
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 17 February 2022
Sec. Molecular Signalling and Pathways
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.805087
This article is part of the Research Topic Protein Degradation, Aggregation, Membrane Trafficking and Exosomes in Neuronal Health and Disease View all 11 articles
 Denisse Sepúlveda1,2,3
Denisse Sepúlveda1,2,3 Marisol Cisternas-Olmedo1,2,3
Marisol Cisternas-Olmedo1,2,3 Javiera Arcos1,2,3
Javiera Arcos1,2,3 Melissa Nassif1,4
Melissa Nassif1,4 René L. Vidal1,2,3*
René L. Vidal1,2,3*Parkinson’s disease (PD) is caused by the degeneration of dopaminergic neurons due to an accumulation of intraneuronal abnormal alpha-synuclein (α-syn) protein aggregates. It has been reported that the levels of exosomal α-syn of neuronal origin in plasma correlate significantly with motor dysfunction, highlighting the exosomes containing α-syn as a potential biomarker of PD. In addition, it has been found that the selective autophagy-lysosomal pathway (ALP) contributes to the secretion of misfolded proteins involved in neurodegenerative diseases. In this review, we describe the evidence that supports the relationship between the ALP and α-syn exosomal secretion on the PD progression and its implications in the diagnosis and progression of this pathology.
Parkinson’s disease (PD) is the second more common neurodegenerative disease globally, and it is associated with age (Dorsey et al., 2018). The PD incidence is estimated from 5 to more than 35 new cases per 100,000 individuals, depending on demographic differences (Poewe et al., 2017). The pathology prevalence ranges from 41/100,000 individuals in the fourth decade of life to more than 1,900/100,000 among those 80 and older (Pringsheim et al., 2014). Have been described that PD is more prevalent in men (1,729/100,000, >65 years) than in women (1,644/100,000) (Pringsheim et al., 2014; Riedel et al., 2016). Although the risk of developing PD is higher in men, women have a higher mortality rate and faster clinical progression (Cerri et al., 2019).
Parkinson’s disease is a complex neurodegenerative disorder clinically characterized by bradykinesia, tremor, rigidity, later postural reflexes instability, and progressive paralysis (Jankovic, 2008). Some non-motor symptoms such as dementia, depression, anxiety, and sleep disorder may precede motor symptoms for more than a decade, affecting several neurotransmitter pathways (Langston, 2006). PD coexists with dementia in over 25% of the cases and depression in over 30% of the cases in some countries (Riedel et al., 2016). As a motor disorder, PD affects patients’ quality of life, making social interaction more difficult and worsening their financial condition due to the high medical expenses associated with the pathology.
Part of the PD symptomatology is caused by the degeneration of dopaminergic (DA) neurons of Substantia Nigra pars compacta (SNpc) and loss of the dopaminergic fibers that innervate the striatum (Dauer and Przedborski, 2003). The appearance of the first symptoms correlates with a 30% loss of dopaminergic neurons (Fearnley and Lees, 1991), indicating degeneration of the neurotransmission integrity in the basal ganglia circuits. Currently, PD has no cure, and the potential treatment to prevent or revert this pathology arises as a substantial challenge (Troncoso-Escudero et al., 2020). Therefore, it is essential to obtain a better understanding of the correlation between associated cellular mechanisms and the clinical features of PD to develop strategies to optimize the prevention, diagnosis, and treatment.
About 10% of PD cases are associated with genetic mutations (Selvaraj and Piramanayagam, 2019), which includes: (i) mutation in the SCNA gene that encodes for the alpha-synuclein protein (α-syn) (Bras et al., 2020), (ii) mutations in the LRRK2 gene, which encodes leucine-rich repeat kinase 2, are a cause of autosomal dominant forms of PD (Zimprich et al., 2004; Tolosa et al., 2020), (iii) heterozygous mutations of the GBA1, encoding for lysosomal enzyme glucocerebrosidase (GCase), are a strong risk factor for PD and can lead to α-syn accumulation (Avenali et al., 2020). While 90% of PD cases are classified as idiopathic, the evidence indicates that the histopathology characteristic of PD is the accumulation of intraneuronal abnormal protein aggregates, including the α-syn protein. These aggregates of the amyloid type are called Lewy bodies and are constituted mainly by oligomers of α-syn and ubiquitin (Spillantini et al., 1998; Schulz-Schaeffer, 2010). Idiopathic cases show an increase of endogenous wild-type α-syn (Golbe et al., 1990; Michell et al., 2005; Shprecher et al., 2018), forming aggregates of this protein that have a toxic effect on dopaminergic neurons, triggering neurodegenerative processes (Petrucelli et al., 2002; Volpicelli-Daley et al., 2016).
Mitochondrial toxins have been identified in epidemiological studies as contributors to “sporadic” PD in humans. In this context, animal and in vitro models used to study PD are based on the administration of neurotoxins, generating oxidative stress and mitochondrial dysfunction. 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat (PQ; 1, 10-dimethyl-4,40-bipyridinium), and rotenone are conventionally used in PD modeling (Chia et al., 2020), as they can be uptaken by DA neurons through dopamine transporters, inhibiting complex I of the mitochondrial electron transport chain, leading to ATP depletion, increasing reactive oxygen species, and ultimately resulting in neuronal death (Betarbet et al., 2002; Devi et al., 2008; Potashkin et al., 2010). PQ, a commonly used herbicide, shares structural similarities with MPP+, the active metabolite of MPTP. PQ crosses the blood-brain barrier, generates reactive oxygen and nitrogen species (ROS/RNS), and causes the loss of SNpc DA neurons in animal models (Castello et al., 2007). In rats, chronic rotenone exposure leads to α-syn aggregation, DA neurodegeneration, and behavioral defects (Hoglinger et al., 2003). This toxin induced the cytosolic accumulation of α-syn through the de novo synthesis, rather than a reduction of degradation by chaperone-mediated autophagy (CMA), suggesting a mechanism independent from lysosomal degradation (Sala et al., 2013). Indeed, rotenone regulates α-syn phosphorylation, reducing protein phosphatase 2A (PP2A) activity (Wang et al., 2016).
Classical pharmacological therapies for PD patients are dopamine precursors as levodopa, L-dopa, and L-3,4-dihydroxyphenylalanine. Other treatments include dopamine agonists such as amantadine, apomorphine, pramipexole, and monoamine oxidase inhibitors (MAO) or catechol-O-methyltransferase (COMT). The sustained administration of these drugs induces a “wearing-off phenomenon” and additional psychomotor, cardio-cerebrovascular, and hormones regulation problems (Cacabelos, 2017). Novel biotherapies, as natural products, should achieve dopaminergic protection to avoid neurodegeneration, enhancing dopaminergic neurotransmission. However, the cellular and molecular events involved in PD must be broadly explored to design and develop efficient treatments (Solayman et al., 2017; Wang et al., 2017; Troncoso-Escudero et al., 2020).
α-syn protein is expressed at high levels in the central nervous system (CNS), specifically neurons. It is found in presynaptic terminals as a monomeric, unfolded, and soluble protein (Maroteaux et al., 1988), bound with high affinity to the membranes of synaptic vesicles (Burre et al., 2010). α-syn was described in neuromuscular junctions (Askanas et al., 2000), suggesting other cellular functions in addition to its activity in the CNS. Although α-syn is enriched in synaptic boutons, which sprout from axons of different neurochemical phenotypes, α-syn is not present in all synaptic terminals. In agreement, not all terminals accumulate the protein in neurodegenerative disorders (Totterdell et al., 2004). Furthermore, the expression of α-syn is not limited to the nervous system. This protein is present in the cerebrospinal fluid (CSF), in plasma (El-Agnaf et al., 2003; Forland et al., 2018), as well as is expressed in the erythropoietic lineage cells (Nakai et al., 2007) and peripheral lymphocytes (Kim et al., 2004).
To clarify the α-syn function, knockout mice for the SCNA gene were generated. Although knockout mice were viable and fertile, with a lack of spontaneous neurodegeneration signs, this model displays alterations in activity-dependent dopamine release from axons in the striatum (Abeliovich et al., 2000). In addition, the triple knockout mice lacking the three variants of syn (α, β, and γ) were generated, showing no neurodegeneration (Greten-Harrison et al., 2010). However, it was possible to observe synapse-structure modifications and a decrease in the synaptic terminal size in an age-dependent manner (Greten-Harrison et al., 2010). Lack of α-syn in the transgenic mice model showed less mobilization of glutamatergic vesicles (Gureviciene et al., 2007) and increased the expression levels of proteins involved in vesicle traffic, such as SNAREs, synapsins, and complexines (Greten-Harrison et al., 2010). In addition, it has been described that the participation of α-syn in vesicle homeostasis is Ca2+-dependent (Lautenschlager et al., 2018). Overall, these data suggest a direct physiologic role of α-syn in synaptic transmission in the CNS, especially in the dopamine system.
Different factors can trigger α-syn aggregation, including point mutations (Narhi et al., 1999; Rutherford et al., 2014), truncations (Li et al., 2005), posttranslational modifications of α-syn (Duda et al., 2000; Fujiwara et al., 2002; Lazaro et al., 2014), and wild-type SCNA gene duplication or triplication (Singleton et al., 2003; Fuchs et al., 2007). An increase in somatic copy number of the SNCA gene in CNS neurons, especially from the SN region, was reported in a cohort of PD patients, contributing to the sporadic α-syn accumulation (Mokretar et al., 2018). However, it is still unknown what prompts the accumulation of wild-type α-syn into toxic aggregates. Evidence indicates that toxic α-syn conformers can act as seeds for the misfolding and aggregation of the native protein. For instance, α-syn preformed fibrils (PFFs), synthetically produced, were added in neuronal cultures and taken up inside cells, recruiting α-syn endogenous into protein aggregates (Luk et al., 2012a,b). The inoculation of PFFs into the brain of young adult A53T SNCA mice (overexpressing mutated human α-syn) generated in vivo aggregates and PD-like symptoms in mice (Luk et al., 2012b). Moreover, species of α-syn isolated from A53T transgenic mice induce aggregation of α-syn in primary neuronal cultures (Colla et al., 2018), indicating a potential trigger role of α-syn aggregates on wild-type soluble α-syn.
