 Yuwei Dai1,2†
Yuwei Dai1,2† Zhuanyi Yang3†
Zhuanyi Yang3† Jialing Guo1,2Haoyu Li3,4Jiaoe Gong5Yuanyuan Xie1,2Bo Xiao1,2
Jialing Guo1,2Haoyu Li3,4Jiaoe Gong5Yuanyuan Xie1,2Bo Xiao1,2 Hua Wang6,7*
Hua Wang6,7* Lili Long1,2,7*
Lili Long1,2,7*- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, China
- 4The Institute of Skull Base Surgery and Neurooncology at Hunan Province, Changsha, China
- 5Department of Neurology, Hunan Children’s Hospital, Changsha, China
- 6Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 7NHC Key Laboratory of Birth Defects Research, Prevention and Treatment (Hunan Provincial Maternal and Child Health Care Hospital), Changsha, China
Aim: De novo DDX3X variants account for 1–3% of unexplained intellectual disability cases in females and very rarely in males. Yet, the clinical and genetic features of DDX3X neurodevelopmental disorder in the Chinese cohort have not been characterized.
Method: A total of 23 Chinese patients (i.e., 22 female and 1 male) with 22 de novo DDX3X deleterious variants were detected among 2,317 probands with unexplained intellectual disability (ID) undertaking whole exome sequencing (WES). The age, sex, genetic data, feeding situation, growth, developmental conditions, and auxiliary examinations of the cohort were collected. The Chinese version of the Gesell Development Diagnosis Scale (GDDS-C) was used to evaluate neurodevelopment of DDX3X patients. The Social Communication Questionnaire (SCQ)-Lifetime version was applied as a primary screener to assess risk for autism spectrum disorder (ASD).
Result: A total of 17 DDX3X variants were novel and 22 were de novo. Missense variants overall were only slightly more common than loss-of-function variants and were mainly located in two functional subdomains. The average age of this cohort was 2.67 (±1.42) years old. The overlapping phenotypic spectrum between this cohort and previously described reports includes intellectual disability (23/23, 100%) with varying degrees of severity, muscle tone abnormalities (17/23, 73.9%), feeding difficulties (13/23, 56.5%), ophthalmologic problems (11/23, 47.8%), and seizures (6/23, 26.1%). A total of 15 individuals had notable brain anatomical disruption (15/23, 65.2%), including lateral ventricle enlargement, corpus callosum abnormalities, and delayed myelination. Furthermore, 9 patients showed abnormal electroencephalogram results (9/23, 39.1%). Hypothyroidism was first noted as a novel clinical feature (6/23, 26.1%). The five primary neurodevelopmental domains of GDDS-C in 21 patients were impaired severely, and 13 individuals were above the “at-risk” threshold for ASD.
Interpretation: Although a certain degree of phenotypic overlap with previously reported cohorts, our study described the phenotypic and variation spectrum of 23 additional individuals carrying DDX3X variants in the Chinese population, adding hypothyroidism as a novel finding. We confirmed the importance of DDX3X as a pathogenic gene in unexplained intellectual disability, supporting the necessity of the application of WES in patients with unexplained intellectual disability.
Introduction
DDX3X (OMIM: 300160) locates in Xp11.4 and encodes a conserved ATP-independent DEAD-box RNA helicase, which is involved in transcription, splicing, RNA transport, and translation (Abdelhaleem, 2005; Garbelli et al., 2011). The DDX3X is composed of 622 amino acid residues containing two functional domains, namely, a helicase ATP-binding domain and a helicase C-terminal domain (Snijders Blok et al., 2015). De novo DDX3X variants account for 1–3% of unexplained intellectual disability (ID) or developmental delay (DD) (Deciphering Developmental Disorders Study., 2017; Maulik et al., 2011) and also perform as a highly plausible pathogenic gene for childhood apraxia of speech (CAS) (Hildebrand et al., 2020). Most cases of DDX3X variants have been reported in females but very rarely in males, and three previous large cohort studies have described heterogeneous clinical manifestations of DDX3X neurodevelopmental disorder, including ID or DD, dystonia, movement disorders, microcephaly, behavioral issues, feeding difficulties in infancy, and seizure (Snijders Blok et al., 2015; Wang et al., 2018; Johnson-Kerner et al., 2020; Lennox et al., 2020). However, the clinical and genetic features of DDX3X neurodevelopmental disorder in the Chinese cohort have not been described yet.
In this study, we elaborated on clinical manifestations of pathogenic variants of DDX3X in 23 patients (i.e., 22 female and 1 male) in the Chinese cohort and explore the association between genotypes and phenotypes.
Materials and Methods
Patients
With the support of the National Key Research and Development Program regarding the birth defect and developmental disorders screening (No. 2019YFC1005100), we collected whole exome sequencing (WES) data on 2,317 patients (1,622 males, 695 females, 5.33 ± 2.10 years old) with unexplained ID or DD and identified 23 DDX3X heterozygous variants in 23 patients by viewing those initial reports of WES. These patients further visited the Xiangya Hospital, Central South University, Hunan Provincial Maternal and Child Health Care Hospital, and Hunan Children’s Hospital between March 2018 and December 2020. Basic demographic information and detailed clinical data, including perinatal conditions, gender, date at birth, family history, genetic data, feeding situation, growth, and developmental conditions, were recorded clearly. Electroencephalography (EEG) and brain MRI were re-reviewed and reanalyzed by two experienced neurological physicians, and they were blind to the genetic results.
Assessment
The Chinese version of the Gesell Development Diagnosis Scale (GDDS-C) was applied to assess the neurodevelopment of infants aged 0–6 years, and each participant calculated separate developmental quotient (DQ) of the five sub-domains, namely, adaptability, gross motor, fine motor, language, and social-emotional response. Based on the full-scale DQ results, the development of infants was classified as follows: normal (DQ ≥ 85), deficient (DQ < 75), and borderline (≥75 ∼ < 85). DQ in any single domain below 75 was considered deficient in this field (You et al., 2019). The GDDS-C was conducted by medical professionals in child health clinics (Yang, 2016).
