 Ivan T. Demchenko1,2
Ivan T. Demchenko1,2 Hagir B. Suliman2Sergey Y. Zhilyaey2Olga S. Alekseeva2Tatyana F. Platonova2Matthew S. Makowski1Claude A. Piantadosi1
Hagir B. Suliman2Sergey Y. Zhilyaey2Olga S. Alekseeva2Tatyana F. Platonova2Matthew S. Makowski1Claude A. Piantadosi1 Heath G. Gasier1*
Heath G. Gasier1*- 1The Duke Center for Hyperbaric Medicine and Environmental Physiology, Duke University Medical Center, Durham, NC, United States
- 2Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, St. Petersburg, Russia
Oxygen breathing at elevated partial pressures (PO2’s) at or more than 3 atmospheres absolute (ATA) causes a reduction in brain γ-aminobutyric acid (GABA) levels that impacts the development of central nervous system oxygen toxicity (CNS-OT). Drugs that increase brain GABA content delay the onset of CNS-OT, but it is unknown if oxidant damage is lessened because brain tissue PO2 remains elevated during hyperbaric oxygen (HBO2) exposures. Experiments were performed in rats and mice to measure brain GABA levels with or without GABA transporter inhibitors (GATs) and its influence on cerebral blood flow, oxidant damage, and aspects of mitochondrial quality control signaling (mitophagy and biogenesis). In rats pretreated with tiagabine (GAT1 inhibitor), the tachycardia, secondary rise in mean arterial blood pressure, and cerebral hyperemia were prevented during HBO2 at 5 and 6 ATA. Tiagabine and the nonselective GAT inhibitor nipecotic acid similarly extended HBO2 seizure latencies. In mice pretreated with tiagabine and exposed to HBO2 at 5 ATA, nuclear and mitochondrial DNA oxidation and astrocytosis was attenuated in the cerebellum and hippocampus. Less oxidant injury in these regions was accompanied by reduced conjugated microtubule-associated protein 1A/1B-light chain 3 (LC3-II), an index of mitophagy, and phosphorylated cAMP response element binding protein (pCREB), an initiator of mitochondrial biogenesis. We conclude that GABA prevents cerebral hyperemia and delays neuroexcitation under extreme HBO2, limiting oxidant damage in the cerebellum and hippocampus, and likely lowering mitophagy flux and initiation of pCREB-initiated mitochondrial biogenesis.
Introduction
Under normal conditions the partial pressure of oxygen (PO2) in the brain is ~30–40 mmHg in rats and humans but breathing hyperbaric oxygen (HBO2) increases it in proportion to the inspired PO2 (Demchenko et al., 2005; Ponce et al., 2012). As consequence, oxidants accumulate and disrupt neurotransmission leading to convulsions resembling epilepsy, neuronal trauma and necrosis, and death, i.e., central nervous system oxygen toxicity (CNS-OT; Piantadosi and Tatro, 1990; Oury et al., 1992; Demchenko and Piantadosi, 2006; D'Agostino et al., 2007). For HBO2 therapy where the inspired PO2 is between 2.0–2.9 atmospheres absolute (ATA) with air intervals, the incidence of seizures is 0.02–0.6% (Smerz, 2004; Costa et al., 2019). In divers who use closed-circuit oxygen rebreathers, the incidence of CNS-OT (signs/symptoms and seizures) is 2.5–7% (Butler and Thalmann, 1986; Arieli et al., 2006). Limited knowledge of how brain cells respond to increased PO2 has prevented expansion of HBO2 indications and oxygen exposure profiles, warranting further study.
Following the discovery of γ-aminobutyric acid (GABA) and its inhibitory function on neuroexcitation in the mammalian CNS (Roberts and Frankel, 1950; Basemore et al., 1957), Wood and Watson (1962) postulated that GABA was involved in oxygen seizures. The hypothesis was supported by data showing that the concentration of brain GABA was reduced in pharmacologically induced convulsions due in part to glutamic acid decarboxylase (GAD) inhibition, and GABA administration offered some level of protection against seizures in animals and humans (Killam and Bain, 1957; Roberts, 1959; Baxter and Roberts, 1960; Gulati and Stanton, 1960). Wood et al. (Wood and Watson, 1963, 1964; Wood et al., 1967, 1969) determined the following: whole brain GABA levels decrease as a function of inspired PO2 above 3 ATA and is due to GAD inhibition, the rate of decline in brain GABA content is related to oxygen seizure latencies, and pretreatment with GABA decreases oxygen seizure incidence, severity and mortality. In the United States, GABA is not prescribed for epilepsy, leading us to test the efficacy of FDA approved antiepileptic drugs in mice exposed to HBO2 at 5 ATA (Demchenko et al., 2019). Of these drugs, GABA enhancers demonstrated the best efficacy in delaying HBO2 seizures. Because seizures cause DNA damage (Cantafora et al., 2014), astrocyte reactivity (Chipres-Tinajero et al., 2021), and increased permeability to the blood–brain barrier (Bargerstock et al., 2014), maintaining brain GABA levels may lessen oxidant damage in brain cells. If, however, cerebral blood flow (CBF) responses to HBO2 are unabated, PO2 will increase and promote oxidant brain injury. This has not been studied in HBO2.
Our objective was to measure GABA’s role in cerebrovascular control and oxidant brain injury. Moreover, since HBO2 damages mitochondria and activates mitochondrial biogenesis (Balentine, 1974; Gutsaeva et al., 2006), we measured activation signaling pathways of mitophagy and mitochondrial biogenesis that are linked to oxidants and inflammation. Focus was placed on the cerebellum and hippocampus due to their susceptibility to oxygen-induced neuronal injury (Balentine, 1982; Gutsaeva et al., 2006). Our approach was to reduce GABA reuptake from the synaptic cleft by inhibiting GABA transporters (GATs) with nipecotic acid (NPA) or tiagabine (TGB). Nipecotic acid is a nonselective inhibitor of brain GATs (1–3), whereas TGB selectively inhibits GAT1 (Nielsen et al., 1991; Kragler et al., 2005). Our hypothesis was that in extreme HBO2, GAT inhibition lessens oxidant injury by delaying neuroexcitation independently of changes in CBF. Given less injury, stimulation of mitochondrial turnover signaling would be reduced.
Materials and methods
All procedures were approved by the Duke University Institutional Animal Care and Use Committee (IACUC) and the Ethical Review Board of the Sechenov Institute of Evolutionary Physiology and Biochemistry Russian Academy of Sciences. Rats were used for cerebrovascular control experiments because physiological stress responses are similar to humans (Goutianos et al., 2015). Mice were used to study oxidant brain injury because their sensitivity to oxygen resembles humans (Marks, 1944).
Experimental protocol in anesthetized rats
Experiments were performed at the Duke Center for Hyperbaric Medicine and Environmental Physiology. Male Sprague Dawley rats weighing 317–367 g (Charles River Laboratories) were anesthetized with IP urethane (750 mg/kg) and α-chloralose (250 mg/kg), placed on a heating pad with rectal thermometer, and ventilated mechanically with 30% oxygen using a small animal respirator (Edco Scientific Inc.). Catheters were inserted into the femoral artery and vein for blood pressure monitoring and drug delivery, respectively. Heads were positioned in a stereotaxic frame (David Kopf Instruments), and two stainless steel screws were placed into the left and right parietal cortexes for electroencephalogram (EEG) recording. CMA 11 microdialysis probes (0.24 mm, CMA Microdialysis AB) and platinum needle electrodes (100 μm) were inserted into the caudate-putamen (striatum) using a micromanipulator and stereotaxic coordinates (Paxinos and Watson, 2007). Microdialysis probes were continuously perfused with artificial cerebral spinal fluid (aCSF) at a rate of 1 μl/min using a CMA microinjection pump (Carnegie Medicine). The platinum electrodes were used for measuring CBF by the hydrogen clearance method (Demchenko et al., 1998). Electrocardiogram (ECG) electrodes were placed bilaterally under the chest skin for measuring heart rate (RR interval). Anesthesia was maintained throughout the experiments by administering one-fourth the initial doses, and pancuronium bromide (500 μg/kg) was provided to inhibit involuntary respiratory movements. The breathing gas was changed to 100% oxygen after baseline measurements, and rats remained at 1 ATA or were compressed to 3, 5, or 6 ATA at a rate of 0.6 ATA/min. Temperature, relative humidity and CO2 were maintained at 23–25°C, 60 and 0.05%, respectively.
For measurement of interstitial amino acids, rats were exposed to 1 (n = 8), 3 (n = 7), 5 (n = 8), and 6 ATA (n = 20) oxygen for 75 min. After a 60 min stabilization period with aCSF infusion, baseline dialysate samples were collected in vials containing 1% perchloric acid before and every 15 min during exposures using a CMA 142 Microfraction Collector (CMA Microdialysis AB). In another group of rats exposed to 6 ATA oxygen (n = 8), aCSF was changed to a mixture of aCSF + 70 μM NPA (MilliporeSigma, 656356) after baseline sampling. Amino acids (aspartate, GABA, glutamate, glutamine, glycine, and serine) in the dialysate were measured by o-phthalaldehyde derivatization and HPLC with electrochemical detection (Donzanti and Yamamoto, 1988).
For measurement of cardio-and cerebrovascular responses in HBO2, rats (n = 16) were exposed to 6 ATA oxygen for 75 min. After a 60 min stabilization period and baseline recording of heart rate, arterial blood pressure, EEG and striatal CBF, rats were injected with 7 μl of aCSF (n = 8) or aCSF + TGB (MilliporeSigma, 1667280) in 5% DMSO (0.34 μmol, n = 8) into the lateral ventricle 30 min before HBO2. Measurements were performed every 15 min in HBO2.
Experimental protocol in conscious rats
Experiments were performed at the Sechenov Institute of Evolutionary Physiology and Biochemistry. Male Sprague Dawley rats weighing 301–349 g were procured from Pushkino Animal-Breeding Facility. One week prior to HBO2, rats were anesthetized with IP pentobarbital (50 mg/kg). A cannula was inserted into the lateral ventricle for drug delivery and secured with acrylic dental cement and two stainless steel anchor screws (EEG in a subset of animals). In 14 rats, PE-50 tubing containing 0.9% NaCl + 2.5% glucose + 300 IU/ml heparin was inserted into the right carotid artery toward the aorta, secured, and tunneled subcutaneously to the back of the neck. Rats were provided penicillin (30,000 IU/kg/day), and the catheter was flushed daily with saline. On experimental days, 7 μl of aCSF (n = 14), TGB (0.34 μmol, n = 11), or NPA (1.1 μmol, n = 14) was administered over 2 min with a Hamilton micro syringe 30 min before exposure to HBO2 at 5 ATA. Rats were placed in a pressure chamber (100 L) and the oxygen pressure was increased to 5 ATA at a rate of 1 ATA/min. Temperature, relative humidity, and CO2 levels were maintained similarly as above. The catheterized rats (aCSF, n = 7 and TGB, n = 7) were lightly restrained in a hammock for arterial blood pressure and heart rate (calculated from arterial blood pressure pulse) monitoring. Animals were monitored with a camera and exposures were terminated upon the appearance of seizures, EEG spikes in the lightly restrained rats, or up to 90 min.