Nevertheless, not only protein interactions determine the status of α-syn aggregation. α-syn contains a lipid-binding domain that allows its binding to vesicles at the presynaptic terminal (Lashuel et al., 2013). However, in pathological conditions or modified lipids composition, this interaction can potentiate conformational changes in α-syn protein, prompting it to aggregation (Marschallinger et al., 2020). Recently, it was reported that the lipid alteration in membrane compartments, as instability of lipid raft microdomains, promoted by aging, and neurotoxins, as the MPTP, could affect α-syn aggregation (Galvagnion, 2017; Canerina-Amaro et al., 2019). Caveolins, a subgroup of lipid rafts, act as scaffolding proteins that recruit other proteins and lipids, leading to colocalization and interaction of proteins involved in vesicular transport, signal transduction, and receptor trafficking (Hanzal-Bayer and Hancock, 2007). The central protein controlling caveolae formation is caveolin-1 (Cav-1). Cav-1 is widely expressed in the central and peripheral nervous systems (Boulware et al., 2007), regulating neurotrophin signaling pathways and synaptic remodeling (Bilderback et al., 1999; Suzuki et al., 2004). In addition, Cav-1 modulates neurotransmitter receptor signaling (Bhatnagar et al., 2004; Francesconi et al., 2009).
aveolin-1 is also involved in the aging process. Since Cav-1 expression is upregulated in old rat brain and aged human cortex (Park et al., 2000; Kang et al., 2006), suggesting that overexpression of Cav-1 may induce aging phenotypes (Wheaton et al., 2001; Lee et al., 2015). Some evidence suggests that scaffold proteins such as Cav-1 may be involved in the pathogenesis of several neurodegenerative disorders, including PD (Hashimoto et al., 2003; Benarroch, 2007). Age-related expression of Cav-1 may affect the cell-to-cell transmission of α-syn, contributing to the pathogenesis of PD (Ha et al., 2021). Cav-1 overexpression facilitated the uptake of α-syn into neurons and the formation of additional Lewy body-like inclusion bodies (Ha et al., 2021). Immunoprecipitation experiments demonstrated that the double mutant alpha-synuclein protein (A30P/A53T) interacts with Cav-1 present in both cytoplasmic and inner membrane extracts of the mouse brain, suggesting that the double mutation of α-syn increases the affinity for Cav-1 in the cytosol. These results suggest a direct interaction between Cav-1 and α-syn under non-physiological conditions (α-syn overexpressed or α-syn mutated) (Madeira et al., 2011). Furthermore, colocalization experiments using SH-SY5Y cells demonstrate that α-syn and caveolin interact directly and mediate endocytosis and colocalize to a lesser extent along the endocytosis pathway with early endosome antigen 1 (EEA1) and Rab7-positive late endosomes (Fakhree et al., 2021). EEA1 is an early endosomal Rab5 effector protein that has been implicated in the docking of incoming endocytic vesicles before fusion with early endosomes (Fakhree et al., 2021), and Rab7, a member of the Rab family of small GTPases, is a ubiquitously expressed protein that plays a vital role in the regulation of the trafficking, maturation, and fusion of endocytic and autophagic vesicles (McCray et al., 2010).
A recent work using human iPSC-derived cerebral organoids found that 3D-cultures from donors carrying homozygous APOE4 allele presented aggregation of α-syn, loss of synaptic integrity, and impairment on lipids metabolism, resulting in accumulation of lipid droplets (Zhao et al., 2021). APOE4 isoform is a known risk factor for late-onset Alzheimer’s disease (AD) development. In this study, researchers also reported a boosted interaction of APOE4 itself with α-syn in postmortem brain samples from Lewy bodies disease patients (Zhao et al., 2021), confirming a link between lipid metabolism and α-syn aggregation. As previously mentioned, heterozygosis variants in the gene encoding GCase (GBA1) represent a significant PD genetic risk factor (Avenali et al., 2020). Indeed, about 10% of PD patients present mutations in the gene that codifies to GCase (Sidransky et al., 2009). GCase is a lysosomal enzyme that catalyzes the hydrolysis of the glycolipid glucosylceramide. Homozygous mutations in GBA1 cause Gaucher’s disease, the most prevalent recessively inherited lysosomal lipid storage disease, characterized by neurodegeneration and peripheral symptoms (Han et al., 2020). GBA1 variants associated with PD present a decreased enzymatic activity, resulting in the accumulation of the substrate glucosylceramide, as shown in CSF samples from PD patients (Huh et al., 2021). Accumulated glucosylceramides were reported to promote wild-type α-syn aggregation in in vitro studies (Taguchi et al., 2017). Of note, a small-molecule modulator (activator) of GCase reduces pathological α-syn aggregates and restores lysosomal function in PD patient midbrain neurons (Mazzulli et al., 2016b). In a bilateral correlation, α-syn aggregation also causes impairment on GCase activity and lysosomal dysfunction (see below).
The overexpression of α-syn selectively induced apoptotic programmed cell death in primary dopamine neurons (Zhou et al., 2000), neuroblastoma cell lines, and hippocampal primary neurons (Mahul-Mellier et al., 2015). The causal relationship between α-syn aggregation and cellular toxicity was investigated by assessing the effect of inhibiting fibrillization on α-syn-induced cell death. It was reported that exogenous α-syn fibrils bind to the plasma membrane and act as nucleation sites for the formation of endogenous α-syn fibrils, promoting the accumulation and internalization of the aggregates that finally turn on the activation of both the extrinsic and intrinsic apoptotic cell death pathways in cellular models (Mahul-Mellier et al., 2015).
It has been described that secreted α-syn can be internalized by neighboring cells via endocytosis (Desplats et al., 2009), demonstrating the cell-to-cell transmission of α-syn accumulation and providing evidence of the pathological mechanism to explain PD progression and other synucleinopathies (Desplats et al., 2009; Hansen et al., 2011). Recent works also showed α-syn transference cell-to-cell through the formation of tunneling nanotubes (TNTs; Abounit et al., 2016; Dieriks et al., 2017). The α-syn uptake by cells depends on the fibrillization (Luk et al., 2009) and oligomeric (Lee et al., 2008) state of α-syn. Oligomers of α-syn have more significant cytotoxicity in recipient cells than soluble monomers of α-syn (Desplats et al., 2009; Emmanouilidou et al., 2010). It was reported that a single intrastriatal injection of synthetic α-syn fibrils initiates a pathological α-syn transmission sufficient to cause PD-like neurodegeneration in non-transgenic mice (Luk et al., 2012b). Furthermore, extracellular α-syn has been shown to activate microglia and astroglia, enhancing neurodegeneration, indicating a cell non-autonomous mechanism (Zhang et al., 2005; Klegeris et al., 2006).
An abundance of synaptic vesicle-related proteins like CD9 (exosomes), Clathrin, AP-2 complex, and dynamin (clathrin-mediated endocytosis), dynein, dynactin, and spectrin (retrograde transport), synaptosomal-associated protein 25, vesicle-associated membrane protein 2, and syntaxin-1 (synaptic vesicle fusion) are present in α-syn-containing protein inclusions purified from post mortem brain tissues from dementia with Lewy bodies (DLB) patients (McCormack et al., 2019). Different models of intercellular transmission of α-syn, not mutually exclusive, have been proposed, such as the α-syn cellular release, movement, and uptake, by different mechanisms, including exocytosis, exosomes, TNTs, glymphatic flow, and endocytosis (Valdinocci et al., 2017). In this regard, different types of vesicles are released from the cells depending on the metabolic and homeostatic cellular status into the extracellular space (extracellular vesicles, EVs), such as exosomes. EVs act as a shuttle for cargo delivery between cells, participate in cell-to-cell communication, and have a potential pathogenic role in the cell-to-cell transmission of toxic aggregated proteins in a neurodegenerative disease context.
Exosomes are small vesicles (40–100 nm in diameter) released into the extracellular space by various cell types, including neurons, astrocytes, microglia, and lymphocytes. Exosomes are generated from multivesicular bodies (MVB) that, after fusion with the plasma membrane, releases intraluminal vesicles – exosomes – containing membrane components, proteins, lipids, and microRNAs (Kowal et al., 2014; Hessvik and Llorente, 2018). Moreover, this EV population can be detected in body fluids such as blood, urine, and CSF (Thery et al., 2006; Keller et al., 2011).
Increasing evidence suggests that the secretion of α-syn, and oligomeric species, is associated with membrane vesicles, as exosomes (Alvarez-Erviti et al., 2011; Danzer et al., 2012; Emmanouilidou and Vekrellis, 2016). The secretion of exosomal α-syn is a calcium-dependent mechanism (Emmanouilidou et al., 2010). The mechanism of exosomes internalization is not entirely decoded, and it seems to depend on the type of recipient cells (Fruhbeis et al., 2013; Nanbo et al., 2013; Svensson et al., 2013; Tian et al., 2014). The pathways caveolin-dependent, clathrin-dependent, and macropinocytosis are not involved in the internalization of exosome-associated oligomeric α-syn (Delenclos et al., 2017). Furthermore, heparin sulfate proteoglycans (HSPGs), transmembrane, and lipid-anchored cell surface receptors modulate the internalization exosomes containing Aβ monomer (Kanekiyo et al., 2011) and α-syn recombinant fibrils (Holmes et al., 2013). However, in contradictory results, another group reported that the deficiency of HSPG did not attenuate the up-taking of α-syn exosomes (Delenclos et al., 2017), suggesting that this pathway is not critical for the α-syn oligomers internalization.
Neurons secrete α-syn by non-canonical cellular pathways that may involve the participation of chaperones UPS19 and DNAJ/HSC70 complex (Lee et al., 2016; Bieri et al., 2018). Although the mechanism of exosomal α-syn secretion and uptake has not been elucidated, it is more apparent that the secreted vesicular α-syn is readily internalized compared to free α-syn oligomers (Delenclos et al., 2017; Gustafsson et al., 2018), conferring toxicity on the neighboring cells (Emmanouilidou et al., 2010; Danzer et al., 2012).
Exosomes may provide a catalytic environment for nucleation of α-syn aggregation (Grey et al., 2015). Vesicles containing-α-syn have been shown to increase the oligomerization status of α-syn (Lee et al., 2005; Grey et al., 2015), and oligomers negatively impact cellular health more than monomers. α-syn species with presumably lost physiological functions or altered aggregation properties may shift the cellular processing toward vesicular secretion. Fluorescent protein tags on the N-terminus of α-syn alter intracellular dynamics (Goncalves and Outeiro, 2013) and induce vesicular secretion (Jang et al., 2010). N-terminal protein tags on α-syn lead to altered membrane-binding properties and may form particularly pathogenic and stable forms of aggregated α-syn that could increase cell-to-cell spreading (Gustafsson et al., 2018).
A minor fraction (0.1–2%) of secreted α-syn are associated with EVs, whereas most of the protein can be found free in the extracellular space (Danzer et al., 2012; Shi et al., 2014). Even though the EV-associated fraction of extracellular α-syn is slight, such vesicles are considered biologically active (van Niel et al., 2006) and molecules in this environment could be more efficiently delivered to other cells (Subra et al., 2010). Interestingly, the exosomal α-syn levels of neuronal origin in plasma correlate significantly with motor dysfunction, a parameter of the severity of the disease (Shi et al., 2014), highlighting the exosomes containing α-syn as a potential biomarker of PD (Figure 1A). α-syn and DJ-1, also known as Parkinson’s disease protein 7 (PARK7), an antioxidant, transcriptional co-activator, and molecular chaperone, presence in plasma neural-derived exosomes were significantly higher in PD patients (Zhao et al., 2018). Recently, it was described that exosomes derivated from saliva also contain α-syn and may be used as a potential biomarker in PD (Cao et al., 2019).
