 Francesco Antonica
Francesco Antonica Giuseppe Aiello
Giuseppe Aiello Alessia Soldano
Alessia Soldano Luana Abballe
Luana Abballe Evelina Miele
Evelina Miele Luca Tiberi
Luca Tiberi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 30 May 2022
Sec. Methods and Model Organisms
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.818696
This article is part of the Research Topic Model Systems to Study the Mechanisms of Neural Development and Disease View all 12 articles
Brain tumors are a large and heterogeneous group of neoplasms that affect the central nervous system and include some of the deadliest cancers. Almost all the conventional and new treatments fail to hinder tumoral growth of the most malignant brain tumors. This is due to multiple factors, such as intra-tumor heterogeneity, the microenvironmental properties of the human brain, and the lack of reliable models to test new therapies. Therefore, creating faithful models for each tumor and discovering tailored treatments pose great challenges in the fight against brain cancer. Over the years, different types of models have been generated, and, in this review, we investigated the advantages and disadvantages of the models currently used.
Primary malignant brain tumors remain among the deadliest form of cancers despite the deeper understanding of their tumorigenic processes, acquired during the recent years, and the multimodality therapeutic approach (Aldape et al., 2019). Brain tumors are also the most common solid tumor in children and are the leading cause of cancer-related morbidity and mortality in this population. An important shared feature of this heterogeneous group of diseases is the unique biology of the brain and its microenvironment, which represents a further degree of complexity in understanding the underlying biological mechanisms and in generating and delivering effective therapies. To this end, genetically engineered mouse models (GEMM) have been widely used in the field of cancer research as they allow to study mechanisms of tumorigenesis and tumor biology in a physiological context (Li and Langhans, 2021). However, most of these models cannot fully recapitulate the human tumor heterogeneity and show great limitations for preclinical drug testing (Gould et al., 2015; Aldape et al., 2019). Patient-derived xenografts (PDX) have been generated to overcome these limitations and to resemble human cancer more closely. These models retain patient mutational heterogeneity and, to date, have been considered one of the most reliable models for preclinical tests (Brabetz et al., 2018; Smith et al., 2020). However, PDXs cannot be used to address mechanisms of tumorigenesis, as they are already derived from tumor tissue. Human-induced pluripotent stem cell (hiPSC)-derived organoids solve the latter limit and represent a great advance for the understanding of tumorigenesis and for the development of new therapeutic strategies (Bian et al., 2018; Ogawa et al., 2018; Ballabio et al., 2020). In this review, our aim is to describe the advantages and disadvantages of current and developing models for brain tumors with the highest incidence (meningiomas) or morbidity and mortality (gliomas, medulloblastoma, and ependymomas) in adulthood and childhood that could lead to new therapeutic strategies (Figure 1).
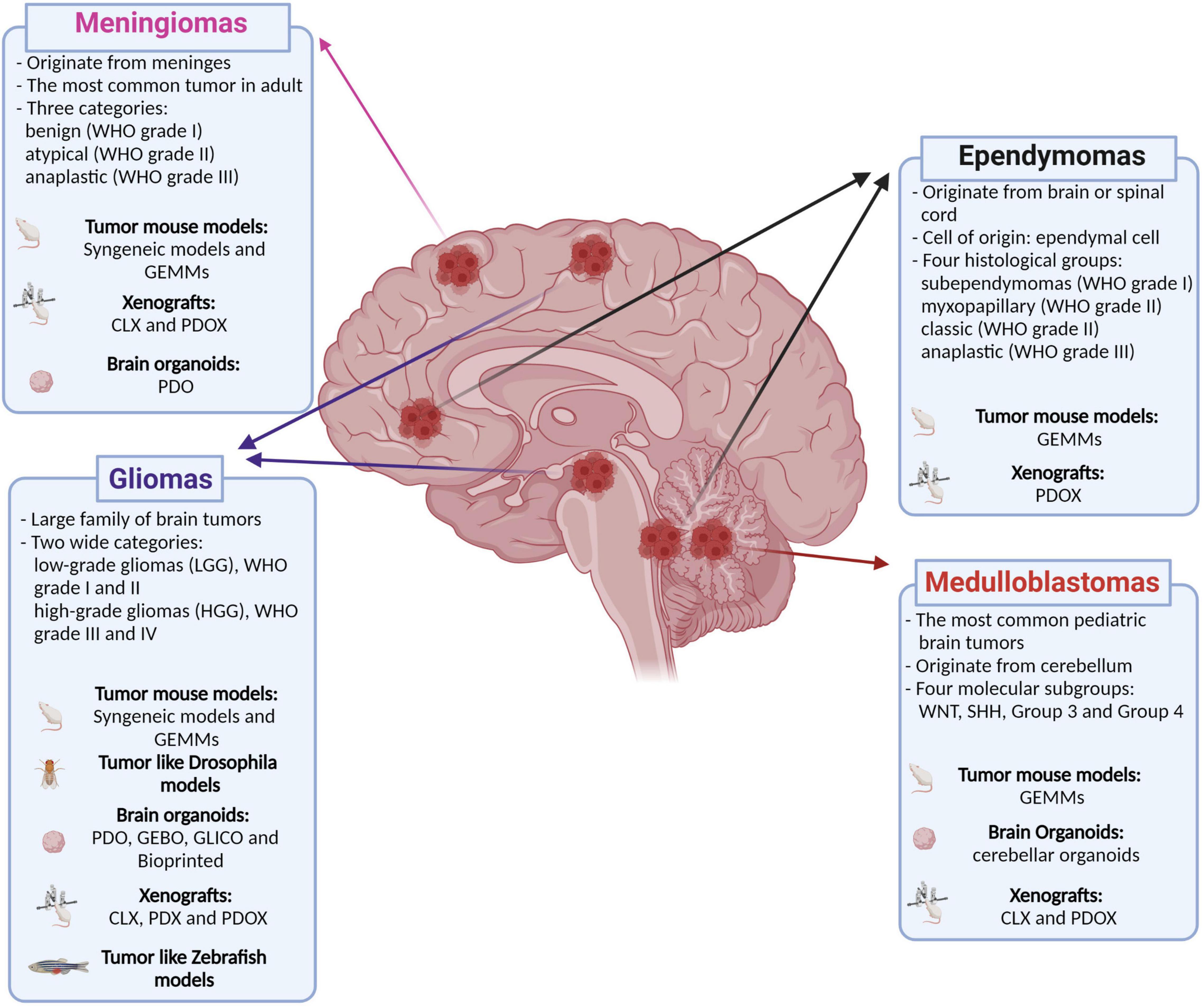
Figure 1. Classification of the main brain tumors discussed in this review and their relative available models. Genetically engineered mouse models (GEMMs); patient-derived organoids (PDO); genetically engineered brain organoids (GEBO); co-culturing GBM-derived GSCs with brain organoids (GLICO); cell lines xenografts (CLX); patient-derived xenografts (PDX); patient-derived orthotopic xenografts (PDOX). Created with BioRender.com.
Gliomas are a large family of brain tumors affecting adult as well as pediatric patients. Gliomas can be subdivided into two wide categories, low- and high-grade gliomas, based on their aggressiveness (Omuro and DeAngelis, 2013). The low-grade tumors (LGGs, Grades I and II according to the World Health Organization – WHO) are characterized by slower growth and infiltration compared with the high-grade (WHO Grades III and IV) (Louis et al., 2021). Nevertheless, the proper grade and the classification of the different gliomas rely not only on their histological features but also on the molecular characteristics such as genes found altered (i.e., IDH1, ATRX, TP53, CDK2A, BRAF, FGFR1, and PDGFRA) and on the methylation profile. In 2021, an updated edition of the WHO Classification of Tumors of the Central Nervous System was published that redefined all the pre-existing tumor classification (i.e., subclassification in adult vs. pediatric) and added new entities (i.e., diffuse glioma G34-mutant) (Louis et al., 2021). While LGGs show a better prognosis due to their low proliferation rate and infiltration, high-grade gliomas (HGGs) remain very challenging to cure with a median survival of less than 15 months in the case of the more aggressive glioblastoma multiforme (GBM), and diffuse midline glioma, H3 K27-altered in pediatrics. Indeed, the invasive nature of GBM makes surgical resection difficult; relapse occurs rapidly after treatments, and the high intra-tumor heterogeneity increases the difficulties in establishing effective drugs. The heterogeneity within GBM tumors has been identified at several levels, such as genetic, transcriptional, and DNA methylation. Since 2010, GBM has begun to be classified into 4 subtypes: proneural, neural, classical, and mesenchymal (Verhaak et al., 2010). This classification relied on peculiar aberrations and gene expression on specific genes, such as PDGFRA/IDH1, EGFR, and NF1. Afterwards, the improvement of high-throughput sequencing as well as the development of new profiling method (i.e., DNA methylation) has allowed to better stratify all the 4 canonical GBM subtypes (Zhang et al., 2020). In addition, it was shown how the prognosis can be closely correlated with the molecular subtype; for example, the proneural GBM has been shown to have a better prognosis than mesenchymal GBM (Phillips et al., 2006). The poorest prognosis of mesenchymal GBM is characterized by over-expression of genes related to angiogenesis and cell invasion (Phillips et al., 2006). Finally, using scRNA-seq technique has been defined that GBM has a peculiar feature of intra-tumoral heterogeneity. Indeed, several studies have shown that the different GBM molecular subtypes can co-exist within the same tumor (Patel et al., 2014; Darmanis et al., 2017; Neftel et al., 2019). This last aspect makes challenging to define which therapy would be more appropriate for each patient. Due to the inner complexity and aggressiveness, the development of reliable models for HGGs and GBM remains a crucial challenge in the tumor biology field, and several efforts have been conducted to generate in vitro and in vivo models, aiming to set up the best experimental strategy for investigating new therapeutic approaches (Ho et al., 2014; Hicks et al., 2021). On the other hand, low-grade gliomas, due to their low proliferative features, make challenging to culture them, as cell lines or organoids or to have a proper tumor growth in a reasonable period in a mouse model, and, for these reasons, are less investigated. Finally, the recent fine re-classification of the different glioma based on their genetic should be taken in consideration, for example, for the future creation of new genetic models.
In the context of gliomas, different types of in vivo models have been generated until now: xenografts including CLX (cell lines xenografts) and PDX (patient-derived xenografts), syngeneic, GEMM, and Drosophila melanogaster.
Xenograft models have been generated by injecting patient-derived cells or established cell lines into the mouse brain (i.e., U251, U87, A172, and T98G) (Table 1) (Fareh et al., 2012; Jandial et al., 2018; Hicks et al., 2021). Nevertheless, all the models based on cell lines lack the heterogeneity present in the human tumor due to the selection occurring in cells when cultured. A further step was taken when glioma stem-like cells (GSC) were isolated and maintained in vitro in 2D or in spheroid culture conditions. Indeed, CD133/PROMININ-1 positive cells can be isolated from primary tumors, cultured and propagated as spheres, and finally grafted in mice (Singh et al., 2004; Yi et al., 2016). Xenograft models can also be obtained by primary tumors (Sasaki et al., 2001; Joo et al., 2013; Kerstetter-Fogle et al., 2020) (Figure 2A and Table 1). These models, called PDX, have the enormous advantage of being directly generated from tumor cells, thus maintaining the features of the original neoplasm, including its cellular composition that will grow in the murine microenvironment (Shu et al., 2008). Although xenograft models are considered a reliable scenario and, in some cases, the closest to humans, they have the important weakness of being devoid of the host’s immune system, as they are generated in immune-compromised mice. More in detail, depending on the immunocompromised mouse strain used, the lack of adaptive or innate immune system does not allow investigating the interaction between tumor and immune system. A possible solution has been recently proposed by xenografting tumor cells into the telencephalon ventricle of wild-type mouse embryos, where tumors persist in postnatal life (Hoffmann et al., 2020). Furthermore, the growth of GBM cells in the brain of nude mice, coupled with the possibility of genetically labeling them with barcodes, has provided a novel approach to trace different tumor clones in vivo. Indeed, Lan et al. (2017) using the patient-derived orthotopic xenografts (PDOX)-based approach showed the contribution of the different cancer cells in tumor formation, aggression, and therapy response. The possibility to track patient-derived GBM cell behavior in vivo with the barcoding approach has suggested the existence of a proliferative hierarchy, contributing to the tumor re-growth with a slow-cycling cancer stem cell subpopulation at its apex (Lan et al., 2017). On the other hand, PDXs have been also shown to be a valid approach for in vivo biobanking of pediatric brain tumors. Indeed, the xenografted tumor tissue closely recapitulated the histology, genetics, and drug sensitivity of the original tumor (Brabetz et al., 2018; Smith et al., 2020). Finally, orthotopic xenografts represent a valuable system to test the tumorigenicity of glioma-related mutations. In particular, the introduction of H3.3-K27M mutation in neural stem cells (NSCs) makes such cells capable to induce tumors formation upon transplantation in the mouse brain (Haag et al., 2021). Furthermore, distinct mutations in H3.3 trigger a tumoral phenotype, depending whether the NSCs derived from forebrain or brainstem modeling the possible different origins of pediatric high-grade glioma (Bressan et al., 2021).