Experimental protocol in conscious mice
Experiments were performed at the Duke Center for Hyperbaric Medicine and Environmental Physiology. Male C57BL/6 J mice (n = 57) aged 8–10 weeks (~25–30 g) were procured from The Jackson Laboratory. Mice were assigned to air vehicle (n = 11), air TGB (n = 11), HBO2 vehicle (n = 17), and HBO2 TGB (n = 18). Mice were administered vehicle (0.9% sodium chloride) or TGB (4.8 mg/kg) IP in a volume of 5 μl per g body weight 30 min before air or HBO2 exposures. This TGB dose was selected based on our previous work showing extended seizure latencies by a factor of 3 over controls (Demchenko et al., 2019). Up to five mice at a time were placed individually in plastic cylinders (22 cm in length and 11.5 cm in diameter). The cylinders were flooded with 100% oxygen for 5 min before compression to 5 ATA oxygen at 0.75 ATA per min. The chamber temperature was maintained between 23 and 25°C. After 30 min at 5 ATA, mice were decompressed to sea level at 0.75 ATA per min.
Because brain GABA levels peak ~40 min after IP injections (Fink-Jensen et al., 1992), air and HBO2 mice were staggered by 30 min to ensure euthanasia (isoflurane) and brain harvest was completed quickly. Brains were flash frozen in liquid N2 or sectioned (sagittal), placed in tissue embedding cassettes and 10% formalin for 24 h before transferring to 70% ETOH and refrigerating at 4°C. Samples were sent to the Duke Substrate Services Core & Research Support for paraffin embedding and slide preparation. In some mice after brain harvest, blood was collected from the abdominal aorta using a 1 ml syringe with a 23-gauge needle and placed in 0.6 ml serum separator tubes (BD Vacutainer®). After 30 min at room temperature, samples were centrifuged at 6,000 g for 90 s. Serum was transferred to Eppendorf vials and frozen at −80°C. Serum S100 calcium-binding protein B (S100B) was quantified using an ELISA assay (LSBio, LS-F5980).
Immunofluorescence
Slides from mice that matched group mean seizure latencies in HBO2, and random air mice were incubated with primary antibodies diluted in phosphate-buffered saline (PBS) overnight, washed 2 in PBS for 10 min, incubated in secondary antibodies diluted in PBS for 1 h, and washed 2 in PBS for 5 min. Primary antibodies included ATP5A (ATP synthase F1 subunit α; Abcam, Ab14748), citrate synthase (GeneTex, GTX110624), focal adhesion kinase family interacting protein of 200 kD (FIP200, Invitrogen™, PA528563), cAMP response element-binding protein (CREB, Santa Cruz, sc-186), phosphorylated (p)-CREB at Ser-133 (Santa Cruz, sc-7978), glial fibrillary acidic protein (GFAP, Booster Immunoleader, MA1045), heme oxygenase 1 (HO-1, Enzo, ADI-SPA-896F), LC3A/B (LC3-II; Cell Signaling, 4108), PTEN-induced kinase 1 (PINK1, Abcam, ab23707), and 8-hydroxy-2’deoxyguanosine (8-OHdG, Santa Cruz, Sc66036). Antibodies were diluted 1:400 except LC3A/B (1:100). Goat anti-rabbit IgG (Alex Fluor™ 488) and goat anti-mouse IgG (Alex Fluor™ 568) secondary antibodies were purchased from ThermoFisher Scientific and diluted 1:400. All incubations were performed at room temperature. Coverslips were mounted using ProLong™ Gold Antifade Mountant with DAPI (Invitrogen™, P36935) and stored at 4°C until imaging. From three animals/group, six regions were imaged at 60 magnification. Multichannel images were captured from each section using a Nikon Eclipse 50i microscope with a DS-Ri2 color CMOS camera and Nikon Plan Fluor objectives. The signal intensity in collected images were compared to the signal of negative controls and used to determine exposure times and prevent false positives. Nikon NIS-Elements AR software v5.30 was used for quantification.
RT-qPCR
Total RNA was isolated from brain tissue using RNAqueous-4 PCR kits (ThermoFisher Scientific). Following DNase treatment (4 units for 1 h at 37°C) and inactivation, cDNA was prepared with a high-capacity cDNA archive kit (Applied Biosystems). The following TaqMan® primers were purchased from ThermoFisher Scientific: autophagy-related protein 9A (Atg9a, Mm01264420_m1), Fip200 (Mm00456545_m1), HO-1 (Hmox1, Mm00516006_m1), nuclear respiratory factor 1 (Nrf1, Mm00447996_m1), peroxisome proliferator-activated receptor γ coactivator 1-α (Ppargc1a, Mm01208836_g1), superoxide dismutase 2 (Sod2, Mm00449726_m1), mitochondrial transcription factor A (Tfam, Mm00447485_m1), and 18S ribosomal RNA (RN18S, Mm03928990_g1). All reactions were completed on a StepOnePlus Real-Time PCR System (Applied Biosystems) for 40 cycles. Data were analyzed using the Fold change assay (DataAssist, v3.01, Applied Biosystems) after normalizing to 18S in each sample and control (air vehicle).
Data monitoring and statistical analysis
Heart rate, arterial blood pressure, striatal CBF, and EEG were recorded and analyzed using WinDaq software and DI-200 data acquisition hardware (DATAQ Instruments) or with LabScribe 2 software on iWorx IX-228/S hardware (iWorx Systems). Interstitial amino acid levels measured at 1, 3, 5, and 6 ATA oxygen were analyzed using a one-way repeated measures ANOVA. Linear regression was used to determine the relationship between GABA and inspired PO2. A two-way repeated measures ANOVA was used to determine the effects of exposure (air and HBO2), treatment (aCSF and NPA or TGB), and interaction (exposure treatment) on interstitial GABA levels, heart rate, mean arterial blood pressure, and striatal CBF in anesthetized and conscious rats exposed to 6 and 5 ATA oxygen, respectively. Seizure latencies in rats and mice were compared using a t-test and one-factor ANOVA. For all measures in conscious mice, a two-factor ANOVA was used to determine the effects of exposure (air and HBO2), treatment (vehicle and TGB) and interaction (exposure treatment). A Bonferroni t-test was used in post hoc analysis. For immunofluorescence data, variances were unequal (Brown-Forsythe) for all measurements except for GFAP in the hippocampus and HO-1 in the cerebellum. In these instances, analysis was performed on transformed data (log or square root). Data were analyzed with SigmaPlot 14.0 (Systat Software Inc.). Values are means ± SD. A p < 0.05 was considered statistically significant.
Results
Inspired PO2 modulates brain interstitial GABA levels
Changes in striatal interstitial GABA and other amino acids (aspartate, glutamate, glutamine, glycine, and serine) were measured in anesthetized rats exposed to HBO2 at 6 ATA for 75 min (Table 1). The striatum is the largest structure in the basal ganglia and central for coordinating behavior and motor function, and contains both GABAA and GABAB receptors (Lacey et al., 2005; Mathew et al., 2010; Girault, 2012). The concentration of GABA progressively declined in HBO2 to 41% of pre-exposure values. After 75 min, serine and glutamine levels decreased by 26 and 17%, respectively. Aspartate, glycine and glutamate content remained stable in HBO2. We also measured the effect of inspired PO2 from 0.3 to 6 ATA on extracellular GABA levels (Figure 1A). At 3 ATA, GABA content initially increased and fell thereafter, reaching significance at 75 min. Increasing the inspired PO2 to 5 and 6 ATA led to faster and greater declines in GABA levels, mirroring the responses in brain PO2 (Demchenko et al., 2005).

Table 1. Interstitial amino acids measured in the striatum of rats exposed to HBO2 at 6 ATA.
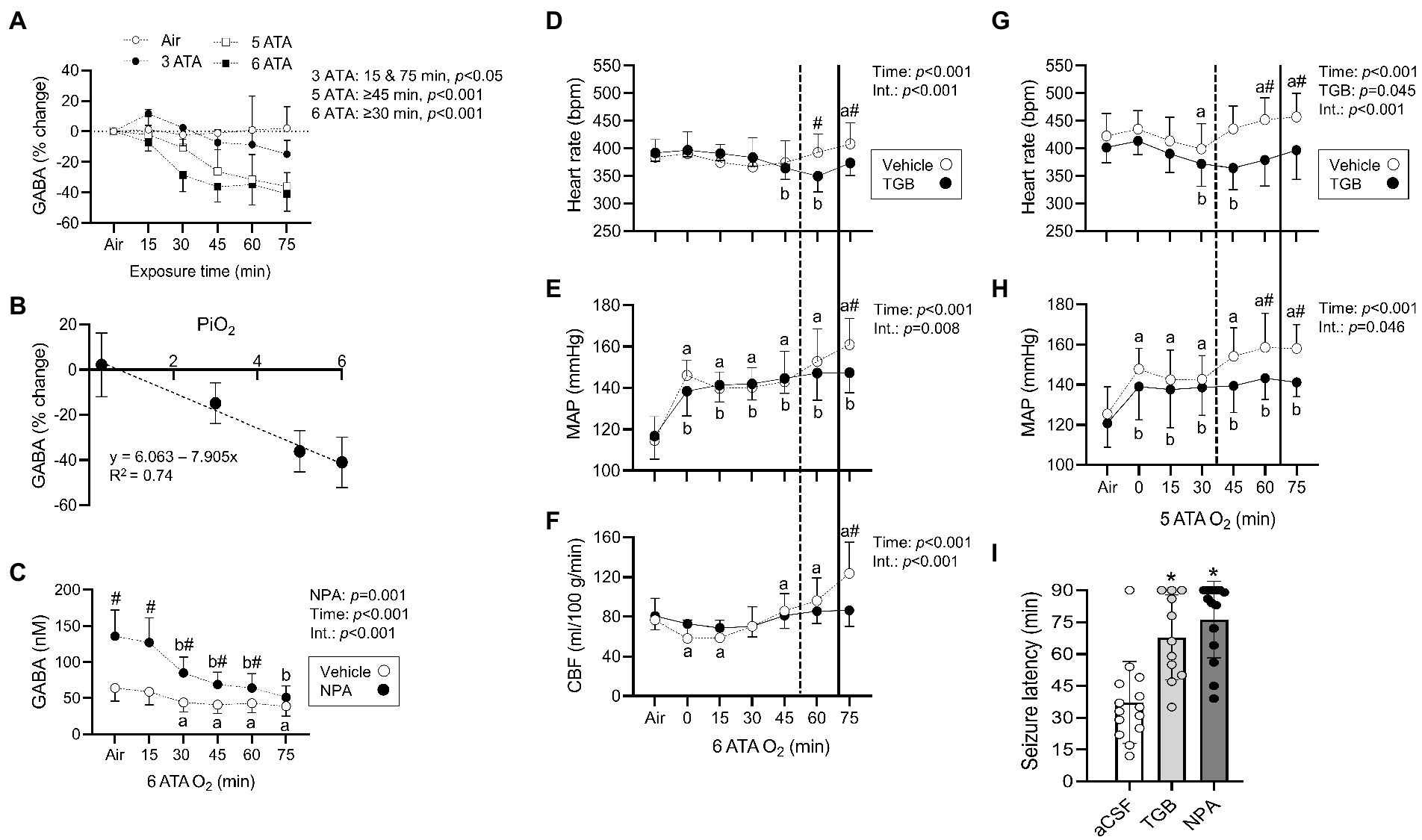
Figure 1. Striatal interstitial γ-aminobutyric acid (GABA) levels, cardiovascular and cerebral blood flow (CBF) responses in rats exposed to extreme HBO2. (A) Changes in the concentration of GABA measured before and every 15 min in air, 3, 5, and 6 atmospheres absolute (ATA) oxygen (n = 7–8/group). (B) The relationship between changes in GABA levels (75 min to Air) and the inspired PO2 (PiO2). (C) Concentration of GABA measured before and every 15 min in HBO2 at 6 ATA in rats perfused with aCSF (CON) or aCSF + NPA (70 μM) into the caudate putamen (n = 8/group). (D–F) Heart rate, mean arterial blood pressure (MAP), and CBF were measured in anesthetized rats before and during exposure to 6 ATA oxygen (n = 8–10/group). (G,H) Heart rate and MAP were measured in conscious rats before and during exposure to 5 ATA oxygen (n = 7–8/group). In both experiments, rats were injected with aCSF (CON) or aCSF + TGB in 5% DMSO (0.34 μmol/7 μl) into the lateral ventricle 30 min before HBO2. (D–H) Vertical dotted (aCSF) and bold (TGB) lines represent mean seizure latencies determined from EEG recordings. (I) Mean seizure latency in conscious rats injected with aCSF, aCSF + TGB in 5% DMSO (0.34 μmol/7 μl), or aCSF + NPA in 5% DMSO (1.1 μmol/7 μl) into the lateral ventricle 30 min before HBO2 at 5 ATA (n = 11–14/group). Values are means ± SD. Time dependent changes in aaCSF and bNPA or TGB groups, p < 0.05. #Between group differences, p < 0.05. *Significantly different from aCSF rats, p < 0.001.