The Social Communication Questionnaire (SCQ) Lifetime version was a brief, 40-item, parent-report clinical tool, which had been widely used as a primary screener to assess risk for autism spectrum disorder (ASD). It was based on a semi-structured parent interview conducted by a trained clinician or researcher. Each item in SCQ required a dichotomous “yes”/“no” response, and each item received a value of 1 point for abnormal behavior. Complete developmental history was needed to be the reference. The caregivers would indicate whether behaviors of Questions 2–19 had ever been presented and whether behaviors of Questions 20–40 were presented at the age 4 or evaluated these behaviors in the past half a year if the child was aged less than 4. Scores above the cutoff of 12 suggested individuals were above the “at-risk” threshold for ASD, and further extended evaluations should be undertaken (Marvin et al., 2017).
Differences between average scores on 2 scales of this cohort and respective cutoff value were statistically evaluated using a one-sample t-test, p-values less than 0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001) were considered significant.
Genetic Analysis
We reanalyzed trio- or single WES data of all probands and their biological parents (19 for trio-WES). Sanger sequencing was conducted to validate whether the variant was de novo. The DDX3X transcript was referenced (NM_001193416.2, GRCh37/hg19). Sequenced reads were aligned to GRCh37/hg19 using the Burrows-Wheeler Aligner (BWA) (v.0.7.12) with default parameters. SAMtools (v0.1.12) was used to call the variants and the RefSeq Genomes database. The Genome Analysis Tool Kit (GATK 3.5) was used for local realignment and base quality score recalibration. Synonymous changes and single-nucleotide polymorphisms with a minor allele frequency greater than 5% were removed.1 Variant pathogenicity was interpreted based on the American College of Medical Genetics (ACMG) guidelines published in 2015 (Richards et al., 2015). The Genome Aggregation Database (GnomAD) was used to annotate the variants. Pathogenicity of the identified variants was predicted using several in silico predictors, including Polymorphism Phenotyping version 2 (Polyphen-2),2 Protein Variation Effect Analyzer (PROVEAN),3 Combined Annotation Dependent Depletion (CADD),4 and Sorting Intolerant From Tolerant (SIFT).5 Silico analysis data for missense DDX3X variants was shown in Supplementary material. Screening of neonatal genetic and metabolic diseases as a routine procedure was performed on all probands when they were born.
Ethical Issues
This research was approved by the Ethics Committee of XiangYa Hospital, Central South University (Location: Hunan Province, P.R. China, Approval No.: 2019030496). Written consents for inclusion in this study and rights to use portraits of each proband were obtained from parents of all participants.
Results
Genomic Analysis