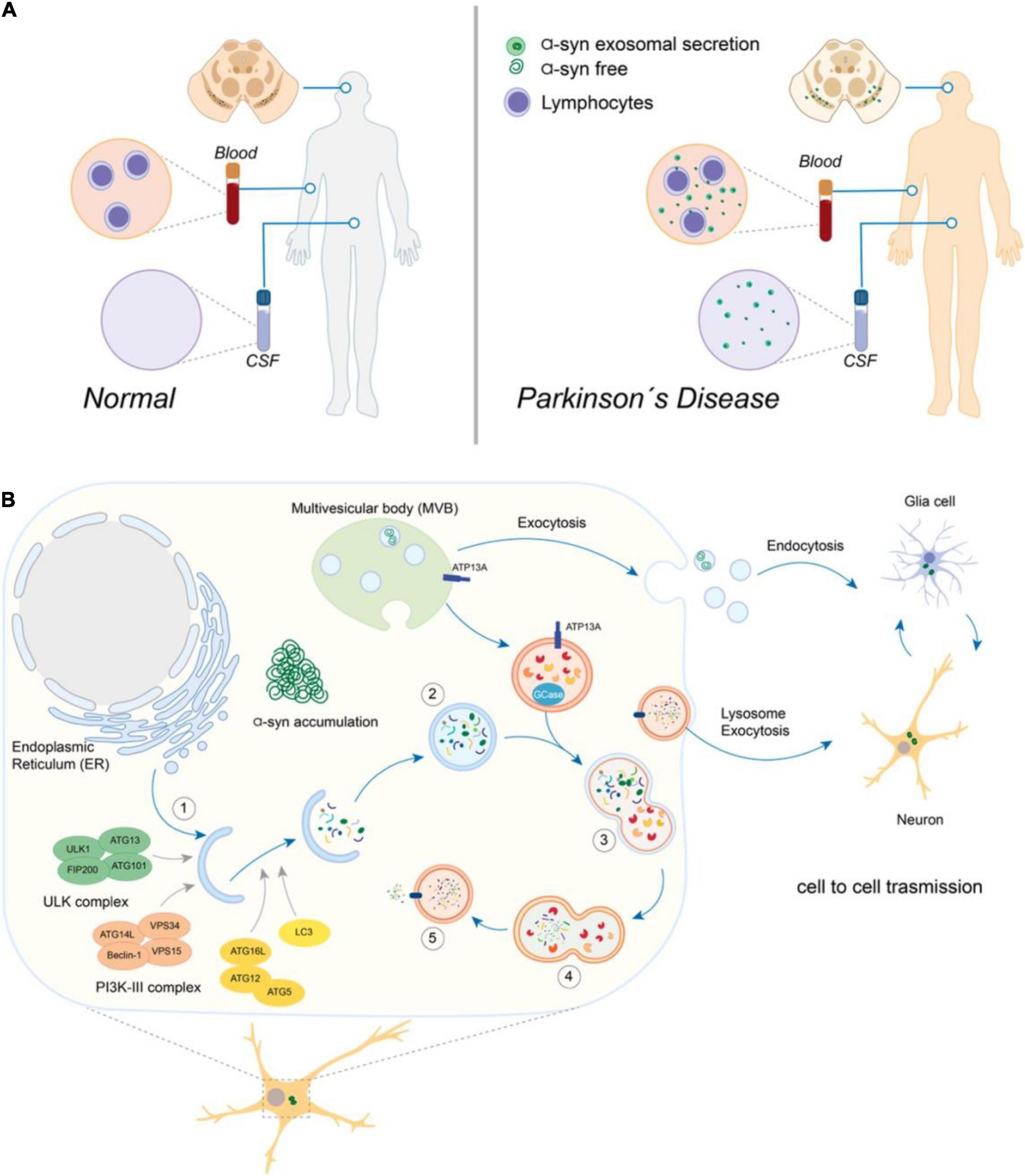
Figure 1. Crosstalk between the autophagy-lysosomal pathway (ALP) and the exosomes secretion in Parkinson’s disease. (A) The presence of soluble free or exosomes-containing α-synuclein (α-syn) derived from Substantia Nigra can be detected in cerebrospinal fluid (CSF) and blood samples from Parkinson’s disease patients, potentially contributing to the early disease’s diagnosis and progression monitoring. (B) Overview of the autophagy-lysosomal pathway (ALP) and exosomes secretion. In (1), the formation of the initial membrane that will originate the double-vesicle autophagosome (2) depends on several complex proteins’ actions (shown in different colors). (3) The fusion of autophagosomes with lysosomes is a final step of the pathway, originating the autolysosome (4), where the substrates are finally degraded into their monomeric components that can be recycled back to the cytosol (5). Multivesicular bodies (MVB) originate the exosomes vesicles, which are secreted by exocytosis and participate in the cell-to-cell transmission of α-syn (neurons and glia cells).
Is the secretion of exosomal α-syn an intercellular transmission mechanism that increases toxicity in the brain?, or could it be a cellular protective response against the intracellular accumulation of α-syn? This response has not been elucidated, and there is controversial literature about it. In this context, it has been described that exosomes isolated from brain tissue of patients with DLB injected into the brain of wild-type mice generate the misfolding of the endogenous α-syn protein (Ngolab et al., 2017). Notable, CSF exosomes from PD patients induce oligomerization of α-syn in a reporter cell line in a dose-dependent manner (Stuendl et al., 2016). About the secretion and uptake of α-syn via EVs in cultured cells, it has been reported that disease-causing mutants, as A53T α-syn, displayed increased association with EVs (Gustafsson et al., 2018). It has been described that γ-syn, another protein family member of synucleins, can be oxidized and initiate α-syn aggregation. γ-syn secreted in exosomes from neuronal cells can be transmitted to glial cells and cause the aggregation of intracellular proteins (Surgucheva et al., 2012).
Interestingly, pramipexole, an agonist of the dopamine receptor family, is used as a treatment for PD patients. After 12 weeks of pharmacological therapy, the patient’s motor performance was statistically improved, and the α-syn content in serum exosomes was lower after the treatment (Luo et al., 2016). Although these results propose a correlation between the α-syn content in serum exosomes and motor symptoms, the mechanism to explain it is still unknown.
Not only neurons would participate in the transmission of α-syn exosomal since microglia also can capture exosomes from the plasma of patients with PD. α-syn induces an increase of exosomal secretion by microglia, and these exosomes showed a high level of MHC class II molecules and TNF-α (Chang et al., 2013). More recently, it was described that the secretion of exosomal human α-syn from the microglia could facilitate its aggregation (Guo et al., 2020) and propagation, possibly through dysregulation of autophagy (Xia et al., 2019).
On the other side, on physiological conditions, it is possible to detect monomers of α-syn, which are degraded by the ubiquitin-proteasome system (UPS; Bennett et al., 1999) and the chaperone-mediated autophagy (CMA; Cuervo et al., 2004; Vogiatzi et al., 2008; Mak et al., 2010). Oligomers, however, are efficiently degraded by the autophagy-lysosomal pathway (ALP; Ebrahimi-Fakhari et al., 2011). Both processes, exosomes secretion of α-syn and degradation by ALP, occur in a coordinate balance (Fussi et al., 2018; Figure 1B). Nevertheless, the mechanism involved in the balance of autophagy and α-syn exosomal secretion in neurons has not been elucidated.
Autophagy (derived from the Greek words for “self” and “eating”) is an evolutionarily conserved lysosomal pathway that digests long-lived proteins, protein aggregates, stress RNA granules, and abnormal cytoplasmic organelles. Based on the type of substrate, mode of cargo recognition, transport, and delivery to the lysosome, three types of autophagy have been described: microautophagy, CMA, and macroautophagy in PD (Martinez-Vicente and Cuervo, 2007; Kenney and Benarroch, 2015). During microautophagy, the cargo is taken into the lysosome or late endosome through its membrane invagination, being quickly degraded in the lysosomal lumen (Frake et al., 2015; Bento et al., 2016). In the CMA, the cargo containing an aminoacidic sequence binds to cytosolic chaperones, is recognized and imported into the lysosomal lumen by a receptor on the lysosomal membrane (Kaushik and Cuervo, 2008). Macroautophagy (hereafter referred to as autophagy or autophagy-lysosomal pathway) is a highly regulated mechanism that forms a double-membrane vesicle called the autophagosome to isolate the cargo that will be degraded (Bento et al., 2016). After maturation, autophagosomes fuse with lysosomes to degrade their content by the activity of lysosomal acid hydrolases (Figure 1B). Lysosomes’ proper function is central to concluding several convergent pathways, including autophagy and endocytosis. Under basal conditions, autophagy is an active quality control process that prevents metabolic and oxidative stress in the cell by degrading aggregated proteins and damaged or dysfunctional organelles. Starvation-induced autophagy is a cellular response to nutrient deprivation that recycles macromolecules to offer substrates for metabolism (Mizushima et al., 2008).
In a pathological context, it has been found that selective autophagy contributes to the clearance of misfolded proteins involved in neurodegenerative diseases such as tau, SOD1, and α-syn (Vidal et al., 2014; Frake et al., 2015). Moreover, evidence has demonstrated a link between PD and mitophagy (selective mitochondrial autophagy). Mitophagy is mediated by binding selective autophagy receptors simultaneously to ubiquitinated proteins in the mitochondria surface and proteins from the autophagy machinery, such as the LC3-II family proteins (Pickles et al., 2018; Conway et al., 2020). The most studied mitophagy pathway is dependent on two proteins, PTEN-induced kinase 1 (PINK1) and Parkin (Narendra et al., 2010). In this pathway, PINK1 accumulates on depolarized mitochondria, triggering the translocation of Parkin from the cytosol, eliciting the ubiquitination of several mitochondrial proteins, including mitofusin 1 and 2 (MFN1 and MFN2), translocase of outer membrane 20 (TOM20), and voltage-dependent anion-selective channel 1 (VDAC1; Bayrhuber et al., 2008; Wang et al., 2011; Sarraf et al., 2013). Notably, mutations in PINK1 and Parkin are associated with familial parkinsonism, while the loss of PINK1 function induces oxidative stress and mitophagy (Valente et al., 2004; Dagda et al., 2009). Moreover, the parkinsonian neurotoxin MPP+ (the active metabolite of MPTP) induces autophagy and mitophagy depending on autophagy proteins ATG5, ATG7, and ATG8, but independently of the protein Beclin 1 (Chu et al., 2007).
Autosomal dominant mutations in the gene LRRK2 encoding the protein leucine-rich repeat kinase 2 are among the most common causing familial PD (Zimprich et al., 2004). Mutations in LRRK2 have been shown to reduce mitochondria trafficking in rat neurons (Godena et al., 2014; Hsieh et al., 2016), impair the mitophagy activity in PD-derived cells (Bonello et al., 2019; Wauters et al., 2020), and increase aggregation of α-syn in mice models and human iPSC-derived dopaminergic neurons (Bieri et al., 2019). In a recent work, researchers studied α-syn spreading levels in CSF from patients carrying different DLB and PD mutations using an α-syn real-time amplification assay (Brockmann et al., 2021). Interestingly, CSF samples from patients harboring mutations in PINK1 or Parkin did not show positive α-syn seeding activity. However, CFS samples from LRRK2 PD patients (78%) showed an elevated α-syn positivity, only exceeded by CSF samples from DLB (100%) or PD (93%) patients carrying GBA1 mutations (Brockmann et al., 2021). Notably, this higher α-syn seeding activity in CSF from GBA1 patients was associated with lower levels of proteins related to α-syn clearance, including autophagy, lysosomal function, and endocytosis pathways in the same samples, suggesting GBA1 mutations promote a negative correlation between α-syn accumulation and degradation pathways, mainly associated with lysosomal dysfunction (Brockmann et al., 2021). This work also found decreased levels of proteins from the UPS and neurosecretion processes in CSF from GBA1-carrying patients, which increase the urge to elucidate the mechanisms involved in this broad reduction in degradation pathways proteome (Brockmann et al., 2021) and to check if this data is reflected in CNS samples.
The failure of the protein quality control systems, especially lysosomal-dependent degradation, promotes the accumulation of α-syn (Desplats et al., 2009). Heterozygous mutations in the GBA1 gene encoding lysosomal enzyme GCase are strong risk factors for PD (Avenali et al., 2020). GCase is an N-glycosylated protein synthesized and transported in vesicles from the ER-to-Golgi apparatus, where it is correctly folded through a maturation process before reaching the lysosomes. α-syn aggregation generated by wild-type SNCA triplication in PD patients iPSC-derived dopaminergic neurons was reported to disrupt the ER-GA trafficking by inhibiting the SNARE protein ykt6 (Cuddy et al., 2019), depleting lysosomes from acid hydrolases, and increasing the accumulation of insoluble immature GCase in ER (Stojkovska et al., 2021). Interestingly, using a pharmacological enhancer of ER proteostasis plus a farnesyltransferase inhibitor (FTI), which restores ykt6 activity, was reestablished GCase maturation and lysosomal activity, becoming a promising therapeutic strategy for future studies in synucleinopathies (Stojkovska et al., 2021).