When GABA content (75 min from air) is plotted as a function of inspired PO2, a linear decrease is observed (Pearson’s r = 0.86, p < 0.001; Figure 1B). To understand the effect of inhibiting GABA transport on interstitial GABA levels, NPA or aCSF were continuously delivered to the striatum before and in HBO2 at 6 ATA (Figure 1C). NPA led to an initial more than 2-fold increase in interstitial GABA levels over rats infused with aCSF, however, levels declined to control values by the end of exposures. These data support an inhibitory effect of increased PO2 on extracellular striatal GABA production (Wood and Watson, 1964; Wood et al., 1967; Gasier et al., 2017).
TGB prevents cerebral hyperemia in HBO2
In HBO2 at 5–6 ATA, a seizure is accompanied by a rise in heart rate, a secondary increase in mean arterial blood pressure and increased CBF (Demchenko et al., 2014). To determine if GABA alters these responses, we administered TGB or aCSF to anesthetized and conscious rats exposed to 6 and 5 ATA of oxygen for 75 min, respectively. In anesthetized rats, TGB prevented tachycardia, hypertension and cerebral hyperemia, and delayed the appearance of EEG spikes by 18 ± 13 min compared to controls (p = 0.013; Figures 1D–F). In addition, electrical discharges were present in only 25% of rats treated with TGB compared to 80% in controls. In conscious rats, TGB was equally efficacious in preventing tachycardia and hypertension (Figures 1G,H), and in extending seizure onset by 31 ± 19 min compared to controls (p < 0.001). To determine if GABA reuptake is primarily through GAT1, we compared seizure latencies in conscious rats administered aCSF, TGB, or NPA and exposed to 5 ATA oxygen for 90 min (Figure 1I). While NPA increased mean seizure latencies by 9 ± 18 min over TGB, mean group differences were not significant. These data indicate that inhibiting GABA reuptake mainly through GAT1 prevents cerebral hyperemia and delays seizures in extreme HBO2.
TGB lessens oxidant injury in the cerebellum and hippocampus
To explore if GAT1 inhibition protects the brain from oxidant injury, mice were pretreated with 0.9% NaCl or TGB before exposure to 5 ATA oxygen for 30 min. This profile caused 82% of control mice to exhibit motor convulsions at a mean time of 11.6 ± 9.4 min, whereas TGB reduced this to 44% (mean latency 23.0 ± 9.5 min; p < 0.001). We assessed oxidant brain injury by measuring nuclear and mitochondrial DNA oxidation and astrocyte reactivity using 8-OHdG and GFAP, respectively (Cantafora et al., 2014; Chipres-Tinajero et al., 2021). HBO2 caused significant nuclear and mitochondrial DNA oxidation (Figures 2A–E) and increased astrocyte reactivity (Figures 3A,B,E) in the cerebellum and hippocampus. As an indicator of blood–brain barrier integrity (Kanner et al., 2003), we measured serum S100B and observed a 28% increase in HBO2 exposed mice independent of treatment (Air, 48 ± 5 pg./ml vs. HBO2, 61 ± 9 pg./ml; p < 0.001). TGB pretreatment decreased the oxidant injury in the cerebellum and hippocampus.
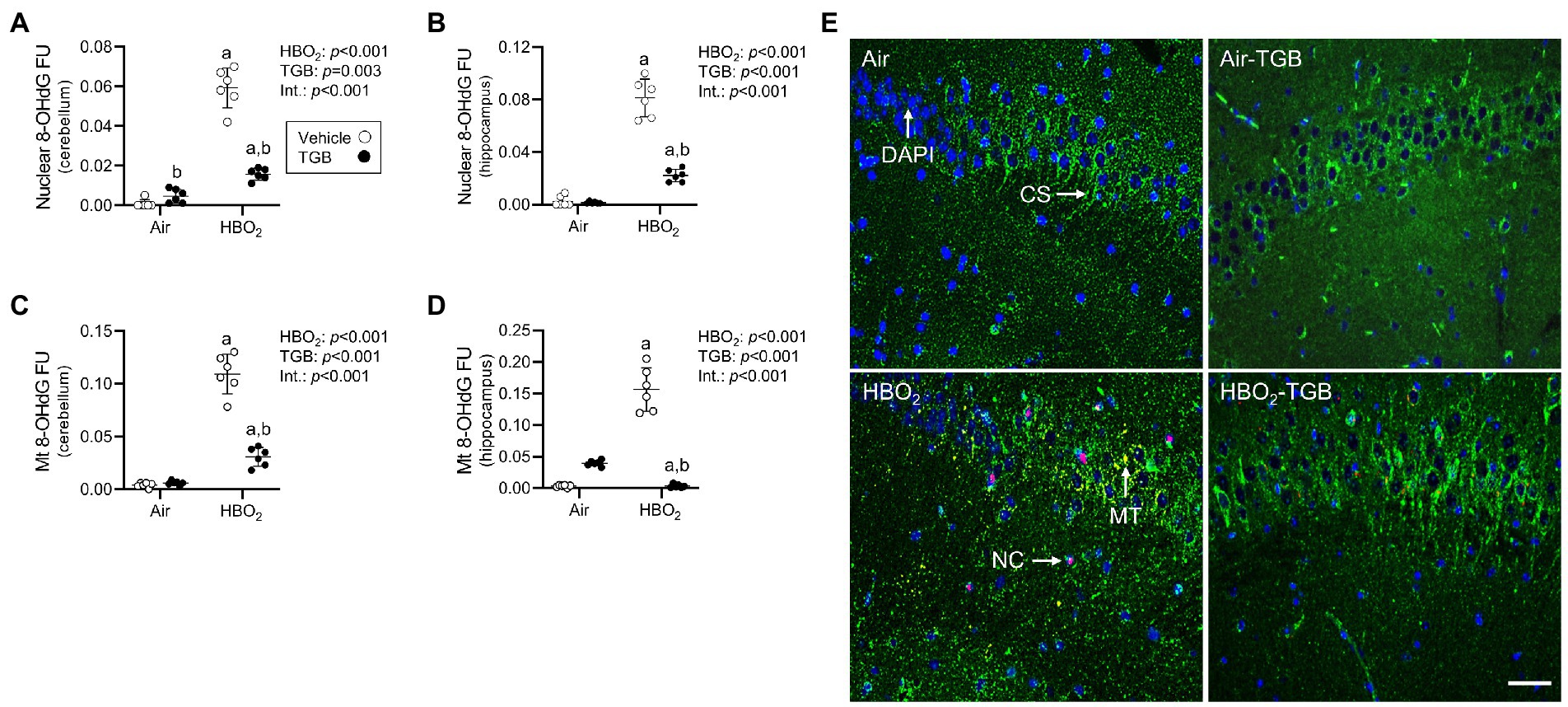
Figure 2. Nuclear and mitochondrial DNA oxidation in mice exposed to extreme HBO2. Mice were pretreated with 0.9% NaCl or TGB (4.8 mg/kg body weight) 30 min before exposure to air or 5 ATA oxygen for 30 min. (A–D) Quantification of nuclear and mitochondrial 8-OHdG in the cerebellum and hippocampus. (E) Representative images from the hippocampus. 8-OHdG (red), citrate synthase (CS, green), and DAPI (blue). 8-OHdG merged with DAPI indicates nuclear oxidation (NC). 8-OHdG merged with citrate synthase indicates mitochondrial oxidation (MT). Scale bar = 100 μm. Main effect of aHBO2 and bTGB, p < 0.001.