Among 2,317 individuals studied by WES who had unexplained ID, 23 deleterious variants in DDX3X were detected, 22 females were found to carry de novo variants in DDX3X, and 1 male was identified to carry an inherited variant in DDX3X from his asymptomatic mother. Furthermore, 17 were novel variants, and the remaining 6 variants were reported previously [c.1595C > T (Lennox et al., 2020); c.136C > T (Snijders Blok et al., 2015); c.865-1G > A (Wang et al., 2018); c.1703C > T (Snijders Blok et al., 2015); c.1463G > A (Lennox et al., 2020); c.1678_1680del (Snijders Blok et al., 2015)]. Of the 23 identified variants in DDX3X, 11 were missense variants, 2 were in-frame deletions, 2 were splice site variants, 5 were frameshift variants, and 3 were nonsense variants (Figure 1A). According to the guidelines set out by the ACMG, 22 variants were interpreted as pathogenic or likely pathogenic variants, and 1 variant was of uncertain significance (VUS) (Table 1).
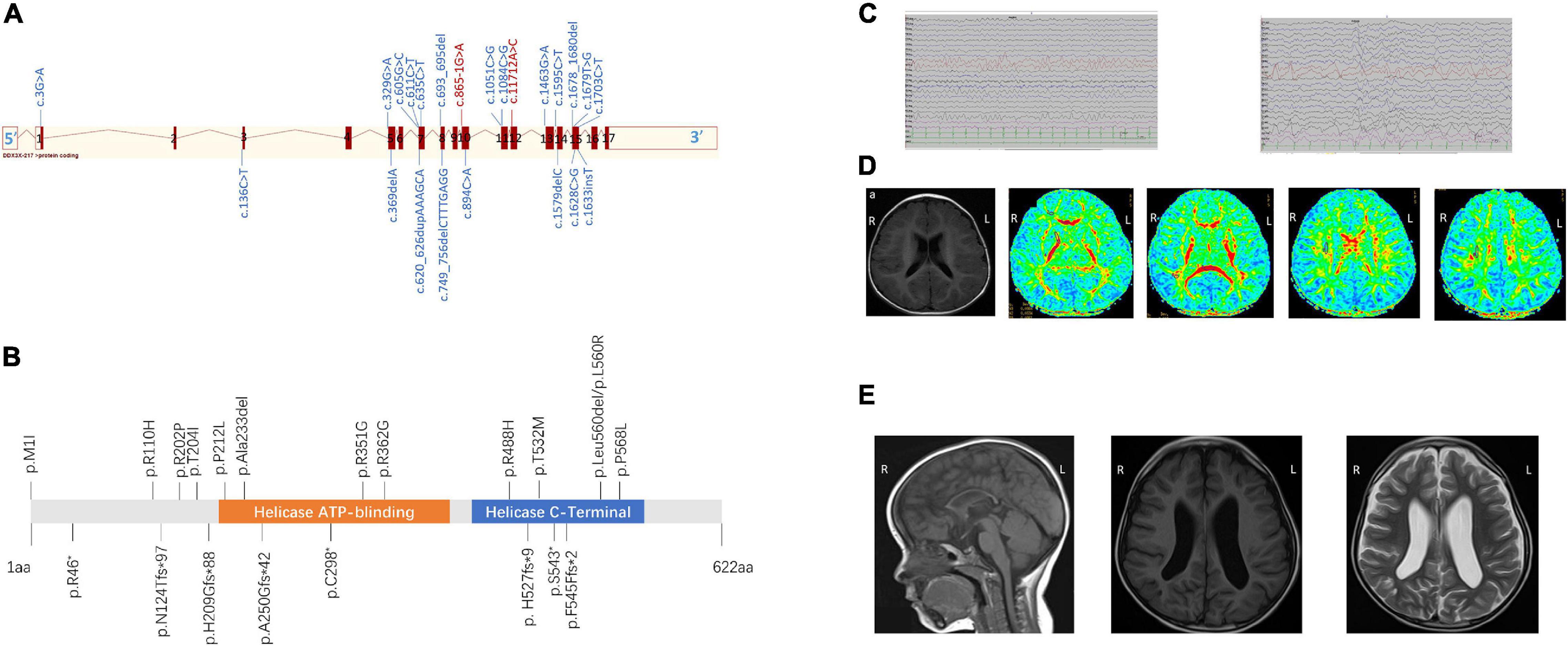
Figure 1. (A) Schematic view of the DDX3X exon structure based on NM_001193416. Red blocks represent exons, and the exon number is listed on each exon. cDNA change is listed for each variant. Splice site mutations are shown in red font. (B) Location of amino acid substitutions in DDX3X (NM_ 001193416.2). Missense and in-frame deletions (top, 13), frameshift, and non-sense variants (bottom, 8). DDX3X contains two subdomains, a helicase ATP-binding domain (orange bar) and a helicase C-terminal domain (blue bar). (C) The latest electroencephalogram (EEG) and of Female 17. Abnormal EEG presentation, multiple slow waves in bilateral occipital lobes. (D) Brain magnetic resonance imaging (MRI) of Female 19 at the age of 1 year and 1 month (A–E) Axial position T1-weighted images show normal sulci and gyri, Axial position diffusion tensor imaging (DTI) images show delayed of white matter of frontal lobe and centrum semiovale myelination. Numbers 1–8 represent genu of the corpus callosum, white matter of the frontal lobe, anterior limb of the internal capsule, posterior limb of the internal capsule, splenium of the corpus callosum, occipital lobe, and centrum semiovale (7 and 8), respectively. (E) Sagittal image shows diffuse thinning of the corpus callosum of Female 13 at the age of 1 year and 3 months (A). MRI (B) T1 and (C) T2 axial slices showed widened bilateral lateral ventricles of Female 6 at the age of 3 years and 10 months.

Table 1. Clinical interpretation of variants detected in DDX3X by the ACMG guideline.
All 23 identified variants were likely to cause changes in the DDX3X protein, 6 of which were in the helicase ATP-binding domain (i.e., p.Ala233del, p.P212L, p.R351G, p.R362G, p.A250Gfs*42, and p.C298* while 8 were in the helicase C-terminal domain (i.e., p.R488H, p.T532M, p.Leu560del, p.L560R, p.P568L, p. H527fs*9, p.S543*, and p.F545Ffs*2) (Figure 1B).
Clinical Features
Table 2 shows the clinical features of 23 participants. The average age of this cohort was 2.67 (±1.42) years old. In the cohort of 23 patients with DDX3X variants, all of them meet the criteria for ID or DD (23/23, 100%), ranging from mild to severe. Muscle tone abnormalities (17/23, 73.9%), including isolated hypotonia, hypertonia, or mixture of hypertonia and hypotonia, microcephaly (9/23, 39.1%), feeding difficulties, or low weight gain (13/23, 56.5%), associated with ophthalmologic problems (11/23, 47.8%), were the most typical clinical characteristics. Movement disorders (9/23, 39.1%), seizures (6/23, 26.1%), behavior issues (5/23, 21.7%), cardiac abnormalities (3/23, 13.0%), and hearing impairment (2/23, 8.7%) were observed in this cohort. Furthermore, six patients presented with hypothyroidism (6/23, 26.1%).
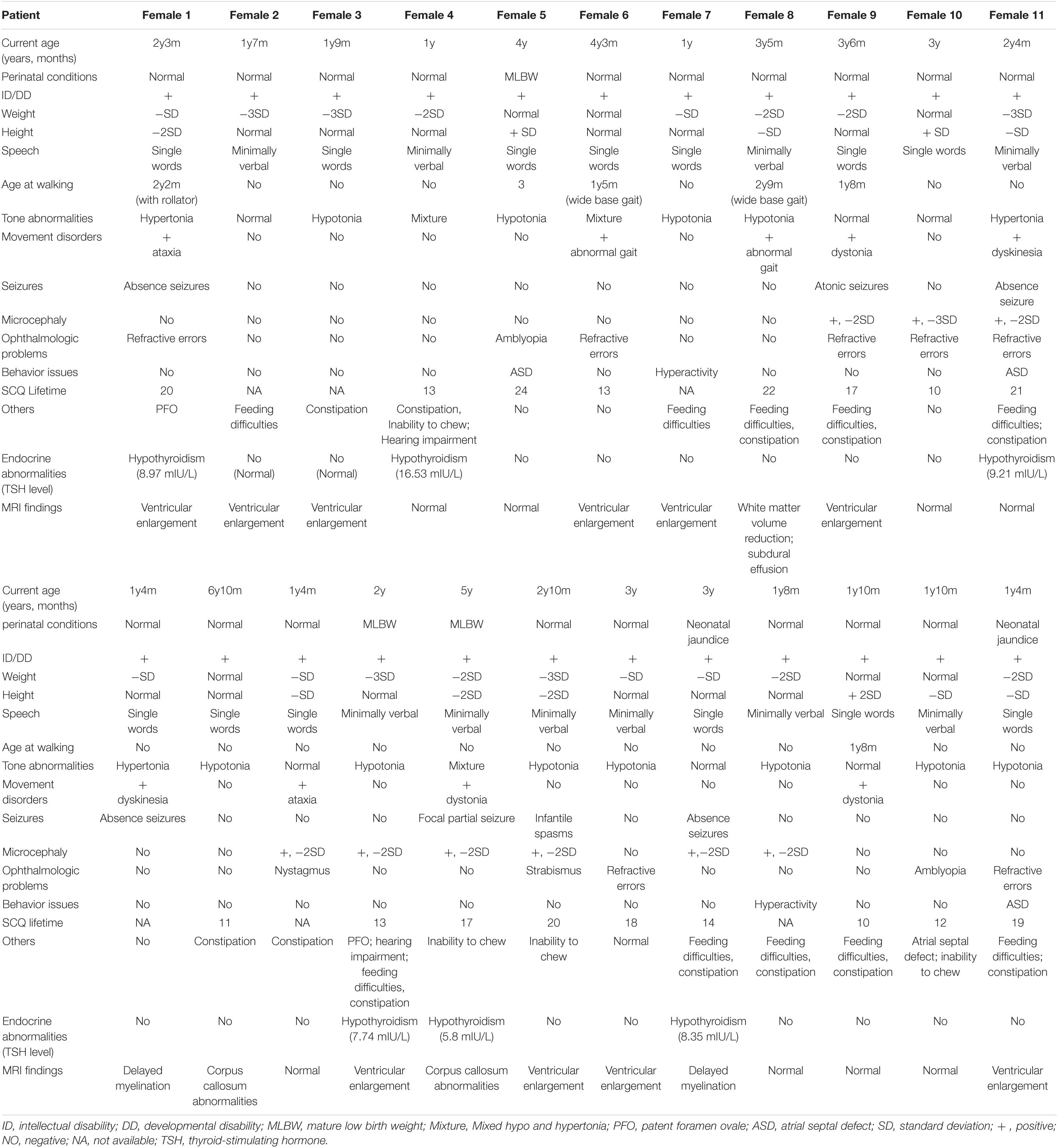
Table 2. Summary of demographic information and phenotypic features in 23 patients with DDX3X variants.
