 Brendan Hoffe
Brendan Hoffe Matthew R. Holahan
Matthew R. Holahan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 24 February 2022
Sec. Molecular Signalling and Pathways
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.831825
This article is part of the Research Topic Molecular Mechanisms of Glutamatergic Synapse Function and Dysfunction View all 14 articles
The biological response of brain tissue to biomechanical strain are of fundamental importance in understanding sequela of a brain injury. The time after impact can be broken into four main phases: hyperacute, acute, subacute and chronic. It is crucial to understand the hyperacute neural outcomes from the biomechanical responses that produce traumatic brain injury (TBI) as these often result in the brain becoming sensitized and vulnerable to subsequent TBIs. While the precise physical mechanisms responsible for TBI are still a matter of debate, strain-induced shearing and stretching of neural elements are considered a primary factor in pathology; however, the injury-strain thresholds as well as the earliest onset of identifiable pathologies remain unclear. Dendritic spines are sites along the dendrite where the communication between neurons occurs. These spines are dynamic in their morphology, constantly changing between stubby, thin, filopodia and mushroom depending on the environment and signaling that takes place. Dendritic spines have been shown to react to the excitotoxic conditions that take place after an impact has occurred, with a shift to the excitatory, mushroom phenotype. Glutamate released into the synaptic cleft binds to NMDA and AMPA receptors leading to increased Ca2+ entry resulting in an excitotoxic cascade. If not properly cleared, elevated levels of glutamate within the synaptic cleft will have detrimental consequences on cellular signaling and survival of the pre- and post-synaptic elements. This review will focus on the synaptic changes during the hyperacute phase that occur after a TBI. With repetitive head trauma being linked to devastating medium – and long-term maladaptive neurobehavioral outcomes, including chronic traumatic encephalopathy (CTE), understanding the hyperacute cellular mechanisms can help understand the course of the pathology and the development of effective therapeutics.
The synapse is regarded as the fundamental site of communication between cells of the nervous system and synaptic dysfunction may represent a choke point for the multitude of upstream factors and downstream responses that lead to neuron atrophy or death. Synaptic malleability is a key component to behavior and cognition, with the formation, reshaping and strengthening of synapses being a key feature underlying learning and memory (Bourne and Harris, 2007; González-Tapia et al., 2016; Noye Tuplin and Holahan, 2019). Prolonged pathological changes at the synaptic level have been shown to play a role in several psychiatric and neurodegenerative diseases, such as Alzheimer’s disease, Huntington’s disease, Fragile X syndrome and schizophrenia (Calabrese et al., 2006; Herms and Dorostkar, 2016).
Over the past 15 years there has been an increase in attention to the relationship between traumatic brain injury (TBI), Alzheimer’s disease and chronic traumatic encephalopathy (Omalu et al., 2005; McKee et al., 2013; Goldstein et al., 2014; Albayram et al., 2020). Given that TBI can be regarded as a singular event (Wojnarowicz et al., 2017), the cellular processes that take place at the synapse dictate how the cell responds to the change in environment. These cellular processes can be categorized into four main time phases: hyperacute, acute, subacute, and chronic (Guerriero et al., 2015; MacFarlane and Glenn, 2015). While much research exists on the changes occurring days to months after TBI, there is comparatively little research investigating the hyperacute synaptic changes occurring within minutes to hours. These short-term changes could reveal the pathological mechanisms that are responsible for the later stages of neurodegeneration which may help shed light on potential therapeutic routes for neurodegenerative diseases associated with head trauma. This review will focus on and highlight the synaptic dysfunction during the hyperacute phase post-TBI, combining crucial findings from both in vitro and in vivo data to better understand what occurs at the synapse within minutes to hours after an impact has occurred.
The timeline of cellular responses that occur after brain impact spans four phases: hyperacute (minutes to hours), acute (hours to several days), subacute (several days to weeks) and chronic (months and beyond; Guerriero et al., 2015; MacFarlane and Glenn, 2015). During the hyperacute phase, affected neurons undergo substantial and abnormal electrical and cellular activity leading to maladaptive remodeling, potentially as an attempt to regain homeostasis following impact. While the affected cells work to re-establish internal homeostasis during each phase, if damage were to remain unresolved (ex. further damage, or severity of initial damage) the pathology continues and moves into the next phase. As each phase progresses to the next, the damage severity increases. This can ultimately lead to the major neurodegeneration observed in pathologies associated with head trauma (Figure 1).
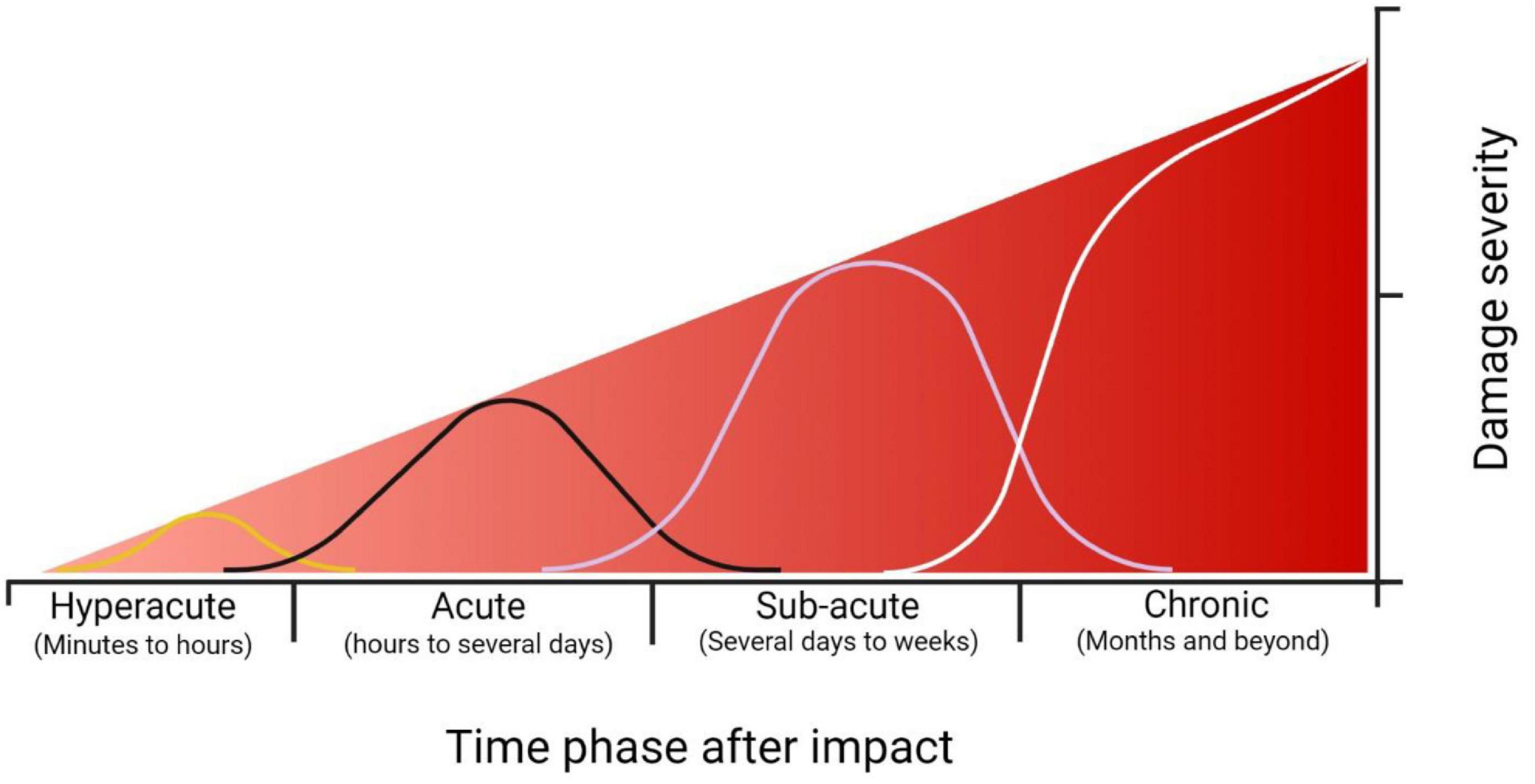
Figure 1. Timescale representation of the severity of damage following impact. Each time phase has the capacity to re-establish homeostasis through cellular repair or some form of treatment. If the cellular homeostasis is unresolved, however, the damage severity increases as time progresses. At the hyperacute time phase (minutes to hours) the damage severity is low, consisting of focal changes at the synaptic level such as hyperexcitability, increased membrane permeability (either through membrane disruption or receptor activity), and onset of excitotoxic conditions. During the acute phase (hours to several days), damage consists of further receptor alterations, activation of proteases and apoptotic pathways, mitochondrial dysfunction, and prolonged excitotoxic conditions. During the subacute phase (several days to weeks), regional deficits within the brain are observed due to cellular dysfunction, prolonged neuroinflammation, and onset of phosphorylated tau accumulation. During the chronic phase (months and beyond), major behavioral deficits and neuropsychiatric conditions are observed, including, but not limited to, impaired learning and memory, depression, and prolonged headaches. The neuropathology of chronic traumatic encephalopathy is also observed during this time (Guerriero et al., 2015; MacFarlane and Glenn, 2015). Created with BioRender.com.