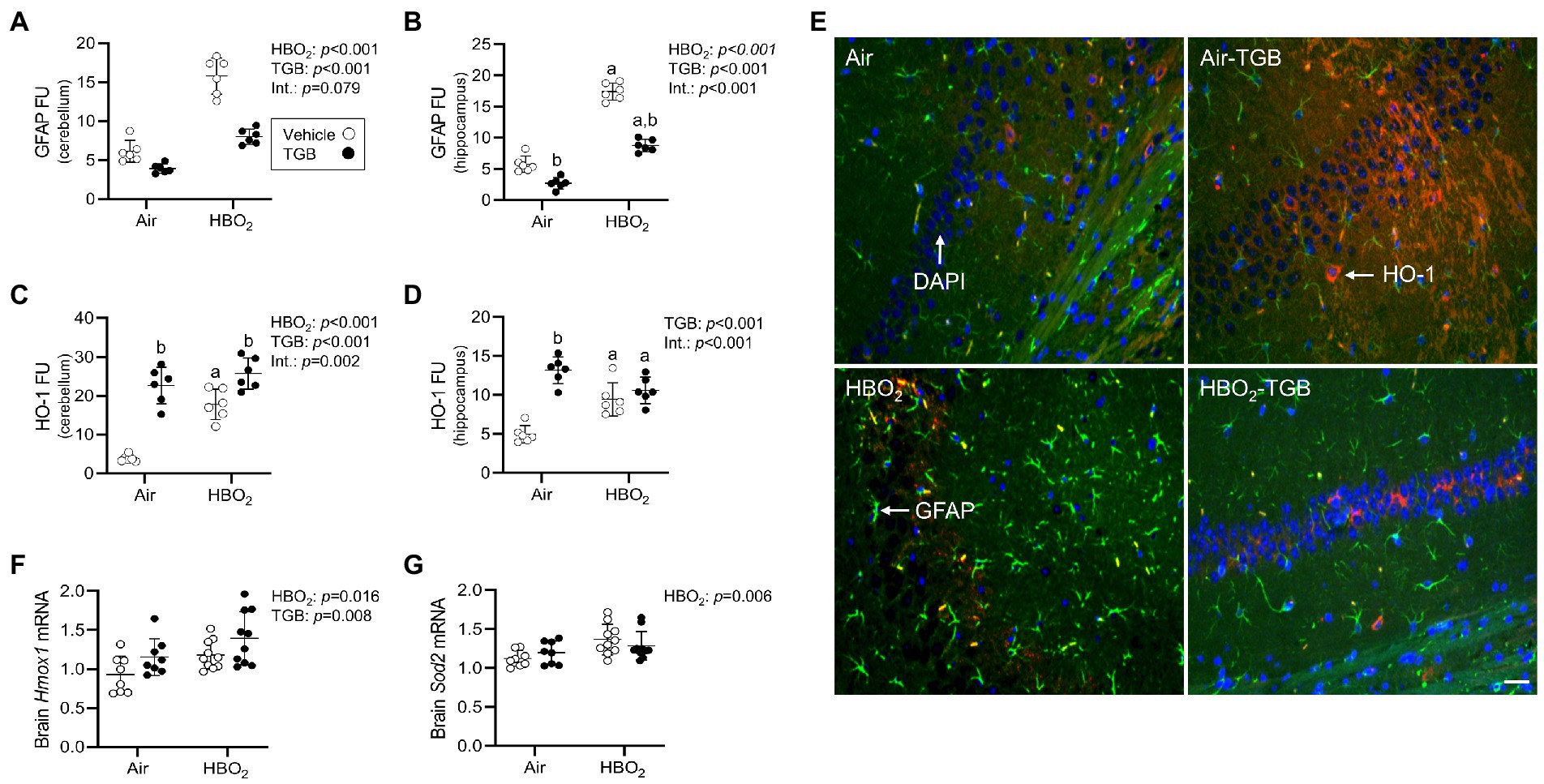
Figure 3. Reactive astrocytes and antioxidant responses in mice exposed to extreme HBO2. Mice were pretreated with 0.9% NaCl or TGB (4.8 mg/kg body weight) 30 min before exposure to air or 5 ATA oxygen for 30 min. (A–D) Quantification of glial fibrillary acid protein (GFAP) and heme oxygenase-1 (HO-1) in the cerebellum and hippocampus. (E) Representative images from the hippocampus. HO-1 (red), GFAP (green), and DAPI (blue). Scale bar = 20 μm. (F,G) Hmox1 and mitochondrial superoxide dismutase (Sod2) mRNA expression in brain homogenates (n = 8–10/group). Main effect of aHBO2 and bTGB, p < 0.05.
A compensatory response to increased oxidant stress is transcriptional activation of antioxidant and anti-inflammatory genes mediated by the nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor (Zhang and Hannink, 2003; Nguyen et al., 2005; Suliman et al., 2017). After HBO2, HO-1 protein expression in the cerebellum and hippocampus were increased (Figures 3C–E), as were Hmox1 and Sod2 mRNA expression in brain homogenates (Figures 3F,G). TGB further increased HO-1 protein expression in the cerebellum. This indicates that GAT1 inhibition reduces oxidant brain damage and activates HO-1, but does not abolish it since DNA oxidation, astrocyte reactivity, and serum S100B remained elevated over air-control mice.
TGB influences oxidant-stress mediated mitophagy and mitochondrial biogenesis signaling in HBO2
In the same mice, we explored activation of mitophagy and mitochondrial biogenesis. During cellular stress and mitochondrial damage, an initial response is for PINK1 to accumulate on the outer mitochondrial membrane (Narendra et al., 2010), resulting in recruitment and phosphorylation of ubiquitin and the E3 ubiquitin ligase Parkin, a critical step in activation of PINK1/Parkin-dependent mitophagy (Matsuda et al., 2010; Kane et al., 2014). HBO2 enhanced PINK1 expression in the cerebellum and hippocampus and TGB led to a further increase in the cerebellum (Figures 4A–C). Phagophore formation is required to recognize damaged mitochondria, a FIP200 dependent process that includes configuration of the UNC-51-like kinase (ULK1) initiation complex (Hara et al., 2008). HBO2 increased FIP200 in the cerebellum and hippocampus (Figures 5A–C) and mRNA in brain homogenates (Figure 5D). HBO2 also increased Atg9a mRNA in brain homogenates (Figure 5E), which is required for organized PINK1/Parkin dependent mitophagy and phagophore growth (Lahiri and Klionsky, 2021). TGB increased FIP200 in both regions, and augmented levels above HBO2 controls in the cerebellum. The expanded phagophore is coated with LC3 that is converted to LC3-I and conjugated to LC3-II, an autophagosome marker that fuses with lysosomes (Kabeya et al., 2000). HBO2 increased LC3-II in mitochondria, and TGB attenuated this response (Figures 6A–C). These data indicate HBO2 activates the classical PINK1 dependent mitophagy pathway, and TGB reduces LC3-II accumulation despite an increase in activation signaling primarily in the cerebellum.
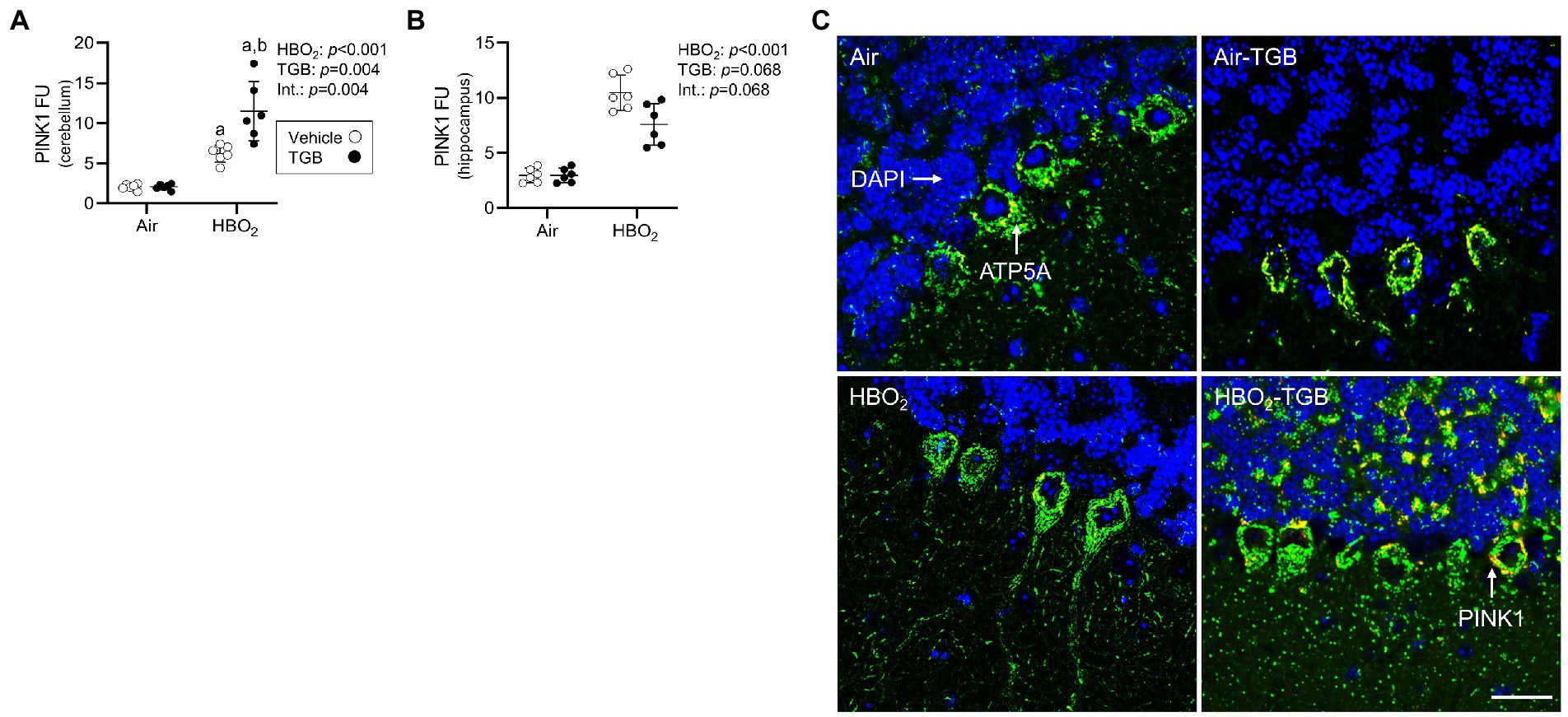
Figure 4. Mitophagy initiation in mice exposed to extreme HBO2. Mice were pretreated with 0.9% NaCl or TGB (4.8 mg/kg body weight) 30 min before exposure to air or 5 ATA oxygen for 30 min. (A,B) Quantification of PTEN-induced kinase 1 (PINK1) in the cerebellum and hippocampus. (C) Representative images from cerebellum. PINK1 (red), ATP synthase F1 subunit α (green) and DAPI (blue). Scale bar = 100 μm. Main effect of aHBO2 and bTGB, p < 0.05.