Recent works have suggested that the secretion of exosomes containing α-syn could result as a protection mechanism against the blockage of autophagy-dependent α-syn clearance (Fussi et al., 2018). In particular, the silencing of ATG5, a key protein involved in the extension of the phagophore membrane in autophagic vesicles (Pyo et al., 2005), increases the secretion of α-syn via exosomes, which are associated with a decrease in cell death α-syn induced (Fussi et al., 2018). In accordance, the inhibition of lysosomal function in α-syn overexpressing neural cell lines generated an increase of exosomal secretion of α-syn, promoting a cell-to-cell transfer of α-syn (Alvarez-Erviti et al., 2011). Other evidence also shows that the ALP inhibition reduces intracellular α-syn while increasing the secretion of smaller oligomers, exacerbating the uptake, inflammation, and cellular damage (Poehler et al., 2014). Moreover, has been reported a secretion of aggregated α-syn by exosomes and Rab11a-associated pathways and by membrane shedding (Poehler et al., 2014). It was confirmed that the ALP inhibition promotes the release and transmission of α-syn via EVs with a hybrid autophagosome-exosome phenotype, increasing the ratio of extracellular α-syn/intracellular α-syn and its association with EV in neuronal cells (Minakaki et al., 2018). GCase loss-of-function was also associated with α-syn secretion. Studies in transgenic mice harboring the human mutation A53T in SNCA found that inhibition of GCase increases the exosome-associated α-syn oligomers release (Papadopoulos et al., 2018). The plasma exosomal/total α-syn ratio is associated with GCase activity, and it correlates with severity (motor deficiency) in PD patients (Cerri et al., 2018; Johnson et al., 2020), proposing the link between lysosomal dysfunction with increased exosome secretion. Similar results were reported in fibroblasts derived from PD patients with or without GBA1, in which defective GCase activity increased the release of exosomes (Cerri et al., 2021). Isolated exosomes from these cells caused increased levels of phospho-α-syn in SH-SY5Y recipient cells, overexpressing wild-type α-syn (Cerri et al., 2021). Interesting, this effect was not due to a seeding effect since fibroblasts are α-syn-free. The researchers hypothesize that fibroblast-derived from patients harboring GBA1 mutations promote changes in the lipid composition of recipient cells, which may account for the increased phospho-α-syn, posttraductional modification that increases the formation of insolubleα-syn forms (Canerina-Amaro et al., 2019). Besides releasing exosomes, MVBs can be eliminated through the ALP by a direct fusion with lysosomes or autophagosomes (Fader et al., 2008; Vanlandingham and Ceresa, 2009; Szatmari et al., 2014; Teixeira et al., 2021). α-syn itself can disturb the ALP activity, promoting potential positive feedback to its secretion. Notably, α-syn fibrils have been shown to impair lysosomes’ morphology from inside the organelle lumen, reducing the ALP-dependent clearance of aggregates and defective organelles. Moreover, lysosomes filled with α-syn fibrils can be transferred to neighboring cells through TNTs or secretion vesicles, contributing to the disease’s spread (Dilsizoglu Senol et al., 2021). There is evidence for a loop between the lysosome and α-syn proteoforms (Wildburger et al., 2020).
Some genes encoding proteins involved in intracellular vesicle trafficking and lysosome transport are risk genes associated with PD (Abeliovich and Gitler, 2016; Mazzulli et al., 2016a). For example, Kufor-Rakeb syndrome (KRS) is caused by an autosomal recessive mutation in the PARK9 gene encoding ATP13A2 (transmembrane lysosomal type 5 P-type ATPase protein) characterized by juvenile-onset parkinsonism. Interestingly, a mutation in this gene was described in a Chilean family patient for the first time (Ramirez et al., 2006). PARK9 encodes a lysosomal ATPase involved in cation homeostasis, and its loss of function leads to lysosomal dysfunction (Gitler et al., 2009; Kong et al., 2014; Tsunemi et al., 2014). Interestingly, in Caenorhabditis elegans and dopamine cell culture models, it was described that ATP13A2 would have a protective role against the accumulation of misfolded α-syn and cellular toxicity (Gitler et al., 2009). However, the overexpression of ATP13A2 increases the release of exosomes, promoting the secretion of α-syn in primary cortical neurons (Tsunemi et al., 2014). The evidence suggests that the ATP13A2 protein regulates the release of α-syn via EVs through the modification of the biogenesis of exosomes by a functional interaction with the lysosomal sorting complex required for transport (ESCRT; Tsunemi et al., 2014). Additionally, the enhanced secretion of exosome-associated α-syn may explain the increased viability in neurons of the SNpc in sporadic PD patients by overexpressing ATP13A2 (Kong et al., 2014). A recent work has shown that PD mutations in ATP13A2 increase α-syn intracellular accumulation by impairing lysosome exocytosis using iPSC-derived neurons from PD patients (Tsunemi et al., 2019). The mechanism by ATP13A2 modulates lysosomal exocytosis is by mediating Ca2+ homeostasis in these organelles. Interestingly, a pharmacological agonist of the lysosomal Ca2+ channel, TRPML1, recovers lysosomal exocytosis, correcting α-syn secretion defects and decreasing intracellular accumulation in ATP13A2 patient neurons (Tsunemi et al., 2019).
What is the contribution of neighboring cells of neurons in PD? Microglia isolated from adult mice, in contrast to microglia from young mice, display phagocytosis deficits of free and exosome-associated α-syn oligomers (Bliederhaeuser et al., 2016). The neuronal α-syn secreted by exosomes or lysosomal exocytosis is partially endocytosed by astrocytes, which contribute to reducing the α-syn spread between neurons. Indeed, degradation of α-syn is more efficient in astrocytes than neurons (Tsunemi et al., 2020). However, the iPSC-derived astrocytic protection against the α-syn accumulation and propagation is partially lost by ATP13A2 mutations, resulting in the increased accumulation and propagation of α-syn between neurons (Tsunemi et al., 2020), suggesting that astrocytic lysosomal dysfunction indirectly contributes to the α-syn neuronal pathology.
Moreover, impaired biogenesis of MVBs by a dominant-negative mutant of vacuolar protein sorting 4 (VPS4) interferes with the lysosomal targeting of α-syn and facilitates α-syn secretion (Hasegawa et al., 2011). The hypersecretion of α-syn in VPS4-defective cells was restored by the functional disruption of recycling endosome regulator Rab11a. VPS4, a master regulator of MVBs sorting, may serve as a determinant of lysosomal targeting or extracellular secretion of α-syn (Hasegawa et al., 2011). Another member of the endosomal protein sorting, VPS35, is also associated with PD (Vilarino-Guell et al., 2011). VPS35 is part of the retromer complex, which mediates the endosome-to-Golgi recovery of membrane proteins. The VPS35 D620N mutation causes an autosomal-dominant form of PD, and the cells expressing the mutant form have impaired autophagy. The defects in autophagy can be explained in part by the abnormal traffic of the transmembrane autophagy protein ATG9A (Zavodszky et al., 2014).
Another traffic protein associated with PD is Secretory Carrier Membrane Protein 5 (SCAMP5), a regulator of membrane trafficking enriched in the brain, identified as an autophagy inhibitor that promotes exosomal secretion of α-syn (Yang et al., 2017). SCAMP5 is a novel coordinator of autophagy and exosome secretion, induced under protein stress by Bafilomycin A1 to clear toxic proteins via the exosomes rather than ALP (Yang et al., 2017).
All these results support the idea that exists a connection between autophagy, lysosomal homeostasis, and α-syn exosomes secretion on the PD progression (Xu et al., 2018). Moreover, autophagy modification (gain and loss function) impacts exosomes released into the extracellular space in vitro (Hu et al., 2020). Although, the full mechanism underlying these processes in vivo and the effect on the progression of the PD remains poorly understood (Figure 2). Overall, several lines of evidence propose that a correction in lysosomal function can boost dopaminergic neurons’ survival in PD, avoiding α-syn aggregation. However, it is open to whether the increase in the α-syn secretion, by exosomes or other vesicles kinds, can result in a progression spread of the disease in an in vivo long-term study.
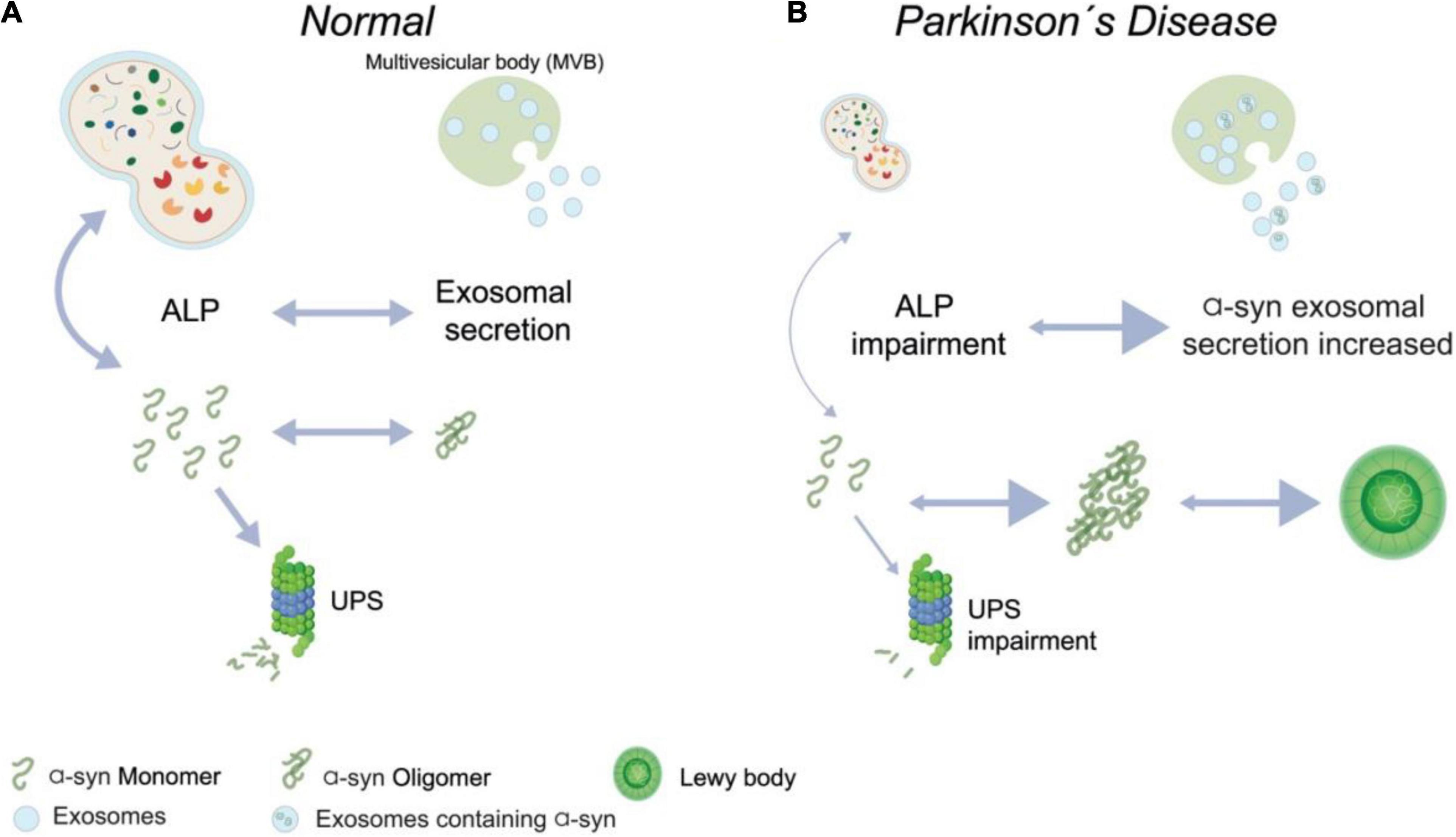
Figure 2. Contribution of autophagy-lysosomal pathway and α-syn secreted in Parkinson’s disease. (A) In normal conditions, it is possible to detect monomers of alpha-synuclein (α-syn), which are degraded by Ubiquitin Proteasome System (UPS), and oligomers of α-syn are efficiently degraded by Autophagy-Lysosomal (ALP) pathway or can be secreted by exosomes. Both degradation and secretion processes occur in a coordinate balance. (B) On Parkinson’s Disease condition, abundant oligomers and fibrils of α-syn are formed, and it less degraded by autophagy due to an impairment of this pathway, and it is possible to observe an increased secretion of α-syn by exosomes.