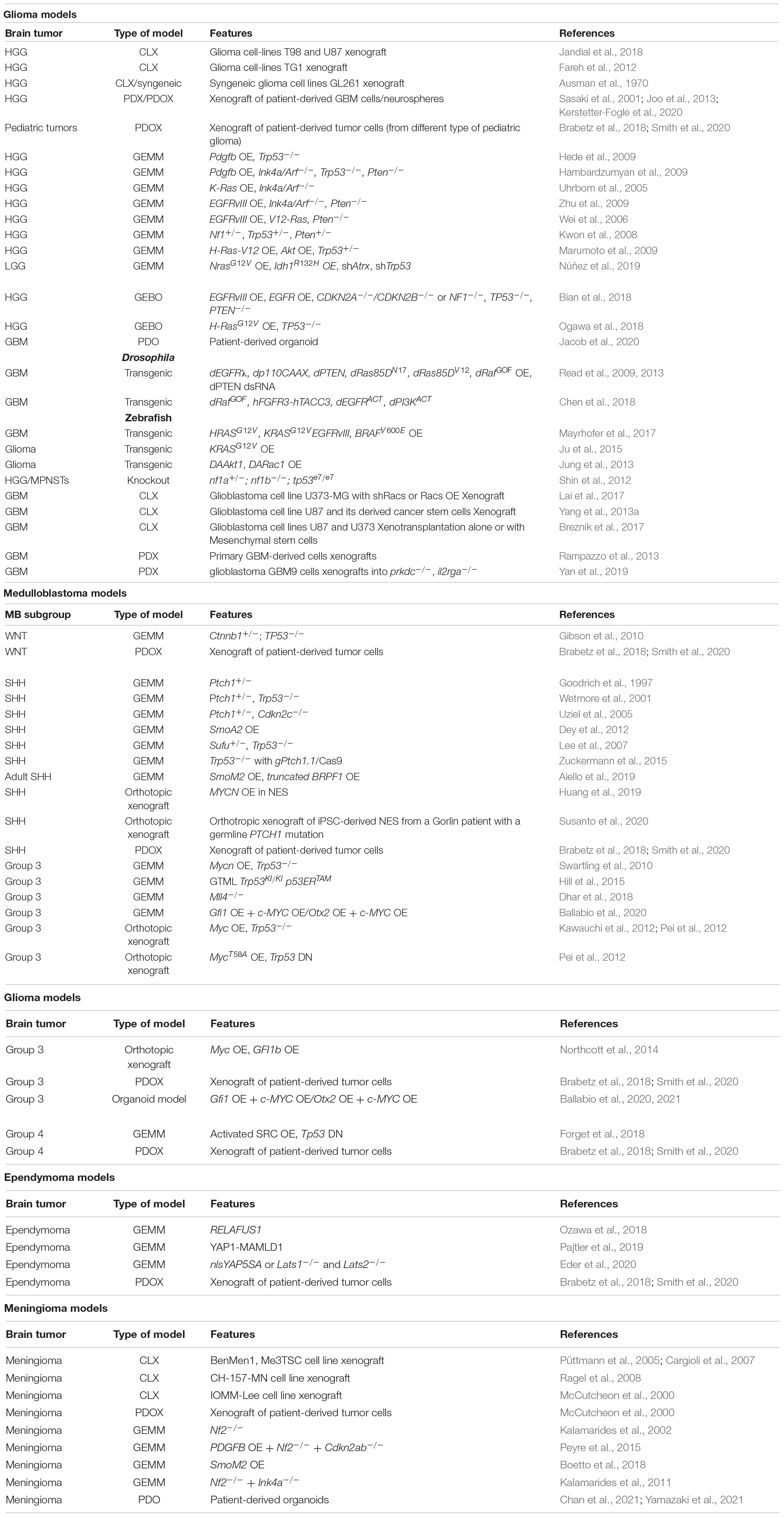
Table 1. An example of different types of brain tumor models and their relative features.
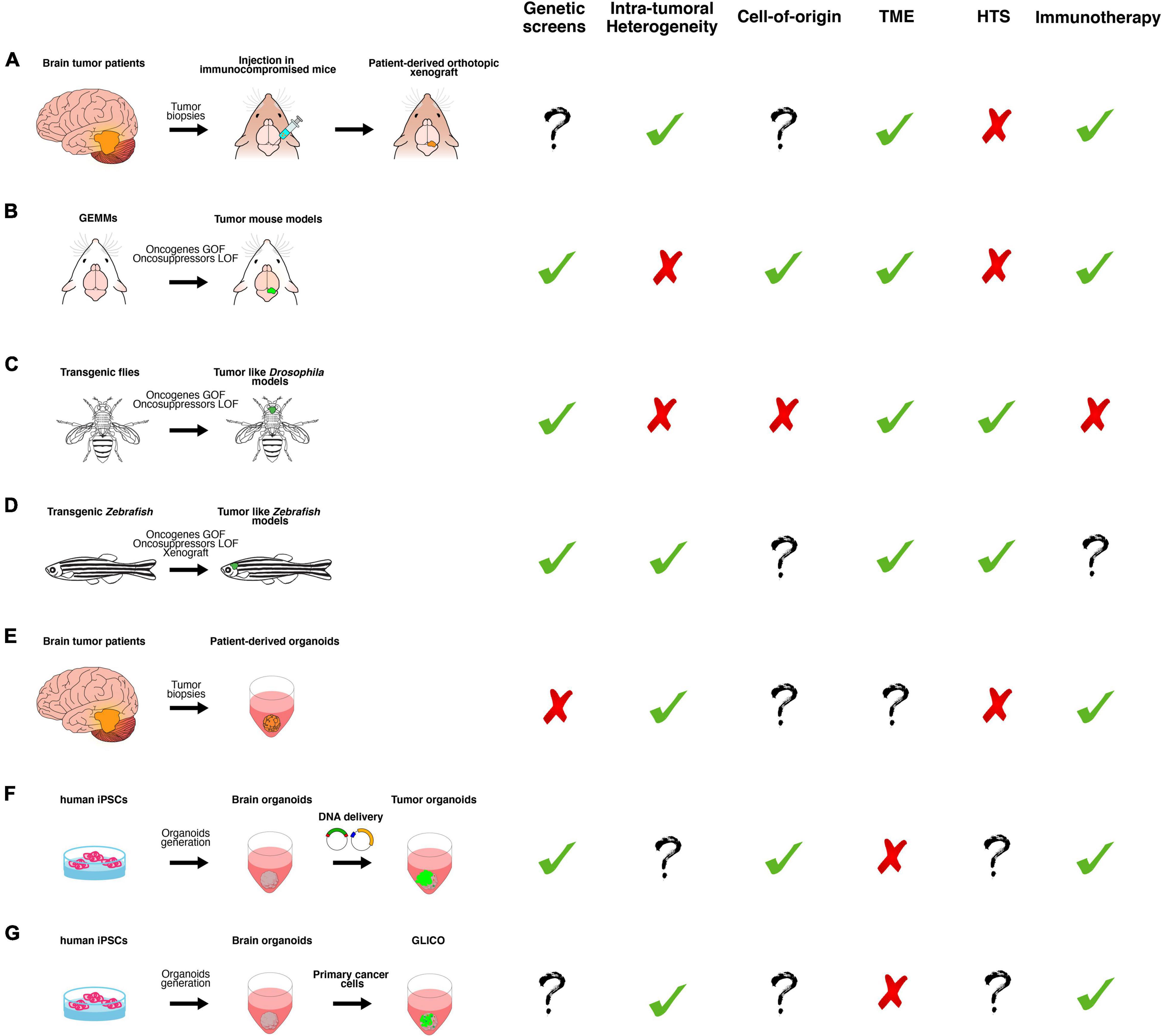
Figure 2. A schematic overview of the main classes of in vitro and in vivo preclinical brain cancer models and their relative applications. (A) Brain cancer tissue surgically resected from patients is directly transplanted in the brain of immunocompromised mice, producing patient-derived orthotopic xenograft models (PDOX). (B) Genetically engineered mouse models (GEMMs) in which the tumor formation is induced by gain or loss of function of oncogenes or oncosuppressors, respectively. They can be generated by breeding animals that carry germline mutations or injecting virus or plasmids, harboring the gene of interest. (C) Transgenic flies in which the tumor-like phenotype is determined by gain or loss of function of oncogenes or oncosuppressors, respectively, in a time- and tissue-specific manner. (D) Transgenic Zebrafish in which the tumor-like phenotype is determined by gain or loss of function of oncogenes or oncosuppressors, respectively, in a time- and tissue-specific manner. Xenotransplantation of human glioma cells in zebrafish. (E) Brain cancer tissue surgically resected from patients is directly cultured in 3D culture as patient-derived organoids (PDO). (F) Tumor organoids can be generated by gain or loss of function of oncogenes or oncosuppressors, respectively, in cerebral or cerebellar organoids derived from human-induced pluripotent stem cells (iPSCs). (G) GLICO can be generated by co-culturing primary cancer cells with cerebral organoids. Based on published works, we summarized the possible usages of each class of the models in the following applications: genetic screens (i.e., testing the function of new genes in tumor formation, progression and aggressiveness); investigation of intra-tumoral heterogeneity, cell of origin and tumor microenvironment (TME); high-throughput screening (HTS) of new chemotherapeutic drugs and, finally, for testing new immunotherapy approaches.
Several years ago, an interesting approach of inducing brain cancer in mice was developed, which consists of injecting ethyl-nitrosourea into the placenta of pregnant females between E15 and E18 (Thomas et al., 1994), or injecting methylcholantrene directly in the brain (Seligman et al., 1939). Both treatments with carcinogens lead to the formation of GBM-like tumors, from which cell lines are derived and, in turn, used to generate syngeneic allograft serially grafting (Ausman et al., 1970; Kaye et al., 1986). These models, contrary to PDOX where immunocompromised mice are used, allow to study the interaction between tumor-immune microenvironment and give the possibility to test immunotherapies. On the other hand, these models do not recapitulate several aspects of HGGs and GBMs, such as infiltration and histology. Recently, injection of engineered NSCs (Nf1 and Pten KO + EGFRvIII overexpression) into immunocompetent syngeneic mouse strain has been used to propose an epigenetic-driven mechanism exploited by GBM stem cells to evade immune system (Gangoso et al., 2021).
The genetically engineered mouse model (GEMM) represents one of the most reliable in vivo models to study whether specific genetic alterations are responsible for tumor initiation and progression (Figure 2B). The analysis of the genetic landscape of HGG and GBM led to the identification of the most frequently mutated or amplified genes, and specific GEMMs have been created based on this knowledge. Such genetic alterations include gene amplification (i.e., PDGFA/B), gain-of-function activating mutations (i.e., KRAS, HRAS, EGFR, NRAS, and PDGFRA) and loss-of-function mutations/gene deletions (i.e., TP53, CDKN2A, PTEN, NF1, ATRX, AKT, IDH1, H3F3A, and INK4a) (Table 1) (Uhrbom et al., 2005; Wei et al., 2006; Kwon et al., 2008; Hambardzumyan et al., 2009; Hede et al., 2009; Marumoto et al., 2009; Zhu et al., 2009; Núñez et al., 2019; Akter et al., 2021; Hicks et al., 2021). However, GEMMs also have several limitations. They are time-consuming and tumors do not always recapitulate the intra-tumor heterogeneity observed in patients (Reilly, 2009; Day et al., 2015).
Cre-lox is a gene-editing technology that allows site-specific recombination between sequences called Lox sites using the enzyme Cre recombinase. The action of Cre can be spatio-temporally controlled by driving its expression in certain cell types via specific promoters or Tamoxifen administration (when Cre is fused to ERT2). For example, this system has been used to generate mice that develop tumors by the introduction of the EGFRvIII genetic variant (Zhu et al., 2009), shedding light on the impact on GBM tumorigenesis of wild type and mutant forms of EGFR. The genetic knocking out of Nf1, Trp53, and Pten is also able to induce glioma in the mouse brain. Furthermore, the Cre-Lox approach helps to investigate the putative cell of origin by selective gene loss in specific lineage using peculiar promoters (Llaguno et al., 2009, 2015, 2019). The penetrance of brain tumor formation changes with the differentiation state of the cell, as observed in transgenic mice where tamoxifen-inducible Cre was expressed in neural stem cells (i.e., Nestin-CreERT2) (Llaguno et al., 2009), bipotential progenitors (i.e., Ascl1-CreERTM), oligodendrocyte progenitors (i.e., NG2-creERTM) (Llaguno et al., 2015) and immature (NeuroD1-creERT2) or post-mitotic neurons (CamKIIα-creERTM) (Llaguno et al., 2019). Indeed, only neuronal stem cells (Nestin+ or GFAP+ cells) and early progenitors (Ascl1+ or NG2+ cells) that are at the top of the differentiation hierarchy are susceptible to cancer formation, while tumorigenesis is abolished in immature or post-mitotic neurons (NeuroD1+ or CamKIIα+ cells). Such an experimental model can be easily applied to the study of other tumors such as medulloblastoma and organoid models (described in the next sections).
Another method to generate GEMMs that overexpress or inactivate/silence the genes responsible for gliomagenesis is to insert the transgenes of interest into the genome, using the transposon system. The sleeping beauty (SB) transposon-based glioma mouse model was created, overexpressing PDGF-A and silencing Nf1 and Trp53 (Sumiyoshi et al., 2018). The same approach was used to test the role of Atrx silencing in brain tumors induced by overexpression of Nras and Trp53 silencing (Koschmann et al., 2016). The possibility to integrate different constructs and deliver the DNA in the newborn mouse brain offers the advantage to test the tumor-inducing function of several genes in a less costly and time-consuming way.
One of the most popular systems to generate GEMM to induce brain tumors is the RCAS-TVA method. RCAS-TVA is an efficient viral gene delivery system, consisting of RCAS (replication-competent avian sarcoma) viruses that carry the genes of interest and can only infect cells expressing the receptor TVA (tumor virus A). These cells were, indeed, previously engineered to express the tTA receptor under the control of specific cell lineage promoters, such as Nestin, Gfap (Holland et al., 1998), and CNPase (Lindberg et al., 2009). This allows studying whether specific oncogenes can induce tumor in a defined population and different anatomical regions. Of note, the RCAS-TVA approach has revealed how GBM can be induced by the EGFR mutant with loss of Ink4a or Trp53 and that they arise more easily from Nestin cells than Gfap ones (Lindberg et al., 2009). The same approach has been used to model the low-grade brain tumor pilocytic astrocytoma (Gronych et al., 2011).
Globally, the GEMMs have the advantage to model GBM and HGG in mice with an intact immune system, allowing to test immunotherapies and to investigate the interaction between neoplastic and immune cells. Furthermore, GEMMs allow studying the impact of the different mutations on tumor progression and response to treatments. On the other hand, GEMMs have also serious and important pitfalls such as the lack of intra-tumor heterogeneity and the diversity of blood–brain barrier (BBB) in the mouse brain compared to the human one (Aday et al., 2016), thus affecting the treatment delivery.
Drosophila melanogaster is a powerful genetic model that can be successfully used to study cancer biology (Figure 2C and Table 1). Its intrinsic peculiarity allows overcoming the downsides of other models, such as tissue cultures and mice. In fact, the signalings involved in cancer are conserved in Drosophila (Reiter et al., 2001; Yamamoto et al., 2014), and a wide range of genetic tools exists to perform large-scale genetic screens. Drosophila represents also a powerful platform to perform pharmacological screens; in fact, compared to cell or organoid cultures, it represents a whole animal system that allows testing of a large number of compounds simultaneously in a high throughput fashion (Bell et al., 2009; Willoughby et al., 2013). Compared to the different techniques used to generate mouse models of brain tumors, fly gliomas have been studied by producing genetically modified Drosophila strains that carry alterations in the key pathways affected in human patients.
