 Ying Yang1
Ying Yang1 Xueyang Niu1
Xueyang Niu1 Miaomiao Cheng1
Miaomiao Cheng1 Qi Zeng2
Qi Zeng2 Jie Deng3
Jie Deng3 Xiaojuan Tian3
Xiaojuan Tian3 Yi Wang4Jing Yu5Wenli Shi6Wenjuan Wu7Jiehui Ma8Yufen Li9
Yi Wang4Jing Yu5Wenli Shi6Wenjuan Wu7Jiehui Ma8Yufen Li9 Xiaoling Yang1Xiaoli Zhang10
Xiaoling Yang1Xiaoli Zhang10 Tianming Jia10
Tianming Jia10 Zhixian Yang1
Zhixian Yang1 Jianxiang Liao2Yan Sun5
Jianxiang Liao2Yan Sun5 Hong Zheng6
Hong Zheng6 Suzhen Sun7
Suzhen Sun7 Dan Sun8
Dan Sun8 Yuwu Jiang1
Yuwu Jiang1 Yuehua Zhang1*
Yuehua Zhang1*
- 1Department of Pediatrics, Peking University First Hospital, Beijing, China
- 2Department of Neurology, Shenzhen Children’s Hospital, Shenzhen, China
- 3Department of Neurology, Beijing Children’s Hospital, Capital Medical University, Beijing, China
- 4Department of Neurology, National Children’s Medical Center, Children’s Hospital of Fudan University, Shanghai, China
- 5Department of Neurology, Children’s Hospital of Xinjiang Uygur Autonomous Region, Xinjiang Hospital of Beijing Children’s Hospital, Ürümqi, China
- 6Department of Pediatrics, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, China
- 7Department of Neurology, Hebei Children’s Hospital, Shijiazhuang, China
- 8Department of Neurology, Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 9Department of Pediatrics, Linyi People’s Hospital, Linyi, China
- 10Department of Pediatrics, The Third Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Objective: This study aimed to obtain a comprehensive understanding of the genetic and phenotypic aspects of GABRG2-related epilepsy and its prognosis and to explore the potential prospects for personalized medicine.
Methods: Through a multicenter collaboration in China, we analyzed the genotype-phenotype correlation and antiseizure medication (ASM) of patients with GABRG2-related epilepsy. The three-dimensional protein structure of the GABRG2 variant was modeled to predict the effect of GABRG2 missense variants using PyMOL 2.3 software.
Results: In 35 patients with GABRG2 variants, 22 variants were de novo, and 18 variants were novel. The seizure onset age was ranged from 2 days after birth to 34 months (median age: 9 months). The seizure onset age was less than 1 year old in 22 patients (22/35, 62.9%). Seizure types included focal seizures (68.6%), generalized tonic-clonic seizures (60%), myoclonic seizures (14.3%), and absence seizures (11.4%). Other clinical features included fever-sensitive seizures (91.4%), cluster seizures (57.1%), and developmental delay (45.7%). Neuroimaging was abnormal in 2 patients, including dysplasia of the frontotemporal cortex and delayed myelination of white matter. Twelve patients were diagnosed with febrile seizures plus, eleven with epilepsy and developmental delay, two with Dravet syndrome, two with developmental and epileptic encephalopathy, two with focal epilepsy, two with febrile seizures, and four with unclassified epilepsy. The proportions of patients with missense variants in the extracellular region and the transmembrane region exhibiting developmental delay were 40% and 63.2%, respectively. The last follow-up age ranged from 11 months to 17 years. Seizures were controlled in 71.4% of patients, and 92% of their seizures were controlled by valproate and/or levetiracetam.
Conclusion: The clinical features of GABRG2-related epilepsy included seizure onset, usually in infancy, and seizures were fever-sensitive. More than half of the patients had cluster seizures. Phenotypes of GABRG2-related epilepsy were ranged from mild febrile seizures to severe epileptic encephalopathies. Most patients with GABRG2 variants who experienced seizures had a good prognosis. Valproate and levetiracetam were effective treatments for most patients.
Introduction
Epilepsy is characterized by an enduring predisposition to generate epileptic seizures. Numerous genetic defects underlying different forms of epilepsy have been identified with most of these genes encoding ion channel proteins (Surguchov et al., 2017). GABRG2 (OMIM:137164) resides on chromosome 5q34 and is a member of the gamma-aminobutyric acid-A (GABAA) receptor gene family of heteromeric pentameric ligand-gated ion channels. GABAA receptor subunits contains a extracellular N-terminus, four transmembrane domains, a small extracellular loop between the second and third transmembrane domains and a larger intracellular loop between the third and the fourth transmembrane domains (Figure 1). The first variant in GABRG2 (p.R43Q) was reported to be related to genetic epilepsy with febrile seizures (GEFS+) in 2001 in an Australian family (Wallace et al., 2001). In the same year, Baulac et al. (2001) identified a GABRG2 heterozygous missense variant (K328M) in a 3-generation French family with variable seizure phenotypes and most consistent with GEFS+. With the broad application of next-generation sequencing (NGS) in patients with epilepsy, pathogenic variants of GABRG2 related to developmental and epileptic encephalopathy 74 (DEE74, OMIM:618396) were reported. To our knowledge, no detailed descriptions of the phenotypic spectrum of patients with epilepsy carrying GABRG2 variants are available. Therefore, this study aimed to determine the phenotypic spectrum of epilepsy patients carrying GABRG2 variants and the prognosis of patients in a Chinese cohort from multicenter.
