 Plinio Casarotto1
Plinio Casarotto1 Eero Castrén
Eero Castrén
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 02 November 2022
Sec. Molecular Signalling and Pathways
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.1032224
This article is part of the Research Topic Implications of Neurotrophins and Their Receptors in the Pathophysiology and Treatments of Depression and Addiction View all 6 articles
Brain-derived neurotrophic factor (BDNF) signaling through its receptor TrkB has for a long time been recognized as a critical mediator of the antidepressant drug action, but BDNF signaling has been considered to be activated indirectly through the action of typical and rapid-acting antidepressants through monoamine transporters and glutamate NMDA receptors, respectively. However, recent findings demonstrate that both typical and the fast-acting antidepressants directly bind to TrkB and thereby allosterically potentiate BDNF signaling, suggesting that TrkB is the direct target for antidepressant drugs. Increased TrkB signaling particularly in the parvalbumin-expressing interneurons orchestrates iPlasticity, a state of juvenile-like enhanced plasticity in the adult brain. iPlasticity sensitizes neuronal networks to environmental influences, enabling rewiring of networks miswired by adverse experiences. These findings have dramatically changed the position of TrkB in the antidepressant effects and they propose a new end-to-end model of the antidepressant drug action. This model emphasizes the enabling role of antidepressant treatment and the active participation of the patient in the process of recovery from mood disorders.
Soon after its discovery, it was recognized that the synthesis of brain-derived neurotrophic factor (BDNF) is regulated by neuronal activity (Zafra et al., 1990; Isackson et al., 1991; Dugich-Djordjevic et al., 1992). This finding laid foundation for the subsequent recognition of BDNF as the critical mediator of activity-dependent neuronal plasticity and connectivity during development as well as in the adult brain (Thoenen, 1995; Poo, 2001). As the limbic seizures used in early studies to induce BDNF mRNA expression in mice resembled seizures induced by the electroconvulsive shock therapy (ECT), these findings raised interest into a possibility that BDNF might be involved in the antidepressant mechanisms of ECT. Indeed, Ron Duman’s group found that ECT-like treatment in rats strongly increased the expression mRNA for BDNF as well as for its cognate receptor TrkB (neurotrophic tyrosine kinase receptor, NTRK2) in the hippocampus and cortex (Nibuya et al., 1995). Unexpectedly, they found that chronic treatment with antidepressant drugs also increase the expression of BDNF mRNA, albeit at lower level (Nibuya et al., 1995). Subsequent studies showed that BDNF injected into the midbrain region or hippocampus produces antidepressant-like effects in rodents (Siuciak et al., 1997; Shirayama et al., 2002). These pioneering findings led to the proposal of a central role of neurotrophic factors in the mechanisms of antidepressant drugs (Duman et al., 1997; Altar, 1999).
Another line of research that led to the recognition of the role of BDNF and TrkB in the mechanisms of antidepressant drug action is related to neuronal plasticity. It has been known for decades that the clinical effects of antidepressants appear after a delay of several weeks, although the biochemical effects of these drugs take place within minutes or hours. One potential explanation for this delay was that some kind of physical, time-consuming event might be required for the clinical effect to appear, and neuronal plasticity that involves physical growth and pruning was a natural candidate (Castrén, 2005). As BDNF signaling through TrkB is a key mediator of activity-dependent plasticity, BDNF was an excellent candidate involved in such a gradual growth process. Indeed, it was shown that antidepressant drugs reactivate a state of juvenile-like plasticity in the adult brain, a state that is called iPlasticity (Castrén, 2005; Umemori et al., 2018; Branchi and Giuliani, 2021). iPlasticity was first demonstrated as the reactivation of ocular dominance plasticity in the visual cortex (Maya Vetencourt et al., 2008), which is the classical model of developmental neuronal plasticity. Subsequent studies have demonstrated that antidepressants produce iPlasticity also in mood-relevant networks, such as the fear extinction and aggression control circuitries (Karpova et al., 2011; Mikics et al., 2018).
The finding that antidepressant treatments promote the proliferation and survival of newly-born neurons in the rodent hippocampal dentate gyrus further supported a role of long-lasting plastic changes in the antidepressant action (Malberg and Duman, 2003; Malberg et al., 2021). However, it later turned out that neuronal plasticity and BDNF signaling are also required for the rapid antidepressant effects of ketamine (Autry et al., 2011; Duman and Li, 2012; Liu et al., 2012), which undermined the role of plasticity as the explanation for the delay in the action of typical antidepressants. Indeed, this delay still remains a mystery. Together these two lines of research, activity-dependent BDNF regulation and its role in neuronal plasticity, laid foundation for the recognition of the critical role for BDNF-TrkB signaling in the mechanisms of antidepressant drug action (Duman et al., 1997; Nestler et al., 2002; Duman and Monteggia, 2006; Castrén and Monteggia, 2021).
Essentially all antidepressant treatments tested so far have proven to increase the expression of BDNF mRNA and in most cases also BDNF protein levels. These treatments include typical antidepressants, including classical tricyclic, monoamine oxidase inhibitors, and serotonin-selective antidepressants (SSRI) (Nibuya et al., 1995; Duman et al., 1997; Altar, 1999; Russo-Neustadt et al., 1999; Coppell et al., 2003; Jacobsen and Mork, 2004; Duman and Monteggia, 2006; Calabrese et al., 2007, 2011), the rapid-acting antidepressants ketamine (Li et al., 2010; Autry et al., 2011; Autry and Monteggia, 2012; Lepack et al., 2014) and scopolamine (Wohleb et al., 2017), lithium (Jacobsen and Mork, 2004) as well as ECT (Nibuya et al., 1995; Jacobsen and Mork, 2004) and vagus nerve stimulation (Follesa et al., 2007; Biggio et al., 2009; Carreno and Frazer, 2014). Some authors have not found increases in BDNF with all antidepressants (Jacobsen and Mork, 2004), but doses, length of treatment and brain regions investigated may have contributed to this. BDNF mRNA levels are rapidly increase after ketamine and ECT (Nibuya et al., 1995; Autry et al., 2011), but several days of treatment with typical antidepressants are needed for the increase in BDNF mRNA and protein levels (Nibuya et al., 1995). The increase in BDNF mRNA levels by antidepressants may be produced by decrease in histone deacetylation at BDNF promoter regions (Russo-Neustadt et al., 2001; Dias et al., 2003; Tsankova et al., 2006; Karpova, 2014).