The promotion of excitotoxic conditions requires prolonged release and presence of glutamate within the synaptic cleft, leading to overactivation of N-Methyl-d-aspartate receptors (NMDAr) and of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAr) on the post-synaptic membrane. In vitro neuronal stretch models have demonstrated an increase in membrane permeability within minutes after stretch through dysregulated ionic channels or the membrane physically rupturing (Smith et al., 1999; Weber, 2012; Siedler et al., 2014). Wolf et al. (2001) found that within minutes following stretch strain, an increase in intracellular Ca2+ was observed with levels that continued to rise during the time course of the experiment (1-20 min). This increase was dependent on the influx of Na+ as blockade of the voltage gated Na+ channels with tetrodotoxin inhibited this rise in intracellular Ca2+ (Wolf et al., 2001).
Increased intracellular Ca2+ in the pre-synaptic neuron prime the readily releasable pool of synaptic vesicles and the SNARE complex that mediates the fusion of vesicles to the membrane wall via various docking proteins (Hackett and Ueda, 2015; Kaeser and Regehr, 2017; Chen et al., 2021). Carlson et al. (2016) found that 6 h after cortical impact to rodents, there was a noticeable increase in SNAP-25 complex, as well as syntaxin, that steadily decreased over a 2-week period. This is of importance as SNAP-25 and syntaxin are two of the three main SNARE proteins responsible for vesicle-membrane fusion at the pre-synaptic terminal (Kaeser and Regehr, 2017; Chen et al., 2021). Ahmed et al. (2012) found that within 30 min following stretch damage to Drosophila motor neurons, there were elevated levels of Synaptotagmin, a Ca2+ sensitive vesicle trafficking protein that interacts with SNAP-25, within the pre-synaptic terminal. These synaptic changes, along with changes to the post-synaptic neuron, are summarized in Figure 2.
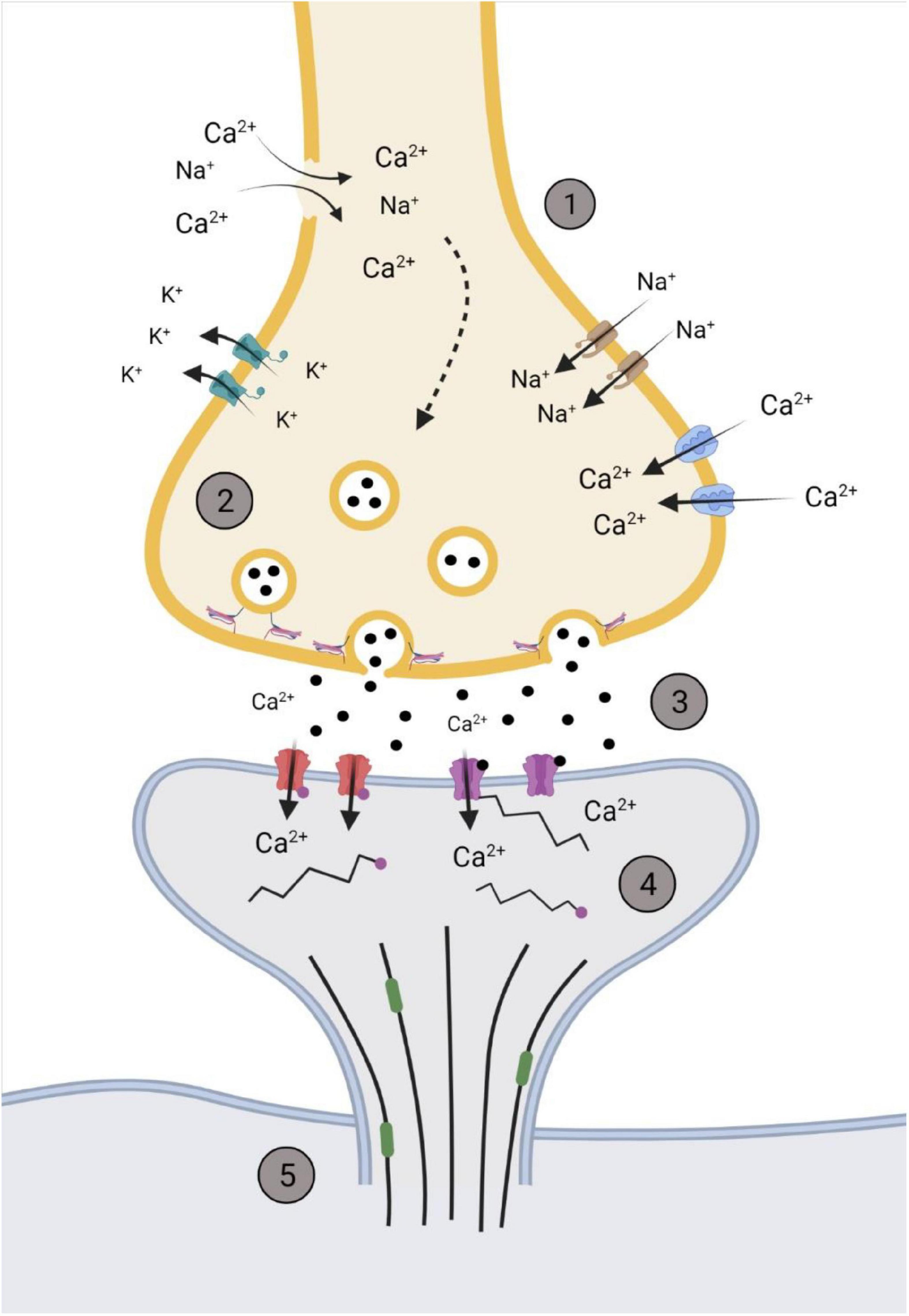
Figure 2. Representation of the synapse within the hyperacute phase after impact. (1) Membrane permeability increases soon after impact through the influx/efflux of ions via voltage gated channels or rupturing of the pre-synaptic membrane. (2) The increase in Ca2+ primes glutamate filled vesicles and increases the rate of fusion and release of glutamate. (3) Increased and sustained release of glutamate (black dots) into the synaptic cleft activates NMDAr NR2B subunits (purple) and increases post-synaptic concentrations of Ca2+. This activates various kinases that phosphorylates AMPA GluR1 subunits (red), further promoting the influx of Ca2+. (4) Post-synaptic cytoskeleton alterations occur through phosphorylation (pink dots), with disinhibition of remodeling proteins, such as end-binding protein 3 (green), occurring to allow for the restructuring of the dendritic spine cytoskeleton. (5) Downstream cascades from the influx of Ca2+alter microtubule dynamics within the cytoskeleton of the neuron. Created with BioRender.com.
Neuroinflammation plays a significant role in modulating the damage after brain injuries, with the activation of numerous proinflammatory cytokines such as interleukin-1, 6, tumor necrosis factor-α (TNF- α), and interferon-γ reported within the hyperacute phase following injury (Patterson and Holahan, 2012; Smith et al., 2012; Xiong et al., 2018). Recently, attention toward molecular recognition of damage has been gaining interest as possible mechanisms for signaling the immune response after injury. Toll-like receptors (TLR) are a class of pattern recognition receptors that activate and mediate the production of cytokines involved in proinflammatory response (Vaure and Liu, 2014). TLR-4, specifically, has been shown to influence neuronal excitability through cytokine-enhanced NMDAr currents in hippocampal cultures (Balosso et al., 2014). In the context of TBI, however, TLR-4 enhances neuronal excitability through upregulation of Ca2+-permeable GluR1, and not NMDAr, hours after impact (Li et al., 2015; Korgaonkar et al., 2020). Using the fluid percussion injury (FPI) model in juvenile rats, Li et al. (2015) demonstrated elevated neuronal expression of TLR-4 within the hippocampus 4 h after impact, with a peak at 24 h, then returning to similar levels as 4 h at 3 days post-impact. This early increase in TLR-4 expression was accompanied with enhanced non-NMDA excitatory postsynaptic currents in granule cell and mossy cells at 3 days. The contribution of TLR-4 to increased neuronal excitability is through the downstream effector of nuclear factor kappa B (NF-κB; Vaure and Liu, 2014). In neurons, increased synaptic expression of NF-κB has been shown to promote excitatory synapses and spinogenesis through NMDAr regulated Ca2+ influx (Boersma et al., 2011). In glial cells, TLR-4-activated NF-κB has downstream effects on the upregulation on proinflammatory factors such as interferon-beta (IFN-β) and TNF-α (Vaure and Liu, 2014). Overproduction of these factors in microglia have been shown to promote excitotoxic conditions by downregulating excitatory amino acid transporter-2, thus reducing glutamate reuptake from astrocytes (Sitcheran et al., 2005). Another mechanism includes the TNF-α induced increase in AMPAr lacking GluR2, increasing the Ca2+ permeability (Stellwagen et al., 2005; for review see Olmos and Lladó, 2014).