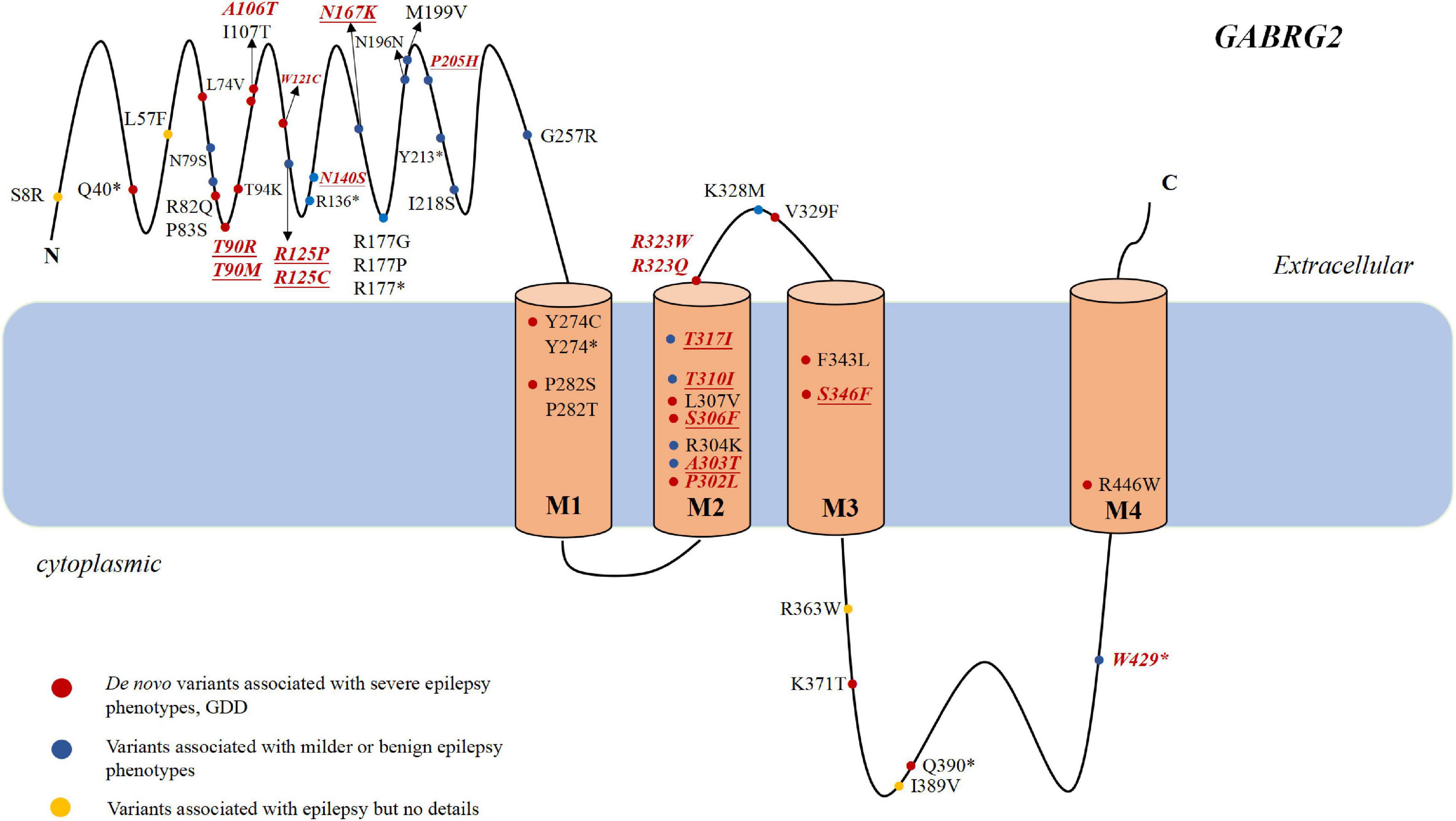
Figure 1. The genotype and phenotype of GABRG2 pathogenic variants in patients with epilepsy. Representation of the GABAA receptor protein structure and the location of GABRG2 variants previously described in the literature (marked in black) and identified in our cohort (marked in red and italics). The novel variants reported for the first time in the present study are indicated in underscored. The red dots indicate severe phenotypes, including epileptic encephalopathy and developmental delay along with epilepsy. The blue dots indicate mild phenotypes, including febrile seizure plus and GEFS+. The yellow dots indicate the variants associated with epilepsy but no details are available. *Represents the nonsense variant.
Materials and Methods
Participants
Genetic testing was conducted in all patients diagnosed with epilepsy without acquired factors (e.g., perinatal brain injury, traumatic brain injury, and central nervous system infections). Thirty-five patients with epilepsy and GABRG2 variants who attended the Pediatric Department of Peking University First Hospital from January 2008 to August 2021 were included in this study, including 22 males and 13 females. Clinical information about the age at seizure onset, seizure frequency, seizure types, developmental milestones, neurological status, family history, the results of accessory examinations including electroencephalogram (EEG) and brain magnetic resonance imaging (MRI), and treatment were collected in the clinic. Brain MRI and video EEG were reviewed by neuroradiologists and neurophysiologists, respectively. All patients identified with GABRG2 variants were followed in outpatient settings or by telephone at least every 3 months.
The Ethics Committee of Peking University First Hospital and the institutional review boards of collaborating groups approved the project (approval number 2012[453]). Written informed consent was obtained from the parents of all patients. The study was performed in accordance with the ethical standards established in the 1964 Declaration of Helsinki and its later amendments.
Genetic Analysis
GABRG2 variations (NM_000816, GRCh37/hg19) were determined using targeted next-generation sequencing of epilepsy (epilepsy gene panel) or whole-exome sequencing. Synonymous variants and single nucleotide polymorphisms with minor allele frequencies greater than 5% were removed1. Functional consequences were predicted by Mutation Taster2, Polyphen-23, and PROVEAN4. The pathogenicity of variants was evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). Sanger sequencing was used to verify variations and identify the inheritance of variants.
Structural Modeling
Three-dimensional protein structure were modeled to predict the effect of GABRG2 missense variants using PyMOL 2.3 software, and the pathogenicity of candidate variants was evaluated.
Results
Genetic Analysis
Our cohort of 35 patients carried 24 unique GABRG2 variants, including 27 missense variants, 5 splicing variants, 1 nonsense variant, 1 frameshift variant, and 1 small deletion variant (Table 1 and Supplementary Table 1). Eighteen variants were novel (p.T90M, p.W121C, p.R125P, p.R125C, p.N140S, p.N167K, p.P205H, c.631 + 4A > G, c.631 + 5G > T, c.922 + 1G > T, p.A303T, p.S306F, p.T310I, p.T317I, c.1128 + 5G > A, p.C382Sfs*57, c.1249-7C > T, and exon1-11 deletion). Six variants (p.T90R, p.A106T, p.P302L, p.R323W, p.R323Q and p.W429X) have been previously reported (Sun et al., 2008; Shen et al., 2017; Zou et al., 2017; Hernandez et al., 2021). Variants T310I, A106T, and R323W recurred twice, and R323Q recurred nine times, respectively. Twenty-two patients carried de novo variants and 13 patients had inherited variants. None of these variants were found in the Genome Aggregation Database.
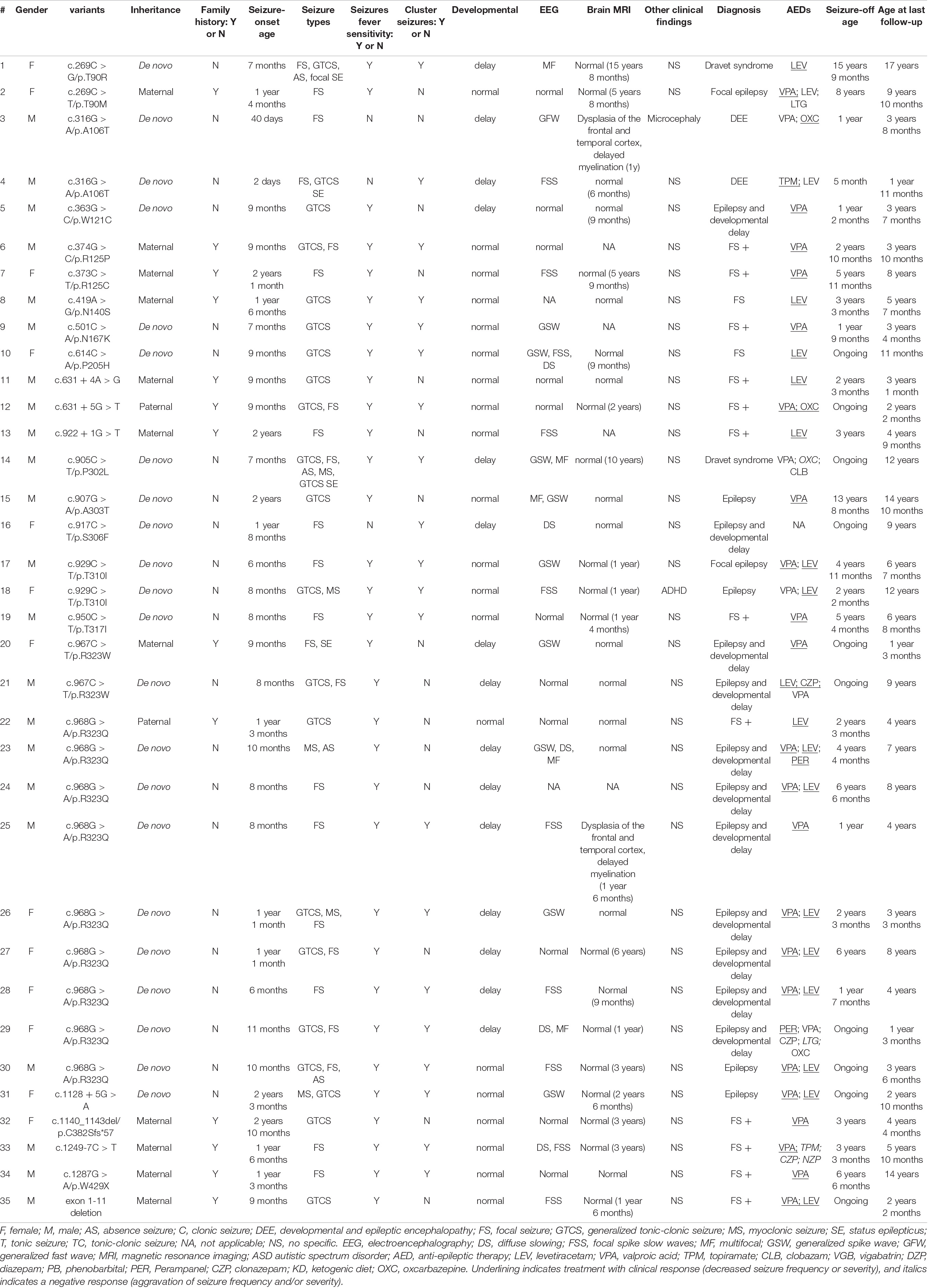
Table 1. The phenotype and genotype of 35 patients with GABRG2 variants in our cohort.
