 Pan-Yue Deng
Pan-Yue Deng Oshri Avraham
Oshri Avraham Valeria Cavalli2,3,4*
Valeria Cavalli2,3,4* Vitaly A. Klyachko
Vitaly A. Klyachko
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 22 December 2021
Sec. Neuroplasticity and Development
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.796053
This article is part of the Research Topic Pathological Hyperactivity and Hyperexcitability in the Central Nervous System View all 10 articles
Sensory hypersensitivity and somatosensory deficits represent the core symptoms of Fragile X syndrome (FXS). These alterations are believed to arise from changes in cortical sensory processing, while potential deficits in the function of peripheral sensory neurons residing in dorsal root ganglia remain unexplored. We found that peripheral sensory neurons exhibit pronounced hyperexcitability in Fmr1 KO mice, manifested by markedly increased action potential (AP) firing rate and decreased threshold. Unlike excitability changes found in many central neurons, no significant changes were observed in AP rising and falling time, peak potential, amplitude, or duration. Sensory neuron hyperexcitability was caused primarily by increased input resistance, without changes in cell capacitance or resting membrane potential. Analyses of the underlying mechanisms revealed reduced activity of HCN channels and reduced expression of HCN1 and HCN4 in Fmr1 KO compared to WT. A selective HCN channel blocker abolished differences in all measures of sensory neuron excitability between WT and Fmr1 KO neurons. These results reveal a hyperexcitable state of peripheral sensory neurons in Fmr1 KO mice caused by dysfunction of HCN channels. In addition to the intrinsic neuronal dysfunction, the accompanying paper examines deficits in sensory neuron association/communication with their enveloping satellite glial cells, suggesting contributions from both neuronal intrinsic and extrinsic mechanisms to sensory dysfunction in the FXS mouse model.
Fragile X syndrome (FXS) is the leading monogenetic cause of intellectual disability (ID) and autism. This disorder most commonly results from transcriptional silencing of the Fmr1 gene causing loss of expression of Fragile X Mental Retardation Protein (FMRP) (Penagarikano et al., 2007). Individuals with FXS typically present with cognitive dysfunction, learning deficits, social and behavioral problems, neurological deficits, and morphological abnormalities. Among most prevalent FXS deficits is hypersensitivity to sensory stimuli, including auditory, visual, and tactile stimuli. Increasing evidence suggests that sensory hypersensitivity may lead to behavioral alterations such as poor eye contact, anxiety, and impaired social interactions (Rais et al., 2018).
Individuals with FXS commonly exhibit somatosensory deficits, such as hypersensitivity to touch (Cascio, 2010), Self-injurious behaviors in Fragile X individuals (Arron et al., 2011; Crawford et al., 2019) are also indicative of abnormal pain perception. Impaired pain induction and perception are also observed in the FXS mouse model, including reduced induction of neuropathic pain (Ramirez-Lopez et al., 2021), and insensitivity to visceral pain (Yang et al., 2020). These and other sensory deficits have been largely attributed to alterations in cortical sensory processing with a wide range of excitability deficits identified in somatosensory cortex of FXS models at neuronal, circuit, and network levels (Contractor et al., 2015). For example, Fmr1 KO mice have abnormal encoding of tactile stimulation frequency and enlarged receptive fields in the somatosensory cortex (Juczewski et al., 2016). However, recent studies in other monogenetic models of autism suggest that many core cognitive and sensory deficits may arise from an earlier abnormality in sensory inputs that drive subsequent abnormal development of cortical circuits (Orefice et al., 2016, 2019). A hyperexcitable state of somatosensory neurons has been suggested to be a part of the core developmental pathology in autism models, leading to region-specific brain abnormalities during the critical period (Orefice et al., 2019). Indeed, the formation of the cortical receptive fields depends on sensory experience (Allen et al., 2003). The enlarged receptive fields in Fmr1 KO mice and altered perception of sensory information may be a consequence, in part, of altered sensory inputs during development. Yet little is known about alterations in the peripheral neural system that receives the primary sensory inputs. Research in Fragile X field has almost exclusively focused on central defects in processing of somatosensory information and dysfunction of the central neurons and circuits. Whether dysfunction of peripheral sensory neurons is present and contributes to FXS pathophysiology remains largely unexplored.
Peripheral sensory neurons in the dorsal root ganglia (DRG) play critical roles in receiving direct sensory information from the environment and conveying it to the central nervous system (CNS). Structurally, these are pseudo-unipolar neurons with one axon projecting into peripheral nerve and the other axon ascending in the dorsal root and spinal cord. Sensory neurons express FMRP, which localize to the soma and axons (Price et al., 2006). While the gross development of DRG is normal in the absence of FMRP (Price and Melemedjian, 2012), there is evidence for functional defects in sensory neurons including an increased surface expression of voltage-gated calcium channels leading to increased somatic glutamate release (Ferron et al., 2014, 2020). However, whether and how excitability of peripheral sensory neurons is altered by the loss of FMRP remains unexplored.
In this study we performed recordings from the DRG neurons isolated from adolescent mice in a short-term culture to examine changes in sensory neuron excitability caused by loss of FMRP.
Firing patterns of sensory neurons situated in the DRG convey information from external and internal environment of the body to the CNS. Thus, these neurons play a critical role in transducing sensory information to neuronal signals. Accordingly, we first asked how excitability of sensory neurons is affected in Fmr1 KO mice. Whole-cell recordings were performed in short-term cultures of sensory neurons, as described (Avraham et al., 2020). Neurons were separated for analysis by size into two groups with a cutoff at 30 μm diameter (Yousuf et al., 2019) into small/medium vs. large diameter neurons. We found that most of the small/medium diameter neurons [mean diameter: 19.14 ± 0.53 μm (WT), 19.84 ± 0.54 μm (KO)] in the short-term DRG cultures exhibit tonic action potential (AP) firing (multiple APs fired in a stimulus intensity-dependent manner, Figure 1A), whereas all tested large neurons (>30 μm) show phasic AP firing (a single AP fired regardless of stimulus intensity, data not shown). To better understand how and to what extent the excitability of somatosensory neurons is affected in Fmr1 KO mice, we therefore used the small/medium diameter sensory neurons as a model neuron. In our culture conditions, the majority of the cells we recorded from are small/medium diameter IB4-positive neurons (Avraham et al., 2020). IB4 positive neurons represent non-peptidergic C- and Aδ-nociceptive neurons and some Aδ-low threshold mechanoreceptors (Wang et al., 1998; Li et al., 2011; Olson et al., 2016), a subset of which detect gentle touch (Liu et al., 2007; Olson et al., 2016).