As mentioned, oligomers and fibrils α-syn degradation is mediated by autophagy, connecting the role of lysosomes to the etiology/progression of PD (Webb et al., 2003; Lee et al., 2004). Concerning the above, the expression of genes from the autophagy pathway [UNC- 51- like kinase (ULK) 3, autophagy-related (Atg) 2A, Atg4B, Atg5, Atg16L1, and histone deacetylase 6] were evaluated in peripheral blood mononuclear cells (PBMCs) of patients with PD. Researchers observed a decrease in the expression of autophagy regulatory components in patients with PD, while they reported an increase of α-syn protein levels in PBMCs compared to controls (Miki et al., 2018). However, the comprehensive mechanisms of the dynamic interaction of ALP-MVBs for the secretion of α-syn via EVs have not been fully elucidated. It has been recently described that a significant percentage of the proteins detected in tissue-purified Lewy bodies from DLB patients and cytoplasmic glial inclusions (CGI) of oligodendrocytes from multiple systemic atrophy (MSA) patients are synaptic vesicle proteins, including CD9 associated with exosomes (McCormack et al., 2019). This fact suggests that the misfolding or accumulation of α-syn, characteristic of synucleinopathies, contributes to the vesicle-mediated transport of these protein inclusions (McCormack et al., 2019). A recent study demonstrated that the inhibition of dynamin-related protein 1 (Drp1) improved both mitochondrial function and autophagic flux in experimental models of α-syn (Fan et al., 2019). The following key step is to determine if the Drp1 inhibition confers neuroprotection through the abolished autophagic impairment induced by α-syn in in vivo models of PD.
Several pharmacological agents targeting ALP components, especially lysosomal function, are active research topics in preclinical and clinical phases to PD and other synucleinopathies. They include the previously mentioned FTI, which restores the SNARE ykt6 activity, reestablishing lysosomal hydrolases maturation, and finally, the lysosomal activity (Stojkovska et al., 2021) pharmacological agonists of lysosomal Ca2+ channel, TRPML1, which has been shown to restore lysosomal exocytosis, enhancing α-syn secretion and decreasing accumulation in ATP13A2 patient iPSC-derived neurons (Tsunemi et al., 2019); or the ambroxol, a cough syrup approved by the FDA since 1971, which has been reported to reduce α-synuclein levels in vitro and in vivo (Migdalska-Richards et al., 2016), and to increase GCase expression and activity (Migdalska-Richards et al., 2017). Ambroxol was shown to restore lysosomal exocytosis (Magalhaes et al., 2018) and promote ER folding. Recently, ambroxol treatment in PD patients harboring or not GBA1 mutations has shown promising results regarding α-syn secretion in CSF (Mullin et al., 2020). However, although these drugs are promising hopes, they need to be tested in different mutations associated with PD, considering the broad clinical and physiological variability between them (Cerri et al., 2018; Johnson et al., 2020).
This review summarized the antecedents that demonstrate altered autophagy in PD and some evidence that proposes a link between ALP components and the exosomal secretion of α-syn. To elucidate the mechanisms that explain the relation between ALP and the secretion of EVs in PD is still a field in study. However, some clues of the crosstalk between exosomes and autophagy have been proposed [review in Xu et al. (2018), Gudbergsson and Johnsen (2019), Buratta et al. (2020), Xing et al. (2021)].
The origin of extracellular vesicles can offer additional information. For example, mitochondrial-derived vesicles are a candidate as biomarkers in body fluids of PD patients may provide clues to understand the association between mitochondrial dysfunction and systemic inflammation in PD (Picca et al., 2019). Interesting proposals are in the therapy field based on autophagic degradation and exosomal secretion. The development of an α-syn nano-scavenger for PD capable of stimulating nuclear translocation of TFEB (master regulator of autophagy), promoting autophagy and calcium-dependent exosome secretion for the clearance of α-syn (Liu et al., 2020). While α-syn expression can be reduced by antisense oligonucleotides (ASOs), the big challenge is delivering ASOs efficiently and safely into the neurons. Exosomes can be a safe and highly effective ASO delivery method (Yang et al., 2021). Promising ALP and exosomal secretion research are developed on in vitro models. However, further validations in animal models and physiology-related conditions are required.
Recently, in vitro study reported that proteins from SARS-CoV-2, which causes COVID-19, interact with α-syn, speeding up the formation of amyloid plaques (Semerdzhiev et al., 2021) open an interesting research field associating virus infections with PD or other neurodegenerative diseases development.
DS, MC-O, JA, MN, and RV wrote and edited the manuscript. DS and RV prepared the figures. DS, MN, and RV planned the manuscript. All authors contributed equally to the critical reading of the final manuscript, including figures.
This work was directly funded by the FONDAP program 15150012, Millennium Institute P09-015-F, and FONDECYT 1191003 (RV).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abeliovich, A., and Gitler, A. D. (2016). Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216. doi: 10.1038/nature20414
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016). Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
Askanas, V., Engel, W. K., Alvarez, R. B., McFerrin, J., and Broccolini, A. (2000). Novel immunolocalization of alpha-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J. Neuropathol. Exp. Neurol. 59, 592–598. doi: 10.1093/jnen/59.7.592
Avenali, M., Blandini, F., and Cerri, S. (2020). Glucocerebrosidase defects as a major risk factor for Parkinson’s disease. Front. Aging Neurosci. 12:97. doi: 10.3389/fnagi.2020.00097
Bayrhuber, M., Meins, T., Habeck, M., Becker, S., Giller, K., Villinger, S., et al. (2008). Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 105, 15370–15375. doi: 10.1073/pnas.0808115105
Benarroch, E. E. (2007). Lipid rafts, protein scaffolds, and neurologic disease. Neurology 69, 1635–1639. doi: 10.1212/01.wnl.0000279590.22544.c3
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Bento, C. F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., et al. (2016). Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713. doi: 10.1146/annurev-biochem-060815-014556
Betarbet, R., Sherer, T. B., and Greenamyre, J. T. (2002). Animal models of Parkinson’s disease. Bioessays 24, 308–318. doi: 10.1002/bies.10067
Bhatnagar, A., Sheffler, D. J., Kroeze, W. K., Compton-Toth, B., and Roth, B. L. (2004). Caveolin-1 interacts with 5-HT2A serotonin receptors and profoundly modulates the signaling of selected Galphaq-coupled protein receptors. J. Biol. Chem. 279, 34614–34623. doi: 10.1074/jbc.M404673200
Bieri, G., Brahic, M., Bousset, L., Couthouis, J., Kramer, N. J., Ma, R., et al. (2019). LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 137, 961–980. doi: 10.1007/s00401-019-01995-0
Bieri, G., Gitler, A. D., and Brahic, M. (2018). Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 109(Pt B), 219–225. doi: 10.1016/j.nbd.2017.03.007
Bilderback, T. R., Gazula, V. R., Lisanti, M. P., and Dobrowsky, R. T. (1999). Caveolin interacts with Trk A and p75(NTR) and regulates neurotrophin signaling pathways. J. Biol. Chem. 274, 257–263. doi: 10.1074/jbc.274.1.257
Bliederhaeuser, C., Grozdanov, V., Speidel, A., Zondler, L., Ruf, W. P., Bayer, H., et al. (2016). Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 131, 379–391. doi: 10.1007/s00401-015-1504-2
Bonello, F., Hassoun, S. M., Mouton-Liger, F., Shin, Y. S., Muscat, A., Tesson, C., et al. (2019). LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 28, 1645–1660. doi: 10.1093/hmg/ddz004
Boulware, M. I., Kordasiewicz, H., and Mermelstein, P. G. (2007). Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J. Neurosci. 27, 9941–9950. doi: 10.1523/JNEUROSCI.1647-07.2007
Bras, I. C., Dominguez-Meijide, A., Gerhardt, E., Koss, D., Lazaro, D. F., Santos, P. I., et al. (2020). Synucleinopathies: where we are and where we need to go. J. Neurochem. 153, 433–454. doi: 10.1111/jnc.14965
Brockmann, K., Quadalti, C., Lerche, S., Rossi, M., Wurster, I., Baiardi, S., et al. (2021). Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 9:175. doi: 10.1186/s40478-021-01276-6
Buratta, S., Tancini, B., Sagini, K., Delo, F., Chiaradia, E., Urbanelli, L., et al. (2020). Lysosomal exocytosis, exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int. J. Mol. Sci. 21:2576. doi: 10.3390/ijms21072576
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Cacabelos, R. (2017). Parkinson’s disease: from pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 18:551. doi: 10.3390/ijms18030551
Canerina-Amaro, A., Pereda, D., Diaz, M., Rodriguez-Barreto, D., Casanas-Sanchez, V., Heffer, M., et al. (2019). Differential aggregation and phosphorylation of alpha synuclein in membrane compartments associated with Parkinson disease. Front. Neurosci. 13:382. doi: 10.3389/fnins.2019.00382
Cao, Z., Wu, Y., Liu, G., Jiang, Y., Wang, X., Wang, Z., et al. (2019). alpha-Synuclein in salivary extracellular vesicles as a potential biomarker of Parkinson’s disease. Neurosci. Lett. 696, 114–120. doi: 10.1016/j.neulet.2018.12.030
Castello, P. R., Drechsel, D. A., and Patel, M. (2007). Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J. Biol. Chem. 282, 14186–14193. doi: 10.1074/jbc.M700827200
Cerri, S., Ghezzi, C., Ongari, G., Croce, S., Avenali, M., Zangaglia, R., et al. (2021). GBA mutations influence the release and pathological effects of small extracellular vesicles from fibroblasts of patients with Parkinson’s disease. Int. J. Mol. Sci. 22:2215. doi: 10.3390/ijms22042215
Cerri, S., Ghezzi, C., Sampieri, M., Siani, F., Avenali, M., Dornini, G., et al. (2018). The exosomal/total alpha-synuclein ratio in plasma is associated with glucocerebrosidase activity and correlates with measures of disease severity in PD patients. Front. Cell Neurosci. 12:125. doi: 10.3389/fncel.2018.00125
Cerri, S., Mus, L., and Blandini, F. (2019). Parkinson’s disease in women and men: what’s the difference? J. Parkinsons Dis. 9, 501–515. doi: 10.3233/JPD-191683
Chang, C., Lang, H., Geng, N., Wang, J., Li, N., and Wang, X. (2013). Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci. Lett. 548, 190–195. doi: 10.1016/j.neulet.2013.06.009
