Hikita, T., Taya, S., Uraguchi-Asaki, J., Toyo-oka, K., Wynshaw-Boris, A., et al. (2008). Identification of YWHAE, a gene encoding 14-3-3epsilon, as a possible susceptibility gene for schizophrenia. Hum. Mol. Genet. 17, 3212–3222. doi: 10.1093/hmg/ddn217
Iritani, S., Sekiguchi, H., Habuchi, C., Hikita, T., Taya, S., Kaibuchi, K., et al. (2010). Immunohistochemical study of vesicle monoamine transporter 2 in the hippocampal region of genetic animal model of schizophrenia. Synapse 64, 948–953. doi: 10.1002/syn.20846
Jablensky, A. (2000). Epidemiology of schizophrenia: the global burden of disease and disability. Eur. Arch. Psychiatry Clin. Neurosci. 250, 274–285. doi: 10.1007/s004060070002
Jaehne, E. J., Ramshaw, H., Xu, X., Saleh, E., Clark, S. R., Schubert, K. O., et al. (2015). In-vivo administration of clozapine affects behaviour but does not reverse dendritic spine deficits in the 14-3-3zeta KO mouse model of schizophrenia-like disorders. Pharmacol. Biochem. Behav. 138, 1–8. doi: 10.1016/j.pbb.2015.09.006
Jia, Y., Yu, X., Zhang, B., Yuan, Y., Xu, Q., Shen, Y., et al. (2004). An association study between polymorphisms in three genes of 14-3-3 (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein) family and paranoid schizophrenia in northern Chinese population. Eur. Psychiatry 19, 377–379. doi: 10.1016/j.eurpsy.2004.07.006
Johnson, C., Crowther, S., Stafford, M. J., Campbell, D. G., Toth, R., and MacKintosh, C. (2010). Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J. 427, 69–78. doi: 10.1042/BJ20091834
Jones, Z. B., Zhang, J., Wu, Y., and Zhou, Y. (2021). Inhibition of 14-3-3 proteins alters neural oscillations in mice. Front. Neural Circuits 15:647856. doi: 10.3389/fncir.2021.647856
Kane, J. M., Kishimoto, T., and Correll, C. U. (2013). Non-adherence to medication in patients with psychotic disorders: epidemiology, contributing factors and management strategies. World Psychiatry 12, 216–226. doi: 10.1002/wps.20060
Kido, M., Nakamura, Y., Nemoto, K., Takahashi, T., Aleksic, B., Furuichi, A., et al. (2014). The polymorphism of YWHAE, a gene encoding 14-3-3epsilon, and brain morphology in schizophrenia: a voxel-based morphometric study. PLoS One 9:e103571. doi: 10.1371/journal.pone.0103571
Kim, D. E., Cho, C. H., Sim, K. M., Kwon, O., Hwang, E. M., Kim, H. W., et al. (2019). 14-3-3Gamma haploinsufficient mice display hyperactive and stress-sensitive behaviors. Exp. Neurobiol. 28, 43–53. doi: 10.5607/en.2019.28.1.43
Lee, G. S., Zhang, J., Wu, Y., and Zhou, Y. (2021). 14-3-3 proteins promote synaptic localization of N-methyl d-aspartate receptors (NMDARs) in mouse hippocampal and cortical neurons. PLoS One 16:e0261791. doi: 10.1371/journal.pone.0261791
Legge, S. E., Santoro, M. L., Periyasamy, S., Okewole, A., Arsalan, A., and Kowalec, K. (2021). Genetic architecture of schizophrenia: a review of major advancements. Psychol. Med. 51, 2168–2177. doi: 10.1017/S0033291720005334
Liu, D., Bienkowska, J., Petosa, C., Collier, R. J., Fu, H., and Liddington, R. (1995). Crystal structure of the zeta isoform of the 14-3-3 protein. Nature 376, 191–194. doi: 10.1038/376191a0
Liu, J., Zhou, G., Ji, W., Li, J., Li, T., Wang, T., et al. (2011). No association of the YWHAE gene with schizophrenia, major depressive disorder or bipolar disorder in the Han Chinese population. Behav. Genet. 41, 557–564. doi: 10.1007/s10519-010-9426-1
Mentzel, T. Q., Lieverse, R., Bloemen, O., Viechtbauer, W., van Harten, P. N., and Risk Genetic and Investigators Outcome of Psychosis. (2017). High incidence and prevalence of drug-related movement disorders in young patients with psychotic disorders. J. Clin. Psychopharmacol. 37, 231–238. doi: 10.1097/JCP.0000000000000666
Middleton, F. A., Peng, L., Lewis, D. A., Levitt, P., and Mirnics, K. (2005). Altered expression of 14-3-3 genes in the prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology 30, 974–983. doi: 10.1038/sj.npp.1300674
Moghaddam, B., and Javitt, D. (2012). From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37, 4–15. doi: 10.1038/npp.2011.181
Morales, D., Skoulakis, E. C., and Acevedo, S. F. (2012). 14-3-3s are potential biomarkers for HIV-related neurodegeneration. J. Neurovirol. 18, 341–353. doi: 10.1007/s13365-012-0121-2