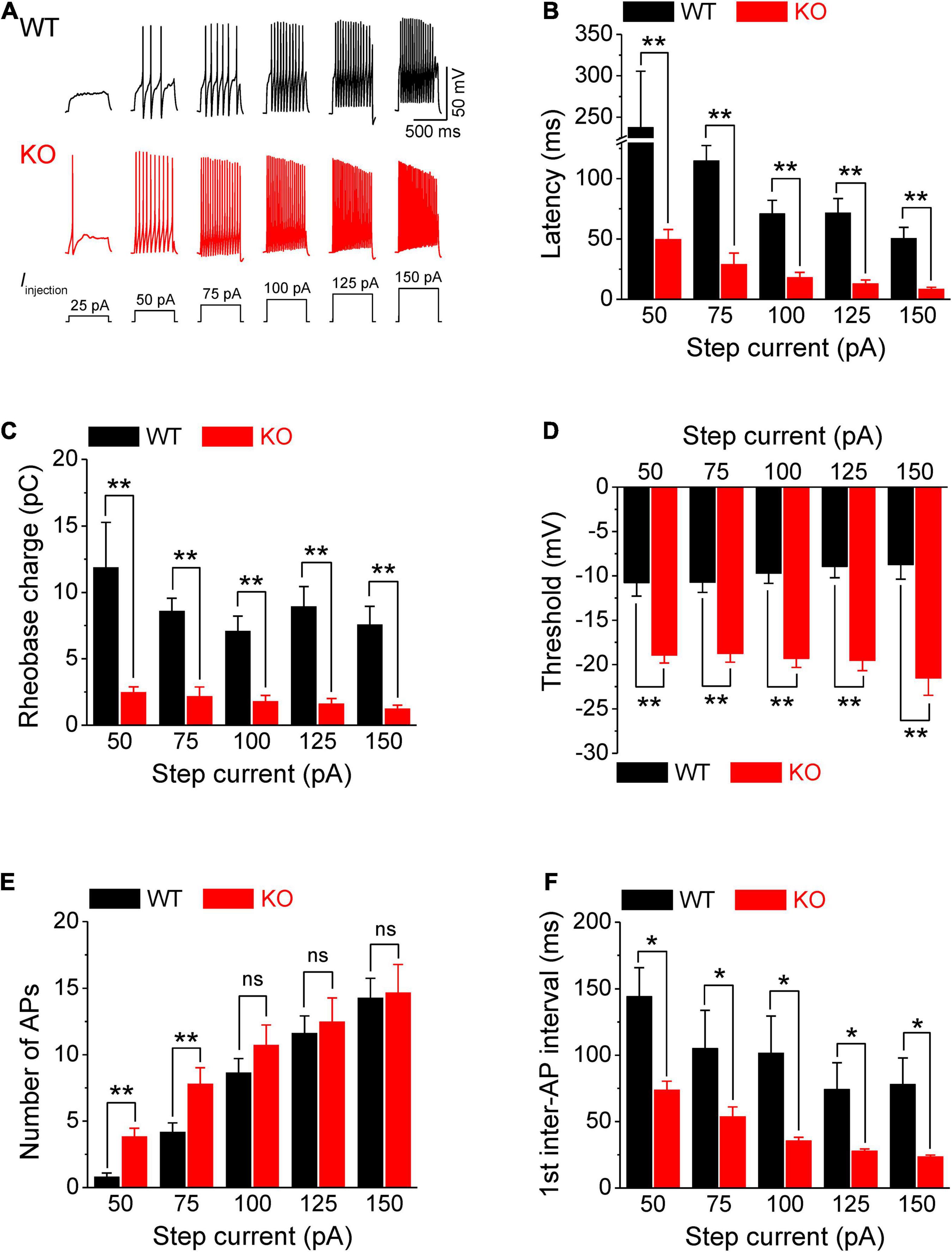
Figure 1. Increased excitability of sensory neurons in Fmr1 KO mice. (A) Sample AP traces evoked by step-current injection. Note, that for a 25-pA step, there was no AP observed in any of tested WT neurons, and in ∼50% of KO neurons. Thus, in the following panels we analyzed AP parameters starting from 50 pA step. (B–F) Analysis of AP parameters in measurements from panel (A). Latency to the first AP (B), rheobase charge transfer (C), threshold (D), number of AP fired (E), and first inter-AP interval (F) in WT and Fmr1 KO neurons. T-test; *p < 0.05; **p < 0.01; ns, not significant.
Action potentials were evoked by multi-step current injections (from 25 to 150 pA with a step size of 25 pA, Figure 1A). Only the first APs were used to determine AP latency, threshold, and rheobase to avoid AP parameters being affected by cumulative Na+ channel inactivation during subsequent APs. We found that excitability of sensory neurons was markedly increased in the absence of FMRP, as evident by decreased latency to the first AP (p < 0.01, Figure 1B, all values here and throughout are summarized in Supplementary Table 1), lower voltage threshold (p < 0.01, Figure 1D), and reduced rheobase charge transfer (p < 0.01, Figure 1C) which represents a measure of current threshold. Sensory neurons in Fmr1 KO mice also fired more APs at lower stimulus intensity steps (p < 0.01, Figure 1E), and had a shortened inter-AP interval (p < 0.05, Figure 1F). Unlike excitability changes observed in many central neurons (Contractor et al., 2015) we observed no significant changes in AP rising and falling time, AP peak potential, and AP amplitude, as well as in AP duration (Figure 2). It is noteworthy that the threshold and rheobase values were largely independent of step current intensities within genotypes, indicating that these are reliable parameters for evaluating neuronal excitability.
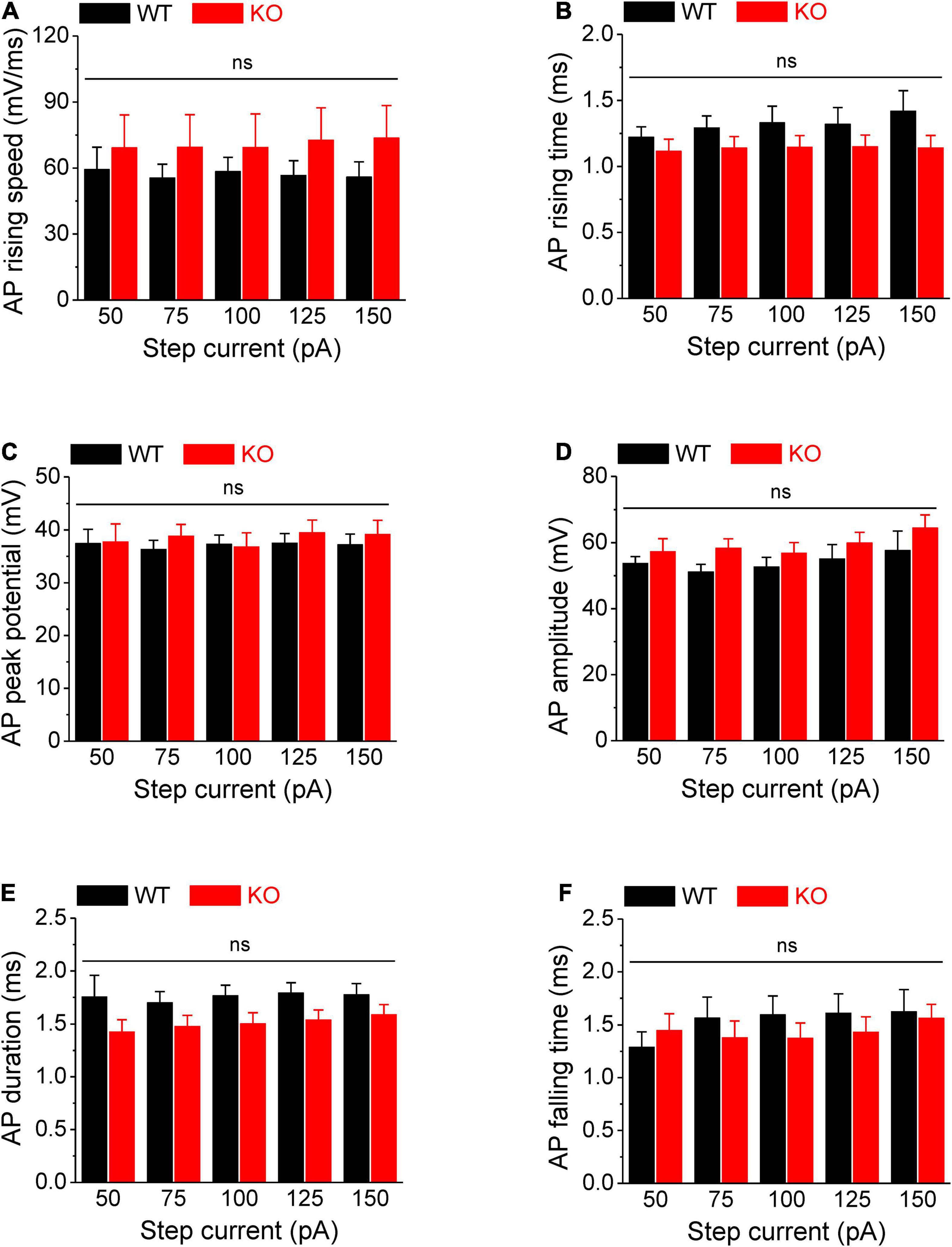
Figure 2. No changes in AP rising and falling time, duration, and amplitude in Fmr1 KO neurons. (A–F) Loss of FMRP did not affect AP rising speed (A), 10–90% rising time (B), AP peak potential (C), AP amplitude (D), AP duration (E), and 90–10% AP falling time (F). T-test; ns, not significant.