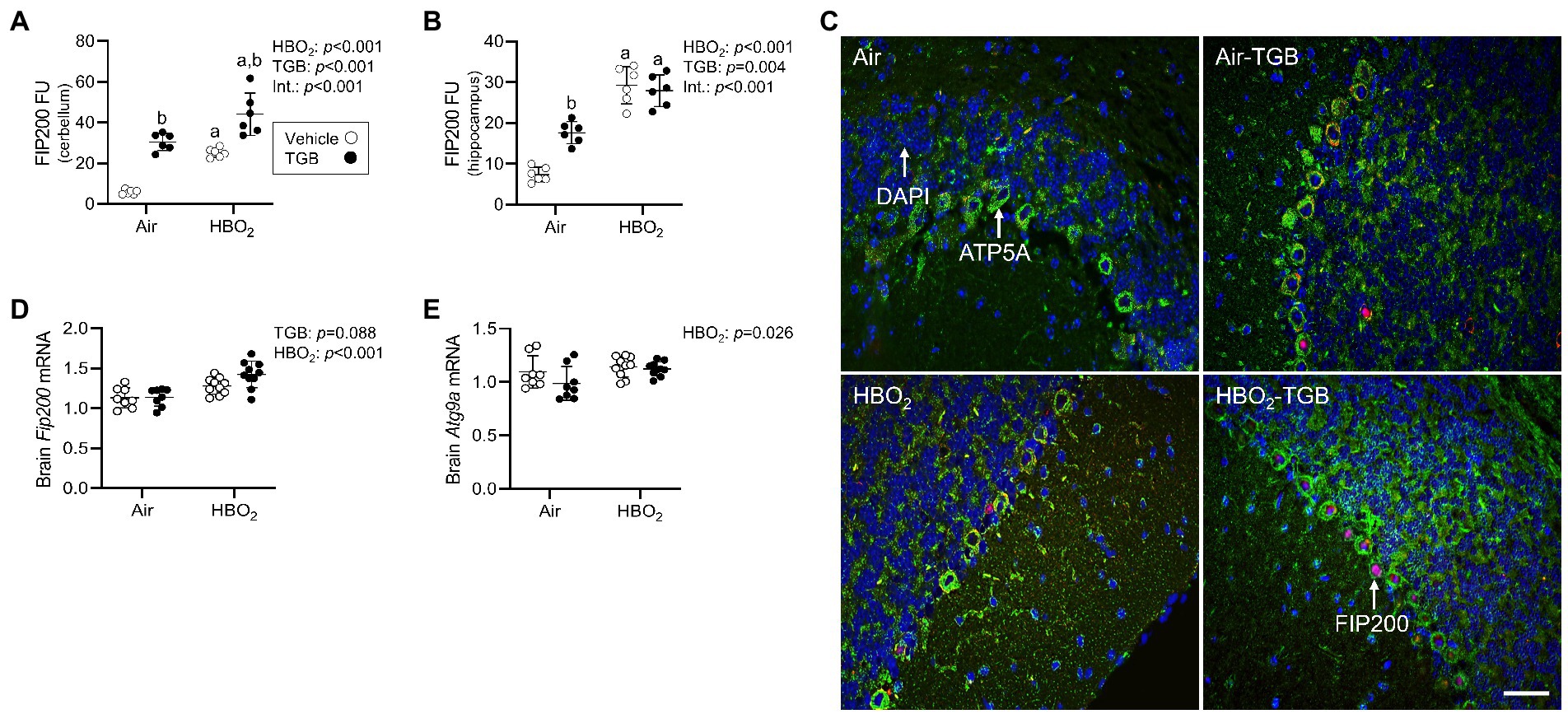
Figure 5. Phagophore formation in mice exposed to extreme HBO2. Mice were pretreated with 0.9% NaCl or TGB (4.8 mg/kg body weight) 30 min before exposure to air or 5 ATA oxygen for 30 min. (A,B) Quantification of focal adhesion kinase family interacting protein of 200 kD (FIP200) in the cerebellum and hippocampus. (C) Representative images from cerebellum. FIP200 (red), ATP synthase F1 subunit α (green) and DAPI (blue). Scale bar = 20 μm. (D,E) Fip200 and autophagy-related protein 9A (Atg9a) mRNA expression in brain homogenates (n = 8–10/group). Main effect of aHBO2 and bTGB, p < 0.01.
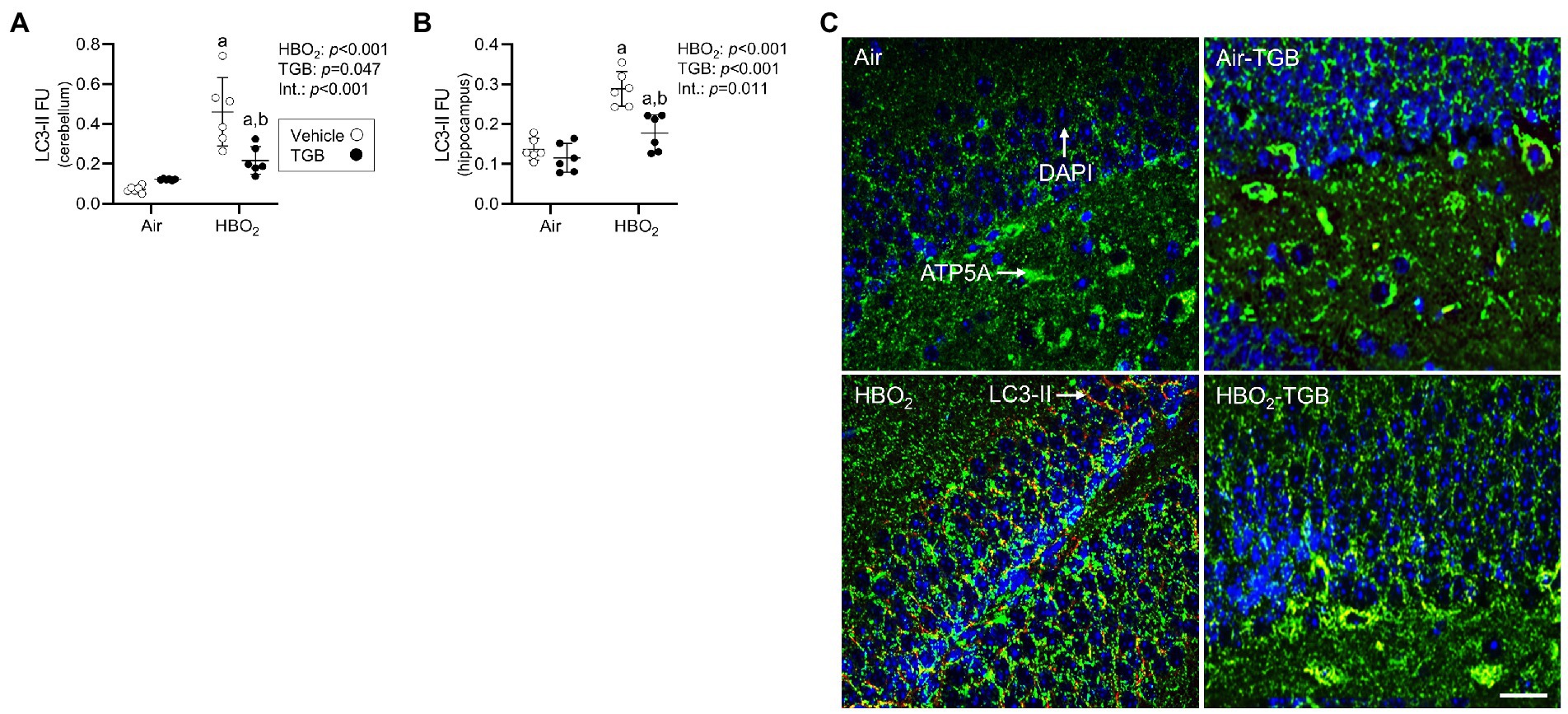
Figure 6. Autophagosome formation in mice exposed to extreme HBO2. Mice were pretreated with 0.9% NaCl or TGB (4.8 mg/kg body weight) 30 min before exposure to air or 5 ATA oxygen for 30 min. (A,B) Quantification of LC3-II in the cerebellum and hippocampus. (C) Representative images from hippocampus. LC3-II (red), ATP synthase F1 subunit α (green) and DAPI (blue). LC-II merged with ATP5A indicates mitophagy. Scale bar = 100 μm. Main effect of aHBO2 and bTGB, p < 0.05.
In order to maintain a healthy mitochondrial volume density, mitochondrial biogenesis must ensue. To determine if this process is stimulated by our HBO2 protocol, we measured levels of p-CREB at Ser-133. Phosphorylated CREB transcriptionally activates peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α; Sheng et al., 2012). PGC-1α and nuclear NRF1 transcriptionally co-activate TFAM, which is required for mitochondrial DNA replication and transcription (Wu et al., 1999). After HBO2, pCREB levels increased in the cerebellum and hippocampus (Figures 7A–C), along with Ppargc1a, Nrf1, and Tfam mRNA in brain homogenates (Figures 7D–F). TGB increased pCREB protein expression independent of exposure and reduced the HBO2-induced increase in both regions. These data imply HBO2 stimulates pCREB mediated biogenesis signaling and TGB attenuates the response.
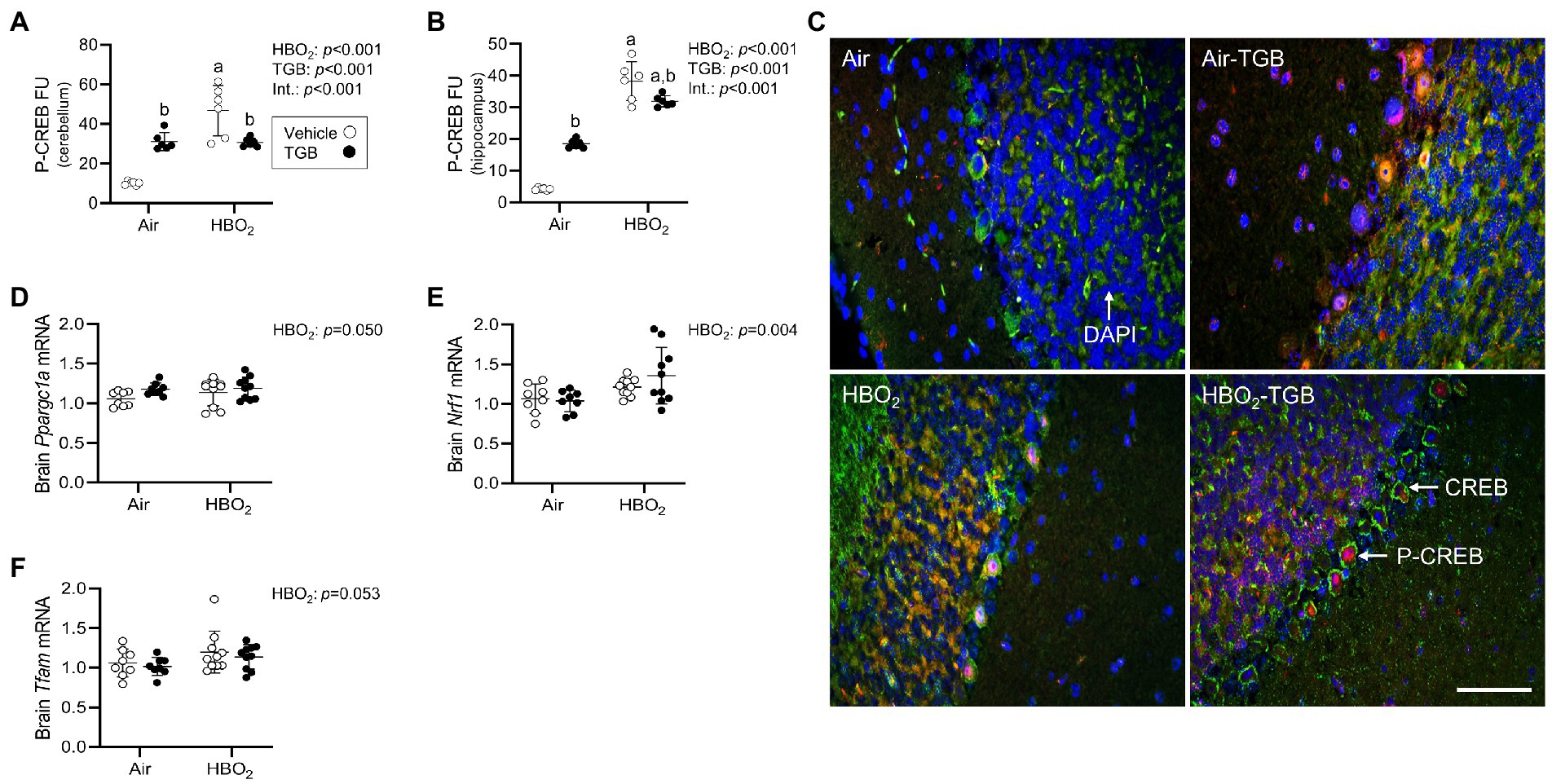
Figure 7. Initiation of mitochondrial biogenesis signaling in mice exposed to extreme HBO2. Mice were pretreated with 0.9% NaCl or TGB (4.8 mg/kg body weight) 30 min before exposure to air or 5 ATA oxygen for 30 min. (A,B) Quantification of phosphorylated cAMP response element-binding protein at Ser-133 (pCREB) in the cerebellum and hippocampus. (C) Representative images from cerebellum. pCREB (red), CREB (green), and DAPI (blue). Scale bar = 20 μm. (D–F) Peroxisome proliferator-activated receptor gamma coactivator 1-α (Ppargc1a), nuclear respiratory factor 1 (Nrf1) and mitochondrial transcription factor A (Tfam) mRNA expression in brain homogenates (n = 8–10/group). Main effect of aHBO2 and bTGB, p < 0.05.