Many of the key signalings perturbed in GBM, such as EGFR signaling and the phosphatidyl-inositol-3-kinase (PI3K) pathway (Brennan et al., 2013), are conserved in Drosophila. Moreover, single functional orthologs exist for most of these genes, such as EGFR (dEGFR), PIK3CA (dp110), PTEN (dPTEN), RAS (dRas), RAF (dRaf), and AKT (dAkt). Glia-specific co-activation of EGFR and PI3K signaling in the fruit fly results in diffuse glial neoplasia in the larva (Read et al., 2009, 2013). This model has been used to perform a kinome-wide genetic screen that identified new genes involved in glioma development (Read et al., 2013). Interestingly, most of the modifiers have orthologs previously connected to GBM, while 16 appeared to be new modifiers, such as dRIOK1 and dRIOK2. Overexpression of mutant dEGFR and dp110 in precise time windows during adult life (Chi et al., 2018) also leads to brain enlargement and extensive glial expansion that ultimately results in a shorter lifespan and defective neural behaviors. Chen et al. (2018) established both high- and low-grade glioma models in the fruit fly by expressing constitutively active Drosophila Raf (dRafGOF), human FGFR3-TACC3 fusion gene, or constitutively active Drosophila EGFR and PI3K (dEGFRACT; dPI3KACT) in glia. Interestingly, the gliomas exhibited an increase of tissue stiffness compared with non-transformed brains, similar to the gradual tissue stiffening observed in human LGGs compared to HGGs (Miroshnikova et al., 2016) mediated by the Ion Channel dPiezo (Chen et al., 2018). The importance of tissue stiffness in glioma has also been demonstrated by Kim et al. (2014); through a genetic screening, they identified Lox, a Lysyl oxidase involved in extracellular matrix stiffness, as a potential mediator of neoplastic glial migration dependent on the pan-glial PDGF receptor (Pvr).
Drosophila has largely contributed to the understanding of the molecular mechanism underlying asymmetric cell division, an intrinsic property of cancer stem cells, leading to the first discoveries connecting this process to tumorigenesis (Caussinus and Gonzalez, 2005; Gómez-López et al., 2013). The previously described Drosophila GBM models driven by RTK-Ras have been used to identify and study transcription factors altered in GSCs. For example, Cheng et al. (2016) described the role of the transcription factor FOXD1 and of its target ALDH14A3, whose expression is altered in patient-derived GSCs. Another example of a transcription factor whose expression is altered in patient-derived GSCs is the Achaete-scute homolog 1 (ASCL1), an ortholog of the Drosophila achaete. Using the Drosophila GBM model described before (Read et al., 2009), it has been demonstrated that overexpression of fly Achaete or human ASCL1 reduced tumor size and proliferation, and induced a switch from glial to neuronal fate (Park et al., 2017). Interestingly, the ASCL1 role is evolutionarily conserved; in fact, ASCL1 overexpression efficiently reduces growth capacity of proneural human cancer stem cells-derived models of GBM and promotes a lineage switch, activating the neuronal fate and repressing the glial one (Narayanan et al., 2019; Azzarelli et al., 2022).
Therefore, Park et al. (2017) showed the usefulness of Drosophila GBM models for understanding how transcription factors involved in differentiation processes can affect GBM formation and gliomagenesis. YAP (yes-associated protein, also known as YAP1) and TAZ (transcriptional co-activator with PDZ-binding motif) are Hippo pathway effectors involved in the control of stem cells fate (proliferation/differentiation). Using the previously described Pten-RNAi/RASV12 overexpression-induced GBM model (Cheng et al., 2016), Minata et al. (2019) demonstrated that Tep1 (CD109 in mammals) loss in glioma cells reduces Yki (the Drosophila YAP/TAZ ortholog) and attenuates gliomagenesis. High levels of CD109 have been reported in GBM samples, and this work clarified its role in clonogenicity, tumor initiation, and radio-resistance of GBM. The data obtained both in human and Drosophila samples (Gangwani et al., 2020) suggest a conserved oncogenic signaling of CD109 through the YAP/TAZ pathway. Understanding the role of this signaling in Drosophila is extremely relevant to human pathology as YAP/TAZ have been shown to be required for GSCs plasticity and for GBM initiation due to their ability to prevent GSC differentiation. Finally, Hakes and Brand (2020) clarified the contribution of neural lineages to GBM by studying the orphan nuclear receptor TLX, which correlates with a poor prognosis in patients with GBM (Park et al., 2010). TLX is required for a linage-specific (Type II) NSCs identity and progression during development and, when overexpressed, can induce tumor formation by inducing a switch in NSC identity, a block of differentiation and reversion of intermediate neural progenitors (INP)s to NSC fate.
In conclusion, Drosophila has been proved very helpful in studying the signaling pathway, and the molecular mechanisms underlying GBM development and the finding obtained using this model are of great relevance for the understanding of human pathogenesis. However, Drosophila still maintains many limitations in mimicking the human microenvironment. Indeed, the absence of an adaptive immune system as well as the differences in the ability of the immune cells to infiltrate the brain tissues limits the possibility to study the immune-tumor interaction. Moreover, due to the differences between humans and Drosophila development, this model does not allow to fully recapitulate human brain tumor heterogeneity and pathology.
Zebrafish is a well-established and robust model for studying cancer pathology and its fit for rapid and efficient screening of new treatments. Thanks to its short embryonic developmental time, small size, and its transparency, coupled with conservation of genetic mechanisms underlying biological processes and the possibility to transplant human cells; it represents an important model for gliomas studies (Figure 2D and Table 1). Compared to Drosophila, where modeling of brain tumor is achieved solely by production of transgenic models, zebrafish models of glioma are produced using different techniques.
Gliomas can be induced in zebrafish by activation of the EGFR/RAS/ERK/AKT pathway via overexpression (a zic4 enhancer) of several oncogenes, such as KRASV12, EGFRvIII, among others (Mayrhofer et al., 2017). Analysis of global RNA expression established that obtained brain tumors resemble GBMs of the mesenchymal subtype, with a strong YAP component. Similarly, overexpression of KRASG12V in a putative neural stem and/or progenitor cells induced brain tumorigenesis (Ju et al., 2015) and overexpression of dominant-active human Akt1 (DAAkt1) or Rac1G12V (DARac1) (Ptf1a promoter)-induced gliomas of various histological grades, frequently infiltrated (Jung et al., 2013). Different tumor-initiating cells affect the tumor type; in fact, overexpression of KRASG12V under the control of the krt5 gene promoter induced low frequency brain tumors in the ventricular zones (VZ) that resemble malignant peripheral nerve sheath tumors (MPNSTs) (Ju et al., 2015). In contrast, overexpression of KRASG12V under the control of the gfap gene promoter induced brain tumors characterized by prominent activation of the canonical RAS-RAF-ERK pathway in both VZs and brain parenchyma at higher frequency (Ju et al., 2015). This study demonstrated that zebrafish could be explored to study cellular origins and molecular mechanisms. Ju et al. (2014) also developed the first animal model of gliomagenesis driven by Sonic Hedgehog (Shh) by overexpressing Smoothened (Smoa1) under the krt5 neural promoter. Transgenic zebrafish models have also been used to study the macrophage infiltration and contribution to tumor growth. Two publications clarified that expression of the human AKT1 oncogene in neural cells leads to tumor formation and significant increase in the macrophage and microglia populations that showed tumor-promoting functions (Chia et al., 2018). In particular, this is due to infiltration of macrophages from the peripheral area into the brain mediated via Sdf1b-Cxcr4b signaling. Cancer cells exploit the mechanisms used to mediating neuro-microglial via P2ry12 activation to promote their own proliferation (Chia et al., 2019). Finally, several transgenic models expressing IDH1 mutations were used to evaluate their roles in tumor development, but none of them led to glioma development, suggesting that further mutations are required (Gao et al., 2018).
Several strategies have been used to produce knock out models to study the involvement of endogenous genes in glioma formation. Shin et al. (2012) used targeting-induced local lesions in genomes (TILLING) strategy to generate several null alleles of nf1a and nf1b. Thanks to these transgenic strains - they demonstrated that loss of nf1 is involved in tumorigenesis; in fact, adult nf1a+/−; nf1b–/–; tp53e7/e7 animals show an earlier onset and increased penetrance of HGGs and MPNSTs (Shin et al., 2012). The same group also induced the CRISPR/Cas9-mediated knockout of atrx, a known tumor suppressor gene in sarcomas, in the nf1/p53-deficient zebrafish, to study its contribution to malignant growth (Oppel et al., 2019). Moreover, knock out models have also been used in combination with transgenesis to study the connection between the immune system and tumor growth, as described above (Chia et al., 2019).
The MO are antisense oligonucleotides that bind to complementary target mRNAs and block their translation, acting similarly to small interfering RNAs (siRNAs) and short hairpin RNAs (shRNAs), or alter the pre-mRNA splicing. Gliomas and, in particular, glioblastomas are tumors characterized by a high level of vascularization, and their growth depends on the formation of new blood vessels (Ahir et al., 2020). MO have been used in zebrafish to study the role of genes in angiogenesis and their contribution to GBM growth. For example, this has been done with Ephrin-B3 (ephrinb3-like in zebrafish), highly expressed in GBM and acting as a survival factor (Royet et al., 2017) and with PlexA, also highly expressed in GBM and in tumor-associated blood vessels in patient biopsies (Jacob et al., 2016).
Xenotransplantation of human glioma cells in zebrafish is a powerful technique to study tumor growth and invasion. In fact, the transplanted cells not only survive but are also able to migrate and interact with the host environment. Zebrafish offers several advantages over other xenograft models: firstly, it lacks an adaptive immune system in early stages of life (before 30 days post-fertilization, pfs) (Lam et al., 2004); therefore, no use of immunocompromised animals is needed at these stages. Moreover, the presence of many genetic tools, together with the small size and the transparency of zebrafish, allows live and high-resolution imaging of the transplanted cells (Pudelko et al., 2018; Vargas-Patron et al., 2019). Xenotransplantation can be performed at different injection sites: glioma/glioblastoma cell lines as U87 and U373T have been injected in the yolk sack at the stage 48-h pfs and analyzed for several parameters such as survival, proliferation, and invasion (Yang et al., 2013a; Lai et al., 2017). These studies led to the characterization of the role of genes such as Rac, MMP-9 in glioma aggressiveness/invasiveness, or the role of TGF-β1 in enhancing tumor-induced angiogenesis (Yang et al., 2013b).
Injections have also been performed in the zebrafish brain, ranging between 48- and 72-h pfs. Two studies used modified U-87 glioma cells lines to characterize the role of the KMT2A-NOTCH regulatory cascade (Huang et al., 2017) and of RECQ1 Helicase (Vittori et al., 2017) in glioma proliferation. Other groups used xenografts injection in the brain to study microenvironment and tumor migration/invasion; Breznik et al. (2017) investigated the role of mesenchymal stem cells (MSCs) in modulation of glioblastoma cells invasion by xenotransplantation of a mixture of MSC and U87 and U373 GBM cells. Xenotransplant into the brain (7 days pfs) was used to study differentiation of primary GBM cells induced by treatment with Wnt ligands, or overexpression of β-catenin (Rampazzo et al., 2013). Recently, it has been reported an optically clear mutant zebrafish (prkdc−/−, il2rga−/−) that lacks adaptive and natural killer immune cells that have been used to graft GBM9 glioblastoma cells intraperitoneally at 2 months old, and seems to be a promising model for developing personalized therapeutic approaches (Yan et al., 2019).
In conclusion, zebrafish overcomes many drawbacks of murine models, in terms of tumor live visualization, late development of the immune response, and presence of many genetic tools for manipulation and of the Drosophila models, especially thanks to the higher degree of physiological similarity to the mammalian models. However, open questions remain on pharmacokinetic studies for accurate drug delivery, dosing, and metabolism in zebrafish due to the divergent physiological features (such as body temperature and organ systems) (Kirchberger et al., 2017; Casey and Stewart, 2020).
Organoid models represent the most advanced approach, combining the most recent techniques of 3D culturing, 3D printing, and bioengineering (Rodrigues et al., 2021). There are two main cellular sources for deriving tumor organoids: the patient tumor biopsies (Figure 2E and Table 1) and pluripotent stem cells derived brain organoids (Figure 2F and Table 1).
The first glioblastoma organoids were generated by Hubert et al. in 2016. Contrary to the classical spheroids formed by one cell type, they created more complex organoids starting from xenografts, GEMMs, and patient biopsies (Hubert et al., 2016). They observed that such cells cultured using Matrigel-based 3D culture methods formed organoids, recapitulating many GBM features, including a hypoxic gradient and resistance to radiation. Recently, Jacob et al. (2020) have established a growth factors-free chemically defined medium-based protocol to derive, expand, and cryopreserve GBM organoids, starting from tumor biopsies, so called PDOs (patient-derived glioblastoma organoids). The cellular composition, as well as the transcriptional profiling, confirmed the high similarity between PDOs and the original tumor. Such similarities were also observed in the response to treatment with radiations and temozolomide, and the infiltrative nature was observed upon xenograft in nude mice. Organoids were also generated from glioma of different grades (Grades II, III, and IV) but, despite some of the original tumor features, are retained; they survive in culture for a limited time, making the biobanking and expansion quite challenging. Nevertheless, Golebiewska et al. (2020) were able to successfully generate a living collection of PDOs that maintains most of the genetic, molecular, and phenotypical features of the primary tumor and a similar response to therapy. They also analyzed the DNA methylation patterns, currently considered the gold standard for the correct brain tumor diagnosis and subtyping, which revealed a good correlation between primary tumor and the respective-derived PDO. Finally, PDOs have been shown to maintain some of the immune cells (i.e., microglia and T-cell) during the 1st weeks in culture, opening the possibility to study their interaction with tumor cells (Jacob et al., 2020). Nevertheless, culture conditions need to be improved to ensure a stable expansion of the immune compartment in long-term culture.