Seizure Types
In 35 children with epilepsy carrying GABRG2 variants, the seizure onset age ranged from 2 days to 34 months of age (median age: 9 months). The seizure onset age was less than 1 year old in 22 patients (22/35, 62.9%). Seizure types were predominantly focal seizures (24/35, 68.6%). Other seizure types included generalized tonic-clonic seizures (21/35, 60%), myoclonic seizures (5/35, 14.3%), and absence seizures (4/35, 11.4%). A striking feature was the occurrence of fever-sensitive seizures in 32/35 (91.4%) patients. Notably, 57.1% (20/35) patients had a cluster of seizures.
Neurodevelopment and Additional Comorbidities
In our cohort, 15 patients (42.8%, 15/35) experienced developmental delays. Two patients (patients 3, and 4) were unable to raise their heads at the age of 1 year. One patient (patient 29) walked after the age of 2 years. Five patients (patients 1, 14, 18, 24, and 25) walked at approximately the age of 18 months but had poor motor coordination. Patient 1 expressed herself in simple language with no more than four words at the age of 16 years. Patient 13 had a mild speech impediment, and he spoke disfluently at 6 years of age. Patient 14 also had learning difficulties. Eight patients had a mild intellectual disability (patients 5, 16, 20, 21, 23, 26, 27, and 28), and some of them had learning difficulties in school (patients 16, 21, 23, and 27). Microcephaly was observed in 1 patient (patient 3). One patient (patient 18) was diagnosed with attention deficit and hyperactivity disorder (ADHD). Twenty patients showed normal psychomotor development at the last follow-up.
Video Electroencephalogram and Brain Imaging
Thirty-five patients underwent video EEG monitoring for 4–24 h (Table 1). The EEG exhibited diffuse slow background activity in 5 patients. Interictal epileptiform discharges were captured in 24 patients, including multifocal spike-slow waves in 5 patients and focal spike-slow waves in 10 patients. Generalized spike-wave or polyspike wave discharges were observed in 9 patients. Seizures were observed in 7 (20%, 7/35) patients, consisting of focal seizures in 3, absence seizures in 3, and myoclonic seizures in one patient. Eleven patients had a normal EEG.
Brain MRI was abnormal in 2 patients (2/31, 6.5%), including dysplasia of the frontotemporal cortex and delayed myelination of white matter in 2 patients (patient 3 and patient 25). The brain MRI of 29 patients was normal at the last follow-up. The brain MRI results of patient 3 are shown in Figure 2.
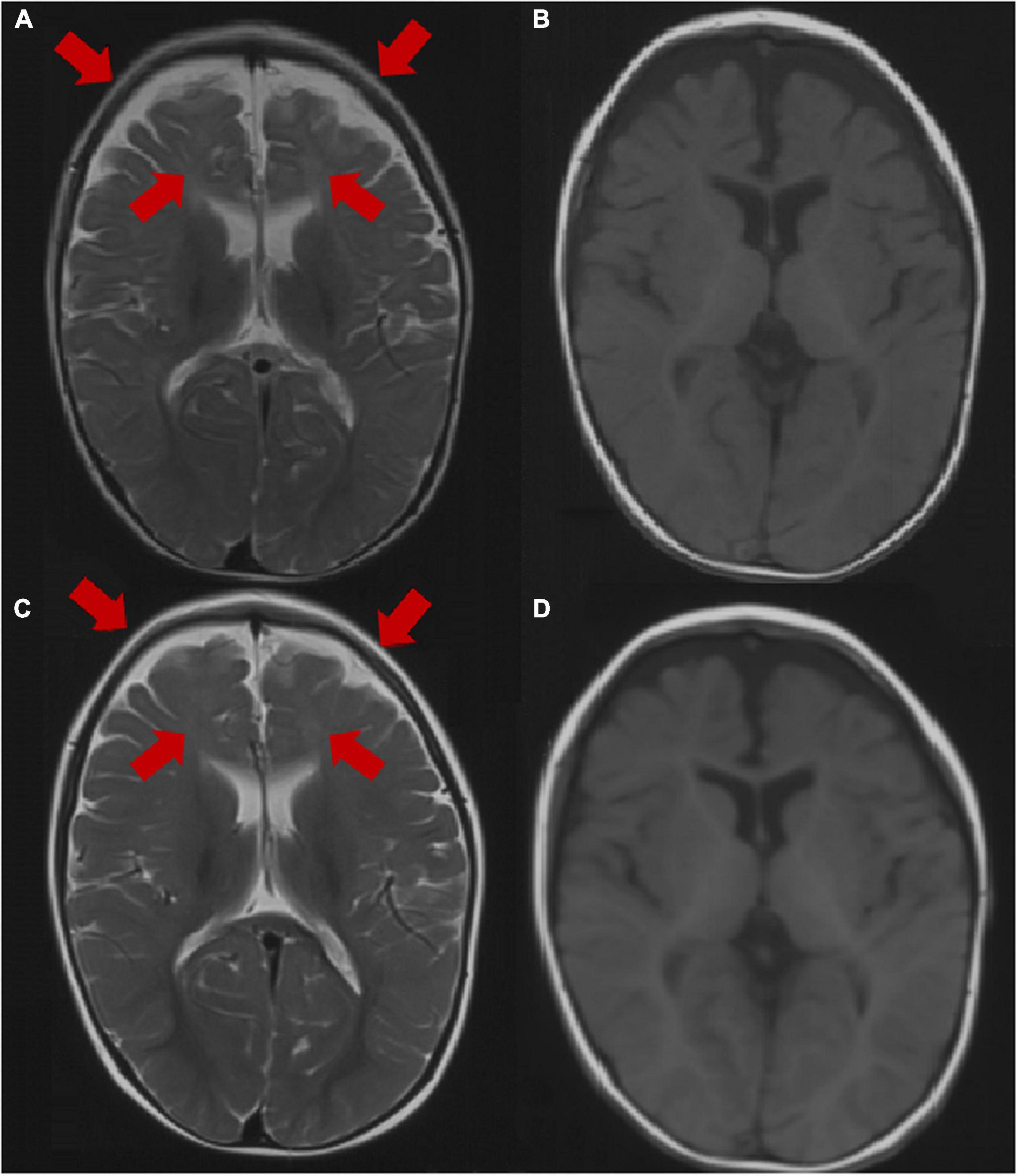
Figure 2. Brain MRI image of patient 3 at the age of 1 year. (A,B) Axial images (T1WI and T2WI) showing dysplasia of the frontal and temporal cortex and delayed myelination. Brain MRI image of patient 3 at the age of 2 years. (C,D) Axial images (T1WI, T2WI) showing dysplasia of the frontal cortex and delayed myelination. The arrow points to the lesion.