Chia, S. J., Tan, E. K., and Chao, Y. X. (2020). Historical perspective: models of Parkinson’s disease. Int. J. Mol. Sci. 21:2464. doi: 10.3390/ijms21072464
Chu, C. T., Zhu, J., and Dagda, R. (2007). Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 3, 663–666. doi: 10.4161/auto.4625
Colla, E., Panattoni, G., Ricci, A., Rizzi, C., Rota, L., Carucci, N., et al. (2018). Toxic properties of microsome-associated alpha-synuclein species in mouse primary neurons. Neurobiol. Dis. 111, 36–47. doi: 10.1016/j.nbd.2017.12.004
Conway, O., Akpinar, H. A., Rogov, V. V., and Kirkin, V. (2020). Selective autophagy receptors in neuronal health and disease. J. Mol. Biol. 432, 2483–2509. doi: 10.1016/j.jmb.2019.10.013
Cuddy, L. K., Wani, W. Y., Morella, M. L., Pitcairn, C., Tsutsumi, K., Fredriksen, K., et al. (2019). Stress-induced cellular clearance is mediated by the SNARE protein ykt6 and disrupted by alpha-synuclein. Neuron 104, 869–884.e11. doi: 10.1016/j.neuron.2019.09.001
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Dagda, R. K., Cherra, S. J. III, Kulich, S. M., Tandon, A., Park, D., and Chu, C. T. (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855. doi: 10.1074/jbc.M808515200
Danzer, K. M., Kranich, L. R., Ruf, W. P., Cagsal-Getkin, O., Winslow, A. R., Zhu, L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7:42. doi: 10.1186/1750-1326-7-42
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. doi: 10.1016/s0896-6273(03)00568-3
Delenclos, M., Trendafilova, T., Mahesh, D., Baine, A. M., Moussaud, S., Yan, I. K., et al. (2017). Investigation of endocytic pathways for the internalization of exosome-associated oligomeric alpha-synuclein. Front. Neurosci. 11:172. doi: 10.3389/fnins.2017.00172
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Dieriks, B. V., Park, T. I., Fourie, C., Faull, R. L., Dragunow, M., and Curtis, M. A. (2017). alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 7:42984. doi: 10.1038/srep42984
Dilsizoglu Senol, A., Samarani, M., Syan, S., Guardia, C. M., Nonaka, T., Liv, N., et al. (2021). alpha-Synuclein fibrils subvert lysosome structure and function for the propagation of protein misfolding between cells through tunneling nanotubes. PLoS Biol. 19:e3001287. doi: 10.1371/journal.pbio.3001287
Dorsey, E. R., Sherer, T., Okun, M. S., and Bloem, B. R. (2018). The emerging evidence of the Parkinson pandemic. J. Parkinsons Dis. 8, S3–S8. doi: 10.3233/JPD-181474
Duda, J. E., Giasson, B. I., Chen, Q., Gur, T. L., Hurtig, H. I., Stern, M. B., et al. (2000). Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am. J. Pathol. 157, 1439–1445. doi: 10.1016/S0002-9440(10)64781-5
Ebrahimi-Fakhari, D., Cantuti-Castelvetri, I., Fan, Z., Rockenstein, E., Masliah, E., Hyman, B. T., et al. (2011). Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 31, 14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011
El-Agnaf, O. M., Salem, S. A., Paleologou, K. E., Cooper, L. J., Fullwood, N. J., Gibson, M. J., et al. (2003). Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–1947. doi: 10.1096/fj.03-0098fje
Emmanouilidou, E., and Vekrellis, K. (2016). Exocytosis and Spreading of Normal and aberrant alpha-Synuclein. Brain Pathol. 26, 398–403. doi: 10.1111/bpa.12373
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Fader, C. M., Sanchez, D., Furlan, M., and Colombo, M. I. (2008). Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 9, 230–250. doi: 10.1111/j.1600-0854.2007.00677.x
Fakhree, M. A. A., Konings, I. B. M., Kole, J., Cambi, A., Blum, C., and Claessens, M. (2021). The localization of alpha-synuclein in the endocytic pathway. Neuroscience 457, 186–195. doi: 10.1016/j.neuroscience.2021.01.017
Fan, R. Z., Guo, M., Luo, S., Cui, M., and Tieu, K. (2019). Exosome release and neuropathology induced by alpha-synuclein: new insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol. Commun. 7:184. doi: 10.1186/s40478-019-0821-4
Fearnley, J. M., and Lees, A. J. (1991). Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5), 2283–2301. doi: 10.1093/brain/114.5.2283
Forland, M. G., Ohrfelt, A., Dalen, I., Tysnes, O. B., Blennow, K., Zetterberg, H., et al. (2018). Evolution of cerebrospinal fluid total alpha-synuclein in Parkinson’s disease. Parkinsonism Relat. Disord. 49, 4–8. doi: 10.1016/j.parkreldis.2018.01.018
Frake, R. A., Ricketts, T., Menzies, F. M., and Rubinsztein, D. C. (2015). Autophagy and neurodegeneration. J. Clin. Invest. 125, 65–74. doi: 10.1172/JCI73944
Francesconi, A., Kumari, R., and Zukin, R. S. (2009). Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J. Neurosci. 29, 3590–3602. doi: 10.1523/JNEUROSCI.5824-08.2009
Fruhbeis, C., Frohlich, D., Kuo, W. P., Amphornrat, J., Thilemann, S., Saab, A. S., et al. (2013). Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11:e1001604. doi: 10.1371/journal.pbio.1001604
Fuchs, J., Nilsson, C., Kachergus, J., Munz, M., Larsson, E. M., Schule, B., et al. (2007). Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916–922. doi: 10.1212/01.wnl.0000254458.17630.c5
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Fussi, N., Hollerhage, M., Chakroun, T., Nykanen, N. P., Rosler, T. W., Koeglsperger, T., et al. (2018). Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 9:757. doi: 10.1038/s41419-018-0816-2
Galvagnion, C. (2017). The role of lipids interacting with alpha-synuclein in the pathogenesis of Parkinson’s disease. J. Parkinsons Dis. 7, 433–450. doi: 10.3233/JPD-171103
Gitler, A. D., Chesi, A., Geddie, M. L., Strathearn, K. E., Hamamichi, S., Hill, K. J., et al. (2009). Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41, 308–315. doi: 10.1038/ng.300
Godena, V. K., Brookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Golbe, L. I., Miller, D. C., and Duvoisin, R. C. (1990). Autosomal dominant lewy-body Parkinson’s disease. Adv. Neurol. 53, 287–292.
Goncalves, S., and Outeiro, T. F. (2013). Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol. Neurobiol. 47, 1081–1092. doi: 10.1007/s12035-013-8406-x
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., et al. (2010). alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U.S.A. 107, 19573–19578. doi: 10.1073/pnas.1005005107
Grey, M., Dunning, C. J., Gaspar, R., Grey, C., Brundin, P., Sparr, E., et al. (2015). Acceleration of alpha-synuclein aggregation by exosomes. J. Biol. Chem. 290, 2969–2982. doi: 10.1074/jbc.M114.585703
Gudbergsson, J. M., and Johnsen, K. B. (2019). Exosomes and autophagy: rekindling the vesicular waste hypothesis. J. Cell Commun. Signal. 13, 443–450. doi: 10.1007/s12079-019-00524-8
Guo, M., Wang, J., Zhao, Y., Feng, Y., Han, S., Dong, Q., et al. (2020). Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 143, 1476–1497. doi: 10.1093/brain/awaa090
Gureviciene, I., Gurevicius, K., and Tanila, H. (2007). Role of alpha-synuclein in synaptic glutamate release. Neurobiol. Dis. 28, 83–89. doi: 10.1016/j.nbd.2007.06.016
Gustafsson, G., Loov, C., Persson, E., Lazaro, D. F., Takeda, S., Bergstrom, J., et al. (2018). Secretion and uptake of alpha-synuclein via extracellular vesicles in cultured cells. Cell Mol. Neurobiol. 38, 1539–1550. doi: 10.1007/s10571-018-0622-5
Ha, T. Y., Choi, Y. R., Noh, H. R., Cha, S. H., Kim, J. B., and Park, S. M. (2021). Age-related increase in caveolin-1 expression facilitates cell-to-cell transmission of alpha-synuclein in neurons. Mol. Brain 14:122. doi: 10.1186/s13041-021-00834-2
Han, T. U., Sam, R., and Sidransky, E. (2020). Small molecule chaperones for the treatment of gaucher disease and GBA1-associated Parkinson disease. Front. Cell Dev. Biol. 8:271. doi: 10.3389/fcell.2020.00271
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Hanzal-Bayer, M. F., and Hancock, J. F. (2007). Lipid rafts and membrane traffic. FEBS Lett. 581, 2098–2104. doi: 10.1016/j.febslet.2007.03.019
Hasegawa, T., Konno, M., Baba, T., Sugeno, N., Kikuchi, A., Kobayashi, M., et al. (2011). The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha-synuclein. PLoS One 6:e29460. doi: 10.1371/journal.pone.0029460
Hashimoto, M., Takenouchi, T., Rockenstein, E., and Masliah, E. (2003). Alpha-synuclein up-regulates expression of caveolin-1 and down-regulates extracellular signal-regulated kinase activity in B103 neuroblastoma cells: role in the pathogenesis of Parkinson’s disease. J. Neurochem. 85, 1468–1479. doi: 10.1046/j.1471-4159.2003.01791.x
Hessvik, N. P., and Llorente, A. (2018). Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 75, 193–208. doi: 10.1007/s00018-017-2595-9
Hoglinger, G. U., Feger, J., Prigent, A., Michel, P. P., Parain, K., Champy, P., et al. (2003). Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J. Neurochem. 84, 491–502. doi: 10.1046/j.1471-4159.2003.01533.x
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Hsieh, C. H., Shaltouki, A., Gonzalez, A. E., Bettencourt da Cruz, A., Burbulla, L. F., St Lawrence, E., et al. (2016). Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19, 709–724. doi: 10.1016/j.stem.2016.08.002
Hu, S. Q., Zhang, Q. C., Meng, Q. B., Hu, A. N., Zou, J. P., and Li, X. L. (2020). Autophagy regulates exosome secretion in rat nucleus pulposus cells via the RhoC/ROCK2 pathway. Exp. Cell Res. 395:112239. doi: 10.1016/j.yexcr.2020.112239
Huh, Y. E., Park, H., Chiang, M. S. R., Tuncali, I., Liu, G., Locascio, J. J., et al. (2021). Glucosylceramide in cerebrospinal fluid of patients with GBA-associated and idiopathic Parkinson’s disease enrolled in PPMI. NPJ Parkinsons Dis. 7:102. doi: 10.1038/s41531-021-00241-3
Jang, A., Lee, H. J., Suk, J. E., Jung, J. W., Kim, K. P., and Lee, S. J. (2010). Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
Johnson, P. H., Weinreb, N. J., Cloyd, J. C., Tuite, P. J., and Kartha, R. V. (2020). GBA1 mutations: prospects for exosomal biomarkers in alpha-synuclein pathologies. Mol. Genet. Metab. 129, 35–46. doi: 10.1016/j.ymgme.2019.10.006
Kanekiyo, T., Zhang, J., Liu, Q., Liu, C. C., Zhang, L., and Bu, G. (2011). Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci. 31, 1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011
Kang, M. J., Chung, Y. H., Hwang, C. I., Murata, M., Fujimoto, T., Mook-Jung, I. H., et al. (2006). Caveolin-1 upregulation in senescent neurons alters amyloid precursor protein processing. Exp. Mol. Med. 38, 126–133. doi: 10.1038/emm.2006.16
Kaushik, S., and Cuervo, A. M. (2008). Chaperone-mediated autophagy. Methods Mol. Biol. 445, 227–244. doi: 10.1007/978-1-59745-157-4_15