Discussion
γ-aminobutyric acid’s main CNS function is to bind post-synaptic GABA receptors that facilitate ion flux and hyperpolarization, depressing excitatory postsynaptic potentials. In HBO2 at or above 3 ATA, GABA production is reduced by oxidant production and modification of GAD, leading to seizures (Wood and Watson, 1963; Wood et al., 1969; Gasier et al., 2017). GABAergic drugs delay seizure onset in HBO2, but its effect on oxidant brain injury is unknown since brain tissue PO2 may not be affected by GABA reuptake. To address this, we exposed anesthetized and conscious rats and mice to 5 and 6 ATA oxygen, which causes rapid development of CNS-OT and oxidant brain injury (Balentine, 1982; Demchenko et al., 2014). Our experiments show two novel findings: First, GAT inhibition increases extracellular GABA content, and prevents cerebral hyperemia. Second, a single exposure to HBO2 that causes oxidant brain injury activates antioxidant and anti-inflammation, mitophagy and mitochondrial biogenesis signaling (Figure 8). TGB attenuates oxidant damage and modifies these responses. Either directly or indirectly, these findings show that GABA’s function extends beyond inhibitory neurotransmission in extreme HBO2.
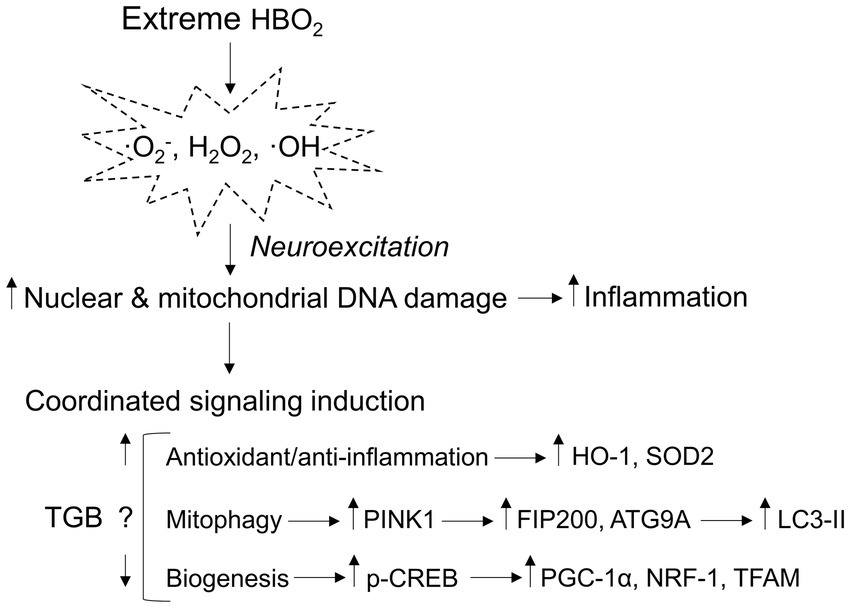
Figure 8. Mitochondria-related signaling responses to extreme HBO2. HBO2 increases brain tissue PO2 leading to amplified production of oxidants, i.e., superoxide (·O2−), hydrogen peroxide (H2O2), and hydroxyl (·OH) that target nuclear and mitochondrial DNA resulting in inflammation. The brain immediately activates antioxidant and anti-inflammation defenses that are linked to mitophagy and mitochondrial biogenesis programs. TGB prevents cerebral hyperemia and a further increase in brain tissue PO2 that delays neuroexcitation. The result is less oxidant damage and inflammation that may reduce mitophagy flux and mitochondrial biogenesis signaling. HO-1, heme oxygenase 1 (antioxidant/anti-inflammation); SOD2, manganese-dependent superoxide dismutase (mitochondrial antioxidant); PINK1, PTEN-induced kinase 1 (mitophagy initiation); FIP200, focal adhesion kinase family interacting protein of 200 kD (phagophore formation); ATG9A, autophagy-related protein 9A (phagophore growth); LC3-II, conjugated microtubule-associated protein 1A/1B-light chain 3 (autophagosome-lysosome fusion); p-CREB, phosphorylated cyclic-AMP responsive element binding protein (mitochondrial biogenesis); PGC-1α, peroxisome proliferator-activated receptor γ coactivator 1-α (mitochondrial biogenesis); NRF-1, nuclear respiratory 1 (transcriptional activation of TFAM); and TFAM, mitochondrial transcription factor A (mitochondrial DNA transcription, replication, and packaging).
Our microdialysis data indicate that interstitial GABA levels in the striatum fall in relation to inspired PO2. This is consistent with Wood et al. (1969) who measured GABA in whole brain homogenates from rats exposed to HBO2 from 4 to 7.5 ATA for 20 min. The mechanism for reduced GABA levels is inhibition of GAD activity caused by a reduction in presynaptic neuronal release and negative feedback inhibition and/or S-nitrosylation of GAD65 (Wood et al., 1967; Green et al., 1987; Gasier et al., 2017). The decrease in extracellular glutamine 75 min into HBO2 is not easily explained because of the complexity in the glutamine-glutamate/GABA cycle. In the brain, glutamine is synthesized only in astrocytes from glutamate and ammonia by glutamine synthetase, an endergonic reaction (Walls et al., 2015). Glutamine is shuttled to glutamatergic and GABAergic neurons where it is reconverted to glutamate/GABA. In times of increased NAD+-to-NADH, glutamate can undergo oxidative deamination, yielding α-ketogluatarate and ammonia (McKenna and Ferreira, 2016). The reaction is catalyzed by glutamate dehydrogenase in mitochondrion. Ammonia levels are reported to increase in the striatum of rodents after HBO2 seizures, and NADH oxidation in the rat brain precedes EEG spikes in extreme HBO2 (Mayevsky et al., 1974; Mialon et al., 1992, 1995). While not statistically significant, extracellular glutamate content decreased by 140 nM from 60 to 75 min in HBO2. Because striatal glucose consumption increases during EEG spikes in rats exposed to HBO2 (Torbati and Lambertsen, 1983), our data may imply oxidative deamination increased to support energy demand and led to decreased glutamine levels. Alternatively, the reduction in extracellular glutamine may be due to glutamine synthetase inhibition or increased neuronal uptake to replenish glutamate (Kanamori and Ross, 2011). The reduction in extracellular serine can be explained by conversion to D-serine or glycine, both of which serve as co-agonists with glutamate to activate neuroexcitatory N-methyl-D-aspartate (NMDA)-type glutamate receptors (Traynelis et al., 2010). Glycine is also an inhibitory neurotransmitter and decreased over time in HBO2 as a result of decreased substrate (serine), hydroxymethyltransferase activity, or postsynaptic receptor binding (Murtas et al., 2020).
Inhibition of GABA reuptake by neurons and astrocytes with NPA resulted in a large increase in extracellular GABA content. Even with continuous infusion, however, the magnitude of decline was greater with NPA (−166%) than controls (−64%). GABA levels were similar after 75 min, at about seizure onset. The dose of NPA used here, 70 μM, is above the IC50 for inhibiting GABA reuptake in cortical neurons (12 μM), astrocytes (16 μM), and striatal synaptosomes (3.6 μM; Mantz et al., 1994; Falch et al., 1999). Plausibly, the rapid decline in extracellular GABA may reflect maximal GABA receptor binding vs. glial reuptake because NPA was infused throughout HBO2 exposures.
In HBO2, CBF is regulated by three principal factors: CO2, nitric oxide (NO), and neurovascular coupling. Here, injecting an IC50 dose of TGB (47 nM) into the lateral ventricle of rats before HBO2 maintained cerebral vascular resistance and prevented cerebral hyperemia. We can only exclude alveolar CO2 as a factor since the rats’ ventilation is maintained during the experiments (Demchenko et al., 1998). Changes in neurovascular coupling, however, may explain why cerebral hyperemia was prevented with TGB. In rat and human studies, CBF and cerebral glucose utilization are reduced when treated with the GABAergic agonist’s muscimol and vigabatrin, respectively (Kelly and McCulloch, 1983; Spanaki et al., 1999). Active electrical discharges were not present in 75% of the rats treated with TGB, supporting intact neurovascular coupling. However, EEG spikes were present in other rats that exhibited different CBF patterns, i.e., one increased by 19% (seizure latency 57 min) and the other decreased by 9% (seizure latency 49 min). Clearly, further study will be needed in this area.
In conscious rats, a single 30 min exposure to 5 ATA oxygen immediately increases neuritic degeneration and mitophagy in the spinal cord (Balentine, 1982). In the brain, this level of PO2 only leads to mild neuronal degeneration within the hippocampus and cerebellum that is not detectable until days after HBO2 (Gutsaeva et al., 2006). The mouse is more susceptible to CNS oxygen toxicity than rats (Wood et al., 1967), as evidenced by a mean seizure latency of 37 min in rats compared with 12 min in mice in this study. Seizure latency correlates with a rise in oxidant production that continues to increase with exposure time (Atochin et al., 2003). The results reported here include nuclear and mitochondrial DNA oxidation and astrocytosis in the cerebellum and hippocampus, and modest opening of the blood brain barrier. The oxidant production triggered activation of antioxidant and anti-inflammatory enzyme systems that are integrated with mitophagy and mitochondrial biogenesis (Piantadosi et al., 2011; Suliman et al., 2017). The signal is hydrogen peroxide (H2O2) generated spontaneously or by the dismutation of superoxide (·Q2−) via SODs and is increased in extreme HBO2 (Piantadosi and Tatro, 1990). The extent and time course of mitochondrial turnover and its influence on cell repair remain unknown.