Antidepressants also promote BDNF release and signaling through TrkB (Castrén and Monteggia, 2021). Consistent with increased BDNF synthesis, typical as well as rapid-acting antidepressants increase TrkB autophosphorylation, which has been used as a proxy for BDNF release and binding to TrkB, and increase downstream signaling pathways activated by TrkB (Saarelainen et al., 2003; Rantamäki et al., 2007; Autry et al., 2011; Lepack et al., 2014). Antidepressants consistently increase the activation of phospholipase γ-1 (PLCγ-1) pathway (Saarelainen et al., 2003; Rantamäki et al., 2007), but the activation of extracellular signal regulated kinase (Erk)-pathway has also been reported to be activated (Duman et al., 2007; Lepack et al., 2016) and the activation of the Erk pathway may play a key role on the ketamine action (Li et al., 2010; Lepack et al., 2016).
The expression of BDNF mRNA and protein have also been investigated in humans with depression and antidepressant treatment. BDNF mRNA and/or protein levels have been reported to be reduced in postmortem brain samples of depressed patients (Dunham et al., 2009; Ray et al., 2011, 2014; Guilloux et al., 2012; Dwivedi, 2013) and suicide victims (Chen et al., 2001; Dwivedi et al., 2003, 2009; Pandey et al., 2008; Dwivedi, 2009; Youssef et al., 2018), and antidepressants restore the reduced levels (Chen et al., 2001). TrkB and phosphorylated TrkB have also been observed to be decreased in suicide victims (Dwivedi et al., 2003, 2009; Tripp et al., 2012). Similarly, serum BDNF levels are reduced in depressed patients (Karege et al., 2002) and successful antidepressant treatment normalizes these reduced levels (Shimizu et al., 2003; Gonul et al., 2005; Karege et al., 2005; Bocchio-Chiavetto et al., 2006, 2010; Yoshimura et al., 2007; Hellweg et al., 2008; Piccinni et al., 2008; Sen et al., 2008; Matrisciano et al., 2009; Molendijk et al., 2011, 2014; Rocha et al., 2016). However, as serum BDNF is derived from platelets (Yamamoto and Gurney, 1990; Radka et al., 1996; Fujimura et al., 2002; Lommatzsch et al., 2005; Naegelin et al., 2018), it is unclear to which extent serum BDNF levels correspond to brain levels (Seifert et al., 2010; Naegelin et al., 2018).
It has been widely considered that the effects of antidepressants on BDNF and TrkB signaling are indirect, mediated by the action of typical and fast-acting antidepressants on serotonin and NMDA-type glutamate receptors, respectively. However, recent findings have questioned the indirect action of antidepressants on BDNF and neuronal plasticity and revealed a direct binding of these drugs to TrkB (Casarotto et al., 2021).
We recently discovered that essentially all antidepressant drugs directly bind to TrkB and thereby allosterically promote TrkB signaling (Casarotto et al., 2021). We first found that labeled fluoxetine and imipramine bind to TrkB and several orthogonal methods verified this binding. A point mutation in the TrkB transmembrane domain (TMD) (TrkB-Y433F) in the amino acids that are predicted to interact with antidepressants abolishes antidepressant binding to TrkB, indicating that fluoxetine binds directly to TrkB. Unexpectedly, we found that not only typical antidepressants, such as SSRIs and tricyclic antidepressants, but also the fast-acting antidepressant ketamine and its active metabolite R,R-hydroxynorketamine (R,R-HNK) (Zanos et al., 2016) directly bind to TrkB, and the effect of ketamine are also lost in the TrkB.Y433F mutants. This mutation as heterozygous abolishes the plasticity-promoting and antidepressant-related behavioral responses of both SSRIs and ketamine both in vitro and in vivo. It is important to note that such heterozygous mutation does not reduce BDNF binding to TrkB and heterozygous mice with this mutation do not show any behavioral phenotype (Biojone et al., 2022). Together these data suggest that direct binding to the TMD of TrkB is the common mechanism of action of both typical and fast-acting antidepressants.
TrkB is a single TMD protein that is activated when the dimeric ligand BDNF induces the dimerization of two TrkB monomers, which leads to TrkB autophosphorylation and signaling. Atomistic simulations of TrkB TMD dimers revealed that TrkB TMD domains cross each other in the plasma membrane (Figure 1A). The positioning of the crisscrossed TrkB TMDs is determined by membrane thickness, which in turn is influenced by cholesterol concentrations. The crisscrossed conformation is stable in membranes with moderate cholesterol concentrations, supporting BDNF signaling, but in thick, cholesterol-rich membranes, such as synaptic membranes, the crisscrossed conformation of TrkB monomers tend to flip to parallel position (Figure 1A). TrkB in this parallel configuration does not appear to be stable and monomers are excluded from synaptic membranes (Suzuki et al., 2004; Pereira and Chao, 2007). Antidepressants bind to the outer crevice of the crossed transmembrane domains, interacting with both monomers. Antidepressant act as a kind of a wedge that stabilizes the crossed monomer configuration of the TrkB dimer, which increases the residence time of TrkB in the synapses, thereby enhancing the probability of BDNF binding and activation of TrkB (Casarotto et al., 2021; Figure 1A).
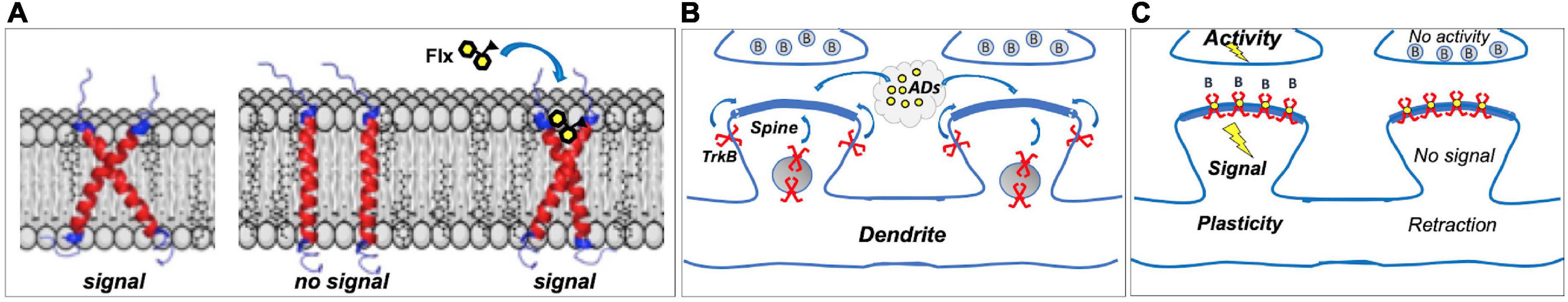
Figure 1. (A) Dimers of TrkB transmembrane domains (TMD) assume a crossed signaling-competent structure; in thick cholesterol-rich synaptic membranes, crossed structure becomes instable, and TrkB is excluded from synapses. Binding of fluoxetine (Flx) to the crossed TMDs acts as a wedge, stabilizing the signaling-competent structure in synaptic membranes. (B) Most TrkBs reside in vesicles or outside synapses; antidepressants (ADs) promote synaptic localization. (C) In Hebbian plasticity, active synapses are stabilized, whereas inactive ones are retracted, and brain-derived neurotrophic factor (BDNF) through TrkB is a critical mediator in this process. Antidepressants act as positive allosteric modulators of TrkB, promoting its localization in synaptic membranes where it can bind BDNF (B) released from stimulated synapses, enhancing TrkB signaling and stabilizing active synapses. Inactive synapses, however, do not release BDNF to activate synaptic TrkB receptors, which gradually leads to spine retraction. Direct TrkB agonist would activate TrkB in both active and inactive synapses, gradually leading to the decline in activity-dependent plasticity.