Keller, S., Ridinger, J., Rupp, A. K., Janssen, J. W., and Altevogt, P. (2011). Body fluid derived exosomes as a novel template for clinical diagnostics. J. Transl. Med. 9:86. doi: 10.1186/1479-5876-9-86
Kenney, D. L., and Benarroch, E. E. (2015). The autophagy-lysosomal pathway: general concepts and clinical implications. Neurology 85, 634–645. doi: 10.1212/WNL.0000000000001860
Kim, S., Jeon, B. S., Heo, C., Im, P. S., Ahn, T. B., Seo, J. H., et al. (2004). Alpha-synuclein induces apoptosis by altered expression in human peripheral lymphocyte in Parkinson’s disease. FASEB J. 18, 1615–1617. doi: 10.1096/fj.04-1917fje
Klegeris, A., Giasson, B. I., Zhang, H., Maguire, J., Pelech, S., and McGeer, P. L. (2006). Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 20, 2000–2008. doi: 10.1096/fj.06-6183com
Kong, S. M., Chan, B. K., Park, J. S., Hill, K. J., Aitken, J. B., Cottle, L., et al. (2014). Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 23, 2816–2833. doi: 10.1093/hmg/ddu099
Kowal, J., Tkach, M., and Thery, C. (2014). Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 29, 116–125. doi: 10.1016/j.ceb.2014.05.004
Langston, J. W. (2006). The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann. Neurol. 59, 591–596. doi: 10.1002/ana.20834
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lautenschlager, J., Stephens, A. D., Fusco, G., Strohl, F., Curry, N., Zacharopoulou, M., et al. (2018). C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 9:712. doi: 10.1038/s41467-018-03111-4
Lazaro, D. F., Rodrigues, E. F., Langohr, R., Shahpasandzadeh, H., Ribeiro, T., Guerreiro, P., et al. (2014). Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 10:e1004741. doi: 10.1371/journal.pgen.1004741
Lee, H. J., Khoshaghideh, F., Patel, S., and Lee, S. J. (2004). Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 24, 1888–1896. doi: 10.1523/JNEUROSCI.3809-03.2004
Lee, H. J., Patel, S., and Lee, S. J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005
Lee, H. J., Suk, J. E., Bae, E. J., Lee, J. H., Paik, S. R., and Lee, S. J. (2008). Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849. doi: 10.1016/j.biocel.2008.01.017
Lee, J. A., Choi, D. I., Choi, J. Y., Kim, S. O., Cho, K. A., Lee, J. B., et al. (2015). Methyl-beta-cyclodextrin up-regulates collagen I expression in chronologically-aged skin via its anti-caveolin-1 activity. Oncotarget 6, 1942–1953. doi: 10.18632/oncotarget.3039
Lee, J. G., Takahama, S., Zhang, G., Tomarev, S. I., and Ye, Y. (2016). Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 18, 765–776. doi: 10.1038/ncb3372
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167. doi: 10.1073/pnas.0406976102
Liu, J., Liu, C., Zhang, J., Zhang, Y., Liu, K., Song, J. X., et al. (2020). A Self-assembled alpha-Synuclein nanoscavenger for Parkinson’s disease. ACS Nano 14, 1533–1549. doi: 10.1021/acsnano.9b06453
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012a). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Kehm, V. M., Zhang, B., O’Brien, P., Trojanowski, J. Q., and Lee, V. M. (2012b). Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 209, 975–986. doi: 10.1084/jem.20112457
Luk, K. C., Song, C., O’Brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056. doi: 10.1073/pnas.0908005106
Luo, H. T., Zhang, J. P., and Miao, F. (2016). Effects of pramipexole treatment on the alpha-synuclein content in serum exosomes of Parkinson’s disease patients. Exp. Ther. Med. 12, 1373–1376. doi: 10.3892/etm.2016.3471
Madeira, A., Yang, J., Zhang, X., Vikeved, E., Nilsson, A., Andren, P. E., et al. (2011). Caveolin-1 interacts with alpha-synuclein and mediates toxic actions of cellular alpha-synuclein overexpression. Neurochem. Int. 59, 280–289. doi: 10.1016/j.neuint.2011.05.017
Magalhaes, J., Gegg, M. E., Migdalska-Richards, A., and Schapira, A. H. (2018). Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 8:1385. doi: 10.1038/s41598-018-19479-8
Mahul-Mellier, A. L., Vercruysse, F., Maco, B., Ait-Bouziad, N., De Roo, M., Muller, D., et al. (2015). Fibril growth and seeding capacity play key roles in alpha-synuclein-mediated apoptotic cell death. Cell Death Differ. 22, 2107–2122. doi: 10.1038/cdd.2015.79
Mak, S. K., McCormack, A. L., Manning-Bog, A. B., Cuervo, A. M., and Di Monte, D. A. (2010). Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 285, 13621–13629. doi: 10.1074/jbc.M109.074617
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
Marschallinger, J., Iram, T., Zardeneta, M., Lee, S. E., Lehallier, B., Haney, M. S., et al. (2020). Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23, 194–208. doi: 10.1038/s41593-019-0566-1
Martinez-Vicente, M., and Cuervo, A. M. (2007). Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 6, 352–361. doi: 10.1016/S1474-4422(07)70076-5
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016b). Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
Mazzulli, J. R., Zunke, F., Isacson, O., Studer, L., and Krainc, D. (2016a). alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. U.S.A. 113, 1931–1936. doi: 10.1073/pnas.1520335113
McCormack, A., Keating, D. J., Chegeni, N., Colella, A., Wang, J. J., and Chataway, T. (2019). Abundance of Synaptic vesicle-related proteins in alpha-Synuclein-containing protein inclusions suggests a targeted formation mechanism. Neurotox. Res. 35, 883–897. doi: 10.1007/s12640-019-00014-0
McCray, B. A., Skordalakes, E., and Taylor, J. P. (2010). Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum. Mol. Genet. 19, 1033–1047. doi: 10.1093/hmg/ddp567
Michell, A. W., Barker, R. A., Raha, S. K., and Raha-Chowdhury, R. (2005). A case of late onset sporadic Parkinson’s disease with an A53T mutation in alpha-synuclein. J. Neurol. Neurosurg. Psychiatry 76, 596–597. doi: 10.1136/jnnp.2004.046425
Migdalska-Richards, A., Daly, L., Bezard, E., and Schapira, A. H. (2016). Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann. Neurol. 80, 766–775. doi: 10.1002/ana.24790
Migdalska-Richards, A., Ko, W. K. D., Li, Q., Bezard, E., and Schapira, A. H. V. (2017). Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 71:e21967. doi: 10.1002/syn.21967
Miki, Y., Shimoyama, S., Kon, T., Ueno, T., Hayakari, R., Tanji, K., et al. (2018). Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 63, 33–43. doi: 10.1016/j.neurobiolaging.2017.11.006
Minakaki, G., Menges, S., Kittel, A., Emmanouilidou, E., Schaeffner, I., Barkovits, K., et al. (2018). Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14, 98–119. doi: 10.1080/15548627.2017.1395992
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Mokretar, K., Pease, D., Taanman, J. W., Soenmez, A., Ejaz, A., Lashley, T., et al. (2018). Somatic copy number gains of alpha-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 141, 2419–2431. doi: 10.1093/brain/awy157
Mullin, S., Smith, L., Lee, K., D’Souza, G., Woodgate, P., Elflein, J., et al. (2020). Ambroxol for the treatment of patients with parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 77, 427–434. doi: 10.1001/jamaneurol.2019.4611
Nakai, M., Fujita, M., Waragai, M., Sugama, S., Wei, J., Akatsu, H., et al. (2007). Expression of alpha-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochem. Biophys. Res. Commun. 358, 104–110. doi: 10.1016/j.bbrc.2007.04.108
Nanbo, A., Kawanishi, E., Yoshida, R., and Yoshiyama, H. (2013). Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 87, 10334–10347. doi: 10.1128/JVI.01310-13
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Narhi, L., Wood, S. J., Steavenson, S., Jiang, Y., Wu, G. M., Anafi, D., et al. (1999). Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 274, 9843–9846. doi: 10.1074/jbc.274.14.9843
Ngolab, J., Trinh, I., Rockenstein, E., Mante, M., Florio, J., Trejo, M., et al. (2017). Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol. Commun. 5:46. doi: 10.1186/s40478-017-0445-5
Papadopoulos, V. E., Nikolopoulou, G., Antoniadou, I., Karachaliou, A., Arianoglou, G., Emmanouilidou, E., et al. (2018). Modulation of beta-glucocerebrosidase increases alpha-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum. Mol. Genet. 27, 1696–1710. doi: 10.1093/hmg/ddy075
Park, W. Y., Park, J. S., Cho, K. A., Kim, D. I., Ko, Y. G., Seo, J. S., et al. (2000). Up-regulation of caveolin attenuates epidermal growth factor signaling in senescent cells. J. Biol. Chem. 275, 20847–20852. doi: 10.1074/jbc.M908162199
Petrucelli, L., O’Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., et al. (2002). Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36, 1007–1019. doi: 10.1016/s0896-6273(02)01125-x
Picca, A., Guerra, F., Calvani, R., Bucci, C., Lo Monaco, M. R., Bentivoglio, A. R., et al. (2019). Mitochondrial-derived vesicles as candidate biomarkers in Parkinson’s disease: rationale, design and methods of the EXosomes in PArkiNson disease (EXPAND) study. Int. J. Mol. Sci. 20:2373. doi: 10.3390/ijms20102373
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Poehler, A. M., Xiang, W., Spitzer, P., May, V. E., Meixner, H., Rockenstein, E., et al. (2014). Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy 10, 2171–2192. doi: 10.4161/auto.36436
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Potashkin, J. A., Blume, S. R., and Runkle, N. K. (2010). Limitations of animal models of Parkinson’s disease. Parkinsons Dis. 2011:658083. doi: 10.4061/2011/658083
Pringsheim, T., Jette, N., Frolkis, A., and Steeves, T. D. (2014). The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 29, 1583–1590. doi: 10.1002/mds.25945
Pyo, J. O., Jang, M. H., Kwon, Y. K., Lee, H. J., Jun, J. I., Woo, H. N., et al. (2005). Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 280, 20722–20729. doi: 10.1074/jbc.M413934200
Ramirez, A., Heimbach, A., Grundemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Riedel, O., Bitters, D., Amann, U., Garbe, E., and Langner, I. (2016). Estimating the prevalence of Parkinson’s disease (PD) and proportions of patients with associated dementia and depression among the older adults based on secondary claims data. Int. J. Geriatr. Psychiatry 31, 938–943. doi: 10.1002/gps.4414
Rutherford, N. J., Moore, B. D., Golde, T. E., and Giasson, B. I. (2014). Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J. Neurochem. 131, 859–867. doi: 10.1111/jnc.12806
Sala, G., Arosio, A., Stefanoni, G., Melchionda, L., Riva, C., Marinig, D., et al. (2013). Rotenone upregulates alpha-synuclein and myocyte enhancer factor 2D independently from lysosomal degradation inhibition. Biomed. Res. Int. 2013:846725. doi: 10.1155/2013/846725