Few years ago, two independent studies showed how genetically modified brain cancer models can also be generated in vitro. Using the same in vivo approach of genes in human gliomas (i.e., HRASG12V and TP53–/–; cMYC overexpression; EGFRvIII and EGFR overexpression and CDKN2A–/–; NF1–/– and PTEN–/– and TP53–/–), cerebral organoids derived from human-induced pluripotent stem cells (hiPSCs) were modified in order to induce neoplastic organoids (Bian et al., 2018; Ogawa et al., 2018) (Figure 2F and Table 1). The addition of fluorescent reporter genes allows to specifically track the modified cells and as they grew. Such neoplastic organoids exhibited a transcriptional profile partially similar to human gliomas, and their aggressiveness was confirmed in vivo upon orthotopic injection into nude mice. However, they did not show strong molecular similarity to patients, and it was not clearly shown whether they also retain intra-tumoral heterogeneity. This represents the major weakness for making this model suitable to investigate the intra-tumoral heterogeneity. On the other hand, these models have a great potential in drug screening by providing “unlimited” mini tumor organoids. They can be genetically modified in a less time-consuming and more cost-effective way than mouse models, allowing for the screening of new genes involved in tumor formation before testing them in vivo. Finally, GEBOs might be a valid model to investigate the cell of origin of glioma as already shown to be feasible for Group 3 Medulloblastoma (Ballabio et al., 2021). Generation of cerebellar as well as cerebral organoids from hiPSCs relies on well-established protocols (Muguruma et al., 2015; Velasco et al., 2019) that generate structures resembling fetal cerebellum or brain also from molecular points of view (Velasco et al., 2019; Nayler et al., 2021).
Cerebral organoids have been also recently used for studying the behavior and invasion of GBM cells. The first approach consisted of co-culturing GBM spheroids with mouse ESC-derived brain organoids, proving the potential of modeling GBM invasion with organoids (da Silva et al., 2018). Similarly, Linkous et al. (2019) described a model of co-culturing GBM-derived GSCs with brain organoids, called GLICO (Figure 2G). With this approach, they were able to detect GBM cells invading normal brain organoids using microtubule structure, similar to what is observed in the GBM cells invading human brain parenchyma (Osswald et al., 2015). Additional studies have shown that this model can be successfully used to investigate invasion (Goranci-Buzhala et al., 2020; Krieger et al., 2020). Furthermore, when GSCs were co-cultured in 3D with organoids starting to differentiate, allowing also to recapitulate the lineage heterogeneity of cancer cells (Pine et al., 2020; Azzarelli et al., 2021). Nevertheless, this phenomenon seemed to be also dependent on the organoid media composition (Azzarelli et al., 2021). The pitfalls of GLICO are those related to being based on cell lines that do not allow investigating cancer in a context where original cell types and microenvironment are maintained (Jacob et al., 2020). Nevertheless, GLICO represents an excellent and valid model for studying cell behavior, such as invasion and cellular plasticity of GSCs.
The concept of bioprinting GBM organoids consists of putting together different cell types to recreate the different tumor regions. For example, it is possible to print freshly dissociated GBM cells on a chip together with vascular endothelial cells using a porcine ECM as “bioink” (Yi et al., 2019). This GBM-on-a-chip approach also allows recreating the hypoxic gradient and the infiltrating region. To increase complexity, GBM stem cells have been bioprinted together with neural stem cells (NSCs), astrocytes, and macrophage. The presence of immune cells allows to study their interaction and effect on the growth and invasion of cancer cells (Tang et al., 2020). Despite these models are very promising, the approach is still in its infancy and, therefore, presents some pitfalls, such as the technology is not yet precise and requires the use of specialized equipment and skilled researchers that, in turn, require significant investments.
To summarize, as for the other models, organoid-based systems have advantages and disadvantages. As an in vitro model, the usage of organoids allows to perform a drug screen and a test for immunotherapy; in addition, organoids can be used for an invasion assay and for testing the function of genes for inducing tumors. On the other hand, such models have disadvantages such as lack of vascular network. In the future, the possibility to induce cancer in “vascularized” brain cancer organoids might overcome this limitation and being used to study in vitro the interaction between cancer cells and blood vessels (Shi et al., 2020; Matsui et al., 2021). Concerning the intra-tumoral heterogeneity, this can be observed only when organoids are generated from tumor biopsies (Jacob et al., 2020). Finally, another challenge in the field remains to create brain cancer organoid with proper tumor microenvironment such as an immune compartment. Indeed, it would be worth establishing co-culture of brain cancer organoids (i.e., PDO) with different types of immune cells derived from patient peripheral blood or tumor biopsies (i.e., tumor infiltrating lymphocytes or tumor-associated macrophages/microglia). Alternatively, following a hiPSC-based approach, GEBOs might be co-cultured with iPSC-derived microglia (Xu et al., 2021).
Medulloblastoma (MB) is the most common malignant brain tumor in childhood that specifically arises in the cerebellum. The highest peak of incidence is during the first decades of life, even though this disease can occur throughout adulthood (Louis et al., 2016). Nowadays, an effective and definitive treatment has not yet been found, and 40% of affected children experience tumor recurrence, while 30% die from MB (Jones et al., 2012). Next-generation techniques have been applied to analyze MB molecular features/profiles. Genome-sequencing and array-based transcriptional profiling allowed the classification of MB into four main molecular subgroups: WNT, SHH, Group 3, and Group 4 (Taylor et al., 2012). The classification depends on a broad variety of macro and micro-genetic aberrations, which define specific cytogenetics, mutational patterns, gene expression signatures, and patient outcomes. Because of its molecular subtyping, here, we give an overview of the different models developed for each MB subgroup.
The gold GEMM of WNT MB comes from the efforts of Gibson et al. (2010), hypothesizing, for the first time, the cell of origin of the tumor. Human WNT MB are distributed within the IV ventricle and infiltrate the brainstem. Indeed, genes marking human WNT MB are more frequently expressed in the lower rhombic lip (LRL) and embryonic dorsal brainstem. For this reason, Gibson and collaborators generated Blbp-Cre+; Ctnnb1+/lox(ex3); Tp53flx/flx mice, characterized by a dominant mutation of the beta-catenin gene and loss of the tumor suppressor Tp53 in Blbp+ cells, which are Olig3+ progenitor cells in the LRL and progenitor cell populations across the hindbrain (including the cerebellar ventricular zone) (Gibson et al., 2010) (Figure 2B and Table 1). Blbp-Cre+; Ctnnb1+/lox(ex3); Tp53flx/flx mice developed MB, recapitulating the anatomy and gene expression profiles of human WNT MB, as well as the aberrant vasculature that interferes in the blood brain barrier formation. A damaged blood brain barrier could increase the drug’s efficacy, thus explaining the good prognosis and excellent response to chemotherapy compared to the other MB subgroups. These data indicate the similarities between the GEMMs and the human patients, showing their importance in defining new therapeutic approaches (Phoenix et al., 2016). A tumor suppressor role has been recently hypothesized for DDX3X (Patmore et al., 2020), often found mutated in WNT MB. DDX3X regulates hindbrain patterning, and its loss removes lineage restriction toward tumor formation, allowing for the onset of WNT MB from either lower or upper rhombic lips.
The first GEMM for SHH MB was a Ptch1+/− model, where aberrant SHH pathway activation leads to uncontrolled proliferation of cerebellar granule neuron progenitors (CGNPs) (Goodrich et al., 1997). Indeed, granule lineage identity is a prerequisite for the SHH MB onset, as it has been demonstrated by the conditional Ptch1 knockout in unipotent (Math1+) CGNPs (Schüller et al., 2008; Yang et al., 2008) and through the deregulation of different effectors of the SHH pathway, such as Smoothened and Sufu (Lee et al., 2007; Dey et al., 2012) (Figure 2B and Table 1). Based on mutations found in human SHH MB, a plethora of GEMMs have been generated using different approaches (Wetmore et al., 2001; Uziel et al., 2005) (listed in Table 1). A subgroup of patients with SHH MB shows somatic loss-of-function mutations of the transcriptional corepressor BCOR. Interestingly, BCOR is involved in MB formation (Tiberi et al., 2014; Kutscher et al., 2020) with possible therapeutic implications in BCOR mutant SHH MB. Zuckermann et al. (2015) have shown an interesting method to validate the oncogenic role of mutations found in human patients by performing postnatal somatic CRISPR/Cas-mediated deletion of tumor suppressor genes using a polyethylenimines-mediated in vivo transfection into the mouse neonatal cerebellum. Indeed, Ptch1 CRISPR/Cas-mediated deletion in mice characterized by homozygous deletion of Trp53 recapitulates MB (Zuckermann et al., 2015). SHH MB can be obtained by different cell types, other than CGNPs, such as cochlear granule neuron progenitors (CNPs) (Grammel et al., 2012). Recently, we have proposed postmitotic granule neurons as a possible origin for the human adult SHH MB. These cells can be reprogramed and give rise to tumors in the cerebellum of mice resembling human adult SHH MB (Aiello et al., 2019). In particular, the co-expression of mutant Brfp1 and SmoM2 alters chromatin accessibility of stem/progenitor-related genes, thus reprogramming the precursor cell properties and favoring the adult SHH MB tumorigenesis. However, whether the de-differentiation process of granule neurons is a key event in BRPF1-mutated SHH MB is still unknown and needs further investigation. Additionally, SHH GEMMs were brought to the discoveries of new important pathological features. Indeed, single-cell RNA sequencing on murine Shh MB treated with vismodegib (a Smo inhibitor) revealed some cell types (Sox2+ and Myod1+) resistant to treatment (Ocasio et al., 2019). A rare Sox2+ quiescent cell population, enriched after antimitotic chemotherapy, and Smoothened inhibition are thought to be responsible for tumor relapse (Vanner et al., 2014). Furthermore, the tumor sustains itself by shaping its surroundings to make it conducive to growth (Yao et al., 2020). CGNPs are, indeed, able to trans-differentiate into tumor astrocytes that sustain tumor progression by activating microglia via IL-4 production. Microglia, in turn, produce IGF-1 that promotes tumor progression. Among the mechanisms driving tumor resistance to conventional therapies and relapse, it has been shown the presence of a stem cell niche within SHH MB, namely cancer stem cells (CSCs), which express the stemness marker Nanog under Hh/Gli transcriptional regulation (Po et al., 2010; Miele et al., 2017; Abballe and Miele, 2021).
Modeling of Group 3 MB is more challenging due to the intratumoral heterogeneity and the similarities to Group 4 in the mutational profile. For years, the GTML mouse model has been considered as the golden standard model for Group 3 MB. This transgenic mouse is characterized by a Tet system that allows the expression of both MYCN and Luciferase under the control of the Glutamate transporter 1 (Glt1) promoter, reported to be expressed in hindbrain progenitors (Swartling et al., 2010). Since several high-risk Group 3 human patients showing relapse after treatment are characterized by MYC amplification and TP53 inactivating mutation, this model was exploited to verify the interaction between P53 and MYCN. GTML/Trp53KI/KI mice produce a more aggressive tumor that recapitulates the human large cell/anaplastic (LCA) histology and relapse (Hill et al., 2015). However, GTML-derived MB neurospheres show, in a small percentage, SHH-dependent features, thus not fully recapitulating the Group 3 MB human scenario (Swartling et al., 2012).
Due to the high diversity in the mutational landscape of Group 3 MB, we developed an in vivo screen approach to test putative tumor driver mutations among hits identified by exome sequencing and microarray data of human Group 3 patients with MB.
Either overexpressing oncogenes with the PiggyBac transposon system or by CRISPR/Cas9-mediated deletion of tumor suppressor genes, we found that Gfi1 + c-MYC and Otx2 + c-MYC genes overexpressions were able to mimic the histological and transcriptional profile of human Group 3 MB (Ballabio et al., 2020) (Figure 2B and Table 1). Moreover, the Otx2 + c-MYC combination of genes generated metastasis, recapitulating the malignancy of the tumor. Chromatin modifiers such as MLL4 (alias for KMT2D) are often mutated in Group 3 patients with MB, as already mentioned, and whether these could have a causative role in tumor formation has been tested. Nestin-cre; Mll4flox/flox mice lead to the downregulation of several tumor suppressors, such as Dnmt3 and Bcl6, and to Group 3 MB formation (Dhar et al., 2018). Despite the extensive genomic characterization, it is still unknown what the developmental origins of Group 3 MB are. We have hypothesized a critical role of Notch1 in tumor formation, claiming that its expression level in different progenitor cells impairs their competence in inducing MB. In particular, S100b+ cells show a higher level of Notch1 compared to Math1+ progenitor cells, and are able to initiate Group 3 MB upon Gfi1 + c-MYC overexpression, while Math1+ and Sox2+ cells do not initiate tumorigenesis (Ballabio et al., 2021). The overexpression of MYC in isolated TP53–/– CGNPs and their following orthotopic transplantation in nude mice is able to mimic some of the Group 3 MB clinical features, strongly suggesting their cooperation in determining a more aggressive behavior (Kawauchi et al., 2012; Pei et al., 2012). However, Group 3 MB features can be also obtained by passing the TP53 loss that is not often found within Group 3 tumors. Indeed, it has been shown that the overexpression of c-Myc, together with GFI1/GFI1B activation, allows to recapitulate Group 3 MB tumorigenesis (Northcott et al., 2014; Vo et al., 2017). A list of G3 GEMMs is presented in Table 1.
The Group 4 MB is the least explored and understood among the subgroups. A comparative analysis of protein phosphorylation levels between Group 3 and Group 4 leads to hypothesize a role of the receptor tyrosine kinase (RTK) pathway in the Group 4 MB onset; these findings were validated by the enrichment at the mRNA level of ERBB4, a well-established RTK member (Forget et al., 2018). SRC is a key regulator of the RTK pathway and was found upregulated at both mRNA and protein levels in Group 4. Moreover, in utero electroporation in the fourth ventricle of E13.5 mouse embryos of the active form of SRC, together with a dominant negative form of TP53, leads to Group 4 MB formation (Figure 2B and Table 1). However, TP53 seems not to be mutated in Group 4 MB, but iso-chromosome 17q is frequently present in these patients. SRC-driven tumors present distinct molecular features from MYC-derived tumors. Since ERBB4 and SRC were detectable in the nuclear transitory zone (NTZ) on embryonic Day 13.5 (E13.5), Forget et al. (2018) speculated that their expression could reflect Group 4 developmental origin. ScRNAseq analysis on human Group 4 MB indicated an enrichment of markers involved in neuronal differentiation, such as ENO2, SYT11, TUBB3, and MAP2, or in glutamatergic lineage specification, such as EOMES and LMX1A. These factors have been implicated in defining neuronal fates of unipolar brush cells (UBC) and glutamatergic cerebellar nuclei (GluCN) in the embryonic upper rhombic lip (Englund et al., 2006; Chizhikov et al., 2010; Hovestadt et al., 2019). Lin et al. (2016) found enrichment of super-enhancer activation of LMX1A, TBR2, and LHX2, which could support origin from progenitors in the embryonic upper rhombic lip. However, the developmental origin of Group 4 is still an open question, and further studies must be conducted to clarify the involvement of UBC, GluCN, or other cell types in the Group 4 MB onset. The identification of the cell of origin is a crucial step for the development of faithful and proper models for Group 4 MB.