We further confirmed the increased excitability of sensory neurons in Fmr1 KO mice by examining AP parameters in a ramp-current evoked AP traces, using a previously reported approach (Deng and Klyachko, 2016a,b; Deng et al., 2019). As expected, we found that excitability of DRG neurons was indeed increased in Fmr1 KO mice, as evident by a significantly larger number of APs fired in KO neurons (p = 0.014; Figures 3A,C); a large hyperpolarizing shift of threshold potential (p = 0.0015; Figures 3A–C), and reduced AP rheobase (p < 0.0001; Figure 3C).
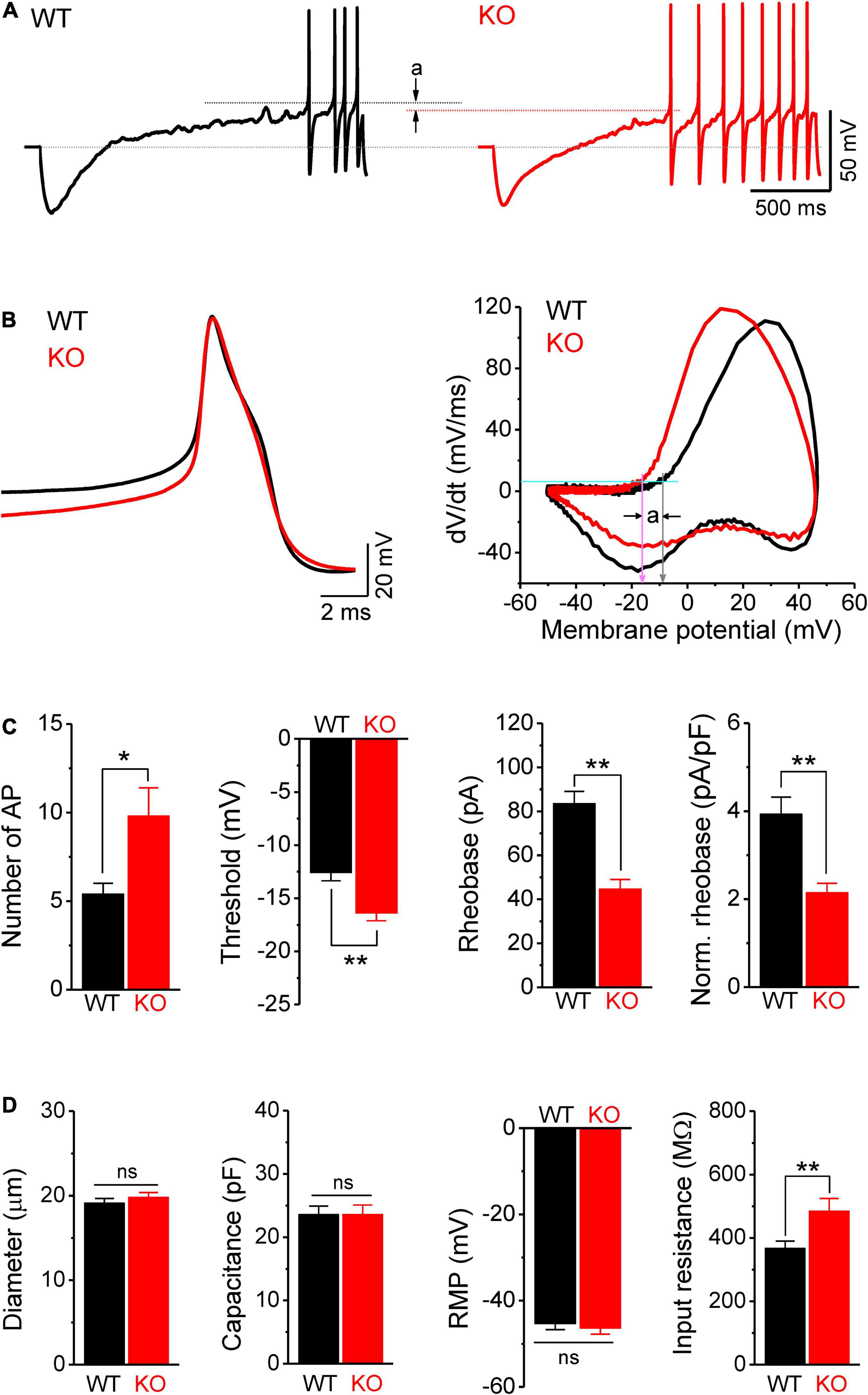
Figure 3. Hyperexcitability of sensory neurons in Fmr1 KO mice is associated with increased input resistance. (A) Sample traces of ramp-evoked APs. Note the differences in number of APs and threshold (a) between WT and KO neurons. Short dot-lines indicate the threshold levels of WT (black) and KO (red) neurons. Long dot-line is the resting membrane potential (RMP) level. (B) The first APs from panel (A) and their corresponding phase plots, indicating decreased AP threshold in Fmr1 KO mice (a). Cyan line is the membrane depolarizing speed of 5 mV/ms, the corresponding voltages of its intersections with phase plot were thresholds for WT (gray arrow) and KO (pink arrow) neurons. (C) Summarized data for the number of APs, threshold, rheobase, and membrane capacitance-normalized rheobase for the measurements in panel (A). (D) Summarized data for cell size, membrane capacitance, RMP, and membrane input resistance for the measurements in panel (A). T-test; *p < 0.05; **p < 0.01; ns, not significant.
Together, these observations demonstrate a state of marked hyperexcitability of peripheral sensory neurons in the absence of FMRP.
Because the intrinsic membrane properties play a major role in setting neuronal excitability, we examined the resting membrane potential (RMP), cell size, membrane capacitance, and input resistance of sensory neurons in Fmr1 KO and WT animals. While no significant differences were observed in cell size, capacitance, and RMP between genotypes (Figure 3D), we found a significant increase in input resistance in Fmr1 KO neurons (p = 0.0097; Figure 3D). Increased input resistance is consistent with the reduced rheobase in the absence of FMRP and may thus be a direct cause of hyperexcitability of KO neurons.
To understand the mechanisms of these excitability defects, we first considered that absence of significant changes in RMP or AP waveform suggests that the voltage-gated Na+ and K+ channels active near RMP or above threshold are unlikely to be strongly affected in sensory neurons of Fmr1 KO mice. Thus, the changes in input resistance may result from alterations in some voltage-dependent sub-threshold conductance, such as M current (carried by Kv7 channels) and/or h current (carried by HCN channels) that are abundantly expressed in sensory neurons (de Moraes et al., 2017). We examined contributions from Kv7 channels and found that the Kv7 channel blocker XE991 (10 μM) failed to abolish the differences between genotypes in either the voltage threshold (p = 0.012, Supplementary Figure 1A), or in the rheobase charge transfer (p < 0.0001; Supplementary Figure 1B). This observation suggests that Kv7 channels are unlikely to underlie excitability changes observed in Fmr1 KO neurons.