Diagnosis of the Phenotype
Twelve of 35 patients were diagnosed with FS+. Eleven patients were diagnosed with developmental delays and epilepsy, and their seizure onset ages ranged from 6 to 20 months. Two patients were diagnosed with Dravet syndrome. They had various seizure types including focal seizures, GTCS, myoclonic seizures, atypical absence seizures, and seizures with fever-sensitivity. Two patients (patients 3 and 4) were diagnosed with DEE, and their seizure onset ages were 2 days and 40 days. Two patients (patients 2 and 17) were diagnosed with focal epilepsy. Two patients (patients 8 and 10) had FS, one of whom inherited the variant from his mother (patient 8), and his mother had FS in childhood. Four patients (patients 15, 18, 30, and 31) were diagnosed with unclassified epilepsy.
Structural Alterations in the GABRG2 Protein and Genotype-Phenotype Correlation of GABRG2 Variants
All GABRG2 variants, including those in reported and our cohort, were analyzed to explore the mechanism underlying genotype-phenotype correlations. To date, 58 patients with 40 different variants have been reported, including 25 missense, 2 splicing, and 13 destructive variants (7 frameshift, 5 nonsense variants, and 1 small deletion variant). Their clinical characteristics are listed in Table 2. In our cohort of 35 patients, 23 unique variants were identified, including 16 missense variants, 4 splicing variants, 1 nonsense variant, 1 frameshift variant, and 1 small deletion variant. Overall, including our data and those from the literature, we reviewed data from 63 distinct variants identified in 93 unrelated patients (Figure 1 and Tables 1, 2). The variants included 41 missense variants (41/63, 65.1%), eight frameshift variants (8/63, 12.7%), six nonsense variants (6/63, 9.5%), six splicing variants (6/63, 9.5%), and two small deletion variants (2/63, 3.2%).
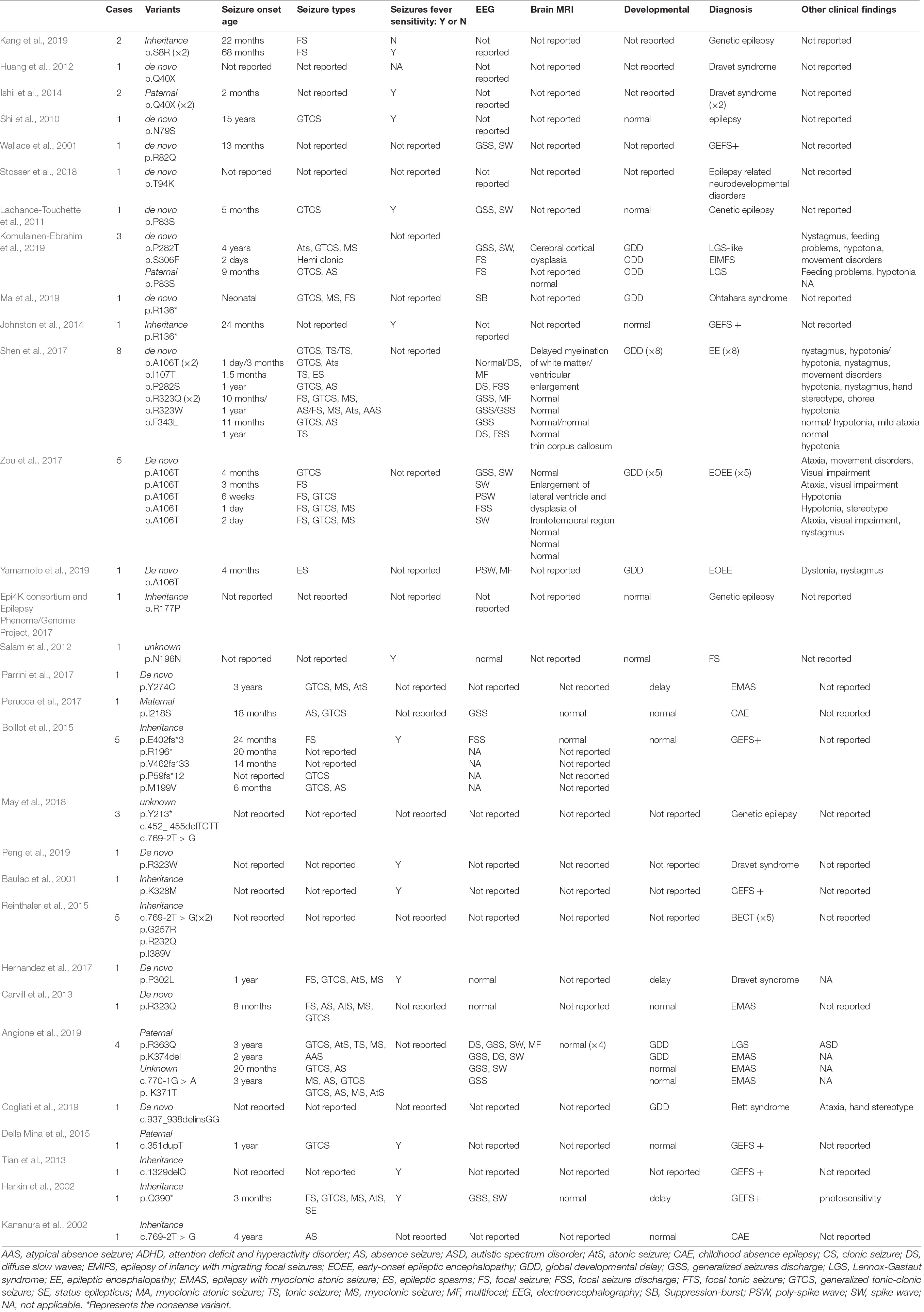
Table 2. The genotype and phenotype of 58 patients reported in literature with GABRG2 variants related to epilepsy.
Most variants (42/63, 66.7%) were located in the extracellular region and transmembrane region of the protein. Two were located at amino acid position 323 (p.R323Q and p.R323W), representing a potential variant hotspot. The p.R323Q variant was detected in 11 patients (patients 22–30, Carvill et al., 2013; Shen et al., 2017), whereas the p.R323W variant was found in 3 patients (patients 20–21, Peng et al., 2019). In the literature, the phenotypes of some patients with epilepsy carrying GABRG2 variants were not described in detail; therefore, we were only able to analyze the genotype and phenotype correlations in our cohort. In our cohort, seven recurrent variants are located in the transmembrane region of the channel, including p.P302L, p.A303T, p.T310I, p.T317I, p.R323Q, p.R323W, and p.S346F (Figure 1). Six of them (p.P302L, p.A303T, p.T310I, p.T317I, p.R323Q, and p.R323W) are located in the M2 domain. The p.S346F variant was located in the M3 region. Nine variants were located toward the extracellular region, including p.T90R, p.T90M, p.A106T, p.W121C, p.R125P, p.R125C, p.N140S, p.N167K, and p.P205H. The p.R429X variant is located in the inner part of the cytoplasmic domain. The proportions of patients with variants in the extracellular region and in the transmembrane region who experienced developmental delay were 40% (4/10) and 63.2% (12/19), respectively.