As previously stated, mouse models show some limitations due to the different biological contexts and cannot fully recapitulate the complexity of human tumors (Gould et al., 2015). For this reason, PDX models emerged as an important tool to investigate subtype specific features of different pediatric brain tumors. A study conducted by the Children’s Oncology Group set the generation of 30 orthotopic pediatric brain tumor PDX models (Brabetz et al., 2018). An important effort in generating PDX models has been also achieved by the St. Jude Children’s Research Hospital by the generation of 37 novel orthotopic PDX models derived from patients with pediatric brain tumor (Smith et al., 2020). These new models include all the four MB subgroups (WNT, SHH, Group 3, and Group 4) that have been shown to maintain histological features of the original tumors and to genetically match the patients’ tumors. DNA methylation, transcriptional and histological analyses at early and late passages demonstrated the reliability of PDX models, providing a useful and valid resource to study cancer biology and to test novel and tailored therapeutic approaches.
Recently, it has been generated a humanized mouse model based on the orthotopic transplantation of human neuroepithelial stem cells (NES) (Huang et al., 2019). NES are multipotent stem cells able to differentiate into neurons with hindbrain identity. Orthotopic transplantation of NES transduced with MYCN leads to the formation of a human cancer phenotype as a powerful tool to dissect the processes of tumorigenesis. An alternative method to model SHH MB using human cells was described by Susanto et al. (2020) that performed an orthotopic transplantation of iPSC-derived NES from a Gorlin patient with a germline PTCH1 mutation. They followed tumor development by re-transplanting tumor-isolated NES (tNES) cells in nude mice and identifying LGALS1 as a putative new therapeutic target.
Similar to gliomas, 3D cerebellar organoid models have been shown to be an emerging important tool to study human medulloblastoma. Taking advantage of already-available exome sequencing data and after adapting a previously developed protocol for cerebellar organoid generation (Muguruma et al., 2015; Ballabio et al., 2020), the first organoid model of Group 3 MB has been generated by overexpressing a combination of Gfi1 + c-MYC and Otx2 + c-MYC oncogenes, identified previously as candidate driver genes in the same study by an in vivo mutagenesis screen. These Group 3 MB organoid models more closely mimic the histological, transcriptional, and DNA methylation profile of human Group 3 MB (Ballabio et al., 2020, 2021) (Figure 2B and Table 1), suggesting that they could be a more suitable platform for high-throughput drug testing and development of personalized therapies (Qian et al., 2019). Future studies could be done to develop models for the other MB subtypes by mutagenesis or by engraftment of patient-derived tumor cells. These could then allow us to study with more precision the mechanisms at the origin of the tumor formation and how the mutations found in the patients’ tumors drive tumorigenesis with unprecedented details in a human tissue. This could then lead to the identification of new potential targets for therapies and for diseases that, to date, cannot be tackled. It is, indeed, also possible that the human brain, due to its species-specific differences, could have different susceptibility to disease and brain tumors (Watanabe et al., 2017; Kanton et al., 2019; Eichmüller et al., 2022), a reason why studying mechanisms of tumorigenesis in human tissue becomes essential.
Ependymoma can originate in the brain or spinal cord. In the brain, ependymomas are thought to originate from ependymal cells lining in the ventricles. Histologically, ependymomas are classified into four groups: subependymoma (WHO Grade I), myxopapillary ependymoma and classic ependymoma (WHO Grade II), and anaplastic ependymoma (WHO Grade III), of which classic and anaplastic ependymoma are the most common subtypes in children (Figure 1). Over 90% of pediatric ependymomas arise in the infratentorial and supratentorial regions. Supratentorial (ST) ependymomas in children have two major subgroups: RELA fusion-positive ependymoma and YAP1 fusion-positive ependymoma. Ependymoma mouse models have been generated by RELAFUS1 fusion gene expression in Nestin+, Gfap+ or Blbp+ cells in the mouse brain. These tumors recapitulate some of the histology and the transcriptome panel of human ependymomas (Ozawa et al., 2018). The YAP1-MAMLD1 fusion gene delivered to mice by in utero electroporation drives tumor formation, and tumors share histological and molecular characteristics of human ependymoma (Pajtler et al., 2019). Recently, Eder et al. (2020) have reported that ectopic expression of active nuclear YAP1 (nlsYAP5SA) or conditional deletion of YAP1’s negative regulators Lats1 and Lats2 kinases in neural progenitor cells of the ventricular zone also induced tumors with molecular characteristics of human ependymoma. PDXs derived from pediatric ependymoma showed to maintain a genetic, molecular, and histological similarity to the parental tumors, opening the possibility to have an additional model for investigating and treating such a disease (Brabetz et al., 2018; Smith et al., 2020). No organoid-based models have been reported for human ependymoma (Table 1).
Meningioma is another common type of tumors located in the brain, which originates from the meninges. Meningiomas, representing the most frequent tumor in the adult, are generally benign (WHO Grade I) with a lower percentage classified as atypical (WHO Grade II) and rarely anaplastic (WHO Grade III) (Boetto et al., 2021). The genomic profile of a large cohort of meningiomas has identified alteration in the genes encoding for NF2, SMARCB1, SMARCE1, TRAF7, KLF4, POLR2A, BAP1, and members of the PI3K and Hedgehog pathways (Youngblood et al., 2019). The mouse model of meningiomas consisted in xenograft of immortalized cell lines or patient-derived tumor cells (McCutcheon et al., 2000; Püttmann et al., 2005; Cargioli et al., 2007; Ragel et al., 2008), GEMMs (Kalamarides et al., 2002, 2011; Peyre et al., 2013, 2015; Boetto et al., 2018, 2021) and syngeneic allogenic graft (Boetto et al., 2021) (Table 1). Meningiomas can be induced in GEMMs overexpressing PDGFB in a context of loss of function of Nf2, Cdkn2ab or p16Ink4a or over-expressing only SmoM2 PGDS+ arachnoid cells. Recently, PDO has been generated also from meningioma biopsies (Chan et al., 2021; Yamazaki et al., 2021). Its histological and molecular characterizations positively confirmed the high similarity between the parental tumor and PDOs (Yamazaki et al., 2021).
The main goal of the different models is to be used for the design of new therapeutical approaches and then being translated into clinical practice for glioma treatments. Concerning high-grade glioma (including GBM), the standard chemotherapeutic treatment is based on temozolomide, while lower grade gliomas relies on procarbazine, lomustine, and vincristine or temozolomide. Despite the enormous number of pre-clinical in vivo and in vitro models, the drugs used for treating gliomas (from Grade II to Grade IV) have remained the same over the last decades. Indeed, this negative and low progression of developing new drugs against glioma has also pushed the field to test already approved drugs originally designed for other diseases (Lyne and Yamini, 2021). This approach, called drug repositioning, has the advantage to have drugs already being tested for their safety, making eventually the path to a clinical trial to be faster. Furthermore, this approach could be coupled to the possibility to generate, expand, and Biobank organoids derived from histologically and molecularly different tumors (Golebiewska et al., 2020; Jacob et al., 2020) in order to have a more “personalized” and specific drug screen.
Current treatment for MB consists of surgery, radio- and chemotherapy; nevertheless, after surgery, patients suffer from severe psychosocial, neurocognitive, and neuro-endocrine deficits. Present studies are focused on the development of target therapies that take into account the molecular differences among the subgroups (Northcott et al., 2019; Thompson et al., 2020). In the scenario, the development of in vitro and in vivo models is crucial. In general, despite the models developed for brain cancer are increasing in terms of numbers, complexity, and the degree of similarity to the original tumor, the drugs available for treating the disease are always the same. What has really improved tough is the knowledge of the genetics and biology of the different types of brain cancer. This could be considered the starting point for rethinking about new pharmacological, immunological, and genetic therapies.
Genomic, transcriptomic, and proteomic analyses on human brain tumor patients have greatly increased our knowledge and understanding of the signaling pathways regulating the different cancer subtypes. Through the exploitation of these data, several models have been created to mimic the pathogenesis and to gain knowledge about the molecular mechanisms of the tumors. All the brain tumor models developed so far have shown some pitfalls in the correct modeling of the disease and the human translation of the findings (Figure 2). The first issue is related to the genetics, with all the mouse models and hiPSCs derived-brain organoid, relying only on a few of the genetic abnormalities found in patients with brain tumor. In this direction, it will be worth expanding the combination of genetic abnormalities inducing tumor in mice and organoids performing large genetic screens. Drosophila represents a valid complementary model to be used for performing a “low-cost” initial genetic screen for a new putative cancer driver. Secondly, the drug screen platforms used so far have mostly relied on 2D-cultured tumor cell lines. Therefore, the use of hiPSCs-derived brain organoid represents a unique platform for medium/high throughput screening of new molecules to hinder tumor progression. On the other hand, organoid cultures have some limitations. The lack of vascularization leads to reduced oxygenation and access to nutrients, with the consequent formation of an inner necrotic core in the tissue when cultures are kept for long-term studies. The absence of immune cells and microglia makes the studies of tumor-microenvironment preferable in vivo. Some limitations can be overcome at the current state-of-the-art by transplanting human organoids in the mouse brain (Mansour et al., 2018; Ballabio et al., 2020; Bhaduri et al., 2020). Furthermore, the ability to cultivate the original human primary tumor with PDO will be a breakthrough that will provide the possibility to personalize the treatments. Recently, patient-derived xenografts (PDXs) have been shown to be the most advanced model, recapitulating the human disease. These models might be used to model frequent as well as rare childhood brain tumor entities, including HGG, MB, anaplastic ependymoma, atypical teratoid rhabdoid tumor, and embryonal tumor (Brabetz et al., 2018; Smith et al., 2020). PDOX models will be valuable platforms for evaluating novel therapies and conducting pre-clinical trials to accelerate progress in the treatment of pediatric brain tumors. However, the usage of immunocompromised mice does not allow to investigate the interaction between tumor cells and immune system. Moreover, since PDO and PDX are derived from tumor biopsies, they should not be considered suitable for investigating the neoplastic transformation of brain cells. In this review, we depicted an overview of the in vitro and in vivo brain tumor models used in the past together with the most technologically advanced ones. Considering the entire pros and cons described in this review, the best model system must be chosen, depending on the specific biological question that needs to be addressed.
FA, GA, AS, and LT made substantial contributions to researching data for the article, to discussions of content, and to writing the manuscript. EM and LA reviewed and edited the manuscript before submission. All authors contributed to the article and approved the submitted version.
LT gratefully acknowledges financial support from the Armenise-Harvard Foundation, AIRC, CARITRO, Pezcoller Foundation and EMBO. FA has received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska Curie, with grant agreement No. 844677, and Fondazione Umberto Veronesi. GA was supported by a FIRC-AIRC fellowship for Italy. EM was supported by Italian Ministry of Health, Ricerca Finalizzata, No. GR-2018-12367328. AS and LA were supported by Fondazione Umberto Veronesi.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all the members of the Armenise-Harvard Laboratory of Brain Disorders and Cancer for helpful discussions.
Abballe, L., and Miele, E. (2021). Epigenetic modulators for brain cancer stem cells: implications for anticancer treatment. World J. Stem Cells 13, 670–684. doi: 10.4252/wjsc.v13.i7.670
Aday, S., Cecchelli, R., Hallier-Vanuxeem, D., Dehouck, M. P., and Ferreira, L. (2016). Stem cell-based human blood–brain barrier models for drug discovery and delivery. Trends Biotechnol. 34, 382–393. doi: 10.1016/j.tibtech.2016.01.001
Ahir, B. K., Engelhard, H. H., and Lakka, S. S. (2020). Tumor development and angiogenesis in adult brain tumor: glioblastoma. Mol. Neurobiol. 57, 2461–2478. doi: 10.1007/s12035-020-01892-8
Aiello, G., Ballabio, C., Ruggeri, R., Fagnocchi, L., Anderle, M., Morassut, I., et al. (2019). Truncated BRPF1 cooperates with smoothened to promote adult Shh medulloblastoma. Cell Rep. 29, 4036.e10–4052.e10.
Akter, F., Simon, B., de Boer, N. L., Redjal, N., Wakimoto, H., and Shah, K. (2021). Pre-clinical tumor models of primary brain tumors: challenges and opportunities. Biochim. Biophys. Acta. Rev. Cancer 1875:188458. doi: 10.1016/j.bbcan.2020.188458
Aldape, K., Brindle, K. M., Chesler, L., Chopra, R., Gajjar, A., Gilbert, M. R., et al. (2019). Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 16, 509–520. doi: 10.1038/s41571-019-0177-5
Ausman, J. I., Shapiro, W. R., and Rall, D. P. (1970). Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res. 30, 2394–2400.