Intellectual Disability or Developmental Delay
Only 2 patients (i.e., Female 14 and Female 18) could raise their heads at the age of 3 months, and 20 patients (except Female 7, Female 10, and Female 21) could not walk independently before the age of 2 years, all of them had poor motor coordination. All parents complained their children showed poor performance in language or speech function. Nearly 50% could express their simple needs in no more than four words and only say several single words. The two eldest individuals above 5 years (i.e., Female 13 and Female 16) had not developed speech ability but could just follow simple instructions.
Seizures and Electroencephalography Monitoring
All of them underwent scalp EEG monitoring, and 9 of them showed abnormal profiles. Slow background activity was observed in 7 out of 23 patients. Focal epileptiform discharges were detected in two patients, and generalized spike waves and sharp waves were detected in four patients. Hyperarrhythmia, associated with multifocal epileptiform discharges, was prevalent in one patient. In 6 individuals with seizures (i.e., Female 1, Female 9, Female 11, Female 16, Female 17, and Female 19), their age at the onset of seizures ranged from 5 to 14 months. Atonic seizures occurred in 1, absence seizures in 3, focal partial seizures in 1, and infantile spasms in 1. Female 17 was diagnosed with infantile spasms induced by fever at the age of 5 months and recurred at the age of 13 months. They had a good response to antiepileptic drugs and no seizures in 6 months. Figure 1C shows the latest EEG of Female 17.
Feeding Difficulties
Among the 13 individuals with feeding difficulties or low weight in our cohort, 8 of their parents reflected that they were intolerant of lactose and allergic to multiple high-protein food sources. Constipation and chewing weakness were common manifestations in 10 cases. Furthermore, 7 of 13 individuals were vitamin B-deficient, but none of them was hyperhomocysteinemia.
Endocrine Abnormalities
Thorough endocrine hormone examinations were conducted in 23 patients because of poor growth and neurocognitive development. None of them showed abnormal sex hormone levels. Furthermore, six patients had lower levels of thyroid hormones (THs), and thyroid ultrasound showed normally located thyroid glands. They all received TH supplementation.
Magnetic Resonance Imaging Findings
All patients completed MRI at different ages. A total of 15 individuals had notable brain anatomical disruption in which, 10/15 had a lateral ventricle enlargement, 2/15 had corpus callosum abnormalities, 2/15 had delayed myelination, and 1/15 had white-matter volume reduction. Figure 1D shows brain MRI of Female 19, and (Figure 1E) shows brain MRI of Female 6 and Female 13.
Assessments
A total of 21 patients have undergone GDDS-C and were counted DQ in five separate domains. The highest scored domain was “gross motor” (56.1 ± 17.2), and the lowest scored domain was “language” (49.1 ± 13.2). The remaining scored domains were “fine motor” (56.1 ± 17.2), “personal-social” (56.0 ± 14.2), and “adaptability” (52.7 ± 12.7) successively. Scores in all 5 domains were significantly below the critical value (75), in which p-value was < 0.01. DQ of each participant is listed in Figure 2.