Essentially all the plasticity-related or antidepressant-like structural and behavioral responses to both fluoxetine and ketamine that we have tested so far are lost in heterozygous mice carrying the TrkB-Y433F mutation (Casarotto et al., 2021), which demonstrates that both of these antidepressants act by binding to the TrkB TMD. These responses include increased survival of newborn hippocampal neurons, promotion of ocular dominance plasticity, enhancement of object location memory, reduction of immobility in the forced swimming test and facilitation of fear extinction (Casarotto et al., 2021). It is important to note that these mice normally respond to BDNF and do not show any baseline behavioral deficits (Biojone et al., 2022), indicating that these behavioral effects are not mediated by any loss-of-function effects of BDNF responses, but are mediated by the inability of TrkB in these mice to bind antidepressants.
Although antidepressants bind to TrkB, they do not activate it on their own. Instead, they stabilize a configuration of TrkB dimers that promote the binding of BDNF, thereby allosterically potentiating the effects of BDNF onto TrkB (Figure 1). This is of physiological importance, since the effects of antidepressants as allosteric BDNF potentiators are confined to active synapses where BDNF is being released, whereas direct TrkB agonists would promote the stabilization of both active and inactive synapses. Therefore, antidepressant-potentiated BDNF signaling preserves and facilitates activity-dependent plasticity, which is a critical feature in both developmental and Hebbian plasticity (Thoenen, 1995; Park and Poo, 2013).
The finding that antidepressants bind to TrkB links them directly to synaptic plasticity, but it has remained unclear how potentiated TrkB activity is translated into plasticity at a network level. Our recent work indicates that TrkB receptors specifically expressed on the parvalbumin (PV)-containing interneurons are critical in this regard. PV neurons have already been implicated in neuronal plasticity: their maturation coincides with the closure of critical periods of early life plasticity and inhibition mediated by PV neurons is reduced during iPlasticity induced by antidepressants or other treatments in the adult brain (Maya Vetencourt et al., 2008; Sale et al., 2010; Reh et al., 2020). We found that the antidepressant fluoxetine fails to induce iPlasticity in mice with reduced expression of TrkB in PV interneurons (Winkel et al., 2021). Conversely, activation of a light-sensitive TrkB (optoTrkB) specifically in the PV cells rapidly orchestrates a state of iPlasticity. Remarkably, optoTrkB activation replicated all the measures of iPlasticity induced by fluoxetine, including the reactivation of ocular dominance plasticity in the visual cortex (Winkel et al., 2021). The state induced by optoTrkB activation is characterized by a reduction in the excitability of PV cells produced by reduced expression and activity of Kv3.1 potassium channels and reduced output of inhibition to pyramidal neurons (Winkel et al., 2021). This leads to disinhibition of cortical pyramidal neurons and increased gamma oscillations, which in turn facilitates plasticity and underlies iPlasticity. It is remarkable that while activation of TrkB increases excitability of pyramidal neurons (Figurov et al., 1996), it reduces excitability of PV interneurons (Winkel et al., 2021), which is at least partially produced by the PV-cell specific expression of the Kv3.1 potassium channels. Therefore, activation of TrkB simultaneously in excitatory and inhibitory neurons do not counteract each other, but synergize, as the inhibition onto excitatory neurons is suppressed. It is important to note that as a consequence of TrkB activation, the PV interneurons are not simply shut down, which may result in uncontrolled excitability and seizures, but TrkB activity orchestrates a new state of PV cell activity that facilitates cortical plasticity in a controlled manner.
Antidepressants influence TrkB activity in PV neurons also through other mechanisms. We recently found that antidepressants disrupt the interaction between TrkB and the protein tyrosine phosphatase sigma (PTPσ) (Lesnikova et al., 2021). PTPσ interacts with TrkB and, when activated, restricts TrkB phosphorylation. Antidepressant-induced disruption of TrkB-PTPσ interaction therefore releases TrkB from this inhibitory control, promoting its activity. Interestingly, PTPσ is a receptor for chondroitin sulfate proteoglycans (Shen et al., 2009) that are the main constituents of perineuronal nets (PNN) that encase PV interneurons in the adult brain. It has been long known that disruption of PNNs by chondroitinase activates iPlasticity (Pizzorusso et al., 2002; Gogolla et al., 2009; Fawcett et al., 2019). PNN disruption is expected to reduce the activity of PTPσ and thereby facilitate TrkB activity. Indeed, we found that chondroitinase treatment fails to activate iPlasticity in mice with reduced expression of TrkB in PV interneurons (Lesnikova et al., 2021), demonstrating that TrkB activity in the PV cells is necessary for iPlasticity induced not only by antidepressants, but also by PNN disruption.
We have further found that antidepressant treatment also disrupts the interaction between TrkB and the adaptor protein complex-2 (AP-2) that is a critical mediator of endocytosis (Fred et al., 2019). Consequently, TrkB endocytosis is inhibited, which leads to increased plasma membrane localization of TrkB, thereby facilitating BDNF signaling.
Taken together, our recent findings show that antidepressants facilitate the ability of BDNF to activate TrkB receptors in PV interneurons through several distinct mechanisms: by directly binding to TrkB and allosterically increasing BDNF signaling (Casarotto et al., 2021); by inhibiting the dephosphorylation of TrkB through PTPσ; and by reducing TrkB endocytosis by disrupting the binding of AP-2 to TrkB. Together these mechanisms underlie the controlled disinhibition of pyramidal networks underlying iPlasticity and explain why TrkB in PV cells is particularly important for iPlasticity.