Neal, C. L., and Yu, D. (2010). 14-3-3zeta as a prognostic marker and therapeutic target for cancer. Expert Opin. Ther. Targets 14, 1343–1354. doi: 10.1517/14728222.2010.531011
Nilsson, J., Gobom, J., Sjodin, S., Brinkmalm, G., Ashton, N. J., Svensson, J., et al. (2021). Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement. 13:e12179. doi: 10.1002/dad2.12179
Qiao, H., Foote, M., Graham, K., Wu, Y., and Zhou, Y. (2014). 14-3-3 proteins are required for hippocampal long-term potentiation and associative learning and memory. J. Neurosci. 34, 4801–4808. doi: 10.1523/JNEUROSCI.4393-13.2014
Qing, Y., Sun, L., Yang, C., Jiang, J., Yang, X., Hu, X., et al. (2016). Dysregulated 14-3-3 Family in peripheral blood leukocytes of patients with schizophrenia. Sci. Rep. 6:23791. doi: 10.1038/srep23791
Ramsey, A. J., Milenkovic, M., Oliveira, A. F., Escobedo-Lozoya, Y., Seshadri, S., Salahpour, A., et al. (2011). Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 108, 5795–5800. doi: 10.1073/pnas.1012621108
Ramshaw, H., Xu, X., Jaehne, E. J., McCarthy, P., Greenberg, Z., Saleh, E., et al. (2013). Locomotor hyperactivity in 14-3-3zeta KO mice is associated with dopamine transporter dysfunction. Transl. Psychiatry 3:e327. doi: 10.1038/tp.2013.99
Rivero, G., Gabilondo, A. M., Garcia-Sevilla, J. A., La Harpe, R., Morentin, B., and Meana, J. J. (2015). Up-regulated 14-3-3beta and 14-3-3zeta proteins in prefrontal cortex of subjects with schizophrenia: effect of psychotropic treatment. Schizophr. Res. 161, 446–451. doi: 10.1016/j.schres.2014.12.014
Schubert, K. O., Focking, M., and Cotter, D. R. (2015). Proteomic pathway analysis of the hippocampus in schizophrenia and bipolar affective disorder implicates 14-3-3 signaling, aryl hydrocarbon receptor signaling, and glucose metabolism: potential roles in GABAergic interneuron pathology. Schizophr. Res. 167, 64–72. doi: 10.1016/j.schres.2015.02.002
Sekiguchi, H., Iritani, S., Habuchi, C., Torii, Y., Kuroda, K., Kaibuchi, K., et al. (2011). Impairment of the tyrosine hydroxylase neuronal network in the orbitofrontal cortex of a genetically modified mouse model of schizophrenia. Brain Res. 1392, 47–53. doi: 10.1016/j.brainres.2011.03.058
Steinacker, P., Schwarz, P., Reim, K., Brechlin, P., Jahn, O., Kratzin, H., et al. (2005). Unchanged survival rates of 14-3-3gamma knockout mice after inoculation with pathological prion protein. Mol. Cell Biol. 25, 1339–1346. doi: 10.1128/MCB.25.4.1339-1346.2005
Takahashi, T., Nakamura, Y., Nakamura, Y., Aleksic, B., Takayanagi, Y., Furuichi, A., et al. (2014). The polymorphism of YWHAE, a gene encoding 14-3-3epsilon, and orbitofrontal sulcogyral pattern in patients with schizophrenia and healthy subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 51, 166–171. doi: 10.1016/j.pnpbp.2014.02.005
Takahashi, Y. (2003). The 14-3-3 proteins: gene, gene expression, and function. Neurochem. Res. 28, 1265–1273. doi: 10.1023/a:1024296932670
Taya, S., Shinoda, T., Tsuboi, D., Asaki, J., Nagai, K., Hikita, T., et al. (2007). DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 27, 15–26. doi: 10.1523/JNEUROSCI.3826-06.2006
Torrico, B., Anton-Galindo, E., Fernandez-Castillo, N., Rojo-Francas, E., Ghorbani, S., Pineda-Cirera, L., et al. (2020). Involvement of the 14-3-3 gene family in autism spectrum disorder and schizophrenia: genetics, transcriptomics and functional analyses. J. Clin. Med. 9:1851. doi: 10.3390/jcm9061851
Toyooka, K., Muratake, T., Tanaka, T., Igarashi, S., Watanabe, H., Takeuchi, H., et al. (1999). 14-3-3 protein eta chain gene (YWHAH) polymorphism and its genetic association with schizophrenia. Am. J. Med. Genet. 88, 164–167. doi: 10.1002/(sici)1096-8628(19990416)88:2<164::aid-ajmg13>3.0.co;2-3
Toyo-oka, K., Shionoya, A., Gambello, M. J., Cardoso, C., Leventer, R., Ward, H. L., et al. (2003). 14-3-3Epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat. Genet. 34, 274–285. doi: 10.1038/ng1169