Next, we examined the HCN channel activity in sensory neurons. HCN channels are activated by a membrane hyperpolarization, and cells with active HCN channels are characterized by voltage sag in current clamp recordings in response to a hyperpolarizing current. As expected, negative current injection produced marked voltage sags in WT neurons (Figure 4A). Most importantly, the voltage sag in Fmr1 KO neurons was significantly decreased compared to WT in all tested steps for both absolute values and sag ratios (p < 0.01 for all steps; Figures 4A–C). These differences were not due to the basal RMP differences between genotypes (p = 0.52, Supplementary Table 1).
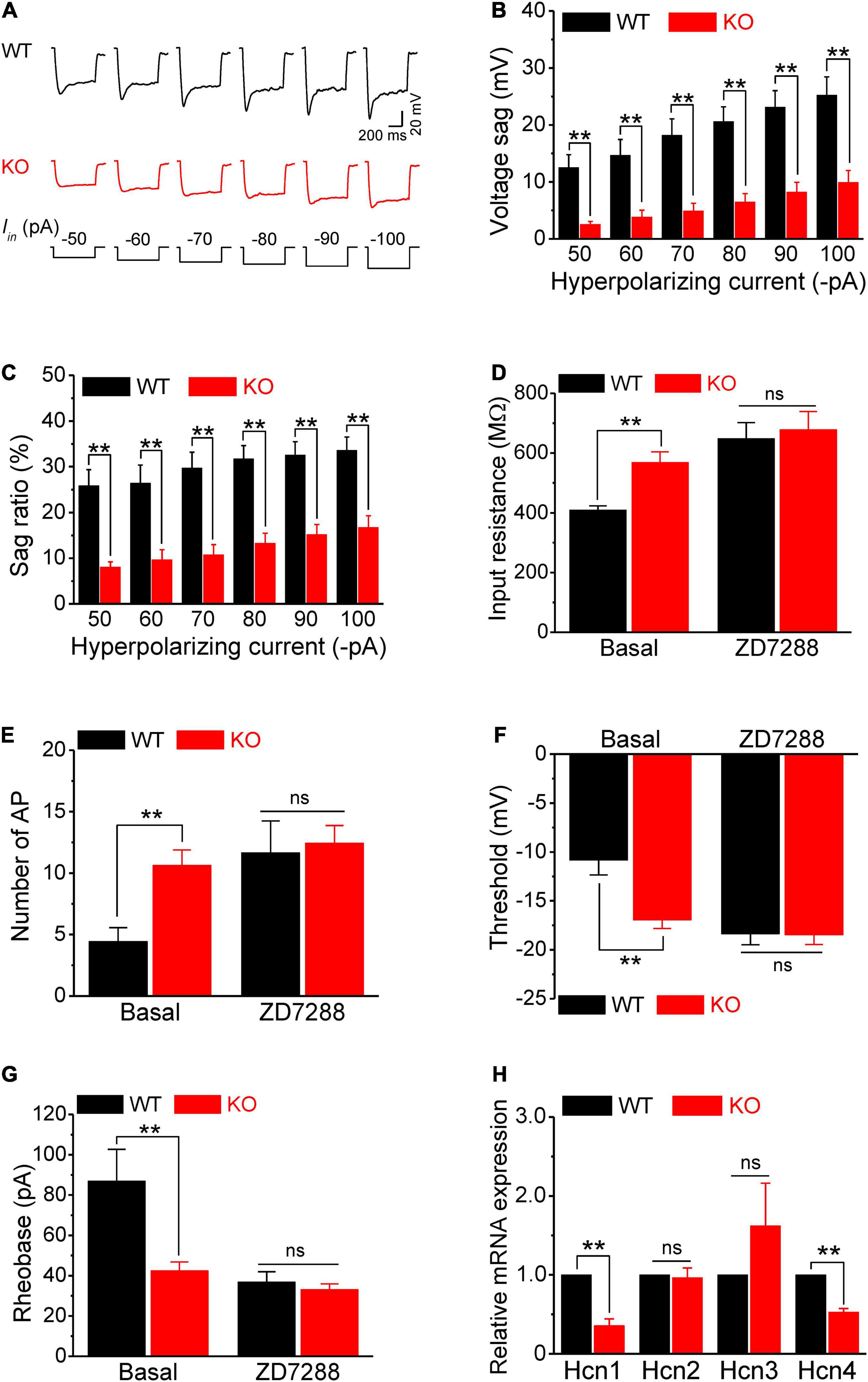
Figure 4. HCN channel dysfunction causes hyperexcitability of sensory neurons in Fmr1 KO mice. (A) Example traces of hyperpolarization-induced voltage sag in sensory neurons. Iin is the injected hyperpolarizing current with intensities indicated in the lower panel. (B,C) Quantification of voltage sag in WT and KO neurons in absolute values (B) and as a sag ratio (C) for all tested hyperpolarizing current levels. (D) A selective HCN channel blocker ZD7288 abolished the differences in input resistance between genotypes. (E–G) HCN channel blocker ZD7288 abolished the differences in the number of APs fired (E), threshold (F), and rheobase (G) between genotypes. (H) qPCR analysis of the relative mRNA expression of Hcn1–4 in Fmr1 KO compared to WT DRGs. N = 3 biologically independent animals. T-test; **p < 0.01; ns, not significant.
Given that the HCN channels contribute significantly to input resistance, these results suggest that the reduced HCN channel function might be the major cause of hyperexcitability in sensory neurons of Fmr1 KO mice. If this is the case, inhibition of HCN channels should eliminate the differences in input resistance and in excitability of sensory neurons between genotypes. In line with this prediction, the HCN channel blocker ZD7288 (10 μM), which potently blocks all HCN channels without preference for a specific HCN subunit, abolished the difference in input resistance between WT and KO neurons (p = 0.73; Figure 4D). Most importantly, ZD7288 also abolished differences in sensory neuron excitability between WT and KO neurons, including the number of AP fired (p = 0.78; Figure 4E), AP threshold (p = 0.94; Figure 4F), as well as rheobase (p = 0.50; Figures 4G,H). Together, these results confirmed that reduced HCN channel function causes increased input resistance, which in turn leads to hyperexcitability of sensory neurons in Fmr1 KO mice.
Fragile X mental retardation protein regulates expression and activity of multiple ion channels (Deng and Klyachko, 2021) and HCN channels are a known target of FMRP translational control (Darnell et al., 2011; Brager et al., 2012; Zhang et al., 2014; Orefice et al., 2016, 2019). We thus examined by quantitative PCR (qPCR) if Hcn channel expression was altered in sensory neurons of Fmr1 KO mice. We used whole DRG for these experiments, since HCN channels are selectively expressed in neurons in the DRG (Moosmang et al., 2001; Biel et al., 2009), with HCN1, HCN2, and to a lesser extent HCN4 being most abundant (Moosmang et al., 2001; Chaplan et al., 2003). We found that mRNA levels of Hcn1 and Hcn4 were significantly reduced in Fmr1 KO DRG (Hcn1, p = 0.0018; Hcn4, p = 0.0004) (Figure 4H) while levels of Hcn2 and Hcn3 were not strongly affected (Hcn2, p = 0.78; Hcn3, p = 0.32) (Figure 4H). Together with the above results, this observation suggests that neuronal hyperexcitability in the absence of FMRP is caused by reduced HCN channel expression in sensory neurons.
Sensory hypersensitivity in FXS has thus far been largely attributed to sensory processing abnormalities in the brain circuits (Contractor et al., 2015). Our results revealed a contribution to sensory abnormalities from peripheral deficits in the FXS mouse model. We found a hyperexcitable state of peripheral sensory neurons characterized by markedly increased AP frequency and reduced threshold caused by loss of FMRP. Altered sensory neuron excitability in Fmr1 KO mice arises, at least in part, from intrinsic neuronal mechanisms involving increased input resistance caused by HCN channel dysfunction.