Previous studies have already established the critical role for BDNF-TrkB signaling in the antidepressant action (Castrén and Monteggia, 2021), but TrkB signaling has been seen as a secondary effect downstream of antidepressant binding to their various effector molecules, such as serotonin and noradrenaline transporters and NMDA receptors. New findings now propose a new end-to-end model of the antidepressant drug action. In this model, antidepressant drugs directly bind to TrkB receptors with low, but clinically meaningful affinity and thereby allosterically promote BDNF signaling in the plasma membranes of active synapses (Casarotto et al., 2021; Figure 1). Through intracellular signaling pathways downstream of TrkB, BDNF synthesis is increased and the translocation of AMPA-type glutamate at plasma membranes are increased. Activation of TrkB receptors particularly in PV-positive interneurons orchestrates a state of reduced activity of PV interneurons, which leads to disinhibition of pyramidal networks, turning on iPlasticity, a state of enhanced plasticity in the cortical networks (Winkel et al., 2021). iPlasticity sensitizes cortical networks to environmental experiences and facilitates rewiring of malfunctioning networks (Umemori et al., 2018; Branchi and Giuliani, 2021), leading to better adaptation to environment and mood recovery. Although a lot of research is needed for many details, this model provides a new framework for the understanding of the antidepressant action.
In the updated network model of depression, the initial event is binding of an antidepressant molecule to TrkB (Casarotto et al., 2021; Figure 1A). The finding that several different antidepressants, seemingly belonging to different chemical classes, such as SSRIs, tricyclic antidepressants, monoamine oxidase inhibitors, and also the rapid-acting antidepressants ketamine and R,R-HNK all bind to TrkB was unexpected. A recent finding failed to find interactions between R,R-HNK and TrkB or any other proteins (Bonaventura et al., 2022), however, concentrations of R,R-HNK tested may have been too low to detect binding to TrkB. Furthermore, in spite of promising preclinical findings (Hess et al., 2022) [but also see Shirayama and Hashimoto (2018)], whether R,R-HNK produces clinical antidepressant effects have been questioned (Farmer et al., 2020) and remain to be determined in clinical trials. If these findings are confirmed, they will overturn the dogma of the critical role of monoamines in the antidepressant drug action. However, it is clear that different antidepressants still bind to monoamine transporters and NMDA receptors and their contribution to the clinical outcome should become an active area of research. For example, the increased positive emotional bias seen early on during the SSRI treatment is likely mediated by serotonin and may play a significant role on the outcome (Harmer et al., 2004, 2017). With improved characterization of the binding site in TrkB, novel potential antidepressants with higher affinity to TrkB should be searched for.
One of the most unexpected aspects of the model of the critical role of TrkB binding in the antidepressant action is that a common binding site would mediate the effects of both fast and slow-acting antidepressants. It should be noted, however, that many different antidepressants reach higher than micromolar brain concentrations at the steady state, which is compatible with binding to TrkB (Renshaw et al., 1992; Karson et al., 1993; Bolo et al., 2000; Henry et al., 2000; Johnson et al., 2007). Intriguingly, it takes several weeks of continuous treatment to reach these micromolar fluoxetine concentrations (Karson et al., 1993). As the brain distribution of other SSRIs (Bolo et al., 2000; Henry et al., 2000) and also tricyclic antidepressants resemble that of fluoxetine (Daniel, 2003; Erb et al., 2016), these observations suggest the tantalizing hypothesis that gradual accumulation of antidepressants into brain at concentrations sufficient for binding to a low-affinity site, such as TrkB, may contribute to the slow onset (Kornhuber et al., 1995). In contrast, infusion of ketamine produces micromolar brain concentrations rapidly (Zanos et al., 2018), which is consistent with rapid onset of action. Therefore, although more research in this domain is needed, kinetic differences in the access of antidepressants to TrkB may be at least one factor influencing the delayed onset of action of typical antidepressants.
The action of antidepressants on network function helps to explain some discrepancies found in the behavioral responses to antidepressants. While administration of BDNF and TrkB agonists produce antidepressant-like effects on the cortex and hippocampus (Shirayama et al., 2002; Liu et al., 2010; Zhang et al., 2015a,b), TrkB antagonist ANA-12 paradoxically also produces antidepressant-like responses (Cazorla et al., 2011; Shirayama et al., 2015; Zhang et al., 2015a,b). Direct injection of ANA-12 to nucleus accumbens replicates the antidepressants effects, which is consistent with earlier studies showing that BDNF injection into this region produces a depression-like phenotype (Eisch et al., 2003). Therefore, TrkB activation does not produce antidepressant effects per se, but by enhancing plasticity promote the action of the particular network, which may ameliorate but also aggravate depression (Branchi and Giuliani, 2021).
The updated network hypothesis of antidepressant action is in many aspects very different from the traditional monoamine hypothesis. Although there is some evidence to suggest that BDNF signaling might be compromised in depression (Castrén and Monteggia, 2021), this model does not suggest that antidepressants act simply by restoring reduced BDNF signaling, but it emphasizes the role of BDNF-mediated plasticity that allows reorganization of networks through coherent activity provided by external and internal environment (Castrén, 2005; Branchi and Giuliani, 2021; Figures 1B,C). Such environmental activity can be enriched and guided by therapy or rehabilitation. However, although neuronal plasticity facilitates adaptation to changing environmental conditions, adaptation is not necessarily a positive phenomenon, but can become maladaptive if guides by an adverse environment (Alboni et al., 2017; Chiarotti et al., 2017; Branchi and Giuliani, 2021). Therefore, the model emphasizes that while antidepressants through facilitated plasticity enable recovery, they do not themselves cure depression, but active participation of the patient is required in the recovery process empowered by antidepressants.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This original work in the Castrén lab has been supported by the European Research Council (#322742), the Sigrid Jusélius Foundation, the Jane and Aatos Erkko Foundation, and the Academy of Finland (#307416, #327192, and # 347358).