Patient 3 and patient 4 carried the same GABRG2 variant (p.A106T) that was located in the extracellular region. Both patients presented with DEE. However, a patient with the R125P variant near the A106T variant was diagnosed with FS+. The R323Q and R323W variants (potential variant hotspots) are located in the transmembrane region. The molecular effect of the missense variants was further analyzed by protein modeling using PyMOL 2.3. Residue A106 originally formed one hydrogen bond with V104. When alanine 106 was replaced by threonine, the hydrogen bonds with I107 and S325 were reestablished. Residue R125 originally formed three hydrogen bonds with D123, two with L81, and one each with Y77 and D78. When arginine 125 was replaced with proline acid, the hydrogen bonds with Y77, D78, L81 and D123 were all destroyed. Residue R323 originally formed one hydrogen bond with residues P327, V329 and D336. In contrast, when arginine 323 was replaced by tryptophan, the hydrogen bonds with residues P327, V329 and D336 were all destroyed, and only one hydrogen bond was retained with L326. Additionally, when arginine 323 was replaced by glutamine, the hydrogen bonds with residues P327, and V329 were destroyed, and only one hydrogen bond was retained with D336 (Figure 3).
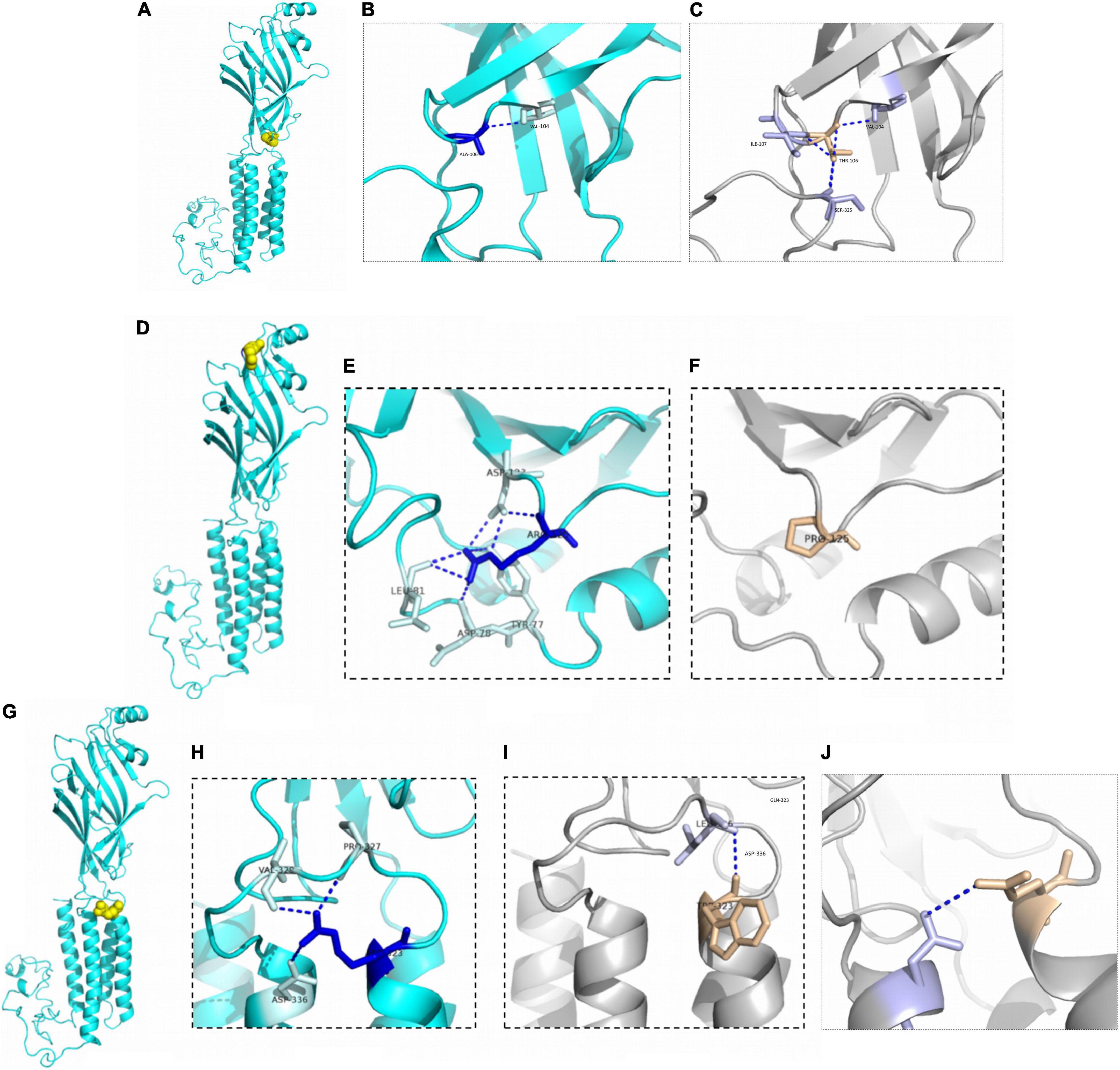
Figure 3. Structural modeling of GABRG2 variants (A106T, R125P, R323W, and R323Q). (A) The position of the 106th amino acid in the subunit (yellow). (B) Wild-type 106A forms ionic bonds with surrounding amino acid residues. (C) The number of ionic bonds between variant 125T amino acid residues and surrounding amino acid residues is increased. (D) The position of the 125th amino acid in the subunit (yellow). (E) Wild-type 125R forms ionic bonds with surrounding amino acid residues. (F) The number of ionic bonds between variant 125P amino acid residues and surrounding amino acid residues is reduced. (G) The position of the 323rd amino acid in the subunit (yellow). (H) Wild-type 323R forms ionic bonds with surrounding amino acid residues. (I) Change in forces between variant 323W amino acid residues and adjacent α helix. (J) Change in forces between variant 323Q amino acid residues.
Treatment and Follow-up
Treatment information was available for 35 patients. The final follow-up age ranged from 11 months to 17 years old. At the last follow-up, 25 patients (71.4%, 25/35) were seizure-free for 1 year to 7.5 years. Effective ASMs in terms of seizure freedom included valproate monotherapy (n = 9), levetiracetam monotherapy (n = 6), perampanel monotherapy (n = 1), valproate and levetiracetam combination therapy (n = 5), multitherapy (valproate, topiramate and perampanel combination therapy, n = 1), and biotherapy other than valproate combine with levetiracetam in 5 patients (patients 2, 3, 4, 12, and 33).
Ten patients still had seizures. One patient (patient 14) was diagnosed with Dravet syndrome. During the last follow-up visit, he still had frequent eyelid myoclonic seizures at the age of 10 years and 6 months. He showed no response to three different ASMs (valproate, levetiracetam, and clobazam). One patient (patient 29) with intractable epilepsy and developmental delay tried three ASMs (valproate, lamotrigine, and oxcarbazepine), but she still experienced cluster focal seizures. When she was treated with perampanel, and the seizure frequency was markedly reduced. Her afebrile seizures were controlled, but she still experienced fever-induced seizures. Patient 16 did not use ASMs due to poor parental compliance, and the seizures were not controlled. For the 7 other patients, the follow-up was too short to evaluate the effect of medication.