While not directly in the scope of this review paper, the role of γ-aminobutyric acid (GABA) in the promotion of excitotoxic conditions is important. Immediately following FPI (i.e., within minutes of impact to tissue fixation), Toth et al. (1997) demonstrated that large basket-like cells within the granular layer of the rat hippocampus were damaged, as evident from positive Gallyas silver staining. The neighboring granule cells, however, showed no signs of damage indicating that, due to their size, these GABAergic interneurons were at more risk of damage compared to the tightly packed granule cells. Further, the authors note that the damage to GABAergic interneurons immediately after impact can further promote the state of hyperexcitability as there is limited inhibitory signals within this region of the hippocampus (Toth et al., 1997). More recently, studies have shown further downregulation of GABAergic subunits within the rat hippocampus, particularly α1, α2, γ2, and δ, 6 h following FPI (Raible et al., 2012; Drexel et al., 2015). Interestingly, both groups found upregulation of α4 subunits, with Raible et al. (2012) showing increase in the ipsilateral side, but not contralateral, and Drexel et al. (2015) showing increase in both ipsilateral and contralateral to region of impact. Considering how the α1, α2 and γ2 GABA subunits regulated phasic inhibition, and the α4 subunit regulates tonic inhibition, the increase in α4 subunit expression could be an indication of a compensatory mechanism to counteract the hyperexcited state occurring after impact (Drexel et al., 2015).
Excitotoxic conditions arise from the prolonged release of glutamate into the synaptic cleft with subsequent binding to and activation of post-synaptic receptors. Having established an increase in membrane permeability, which leads to influx of Na+ and Ca2+, as well as the efflux of K+ (MacFarlane and Glenn, 2015), one would assume that the dysregulation of ions across the membrane could be enough to trigger aberrant depolarizations and produce a hyperactive state shortly after trauma. However, much like the dynamics of the pre-synaptic terminal, there remains limited research into the hyperacute phase of TBI (Cohen et al., 2007). This could be in part due to the complexity of techniques from one study to another, or experimental limitations associated with electrophysiological recordings from slice preparations in such a protracted timeframe. Nonetheless, there have been a handful of studies looking at the electrophysiological properties that occur within the hyperacute phase following TBI.
Using microelectrode arrays to measure neural population activity following cortical compression, Ding et al. (2011) found a time-dependent shift in electrical activity after compression. Within minutes of injury, there was a depression in cortical activity, with a gradual rise in excitability within an hour post injury, eventually resulting in cortical hyperexcitability by the 2-h mark. Similarly, using an in vitro stretch model, it was shown that within 30 min there was a reduced occurrence of spontaneous action potentials in neurons that were damaged (Magou et al., 2015). Interestingly, the authors found that in the adjacent neurons that did not experience stretch damage, there was an increase in action potentials generated within that timeframe. This could potentially indicate that due to imbalance of ionic homeostasis immediately after trauma, the depression of electrical activity could act as a primer for the hyperexcitability experienced shortly after (Berger et al., 2008; Ding et al., 2011). This enhanced activity in adjacent neurons could propagate hyperexcitatory conditions, potentially initiating the secondary, maladaptive intracellular pathways associated with TBI.
The release of ions following injury has also been shown to be enhanced within 24 h after injury and alter electrophysiological properties. In a rather paradoxical way, D’Ambrosio et al. (1998) demonstrated a loss of long term potentiation (LTP), but not long term depression (LTD), in hippocampal slice recordings, 24 h after FPI in rats. They found that rats who received FPI had higher thresholds required to evoke population spikes within the CA1. However, previous work from Katayama et al. (1990) demonstrated, in vivo, a massive increase in extracellular K+ following FPI resulting in neuronal hyperexcitability. D’Ambrosio et al. (1998) attribute their opposite finding to differences in the microenvironments between in vivo and in vitro settings. They note that the elevated efflux of K+ abolishes LTP as the sudden overactivation of NMDAr and firing of the post-synaptic neuron leaves the neuron unable to undergo further potentiation after injury; something not reproducible in an in vitro setting (D’Ambrosio et al., 1998). Interestingly, a year later D’Ambrosio et al. (1999) demonstrated altered neuronal activity due to excessive accumulation of extracellular K+ 24 h after FPI in hippocampal slices. This accumulation led to a hyperexcited state in CA3 neurons, as evident by the higher percentage of burst discharges in the post-FPI slices. The authors also note a significant loss of inward rectifying K+ currents from recruited glia cells. These findings indicate a disruption to mechanisms responsible for maintaining extracellular homeostasis, such as the decreased K+ clearance from glia cells plus the prolonged release of K+ from hyperactive neurons (D’Ambrosio et al., 1999).
A report by Ross and Soltesz (2000) examined the electrophysiological properties of interneurons in the dentate gyrus after lateral FPI. Within the dentate gyrus, there was an observed increase of spontaneous interneuron firing rates due to Na+/K+-ATPase-related depolarizations. This increase in interneuron firing rates after impact led to increased frequency and amplitude of spontaneous inhibitory postsynaptic potentials in dentate gyrus granule cells. It is important to note that this increase in spontaneous inhibitory postsynaptic potentials occurs simultaneously to a decrease in miniature inhibitory postsynaptic current frequency. The authors describe this post-FPI increase in interneuron depolarization and increased GABAergic transmission could increase the efficacy of incoming excitatory signals to further promote interneuron firing after impact. This rather paradoxical mechanism has been reported in previous models of hyperexcitability, such as kindling seizures, chronic stress-induced seizures, and febrile seizures (Nusser et al., 1998; Chen et al., 1999; MacKenzie and Maguire, 2015).
Using animal models of TBI, it has been shown that extracellular glutamate levels rise drastically within minutes after impact and remained elevated for hours (Faden et al., 1989; Folkersma et al., 2011; Xiong et al., 2019). Along the post-synaptic membrane, AMPAr and NMDAr become over-activated from the increased presence of glutamate within the synaptic cleft. The GluR1 subunit of AMPAr plays a crucial role in LTP and membrane conductance under normal conditions; however, they have been implicated in progressing neurodegenerative diseases under pathological conditions (Spaethling et al., 2012). Unlike the GluR2 subunit, which limits membrane permeability of Ca2+, phosphorylated GluR1, via Ca2+/calmodulin-dependent protein kinase II (CaMKII) and PKC, increases membrane permeability to Ca2+ and promotes the upregulation of GluR1 insertion into the membrane and the influx of Ca2+ (Isaac et al., 2007). In the context of TBI, in vitro stretch models reveal an increase in GluR1 phosphorylation within hours of injury and increased levels of intracellular Ca2+ (Spaethling et al., 2008, 2012). Phosphorylation of GluR1 is mediated through NMDAr activity, specifically the NR2B subunit activity, as blocking the NR2B subunit blocked the rise in Ca2+ levels and reduced levels of phosphorylated GluR1 in stretched neurons (Spaethling et al., 2012). This has been observed in animal models of TBI, with increased CaMKII autophosphorylation and GluR1 observed within the hippocampus as early as 15 min after using weighted drop controlled cortical impact (CCI; Schumann et al., 2008) and 1 h post FPI (Atkins et al., 2006).
Calcium influx via the activation of NMDAr is crucial for synaptic plasticity and activating various biochemical pathways responsible for neuronal physiology. However, sustained activation of NMDAr promotes pathological secondary cascades which underlie the onset of neurodegenerative diseases (Kalia et al., 2008; Shohami and Biegon, 2014). Within the hyperacute phase of TBI, phosphorylated NR2B levels begin to rise and promote the upregulation of other NMDAr subunits, such as NR1 and NR2A (Schumann et al., 2008). The NR2B subunit is responsible for high influxes on Ca2+ into the neuron, with dysregulation of this subunit leading to excitotoxic conditions (Arnsten et al., 2021). In cultured neurons, the loss of functional connectivity is primarily mediated through activation of NR2B subunit (Patel et al., 2014). The NR2B subunit appears to be the focal subunit of potential neurotoxic effects, as administration of inhibiting kinases involved in regulating the receptor reduced the elevated levels of phosphorylated NR2B and improved long term neurological function after trauma (Schumann et al., 2008; Zhou and Sheng, 2013; Patel et al., 2014).
Dendritic spines are the primary site for excitatory synaptic communication between two neurons and their morphology can greatly dictate neural function. Dendritic spines are dynamic in their morphology, constantly shifting between phenotypes depending on the surrounding environment or internal cellular conditions (Hering and Sheng, 2001). The constant remodeling and development of spines is termed spinogenesis with these changes taking place in the time frame of seconds to minutes to days to weeks (Calabrese et al., 2006). The remodeling and retracting of spines is a highly conserved trait across species, with evidence to show spinogenesis rates changing during learning and memory (Tønnesen and Nägerl, 2016; Luengo-Sanchez et al., 2018), disease progression (Calabrese et al., 2006; Aguayo et al., 2018; Jamjoom et al., 2021) and naturally with age (Benavides-Piccione et al., 2013).