Azzarelli, R., McNally, A., Dell’Amico, C., Onorati, M., Simons, B., and Philpott, A. (2022). ASCL1 phosphorylation and ID2 upregulation are roadblocks to glioblastoma stem cell differentiation. Sci. Rep. 12:2341. doi: 10.1038/s41598-022-06248-x
Azzarelli, R., Ori, M., Philpott, A., and Simons, B. D. (2021). Three-dimensional model of glioblastoma by co-culturing tumor stem cells with human brain organoids. Biol. Open 10:bio056416. doi: 10.1242/bio.056416
Ballabio, C., Anderle, M., Gianesello, M., Lago, C., Miele, E., Cardano, M., et al. (2020). Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 11, 1–18. doi: 10.1038/s41467-019-13989-3
Ballabio, C., Gianesello, M., Lago, C., Okonechnikov, K., Anderle, M., Aiello, G., et al. (2021). Notch1 switches progenitor competence in inducing medulloblastoma. Sci. Adv. 7:eabd2781. doi: 10.1126/sciadv.abd2781
Bell, A. J., McBride, S. M. J., and Dockendorff, T. C. (2009). Flies as the ointment: drosophila modeling to enhance drug discovery. Fly (Austin) 3, 39–49. doi: 10.4161/fly.3.1.7774
Bhaduri, A., Andrews, M. G., Mancia Leon, W., Jung, D., Shin, D., Allen, D., et al. (2020). Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148. doi: 10.1038/s41586-020-1962-0
Bian, S., Repic, M., Guo, Z., Kavirayani, A., Burkard, T., Bagley, J. A., et al. (2018). Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 15, 631–639. doi: 10.1038/s41592-018-0070-7
Boetto, J., Apra, C., Bielle, F., Peyre, M., and Kalamarides, M. (2018). Selective vulnerability of the primitive meningeal layer to prenatal Smo activation for skull base meningothelial meningioma formation. Oncogene 37, 4955–4963. doi: 10.1038/s41388-018-0328-7
Boetto, J., Peyre, M., and Kalamarides, M. (2021). Mouse models in meningioma research: a systematic review. Cancers 13:3712. doi: 10.3390/cancers13153712
Brabetz, S., Leary, S. E. S., Gröbner, S. N., Nakamoto, M. W., Şeker-Cin, H., Girard, E. J., et al. (2018). A biobank of patient-derived pediatric brain tumor models. Nat. Med. 24, 1752–1761. doi: 10.1038/s41591-018-0207-3
Brennan, C. W., Verhaak, R. G. W., McKenna, A., Campos, B., Noushmehr, H., Salama, S. R., et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. doi: 10.1016/j.cell.2013.09.034
Bressan, R. B., Southgate, B., Ferguson, K. M., Blin, C., Grant, V., Alfazema, N., et al. (2021). Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell 28, 877.e9–893.e9. doi: 10.1016/j.stem.2021.01.016
Breznik, B., Motaln, H., Vittori, M., Rotter, A., and Turnšek, T. L. (2017). Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 8, 25482–25499. doi: 10.18632/oncotarget.16041
Cargioli, T. G., Ugur, H. C., Ramakrishna, N., Chan, J., Black, P. M., and Carroll, R. S. (2007). Establishment of an in vivo meningioma model with human telomerase reverse transcriptase. Neurosurgery 60, 750–760. doi: 10.1227/01.NEU.0000255397.00410.8F
Casey, M. J., and Stewart, R. A. (2020). Pediatric cancer models in zebrafish. Trends Cancer 6, 407–418. doi: 10.1016/j.trecan.2020.02.006
Caussinus, E., and Gonzalez, C. (2005). Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat. Genet. 37, 1125–1129. doi: 10.1038/ng1632
Chan, H. S. C., Ng, H. K., Chan, A. K. Y., Cheng, S. H., Chow, C., Wong, N., et al. (2021). Establishment and characterization of meningioma patient-derived organoid. J. Clin. Neurosci. 94, 192–199. doi: 10.1016/j.jocn.2021.10.035
Chen, X., Wanggou, S., Bodalia, A., Zhu, M., Dong, W., Fan, J. J., et al. (2018). A feedforward mechanism mediated by mechanosensitive ion channel PIEZO1 and tissue mechanics promotes glioma aggression. Neuron 100, 799.e7–815.e7. doi: 10.1016/j.neuron.2018.09.046
Cheng, P., Wang, J., Waghmare, I., Sartini, S., Coviello, V., Zhang, Z., et al. (2016). FOXD1-ALDH1A3 signaling is a determinant for the self-renewal and tumorigenicity of mesenchymal glioma stem cells. Cancer Res. 76, 7219–7230. doi: 10.1158/0008-5472.CAN-15-2860
Chi, K.-C., Tsai, W.-C., Wu, C.-L., Lin, T.-Y., and Hueng, D.-Y. (2018). An adult drosophila glioma model for studying pathometabolic pathways of gliomagenesis. Mol. Neurobiol. 56, 4589–4599. doi: 10.1007/s12035-018-1392-2
Chia, K., Keatinge, M., Mazzolini, J., and Sieger, D. (2019). Brain tumours repurpose endogenous neuron to microglia signalling mechanisms to promote their own proliferation. Elife 8:e46912. doi: 10.7554/eLife.46912
Chia, K., Mazzolini, J., Mione, M., and Sieger, D. (2018). Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. Elife 7:e31918. doi: 10.7554/eLife.31918
Chizhikov, V. V., Lindgren, A. G., Mishima, Y., Roberts, R. W., Aldinger, K. A., Miesegaes, G. R., et al. (2010). Lmx1a regulates fates and location of cells originating from the cerebellar rhombic lip and telencephalic cortical hem. Proc. Natl. Acad. Sci. U.S.A. 107, 10725–10730. doi: 10.1073/pnas.0910786107
da Silva, B., Mathew, R. K., Polson, E. S., Williams, J., and Wurdak, H. (2018). Spontaneous glioblastoma spheroid infiltration of early-stage cerebral organoids models brain tumor invasion. SLAS Discov. 23, 862–868. doi: 10.1177/2472555218764623
Darmanis, S., Sloan, S. A., Croote, D., Mignardi, M., Chernikova, S., Samghababi, P., et al. (2017). Single-Cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21, 1399–1410. doi: 10.1016/j.celrep.2017.10.030
Day, C., Merlino, G., and Van Dyke, T. (2015). Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53. doi: 10.1016/j.cell.2015.08.068
Dey, J., Ditzler, S., Knoblaugh, S. E., Hatton, B. A., Schelter, J. M., Cleary, M. A., et al. (2012). A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol. Cell. Biol. 32, 4104–4115. doi: 10.1128/MCB.00862-12
Dhar, S., Zhao, D., Lin, T., Gu, B., Pal, K., Wu, S. J., et al. (2018). MLL4 Is Required to maintain broad H3K4me3 peaks and super-enhancers at tumor suppressor genes. Mol. Cell 70, 825.e6–841.e6. doi: 10.1016/j.molcel.2018.04.028
Eder, E., Roncaroli, F., Domart, M. C., Horswell, S., Andreiuolo, F., Flynn, H. R., et al. (2020). YAP1/TAZ drives ependymoma-like tumour formation in mice. Nat. Commun. 11:2380.
Eichmüller, O. L., Corsini, N. S., Vértesy, A., Morassut, I., Scholl, T., Gruber, V.-E., et al. (2022). Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science 375:eabf5546. doi: 10.1126/science.abf5546
Englund, C., Kowalczyk, T., Daza, R. A. M., Dagan, A., Lau, C., Rose, M. F., et al. (2006). Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J. Neurosci. 26, 9184–9195. doi: 10.1523/JNEUROSCI.1610-06.2006
Fareh, M., Turchi, L., Virolle, V., Debruyne, D., Almairac, F., de-la-Forest Divonne, S., et al. (2012). The miR 302-367 cluster drastically affects self-renewal and infiltration properties of glioma-initiating cells through CXCR4 repression and consequent disruption of the SHH-GLI-NANOG network. Cell Death Differ. 19, 232–244. doi: 10.1038/cdd.2011.89
Forget, A., Martignetti, L., Puget, S., Calzone, L., Brabetz, S., Picard, D., et al. (2018). Aberrant ERBB4-SRC signaling as a hallmark of Group 4 medulloblastoma revealed by integrative phosphoproteomic profiling. Cancer Cell 34, 379.e7–395.e7. doi: 10.1016/j.ccell.2018.08.002
Gangoso, E., Southgate, B., Bradley, L., Rus, S., Galvez-Cancino, F., McGivern, N., et al. (2021). Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184, 2454.e26–2470.e26. doi: 10.1016/j.cell.2021.03.023
Gangwani, K., Snigdha, K., and Kango-Singh, M. (2020). Tep1 regulates Yki activity in neural stem cells in drosophila glioma model. Front. Cell Dev. Biol. 8:306. doi: 10.3389/fcell.2020.00306
Gao, Y., de Wit, M., Struys, E. A., van der Linde, H. C. Z., Salomons, G. S., Lamfers, M. L. M., et al. (2018). IDH1-mutated transgenic zebrafish lines: an in-vivo model for drug screening and functional analysis. PLoS One 13:e0199737. doi: 10.1371/journal.pone.0199737
Gibson, P., Tong, Y., Robinson, G., Thompson, M. C., Currle, D. S., Eden, C., et al. (2010). Subtypes of medulloblastoma have distinct developmental origins. Nature 468, 1095–1099. doi: 10.1038/nature09587
Golebiewska, A., Hau, A.-C., Oudin, A., Stieber, D., Yabo, Y. A., Baus, V., et al. (2020). Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 140, 919–949. doi: 10.1007/s00401-020-02226-7
Gómez-López, S., Lerner, R. G., and Petritsch, C. (2013). Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell. Mol. Life Sci. 71, 575–597. doi: 10.1007/s00018-013-1386-1
Goodrich, L. V., Milenkoviæ, L., Higgins, K. M., and Scott, M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113. doi: 10.1126/science.277.5329.1109
Goranci-Buzhala, G., Mariappan, A., Gabriel, E., Ramani, A., Ricci-Vitiani, L., Buccarelli, M., et al. (2020). Rapid and efficient invasion assay of glioblastoma in human brain organoids. Cell Rep. 31:107738. doi: 10.1016/j.celrep.2020.107738
Gould, S. E., Junttila, M. R., and de Sauvage, F. J. (2015). Translational value of mouse models in oncology drug development. Nat. Med. 21, 431–439. doi: 10.1038/nm.3853
Grammel, D., Warmuth-Metz, M., von Bueren, A. O., Kool, M., Pietsch, T., and Kretzschmar, H. A. (2012). Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 123, 601–614. doi: 10.1007/s00401-012-0961-0
Gronych, J., Korshunov, A., Bageritz, J., Milde, T., Jugold, M., Hambardzumyan, D., et al. (2011). An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J. Clin. Invest. 121, 1344–1348. doi: 10.1172/JCI44656
Haag, D., Mack, N., Benites Goncalves da Silva, P., Statz, B., Clark, J., Tanabe, K., et al. (2021). H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell 39, 407.e13–422.e13. doi: 10.1016/j.ccell.2021.01.005
Hakes, A. E., and Brand, A. H. (2020). Tailless/TLX reverts intermediate neural progenitors to stem cells driving tumourigenesis via repression of asense/ASCL1. Elife 9:e53377. doi: 10.7554/eLife.53377
Hambardzumyan, D., Amankulor, N. M., Helmy, K. Y., Becher, O. J., and Holland, E. C. (2009). Modeling adult gliomas using RCAS/t-va technology. Transl. Oncol. 2, 89–95. doi: 10.1593/tlo.09100
Hede, S.-M., Hansson, I., Afink, G. B., Eriksson, A., Nazarenko, I., Andrae, J., et al. (2009). GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 57, 1143–1153. doi: 10.1002/glia.20837
Hicks, W., Bird, C. E., Traylor, J. I., Shi, D. D., Ahmadieh, T. Y. E., and Richardson, T. E. (2021). Contemporary mouse models in glioma research. Cells 10:712. doi: 10.3390/cells10030712
Hill, R. M., Kuijper, S., Lindsey, J. C., Petrie, K., Schwalbe, E. C., Barker, K., et al. (2015). Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 27, 72–84. doi: 10.1016/j.ccell.2014.11.002
Ho, V. K. Y., Reijneveld, J. C., Enting, R. H., Bienfait, H. P., Robe, P., Baumert, B. G., et al. (2014). Changing incidence and improved survival of gliomas. Eur. J. Cancer 50, 2309–2318. doi: 10.1016/j.ejca.2014.05.019
Hoffmann, N., Fernández, V., Cruz Pereira, R., Rancati, S., Pelizzoli, R., and De Pietri Tonelli, D. (2020). A Xenotransplant model of human brain tumors in wild-type mice. iScience 23:100813. doi: 10.1016/j.isci.2019.100813
Holland, E. C., Hively, W. P., DePinho, R. A., and Varmus, H. E. (1998). A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 12:3675. doi: 10.1101/gad.12.23.3675
Hovestadt, V., Smith, K. S., Bihannic, L., Filbin, M. G., Shaw, M. K. L., Baumgartner, A., et al. (2019). Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 572, 74–79. doi: 10.1038/s41586-019-1434-6
Huang, M., Tailor, J., Zhen, Q., Gillmor, A. H., Miller, M. L., Weishaupt, H., et al. (2019). Engineering genetic predisposition in human neuroepithelial stem cells recapitulates medulloblastoma tumorigenesis. Cell Stem Cell. 25, 433.e7–446.e7. doi: 10.1016/j.stem.2019.05.013
Huang, Y. C., Lin, S. J., Shih, H. Y., Chou, C. H., Chu, H. H., Chiu, C. C., et al. (2017). Epigenetic regulation of NOTCH1 and NOTCH3 by KMT2A inhibits glioma proliferation. Oncotarget 8, 63110–63120. doi: 10.18632/oncotarget.18668
Hubert, C. G., Rivera, M., Spangler, L. C., Wu, Q., Mack, S. C., Prager, B. C., et al. (2016). A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 76, 2465–2477. doi: 10.1158/0008-5472.CAN-15-2402
Jacob, F., Salinas, R. D., Zhang, D. Y., Nguyen, P. T. T., Schnoll, J. G., Wong, S. Z. H., et al. (2020). A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188.e22–204.e22. doi: 10.1016/j.cell.2019.11.036
Jacob, L., Sawma, P., Garnier, N., Meyer, L. A. T., Fritz, J., Hussenet, T., et al. (2016). Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 7, 57851–57865. doi: 10.18632/oncotarget.11072
Jandial, R., Neman, J., Lim, P. P., Tamae, D., Kowolik, C. M., Wuenschell, G. E., et al. (2018). Inhibition of GLO1 in glioblastoma multiforme increases DNA-AGEs, stimulates RAGE expression, and inhibits brain tumor growth in orthotopic mouse models. Int. J. Mol. Sci. 19:406. doi: 10.3390/ijms19020406
Jones, D., Jäger, N., Kool, M., Zichner, T., Hutter, B., Sultan, M., et al. (2012). Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105. doi: 10.1038/nature11284
Joo, K. M., Kim, J., Jin, J., Kim, M., Seol, H. J., Muradov, J., et al. (2013). Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 3, 260–273. doi: 10.1016/j.celrep.2012.12.013
Ju, B., Chen, W., Orr, B. A., Spitsbergen, J. M., Jia, S., Eden, C. J., et al. (2015). Oncogenic KRAS promotes malignant brain tumors in zebrafish. Mol. Cancer 14:18. doi: 10.1186/s12943-015-0288-2
Ju, B., Chen, W., Spitsbergen, J. M., Lu, J., Vogel, P., Peters, J. L., et al. (2014). Activation of Sonic hedgehog signaling in neural progenitor cells promotes glioma development in the zebrafish optic pathway. Oncogenesis 3:e96. doi: 10.1038/oncsis.2014.10
Jung, I. H., Leem, G. L., Jung, D. E., Kim, M. H., Kim, E. Y., Kim, S. H., et al. (2013). Glioma is formed by active Akt1 alone and promoted by active Rac1 in transgenic zebrafish. Neuro. Oncol. 15, 290–304. doi: 10.1093/neuonc/nos387
Kalamarides, M., Niwa-Kawakita, M., Leblois, H., Abramowski, V., Perricaudet, M., Janin, A., et al. (2002). Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 16, 1060–1065. doi: 10.1101/gad.226302
Kalamarides, M., Stemmer-Rachamimov, A. O., Niwa-Kawakita, M., Chareyre, F., Taranchon, E., Han, Z.-Y., et al. (2011). Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 30, 2333–2344. doi: 10.1038/onc.2010.609
Kanton, S., Boyle, M. J., He, Z., Santel, M., Weigert, A., Sanchís-Calleja, F., et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. doi: 10.1038/s41586-019-1654-9
Kawauchi, D., Robinson, G., Uziel, T., Gibson, P., Rehg, J., Gao, C., et al. (2012). A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21, 168–180. doi: 10.1016/j.ccr.2011.12.023
Kaye, A. H., Morstyn, G., Gardner, I., and Pyke, K. (1986). Development of a Xenograft glioma model in mouse brain. Cancer Res. 46, 1367–1373.