Maintaining vascular resistance and extending seizure latencies should reduce oxidant production, but not eliminate it because brain tissue PO2 is increased 15-fold in HBO2 at 5 ATA and rises to over 1,000 mmHg during the appearance of EEG spikes (Demchenko et al., 2005). Indeed, TGB reduced nuclear and mitochondrial DNA oxidation and astrocytosis in the cerebellum and hippocampus. Greater HO-1 protein expression may explain the reduction in astrocyte activation found during HBO2. In support of this, Godai and Moriyama (2022) reported pregabalin or gabapentin increased HO-1 and decreased GFAP mRNA expression in the spinal dorsal horn of mice with nerve injury. However, the effects were abolished when the HO-1 inhibitor tin protoporphyrin IX was used. While these drugs do not bind GABA receptors, they do effect GABAergic transmission. In the hippocampus, TGB prevented mitochondrial DNA oxidation and normalized HO-1 protein expression in HBO2, suggesting that mitochondrial damage was less than in the cerebellum, thus reducing initiation of mitophagy. The pattern of PINK1, FIP200 and LC3-II in the hippocampus supports this notion. However, TGB also reduced the expression of LC3-II in the cerebellum despite increased PINK1 and FIP200 protein expression. Reduced LC3-II may also indicate protein degradation within lysosomes. Thus, TGB may reduce oxidant damage and mitochondrial turnover requirements. Reduction of excess pCREB protein expression in HBO2 mice treated with TGB strengthens this assertion. The increase in pCREB in TGB treated control mice may be due increased GABA, which is reported to increase pCREB levels (Auger et al., 2001). Also, pCREB has other functions that include anti-inflammation and neurogenesis (Jagasia et al., 2009; Li et al., 2018). The increased HO-1 and reduced GFAP protein levels in TGB treated mice along with the limited duration of our experiments favors an anti-inflammatory mechanism.
In summary, we show for the first time that GAT inhibition modifies the cerebrovascular responses to extreme HBO2 that serve to protect against oxidant brain injury. HBO2 initiates mitophagy and pCREB mediated mitochondrial biogenesis signaling. The antiepileptic drug TGB maintains CBF and delays neuroexcitation, which perhaps lowers the requirements for mitochondrial turnover. The time course for brain injury repair, and whether TGB shortens recovery time remain unknown. This study expands our knowledge of the therapeutic function of GABAergic signaling that could be extended to other conditions such as epilepsy, stroke, and brain trauma.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by The Duke University Institutional Animal Care and Use Committee (IACUC) and the Ethical Review Board of the Sechenov Institute of Evolutionary Physiology and Biochemistry Russian Academy of Sciences.
Author contributions
ID and CP developed the experimental design. ID, CP, and HG directed the overall research. ID, HS, SZ, OA, TP, MM, and HG performed the experiments. ID, HS, MM, and HG analyzed the data and prepared the figures. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by the Office of Naval Research, Award #: N00014-18-1-2702 (CP and HG), the Russian Science Foundation, Award #: 22-25-00539 (ID), and the National Institutes of Health, Award #: T32 HL160494-01 (MM).
Acknowledgments
The authors extend their gratitude to Martha Salinas, Kristine Porter, Peltzer Doar III, and Robert Brown (Duke University) for their technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arieli, R., Shochat, T., and Adir, Y. (2006). CNS toxicity in closed-circuit oxygen diving: symptoms reported from 2527 dives. Aviat. Space Environ. Med. 77, 526–532.
Atochin, D. N., Demchenko, I. T., Astern, J., Boso, A. E., Piantadosi, C. A., and Huang, P. L. (2003). Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J. Cereb. Blood Flow Metab. 23, 1219–1226. doi: 10.1097/01.WCB.0000089601.87125.E4
Auger, A. P., Perrot-Sinal, T. S., and McCarthy, M. M. (2001). Excitatory versus inhibitory GABA as a divergence point in steroid-mediated sexual differentiation of the brain. Proc. Natl. Acad. Sci. U. S. A. 98, 8059–8064. doi: 10.1073/pnas.131016298
Balentine, J. D. (1974). Ultrastructural pathology of hyperbaric oxygenation in the central nervous system. Observations in anterior horn gray matter. Lab. Investig. 31, 580–592.
Bargerstock, E., Puvenna, V., Iffland, P., Falcone, T., Hossain, M., Vetter, S., et al. (2014). Is peripheral immunity regulated by blood-brain barrier permeability changes? PLoS One 9:e101477. doi: 10.1371/journal.pone.0101477
Basemore, A. W., Elliot, K. A., and Florey, E. (1957). Isolation of factor I. J. Neurochem. 1, 334–339. doi: 10.1111/j.1471-4159.1957.tb12090.x
Baxter, C. F., and Roberts, E. (1960). Demonstration of thiosemicarbazide-induced convulsions in rats with elevated brain levels of gamma-aminobutyric acid. Proc. Soc. Exp. Biol. Med. 104, 426–427. doi: 10.3181/00379727-104-25861
Butler, F. K. Jr., and Thalmann, E. D. (1986). Central nervous system oxygen toxicity in closed circuit scuba divers II. Undersea Biomed. Res. 13, 193–223.
Cantafora, E., Giorgi, F. S., Frenzilli, G., Scarcelli, V., Busceti, C. L., Nigro, M., et al. (2014). Region-specific DNA alterations in focally induced seizures. J. Neural Transm. (Vienna) 121, 1399–1403. doi: 10.1007/s00702-014-1211-5
Chipres-Tinajero, G. A., Nunez-Ochoa, M. A., and Medina-Ceja, L. (2021). Increased immunoreactivity of glutamate receptors, neuronal nuclear protein and glial fibrillary acidic protein in the hippocampus of epileptic rats with fast ripple activity. Exp. Brain Res. 239, 2015–2024. doi: 10.1007/s00221-021-06108-6
Costa, D. A., Ganilha, J. S., Barata, P. C., and Guerreiro, F. G. (2019). Seizure frequency in more than 180,000 treatment sessions with hyperbaric oxygen therapy—a single Centre 20-year analysis. Div. Hyperb. Med. 49, 167–174. doi: 10.28920/dhm49.3.167-174
D'Agostino, D. P., Putnam, R. W., and Dean, J. B. (2007). Superoxide (*O2-) production in CA1 neurons of rat hippocampal slices exposed to graded levels of oxygen. J. Neurophysiol. 98, 1030–1041. doi: 10.1152/jn.01003.2006
Demchenko, I. T., Boso, A. E., Natoli, M. J., Doar, P. O., O'Neill, T. J., Bennett, P. B., et al. (1998). Measurement of cerebral blood flow in rats and mice by hydrogen clearance during hyperbaric oxygen exposure. Undersea Hyperb. Med. 25, 147–152.
Demchenko, I. T., Gasier, H. G., Zhilyaev, S. Y., Moskvin, A. N., Krivchenko, A. I., Piantadosi, C. A., et al. (2014). Baroreceptor afferents modulate brain excitation and influence susceptibility to toxic effects of hyperbaric oxygen. J. Appl. Physiol. 117, 525–534. doi: 10.1152/japplphysiol.00435.2014
Demchenko, I. T., Luchakov, Y. I., Moskvin, A. N., Gutsaeva, D. R., Allen, B. W., Thalmann, E. D., et al. (2005). Cerebral blood flow and brain oxygenation in rats breathing oxygen under pressure. J. Cereb. Blood Flow Metab. 25, 1288–1300. doi: 10.1038/sj.jcbfm.9600110
Demchenko, I. T., and Piantadosi, C. A. (2006). Nitric oxide amplifies the excitatory to inhibitory neurotransmitter imbalance accelerating oxygen seizures. Undersea Hyperb. Med. 33, 169–174.
Demchenko, I. T., Zhilyaev, S. Y., Alekseeva, O. S., Krivchenko, A. I., Piantadosi, C. A., and Gasier, H. G. (2019). Increased antiseizure effectiveness with tiagabine combined with sodium channel antagonists in mice exposed to hyperbaric oxygen. Neurotox. Res. 36, 788–795. doi: 10.1007/s12640-019-00063-5
Donzanti, B. A., and Yamamoto, B. K. (1988). An improved and rapid HPLC-EC method for the isocratic separation of amino acid neurotransmitters from brain tissue and microdialysis perfusates. Life Sci. 43, 913–922. doi: 10.1016/0024-3205(88)90267-6
Falch, E., Perregaard, J., FrLlund, B., SŁkilde, B., Buur, A., Hansen, L. M., et al. (1999). Selective inhibitors of glial GABA uptake: synthesis, absolute stereochemistry, and pharmacology of the enantiomers of 3-hydroxy-4-amino-4,5,6,7-tetrahydro-1,2-benzisoxazole (exo-THPO) and analogues. J. Med. Chem. 42, 5402–5414. doi: 10.1021/jm9904452
Fink-Jensen, A., Suzdak, P. D., Swedberg, M. D., Judge, M. E., Hansen, L., and Nielsen, P. G. (1992). The gamma-aminobutyric acid (GABA) uptake inhibitor, tiagabine, increases extracellular brain levels of GABA in awake rats. Eur. J. Pharmacol. 220, 197–201. doi: 10.1016/0014-2999(92)90748-S
Gasier, H. G., Demchenko, I. T., Tatro, L. G., and Piantadosi, C. A. (2017). S-nitrosylation of GAD65 is implicated in decreased GAD activity and oxygen-induced seizures. Neurosci. Lett. 653, 283–287. doi: 10.1016/j.neulet.2017.05.067
Girault, J. A. (2012). Integrating neurotransmission in striatal medium spiny neurons. Adv. Exp. Med. Biol. 970, 407–429. doi: 10.1007/978-3-7091-0932-8_18
Godai, K., and Moriyama, T. (2022). Heme oxygenase-1 in the spinal cord plays crucial roles in the analgesic effects of pregabalin and gabapentin in a spared nerve-injury mouse model. Neurosci. Lett. 767:136310. doi: 10.1016/j.neulet.2021.136310
Goutianos, G., Tzioura, A., Kyparos, A., Paschalis, V., Margaritelis, N. V., Veskoukis, A. S., et al. (2015). The rat adequately reflects human responses to exercise in blood biochemical profile: a comparative study. Phys. Rep. 3:e12293. doi: 10.14814/phy2.12293
Green, A. R., Metz, A., Minchin, M. C., and Vincent, N. D. (1987). Inhibition of the rate of GABA synthesis in regions of rat brain following a convulsion. Br. J. Pharmacol. 92, 5–11. doi: 10.1111/j.1476-5381.1987.tb11288.x
Gulati, O. D., and Stanton, H. C. (1960). Some effects on the central nervous system of gamma-amino-n-butyric acid (GABA) and certain related amino acids administered systemically and intracerebrally to mice. J. Pharmacol. Exp. Ther. 129, 178–185.