Discussion
Pathogenic GABRG2 variants have been reported in patients with epilepsy, developmental delays and behavioral disorders. The phenotypic spectrum of GABRG2 variants extends to the pharmacoresistant epilepsies, including Dravet syndrome and developmental epileptic encephalopathies (DEEs) (Harkin et al., 2002; Shen et al., 2017; Oyrer et al., 2018). Recently, GABRG2 was included in DEE74 (OMIM: 618396). The pathogenic variants in GABRG2 (p.Q40X) were first reported in a patient with Dravet syndrome in Kanaumi et al. (2004). In 2013, Carvill (Carvill et al., 2013) reported a male patient with epilepsy presenting myoclonic-atonic seizures (EMAS) who carried a GABRG2 variant (p.R323Q) and experienced multiple seizure types, including GTCS, absence seizures, atonic seizures, myoclonic seizures, and tonic-clonic seizures. Additionally, Zou et al. (2017) identified 5 patients with seizures carrying the same GABRG2 de novo variants (p.A106T) associated with DEE. To date, GABRG2 variants have been reported in 58 patients who presented with epilepsy in multiple epilepsy centers (Table 2). However, the largest number of patients evaluated in a single study was only 8. To date, systematic research on the phenotypic spectrum and prognosis of GABRG2 variants related to epilepsy is lacking. Here, we identified 31 novel and 4 previously reported GABRG2 variants in patients with epilepsy (Sun et al., 2008; Shen et al., 2017; Zou et al., 2017). The phenotypic spectrum and prognosis of patients with GABRG2-related epilepsy were further studied.
Among the 35 patients with GABRG2 variants in our cohort, the seizure onset age in 62.9% of patients (22/35) was during the 1st year of life, and the seizure onset ages of the remaining patients ranged from 1 year and 1 month to 2 years and 10 months. In previous reports, the seizure onset age ranged from birth to 15 years (median age: 1 year) (Table 2). This observation indicated that the seizure onset age of GABRG2-related epilepsy occurred mainly before 1 year of age. In our cohort, we identified several key phenotypic features as part of the GABRG2-related disease spectrum; 91.4% of patients had fever-sensitive seizures, 68.6% of patients had focal seizures, and most of the patients (71.4%) were seizure-free after ASMs treatment.
Seizures with fever sensitivity have been reported in some patients; however, the percentage of patients presenting this feature had not been delineated. In previous reports, 47.4% (18/38) of patients with GABRG2 variants experienced seizures associated with fever-sensitivity (Table 2). Fever sensitivity was not reported in the remaining patients. Boillot et al. (2015) reported 5 families with inherited GABRG2 variants who had febrile seizures and temporal lobe epilepsy. One patient (p.P302L) who experienced febrile seizures, multiple seizure types, and early psychomotor and language developmental delay was diagnosed with Dravet syndrome (Hernandez et al., 2017).
We observed developmental delay in 42.8% (15/35) of patients with GABRG2 variants, while 66.7% (24/36) of the previously published patients (available data) had developmental delay (Table 2). Most of the patients in our study were still young, and the proportion of patients with developmental delay should be further studied in a large cohort. However, Komulainen-Ebrahim et al. (2019) reported 3 patients with GABRG2 variants presenting with global developmental delay. Eight patients with de novo GABRG2 variants associated with epileptic encephalopathies were reported by Shen et al. (2017). In our cohort, the clinical phenotypes of patients with normal development were mostly diagnosed with FS and FS+. The patients with developmental delay mostly had an early seizure onset, and multiple seizure types, some of which were diagnosed with developmental and epileptic encephalopathy.
In our study, no specific EEG pattern in the seizure evolution was observed in the patients with GABRG2 variants. Interictal EEG performed in patients with epileptic encephalopathy patients, showed epileptiform discharges, including focal and multifocal spike waves, generalized spike waves and polyspike waves. Of the 58 patients reported previously, 51.7% of patients (30/58) were monitored for epileptiform discharges in interictal EEG. One patient with variant p. R136* presented a suppression burst pattern on interictal EEG (Ma et al., 2019). In our cohort, 33.3% (11/33) of patients had a normal EEG, which may be related to the benign phenotype of most children. However, only 13.3% (4/30) of patients had a normal EEG in the published cohort, and the difference may be related to the patients recruited from different research centers; the number of patients must be further expanded for an in-depth analysis. Brain MRI of patients with GABRG2 variants was usually normal. In our cohort, the brain MRI of 2 patients (patients 3 and 25) was abnormal. These two patients had severe developmental delays. Their phenotypes included DEE, developmental delay and epilepsy. In the published studies, only 22.7% (5/22) of patients had an abnormal MRI: delayed myelination of white matter in one, ventricular enlargement in one, thin corpus callosum in one, and all with epileptic encephalopathy (Shen et al., 2017); enlargement of lateral ventricles and dysplasia of the frontotemporal region in one patient with early-onset epileptic encephalopathy (Zou et al., 2017); and cerebral cortical dysplasia in one patient with an LGS-like syndrome (Komulainen-Ebrahim et al., 2019). All these five patients had a global developmental delay.
Several studies have documented the phenotypic heterogeneity of GABRG2-related epilepsy. Of the 58 patients with GABRG2 variants reported previously, the epilepsy phenotypes were varied (Table 2). The phenotypic spectrum observed for GABRG2 variants, ranging from febrile seizures to epileptic encephalopathy, is similar to those of the other GABAA receptor genes GABRA1, GABRB2, and GABRB3 (Johannesen et al., 2016; Moller et al., 2017; Yang et al., 2020, 2021). However, the prognosis of patients with GABRG2-related epilepsy is better than that of patients carrying variants in the other three genes. In our cohort, 57.1% (20/35) of patients presented with milder phenotypes, including febrile seizures, febrile seizures plus, focal epilepsy and unclassified epilepsy with normal development. Additionally, 38.1% (16/42) of patients manifested milder phenotypes than those in the previously published cohort (Table 2).
The mechanism of the phenotypic variation caused by GABAA receptor family genes is unclear. From the perspective of biological functions, the γ2 subunit (gene GABRG2) plays a critical role in GABAA receptor trafficking and localization at the postsynapse and yields benzodiazepine-sensitive and Zn2+-insensitive GABAA receptors (Alldred et al., 2005; Kang and Macdonald, 2016). GABRG2 functions as a part of GABAA receptor complexes, potentially explaining the variability of the clinical phenotypes. Further analysis showed that the GABRG2 missense variants associated with severe epilepsy phenotypes were mainly clustered in the transmembrane region from the M1 region to the M3 domain. However, the variants located in the extracellular region were associated with phenotypes ranging from mild to severe. Moreover, few variants were located in the cytoplasmic region.
Bioinformatics analysis supported the deleterious effects of the p.R125P and p.R323W variants. The number of ion bonds between amino acid residues decreased in p.R125P according to the structural modeling. These changes may partially be responsible for the instability of protein structure. Arginine is an amino acid that increases the stability of proteins to a certain extent, and the variant at this site may reduce the stability of proteins (Cai et al., 2018). In our cohort, patient 6 with the R125P variant manifested febrile seizures plus. Nine patients were detected with the p.R323Q variant of GABRG2. Two patients were detected with the p.R323W variant of GABRG2. However, their phenotypes were different. The p.R323W and p.R323Q variants were located in the transmembrane domain. According to cryo-EM, the transmembrane domain is composed of a five-fold symmetric of the α-helix structure (Laverty et al., 2019). Both the R323W and R323Q variants change the ionic bonds with the adjacent α-helix according to structural modeling. Structural studies have shown that the transmembrane domain of the GABA receptor has larger flexibility (Zhu et al., 2018), and the variant in the adjacent α-helix also presents local flexibility in the transmembrane domain in some aspects. This genetic study may provide potential prospects for personalized medicine. The precise mechanism of GABRG2-related epilepsy is complex and requires further study in the future. The lack of functional studies limits our understanding of the impact of variants on proteins (loss or gain function).