In their 1970 paper, Peters and Kaiserman-Abramof (1970) described the three main categories of spine type: thin, stubby and mushroom. Thin spines represent a transitory phenotype that are primed to develop into mushroom-type spines through repeated synaptic activity (Pchitskaya and Bezprozvanny, 2020). Stubby spines represent immature spines and are less likely to be a place of synaptic communication between neurons (Helm et al., 2021). Mushroom spines are considered to represent an excitatory synapse and are associated with synaptic plasticity achieved through LTP (Jaworski et al., 2009; Helm et al., 2021). Using modern neuronal reconstruction, Luengo-Sanchez et al. (2018) characterized the spine morphology in human cortical pyramidal neurons using three dimensional meshes on individual spines and quantitative features such as, but not limited to, height, growth direction of the spine, and volume. This group was able to categorize dendritic spines into six distinct clusters based on the Bayesian information criterion, which contained sub-sets of spine shapes based on the authors described criterion. Bokota et al. (2016) produced 10 distinct spine clusters using automatic three-dimensional dendritic spine reconstruction, with each cluster containing a range of spine morphology subsets. While modern imaging techniques show that spine classification exists on a continuum at any one time (Rodriguez et al., 2008; Bokota et al., 2016; Luengo-Sanchez et al., 2018), the categories set forth by Peters and Kaiserman-Abramof (1970) are still widely used to classify spines when performing histological analyses (Figure 3).
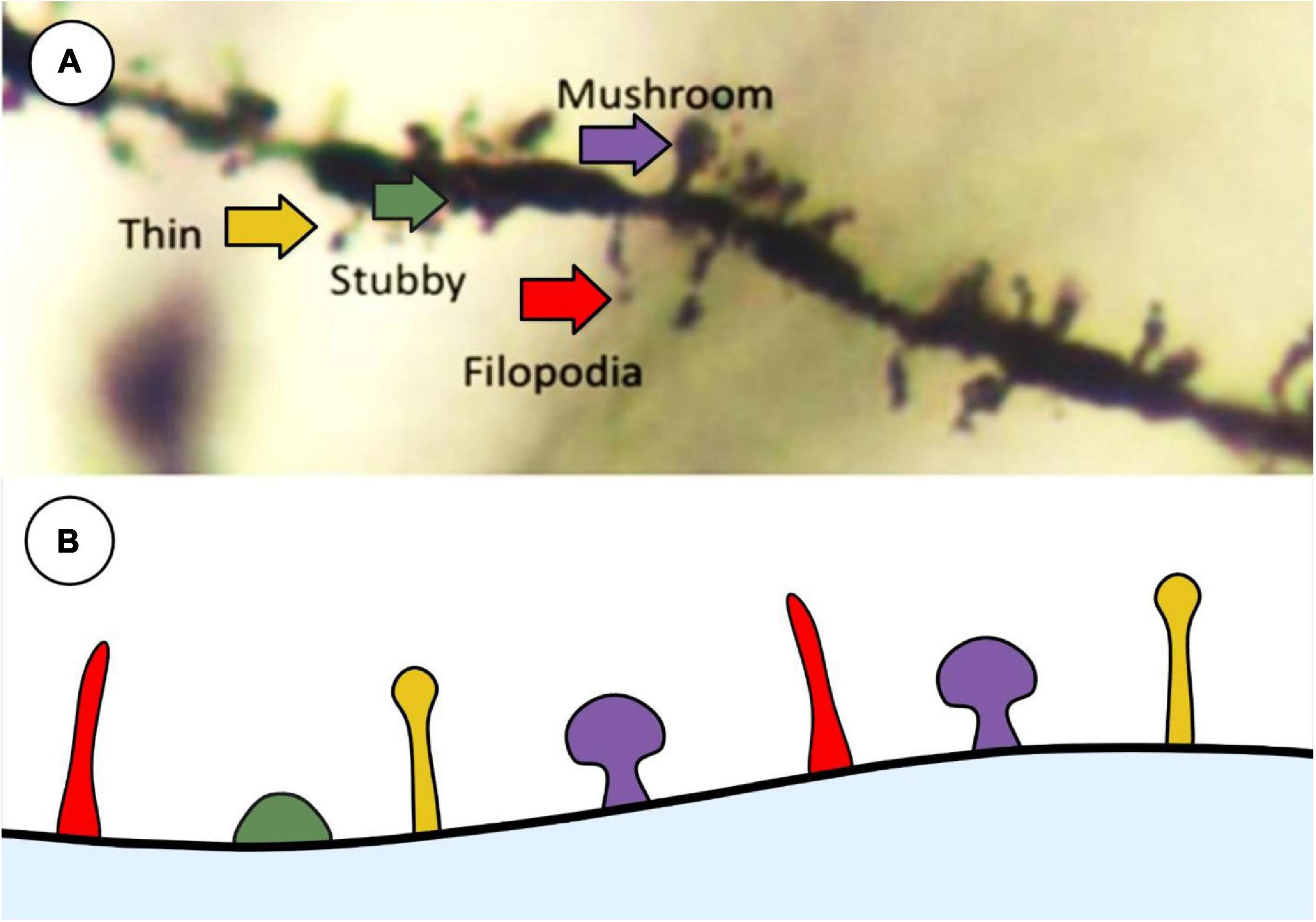
Figure 3. Representative image of main spine types that are commonly used in histological analysis described by Peters and Kaiserman-Abramof (1970). (A) Golgi-stained neuron showcasing the 4 main types of spines: thin (yellow), stubby (green), filopodia (red), and mushroom (purple). (B) Schematic representation of the types of spines to illustrate differences between shapes. Created with BioRender.com.
The cytoskeletal filaments within the spine head are made up of an intricate network of actin filaments that form the basis for morphological spine remodeling. These filaments, along with the molecular machinery involved in the dynamics of the spine cytoskeleton, act independently from the machinery in neighboring spines (Newpher and Ehlers, 2009). The ultrastructural changes within a single dendritic spine involve the recruitment of several protein kinase families that regulate actin filament dynamics within the spine head (Meng et al., 2003; Jaworski et al., 2009; Aguayo et al., 2018; Reza-Zaldivar et al., 2020). Lim-kinase (LIMK), for example, is a major family of protein kinases that regulate the shape of dendritic spines and their function and plays a major role in LTP (Meng et al., 2003). Through the influx of Ca22+ via NMDAr, CaMKI, and CaMKII stimulate the activity of Rac1. The stimulation of Rac1 has downstream effects of promoting LIMK-1 activity which ultimately leads to actin polymerization and the maintenance of spine morphology [for full review see Saneyoshi et al. (2010)]. The rate in which LIMK-1 is activated and suppressed is achieved by brain derived neurotrophic factor (BDNF) and microRNA-134 (miRNA-134), respectfully, however the exact mechanisms in which this operates is not well understood (Schratt et al., 2006; Saneyoshi et al., 2010; Reza-Zaldivar et al., 2020).
In addition to the regulation of dendritic spine actin, the activity along the synaptic membrane and the scaffolding proteins that make up the post-synaptic density (PSD) regulate cytoskeletal dynamics of the spine. The PSD is roughly 50 nm wide and is enriched in a wide variety of proteins responsible for synaptic integrity and regulating what enters the neuron (Harris and Kater, 1994). One of the most important synaptic proteins responsible for synaptic stability is PSD-95. PSD-95 is a scaffolding protein responsible for the regulation of AMPAr and NMDAr, as well as neuroligins and neurexins responsible for anchoring the pre-synaptic and post-synaptic terminals together (Keith and El-Husseini, 2008; Jeong et al., 2019). Knockdown models of PSD-95 have shown decreased spine numbers, altered formation of functional synapses and decreased rates of spinogenesis (Jeong et al., 2019). The overexpression of PSD-95 promotes development of excitatory synapses along the dendrite (Levinsoni et al., 2005).