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Schulz-Schaeffer, W. J. (2010). The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies. Park. Dis. Park. Dis. Dementia Acta Neuropathol. 120, 131–143. doi: 10.1007/s00401-010-0711-0
Selvaraj, S., and Piramanayagam, S. (2019). Impact of gene mutation in the development of Parkinson”s disease. Genes Dis. 6, 120–128. doi: 10.1016/j.gendis.2019.01.004
Semerdzhiev, S. A., Fakhree, M. A. A., Segers-Nolten, I., Blum, C., and Claessens, M. (2021). Interactions between SARS-CoV-2 N-protein and alpha-Synuclein accelerate amyloid formation. ACS Chem. Neurosci. 13, 143–150. doi: 10.1021/acschemneuro.1c00666
Shi, M., Liu, C., Cook, T. J., Bullock, K. M., Zhao, Y., Ginghina, C., et al. (2014). Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 128, 639–650. doi: 10.1007/s00401-014-1314-y
Shprecher, D. R., Adler, C. H., Zhang, N., Hentz, J. G., Serrano, G. E., Dugger, B. N., et al. (2018). Predicting alpha-synuclein pathology by REM sleep behavior disorder diagnosis. Parkinsonism Relat. Disord. 55, 92–96. doi: 10.1016/j.parkreldis.2018.05.020
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Solayman, M., Islam, M. A., Alam, F., Khalil, M. I., Kamal, M. A., and Gan, S. H. (2017). Natural products combating neurodegeneration: Parkinson’s disease. Curr. Drug Metab. 18, 50–61. doi: 10.2174/1389200217666160709204826
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Stojkovska, I., Wani, W. Y., Zunke, F., Belur, N. R., Pavlenko, E. A., Mwenda, N., et al. (2021). Rescue of alpha-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 110, 436–451. doi: 10.1016/j.neuron.2021.10.032
Stuendl, A., Kunadt, M., Kruse, N., Bartels, C., Moebius, W., Danzer, K. M., et al. (2016). Induction of alpha-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 139(Pt 2), 481–494. doi: 10.1093/brain/awv346
Subra, C., Grand, D., Laulagnier, K., Stella, A., Lambeau, G., Paillasse, M., et al. (2010). Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 51, 2105–2120. doi: 10.1194/jlr.M003657
Surgucheva, I., Sharov, V. S., and Surguchov, A. (2012). gamma-Synuclein: seeding of alpha-synuclein aggregation and transmission between cells. Biochemistry 51, 4743–4754. doi: 10.1021/bi300478w
Suzuki, S., Numakawa, T., Shimazu, K., Koshimizu, H., Hara, T., Hatanaka, H., et al. (2004). BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J. Cell Biol. 167, 1205–1215. doi: 10.1083/jcb.200404106
Svensson, K. J., Christianson, H. C., Wittrup, A., Bourseau-Guilmain, E., Lindqvist, E., Svensson, L. M., et al. (2013). Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 288, 17713–17724. doi: 10.1074/jbc.M112.445403
Szatmari, Z., Kis, V., Lippai, M., Hegedus, K., Farago, T., Lorincz, P., et al. (2014). Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol. Biol. Cell 25, 522–531. doi: 10.1091/mbc.E13-10-0574
Taguchi, Y. V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., et al. (2017). Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 37, 9617–9631. doi: 10.1523/JNEUROSCI.1525-17.2017
Teixeira, M., Sheta, R., Idi, W., and Oueslati, A. (2021). Alpha-Synuclein and the endolysosomal system in parkinson’s disease: guilty by association. Biomolecules 11:1333. doi: 10.3390/biom11091333
Thery, C., Amigorena, S., Raposo, G., and Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 3:Unit 3.22. doi: 10.1002/0471143030.cb0322s30
Tian, T., Zhu, Y. L., Zhou, Y. Y., Liang, G. F., Wang, Y. Y., Hu, F. H., et al. (2014). Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 289, 22258–22267. doi: 10.1074/jbc.M114.588046
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. doi: 10.1038/s41582-019-0301-2
Totterdell, S., Hanger, D., and Meredith, G. E. (2004). The ultrastructural distribution of alpha-synuclein-like protein in normal mouse brain. Brain Res. 1004, 61–72. doi: 10.1016/j.brainres.2003.10.072
Troncoso-Escudero, P., Sepulveda, D., Perez-Arancibia, R., Parra, A. V., Arcos, J., Grunenwald, F., et al. (2020). On the right track to treat movement disorders: promising therapeutic approaches for Parkinson’s and Huntington’s disease. Front. Aging Neurosci. 12:571185. doi: 10.3389/fnagi.2020.571185
Tsunemi, T., Hamada, K., and Krainc, D. (2014). ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 34, 15281–15287. doi: 10.1523/JNEUROSCI.1629-14.2014
Tsunemi, T., Ishiguro, Y., Yoroisaka, A., Valdez, C., Miyamoto, K., Ishikawa, K., et al. (2020). Astrocytes protect human dopaminergic neurons from alpha-Synuclein accumulation and propagation. J. Neurosci. 40, 8618–8628. doi: 10.1523/JNEUROSCI.0954-20.2020
Tsunemi, T., Perez-Rosello, T., Ishiguro, Y., Yoroisaka, A., Jeon, S., Hamada, K., et al. (2019). Increased lysosomal exocytosis induced by lysosomal Ca(2+) channel agonists protects human dopaminergic neurons from alpha-Synuclein toxicity. J. Neurosci. 39, 5760–5772. doi: 10.1523/JNEUROSCI.3085-18.2019
Valdinocci, D., Radford, R. A., Siow, S. M., Chung, R. S., and Pountney, D. L. (2017). Potential modes of intercellular alpha-Synuclein transmission. Int. J. Mol. Sci. 18:469. doi: 10.3390/ijms18020469
Valente, E. M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A. E., Caputo, V., et al. (2004). PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 56, 336–341. doi: 10.1002/ana.20256
van Niel, G., Porto-Carreiro, I., Simoes, S., and Raposo, G. (2006). Exosomes: a common pathway for a specialized function. J. Biochem. 140, 13–21. doi: 10.1093/jb/mvj128
Vanlandingham, P. A., and Ceresa, B. P. (2009). Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 284, 12110–12124. doi: 10.1074/jbc.M809277200
Vidal, R. L., Matus, S., Bargsted, L., and Hetz, C. (2014). Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 35, 583–591. doi: 10.1016/j.tips.2014.09.002
Vilarino-Guell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. doi: 10.1016/j.ajhg.2011.06.001
Vogiatzi, T., Xilouri, M., Vekrellis, K., and Stefanis, L. (2008). Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556. doi: 10.1074/jbc.M801992200
Volpicelli-Daley, L. A., Kirik, D., Stoyka, L. E., Standaert, D. G., and Harms, A. S. (2016). How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J. Neurochem. 139 Suppl 1, 131–155. doi: 10.1111/jnc.13627
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wang, Y., Liu, J., Chen, M., Du, T., Duan, C., Gao, G., et al. (2016). The novel mechanism of rotenone-induced alpha-synuclein phosphorylation via reduced protein phosphatase 2A activity. Int. J. Biochem. Cell Biol. 75, 34–44. doi: 10.1016/j.biocel.2016.03.007
Wang, Z. Y., Liu, J. Y., Yang, C. B., Malampati, S., Huang, Y. Y., Li, M. X., et al. (2017). Neuroprotective natural products for the treatment of Parkinson’s disease by targeting the autophagy-lysosome pathway: a systematic review. Phytother. Res. 31, 1119–1127. doi: 10.1002/ptr.5834
Wauters, F., Cornelissen, T., Imberechts, D., Martin, S., Koentjoro, B., Sue, C., et al. (2020). LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222. doi: 10.1080/15548627.2019.1603548
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Wheaton, K., Sampsel, K., Boisvert, F. M., Davy, A., Robbins, S., and Riabowol, K. (2001). Loss of functional caveolae during senescence of human fibroblasts. J. Cell Physiol. 187, 226–235. doi: 10.1002/jcp.1071
Wildburger, N. C., Hartke, A. S., Schidlitzki, A., and Richter, F. (2020). Current evidence for a bidirectional loop between the lysosome and alpha-synuclein proteoforms. Front. Cell Dev. Biol. 8:598446. doi: 10.3389/fcell.2020.598446
Xia, Y., Zhang, G., Han, C., Ma, K., Guo, X., Wan, F., et al. (2019). Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 10:174. doi: 10.1038/s41419-019-1404-9
Xing, H., Tan, J., Miao, Y., Lv, Y., and Zhang, Q. (2021). Crosstalk between exosomes and autophagy: a review of molecular mechanisms and therapies. J. Cell Mol. Med. 25, 2297–2308. doi: 10.1111/jcmm.16276
Xu, J., Camfield, R., and Gorski, S. M. (2018). The interplay between exosomes and autophagy - partners in crime. J. Cell Sci. 131:jcs215210. doi: 10.1242/jcs.215210
Yang, J., Luo, S., Zhang, J., Yu, T., Fu, Z., Zheng, Y., et al. (2021). Exosome-mediated delivery of antisense oligonucleotides targeting alpha-synuclein ameliorates the pathology in a mouse model of Parkinson’s disease. Neurobiol. Dis. 148:105218. doi: 10.1016/j.nbd.2020.105218
Yang, Y., Qin, M., Bao, P., Xu, W., and Xu, J. (2017). Secretory carrier membrane protein 5 is an autophagy inhibitor that promotes the secretion of alpha-synuclein via exosome. PLoS One 12:e0180892. doi: 10.1371/journal.pone.0180892
Zavodszky, E., Seaman, M. N., Moreau, K., Jimenez-Sanchez, M., Breusegem, S. Y., Harbour, M. E., et al. (2014). Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 5:3828. doi: 10.1038/ncomms4828
Zhang, W., Wang, T., Pei, Z., Miller, D. S., Wu, X., Block, M. L., et al. (2005). Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542. doi: 10.1096/fj.04-2751com
Zhao, J., Lu, W., Ren, Y., Fu, Y., Martens, Y. A., Shue, F., et al. (2021). Apolipoprotein E regulates lipid metabolism and alpha-synuclein pathology in human iPSC-derived cerebral organoids. Acta Neuropathol. 142, 807–825. doi: 10.1007/s00401-021-02361-9
Zhao, Z. H., Chen, Z. T., Zhou, R. L., Zhang, X., Ye, Q. Y., and Wang, Y. Z. (2018). Increased DJ-1 and alpha-synuclein in plasma neural-derived exosomes as potential markers for Parkinson’s disease. Front. Aging Neurosci. 10:438. doi: 10.3389/fnagi.2018.00438
Zhou, W., Hurlbert, M. S., Schaack, J., Prasad, K. N., and Freed, C. R. (2000). Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 866, 33–43. doi: 10.1016/s0006-8993(00)02215-0
Keywords: autophagy-lysosomal pathway, α-syn exosomal secretion, Parkinson’s disease progression, biomarker, degradation
Citation: Sepúlveda D, Cisternas-Olmedo M, Arcos J, Nassif M and Vidal RL (2022) Contribution of Autophagy-Lysosomal Pathway in the Exosomal Secretion of Alpha-Synuclein and Its Impact in the Progression of Parkinson’s Disease. Front. Mol. Neurosci. 15:805087. doi: 10.3389/fnmol.2022.805087
Received: 29 October 2021; Accepted: 07 January 2022;
Published: 17 February 2022.
Edited by:
Miguel Diaz-Hernandez, Complutense University of Madrid, SpainReviewed by:
Maria Xilouri, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceCopyright © 2022 Sepúlveda, Cisternas-Olmedo, Arcos, Nassif and Vidal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: René L. Vidal, cmVuZS52aWRhbEB1bWF5b3IuY2w=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.