EC has received lecture fees from Janssen-Cilag.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alboni, S., van Dijk, R. M., Poggini, S., Milior, G., Perrotta, M., Branchi, I., et al. (2017). Fluoxetine effects on molecular, cellular and behavioral endophenotypes of depression are driven by the living environment. Mol. Psychiatry 22, 552–561. doi: 10.1038/mp.2015.142
Altar, C. A. (1999). Neurotrophins and depression. Trends Pharmacol. Sci. 20, 59–61. doi: 10.1016/S0165-6147(99)01309-7
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Monteggia, L. M., et al. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. doi: 10.1038/nature10130
Autry, A. E., and Monteggia, L. M. (2012). Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 64, 238–258. doi: 10.1124/pr.111.005108
Biggio, F., Gorini, G., Utzeri, C., Olla, P., Marrosu, F., Follesa, P., et al. (2009). Chronic vagus nerve stimulation induces neuronal plasticity in the rat hippocampus. Int. J. Neuropsychopharmacol. 12, 1209–1221. doi: 10.1017/S1461145709000200
Biojone, C., Cannarozzo, C., Seiffert, N., Diniz, C., Brunello, C., Casarotto, P., et al. (2022). Mutation in the TRKB cholesterol recognition site that blocks antidepressant binding does not influence the basal or BDNF-stimulated activation of TRKB. bioRxiv [Preprint]. doi: 10.1101/2022.08.26.505413
Bocchio-Chiavetto, L., Bagnardi, V., Zanardini, R., Molteni, R., Nielsen, M. G., Gennarelli, M., et al. (2010). Serum and plasma BDNF levels in major depression: A replication study and meta-analyses. World J. Biol. Psychiatry 11, 763–773. doi: 10.3109/15622971003611319
Bocchio-Chiavetto, L., Zanardini, R., Bortolomasi, M., Abate, M., Segala, M., Gennarelli, M., et al. (2006). Electroconvulsive therapy (ECT) increases serum brain derived neurotrophic factor (BDNF) in drug resistant depressed patients. Eur. Neuropsychopharmacol. 16, 620–624. doi: 10.1016/j.euroneuro.2006.04.010
Bolo, N. R., Hodé, Y., Nédélec, J. F., Lainé, E., Wagner, G., and Macher, J. P. (2000). Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology 23, 428–438. doi: 10.1016/S0893-133X(00)00116-0
Bonaventura, J., Gomez, J. L., Carlton, M. L., Lam, S., Sanchez-Soto, M., Michaelides, M., et al. (2022). Target deconvolution studies of (2R,6R)-hydroxynorketamine: An elusive search. Mol. Psychiatry doi: 10.1038/s41380-022-01673-w [Epub ahead of print].
Branchi, I., and Giuliani, A. (2021). Shaping therapeutic trajectories in mental health: Instructive vs. Permissive causality. Eur. Neuropsychopharmacol. 43, 1–9. doi: 10.1016/j.euroneuro.2020.12.001
Calabrese, F., Molteni, R., Gabriel, C., Mocaer, E., Racagni, G., and Riva, M. A. (2011). Modulation of neuroplastic molecules in selected brain regions after chronic administration of the novel antidepressant agomelatine. Psychopharmacology (Berl) 215, 267–275. doi: 10.1007/s00213-010-2129-8
Calabrese, F., Molteni, R., Maj, P. F., Cattaneo, A., Gennarelli, M., Riva, M. A., et al. (2007). Chronic duloxetine treatment induces specific changes in the expression of BDNF transcripts and in the subcellular localization of the neurotrophin protein. Neuropsychopharmacology 32, 2351–2359. doi: 10.1038/sj.npp.1301360
Carreno, F. R., and Frazer, A. (2014). Activation of signaling pathways downstream of the brain-derived neurotrophic factor receptor, TrkB, in the rat brain by vagal nerve stimulation and antidepressant drugs. Int. J. Neuropsychopharmacol. 17, 247–258. doi: 10.1017/S1461145713000977
Casarotto, P. C., Girych, M., Fred, S. M., Kovaleva, V., Moliner, R., Castrén, E., et al. (2021). Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313.e19. doi: 10.1016/j.cell.2021.01.034
Castrén, E., and Monteggia, L. M. (2021). Brain-Derived neurotrophic factor signaling in depression and antidepressant action. Biol. Psychiatry 90, 128–136. doi: 10.1016/j.biopsych.2021.05.008
Cazorla, M., Premont, J., Mann, A., Girard, N., Kellendonk, C., and Rognan, D. (2011). Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest. 121, 1846–1857. doi: 10.1172/JCI43992
Chen, B., Dowlatshahi, D., MacQueen, G. M., Wang, J. F., and Young, L. T. (2001). Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 50, 260–265. doi: 10.1016/S0006-3223(01)01083-6
Chiarotti, F., Viglione, A., Giuliani, A., and Branchi, I. (2017). Citalopram amplifies the influence of living conditions on mood in depressed patients enrolled in the STAR*D study. Transl. Psychiatry 7:e1066. doi: 10.1038/tp.2017.35
Coppell, A. L., Pei, Q., and Zetterstrom, T. S. (2003). Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology 44, 903–910. doi: 10.1016/S0028-3908(03)00077-7
Daniel, W. A. (2003). Mechanisms of cellular distribution of psychotropic drugs. Significance for drug action and interactions. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 65–73. doi: 10.1016/S0278-5846(02)00317-2
Dias, B. G., Banerjee, S. B., Duman, R. S., and Vaidya, V. A. (2003). Differential regulation of brain derived neurotrophic factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology 45, 553–563. doi: 10.1016/S0028-3908(03)00198-9
Dugich-Djordjevic, M. M., Tocco, G., Lapchak, P. A., Pasinetti, G. M., Najm, I., Hefti, F., et al. (1992). Regionally specific and rapid increases in brain-derived neurotrophic factor messenger RNA in the adult rat brain following seizures induced by systemic administration of kainic acid. Neuroscience 47, 303–315. doi: 10.1016/0306-4522(92)90246-X
Duman, C. H., Schlesinger, L., Kodama, M., Russell, D. S., and Duman, R. S. (2007). A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol. Psychiatry 61, 661–670. doi: 10.1016/j.biopsych.2006.05.047
Duman, R. S., and Li, N. (2012). A neurotrophic hypothesis of depression: Role of synaptogenesis in the actions of NMDA receptor antagonists. Philos. Trans. R Soc. Lond. B Biol. Sci. 367, 2475–2484. doi: 10.1098/rstb.2011.0357
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127. doi: 10.1016/j.biopsych.2006.02.013
Duman, R. S., Heninger, G. R., and Nestler, E. J. (1997). A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597–606. doi: 10.1001/archpsyc.1997.01830190015002
Dunham, J. S., Deakin, J. F., Miyajima, F., Payton, A., and Toro, C. T. (2009). Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. J. Psychiatr. Res. 43, 1175–1184. doi: 10.1016/j.jpsychires.2009.03.008