Valproate and levetiracetam treatment might be suitable for patients harboring GABRG2 variants. In this study, eight patients were effectively treated with valproate and levetiracetam. Previously, few reports described the effect of ASMs on patients with GABRG2 variants. Zou reported that seizures were effectively controlled in two patients with GABRG2 variant (A106T)- related epileptic encephalopathies after treatment with oxcarbazepine (Zou et al., 2017). In our cohort, patient 3 achieved seizure control after receiving treatment with oxcarbazepine. However, oxcarbazepine was ineffective in two patients. One patient with intractable epilepsy had a reduction in seizures after receiving perampanel treatment. Perampanel, is a novel non-competitive α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor antagonist. An imbalance between glutamate and gamma-aminobutyric acid neurotransmitter systems may lead to hyperexcitability (Engelborghs et al., 2000). However, due to the limited number of patients with follow-up data, further studies are needed in the future. In our previous study of other types of GABAA receptor-related epilepsy, we found that patients with GABRB2 and GABRB3 variant-related epilepsy patients also had a good response to valproate and levetiracetam (Yang et al., 2020, 2021). The heterogeneity of the clinical presentations related to the GABRG2 variants makes early diagnosis difficult, and the lack of explanation for this heterogeneity does not currently allow personalized treatment.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Files, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Peking University First Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YY wrote the article under the supervision of YZ. XN, MC, QZ, JD, XT, YW, JY, WS, WW, JM, YL, XY, XZ, TJ, ZY, JL, YS, HZ, SS, DS, and YJ had collected relevant patient information. All authors read and approved the final manuscript.
Funding
This study was supported in part by the National Natural Science Foundation of China (82071451), and Key Research Project of the Ministry of Science and Technology of China (Grant numbers: 2016YFC0904400 and 2016YFC0904401).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to extend our deep appreciation to the patients and their families who participated in this study. We would also like to thank team of staff who assisted with data collection. We would also like to thank Jintong Jia from the Singleron Biotechnologies, Ltd., for assisting with structural modeling using PyMOL2.3. We would also like to thank Dr. Xiaodong Wang from the Cipher Gene, Ltd., for assisting with manuscript editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.809163/full#supplementary-material
Footnotes
- ^ https://gnomad.broadinstitute.org
- ^ https://www.mutationtaster.org/
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://provean.jcvi.org/protein_batch_submit.php?species=human
References
Alldred, M. J., Mulder-Rosi, J., Lingenfelter, S. E., Chen, G., and Luscher, B. (2005). Distinct gamma2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J. Neurosci. 25, 594–603. doi: 10.1523/JNEUROSCI.4011-04.2005
Angione, K., Eschbach, K., Smith, G., Joshi, C., and Demarest, S. (2019). Genetic testing in a cohort of patients with potential epilepsy with myoclonic-atonic seizures. Epilepsy Res. 150, 70–77. doi: 10.1016/j.eplepsyres.2019.01.008
Baulac, S., Huberfeld, G., Gourfinkel-An, I., Mitropoulou, G., Beranger, A., Prud’homme, J. F., et al. (2001). First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet. 28, 46–48. doi: 10.1038/88254
Boillot, M., Morin-Brureau, M., Picard, F., Weckhuysen, S., Lambrecq, V., Minetti, C., et al. (2015). Novel GABRG2 mutations cause familial febrile seizures. Neurol. Genet 1:e35. doi: 10.1212/NXG.0000000000000035
Cai, X., Jiang, H., Zhang, T., Jiang, B., Mu, W., and Miao, M. (2018). Thermostability and Specific-Activity Enhancement of an Arginine Deiminase from Enterococcus faecalis SK23.001 via Semirational Design for l-Citrulline Production. J. Agric. Food Chem. 66, 8841–8850. doi: 10.1021/acs.jafc.8b02858
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O’Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi: 10.1038/ng.2646
Cogliati, F., Giorgini, V., Masciadri, M., Bonati, M. T., Marchi, M., Cracco, I., et al. (2019). Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes. Int. J. Mol. Sci. 20:3621. doi: 10.3390/ijms20153621
Della Mina, E., Ciccone, R., Brustia, F., Bayindir, B., Limongelli, I., Vetro, A., et al. (2015). Improving molecular diagnosis in epilepsy by a dedicated high-throughput sequencing platform. Eur. J. Hum. Genet. 23, 354–362. doi: 10.1038/ejhg.2014.92
Engelborghs, S., D’Hooge, R., and De Deyn, P. P. (2000). Pathophysiology of epilepsy. Acta Neurol. Belg. 100, 201–213.
Epi4K consortium, and Epilepsy Phenome/Genome Project (2017). Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 16, 135–143. doi: 10.1016/S1474-4422(16)30359-3
Harkin, L. A., Bowser, D. N., Dibbens, L. M., Singh, R., Phillips, F., Wallace, R. H., et al. (2002). Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 70, 530–536. doi: 10.1086/338710
Hernandez, C. C., Kong, W., Hu, N., Zhang, Y., Shen, W., Jackson, L., et al. (2017). Altered Channel Conductance States and Gating of GABAA Receptors by a Pore Mutation Linked to Dravet Syndrome. eNeuro 4:ENEURO.251–ENEURO.216. doi: 10.1523/ENEURO.0251-16.2017
Hernandez, C. C., Tian, X., Hu, N., Shen, W., Catron, M. A., Yang, Y., et al. (2021). Dravet syndrome-associated mutations in GABRA1, GABRB2 and GABRG2 define the genetic landscape of defects of GABAA receptors. Brain Commun. 3:fcab033. doi: 10.1093/braincomms/fcab033
Huang, X., Tian, M., Hernandez, C. C., Hu, N., and Macdonald, R. L. (2012). The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol. Dis. 48, 115–123. doi: 10.1016/j.nbd.2012.06.013
Ishii, A., Kanaumi, T., Sohda, M., Misumi, Y., Zhang, B., Kakinuma, N., et al. (2014). Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. 108, 420–432. doi: 10.1016/j.eplepsyres.2013.12.005
Johannesen, K., Marini, C., Pfeffer, S., Moller, R. S., Dorn, T., Niturad, C. E., et al. (2016). Phenotypic spectrum of GABRA1: from generalized epilepsies to severe epileptic encephalopathies. Neurology 87, 1140–1151. doi: 10.1212/WNL.0000000000003087
Johnston, A. J., Kang, J. Q., Shen, W., Pickrell, W. O., Cushion, T. D., Davies, J. S., et al. (2014). A novel GABRG2 mutation, p.R136*, in a family with GEFS+ and extended phenotypes. Neurobiol. Dis. 64, 131–141. doi: 10.1016/j.nbd.2013.12.013
Kananura, C., Haug, K., Sander, T., Runge, U., Gu, W., Hallmann, K., et al. (2002). A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch. Neurol. 59, 1137–1141. doi: 10.1001/archneur.59.7.1137
Kanaumi, T. F. G., Ueno, S., Ishii, A., Haga, Y., Hamachi, A., Yonetani, M., et al. (2004). Possible pathogenesis of severe myoclonic epilepsy in infancy: a novel nonsense mutation of GABRG2 leading to aggregation of GABAA receptors in neurons. Neurol. Asia 9:151.