Gutsaeva, D. R., Suliman, H. B., Carraway, M. S., Demchenko, I. T., and Piantadosi, C. A. (2006). Oxygen-induced mitochondrial biogenesis in the rat hippocampus. Neuroscience 137, 493–504. doi: 10.1016/j.neuroscience.2005.07.061
Hara, T., Takamura, A., Kishi, C., Iemura, S., Natsume, T., Guan, J. L., et al. (2008). FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510. doi: 10.1083/jcb.200712064
Jagasia, R., Steib, K., Englberger, E., Herold, S., Faus-Kessler, T., Saxe, M., et al. (2009). GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J. Neurosci. 29, 7966–7977. doi: 10.1523/JNEUROSCI.1054-09.2009
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kanamori, K., and Ross, B. D. (2011). Chronic electrographic seizure reduces glutamine and elevates glutamate in the extracellular fluid of rat brain. Brain Res. 1371, 180–191. doi: 10.1016/j.brainres.2010.11.064
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kanner, A. A., Marchi, N., Fazio, V., Mayberg, M. R., Koltz, M. T., Siomin, V., et al. (2003). Serum S100beta: a noninvasive marker of blood-brain barrier function and brain lesions. Cancer 97, 2806–2813. doi: 10.1002/cncr.11409
Kelly, P. A., and McCulloch, J. (1983). The effects of the GABAergic agonist muscimol upon the relationship between local cerebral blood flow and glucose utilization. Brain Res. 258, 338–342. doi: 10.1016/0006-8993(83)91162-9
Killam, K. F., and Bain, J. A. (1957). Convulsant hydrazides. I. in vitro and in vivo inhibition of vitamin B6 enzymes by convulsant hydrazides. J. Pharmacol. Exp. Ther. 119, 255–262.
Kragler, A., Hofner, G., and Wanner, K. T. (2005). Novel parent structures for inhibitors of the murine GABA transporters mGAT3 and mGAT4. Eur. J. Pharmacol. 519, 43–47. doi: 10.1016/j.ejphar.2005.06.053
Lacey, C. J., Boyes, J., Gerlach, O., Chen, L., Magill, P. J., and Bolam, J. P. (2005). GABA(B) receptors at glutamatergic synapses in the rat striatum. Neuroscience 136, 1083–1095. doi: 10.1016/j.neuroscience.2005.07.013
Lahiri, V., and Klionsky, D. J. (2021). ATG4-family proteins drive phagophore growth independently of the LC3/GABARAP lipidation system. Autophagy 17, 1293–1295. doi: 10.1080/15548627.2021.1917284
Li, C., Chen, T., Zhou, H., Feng, Y., Hoi, M. P. M., Ma, D., et al. (2018). BHDPC is a novel Neuroprotectant that provides anti-neuroinflammatory and neuroprotective effects by inactivating NF-kappaB and activating PKA/CREB. Front. Pharmacol. 9:614. doi: 10.3389/fphar.2018.00614
Mantz, J., Laudenbach, V., Lecharny, J. B., Henzel, D., and Desmonts, J. M. (1994). Riluzole, a novel antiglutamate, blocks GABA uptake by striatal synaptosomes. Eur. J. Pharmacol. 257, R7–R8. doi: 10.1016/0014-2999(94)90716-1
Marks, H.P. (1944). "Interim Report on Oxygen Intoxication in Animals, and the Effect of Drugs on Sensitivity to Oxygen Intoxication". Hampstead, UK: National Institute for Medical Research.
Mathew, J., Soman, S., Sadanandan, J., and Paulose, C. S. (2010). Decreased GABA receptor in the striatum and spatial recognition memory deficit in epileptic rats: effect of Bacopa monnieri and bacoside-a. J. Ethnopharmacol. 130, 255–261. doi: 10.1016/j.jep.2010.04.043
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Mayevsky, A., Jamieson, D., and Chance, B. (1974). Oxygen poisoning in the unanesthetized brain: correlation of the oxidation-reduction state of pyridine nucleotide with electrical activity. Brain Res. 76, 481–491. doi: 10.1016/0006-8993(74)90824-5
McKenna, M. C., and Ferreira, G. C. (2016). Enzyme complexes important for the glutamate-glutamine cycle. Adv. Neurobiol. 13, 59–98. doi: 10.1007/978-3-319-45096-4_4
Mialon, P., Gibey, R., Bigot, J. C., and Barthelemy, L. (1992). Changes in striatal and cortical amino acid and ammonia levels of rat brain after one hyperbaric oxygen-induced seizure. Aviat. Space Environ. Med. 63, 287–291.
Mialon, P., Joanny, P., Gibey, R., Cann-Moisan, C., Caroff, J., Steinberg, J., et al. (1995). Amino acids and ammonia in the cerebral cortex, the corpus striatum and the brain stem of the mouse prior to the onset and after a seizure induced by hyperbaric oxygen. Brain Res. 676, 352–357. doi: 10.1016/0006-8993(95)00120-F
Murtas, G., Marcone, G. L., Sacchi, S., and Pollegioni, L. (2020). L-serine synthesis via the phosphorylated pathway in humans. Cell. Mol. Life Sci. 77, 5131–5148. doi: 10.1007/s00018-020-03574-z
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Nguyen, T., Sherratt, P. J., Nioi, P., Yang, C. S., and Pickett, C. B. (2005). Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 280, 32485–32492. doi: 10.1074/jbc.M503074200
Nielsen, E. B., Suzdak, P. D., Andersen, K. E., Knutsen, L. J., Sonnewald, U., and Braestrup, C. (1991). Characterization of tiagabine (NO-328), a new potent and selective GABA uptake inhibitor. Eur. J. Pharmacol. 196, 257–266. doi: 10.1016/0014-2999(91)90438-V
Oury, T. D., Ho, Y. S., Piantadosi, C. A., and Crapo, J. D. (1992). Extracellular superoxide dismutase, nitric oxide, and central nervous system O2 toxicity. Proc. Natl. Acad. Sci. U. S. A. 89, 9715–9719. doi: 10.1073/pnas.89.20.9715
Paxinos, G., and Watson, C. (2007). "The Rat Brain in Stereotaxic Coordinates ". 6th Edn. Boston, MA: Elsevier
Piantadosi, C. A., and Tatro, L. G. (1990). Regional H2O2 concentration in rat brain after hyperoxic convulsions. J. Appl. Physiol. 69, 1761–1766. doi: 10.1152/jappl.1990.69.5.1761
Piantadosi, C. A., Withers, C. M., Bartz, R. R., MacGarvey, N. C., Fu, P., Sweeney, T. E., et al. (2011). Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 286, 16374–16385. doi: 10.1074/jbc.M110.207738
Ponce, L. L., Pillai, S., Cruz, J., Li, X., Julia, H., Gopinath, S., et al. (2012). Position of probe determines prognostic information of brain tissue PO2 in severe traumatic brain injury. Neurosurgery 70, 1492–1503. doi: 10.1227/NEU.0b013e31824ce933
Roberts, E. (1959). Inhibition in the Nervous System and Gamma-Aminobutyric Acid. Duarte, CA: Pergamon Press
Roberts, E., and Frankel, S. (1950). Gamma-aminobutyric acid in brain: its formation from glutamic acid. J. Biol. Chem. 187, 55–63. doi: 10.1016/S0021-9258(19)50929-2
Sheng, B., Wang, X., Su, B., Lee, H. G., Casadesus, G., Perry, G., et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J. Neurochem. 120, 419–429. doi: 10.1111/j.1471-4159.2011.07581.x
Smerz, R. W. (2004). Incidence of oxygen toxicity during the treatment of dysbarism. Undersea Hyperb. Med. 31, 199–202.
Spanaki, M. V., Siegel, H., Kopylev, L., Fazilat, S., Dean, A., Liow, K., et al. (1999). The effect of vigabatrin (gamma-vinyl GABA) on cerebral blood flow and metabolism. Neurology 53, 1518–1522. doi: 10.1212/WNL.53.7.1518
Suliman, H. B., Keenan, J. E., and Piantadosi, C. A. (2017). Mitochondrial quality-control dysregulation in conditional HO-1(−/−) mice. JCI Insight 2:e89676. doi: 10.1172/jci.insight.89676
Torbati, D., and Lambertsen, C. J. (1983). Regional cerebral metabolic rate for glucose during hyperbaric oxygen-induced convulsions. Brain Res. 279, 382–386. doi: 10.1016/0006-8993(83)90215-9
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Walls, A. B., Waagepetersen, H. S., Bak, L. K., Schousboe, A., and Sonnewald, U. (2015). The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 40, 402–409. doi: 10.1007/s11064-014-1473-1
Wood, J. D., and Watson, W. J. (1962). Protective action of gamma-aminobutyric acid against oxygen toxicity. Nature 195:296. doi: 10.1038/195296a0
Wood, J. D., and Watson, W. J. (1963). Gamma-aminobutyric acid levels in the brain of rats exposed to oxygen at high pressures. Can. J. Biochem. Physiol. 41, 1907–1913. doi: 10.1139/y63-217
Wood, J. D., and Watson, W. J. (1964). The effect of oxygen on glutamic acid decarboxylase and gamma-aminobutyric acid-alpha-Ketoglutaric acid transaminase activities in rat brain homogenates. Can. J. Physiol. Pharmacol. 42, 277–279. doi: 10.1139/y64-032
Wood, J. D., Watson, W. J., and Ducker, A. J. (1967). Oxygen poisoning in various mammalian species and the possible role of gamma-aminobutyric acid metabolism. J. Neurochem. 14, 1067–1074. doi: 10.1111/j.1471-4159.1967.tb09517.x
Wood, J. D., Watson, W. J., and Murray, G. W. (1969). Correlation between decreases in brain gamma-aminobutyric acid levels and susceptibility to convulsions induced by hyperbaric oxygen. J. Neurochem. 16, 281–287. doi: 10.1111/j.1471-4159.1969.tb10366.x
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cells 98, 115–124. doi: 10.1016/S0092-8674(00)80611-X
Keywords: anti-inflammation, cerebellum, GABA, hippocampus, mitophagy, mitochondrial biogenesis
Citation: Demchenko IT, Suliman HB, Zhilyaey SY, Alekseeva OS, Platonova TF, Makowski MS, Piantadosi CA and Gasier HG (2023) GAT inhibition preserves cerebral blood flow and reduces oxidant damage to mitochondria in rodents exposed to extreme hyperbaric oxygen. Front. Mol. Neurosci. 15:1062410. doi: 10.3389/fnmol.2022.1062410
Edited by:
Katia Aquilano, University of Rome Tor Vergata, ItalyReviewed by:
Ishrat Jabeen, National University of Sciences and Technology (NUST), PakistanSridhar Kannurpatti, Rutgers, The State University of New Jersey, United States
Copyright © 2023 Demchenko, Suliman, Zhilyaey, Alekseeva, Platonova, Makowski, Piantadosi and Gasier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heath G. Gasier, ✉ aGVhdGguZ2FzaWVyQGR1a2UuZWR1