HCN channels are active at rest and play a crucial role in controlling input resistance, and thus neuronal excitability (Shah, 2014). Voltage dependence of HCN channels is regulated by a number of intracellular factors. Other voltage-gated channels have a strong influence on HCN channel activity. The resulting action of HCN channels on membrane excitability in a given cell type is thus determined by a delicate balance of these factors. For example, in the CA1 neurons, through interaction with Kv7 channels, HCN channels can enhance AP firing in response to an EPSP when AP threshold is low and can inhibit AP firing when AP threshold is high (George et al., 2009). The AP threshold of DRG neurons is comparatively high, thus reduction of HCN channel activity in Fmr1 KO neurons is consistent with increased firing. Also, HCN channels have two separate influences on membrane excitability: one is the channel-mediated inward current (i.e., excitatory influence), the second one is a shunting effect (inhibitory influence). The overall effect might be complicated or even “paradoxical” (George et al., 2009). Given that the input resistance is a direct determinant of AP rheobase, our observations of reduced rheobase in Fmr1 KO neurons support the notion that HCN channels in sensory neurons function through the shunting effect to reduce input resistance.
Cell-type specific HCN channel defects have been previously implicated in excitability alterations of central neurons in the FXS models. For example, the elevated HCN1 subunit expression and increased Ih current were found in dendrites of CA1 pyramidal cells from Fmr1 KO mice leading to decreased input resistance and reduced temporal summation (Brager et al., 2012), while the opposite changes in HCN1 expression, Ih and excitability were observed in the layer 5 pyramidal cells of somatosensory cortex (Zhang et al., 2014) or layer 4 stellate cells of Fmr1 KO mice (Booker et al., 2019). Reduced HCN1 expression and decreased Ih were also found in large (mechanosensory) but not small diameter DRG neurons in Shank3 deletion model of Phelan–McDermid syndrome often associated with ASD (Orefice et al., 2016, 2019). Here we observed that Hcn1 and Hcn4 expression is strongly reduced in the DRG of Fmr1 KO mice. Together with the observations that a selective HCN channel blocker abolished differences in all measures of excitability between Fmr1 KO and WT mice, these observations suggest that reduced HCN channel expression is the major cause of hyperexcitability of sensory DRG neurons caused by FMRP loss. Notably, in central neurons, FMRP is also known to exert powerful control over ion channel activity via protein–protein interactions (Brown et al., 2010; Deng et al., 2013, 2019; Deng and Klyachko, 2021). In the case of HCN channels, FMRP can regulate the channel’s surface levels in a tissue-specific manner via protein–protein interactions with the HCN-TRIP8b complexes (Brandalise et al., 2020). Whether this interaction is present in sensory neurons and contribute to neuronal excitability defects remains to be determined.
Interestingly, HCN channel expression and Ih current show age-dependent increase in thalamic neurons (Kanyshkova et al., 2009) which dampens their excitability as these neurons mature. Furthermore, there is evidence of maturation-dependent regulation of HCN channels in spiral ganglia in the auditory pathway (Shen et al., 2018) and immature neurons are known to exhibit higher intrinsic excitability and plasticity (Schmidt-Hieber et al., 2004; Oh et al., 2010). Can hyperexcitability of peripheral sensory neurons be a consequence, in part, of the delayed neuronal maturation in Fmr1 KO mice? The age-dependent changes in excitability have been reported in central neurons of Fmr1 KO mice: the CA3 pyramidal neurons show increased excitability in young Fmr1 KO mice (3–4 weeks) (Deng et al., 2019; Dwivedi et al., 2019), but this was not seen in the older animals (6–8 weeks) (Dwivedi et al., 2019). Further, a delay in neuronal maturation and immature state of dendritic spines is widely documented in central neurons of Fmr1 KO mice (Comery et al., 1997; Harlow et al., 2010; Guo et al., 2015; Moskalyuk et al., 2020), resulting in delayed maturation of local networks (Vislay et al., 2013; Nomura et al., 2017) and a developmental delay in somatosensory map formation (Till et al., 2012). This is also consistent with abnormal neurogenesis and altered differentiation of neural stem cells in Fmr1 KOs, leading to poor neuronal maturation and high gliogenic development (Castren et al., 2005; Telias et al., 2013, 2015). Our single-cell RNA-seq analyses described in detail in the accompanying paper indeed suggest that maturation of sensory neurons in the DRG is delayed/aberrant, as evident in upregulation of progenitor markers and downregulation of neuronal differentiation/neuronal identity markers. Thus, the delayed maturation of sensory neurons in the DRG could be an underlying or contributing factor driving their hyperexcitability.
The increased intrinsic excitability of sensory neurons is only one of complex multifaceted changes that occur in the peripheral sensory system in the absence of FMRP. For example, the excitability changes we observed here will combine with the increased glutamate release from the soma and terminals of these neurons, which occurs independently due to excessive surface expression of N-type calcium channels (Ferron et al., 2014, 2020), further multiplying the excessive output from sensory neurons. Moreover, morphological changes of neuronal processes, such as axon structure or axon initial segment length, which are affected centrally in Fmr1 KO mice (Booker et al., 2020), can also contribute to sensory neuron excitability. Given the long-range projections of the sensory neurons of the DRG, future central and peripheral projection tracing will be needed to define the precise morphological changes in sensory neuron processes. In addition to these intrinsic mechanisms, in the accompanying paper, we describe extrinsic mechanisms that may contribute to sensory deficits due to disruption of the peripheral neurons’ association/communication with their enveloping satellite glial cells. Thus, an interplay of multiple peripheral deficits needs to be considered to fully understand sensory deficits caused by FMRP loss. Notably, our measurements of neuronal excitability are limited to short-term cultures in which neurons do not develop full length long-range projections and do not get enveloped by the satellite glia cells. Thus how the complex interplay between the intrinsic and extrinsic changes influences sensory processing in vivo remains to be elucidated. This includes defining how sensory transduction is affected in the intact DRG and whether distorted cortical maps in Fmr1 KOs (Till et al., 2012) are a consequence of altered sensory receptive fields. Moreover, in vivo measurements will also be needed to define the extent to which peripheral deficits contribute to the abnormal processing of repeating sensory stimuli (Domanski et al., 2019). Such measurements will present a technical challenge because sensory DRG neurons in more intact settings (ex vivo slices or in vivo) are entirely surrounded by the satellite glia coat.
What is the relevance of sensory neuron hyperexcitability to clinical FXS phenotypes? The majority of cells analyzed in our experiments were small/medium diameter IB4-positive nociceptors (Avraham et al., 2020). A previous study showed that loss of FMRP decreases nociceptive sensitization in adult mice, even though the basal nociceptive thresholds were intact (Price et al., 2007). Recent evidence also indicates impaired pain induction and perception in the FXS mouse model, including reduced neuropathic pain (Ramirez-Lopez et al., 2021), and visceral pain (Yang et al., 2020). Future in vivo studies will be needed to determine whether and how increased excitability of nociceptive neurons in adolescent mice we observed here is linked to abnormal pain induction or perception in FXS mice. Notably, IB4-positive sensory neurons also include a subset of mechanoreceptors that detect gentle touch (Liu et al., 2007). Thus, our observations could be relevant to the clinical FXS phenotypes beyond the pain induction/perception, since individuals with Fragile X are known to experience hypersensitivity to touch (Arnett et al., 2014; He et al., 2017).