Kang, J. Q., and Macdonald, R. L. (2016). Molecular Pathogenic Basis for GABRG2 Mutations Associated With a Spectrum of Epilepsy Syndromes, From Generalized Absence Epilepsy to Dravet Syndrome. JAMA Neurol. 73, 1009–1016. doi: 10.1001/jamaneurol.2016.0449
Kang, K. W., Kim, W., Cho, Y. W., Lee, S. K., Jung, K. Y., Shin, W., et al. (2019). Genetic characteristics of non-familial epilepsy. PeerJ 7:e8278. doi: 10.7717/peerj.8278
Komulainen-Ebrahim, J., Schreiber, J. M., Kangas, S. M., Pylkas, K., Suo-Palosaari, M., Rahikkala, E., et al. (2019). Novel variants and phenotypes widen the phenotypic spectrum of GABRG2-related disorders. Seizure 69, 99–104. doi: 10.1016/j.seizure.2019.03.010
Lachance-Touchette, P., Brown, P., Meloche, C., Kinirons, P., Lapointe, L., Lacasse, H., et al. (2011). Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur. J. Neurosci. 34, 237–249. doi: 10.1111/j.1460-9568.2011.07767.x
Laverty, D., Desai, R., Uchanski, T., Masiulis, S., Stec, W. J., Malinauskas, T., et al. (2019). Cryo-EM structure of the human alpha 1 beta 3 gamma 2 GABA(A) receptor in a lipid bilayer. Nature 565, 516–520. doi: 10.1038/s41586-018-0833-4
Ma, X., Yang, F., and Hua, Z. (2019). Genetic diagnosis of neonatal-onset seizures. Genes Dis. 6, 441–447. doi: 10.1016/j.gendis.2019.02.002
May, P., Girard, S., Harrer, M., Bobbili, D. R., Schubert, J., Wolking, S., et al. (2018). Rare coding variants in genes encoding GABA(A) receptors in genetic generalised epilepsies: an exome-based case-control study. Lancet Neurol. 17, 699–708. doi: 10.1016/S1474-4422(18)30215-1
Moller, R. S., Wuttke, T. V., Helbig, I., Marini, C., Johannesen, K. M., Brilstra, E. H., et al. (2017). Mutations in GABRB3: from febrile seizures to epileptic encephalopathies. Neurology 88, 483–492. doi: 10.1212/WNL.0000000000003565
Oyrer, J., Maljevic, S., Scheffer, I. E., Berkovic, S. F., Petrou, S., and Reid, C. A. (2018). Ion Channels in Genetic Epilepsy: from Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 70, 142–173. doi: 10.1124/pr.117.014456
Parrini, E., Marini, C., Mei, D., Galuppi, A., Cellini, E., Pucatti, D., et al. (2017). Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 38, 216–225. doi: 10.1002/humu.23149
Peng, J., Pang, N., Wang, Y., Wang, X. L., Chen, J., Xiong, J., et al. (2019). Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci. Ther. 25, 14–20. doi: 10.1111/cns.12869
Perucca, P., Scheffer, I. E., Harvey, A. S., James, P. A., Lunke, S., Thorne, N., et al. (2017). Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 131, 1–8. doi: 10.1016/j.eplepsyres.2017.02.001
Reinthaler, E. M., Dejanovic, B., Lal, D., Semtner, M., Merkler, Y., Reinhold, A., et al. (2015). Rare variants in gamma-aminobutyric acid type A receptor genes in rolandic epilepsy and related syndromes. Ann. Neurol. 77, 972–986. doi: 10.1002/ana.24395
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17, 405–424. doi: 10.1038/gim.2015.30
Salam, S. M., Rahman, H. M., and Karam, R. A. (2012). GABRG2 gene polymorphisms in Egyptian children with simple febrile seizures. Indian J. Pediatr. 79, 1514–1516. doi: 10.1007/s12098-011-0564-0
Shen, D., Hernandez, C. C., Shen, W., Hu, N., Poduri, A., Shiedley, B., et al. (2017). De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 140, 49–67. doi: 10.1093/brain/aww272
Shi, X., Huang, M. C., Ishii, A., Yoshida, S., Okada, M., Morita, K., et al. (2010). Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. J. Hum. Genet. 55, 375–378. doi: 10.1038/jhg.2010.47
Stosser, M. B., Lindy, A. S., Butler, E., Retterer, K., Piccirillo-Stosser, C. M., Richard, G., et al. (2018). High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 20, 403–410. doi: 10.1038/gim.2017.114
Sun, H. H., Zhang, Y. H., Liang, J. M., Liu, X. Y., Ma, X. W., Wu, H. S., et al. (2008). SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J. Hum. Genet. 53, 769–774. doi: 10.1007/s10038-008-0306-y
Surguchov, A., Surgucheva, I., Sharma, M., Sharma, R., and Singh, V. (2017). Pore-Forming Proteins as Mediators of Novel Epigenetic Mechanism of Epilepsy. Front. Neurol. 8:3. doi: 10.3389/fneur.2017.00003
Tian, M., Mei, D., Freri, E., Hernandez, C. C., Granata, T., Shen, W., et al. (2013). Impaired surface alphabetagamma GABA(A) receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol. Dis. 50, 135–141. doi: 10.1016/j.nbd.2012.10.008
Wallace, R. H., Marini, C., Petrou, S., Harkin, L. A., Bowser, D. N., Panchal, R. G., et al. (2001). Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 28, 49–52. doi: 10.1038/88259
Yamamoto, T., Imaizumi, T., Yamamoto-Shimojima, K., Lu, Y., Yanagishita, T., Shimada, S., et al. (2019). Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev. 41, 776–782. doi: 10.1016/j.braindev.2019.05.007
Yang, Y., Xiangwei, W., Zhang, X., Xiao, J., Chen, J., Yang, X., et al. (2020). Phenotypic spectrum of patients with GABRB2 variants: from mild febrile seizures to severe epileptic encephalopathy. Dev. Med. Child Neurol. 62, 1213–1220. doi: 10.1111/dmcn.14614
Yang, Y., Zeng, Q., Cheng, M., Niu, X., Xiangwei, W., Gong, P., et al. (2021). GABRB3-related epilepsy: novel variants, clinical features and therapeutic implications. J. Neurol. [Online ahead of print] doi: 10.1007/s00415-021-10834-w
Zhu, S., Noviello, C. M., Teng, J., Walsh, R. M. Jr., Kim, J. J., and Hibbs, R. E. (2018). Structure of a human synaptic GABAA receptor. Nature 559, 67–72. doi: 10.1038/s41586-018-0255-3
Keywords: GABRG2, epilepsy, infancy, fever-sensitive, prognosis
Citation: Yang Y, Niu X, Cheng M, Zeng Q, Deng J, Tian X, Wang Y, Yu J, Shi W, Wu W, Ma J, Li Y, Yang X, Zhang X, Jia T, Yang Z, Liao J, Sun Y, Zheng H, Sun S, Sun D, Jiang Y and Zhang Y (2022) Phenotypic Spectrum and Prognosis of Epilepsy Patients With GABRG2 Variants. Front. Mol. Neurosci. 15:809163. doi: 10.3389/fnmol.2022.809163
Received: 04 November 2021; Accepted: 28 January 2022;
Published: 14 March 2022.
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
Irina G. Sourgoutcheva, University of Kansas Medical Center, United StatesCharlotte Dravet, Scientific Institute for Research, Hospitalization and Healthcare (IRCCS), Italy
Svetlana Gataullina, Hôpital Antoine-Béclère, France
Copyright © 2022 Yang, Niu, Cheng, Zeng, Deng, Tian, Wang, Yu, Shi, Wu, Ma, Li, Yang, Zhang, Jia, Yang, Liao, Sun, Zheng, Sun, Sun, Jiang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuehua Zhang, emhhbmd5aGRyQDEyNi5jb20=