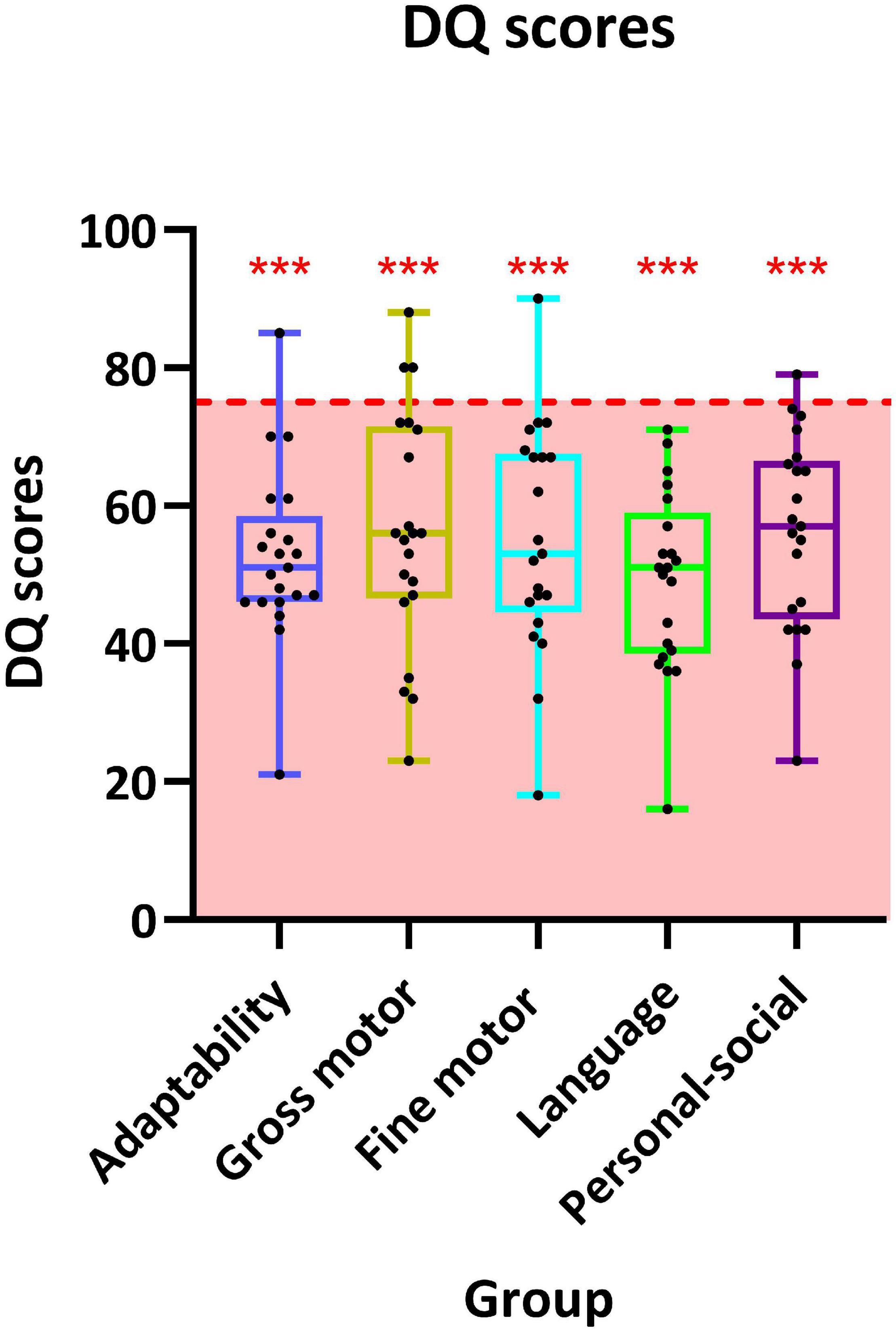
Figure 2. Distribution of developmental quotient (DQ) scores of 19 individuals in the five sub-domains of the Chinese version of Gesell Development Diagnosis Scale (GDDS-C). The interquartile ranges are shown as a box-and-whisker plot. For one-sample t-test, the critical value is 75 (*** represents p < 0.001).
A total of 17 patients completed the SCQ-Lifetime version, and 13 individuals who were above the cutoff value (12) were at the risk of ASD, a comprehensive evaluation for ASD was warranted. Significant deviation was found in the mean score in the SCQ-Lifetime version of 17 participants (16.1 ± 4.4, p = 0.002) compared with the cutoff value.
Discussion
In this report, we described a Chinese cohort of patients with DDX3X variants (n = 23, 22 confirmed de novo), and we are the first study to pinpoint clinical and genetic characteristics in the Chinese population. Among the 23 DDX3X variants in our cohort, 17 were novel variants and 14 variants were located in two functional domains of DDX3X (9 were amino acid variants and 5 were truncating variants). Significant differences in sex composition of DDX3X neurodevelopmental disorder have been noted (22 female and 1 male). ID was considered as a universal feature of DDX3X neurodevelopmental disorder in our study, followed by tone abnormalities, microcephaly, feeding difficulties, and seizures. Major aspects of neural development were assessed and quantified using the GDDS-C, and mean scores of five domains were significantly lower than the critical value of 75 (all p-value < 0.05), and language domain was impaired strikingly. Hypothyroidism was reported in 6 patients with DDX3X variants for the first time. Altogether, our study expanded the clinical and genetic spectrum associated with DDX3X neurodevelopmental disorder and evaluated the degree of developmental delay by a standardized scale. It highlighted that WES was necessary for those unexplained ID individuals.
DDX3X, an RNA-binding protein of the DEAD-box family encoded by the DDX3X gene (Abdelhaleem, 2005), acts as a translational regulator (Lai et al., 2008; Lee et al., 2008), particularly for mRNAs with highly structured 5′ untranslated regions (UTRs) (Chen et al., 2018) and for repeat-associated non-AUG translation (Cheng et al., 2019; Linsalata et al., 2019). DDX3X is also the key component of ribonucleoprotein (RNP) granules composed of mRNA and protein (Huang et al., 2019), a pathological hallmark of many neurodegenerative diseases (Ramaswami et al., 2013). Dd3x plays an indispensable role in mouse embryogenesis, synaptogenesis, and brain development (Chen et al., 2016; Lennox et al., 2020). Dd3x neural stem cells knockout at embryonic day (E) 9.5 in mice hampered brain growth, accompanied by seizures and ataxia (Patmore et al., 2020). Boitnott et al. (2021) generated a Ddx3x haploinsufficient mouse (Ddx3x± female) with construct validity for DDX3X loss-of-function mutations. The Ddx3x± mice showed global development delay and evolved into behavioral anomalies in adulthood (Boitnott et al., 2021).
Comparing with three previously reported cohort studies of DDX3X neurodevelopmental disorder, we found a certain degree of phenotypic overlap but some special points (Snijders Blok et al., 2015; Wang et al., 2018; Lennox et al., 2020; Table 3). ID was still a general phenotypic feature of patients with DDX3X variants in both our study and previous reports. Similarly, compared with healthy controls, the mean score in GDDS-C of the cohort could reflect global DD. The worst performance in the language subdomain of GDDS-C consolidated language impairments as the most prominent clinical feature of DDX3X neurodevelopmental disorder (Johnson-Kerner et al., 2020). Ophthalmologic problems including refractive errors, nystagmus, strabismus, and amblyopia were presented in 11/23 (47.8%) patients. Previous studies have shown that a variety of eye phenotypes including hypoplasia of the eye or absence of one or both eyes in functional studies of ddx3x knockdown in zebrafish, suggesting deleterious variants in DDX3X, may hamper eye function (Snijders Blok et al., 2015; Kellaris et al., 2018). The SCQ-Lifetime version scores indicated an increased risk for ASD in DDX3X neurodevelopmental disorder, suggesting the necessity of screening for behavior problems by trained behavioral pediatricians.