Dwivedi, Y. (2009). Brain-derived neurotrophic factor: Role in depression and suicide. Neuropsychiatr. Dis. Treat. 5, 433–449. doi: 10.2147/NDT.S5700
Dwivedi, Y. (2013). Involvement of brain-derived neurotrophic factor in late-life depression. Am. J. Geriatr. Psychiatry 21, 433–449. doi: 10.1016/j.jagp.2012.10.026
Dwivedi, Y., Rizavi, H. S., Conley, R. R., Roberts, R. C., Tamminga, C. A., and Pandey, G. N. (2003). Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 60, 804–815. doi: 10.1001/archpsyc.60.8.804
Dwivedi, Y., Rizavi, H. S., Zhang, H., Mondal, A. C., Roberts, R. C., Pandey, G. N., et al. (2009). Neurotrophin receptor activation and expression in human postmortem brain: Effect of suicide. Biol. Psychiatry 65, 319–328. doi: 10.1016/j.biopsych.2008.08.035
Eisch, A. J., Bolanos, C. A., De Wit, J., Simonak, R. D., Pudiak, C. M., Nestler, E. J., et al. (2003). Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: A role in depression. Biol. Psychiatry 54, 994–1005. doi: 10.1016/j.biopsych.2003.08.003
Erb, S. J., Schappi, J. M., and Rasenick, M. M. (2016). Antidepressants accumulate in lipid rafts independent of monoamine transporters to modulate redistribution of the G protein. J. Biol. Chem. 291, 19725–19733. doi: 10.1074/jbc.M116.727263
Farmer, C. A., Gilbert, J. R., Moaddel, R., George, J., Adeojo, L., Zarate, C. A. J., et al. (2020). Ketamine metabolites, clinical response, and gamma power in a randomized, placebo-controlled, crossover trial for treatment-resistant major depression. Neuropsychopharmacology 45, 1398–1404. doi: 10.1038/s41386-020-0663-6
Fawcett, J. W., Oohashi, T., and Pizzorusso, T. (2019). The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat. Rev. Neurosci. 20, 451–465. doi: 10.1038/s41583-019-0196-3
Figurov, A., Pozzo-Miller, L. D., Olafsson, P., Wang, T., and Lu, B. (1996). Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the Hippocampus. Nature 381, 706–709. doi: 10.1038/381706a0
Follesa, P., Biggio, F., Gorini, G., Caria, S., Talani, G., Biggio, G., et al. (2007). Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 1179, 28–34. doi: 10.1016/j.brainres.2007.08.045
Fred, S. M., Laukkanen, L., Brunello, C. A., Vesa, L., Goos, H., Castrén, E., et al. (2019). Pharmacologically diverse antidepressants facilitate TRKB receptor activation by disrupting its interaction with the endocytic adaptor complex AP-2. J. Biol. Chem. 294, 18150–18161. doi: 10.1074/jbc.RA119.008837
Fujimura, H., Altar, C. A., Chen, R., Nakamura, T., Nakahashi, T., Tandon, N. N., et al. (2002). Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb. Haemost. 87, 728–734. doi: 10.1055/s-0037-1613072
Gogolla, N., Caroni, P., Luthi, A., and Herry, C. (2009). Perineuronal nets protect fear memories from erasure. Science 325, 1258–1261. doi: 10.1126/science.1174146
Gonul, A. S., Akdeniz, F., Taneli, F., Donat, O., Eker, C., and Vahip, S. (2005). Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. Eur. Arch. Psychiatry Clin. Neurosci. 255, 381–386. doi: 10.1007/s00406-005-0578-6
Guilloux, J. P., Douillard-Guilloux, G., Kota, R., Wang, X., Gardier, A. M., Sibille, E., et al. (2012). Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol. Psychiatry 17, 1130–1142. doi: 10.1038/mp.2011.113
Harmer, C. J., Duman, R. S., and Cowen, P. J. (2017). How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry 4, 409–418. doi: 10.1016/S2215-0366(17)30015-9
Harmer, C. J., Shelley, N. C., Cowen, P. J., and Goodwin, G. M. (2004). Increased positive versus negative affective perception and memory in healthy volunteers following selective serotonin and norepinephrine reuptake inhibition. Am. J. Psychiatry 161, 1256–1263. doi: 10.1176/appi.ajp.161.7.1256
Hellweg, R., Ziegenhorn, A., Heuser, I., and Deuschle, M. (2008). Serum concentrations of nerve growth factor and brain-derived neurotrophic factor in depressed patients before and after antidepressant treatment. Pharmacopsychiatry 41, 66–71. doi: 10.1055/s-2007-1004594
Henry, M. E., Moore, C. M., Kaufman, M. J., Michelson, D., Schmidt, M. E., Renshaw, P. F., et al. (2000). Brain kinetics of paroxetine and fluoxetine on the third day of placebo substitution: A fluorine MRS study. Am. J. Psychiatry 157, 1506–1508. doi: 10.1176/appi.ajp.157.9.1506
Hess, E. M., Riggs, L. M., Michaelides, M., and Gould, T. D. (2022). Mechanisms of ketamine and its metabolites as antidepressants. Biochem. Pharmacol. 197:114892. doi: 10.1016/j.bcp.2021.114892
Isackson, P. J., Huntsman, M. M., Murray, K. D., and Gall, C. M. (1991). BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: Temporal pattern of induction distinct from NGF. Neuron 6, 937–948. doi: 10.1016/0896-6273(91)90234-Q
Jacobsen, J. P., and Mork, A. (2004). The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res. 1024, 183–192. doi: 10.1016/j.brainres.2004.07.065
Johnson, R. D., Lewis, R. J., and Angier, M. K. (2007). The distribution of fluoxetine in human fluids and tissues. J. Anal. Toxicol. 31, 409–414. doi: 10.1093/jat/31.7.409
Karege, F., Bondolfi, G., Gervasoni, N., Schwald, M., Aubry, J. M., and Bertschy, G. (2005). Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol. Psychiatry 57, 1068–1072. doi: 10.1016/j.biopsych.2005.01.008
Karege, F., Perret, G., Bondolfi, G., Schwald, M., Bertschy, G., and Aubry, J. M. (2002). Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 109, 143–148. doi: 10.1016/S0165-1781(02)00005-7
Karpova, N. N. (2014). Role of BDNF epigenetics in activity-dependent neuronal plasticity. Neuropharmacology 76(Pt C), 709–718. doi: 10.1016/j.neuropharm.2013.04.002
Karpova, N. N., Pickenhagen, A., Lindholm, J., Tiraboschi, E., Kulesskaya, N., Castrén, E., et al. (2011). Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 334, 1731–1734. doi: 10.1126/science.1214592
Karson, C. N., Newton, J. E., Livingston, R., Jolly, J. B., Cooper, T. B., Komoroski, R. A., et al. (1993). Human brain fluoxetine concentrations. J. Neuropsychiatry Clin. Neurosci. 5, 322–329. doi: 10.1176/jnp.5.3.322
Kornhuber, J., Retz, W., and Riederer, P. (1995). Slow accumulation of psychotropic substances in the human brain. Relationship to therapeutic latency of neuroleptic and antidepressant drugs? J. Neural Transm. Suppl. 46, 315–323.