Kerstetter-Fogle, A. E., Harris, P. L. R., Brady-Kalnay, S. M., and Sloan, A. E. (2020). Generation of glioblastoma patient-derived intracranial xenografts for preclinical studies. Int. J. Mol. Sci. 21:5113. doi: 10.3390/ijms21145113
Kim, S. N., Jeibmann, A., Halama, K., Witte, H. T., Wälte, M., Matzat, T., et al. (2014). ECM stiffness regulates glial migration in Drosophila and mammalian glioma models. Development 141, 3233–3242. doi: 10.1242/dev.106039
Kirchberger, S., Sturtzel, C., Pascoal, S., and Distel, M. (2017). Quo natas, Danio?-Recent progress in modeling cancer in Zebrafish. Front. Oncol. 7:186. doi: 10.3389/fonc.2017.00186
Koschmann, C., Calinescu, A.-A., Nunez, F. J., Mackay, A., Fazal-Salom, J., Thomas, D., et al. (2016). ATRX loss promotes tumor growth and impairs non-homologous end joining DNA repair in Glioma. Sci. Transl. Med. 8:328ra28. doi: 10.1126/scitranslmed.aac8228
Krieger, T. G., Tirier, S. M., Park, J., Jechow, K., Eisemann, T., Peterziel, H., et al. (2020). Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro. Oncol. 22, 1138–1149. doi: 10.1093/neuonc/noaa091
Kutscher, L., Okonechnikov, K., Batora, N. V., Clark, J., Silva, P. B. G., Vouri, M., et al. (2020). Functional loss of a noncanonical BCOR-PRC1.1 complex accelerates SHH-driven medulloblastoma formation. Genes Dev. 34, 1161–1176. doi: 10.1101/gad.337584.120
Kwon, C.-H., Zhao, D., Chen, J., Alcantara, S., Li, Y., Burns, D. K., et al. (2008). Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 68, 3286–3294. doi: 10.1158/0008-5472.CAN-07-6867
Lai, Y. J., Tsai, J. C., Tseng, Y. T., Wu, M. S., Liu, W. S., Lam, H. I., et al. (2017). Small G protein Rac GTPases regulate the maintenance of glioblastoma stem-like cells in vitro and in vivo. Oncotarget 8, 18031–18049. doi: 10.18632/oncotarget.14949
Lam, S. H., Chua, H. L., Gong, Z., Lam, T. J., and Sin, Y. M. (2004). Development and maturation of the immune system in zebrafish, Danio rerio: a gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 28, 9–28. doi: 10.1016/s0145-305x(03)00103-4
Lan, X., Jörg, D. J., Cavalli, F. M. G., Richards, L. M., Nguyen, L. V., Vanner, R. J., et al. (2017). Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 549, 227–232. doi: 10.1038/nature23666
Lee, Y., Kawagoe, R., Sasai, K., Li, Y., Russell, H. R., Curran, T., et al. (2007). Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 26, 6442–6447. doi: 10.1038/sj.onc.1210467
Li, Z., and Langhans, S. A. (2021). In Vivo and Ex Vivo pediatric brain tumor models: an overview. Front. Oncol. 11:620831. doi: 10.3389/fonc.2021.620831
Lin, C. Y., Erkek, S., Tong, Y., Yin, L., Federation, A. J., Zapatka, M., et al. (2016). Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. doi: 10.1038/nature16546
Lindberg, N., Kastemar, M., Olofsson, T., Smits, A., and Uhrbom, L. (2009). Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28, 2266–2275. doi: 10.1038/onc.2009.76
Linkous, A., Balamatsias, D., Snuderl, M., Edwards, L., Miyaguchi, K., Milner, T., et al. (2019). Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 26, 3203.e5–3211.e5. doi: 10.1016/j.celrep.2019.02.063
Llaguno, S. A., Chen, J., Kwon, C. H., Jackson, E. L., Li, Y., Burns, D. K., et al. (2009). Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15, 45–56. doi: 10.1016/j.ccr.2008.12.006
Llaguno, S. A., Sun, D., Pedraza, A. M., Vera, E., Wang, Z., Burns, D. K., et al. (2019). Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 22, 545–555. doi: 10.1038/s41593-018-0333-8
Llaguno, S. R. A., Wang, Z., Sun, D., Chen, J., Xu, J., Kim, E., et al. (2015). Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell 28, 429–440. doi: 10.1016/j.ccell.2015.09.007
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., et al. (2016). The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. doi: 10.1007/s00401-016-1545-1
Louis, D. N., Perry, A., Wesseling, P., Brat, D. J., Cree, I. A., Figarella-Branger, D., et al. (2021). The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro. Oncol. 23, 1231–1251.
Lyne, S. B., and Yamini, B. (2021). An alternative pipeline for glioblastoma therapeutics: a systematic review of drug repurposing in Glioblastoma. Cancers (Basel) 13:1953. doi: 10.3390/cancers13081953
Mansour, A. A., Gonçalves, J. T., Bloyd, C. W., Li, H., Fernandes, S., Quang, D., et al. (2018). An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441. doi: 10.1038/nbt.4127
Marumoto, T., Tashiro, A., Friedmann-Morvinski, D., Scadeng, M., Soda, Y., Gage, F. H., et al. (2009). Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 15, 110–116. doi: 10.1038/nm.1863
Matsui, T. K., Tsuru, Y., Hasegawa, K., and Kuwako, K. (2021). Vascularization of human brain organoids. Stem Cells 39, 1017–1024. doi: 10.1002/stem.3368
Mayrhofer, M., Gourain, V., Reischl, M., Affaticati, P., Jenett, A., Joly, J. S., et al. (2017). A novel brain tumour model in zebrafish reveals the role of YAP activation in MAPK- and PI3K-induced malignant growth. Dis. Model. Mech. 10, 15–28. doi: 10.1242/dmm.026500
McCutcheon, I. E., Friend, K. E., Gerdes, T. M., Zhang, B. M., Wildrick, D. M., and Fuller, G. N. (2000). Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J. Neurosurg. 92, 306–314. doi: 10.3171/jns.2000.92.2.0306
Miele, E., Po, A., Begalli, F., Antonucci, L., Mastronuzzi, A., Marras, C. E., et al. (2017). β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 17:488. doi: 10.1186/s12885-017-3477-0
Minata, M., Audia, A., Shi, J., Lu, S., Bernstock, J., Pavlyukov, M. S., et al. (2019). Phenotypic plasticity of invasive edge glioma stem-like cells in response to ionizing radiation. Cell Rep. 26, 1893.e7–1905.e7. doi: 10.1016/j.celrep.2019.01.076
Miroshnikova, Y., Mouw, J. K., Barnes, J. M., Pickup, M. W., Lakins, J. N., Kim, Y., et al. (2016). Tissue mechanics promote IDH1-dependent HIF1α-tenascin C feedback to regulate glioblastoma aggression. Nat. Cell Biol. 18, 1336–1345. doi: 10.1038/ncb3429
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., and Sasai, Y. (2015). Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10, 537–550. doi: 10.1016/j.celrep.2014.12.051
Narayanan, A., Gagliardi, F., Gallotti, A. L., Mazzoleni, S., Cominelli, M., Fagnocchi, L., et al. (2019). The proneural gene ASCL1 governs the transcriptional subgroup affiliation in glioblastoma stem cells by directly repressing the mesenchymal gene NDRG1. Cell Death Differ. 26, 1813–1831. doi: 10.1038/s41418-018-0248-7
Nayler, S., Agarwal, D., Curion, F., Bowden, R., and Becker, E. B. E. (2021). High-resolution transcriptional landscape of xeno-free human induced pluripotent stem cell-derived cerebellar organoids. Sci. Rep. 11:12959. doi: 10.1038/s41598-021-91846-4
Neftel, C., Laffy, J., Filbin, M. G., Hara, T., Shore, M. E., Rahme, G. J., et al. (2019). An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835.e21–849.e21. doi: 10.1016/j.cell.2019.06.024
Northcott, P. A., Lee, C., Zichner, T., Stütz, A. M., Erkek, S., Kawauchi, D., et al. (2014). Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434. doi: 10.1038/nature13379
Northcott, P. A., Robinson, G. W., Kratz, C. P., Mabbott, D. J., Pomeroy, S. L., Clifford, S. C., et al. (2019). Medulloblastoma. Nat. Rev. Dis. Prim. 5:11.
Núñez, F. J., Mendez, F. M., Kadiyala, P., Alghamri, M. S., Savelieff, M. G., Garcia-Fabiani, M. B., et al. (2019). IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 11:eaaq1427. doi: 10.1126/scitranslmed.aaq1427
Ocasio, J., Babcock, B., Malawsky, D., Weir, S. J., Loo, L., Simon, J. M., et al. (2019). scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 10, 1–17. doi: 10.1038/s41467-019-13657-6
Ogawa, J., Pao, G. M., Shokhirev, M. N., and Verma, I. M. (2018). Glioblastoma model using human cerebral organoids. Cell Rep. 23, 1220–1229. doi: 10.1016/j.celrep.2018.03.105
Omuro, A., and DeAngelis, L. M. (2013). Glioblastoma and other malignant gliomas: a clinical review. JAMA 310, 1842–1850. doi: 10.1001/jama.2013.280319
Oppel, F., Tao, T., Shi, H., Ross, K. N., Zimmerman, M. W., He, S., et al. (2019). Loss of atrx cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 15:e1008039. doi: 10.1371/journal.pgen.1008039
Osswald, M., Jung, E., Sahm, F., Solecki, G., Venkataramani, V., Blaes, J., et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98. doi: 10.1038/nature16071
Ozawa, T., Arora, S., Szulzewsky, F., Juric-Sekhar, G., Miyajima, Y., Bolouri, H., et al. (2018). A de novo mouse model of C11orf95-RELA fusion-driven ependymoma identifies driver functions in addition to NF-κB. Cell Rep. 23, 3787–3797. doi: 10.1016/j.celrep.2018.04.099
Pajtler, K., Wei, Y., Okonechnikov, K., Silva, P. B. G., Vouri, M., Zhang, L., et al. (2019). YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat. Commun. 10:3914. doi: 10.1038/s41467-019-11884-5
Park, H.-J., Kim, J.-K., Jeon, H.-M., Oh, S.-Y., Kim, S.-H., Nam, D.-H., et al. (2010). The neural stem cell fate determinant TLX promotes tumorigenesis and genesis of cells resembling glioma stem cells. Mol. Cells 30, 403–408. doi: 10.1007/s10059-010-0122-z
Park, N., Guilhamon, P., Desai, K., McAdam, R. F., Langille, E., O’Connor, M., et al. (2017). ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells. Cell Stem Cell 21, 209.e7–224.e7.