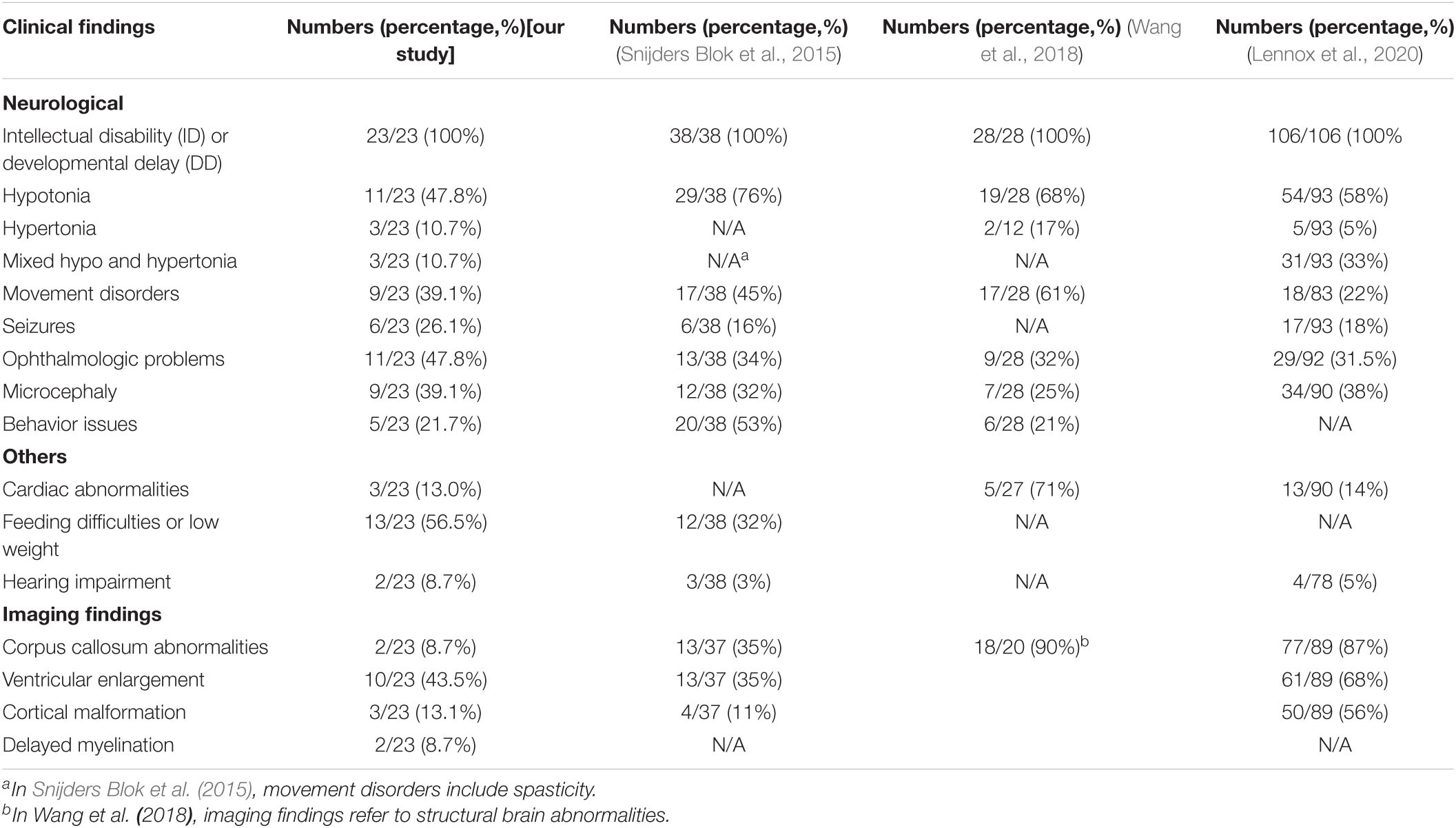
Table 3. Comparison of clinical characteristics in our cohort and three previously published cohorts.
Furthermore, we noticed hypothyroidism in 6 patients. This novel or rare clinical feature was not previously reported in the original description of DDX3X neurodevelopmental disorder. Poor nutrient absorption and feeding difficulty could make them at increased risk of iodine deficiency, which could be an extrinsic factor causing hypothyroidism in these younger children (Bauer and Wassner, 2019). THs play an essential role in the growth and metabolic homeostasis in humans as well as in animals (Prezioso et al., 2018). Triiodothyronine (T3), the active form of thyroid hormone, acts on its nuclear receptor and modulates target gene transcription (Kumar et al., 2015). Even a 25% reduction in DDX3X levels strongly perturbs neurogenesis, suggesting the high dose-dependency of embryonic cortical development to DDX3X (Lennox et al., 2020). Defective RNA metabolism was considered as the potential mechanism through which DDX3X missense variants hamper fetal brain cortical development (Kumar et al., 2015; Lennox et al., 2020). We speculated that TH deficiency may intensify the adverse effect on RNA metabolism caused by DDX3X missense variants. Appropriate TH supplementation in DDX3X patients with hypothyroidism could be worth trying, but overtreatment or prophylactic hormonal therapy should be avoided because the higher dose of TH supplementation could worsen the outcome (Tuhan et al., 2016). Therefore, it is crucial to have a close follow-up in DDX3X patients with hypothyroidism. Hypothyroidism has not been verified in animal models with in vivo depletion of Ddx3x.