Toyo-oka, K., Wachi, T., Hunt, R. F., Baraban, S. C., Taya, S., Ramshaw, H., et al. (2014). 14-3-3epsilon and zeta regulate neurogenesis and differentiation of neuronal progenitor cells in the developing brain. J. Neurosci. 34, 12168–12181. doi: 10.1523/JNEUROSCI.2513-13.2014
Van Everbroeck, B., Boons, J., and Cras, P. (2005). Cerebrospinal fluid biomarkers in Creutzfeldt-Jakob disease. Clin. Neurol. Neurosurg. 107, 355–360. doi: 10.1016/j.clineuro.2004.12.002
van Hemert, M. J., Steensma, H. Y., and van Heusden, G. P. (2001). 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays 23, 936–946. doi: 10.1002/bies.1134
Vawter, M. P., Barrett, T., Cheadle, C., Sokolov, B. P., Wood, W. H. III, Donovan, D. M., et al. (2001). Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res. Bull. 55, 641–650. doi: 10.1016/s0361-9230(01)00522-6
Vega-Quiroga, I., Yarur, H. E., and Gysling, K. (2018). Lateral septum stimulation disinhibits dopaminergic neurons in the antero-ventral region of the ventral tegmental area: role of GABA-A alpha 1 receptors. Neuropharmacology 128, 76–85. doi: 10.1016/j.neuropharm.2017.09.034
Volavka, J., Czobor, P., Sheitman, B., Lindenmayer, J. P., Citrome, L., McEvoy, J. P., et al. (2002). Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am. J. Psychiatry 159, 255–262. doi: 10.1176/appi.ajp.159.2.255
Wachi, T., Cornell, B., Marshall, C., Zhukarev, V., Baas, P. W., and Toyo-oka, K. (2016). Ablation of the 14-3-3gamma protein results in neuronal migration delay and morphological defects in the developing cerebral cortex. Dev. Neurobiol. 76, 600–614. doi: 10.1002/dneu.22335
Wachi, T., Cornell, B., and Toyo-Oka, K. (2017). Complete ablation of the 14-3-3epsilon protein results in multiple defects in neuropsychiatric behaviors. Behav. Brain Res. 319, 31–36. doi: 10.1016/j.bbr.2016.11.016
Wang, H., Zhang, L., Liddington, R., and Fu, H. (1998). Mutations in the hydrophobic surface of an amphipathic groove of 14-3-3zeta disrupt its interaction with Raf-1 kinase. J. Biol. Chem. 273, 16297–16304. doi: 10.1074/jbc.273.26.16297
Wang, H. S., Duan, S. W., Xing, Q. H., Du, J., Li, X. W., Xu, Y. F., et al. (2005). [Association study between NPY and YWHAH gene polymorphisms and schizophrenia]. Yi Chuan Xue Bao 32, 1235–1240.
Wong, A. H., Likhodi, O., Trakalo, J., Yusuf, M., Sinha, A., Pato, C. N., et al. (2005). Genetic and post-mortem mRNA analysis of the 14-3-3 genes that encode phosphoserine/threonine-binding regulatory proteins in schizophrenia and bipolar disorder. Schizophr. Res. 78, 137–146. doi: 10.1016/j.schres.2005.06.009
Wong, A. H., Macciardi, F., Klempan, T., Kawczynski, W., Barr, C. L., Lakatoo, S., et al. (2003). Identification of candidate genes for psychosis in rat models, and possible association between schizophrenia and the 14-3-3eta gene. Mol. Psychiatry 8, 156–166. doi: 10.1038/sj.mp.4001237
Wu, J. Q., Wang, X., Beveridge, N. J., Tooney, P. A., Scott, R. J., Carr, V. J., et al. (2012). Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLoS One 7:e36351. doi: 10.1371/journal.pone.0036351
Xu, X., Jaehne, E. J., Greenberg, Z., McCarthy, P., Saleh, E., Parish, C. L., et al. (2015). 14-3-3zeta deficient mice in the BALB/c background display behavioural and anatomical defects associated with neurodevelopmental disorders. Sci. Rep. 5:12434. doi: 10.1038/srep12434
Yaffe, M. B., Rittinger, K., Volinia, S., Caron, P. R., Aitken, A., Leffers, H., et al. (1997). The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971. doi: 10.1016/s0092-8674(00)80487-0
Zeng, T., and Tan, L. (2018). 14-3-3eta protein: a promising biomarker for rheumatoid arthritis. Biomark. Med. 12, 917–925. doi: 10.2217/bmm-2017-0385
Keywords: 14-3-3 proteins, schizophrenia, knockout mice, animal models, human studies, genetic linkage
Citation: Navarrete M and Zhou Y (2022) The 14-3-3 Protein Family and Schizophrenia. Front. Mol. Neurosci. 15:857495. doi: 10.3389/fnmol.2022.857495
Received: 18 January 2022; Accepted: 17 February 2022;
Published: 14 March 2022.
Edited by:
Daniela Tropea, Trinity College Dublin, IrelandReviewed by:
Annie Andrieux, CEA Grenoble, FranceCopyright © 2022 Navarrete and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhou, WWkuemhvdUBtZWQuZnN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.