Lepack, A. E., Bang, E., Lee, B., Dwyer, J. M., and Duman, R. S. (2016). Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 111, 242–252. doi: 10.1016/j.neuropharm.2016.09.011
Lepack, A. E., Fuchikami, M., Dwyer, J. M., Banasr, M., and Duman, R. S. (2014). BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 18:yu033. doi: 10.1093/ijnp/pyu033
Lesnikova, A., Casarotto, P. C., Fred, S. M., Voipio, M., Winkel, F., Castrén, E., et al. (2021). Chondroitinase and antidepressants promote plasticity by releasing TRKB from dephosphorylating control of PTPσ in parvalbumin neurons. J. Neurosci. 41, 972–980. doi: 10.1523/JNEUROSCI.2228-20.2020
Li, N., Lee, B., Liu, R. J., Banasr, M., Dwyer, J. M., Duman, R. S., et al. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. doi: 10.1126/science.1190287
Liu, R. J., Lee, F. S., Li, X. Y., Bambico, F., Duman, R. S., and Aghajanian, G. K. (2012). Brain-derived neurotrophic factor val66met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry 71, 996–1005. doi: 10.1016/j.biopsych.2011.09.030
Liu, X., Chan, C. B., Jang, S. W., Pradoldej, S., Huang, J., Ye, K., et al. (2010). A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 53, 8274–8286. doi: 10.1021/jm101206p
Lommatzsch, M., Zingler, D., Schuhbaeck, K., Schloetcke, K., Zingler, C., Virchow, J. C., et al. (2005). The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol. Aging 26, 115–123. doi: 10.1016/j.neurobiolaging.2004.03.002
Malberg, J. E., and Duman, R. S. (2003). Cell proliferation in adult hippocampus is decreased by inescapable stress: Reversal by fluoxetine treatment. Neuropsychopharmacology 28, 1562–1571. doi: 10.1038/sj.npp.1300234
Malberg, J. E., Hen, R., and Madsen, T. M. (2021). Adult neurogenesis and antidepressant treatment: The surprise finding by Ron Duman and the field 20 years later. Biol. Psychiatry 90, 96–101. doi: 10.1016/j.biopsych.2021.01.010
Matrisciano, F., Bonaccorso, S., Ricciardi, A., Scaccianoce, S., Panaccione, I., Shelton, R. C., et al. (2009). Changes in BDNF serum levels in patients with major depression disorder (MDD) after 6 months treatment with sertraline, escitalopram, or venlafaxine. J. Psychiatr. Res. 43, 247–254. doi: 10.1016/j.jpsychires.2008.03.014
Maya Vetencourt, J. F., Sale, A., Viegi, A., Baroncelli, L., De Pasquale, R., Maffei, L., et al. (2008). The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 320, 385–388. doi: 10.1126/science.1150516
Mikics, É, Guirado, R., Umemori, J., Tóth, M., Biró, L., Karpova, N. N., et al. (2018). Social learning requires plasticity enhanced by fluoxetine through prefrontal Bdnf-TrkB signaling to limit aggression induced by post-weaning social isolation. Neuropsychopharmacology 43, 235–245. doi: 10.1038/npp.2017.142
Molendijk, M. L., Bus, B. A., Spinhoven, P., Penninx, B. W., Kenis, G., Elzinga, B. M., et al. (2011). Serum levels of brain-derived neurotrophic factor in major depressive disorder: State-trait issues, clinical features and pharmacological treatment. Mol. Psychiatry 16, 1088–1095. doi: 10.1038/mp.2010.98
Molendijk, M. L., Spinhoven, P., Polak, M., Bus, B. A., Penninx, B. W., and Elzinga, B. M. (2014). Serum BDNF concentrations as peripheral manifestations of depression: Evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol. Psychiatry 19, 791–800. doi: 10.1038/mp.2013.105
Naegelin, Y., Dingsdale, H., Sauberli, K., Schadelin, S., Kappos, L., and Barde, Y. A. (2018). Measuring and validating the levels of brain-derived neurotrophic factor in human serum. eNeuro 5, 1–9. doi: 10.1523/ENEURO.0419-17.2018
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of depression. Neuron 34, 13–25. doi: 10.1016/S0896-6273(02)00653-0
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995
Pandey, G. N., Ren, X., Rizavi, H. S., Conley, R. R., Roberts, R. C., and Dwivedi, Y. (2008). Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int. J. Neuropsychopharmacol. 11, 1047–1061. doi: 10.1017/S1461145708009000
Park, H., and Poo, M. M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. doi: 10.1038/nrn3379
Pereira, D. B., and Chao, M. V. (2007). The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J. Neurosci. 27, 4859–4869. doi: 10.1523/JNEUROSCI.4587-06.2007
Piccinni, A., Marazziti, D., Catena, M., Domenici, L., Del Debbio, A., Dell’Osso, L., et al. (2008). Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J. Affect. Disord. 105, 279–283. doi: 10.1016/j.jad.2007.05.005
Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J. W., and Maffei, L. (2002). Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251. doi: 10.1126/science.1072699
Poo, M. M. (2001). Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2, 24–32. doi: 10.1038/35049004
Radka, S. F., Holst, P. A., Fritsche, M., and Altar, C. A. (1996). Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res. 709, 122–301. doi: 10.1016/0006-8993(95)01321-0
Rantamäki, T., Hendolin, P., Kankaanpaa, A., Mijatovic, J., Piepponen, P., Castrén, E., et al. (2007). Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cδ signaling pathways in mouse brain. Neuropsychopharmacology 32, 2152–2162. doi: 10.1038/sj.npp.1301345
Ray, M. T., Shannon Weickert, C., and Webster, M. J. (2014). Decreased BDNF and TrkB mRNA expression in multiple cortical areas of patients with schizophrenia and mood disorders. Transl. Psychiatry 4:e389. doi: 10.1038/tp.2014.26
Ray, M. T., Weickert, C. S., Wyatt, E., and Webster, M. J. (2011). Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 36, 195–203. doi: 10.1503/jpn.100048
Reh, R. K., Dias, B. G., Nelson, C. A., Kaufer, D., Werker, J. F., Hensch, T. K., et al. (2020). Critical period regulation across multiple timescales. Proc. Natl. Acad. Sci. U.S.A. 117, 23242–23251. doi: 10.1073/pnas.1820836117
Renshaw, P. F., Guimaraes, A. R., Fava, M., Rosenbaum, J. F., Pearlman, J. D., Gonzalez, R. G., et al. (1992). Accumulation of fluoxetine and norfluoxetine in human brain during therapeutic administration. Am. J. Psychiatry 149, 1592–1594. doi: 10.1176/ajp.149.11.1592
Rocha, R. B., Dondossola, E. R., Grande, A. J., Colonetti, T., Ceretta, L. B., da Rosa, M. I., et al. (2016). Increased BDNF levels after electroconvulsive therapy in patients with major depressive disorder: A meta-analysis study. J. Psychiatr. Res. 83, 47–53. doi: 10.1016/j.jpsychires.2016.08.004