The basis for synaptic plasticity and LTP involves the movement of ions across the membrane through AMPAr channels and NMDAr channels. NMDAr have been largely implicated in the consequences of excitotoxicity, however there is mounting evidence to suggest the upregulation of the AMPA GluR1 subunit contributes to excitotoxic conditions (for review see Guerriero et al., 2015). AMPAr are the primary fast excitatory receptor within the nervous system, consisting of 4 subunits, GluR1 to GluR4, which form tetrameric complexes that have distinct receptor binding subtypes. Most AMPAr contain the Ca2+-impermeable GluR2 subunit, however a subset of AMPAr contain the Ca2+-permeable GluR1 subunit (Song and Huganir, 2002). Once glutamate is bound, the pore of the receptor opens allowing for cations (Na+ and Ca2+) to move across the membrane resulting in depolarization (Chater and Goda, 2014). NMDAr consist of three main subfamilies, GluN1, N2, N3, with multiple subunits within each family. A combination of subfamilies makes up the NMDAr channel, with GluN1 and N2 subunits pairing together, or GluN2 and N3 together. Acting as a coincidence detector, the opening of NMDAr channels requires the depolarization of the neuron as well as the binding of glutamate followed by the removal of the Mg2+ ion within the membrane-spanning domain of the channel (Paoletti et al., 2013). Once Mg2+ is removed, the NMDAr becomes permeable to Na+ and Ca2+. NMDAr activation and Ca2+ play a crucial role in the LIMK pathway through activating CaMKII and remodeling the actin cytoskeleton of the dendritic spine (Newpher and Ehlers, 2009; Saneyoshi et al., 2010). The influx of Ca2+ into the neuron is required for normal cellular functioning through the activation of various enzymes, protein complexes, and immediate early genes (Paoletti et al., 2013). Too large of an influx of Ca2+, however, has been established as a main factor in the onset of excitotoxic conditions that put the neuron at risk of cell death, and ultimately the onset of various neurodegenerative diseases.
Within the post-synaptic membrane, the synaptic architecture is stabilized primarily through PSD-95 which is regulated through the activity of NR2B subunits (Szydlowska and Tymianski, 2010; Frank et al., 2016; Jeong et al., 2019). PSD-95 has also been shown to restrict the remodeling of dendrites by mediating the activity of end-binding protein 3 (EB3); a protein responsible for growth and microtubule dynamics (Patel et al., 2019). EB3 is a well conserved member of the EB family responsible for restructuring dendritic spine cytoskeleton, maintenance of excitatory synapses, shuttling organelles into dendritic spines, and has shown to be involved in LTP (Jaworski et al., 2009; Penzes et al., 2009). The activity of PSD-95 is regulated by phosphorylation on cyclin-dependent kinase 5 (cdk5; Keith and El-Husseini, 2008). Park et al. (2013) demonstrated an increase in phosphorylated PSD-93, a similar scaffolding protein to PSD-95, several hours following FPI in the rodent cortex. These proteins are considered deactivated once phosphorylated (Nowacka et al., 2020). This could suggest that following TBI, the disruption of PSD-95/93 allows for the remodeling of the dendrite and spine through proteins such as EB3 (Figure 4), as their activity is no longer inhibited (Patel et al., 2019). Very few studies have investigated the remodeling of dendritic spine morphology post-TBI within the hyperacute phase. Pijet et al. (2019) found that 24 h after CCI, dendritic spines in the ipsilateral cortex became shorter and wider, a characteristic of excitatory mushroom-type spines (Jontes and Smith, 2000). The expression of shorter-wider spines was accompanied by an increase in the matrix metalloproteinase-9 (MMP-9). This extracellular protease has a role in synaptic plasticity as it is present in excitatory synapses within the hippocampus, cerebellum and cerebral cortex (for review see Michaluk and Kaczmarek, 2007). Moreover, inhibition of MMP-9 activity has been shown to make spines resistant to excitotoxic damage from TBI as well as post-impact-induced seizures (Michaluk and Kaczmarek, 2007; Hayashi et al., 2009; Pijet et al., 2019). Expression of MMP-9 has been seen to increase 30 min after TBI, and remaining elevated for hours and days (Wang et al., 2000; Truettner et al., 2005; Hayashi et al., 2009; Pijet et al., 2018). While not reported in Pijet et al. (2018), it would be interesting to see if the increase in MMP-9 expression 30 min after CCI would be accompanied by a change in dendritic spine phenotype, similar to what they found a year later at 24 h after impact.
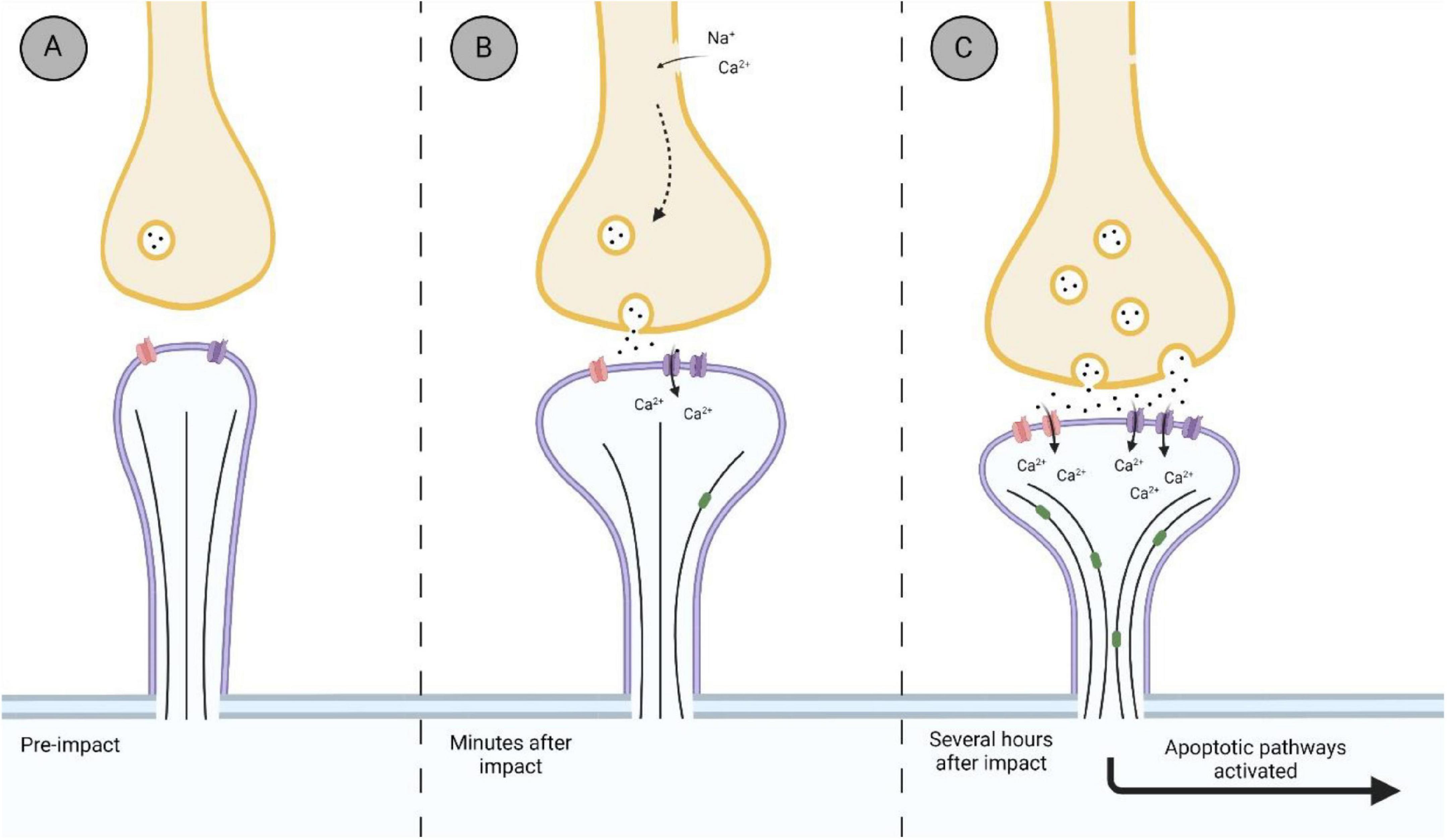
Figure 4. Representation of the biological timeline of the synapse after impact. (A) Thin synapse before an impact. Thin spines are considered a transitory spine phenotype, with the potential to transition in an excitatory synapse. (B) Within minutes after impact, synaptic changes within the hyperacute phase after impact. Membrane ruptures and ionic imbalances promote the hyperexcitability of the synapse, with an influx of Ca2+ into the post-synaptic terminal via NMDAr (purple). (C) Further damage to the synapse due to hyperexcitability and sustained release of glutamate from the pre-synaptic terminal is observed several hours after impact. Increased influx of Ca2+ into the post-synaptic terminal promotes the further upregulation of NMDAr, as well as upregulating GluR1 subunit of AMPAr (red). Disinhibition of synaptic reconstruction proteins, such as EB3 (green), occurs as restricting proteins become inactivated, promoting excitatory mushroom spine phenotype. Hyperexcitability promotes neurodegenerative pathways via overworked mitochondria and damage to the microtubule cytoskeleton of the neuron. Created with BioRender.com.
The large influx of Ca2+ that follows TBI activates various downstream pathophysiological cascades that put the neuron at risk of degeneration. One of these pathways includes alterations to the neuronal microtubule network. Microtubules provide both cytoskeletal structuring to the neuron, as well as act as a “molecular highway” for proteins and organelles to travel throughout the neuron. Calpain is a family of calcium-activated proteases involved in numerous physiological functions ranging from modifications to receptors and channels, activation of apoptotic proteins and regulation of structural proteins (Saatman et al., 2010). The activation of calpain can occur within minutes of injury and be sustained for several days (Saatman et al., 2010). In particular, calpain-2 has been shown to have neurodegenerative consequences several hours post-TBI, as evidence by increased calpain-2 levels correlating with the activation of apoptotic pathways in mice (Wang et al., 2018). Calpain-2 also disrupts microtubule dynamics by phosphorylating tau, the accumulation and aggregation of which has been well documented in the onset of tauopathy’s such as chronic traumatic encephalopathy and Alzheimer’s disease (Goldstein et al., 2012; Baudry and Bi, 2016; for review see McKee et al., 2013).