Many study reports have tried to establish the connection between the severity of clinical phenotypes and the location and type of variants and obtained two main findings (Lennox et al., 2020). First, the same recurrent de novo variants were more likely to have similar phenotypes. Recurrent amino acids changes, including R326, I415, and T532, all could cause polymicrogyria (PMG) (Abdelhaleem, 2005; Tuhan et al., 2016; Lennox et al., 2020). Besides the 11 individuals with PMG, 10 were missense variants and one was in-frame deletion, underscoring a striking association between missense variants and severe cerebral anatomical phenotypes, like PMG or dysgyria (Lennox et al., 2020).
Regrettably, no PMG or dysgyria was observed in our cohort, nor the previously reported DDX3X variants relating with PMG. However, we found that females with missense or in-frame deletion DDX3X variants (10/11, 90.9%) were more likely to have abnormal brain structural MRI compared with those with LOF variants (4/11, 36.3%). This could be interpreted by different pathogenic mechanisms. Many aberrant truncating mRNAs (i.e., frameshift or non-sense variants) might undergo non-sense-mediated RNA decay (NMD) and resulted in a haploinsufficiency effect, while a subset of missense variants could function in a dominant-negative manner (Chen et al., 2020; Lennox et al., 2020). Further investigations about gain-of-function mechanism behind certain missense mutations will be carried out by modeling missense mutations in mice. Multiple malignancies have a solid association with somatic DDX3X variants, like malignant melanoma and medulloblastoma (Phung et al., 2019; Patmore et al., 2020). Even though no malignancy was reported in our cohort yet, regular cancer screening is still quite necessary. Finally, a small sample size and a relatively small number of novel phenotypes, such as hypothyroidism, were the main limitations of this study.
In summary, we identified 23 unrelated Chinese patients with causal variants in DDX3X and expanded the knowledge of these increasingly recognized ID disorders. Our study delineated many clinical characteristics of the Chinese cohort with DDX3X variants, largely overlapping with phenotypic spectrum in previously reported studies, but hypothyroidism was first noted as a novel clinical feature. Overall, missense variants were only slightly more common than loss-of-function variants and were mainly located in two functional subdomains. The DDX3X missense variants may have a certain association with abnormal brain anatomical structures. Given the heterogeneous clinical manifestations and involvement of the nervous system and non-nervous systems, unexplained ID in both males and females should take the use of multigene panels that include DDX3X or WES into consideration.
Data Availability Statement
Sequencing data involved in the study are available through the Genbank repository (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA795095). There are restrictions to the full availability of sequencing data of the research participants due to privacy and ethical/legal issues. The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of XiangYa Hospital, Central South University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YD, ZY, and LL: study design, analysis and revision of the manuscript. YD, ZY, and JGu: follow-up of patient’s information. HL, JGo, and YX: reanalysis of WES data and original draft preparation. YD, BX, HW, and LL: collection of clinical and WES data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2021YFC1005300 and 2019YFC1005100), the National Natural Science Foundation of China (82171454 and 81671300), the Key Research and Development Program of Hunan Province (2022SK2042), the Natural Science Foundation of Hunan Province Project (2020JJ5914 and 2021JJ30389), the NHC Key Laboratory of Birth Defect for Research and Prevention (Hunan Provincial Maternal and Child Health Care Hospital) (KF2020001), and the Scientific Research Project of Hunan Provincial Health Commission (20200475).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the patients and their family members involved in this study for their participation, and the Berry Genomics Co., for their technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.793001/full#supplementary-material
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/projects/SNP
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://provean.jcvi.org
- ^ https://cadd.gs.washington.edu/
- ^ http://sift.jcvi.org
References
Abdelhaleem, M. (2005). RNA helicases: regulators of differentiation. Clin. Biochem. 38, 499–503. doi: 10.1016/j.clinbiochem.2005.01.010
Bauer, A. J., and Wassner, A. J. (2019). Thyroid hormone therapy in congenital hypothyroidism and pediatric hypothyroidism. Endocrine 66, 51–62. doi: 10.1007/s12020-019-02024-6
Boitnott, A., Garcia-Forn, M., Ung, D. C., Niblo, K., Mendonca, D., Park, Y., et al. (2021). Developmental and Behavioral Phenotypes in a Mouse Model of DDX3X Syndrome. Biol. Psychiatr. 90, 742–755. doi: 10.1016/j.biopsych.2021.05.027
Chen, H. H., Yu, H. I., and Tarn, W. Y. (2016). DDX3 Modulates Neurite Development via Translationally Activating an RNA Regulon Involved in Rac1 Activation. J. Neurosci. 36, 9792–9804. doi: 10.1523/JNEUROSCI.4603-15.2016
Chen, H. H., Yu, H. I., Yang, M. H., and Tarn, W. Y. (2018). DDX3 Activates CBC-eIF3-Mediated Translation of uORF-Containing Oncogenic mRNAs to Promote Metastasis in HNSCC. Cancer Res. 78, 4512–4523. doi: 10.1158/0008-5472.CAN-18-0282
Chen, Y., Liu, K. Y., Yang, Z. L., Li, X. H., Xu, R., and Zhou, H. (2020). A de novo DDX3X Variant Is Associated With Syndromic Intellectual Disability: case Report and Literature Review. Front. Pediatr. 8:303. doi: 10.3389/fped.2020.00303
Cheng, W., Wang, S., Zhang, Z., Morgens, D. W., Hayes, L. R., Lee, S., et al. (2019). CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 104, 885.e–898.e. doi: 10.1016/j.neuron.2019.09.003
Deciphering Developmental Disorders Study. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Garbelli, A., Beermann, S., Di Cicco, G., Dietrich, U., and Maga, G. (2011). A motif unique to the human DEAD-box protein DDX3 is important for nucleic acid binding, ATP hydrolysis, RNA/DNA unwinding and HIV-1 replication. PLoS One 6:e19810. doi: 10.1371/journal.pone.0019810
Hildebrand, M. S., Jackson, V. E., Scerri, T. S., Van Reyk, O., Coleman, M., Braden, R. O., et al. (2020). Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology 94, e2148–e2167. doi: 10.1212/WNL.0000000000009441
Huang, M., Tailor, J., Zhen, Q., Gillmor, A. H., Miller, M. L., Weishaupt, H., et al. (2019). Engineering Genetic Predisposition in Human Neuroepithelial Stem Cells Recapitulates Medulloblastoma Tumorigenesis. Cell Stem Cell 25, 433.e–446.e. doi: 10.1016/j.stem.2019.05.013
Johnson-Kerner, B., Snijders Blok, L., Suit, L., Thomas, J., Kleefstra, T., Sherr, E. H., et al. (2020). ““DDX3X-Related Neurodevelopmental Disorder,”,” in GeneReviews(§), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaa, et al. (Seattle (WA): University of Washington).