Fmr1 KO (FVB.129P2-Pde6b+Tyrc–chFmr1tm1Cgr/J; stock #004624) and WT control (FVB.129P2-Pde6b+Tyrc–ch/AntJ; stock #004828) mice on FVB background were obtained from The Jackson Laboratory. Male mice (28- to 30-day old) were used for DRG cultures since male FXS individuals typically have more severe symptoms than do female individuals (Hagerman et al., 2009). Lumbar DRG (L1–L5) were dissected from Fmr1 KO and WT control mice and collected into cold Hank’s balanced salt solution (HBSS) with 5% Hepes, then transferred to warm papain solution and incubated for 20 min in 37°C. DRG’s were washed in HBSS and incubated with collagenase for 20 min in 37°C. Ganglia were then mechanically dissociated to a single cell suspension by triturating in culture medium (Neurobasal medium), with Glutamax, PenStrep, and B-27. Cells were then cultured on 100 μg/ml poly-D-lysine coated cover slips and used for electrophysiological recording 24 h after plating. All animal procedures were in compliance with the NIH Guide for the Care and Use of Laboratory Animals and conformed to Washington University Animal Studies Committee guidelines.
Whole-cell patch-clamp recordings in a current-clamp mode were performed using a MultiClamp 700B amplifier (Molecular Devices) from short-term cultures (24 h after plating) of isolated DRG neurons, visually identified with infrared video microscopy and differential interference contrast optics (Olympus BX51WI). Current-clamp recordings were made with pipette capacitance compensation and bridge-balance compensation. Recordings were conducted at near-physiological temperature (33–34°C). In these conditions, the majority of cells analyzed were small/medium diameter IB4-positive neurons (Avraham et al., 2020). The recording electrodes were filled with the following (in mM): 130 K-gluconate, 10 KCl, 0.1 EGTA, 2 MgCl2, 2 ATPNa2, 0.4 GTPNa, and 10 HEPES, pH 7.3. The extracellular solution contained (in mM): 145 NaCl, 3 KCl, 10 HEPES, 2.5 CaCl2, 1.2 MgCl2, and 7 glucose, pH 7.4 (saturated with 95% O2 and 5% CO2). APs were evoked either by multiple-step-current injection (from 25 to 150 pA with step duration of 600 ms and step size 25 pA) or by a ramp-current injection (0.1 pA/ms) with a hyperpolarizing onset. To determine the number of APs, all APs for each step were counted (step-evoked APs), but for the ramp-evoked APs, only APs within the first 2 s from beginning of the ramp were counted. AP threshold was defined as voltage where the AP rise speed reaches 5 mV/ms. The AP threshold was determined only from the first APs in the trace. For ramp-evoked APs, AP rheobase was determined as current amplitude difference from baseline to threshold point. Rheobase charge transfer was the integration of the current over the time interval, which was from the beginning of the steps (or ramp cross baseline) to the first AP threshold point. AP latency was defined as the time duration from the beginning of step-current to the first AP threshold point. AP duration was defined as the time interval between AP rising and falling parts at a membrane potential of +15 mV level. When the number of APs was more than 2, the first inter-AP interval was defined as the time duration between the peaks of first and second APs. All data were averaged over 5–8 trials for each cell. All chemicals for internal solution and bath solution were from Sigma-Aldrich. The channel blockers ZD7288 and XE991 were from Tocris. Different cells were used to test the effect of blockers (ZD7288 or XE991) to minimize the influence from “washout effect” due to recording time limitations (recordings in DRG cultures have a fast rundown during whole cell recordings).
Resting membrane potential was measured immediately after whole-cell formation. Cell capacitance was determined by the amplifier’s auto whole-cell compensation function with slight manual adjustment to optimize the measurement if needed. Under current-clamp mode, a negative current (−50 pA for 500 ms) was injected every 5 s to assess the input resistance. The voltage difference between baseline and steady state was used to calculate input resistance.
For evaluation of HCN channel activity, hyperpolarization-evoked voltage sag was determined by step-current injection (from −50 to −100 pA with step size −10 pA and duration 600 ms). Sag amplitude was defined as the voltage difference between the lowest point of voltage trace and steady-state part (average 50 ms) immediate before the end of step. Sag ratio was calculated as 100% × (sag amplitude) ÷ (voltage difference between baseline and the lowest point of voltage trace).
Dorsal root ganglia were lysed and total RNA was extracted using Trizol reagent (Thermo Fisher, Cat# 15596026). Next, RNA concentration was determined using a NanoDrop 2000 (Thermo Fisher Scientific). First strand synthesis was then performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). qPCR was performed using PowerUp SYBR Green master mix (Thermo Fisher, Cat# a25742) using 5 ng of cDNA per reaction. Plates were run on a QuantStudio 6 Flex system. Quantification of relative gene expression was performed using an automated software package (QuantStudio, ThermoFisher Scientific) following a standard 2–ΔΔCt method as described (Rao et al., 2013). Briefly, the cycle threshold (Ct) information generated by the qPCR system is directly used to determine relative gene expression in target and reference samples, using a reference gene as the normalizing factor (Rao et al., 2013). The Ct for the mRNA of a housekeeping gene (Rpl13a) was first subtracted from the Ct for the mRNA of the different Hcn isoforms in the same sample to normalize for variation in the amount and quality of mRNA between different samples. This normalization procedure (ΔCt) permits comparison of expression of a gene of interest among different samples. The average ΔCt value from three technical replicates was calculated for each of the biological replicates (n = 3). The final outcome of this quantification was calculated as the fold change of Hcn isoforms expression in the KO samples relative to their expression in the WT samples (ΔΔCt). The relative gene expression is usually set to 1 for reference samples (WT) because ΔΔCt is equal to 0 and therefore 20 is equal to 1 (Rao et al., 2013).
Primer sequences were obtained from PrimerBank or published literature and product size validated using agarose gel electrophoresis.
Rpl13a (PrimerBank ID 334688867c2) Forward Primer AGCCTACCAGAAAGTTTGCTTAC Reverse Primer GCTTCTTCTTCCGATAGTGCATC.
Hcn1 Forward Primer ACATGCTGTGCATTGGTTATGGCG, Reverse PrimerAACAAACATTGCGTAGCAGGTGGC.
Hcn2 Forward Primer ACTTCCGCACCGGCATTGTTATTG, Reverse Primer TCGATTCCCTTCTCCACTATG AGG.
Hcn3 Forward Primer TGGGAACCACTGGTGCACG, Reverse Primer TGAGCGTCTAGCAGATCGAG.
Hcn4 Forward Primer GCATGATGCTTCTGCTGTGTCACT, Reverse Primer TTCACCATGCCATTGATGGACACC.
Data are presented as mean ± SEM. Student’s T-test was used for statistical analysis as appropriate. Significance was set as p < 0.05. The n was number of cells tested. All statistical values and tests used in each experiment are given in Supplementary Table 1 for each panel.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All animal procedures were reviewed and approved by the Washington University School of Medicine Institutional Animal Care and Use Committee (IACUC) under protocol A-3381-01. All experiments were performed in accordance with the relevant guidelines and regulations. All experimental protocols involving mice were approved by the Washington University School of Medicine (protocol #21-0104 and #20-0173). Mice were housed and cared for in the Washington University School of Medicine animal care facility. This facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AALAC) and conforms to the PHS guidelines for Animal Care. Accreditation - 7/18/97, USDA Accreditation: Registration # 43-R-008.
P-YD, OA, VC, and VK conceived and designed the experiments and wrote the manuscript. P-YD and OA performed the experiments and data analysis. VC and VK secured the funding. All authors contributed to the article and approved the submitted version.