Along with activation of neurodegenerative pathways such as calpain-2, the influx of Ca2+ that follows a TBI places stress upon the mitochondria of the neuron (Watts et al., 2015). In data not shown, Griesemer and Mautes (2007) noted an increase in adenosine triphosphate levels 1 h post closed head impact, with a decrease occurring by hour 4. Overworked mitochondria fail to buffer the rise in intracellular Ca2+ levels and as a result damaging reactive oxygen species, such as H2O2, are created (Vercesi et al., 2018). While these secondary pathophysiological pathways take time to develop, the initial onset of the sequelae begins with a moment of disruption shortly after impact. Further investigations into the hyperacute neuronal responses to TBI will be beneficial in the development of possible therapeutic targets and better understanding of how these chronic diseases initiate.
The hyperacute phase that follows TBI has been relatively understudied when it comes to understanding the dynamics of head trauma. The changes at the level of the synapse showcase the disrupted communication between neurons and how it reacts to the change in the environment. These subtle changes at the synapse amplify as further damage occurs, progressing the pathological pathway that is seen in the sub-acute and chronic phases of diseases associated with TBI. Understanding where the biochemical changes begin following head trauma could direct the field of research to the development of effective treatments and therapeutics soon after an impact has occurred.
BH contributed to the research and drafting of manuscript. MRH contributed to the revisions and final approval of manuscript. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aguayo, F. I., Tejos-Bravo, M., Díaz-Véliz, G., Pacheco, A., García-Rojo, G., Corrales, W., et al. (2018). Hippocampal Memory Recovery After Acute Stress: A Behavioral, Morphological and Molecular Study. Front. Mol. Neurosci. 11:283. doi: 10.3389/fnmol.2018.00283
Ahmed, W. W., Li, T. C., Rubakhin, S. S., Chiba, A., Sweedler, J. V., and Saif, T. A. (2012). Mechanical tension modulates local and global vesicle dynamics in neurons. Cell. Mol. Bioengine. 2012, 155–164. doi: 10.1007/s12195-012-0223-1
Albayram, O., Albayram, S., and Mannix, R. (2020). Chronic traumatic encephalopathy—a blueprint for the bridge between neurological and psychiatric disorders. Transl. Psychiatry 10:424. doi: 10.1038/s41398-020-01111-x
Arnsten, A. F. T., Datta, D., and Preuss, T. M. (2021). Studies of aging nonhuman primates illuminate the etiology of early-stage Alzheimer’s-like neuropathology: An evolutionary perspective. Am. J. Primatol. 83:23254. doi: 10.1002/ajp.23254
Atkins, C. M., Chen, S., Alonso, O. F., Dietrich, D., and Hu, B.-R. (2006). Activation of calcium/calmodulin-dependent protein kinases after traumatic brain injury. J. Cereb. Blood Flow Metab. 26, 1507–1518. doi: 10.1038/sj.jcbfm.9600301
Balosso, S., Liu, J., Bianchi, M. E., and Vezzani, A. (2014). Disulfide-containing high mobility group box-1 promotes N-methyl-d-aspartate receptor function and excitotoxicity by activating toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid. Redox Signal. 21, 1726–1740. doi: 10.1089/ars.2013.5349
Baudry, M., and Bi, X. (2016). Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 39, 235–245. doi: 10.1016/j.tins.2016.01.007
Benavides-Piccione, R., Fernaud-Espinosa, I., Robles, V., Yuste, R., and DeFelipe, J. (2013). Age-Based Comparison of Human Dendritic Spine Structure Using Complete Three-Dimensional Reconstructions. Cereb. Cortex 23:1798. doi: 10.1093/CERCOR/BHS154
Berger, M., Speckmann, E. J., Pape, H. C., and Gorji, A. (2008). Spreading depression enhances human neocortical excitability in vitro. Cephalalgia 28, 558–562. doi: 10.1111/j.1468-2982.2008.01556.x
Boersma, M. C. H., Dresselhaus, E. C., de Biase, L. M., Mihalas, A. B., Bergles, D. E., and Meffert, M. K. (2011). A requirement for nuclear factor-κB in developmental and plasticity-associated synaptogenesis. J. Neurosci. 31, 5414–5425. doi: 10.1523/JNEUROSCI.2456-10.2011
Bokota, G., Magnowska, M., Kuśmierczyk, T., Łukasik, M., Roszkowska, M., and Piewczynski, D. (2016). Computational approach to dendritic spine taxonomy and shape transition analysis. Front. Comput. Neurosci. 10:140. doi: 10.3389/fncom.2016.00140
Bourne, J., and Harris, K. M. (2007). Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 17, 381–386. doi: 10.1016/j.conb.2007.04.009
Calabrese, B., Wilson, M. S., and Halpain, S. (2006). Development and regulation of dendritic spine synapses. Physiology 21, 38–47. doi: 10.1152/physiol.00042.2005
Carlson, S. W., Yan, H., Ma, M., Li, Y., Henchir, J., and Dixon, C. E. (2016). Traumatic brain injury impairs soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex formation and alters synaptic vesicle distribution in the hippocampus. J. Neurotrauma 33, 113–121. doi: 10.1089/neu.2014.3839
Chater, T. E., and Goda, Y. (2014). The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 8:401. doi: 10.3389/fncel.2014.00401
Chen, F., Chen, H., Chen, Y., Wei, W., Sun, Y., Zhang, L., et al. (2021). Dysfunction of the SNARE complex in neurological and psychiatric disorders. Pharmacol. Res. 165, 1043–6618. doi: 10.1016/J.PHRS.2021.105469
Chen, K., Baram, T. Z., and Soltesz, I. (1999). Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat. Med. 5, 888–894. doi: 10.1038/11330
Cohen, A. S., Pfister, B. J., Schwarzbach, E., Sean Grady, M., Goforth, P. B., and Satin, L. S. (2007). Injury-induced alterations in CNS electrophysiology. Prog. Brain Res. 161, 143–169. doi: 10.1016/S0079-6123(06)61010-8
D’Ambrosio, R., Maris, D. O., Grady, M. S., Winn, H. R., and Janigro, D. (1998). Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 786, 64–79. doi: 10.1016/S0006-8993(97)01412-1
D’Ambrosio, R., Maris, D. O., Grady, M. S., Winn, H. R., and Janigro, D. (1999). Impaired K+ homeostasis and altered electrophysiological properties of post-traumatic hippocampal gila. J. Neurosci. 19, 8152–8162. doi: 10.1523/jneurosci.19-18-08152.1999
Ding, M. C., Wang, Q., Lo, E. H., and Stanley, G. B. (2011). Cortical excitation and inhibition following focal traumatic brain injury. J. Neurosci. 31, 14085–14094. doi: 10.1523/JNEUROSCI.3572-11.2011
Drexel, M., Puhakka, N., Kirchmair, E., Hörtnagl, H., Pitkänen, A., and Sperk, G. (2015). Expression of GABA receptor subunits in the hippocampus and thalamus after experimental traumatic brain injury. Neuropharmacology 88, 122–133. doi: 10.1016/j.neuropharm.2014.08.023
Faden, A. I., Demediuk, P., Panter, S. S., and Vink, R. (1989). The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800. doi: 10.1126/science.2567056
Folkersma, H., Foster Dingley, J. C., van Berckel, B. N. M., Rozemuller, A., Boellaard, R., Huisman, M. C., et al. (2011). Increased cerebral (R)-[11C]PK11195 uptake and glutamate release in a rat model of traumatic brain injury: A longitudinal pilot study. J. Neuroinflamm. 8:67. doi: 10.1186/1742-2094-8-67
Frank, R. A. W., Komiyama, N. H., Ryan, T. J., Zhu, F., O’Dell, T. J., and Grant, S. G. N. (2016). NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat. Commun. 7:ncomms11264. doi: 10.1038/ncomms11264
Goldstein, L. E., Fisher, A. M., Tagge, C. A., Zhang, X. L., Velisek, L., Sullivan, J. A., et al. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4:134ra60. doi: 10.1126/scitranslmed.3003716
Goldstein, L. E., McKee, A. C., and Stanton, P. K. (2014). Considerations for animal models of blast-related traumatic brain injury and chronic traumatic encephalopathy. Alzheimers. Res. Ther. 6:64. doi: 10.1186/s13195-014-0064-3