Kellaris, G., Khan, K., Baig, S. M., Tsai, I. C., Zamora, F. M., Ruggieri, P., et al. (2018). A hypomorphic inherited pathogenic variant in DDX3X causes male intellectual disability with additional neurodevelopmental and neurodegenerative features. Hum. Genom. 12:11. doi: 10.1186/s40246-018-0141-y
Kumar, P., Mohan, V., Sinha, R. A., Chagtoo, M., and Godbole, M. M. (2015). Histone deacetylase inhibition reduces hypothyroidism-induced neurodevelopmental defects in rats. J. Endocrinol. 227, 83–92. doi: 10.1530/JOE-15-0168
Lai, M. C., Lee, Y. H., and Tarn, W. Y. (2008). The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol. Biol. Cell 19, 3847–3858. doi: 10.1091/mbc.e07-12-1264
Lee, C. S., Dias, A. P., Jedrychowski, M., Patel, A. H., Hsu, J. L., and Reed, R. (2008). Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 36, 4708–4718. doi: 10.1093/nar/gkn454
Lennox, A. L., Hoye, M. L., Jiang, R., Johnson-Kerner, B. L., Suit, L. A., Venkataramanan, S., et al. (2020). Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron 106, 404.e–420.e. doi: 10.1016/j.neuron.2020.01.042
Linsalata, A. E., He, F., Malik, A. M., Glineburg, M. R., Green, K. M., Natla, S., et al. (2019). DDX3X and specific initiation factors modulate FMR1 repeat-associated non-AUG-initiated translation. EMBO Rep. 20:e47498. doi: 10.15252/embr.201847498
Marvin, A. R., Marvin, D. J., Lipkin, P. H., and Law, J. K. (2017). Analysis of Social Communication Questionnaire (SCQ) Screening for Children Less Than Age 4. Curr. Dev. Dis. Rep. 4, 137–144. doi: 10.1007/s40474-017-0122-1
Maulik, P. K., Mascarenhas, M. N., Mathers, C. D., Dua, T., and Saxena, S. (2011). Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 32, 419–436. doi: 10.1016/j.ridd.2010.12.018
Patmore, D. M., Jassim, A., Nathan, E., Gilbertson, R. J., Tahan, D., Hoffmann, N., et al. (2020). DDX3X Suppresses the Susceptibility of Hindbrain Lineages to Medulloblastoma. Dev. Cell 54, 455.e–470.e. doi: 10.1016/j.devcel.2020.05.027
Phung, B., Cieśla, M., Sanna, A., Guzzi, N., Beneventi, G., Ngoc, P., et al. (2019). The X-Linked DDX3X RNA Helicase Dictates Translation Reprogramming and Metastasis in Melanoma. Cell Rep. 27, 3573.e–3586.e. doi: 10.1016/j.celrep.2019.05.069
Prezioso, G., Giannini, C., and Chiarelli, F. (2018). Effect of Thyroid Hormones on Neurons and Neurodevelopment. Horm. Res. Paediatr. 90, 73–81. doi: 10.1159/000492129
Ramaswami, M., Taylor, J. P., and Parker, R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736. doi: 10.1016/j.cell.2013.07.038
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Snijders Blok, L., Madsen, E., Juusola, J., Gilissen, C., Baralle, D., Reijnders, M. R., et al. (2015). Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am. J. Hum. Genet. 97, 343–352. doi: 10.1016/j.ajhg.2015.07.004
Tuhan, H., Abaci, A., Cicek, G., Anik, A., Catli, G., Demir, K., et al. (2016). Levothyroxine replacement in primary congenital hypothyroidism: the higher the initial dose the higher the rate of overtreatment. J. Pediatr. Endocrinol. Metab. 29, 133–138. doi: 10.1515/jpem-2015-0047
Wang, X., Posey, J. E., Rosenfeld, J. A., Bacino, C. A., Scaglia, F., Immken, L., et al. (2018). Phenotypic expansion in DDX3X - a common cause of intellectual disability in females. Ann. Clin. Transl. Neurol. 5, 1277–1285. doi: 10.1002/acn3.622
Yang, Y. (2016). Rating Scales For Children’s Developmental Behavior and Mental Health. Beijing: People’s Medical Publishing House.
Keywords: DDX3X, intellectual disability, DDX3X syndrome, neuronal development, X-linked intellectual disability
Citation: Dai Y, Yang Z, Guo J, Li H, Gong J, Xie Y, Xiao B, Wang H and Long L (2022) Expansion of Clinical and Genetic Spectrum of DDX3X Neurodevelopmental Disorder in 23 Chinese Patients. Front. Mol. Neurosci. 15:793001. doi: 10.3389/fnmol.2022.793001
Received: 11 October 2021; Accepted: 02 February 2022;
Published: 22 March 2022.
Edited by:
Liana Fattore, CNR Neuroscience Institute (IN), ItalyReviewed by:
Taimoor Sheikh, North York General Hospital, CanadaClaude Besmond, INSERM U1163 Institut Imagine, France
Anna Ruiz, Instituto de Investigación e Innovación Parc Taulí (I3PT), Spain
Copyright © 2022 Dai, Yang, Guo, Li, Gong, Xie, Xiao, Wang and Long. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lili Long, bG9uZ2xpbGkxOTgyQDEyNi5jb20=; Hua Wang, d2FuZ2h1YV8yMTNAaG90bWFpbC5jb20=
†These authors have contributed equally to this work