This work was funded in part by the NIH grant R35 NS111596 to VK, and by the NIH grant R01 NS111719 and R35 NS122260 to VC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank members of the Klyachko and Cavalli Lab for valuable discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2021.796053/full#supplementary-material
Allen, C. B., Celikel, T., and Feldman, D. E. (2003). Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat. Neurosci. 6, 291–299. doi: 10.1038/nn1012
Arnett, M. T., Herman, D. H., and McGee, A. W. (2014). Deficits in tactile learning in a mouse model of fragile X syndrome. PLoS One 9:e109116. doi: 10.1371/journal.pone.0109116
Arron, K., Oliver, C., Moss, J., Berg, K., and Burbidge, C. (2011). The prevalence and phenomenology of self-injurious and aggressive behaviour in genetic syndromes. J. Intellect. Disabil. Res. 55, 109–120. doi: 10.1111/j.1365-2788.2010.01337.x
Avraham, O., Deng, P. Y., Jones, S., Kuruvilla, R., Semenkovich, C. F., Klyachko, V. A., et al. (2020). Satellite glial cells promote regenerative growth in sensory neurons. Nat. Commun. 11:4891. doi: 10.1038/s41467-020-18642-y
Biel, M., Wahl-Schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885. doi: 10.1152/physrev.00029.2008
Booker, S. A., Domanski, A. P. F., Dando, O. R., Jackson, A. D., Isaac, J. T. R., Hardingham, G. E., et al. (2019). Altered dendritic spine function and integration in a mouse model of fragile X syndrome. Nat. Commun. 10:4813. doi: 10.1038/s41467-019-11891-6
Booker, S. A., Simoes de Oliveira, L., Anstey, N. J., Kozic, Z., Dando, O. R., Jackson, A. D., et al. (2020). Input-output relationship of CA1 pyramidal neurons reveals intact homeostatic mechanisms in a mouse model of fragile X syndrome. Cell Rep. 32:107988. doi: 10.1016/j.celrep.2020.107988
Brager, D. H., Akhavan, A. R., and Johnston, D. (2012). Impaired dendritic expression and plasticity of h-channels in the fmr1(−/y) mouse model of fragile X syndrome. Cell Rep. 1, 225–233. doi: 10.1016/j.celrep.2012.02.002
Brandalise, F., Kalmbach, B. E., Mehta, P., Thornton, O., Johnston, D., Zemelman, B. V., et al. (2020). Fragile X mental retardation protein bidirectionally controls dendritic Ih in a cell-type specific manner between mouse hippocampus and prefrontal cortex. J. Neurosci. 40, 5327–5340. doi: 10.1523/JNEUROSCI.1670-19.2020
Brown, M. R., Kronengold, J., Gazula, V. R., Chen, Y., Strumbos, J. G., Sigworth, F. J., et al. (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 13, 819–821. doi: 10.1038/nn.2563
Cascio, C. J. (2010). Somatosensory processing in neurodevelopmental disorders. J. Neurodev. Dis. 2, 62–69. doi: 10.1007/s11689-010-9046-3
Castren, M., Tervonen, T., Karkkainen, V., Heinonen, S., Castren, E., Larsson, K., et al. (2005). Altered differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 17834–17839. doi: 10.1073/pnas.0508995102
Chaplan, S. R., Guo, H. Q., Lee, D. H., Luo, L., Liu, C., Kuei, C., et al. (2003). Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J. Neurosci. 23, 1169–1178. doi: 10.1523/jneurosci.23-04-01169.2003
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Contractor, A., Klyachko, V. A., and Portera-Cailliau, C. (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. doi: 10.1016/j.neuron.2015.06.017
Crawford, H., Karakatsani, E., Singla, G., and Oliver, C. (2019). The persistence of self-injurious and aggressive behavior in males with fragile X syndrome over 8 years: a longitudinal study of prevalence and predictive risk markers. J. Autism Dev. Disord. 49, 2913–2922. doi: 10.1007/s10803-019-04002-3
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
de Moraes, E. R., Kushmerick, C., and Naves, L. A. (2017). Morphological and functional diversity of first-order somatosensory neurons. Biophys. Rev. 9, 847–856. doi: 10.1007/s12551-017-0321-3
Deng, P. Y., Carlin, D., Oh, Y. M., Myrick, L. K., Warren, S. T., Cavalli, V., et al. (2019). Voltage-independent SK-channel dysfunction causes neuronal hyperexcitability in the hippocampus of Fmr1 knock-out mice. J. Neurosci. 39, 28–43. doi: 10.1523/JNEUROSCI.1593-18.2018
Deng, P. Y., and Klyachko, V. A. (2016a). Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J. Physiol. 594, 83–97. doi: 10.1113/JP271031
Deng, P. Y., and Klyachko, V. A. (2016b). Increased persistent sodium current causes neuronal hyperexcitability in the entorhinal cortex of Fmr1 knockout mice. Cell Rep. 16, 3157–3166. doi: 10.1016/j.celrep.2016.08.046
Deng, P. Y., and Klyachko, V. A. (2021). Channelopathies in fragile X syndrome. Nat. Rev. Neurosci. 22, 275–289. doi: 10.1038/s41583-021-00445-9
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V., et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2012.12.018
Domanski, A. P. F., Booker, S. A., Wyllie, D. J. A., Isaac, J. T. R., and Kind, P. C. (2019). Cellular and synaptic phenotypes lead to disrupted information processing in Fmr1-KO mouse layer 4 barrel cortex. Nat. Commun. 10:4814. doi: 10.1038/s41467-019-12736-y
Dwivedi, D., Chattarji, S., and Bhalla, U. S. (2019). Impaired reliability and precision of spiking in adults but not juveniles in a mouse model of fragile X syndrome. eNeuro 6:ENEURO.0217-19.2019. doi: 10.1523/ENEURO.0217-19.2019
Ferron, L., Nieto-Rostro, M., Cassidy, J. S., and Dolphin, A. C. (2014). Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat. Commun. 5:3628. doi: 10.1038/ncomms4628
Ferron, L., Novazzi, C. G., Pilch, K. S., Moreno, C., Ramgoolam, K., and Dolphin, A. C. (2020). FMRP regulates presynaptic localization of neuronal voltage gated calcium channels. Neurobiol. Dis. 138:104779. doi: 10.1016/j.nbd.2020.104779
George, M. S., Abbott, L. F., and Siegelbaum, S. A. (2009). HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K(+) channels. Nat. Neurosci. 12, 577–584. doi: 10.1038/nn.2307
Guo, W., Polich, E. D., Su, J., Gao, Y., Christopher, D. M., Allan, A. M., et al. (2015). Fragile X proteins FMRP and FXR2P control synaptic GluA1 expression and neuronal maturation via distinct mechanisms. Cell Rep. 11, 1651–1666. doi: 10.1016/j.celrep.2015.05.013
Hagerman, R. J., Berry-Kravis, E., Kaufmann, W. E., Ono, M. Y., Tartaglia, N., Lachiewicz, A., et al. (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123, 378–390.