González-Tapia, D., Martínez-Torres, N. I., Hernández-González, M., Guevara, M. A., and González-Burgos, I. (2016). Plastic changes to dendritic spines on layer V pyramidal neurons are involved in the rectifying role of the prefrontal cortex during the fast period of motor learning. Behav. Brain Res. 298, 261–267. doi: 10.1016/j.bbr.2015.11.013
Griesemer, D., and Mautes, A. M. (2007). Closed head injury causes hyperexcitability in rat hippocampal CA1 but not in CA3 pyramidal cells. J. Neurotrauma 24, 1823–1832. doi: 10.1089/neu.2006.0237
Guerriero, R. M., Giza, C. C., and Rotenberg, A. (2015). Glutamate and GABA Imbalance Following Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 15:1. doi: 10.1007/s11910-015-0545-1
Hackett, J. T., and Ueda, T. (2015). Glutamate Release. Neurochem. Res. 40, 2443–2460. doi: 10.1007/s11064-015-1622-1
Harris, K. M., and Kater, S. B. (1994). Dendritic spines: Cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 17, 341–371. doi: 10.1146/annurev.ne.17.030194.002013
Hayashi, T., Kaneko, Y., Yu, S. J., Bae, E. K., Stahl, C. E., Kawase, T., et al. (2009). Quantitative analyses of matrix metalloproteinase activity after traumatic brain injury in adult rats. Brain Res. 1280, 172–177. doi: 10.1016/j.brainres.2009.05.040
Helm, M. S., Dankovich, T. M., Mandad, S., Rammner, B., Jähne, S., Salimi, V., et al. (2021). A large-scale nanoscopy and biochemistry analysis of postsynaptic dendritic spines. Nat. Neurosci. 24, 1151–1162. doi: 10.1038/s41593-021-00874-w
Hering, H., and Sheng, M. (2001). Dentritic spines: structure, dynamics and regulation. Nat. Rev. Neurosci. 2, 880–888. doi: 10.1038/35104061
Herms, J., and Dorostkar, M. M. (2016). Dendritic Spine Pathology in Neurodegenerative Diseases. Annu. Rev. Pathol. Mech. Dis. 11, 221–250. doi: 10.1146/annurev-pathol-012615-044216
Isaac, J. T. R., Ashby, M., and McBain, C. J. (2007). The Role of the GluR2 Subunit in AMPA Receptor Function and Synaptic Plasticity. Neuron 54, 859–871. doi: 10.1016/j.neuron.2007.06.001
Jamjoom, A. A. B., Rhodes, J., Andrews, P. J. D., and Grant, S. G. N. (2021). The synapse in traumatic brain injury. Brain 144, 18–31. doi: 10.1093/brain/awaa321
Jaworski, J., Kapitein, L. C., Gouveia, S. M., Dortland, B. R., Wulf, P. S., Grigoriev, I., et al. (2009). Dynamic Microtubules Regulate Dendritic Spine Morphology and Synaptic Plasticity. Neuron 61, 85–100. doi: 10.1016/j.neuron.2008.11.013
Jeong, J., Pandey, S., Li, Y., Badger, J. D., Lu, W., and Roche, K. W. (2019). PSD-95 binding dynamically regulates NLGN1 trafficking and function. Proc. Natl. Acad. Sci. U S A. 116, 12035–12044. doi: 10.1073/pnas.1821775116
Jontes, J. D., and Smith, S. J. (2000). Filopodia, Spines, and the Generation of Synaptic Diversity. Neuron 27, 11–14. doi: 10.1016/s0896-6273(00)00003-9
Kaeser, P. S., and Regehr, W. G. (2017). The readily releasable pool of synaptic vesicles. Curr. Opin. Neurobiol. 43, 63–70. doi: 10.1016/J.CONB.2016.12.012
Kalia, L. V., Kalia, S. K., and Salter, M. W. (2008). NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 7, 742–755. doi: 10.1016/S1474-4422(08)70165-0
Katayama, Y., Becker, D. P., Tamura, T., and Hovda, D. A. (1990). Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 73, 889–900. doi: 10.3171/jns.1990.73.6.0889
Keith, D., and El-Husseini, A. (2008). Excitation control: Balancing PSD-95 function at the synapse. Front. Mol. Neurosci. 1:4. doi: 10.3389/neuro.02.004.2008
Korgaonkar, A. A., Li, Y., Sekhar, D., Subramanian, D., Guevarra, J., Swietek, B., et al. (2020). Toll-like Receptor 4 Signaling in Neurons Enhances Calcium-Permeable α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor Currents and Drives Post-Traumatic Epileptogenesis. Ann. Neurol. 87, 497–515. doi: 10.1002/ana.25698
Levinsoni, J. N., Chéry, N., Huang, K., Wong, T. P., Gerrow, K., Kang, R., et al. (2005). Neuroligins mediate excitatory and inhibitory synapse formation: Involvement of PSD-95 and neueexin-1β in neuroligin-induced synaptic specificity. J. Biol. Chem. 280, 17312–17319. doi: 10.1074/jbc.M413812200
Li, Y., Korgaonkar, A. A., Swietek, B., Wang, J., Elgammal, F. S., Elkabes, S., et al. (2015). Toll-like receptor 4 enhancement of non-NMDA synaptic currents increases dentate excitability after brain injury. Neurobiol. Dis. 74, 240–253. doi: 10.1016/j.nbd.2014.11.021
Luengo-Sanchez, S., Fernaud-Espinosa, I., Bielza, C., Benavides-Piccione, R., Larrañaga, P., and DeFelipe, J. (2018). 3D morphology-based clustering and simulation of human pyramidal cell dendritic spines. PLoS Comput. Biol. 14:1006221. doi: 10.1371/journal.pcbi.1006221
MacFarlane, M. P., and Glenn, T. C. (2015). Neurochemical cascade of concussion. Brain Inj. 29, 139–153. doi: 10.3109/02699052.2014.965208
MacKenzie, G., and Maguire, J. (2015). Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res. 109, 13–27. doi: 10.1016/j.eplepsyres.2014.10.003
Magou, G. C., Pfister, B. J., and Berlin, J. R. (2015). Effect of acute stretch injury on action potential and network activity of rat neocortical neurons in culture. Brain Res. 1624, 525–535. doi: 10.1016/j.brainres.2015.07.056
McKee, A. C., Stein, T. D., Nowinski, C. J., Stern, R. A., Daneshvar, D. H., Alvarez, V. E., et al. (2013). The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64. doi: 10.1093/brain/aws307
Meng, Y., Zhang, Y., Tregoubov, V., Falls, D. L., and Jia, Z. (2003). Regulation of spine morphology and synaptic function by LIMK and the actin cytoskeleton. Rev. Neurosci. 14, 233–240. doi: 10.1515/REVNEURO.2003.14.3.233
Michaluk, P., and Kaczmarek, L. (2007). Matrix metalloproteinase-9 in glutamate-dependent adult brain function and dysfunction. Cell Death Differ. 14, 1255–1258. doi: 10.1038/sj.cdd.4402141
Newpher, T. M., and Ehlers, M. D. (2009). Spine microdomains for postsynaptic signaling and plasticity. Trends Cell Biol. 19, 218–227. doi: 10.1016/j.tcb.2009.02.004
Nowacka, A., Borczyk, M., Salamian, A., Wójtowicz, T., Włodarczyk, J., and Radwanska, K. (2020). PSD-95 Serine 73 phosphorylation is not required for induction of NMDA-LTD. Sci. Rep. 10, 1–11. doi: 10.1038/s41598-020-58989-2
Noye Tuplin, E. W., and Holahan, M. R. (2019). Exploring time-dependent changes in conditioned place preference for food reward and associated changes in the nucleus accumbens. Behav. Brain Res. 361, 14–25. doi: 10.1016/j.bbr.2018.12.031
Nusser, Z., Hájos, N., Somogyi, P., and Mody, I. (1998). Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature 395, 172–177. doi: 10.1038/25999
Olmos, G., and Lladó, J. (2014). Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014:861231. doi: 10.1155/2014/861231
Omalu, B. I., DeKosky, S. T., Minster, R. L., Kamboh, M. I., Hamilton, R. L., and Wecht, C. H. (2005). Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57, 128–133. doi: 10.1227/01.NEU.0000163407.92769.ED
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Park, Y., Luo, T., Zhang, F., Liu, C., Bramlett, H. M., Dalton Dietrich, W., et al. (2013). Downregulation of Src-kinase and glutamate-receptor phosphorylation after traumatic brain injury. J. Cereb. Blood Flow Metab. 33, 1642–1649. doi: 10.1038/jcbfm.2013.121
Patel, M. V., Sewell, E., Dickson, S., Kim, H., Meaney, D. F., and Firestein, B. L. (2019). A Role for Postsynaptic Density 95 and Its Binding Partners in Models of Traumatic Brain Injury. J. Neurotrauma 36, 2129–2138. doi: 10.1089/neu.2018.6291
Patel, T. P., Ventre, S. C., Geddes-Klein, D., Singh, P. K., and Meaney, D. F. (2014). Single-neuron NMDA receptor phenotype influences neuronal rewiring and reintegration following traumatic injury. J. Neurosci. 34, 4200–4213. doi: 10.1523/JNEUROSCI.4172-13.2014