Patel, A. P., Tirosh, I., Trombetta, J. J., Shalek, A. K., Gillespie, S. M., Wakimoto, H., et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (80) 344, 1396–1401. doi: 10.1126/science.1254257
Patmore, D., Jassim, A., Nathan, E., Gilbertson, R. J., Tahan, D., Hoffmann, N., et al. (2020). DDX3X suppresses the susceptibility of hindbrain lineages to medulloblastoma. Dev. Cell 54, 455.e5–470.e5. doi: 10.1016/j.devcel.2020.05.027
Pei, Y., Moore, C. E., Wang, J., Tewari, A. K., Eroshkin, A., Cho, Y. J., et al. (2012). An animal model of MYC-driven medulloblastoma. Cancer Cell 21, 155–167. doi: 10.1016/j.ccr.2011.12.021
Peyre, M., Salaud, C., Clermont-Taranchon, E., Niwa-Kawakita, M., Goutagny, S., Mawrin, C., et al. (2015). PDGF activation in PGDS-positive arachnoid cells induces meningioma formation in mice promoting tumor progression in combination with Nf2 and Cdkn2ab loss. Oncotarget 6, 32713–32722. doi: 10.18632/oncotarget.5296
Peyre, M., Stemmer-Rachamimov, A., Clermont-Taranchon, E., Quentin, S., El-Taraya, N., Walczak, C., et al. (2013). Meningioma progression in mice triggered by Nf2 and Cdkn2ab inactivation. Oncogene 32, 4264–4272. doi: 10.1038/onc.2012.436
Phillips, H. S., Kharbanda, S., Chen, R., Forrest, W. F., Soriano, R. H., Wu, T. D., et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173. doi: 10.1016/j.ccr.2006.02.019
Phoenix, T., Patmore, D. M., Boop, S., Boulos, N., Jacus, M. O., Patel, Y. T., et al. (2016). Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell 29, 508–522. doi: 10.1016/j.ccell.2016.03.002
Pine, A. R., Cirigliano, S. M., Nicholson, J. G., Hu, Y., Linkous, A., Miyaguchi, K., et al. (2020). Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov. 10, 964–979. doi: 10.1158/2159-8290.CD-20-0057
Po, A., Ferretti, E., Miele, E., Smaele, E., Paganelli, A., Canettieri, G., et al. (2010). Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 29, 2646–2658. doi: 10.1038/emboj.2010.131
Pudelko, L., Edwards, S., Balan, M., Nyqvist, D., Al-Saadi, J., Dittmer, J., et al. (2018). An orthotopic glioblastoma animal model suitable for high-throughput screenings. Neuro. Oncol. 20, 1475–1484. doi: 10.1093/neuonc/noy071
Püttmann, S., Senner, V., Braune, S., Hillmann, B., Exeler, R., Rickert, C. H., et al. (2005). Establishment of a benign meningioma cell line by hTERT-mediated immortalization. Lab. Invest. 85, 1163–1171. doi: 10.1038/labinvest.3700307
Qian, X., Song, H., and Ming, G. L. (2019). Brain organoids: advances, applications and challenges. Development 146:dev166074. doi: 10.1242/dev.166074
Ragel, B. T., Elam, I. L., Gillespie, D. L., Flynn, J. R., Kelly, D. A., Mabey, D., et al. (2008). A novel model of intracranial meningioma in mice using luciferase-expressing meningioma cells. Laboratory investigation. J. Neurosurg. 108, 304–310. doi: 10.3171/JNS/2008/108/2/0304
Rampazzo, E., Persano, L., Pistollato, F., Moro, E., Frasson, C., Porazzi, P., et al. (2013). Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 4:e500. doi: 10.1038/cddis.2013.32
Read, R. D., Cavenee, W. K., Furnari, F. B., and Thomas, J. B. (2009). A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 5:e1000374. doi: 10.1371/journal.pgen.1000374
Read, R., Fenton, T. R., Gomez, G. G., Wykosky, J., Vandenberg, S. R., Babic, I., et al. (2013). A kinome-wide RNAi screen in Drosophila Glia reveals that the RIO kinases mediate cell proliferation and survival through TORC2-Akt signaling in glioblastoma. PLoS Genet. 9:e1003253. doi: 10.1371/journal.pgen.1003253
Reilly, K. M. (2009). Brain tumor susceptibility: the role of genetic factors and uses of mouse models to unravel risk. Brain Pathol. 19, 121–131. doi: 10.1111/j.1750-3639.2008.00236.x
Reiter, L. T., Potocki, L., Chien, S., Gribskov, M., and Bier, E. (2001). A systematic analysis of human disease-associated gene sequences in drosophila melanogaster. Genome Res. 11, 1114–1125. doi: 10.1101/gr.169101
Rodrigues, J., Heinrich, M. A., Teixeira, L. M., and Prakash, J. (2021). 3D In Vitro Model (R)evolution: unveiling tumor-stroma interactions. Trends Cancer 7, 249–264. doi: 10.1016/j.trecan.2020.10.009
Royet, A., Broutier, L., Coissieux, M. M., Malleval, C., Gadot, N., Maillet, D., et al. (2017). Ephrin-B3 supports glioblastoma growth by inhibiting apoptosis induced by the dependence receptor EphA4. Oncotarget 8, 23750–23759. doi: 10.18632/oncotarget.16077
Sasaki, A., Ishiuchi, S., Kanda, T., Hasegawa, M., and Nakazato, Y. (2001). Analysis of interleukin-6 gene expression in primary human gliomas, glioblastoma xenografts, and glioblastoma cell lines. Brain Tumor Pathol. 18, 13–21. doi: 10.1007/BF02478920
Schüller, U., Heine, V. M., Mao, J., Kho, A. T., Dillon, A. K., Han, Y. G., et al. (2008). Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-Induced medulloblastoma. Cancer Cell 14, 123–134. doi: 10.1016/j.ccr.2008.07.005
Seligman, A. M., Shear, M. J., and Alexander, L. (1939). Studies in Carcinogenesis: VIII. Experimental production of brain tumors in mice with methylcholanthrene. Am. J. Cancer 37, 364–395.
Shi, Y., Sun, L., Wang, M., Liu, J., Zhong, S., Li, R., et al. (2020). Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 18:e3000705. doi: 10.1371/journal.pbio.3000705
Shin, J., Padmanabhan, A., De Groh, E. D., Lee, J. S., Haidar, S., Dahlberg, S., et al. (2012). Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis. Model. Mech. 5, 881–894. doi: 10.1242/dmm.009779
Shu, Q., Wong, K. K., Su, J. M., Adesina, A. M., Yu, L. T., Tsang, Y. T. M., et al. (2008). Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells 26, 1414–1424. doi: 10.1634/stemcells.2007-1009
Singh, S., Hawkins, C., Clarke, I. D., Squire, J. A., and Hide, J. B. T. (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401. doi: 10.1038/nature03128
Smith, K. S., Xu, K., Mercer, K. S., Boop, F., Klimo, P., DeCupyere, M., et al. (2020). Patient-derived orthotopic xenografts of pediatric brain tumors: a St. Jude resource. Acta Neuropathol. 140, 209–225. doi: 10.1007/s00401-020-02171-5
Sumiyoshi, K., Koso, H., and Watanabe, S. (2018). Spontaneous development of intratumoral heterogeneity in a transposon-induced mouse model of glioma. Cancer Sci. 109, 1513–1523. doi: 10.1111/cas.13579
Susanto, E., Navarro, A. M., Zhou, L., Sundström, A., van Bree, N., Stantic, M., et al. (2020). Modeling SHH-driven medulloblastoma with patient iPS cell-derived neural stem cells. Proc. Natl. Acad. Sci. U.S.A. 117, 20127–20138. doi: 10.1073/pnas.1920521117
Swartling, F. J., Grimmer, M. R., Hackett, C. S., Northcott, P. A., Fan, Q. W., Goldenberg, D. D., et al. (2010). Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 24, 1059–1072. doi: 10.1101/gad.1907510
Swartling, F. J., Savov, V., Persson, A. I., Chen, J., Hackett, C. S., Northcott, P. A., et al. (2012). Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 21, 601–613. doi: 10.1016/j.ccr.2012.04.012
Tang, M., Xie, Q., Gimple, R. C., Zhong, Z., Tam, T., Tian, J., et al. (2020). Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 30, 833–853. doi: 10.1038/s41422-020-0338-1
Taylor, M. D., Northcott, P. A., Korshunov, A., Remke, M., Cho, Y.-J., Clifford, S. C., et al. (2012). Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 123:465. doi: 10.1007/s00401-011-0922-z
Thomas, R. S., Susil, B., and Kola, I. (1994). Activating point mutations of the neu oncogene in schwannomas induced by ethylnitrosourea exposure to day 15 and day 18 fetal rats. Int. J. Oncol. 5, 1219–1225. doi: 10.3892/ijo.5.6.1219
Thompson, E. M., Ashley, D., and Landi, D. (2020). Current medulloblastoma subgroup specific clinical trials. Transl. Pediatr. 9, 157–162. doi: 10.21037/tp.2020.03.03
Tiberi, L., Bonnefont, J., van den Ameele, J., Le Bon, S. D., Herpoel, A., and Bilheu, A. (2014). A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing Sonic Hedgehog signaling. Cancer Cell 26, 797–812. doi: 10.1016/j.ccell.2014.10.021
Uhrbom, L., Kastemar, M., Johansson, F. K., Westermark, B., and Holland, E. C. (2005). Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 65, 2065–2069. doi: 10.1158/0008-5472.CAN-04-3588
Uziel, T., Zindy, F., Xie, S., Lee, Y., Forget, A., Magdaleno, S., et al. (2005). The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 19, 2656–2667. doi: 10.1101/gad.1368605
Vanner, R. J., Remke, M., Gallo, M., Selvadurai, H. J., Coutinho, F., Lee, L., et al. (2014). Quiescent Sox2+Cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 26, 33–47. doi: 10.1016/j.ccr.2014.05.005
Vargas-Patron, L. A., Agudelo-Dueñãs, N., Madrid-Wolff, J., Venegas, J. A., González, J. M., Forero-Shelton, M., et al. (2019). Xenotransplantation of Human glioblastoma in Zebrafish larvae: in vivo imaging and proliferation assessment. Biol. Open 8:bio043257. doi: 10.1242/bio.043257
Velasco, S., Kedaigle, A. J., Simmons, S. K., Nash, A., Rocha, M., Quadrato, G., et al. (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527. doi: 10.1038/s41586-019-1289-x
Verhaak, R. G. W., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M. D., et al. (2010). An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 17:98. doi: 10.1016/j.ccr.2009.12.020
Vittori, M., Breznik, B., Hrovat, K., Kenig, S., and Lah, T. T. (2017). RECQ1 helicase silencing decreases the tumour growth rate of U87 glioblastoma cell xenografts in Zebrafish embryos. Genes (Basel) 8:222. doi: 10.3390/genes8090222
Vo, B. H. T., Li, C., Morgan, M. A., Theurillat, I., Finkelstein, D., Wright, S., et al. (2017). Inactivation of Ezh2 Upregulates Gfi1 and drives aggressive myc-driven group 3 medulloblastoma. Cell Rep. 18, 2907–2917. doi: 10.1016/j.celrep.2017.02.073
Watanabe, M., Buth, J. E., Vishlaghi, N., de la Torre-Ubieta, L., Taxidis, J., Khakh, B. S., et al. (2017). Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep. 21, 517–532. doi: 10.1016/j.celrep.2017.09.047
Wei, Q., Clarke, L., Scheidenhelm, D. K., Qian, B., Tong, A., Sabha, N., et al. (2006). High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 66, 7429–7437. doi: 10.1158/0008-5472.CAN-06-0712
Wetmore, C., Eberhart, D. E., and Curran, T. (2001). Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 61, 513–516.
Willoughby, L. F., Schlosser, T., Manning, S. A., Parisot, J. P., Street, I. P., Richardson, H. E., et al. (2013). An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis. Model. Mech. 6, 521–529. doi: 10.1242/dmm.009985
Xu, R., Boreland, A. J., Li, X., Erickson, C., Jin, M., Atkins, C., et al. (2021). Developing human pluripotent stem cell-based cerebral organoids with a controllable microglia ratio for modeling brain development and pathology. Stem Cell Rep. 16, 1923–1937. doi: 10.1016/j.stemcr.2021.06.011
Yamamoto, S., Jaiswal, M., Charng, W. L., Gambin, T., Karaca, E., Mirzaa, G., et al. (2014). A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159, 200–214. doi: 10.1016/j.cell.2014.09.002
Yamazaki, S., Ohka, F., Hirano, M., Shiraki, Y., Motomura, K., Tanahashi, K., et al. (2021). Newly established patient-derived organoid model of intracranial meningioma. Neuro-Oncology 23, 1936–1948. doi: 10.1093/neuonc/noab155
Yan, C., Brunson, D. C., Tang, Q., Do, D., Iftimia, N. A., Moore, J. C., et al. (2019). Visualizing engrafted human cancer and therapy responses in immunodeficient Zebrafish. Cell 177, 1903.e14–1914.e14. doi: 10.1016/j.cell.2019.04.004
Yang, X. J., Cui, W., Gu, A., Xu, C., Yu, S. C., Li, T. T., et al. (2013a). A novel zebrafish xenotransplantation model for study of glioma stem cell invasion. PLoS One 8:e61801. doi: 10.1371/journal.pone.0061801
Yang, X. J., Chen, G. L., Yu, S. C., Xu, C., Xin, Y. H., Li, T. T., et al. (2013b). TGF-β1 enhances tumor-induced angiogenesis via JNK pathway and macrophage infiltration in an improved zebrafish embryo/xenograft glioma model. Int. Immunopharmacol. 15, 191–198. doi: 10.1016/j.intimp.2012.12.002
Yang, Z. J., Ellis, T., Markant, S. L., Read, T. A., Kessler, J. D., Bourboulas, M., et al. (2008). Medulloblastoma can be initiated by deletion of patched in lineage-restricted progenitors or stem cells. Cancer Cell 14, 135–145. doi: 10.1016/j.ccr.2008.07.003
Yao, M., Ventura, P. B., Jiang, Y., Rodriguez, F. J., Wang, L., Perry, J. S. A., et al. (2020). Astrocytic trans-differentiation completes a multicellular paracrine feedback loop required for medulloblastoma tumor growth. Cell 180, 502.e19–520.e19. doi: 10.1016/j.cell.2019.12.024
Yi, H.-G., Jeong, Y. H., Kim, Y., Choi, Y.-J., Moon, H. E., Park, S. H., et al. (2019). A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 3, 509–519. doi: 10.1038/s41551-019-0363-x
Yi, Y., Hsieh, I.-Y., Huang, X., Li, J., and Zhao, W. (2016). Glioblastoma stem-like cells: characteristics, microenvironment, and therapy. Front. Pharmacol. 7:477. doi: 10.3389/fphar.2016.00477
Youngblood, M. W., Duran, D., Montejo, J. D., Li, C., Omay, S. B., Özduman, K., et al. (2019). Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J. Neurosurg. 133, 1345–1354. doi: 10.3171/2019.8.JNS191266
Zhang, P., Xia, Q., Liu, L., Li, S., and Dong, L. (2020). Current opinion on molecular characterization for GBM classification in guiding clinical diagnosis, prognosis, and therapy. Front. Mol. Biosci. 7:562798. doi: 10.3389/fmolb.2020.562798
Zhu, H., Acquaviva, J., Ramachandran, P., Boskovitz, A., Woolfenden, S., Pfannl, R., et al. (2009). Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 2712–2716. doi: 10.1073/pnas.0813314106
Keywords: organoid, mouse, Drosophila, xenograft, model, cancer, zebrafish
Citation: Antonica F, Aiello G, Soldano A, Abballe L, Miele E and Tiberi L (2022) Modeling Brain Tumors: A Perspective Overview of in vivo and Organoid Models. Front. Mol. Neurosci. 15:818696. doi: 10.3389/fnmol.2022.818696
Received: 19 November 2021; Accepted: 23 March 2022;
Published: 30 May 2022.
Edited by:
Parthiv Haldipur, Seattle Children’s Research Institute, United StatesReviewed by:
Massimo Zollo, University of Naples Federico II, ItalyCopyright © 2022 Antonica, Aiello, Soldano, Abballe, Miele and Tiberi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Tiberi, bHVjYS50aWJlcmlAdW5pdG4uaXQ=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.