Harlow, E. G., Till, S. M., Russell, T. A., Wijetunge, L. S., Kind, P., and Contractor, A. (2010). Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65, 385–398. doi: 10.1016/j.neuron.2010.01.024
He, C. X., Cantu, D. A., Mantri, S. S., Zeiger, W. A., Goel, A., and Portera-Cailliau, C. (2017). Tactile defensiveness and impaired adaptation of neuronal activity in the Fmr1 knock-out mouse model of autism. J. Neurosci. 37, 6475–6487. doi: 10.1523/JNEUROSCI.0651-17.2017
Juczewski, K., von Richthofen, H., Bagni, C., Celikel, T., Fisone, G., and Krieger, P. (2016). Somatosensory map expansion and altered processing of tactile inputs in a mouse model of fragile X syndrome. Neurobiol. Dis. 96, 201–215. doi: 10.1016/j.nbd.2016.09.007
Kanyshkova, T., Pawlowski, M., Meuth, P., Dube, C., Bender, R. A., Brewster, A. L., et al. (2009). Postnatal expression pattern of HCN channel isoforms in thalamic neurons: relationship to maturation of thalamocortical oscillations. J. Neurosci. 29, 8847–8857. doi: 10.1523/JNEUROSCI.0689-09.2009
Li, L., Rutlin, M., Abraira, V. E., Cassidy, C., Kus, L., Gong, S., et al. (2011). The functional organization of cutaneous low-threshold mechanosensory neurons. Cell 147, 1615–1627. doi: 10.1016/j.cell.2011.11.027
Liu, Q., Vrontou, S., Rice, F. L., Zylka, M. J., Dong, X., and Anderson, D. J. (2007). Molecular genetic visualization of a rare subset of unmyelinated sensory neurons that may detect gentle touch. Nat. Neurosci. 10, 946–948. doi: 10.1038/nn1937
Moosmang, S., Stieber, J., Zong, X., Biel, M., Hofmann, F., and Ludwig, A. (2001). Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur. J. Biochem. 268, 1646–1652. doi: 10.1046/j.1432-1327.2001.02036.x
Moskalyuk, A., Van De Vijver, S., Verstraelen, P., De Vos, W. H., Kooy, R. F., and Giugliano, M. (2020). Single-cell and neuronal network alterations in an in vitro model of fragile X syndrome. Cereb. Cortex 30, 31–46. doi: 10.1093/cercor/bhz068
Nomura, T., Musial, T. F., Marshall, J. J., Zhu, Y., Remmers, C. L., Xu, J., et al. (2017). Delayed maturation of fast-spiking interneurons is rectified by activation of the TrkB receptor in the mouse model of fragile X syndrome. J. Neurosci. 37, 11298–11310. doi: 10.1523/JNEUROSCI.2893-16.2017
Oh, M. M., Oliveira, F. A., and Disterhoft, J. F. (2010). Learning and aging related changes in intrinsic neuronal excitability. Front. Aging Neurosci. 2:2. doi: 10.3389/neuro.24.002.2010
Olson, W., Dong, P., Fleming, M., and Luo, W. (2016). The specification and wiring of mammalian cutaneous low-threshold mechanoreceptors. Wiley Interdiscip. Rev. Dev. Biol. 5, 389–404. doi: 10.1002/wdev.229
Orefice, L. L., Mosko, J. R., Morency, D. T., Wells, M. F., Tasnim, A., Mozeika, S. M., et al. (2019). Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cell 178, 867–886.e24. doi: 10.1016/j.cell.2019.07.024
Orefice, L. L., Zimmerman, A. L., Chirila, A. M., Sleboda, S. J., Head, J. P., and Ginty, D. D. (2016). Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166, 299–313. doi: 10.1016/j.cell.2016.05.033
Penagarikano, O., Mulle, J. G., and Warren, S. T. (2007). The pathophysiology of fragile X syndrome. Annu. Rev. Genomics Hum. Genet. 8, 109–129.
Price, T. J., Flores, C. M., Cervero, F., and Hargreaves, K. M. (2006). The RNA binding and transport proteins staufen and fragile X mental retardation protein are expressed by rat primary afferent neurons and localize to peripheral and central axons. Neuroscience 141, 2107–2116. doi: 10.1016/j.neuroscience.2006.05.047
Price, T. J., and Melemedjian, O. K. (2012). Fragile X mental retardation protein (FMRP) and the spinal sensory system. Res. Prob. Cell Differ. 54, 41–59. doi: 10.1007/978-3-642-21649-7_4
Price, T. J., Rashid, M. H., Millecamps, M., Sanoja, R., Entrena, J. M., and Cervero, F. (2007). Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J. Neurosci. 27, 13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007
Rais, M., Binder, D. K., Razak, K. A., and Ethell, I. M. (2018). Sensory processing phenotypes in fragile X syndrome. ASN Neuro 10:1759091418801092. doi: 10.1177/1759091418801092
Ramirez-Lopez, A., Pastor, A., de la Torre, R., La Porta, C., Ozaita, A., Cabanero, D., et al. (2021). Role of the endocannabinoid system in a mouse model of Fragile X undergoing neuropathic pain. Eur. J. Pain 25, 1316–1328. doi: 10.1002/ejp.1753
Rao, X., Huang, X., Zhou, Z., and Lin, X. (2013). An improvement of the 2−ΔΔCT method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 3, 71–85.
Schmidt-Hieber, C., Jonas, P., and Bischofberger, J. (2004). Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429, 184–187. doi: 10.1038/nature02553
Shah, M. M. (2014). Cortical HCN channels: function, trafficking and plasticity. J. Physiol. 592, 2711–2719. doi: 10.1113/jphysiol.2013.270058
Shen, H., Liu, W., Geng, Q., Li, H., Lu, M., Liang, P., et al. (2018). Age-dependent up-regulation of HCN channels in spiral ganglion neurons coincide with hearing loss in mice. Front. Aging Neurosci. 10:353. doi: 10.3389/fnagi.2018.00353
Telias, M., Kuznitsov-Yanovsky, L., Segal, M., and Ben-Yosef, D. (2015). Functional deficiencies in fragile X neurons derived from human embryonic stem cells. J. Neurosci. 35, 15295–15306. doi: 10.1523/JNEUROSCI.0317-15.2015
Telias, M., Segal, M., and Ben-Yosef, D. (2013). Neural differentiation of fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 374, 32–45. doi: 10.1016/j.ydbio.2012.11.031
Till, S. M., Wijetunge, L. S., Seidel, V. G., Harlow, E., Wright, A. K., Bagni, C., et al. (2012). Altered maturation of the primary somatosensory cortex in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 2143–2156. doi: 10.1093/hmg/dds030
Vislay, R. L., Martin, B. S., Olmos-Serrano, J. L., Kratovac, S., Nelson, D. L., Corbin, J. G., et al. (2013). Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J. Neurosci. 33, 7548–7558. doi: 10.1523/JNEUROSCI.2764-12.2013
Wang, H. F., Shortland, P., Park, M. J., and Grant, G. (1998). Retrograde and transganglionic transport of horseradish peroxidase-conjugated cholera toxin B subunit, wheatgerm agglutinin and isolectin B4 from Griffonia simplicifolia I in primary afferent neurons innervating the rat urinary bladder. Neuroscience 87, 275–288. doi: 10.1016/s0306-4522(98)00061-x
Yang, L. K., Lu, L., Feng, B., Wang, X. S., Yue, J., Li, X. B., et al. (2020). FMRP acts as a key messenger for visceral pain modulation. Mol. Pain 16:1744806920972241. doi: 10.1177/1744806920972241
Yousuf, M. S., Noh, M. C., Friedman, T. N., Zubkow, K., Johnson, J. C., Tenorio, G., et al. (2019). Sensory neurons of the dorsal root ganglia become hyperexcitable in a T-cell-mediated MOG-EAE model of multiple sclerosis. eNeuro 6:ENEURO.0024-19.2019. doi: 10.1523/ENEURO.0024-19.2019
Keywords: hyperexcitability, Fragile X syndrome, action potential, sensory neuron, HCN channel
Citation: Deng P-Y, Avraham O, Cavalli V and Klyachko VA (2021) Hyperexcitability of Sensory Neurons in Fragile X Mouse Model. Front. Mol. Neurosci. 14:796053. doi: 10.3389/fnmol.2021.796053
Received: 15 October 2021; Accepted: 17 November 2021;
Published: 22 December 2021.
Edited by:
Michael Telias, University of California, Berkeley, United StatesReviewed by:
Judith Ann Strong, University of Cincinnati, United StatesCopyright © 2021 Deng, Avraham, Cavalli and Klyachko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Cavalli, Y2F2YWxsaUB3dXN0bC5lZHU=; Vitaly A. Klyachko, a2x5YWNoa29Ad3VzdGwuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.