Patterson, Z. R., and Holahan, M. R. (2012). Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front. Cell. Neurosci. 0:58. doi: 10.3389/fncel.2012.00058
Pchitskaya, E., and Bezprozvanny, I. (2020). Dendritic Spines Shape Analysis—Classification or Clusterization? Perspective. Front. Synaptic Neurosci. 12:31. doi: 10.3389/fnsyn.2020.00031
Penzes, P., Srivastava, D. P., and Woolfrey, K. M. (2009). Not Just Actin? A Role for Dynamic Microtubules in Dendritic Spines. Neuron 61, 3–5. doi: 10.1016/j.neuron.2008.12.018
Peters, A., and Kaiserman-Abramof, I. R. (1970). The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am. J. Anat. 127, 321–355. doi: 10.1002/aja.1001270402
Pijet, B., Stefaniuk, M., and Kaczmarek, L. (2019). MMP-9 contributes to dendritic spine remodeling following traumatic brain injury. Neural Plast. 2019, 42–45. doi: 10.1155/2019/3259295
Pijet, B., Stefaniuk, M., Kostrzewska-Ksiezyk, A., Tsilibary, P. E., Tzinia, A., and Kaczmarek, L. (2018). Elevation of MMP-9 Levels Promotes Epileptogenesis After Traumatic Brain Injury. Mol. Neurobiol. 55, 9294–9306. doi: 10.1007/s12035-018-1061-5
Raible, D. J., Frey, L. C., Cruz Del Angel, Y., Russek, S. J., and Brooks-Kayal, A. R. (2012). GABAA receptor regulation after experimental traumatic brain injury. J. Neurotrauma 29, 2548–2554. doi: 10.1089/neu.2012.2483
Reza-Zaldivar, E. E., Hernández-Sápiens, M. A., Minjarez, B., Gómez-Pinedo, U., Sánchez-González, V. J., Márquez-Aguirre, A. L., et al. (2020). Dendritic Spine and Synaptic Plasticity in Alzheimer’s Disease: A Focus on MicroRNA. Front. Cell Dev. Biol. 8:255. doi: 10.3389/fcell.2020.00255
Rodriguez, A., Ehlenberger, D. B., Dickstein, D. L., Hof, P. R., and Wearne, S. L. (2008). Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3:1997. doi: 10.1371/journal.pone.0001997
Ross, S. T., and Soltesz, I. (2000). Selective depolarization of interneurons in the early posttraumatic dentate gyrus: Involvement of the Na+/K+-ATPase. J. Neurophysiol. 83, 2916–2930. doi: 10.1152/jn.2000.83.5.2916
Saatman, K. E., Creed, J., and Raghupathi, R. (2010). Calpain as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics 7, 31–42. doi: 10.1016/j.nurt.2009.11.002
Saneyoshi, T., Fortin, D. A., and Soderling, T. R. (2010). Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr. Opin. Neurobiol. 20, 108–115. doi: 10.1016/j.conb.2009.09.013
Schratt, G. M., Tuebing, F., Nigh, E. A., Kane, C. G., Sabatini, M. E., Kiebler, M., et al. (2006). A brain-specific microRNA regulates dendritic spine development. Nature 439, 283–289. doi: 10.1038/nature04367
Schumann, J., Alexandrovich, G. A., Biegon, A., and Yaka, R. (2008). Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J. Neurotrauma 25, 945–957. doi: 10.1089/neu.2008.0521
Shohami, E., and Biegon, A. (2014). Novel Approach to the Role of NMDA Receptors in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 13, 567–573. doi: 10.2174/18715273113126660196
Siedler, D. G., Chuah, M. I., Kirkcaldie, M. T. K., Vickers, J. C., and King, A. E. (2014). Diffuse axonal injury in brain trauma: Insights from alterations in neurofilaments. Front. Cell. Neurosci. 8:429. doi: 10.3389/fncel.2014.00429
Sitcheran, R., Gupta, P., Fisher, P. B., and Baldwin, A. S. (2005). Positive and negative regulation of EAAT2 by NF-κB: A role for N-myc in TNFα-controlled repression. EMBO J. 24, 510–520. doi: 10.1038/sj.emboj.7600555
Smith, D. H., Wolf, J. A., Lusardi, T. A., Lee, V. M. Y., and Meaney, D. F. (1999). High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J. Neurosci. 19, 4263–4269. doi: 10.1523/jneurosci.19-11-04263.1999
Smith, J. A., Das, A., Ray, S. K., and Banik, N. L. (2012). Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. doi: 10.1016/j.brainresbull.2011.10.004
Song, I., and Huganir, R. L. (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588. doi: 10.1016/S0166-2236(02)02270-1
Spaethling, J. M., Klein, D. M., Singh, P., and Meaney, D. F. (2008). Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 2008, 1207–1216. doi: 10.1089/neu.2008.0532
Spaethling, J., Le, L., and Meaney, D. F. (2012). NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiol. Dis. 46, 646–654. doi: 10.1016/j.nbd.2012.03.003
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Szydlowska, K., and Tymianski, M. (2010). Calcium, ischemia and excitotoxicity. Cell Calcium 47, 122–129. doi: 10.1016/j.ceca.2010.01.003
Tønnesen, J., and Nägerl, U. V. (2016). Dendritic spines as tunable regulators of synaptic signals. Front. Psychiatry 7:101. doi: 10.3389/fpsyt.2016.00101
Toth, Z., Hollrigel, G. S., Gorcs, T., and Soltesz, I. (1997). Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J. Neurosci. 17, 8106–8117. doi: 10.1523/jneurosci.17-21-08106.1997
Truettner, J. S., Alonso, O. F., and Dietrich, W. D. (2005). Influence of therapeutic hypothermia on matrix metalloproteinase activity after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 25, 1505–1516. doi: 10.1038/sj.jcbfm.9600150
Vaure, C., and Liu, Y. (2014). A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 5:316. doi: 10.3389/fimmu.2014.00316
Vercesi, A. E., Castilho, R. F., Kowaltowski, A. J., de Oliveira, H. C. F., de Souza-Pinto, N. C., Figueira, T. R., et al. (2018). Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic. Biol. Med. 129, 1–24. doi: 10.1016/j.freeradbiomed.2018.08.034
Wang, X., Jung, J., Asahi, M., Chwang, W., Russo, L., Moskowitz, M. A., et al. (2000). Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. 20, 7037–7042. doi: 10.1523/jneurosci.20-18-07037.2000
Wang, Y., Liu, Y., Lopez, D., Lee, M., Dayal, S., Hurtado, A., et al. (2018). Protection against TBI-Induced Neuronal Death with Post-Treatment with a Selective Calpain-2 Inhibitor in Mice. J. Neurotrauma 35, 105–117. doi: 10.1089/neu.2017.5024
Watts, L. T., Shen, Q., Deng, S., Chemello, J., and Duong, T. Q. (2015). Manganese-Enhanced Magnetic Resonance Imaging of Traumatic Brain Injury. J. Neurotrauma 32, 1001–1010. doi: 10.1089/neu.2014.3737
Weber, J. T. (2012). Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 3:60. doi: 10.3389/fphar.2012.00060
Wojnarowicz, M. W., Fisher, A. M., Minaeva, O., and Goldstein, L. E. (2017). Considerations for experimental animal models of concussion, traumatic brain injury, and chronic traumatic encephalopathy-these matters matter. Front. Neurol. 8:240. doi: 10.3389/fneur.2017.00240
Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D., and Smith, D. H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 21, 1923–1930. doi: 10.1523/jneurosci.21-06-01923.2001
Xiong, Y., Mahmood, A., and Chopp, M. (2018). Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chinese J. Traumatol. English Ed. 21, 137–151. doi: 10.1016/j.cjtee.2018.02.003
Xiong, Y., Mahmood, A., and Chopp, M. (2019). Remodeling dendritic spines for treatment of traumatic brain injury. Neural Regen. Res. 14, 1477–1480. doi: 10.4103/1673-5374.255957
Keywords: traumatic brain injury, synaptic dysfunction, excitotoxicity, dendritic spine, neurodegeneration
Citation: Hoffe B and Holahan MR (2022) Hyperacute Excitotoxic Mechanisms and Synaptic Dysfunction Involved in Traumatic Brain Injury. Front. Mol. Neurosci. 15:831825. doi: 10.3389/fnmol.2022.831825
Received: 08 December 2021; Accepted: 07 February 2022;
Published: 24 February 2022.
Edited by:
James P. Clement, Jawaharlal Nehru Centre for Advanced Scientific Research, IndiaReviewed by:
Vijayalakshmi Santhakumar, University of California, Riverside, United StatesCopyright © 2022 Hoffe and Holahan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brendan Hoffe, YnJlbmRhbmhvZmZlQGNhcmxldG9uLmNh; Matthew R. Holahan, TWF0dGhld0hvbGFoYW5AQ3VuZXQuQ2FybGV0b24uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.