Russo-Neustadt, A., Beard, R. C., and Cotman, C. W. (1999). Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology 21, 679–682. doi: 10.1016/S0893-133X(99)00059-7
Russo-Neustadt, A., Ha, T., Ramirez, R., and Kesslak, J. P. (2001). Physical activity-antidepressant treatment combination: Impact on brain-derived neurotrophic factor and behavior in an animal model. Behav. Brain Res. 120, 87–95. doi: 10.1016/S0166-4328(00)00364-8
Saarelainen, T., Hendolin, P., Lucas, G., Koponen, E., Sairanen, M., Castrén, E., et al. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 23, 349–357. doi: 10.1523/JNEUROSCI.23-01-00349.2003
Sale, A., Berardi, N., Spolidoro, M., Baroncelli, L., and Maffei, L. (2010). GABAergic inhibition in visual cortical plasticity. Front. Cell. Neurosci. 4:10. doi: 10.3389/fncel.2010.00010
Seifert, T., Brassard, P., Wissenberg, M., Rasmussen, P., Nordby, P., Secher, N. H., et al. (2010). Endurance training enhances BDNF release from the human brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R372–R377. doi: 10.1152/ajpregu.00525.2009
Sen, S., Duman, R., and Sanacora, G. (2008). Serum brain-derived neurotrophic factor, depression, and antidepressant medications: Meta-analyses and implications. Biol. Psychiatry 64, 527–532. doi: 10.1016/j.biopsych.2008.05.005
Shen, Y., Tenney, A. P., Busch, S. A., Horn, K. P., Cuascut, F. X., Flanagan, J. G., et al. (2009). PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 326, 592–596. doi: 10.1126/science.1178310
Shimizu, E., Hashimoto, K., Okamura, N., Koike, K., Komatsu, N., Iyo, M., et al. (2003). Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol. Psychiatry 54, 70–75. doi: 10.1016/S0006-3223(03)00181-1
Shirayama, Y., and Hashimoto, K. (2018). Lack of antidepressant effects of (2R,6R)-hydroxynorketamine in a rat learned helplessness model: Comparison with (R)-ketamine. Int. J. Neuropsychopharmacol. 21, 84–88. doi: 10.1093/ijnp/pyx108
Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S., and Duman, R. S. (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 22, 3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002
Shirayama, Y., Yang, C., Zhang, J.-C., Ren, Q., Yao, W., and Hashimoto, K. (2015). Alterations in brain-derived neurotrophic factor (BDNF) and its precursor proBDNF in the brain regions of a learned helplessness rat model and the antidepressant effects of a TrkB agonist and antagonist. Eur. Neuropsychopharmacol. 25, 2449–2458. doi: 10.1016/j.euroneuro.2015.09.002
Siuciak, J. A., Lewis, D. R., Wiegand, S. J., and Lindsay, R. M. (1997). Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol. Biochem. Behav. 56, 131–137. doi: 10.1016/S0091-3057(96)00169-4
Suzuki, S., Numakawa, T., Shimazu, K., Koshimizu, H., Hara, T., Kojima, M., et al. (2004). BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: Roles in synaptic modulation. J. Cell. Biol. 167, 1205–1215. doi: 10.1083/jcb.200404106
Thoenen, H. (1995). Neurotrophins and neuronal plasticity. Science 270, 593–598. doi: 10.1126/science.270.5236.593
Tripp, A., Oh, H., Guilloux, J. P., Martinowich, K., Lewis, D. A., and Sibille, E. (2012). Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 169, 1194–1202. doi: 10.1176/appi.ajp.2012.12020248
Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., and Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525. doi: 10.1038/nn1659
Umemori, J., Winkel, F., Didio, G., Llach Pou, M., and Castren, E. (2018). iPlasticity: Induced juvenile-like plasticity in the adult brain as a mechanism of antidepressants. Psychiatry Clin. Neurosci. 72, 633–653. doi: 10.1111/pcn.12683
Winkel, F., Ryazantseva, M., Voigt, M. B., Didio, G., Lilja, A., Castrén, E., et al. (2021). Pharmacological and optical activation of TrkB in Parvalbumin interneurons regulate intrinsic states to orchestrate cortical plasticity. Mol. Psychiatry 26, 7247–7256. doi: 10.1038/s41380-021-01211-0
Wohleb, E. S., Gerhard, D., Thomas, A., and Duman, R. S. (2017). Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr. Neuropharmacol. 15, 11–20. doi: 10.2174/1570159X14666160309114549
Yamamoto, H., and Gurney, M. E. (1990). Human platelets contain brain-derived neurotrophic factor. J. Neurosci. 10, 3469–3478. doi: 10.1523/JNEUROSCI.10-11-03469.1990
Yoshimura, R., Mitoma, M., Sugita, A., Hori, H., Okamoto, T., Nakamura, J., et al. (2007). Effects of paroxetine or milnacipran on serum brain-derived neurotrophic factor in depressed patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 1034–1037. doi: 10.1016/j.pnpbp.2007.03.001
Youssef, M. M., Underwood, M. D., Huang, Y. Y., Hsiung, S. C., Liu, Y., Mann, J. J., et al. (2018). Association of BDNF Val66Met polymorphism and brain BDNF levels with major depression and suicide. Int. J. Neuropsychopharmacol. 21, 528–538. doi: 10.1093/ijnp/pyy008
Zafra, F., Hengerer, B., Leibrock, J., Thoenen, H., and Lindholm, D. (1990). Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 9, 3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Gould, T. D., et al. (2016). NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486. doi: 10.1038/nature17998
Zanos, P., Moaddel, R., Morris, P. J., Riggs, L. M., Highland, J. N., Gould, T. D., et al. (2018). Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacol. Rev. 70, 621–660. doi: 10.1124/pr.117.015198
Zhang, J. C., Wu, J., Fujita, Y., Yao, W., Ren, Q., Hashimoto, K., et al. (2015a). Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int. J. Neuropsychopharmacol. 18:yu077. doi: 10.1093/ijnp/pyu077
Keywords: antidepressant (AD), BDNF, TrkB, plasticity, parvalbumin interneurons
Citation: Casarotto P, Umemori J and Castrén E (2022) BDNF receptor TrkB as the mediator of the antidepressant drug action. Front. Mol. Neurosci. 15:1032224. doi: 10.3389/fnmol.2022.1032224
Received: 30 August 2022; Accepted: 17 October 2022;
Published: 02 November 2022.
Edited by:
Deepak Prakash Srivastava, King’s College London, United KingdomReviewed by:
Kenji Hashimoto, Chiba University, JapanCopyright © 2022 Casarotto, Umemori and Castrén. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eero Castrén, ZWVyby5jYXN0cmVuQGhlbHNpbmtpLmZp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.